 Kobey Karamendin1*
Kobey Karamendin1* Temirlan Sabyrzhan1
Temirlan Sabyrzhan1 Aidyn Kydyrmanov1
Aidyn Kydyrmanov1 Yermukhammet Kasymbekov1Yelizaveta Khan1
Yermukhammet Kasymbekov1Yelizaveta Khan1 Sardor Nuralibekov1Simon J. Goodman2
Sardor Nuralibekov1Simon J. Goodman2- 1Department of Virology, Laboratory of Viral Ecology, Research and Production Center for Microbiology and Virology, Almaty, Kazakhstan
- 2School of Biology, Faculty of Biological Sciences, University of Leeds, Leeds, United Kingdom
Bats are known as an important natural reservoir of many viral infections, from which transmission into the human population occurs. This article aimed to examine bats in Kazakhstan for possible circulation of viruses dangerous to humans and other animals. Two hundred sixty samples, including nasal and rectal swabs and blood sera, were collected from bats in southern Kazakhstan. Studies of the bat virome applying high-throughput sequencing revealed the presence of five large families: Circoviridae, Parvoviridae, Retroviridae, Adenoviridae, and Orthoherpesviridae. All of them were found in southern Kazakhstan from Myotis blythii adult individuals of both sexes. Genomic studies of bat herpesviruses have shown that they are potentially representatives of novel species, genetically different from existing ones. Their epidemic potential for humans and other animals requires further research. Serological studies for possible circulation of antibodies to coronaviruses and influenza A viruses have shown negative results, indicating the absence of antibodies in the tested samples. The results demonstrate the need for more profound research to identify the relationship between human and bat viruses in the future.
Introduction
Bats belong to the order Chiroptera. The current total number of bat species worldwide is 1487 (1), which constitutes about 1/5 of all mammals. Bats play a vital role in maintaining healthy ecosystems and biodiversity across the globe. As key species, they provide essential ecosystem services that directly benefit both natural environments and human activities. Insectivorous bats contribute significantly to pest control by consuming vast quantities of insects, including agricultural pests and disease-carrying mosquitoes. Frugivorous and nectar-feeding bats are important pollinators and seed dispersers, particularly in tropical and subtropical regions, facilitating forest regeneration and the reproduction of numerous plant species. These roles highlight their importance as keystone species, sustaining ecosystem balance beyond their often-discussed virological significance and various unsubstantiated human phobias.
Worldwide studies on bat viruses identified more than 130 viral species, including more than 60 anthropozoonotic viruses that are pathogenic for humans, including Ebola, Nipah, and Hendra (2–5). The SARS outbreak that arose in China in 2002 infected more than 8000 people worldwide, and approximately 10% of them died. The role of bats as a natural reservoir of SARS-CoV has been proved then (6). MERS-associated coronavirus was also detected in bats (7).
The use of high-throughput sequencing for metagenomic analysis of viral communities, or viromes, is one of the fastest-growing methods for identifying rare, and novel viruses (8, 9). In recent years, Next Generation Sequencing (NGS) technology allowed researchers to describe a multitude of viruses from bats: hepatitis B virus (10), rotavirus (11), novel bat coronavirus (12) and other important viruses of Nairoviridae (13), Circoviridae, Retroviridae, Orthoherpesviridae, Papillomaviridae (14) and Paramyxoviridae (15) families, indicating that bat species might be associated with a wide diversity of viruses worldwide. It should be noted that the Orthoherpesviridae sequence obtained by Sanger sequencing was used in the work since its sequence was longer than that obtained by the NGS. Past research has indicated that the structure of the virome in bats varies according to both the geographic area and the species of bat.
Kazakhstan has recorded 27 species of bats (16). All of them make up more than 70 percent of the bat population in the territories of the former USSR and represent approximately half of the bat species found in the Palearctic region. This high level of diversity is due to Kazakhstan’s wide range of climatic zones. Almost all types of them are inhabited by specific species for those regions, from the southern mountains and deserts to the northern landscapes (17).
In this regard, bats have become essential for virus screening studies worldwide. The Central Asian region has been little studied for various viral infections spread by bats. This study aimed to identify potential natural foci of various infections common among bats.
Materials and methods
Laboratory studies were carried out in compliance with all bioethical standards in accordance with the instructions of the local ethics commission No.02-09–106 from 25.03.2022, Order of the Minister of Health and Social Development of the Republic of Kazakhstan dated May 27, 2015 No. 392 “On approval of good pharmaceutical practices”, May 29, 2015 No. 415. “On approval of the Rules preclinical studies, requirements for preclinical bases,” as well as the order of the Minister of Agriculture of the Republic of Kazakhstan dated June 29, 2015, No. 7-1/587 “Veterinary (veterinary and sanitary) rules”.
Samples collection, their preliminary processing, and RNA extraction
Bats were sampled by taking nasal and rectal swabs with sterile cotton swabs. These swabs were then stored in vials filled with medium 199 (Gibco, ThermoFisher Scientific, USA) supplemented with antibiotics and bovine serum albumin, and kept in liquid nitrogen (−196°С). Samples were duplicated in DNA/RNA Shield reagent (Zymo, USA) for molecular biological studies and stored at room temperature. Prior to RNA extraction, the samples underwent centrifugation at 3200 rpm for 15 minutes. The supernatants were then passed through 0.45 µm filters (Roth, Germany) and exposed to a combination of nucleases, including Turbo DNAse, DNAse I, and RNAse T1 (ThermoFisher, Lithuania). RNA was then isolated using the QIAamp Viral RNA Mini kit (Qiagen, Germany), following the protocol provided by the manufacturer.
Next generation sequencing
The libraries for high-throughput sequencing were prepared using the QIAseq FX Single Cell RNA Library Kit (Qiagen, Germany) and the NEBNext Ultra DNA Library Prep Kit for Illumina (NEB, USA) following the manufacturers’ instructions. The fragmentation of cDNA to sizes ranging from approximately 450 to 600 base pairs (bp) was achieved through an enzymatic process utilizing the Fragmentase kit (NEB, USA). The libraries’ concentrations were determined on a Qubit 2.0 instrument (Invitrogen, Germany). The libraries’ sizes were determined by the Bioanalyzer 2100 device (Agilent, Germany). Sequencing was performed using the MiSeq Reagent v.3 kit (Illumina, USA) on a MiSeq sequencer (Illumina, USA).
Bioinformatic analyses
Bioinformatic analysis, which consists of assembling all obtained reads into longer contig sequences with subsequent annotation, was performed on a high-performance computer with the installed Geneious Prime program (Biomatters, New Zealand). Homologous sequences were searched using BLAST in the international GenBank database. The data were compared online against the GenBank non-redundant and viral reference databases with both BLASTx and BLASTn. A BLAST hit was considered significant with an E-value threshold of 10e-25. Contigs from bacteria and eukaryotes were omitted from the analysis, and sequences resembling viruses were advanced for further study.
Serology
The blood serum samples were analyzed for antibodies against coronaviruses using an enzyme-linked immunosorbent assay (ELISA) with the SARS-CoV-2 Double Antigen Multi-species ID Vet COVIDA/0520 kit. Additionally, they were tested for antibodies to the influenza A virus with the AID Screen® Influenza A Antibody Competition Multispecies ELISA kit, following the guidelines provided by the manufacturers. In the case of serological testing for coronavirus, a multi-species kit for the detection of antibodies to SARS-CoV-2 was used, which is capable of working with sera from a wide range of animal species sensitive to this pathogen. This study was experimental, and in case of positive results, it was planned to confirm using more specific tests. Since strictly negative results for coronavirus were obtained, serological studies were not continued.
As for serological testing for influenza A, according to information from the ELISA kit manufacturer, this kit is highly sensitive and specific to antibodies to the influenza A virus in susceptible animals. It is known that bats are susceptible to H9 influenza A subtype (18) and supposedly form a specific bat lineage (19). Commercial multi-species ELISA kits, like the validated for the bat species BioChek CK401 AI Multi ELISA kit, are commonly used to detect antibodies to the influenza A virus in bats. In our case, we also used a similar multi-species AID Screen kit from another manufacturer that detects antibodies to Influenza A nucleoprotein, regardless of the virus subtype, including H9, circulating in bats.
Results
In 2023, trips were organized to the southern region of Kazakhstan to collect samples from bats (Figure 1). At the collection sites, 260 samples were collected from 201 individuals of 3 genera: Myotis, Nyctalus (Vespertilionidae family), and Rhinolophus (Rhinolophidae family) in the Chiroptera order. The sizes of the libraries constructed ranged from 250 to 700 nucleotides (nt), fitting the length specifications for sequencing on a MiSeq device with the Reagent kit v.3 (Illumina, USA). A bioinformatics analysis was then conducted on the results from the high-throughput sequencing of bat samples. The obtained contig lengths, indicated in Table 1, were used in analyses. Unfortunately, complete viral sequences were not obtained, and this work will be carried out in the future after the viruses have been propagated on sensitive media. Illumina technology sequencing identified viruses belonging to different families.
Figure 1. Sampling location in Southern Kazakhstan. The sample collection site is indicated by a red asterisk.
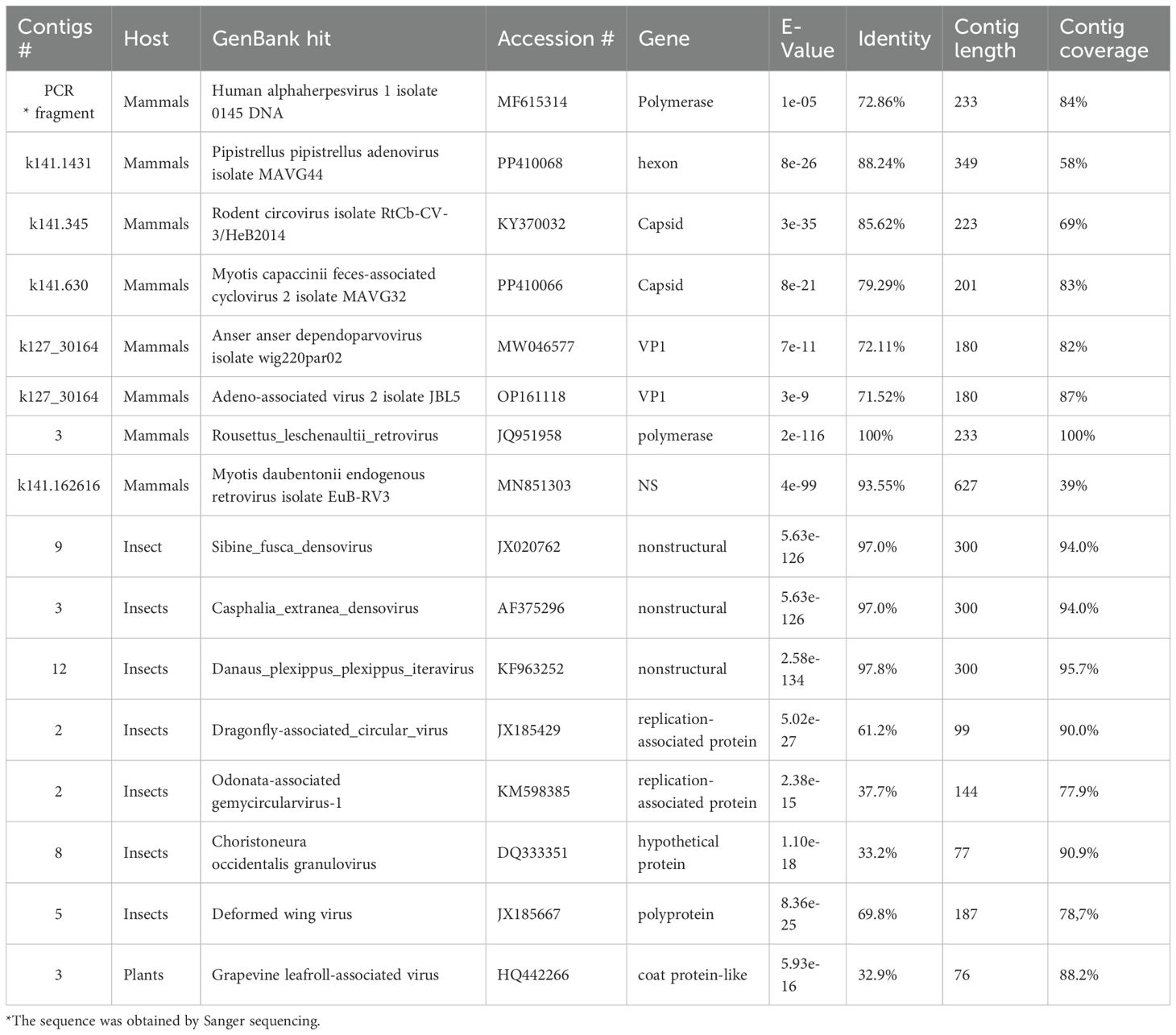
Table 1. BLAST search in GenBank.
The sequences were obtained from hosts classified primarily as mammals and insects, with their closest matches in GenBank listed. Key parameters such as the contig identifier, GenBank hit, accession number, identified gene, E-value, identity percentage, contig length, and coverage are provided. Genomic analysis and identification of viral sequences through BLAST search in GenBank were summarized in Table 1. All the mammalian viral sequences obtained in this research were deposited in GenBank and their accession numbers are shown in Supplementary Table 1.
Mammalian viral families
The viral sequences identified included representatives from five distinct viral families: Orthoherpesviridae, Adenoviridae, Circoviridae, Parvoviridae, and Retroviridae. Notably, all these viral families were detected in adult individuals of both sexes from a single bat species, Myotis blythii, in the Southern Kazakhstan population. Further studies of other bat species in Kazakhstan are required to determine the presence of the detected viral families and possibly others not detected in the Myotis blythii species.
The most concerning finding was the identification of herpesvirus sequences (Orthoherpesviridae family) showing 72.86% similarity to Human alphaherpesvirus 1 isolate 0145 by the 233 nt fragment of DNA polymerase gene. This similarity level indicates a related but distinct virus rather than direct contamination, as human viral contaminants would typically show >95% similarity. Further amino acid sequence analyses have shown that the virus under study is closer (83%) to Bat simplexvirus 1 isolated from frugivorous tropical bats (Pteropodidae and Phyllostomidae bat families) than to human alphaherpesvirus (75.32%) (20, 21). This finding suggests a potentially novel bat herpesvirus with partial genetic similarity to frugivorous bat and human herpesviruses, which aligns with the known pattern of co-evolution between herpesviruses and their mammalian hosts.
The Adenoviridae family was represented by sequences showing the highest similarity (E-value: 8e-26, 88.24% identity by the 349 nt fragment of the hexon gene) to Pipistrellus adenovirus (Vespertilionidae bat family). This virus was previously detected in the common pipistrelle insectivorous bat (22) in Europe, suggesting evolutionary conservation of genomic regions between different bat species from distant parts of the world. Unfortunately, because of their short length, analysis of the available genome fragments of newly discovered bat adenoviruses and herpesviruses in Kazakhstan did not allow the possibility of finding genetic markers associated with the pathogenicity level.
The Circoviridae family was represented by sequences that were 85.62% by the 223 nt fragment of the capsid gene similar to rodent circoviruses, identified in China (23). The presence of circovirus-like sequences in bats has been reported previously in global studies (4, 24, 25), but more similarity to rodent-associated viruses rather than to Myotis capaccinii feces-associated cyclovirus 2 (79.29% by the 201 nt fragment of the capsid gene) (Vespertilionidae bat family) (22), raises interesting questions about the evolution of this viral family in distant species.
Parvoviridae sequences were also identified in the samples, though specific details on the closest matches were limited in the initial analysis. Parvoviruses are commonly found in bat populations worldwide (Myotis, Pipistrellus, Eptesicus, Vespertilio and Nyctalus bat genera), with varying levels of host specificity and potential pathogenicity (5, 25). In this research, contigs (180 nt fragments of the VP1 gene) from the Kazakhstan bats by the were closer to avian Anser anser dependoparvovirus isolate wig220par02 (72.11%) and human Adeno-associated virus 2 isolate JBL5 (71.52%) respectively than to known bat parvoviruses.
Retroviridae was identified through sequences most closely matching Myotis daubentonii endogenous retrovirus (93.55% by the 627 nt fragment of the nonstructural protein gene) (Vespertilionidae bat family) from Germany (5) and Rousettus leschenaultii retrovirus (100% by the 233 nt fragment of the polymerase gene) described in bats of the Pteropodidae family (26) in China. This finding is particularly interesting as Rousettus is a genus of fruit bats not native to Kazakhstan, suggesting either evolutionary conservation of retroviral sequences across different bat genera or potential ancestral relationships between viral strains from geographically distant bat populations.
Insect-associated viral sequences
The second category of viral sequences identified in bat samples showed the highest similarity to insect-associated viruses, likely reflecting the insectivorous diet of the sampled bat species. Densoviruses, which belong to the Parvoviridae family but specifically infect arthropods, were prominently represented. The highest similarity (97.0% by the 300 nt fragment of the NS gene) was to Sibine fusca densovirus and Casphalia extranea densovirus, suggesting recent ingestion of infected prey rather than adaptation of these viruses to bat hosts. A densovirus belonging to the Iteravirus genus, with remarkably high similarity (97.8% identity, also by the 300 nt fragment of the NS gene) to Danaus plexippus iteravirus, was also detected. This high percentage identity suggests recent consumption of infected lepidopteran prey, as Danaus plexippus (monarch butterfly) is not native to Kazakhstan, but related lepidopteran species carrying similar viruses may be present in the region. Densoviruses have previously been found in bats, and it has also been suggested that insect viruses in bats are of alimentary origin (27).
Circular virus of the Circoviridae family sequences were 61.2% and 37.7% similar to Dragonfly-associated circular virus and Odonata associated gemycircularvirus respectively (99–144 nt fragments of the Rep gene), further supporting the dietary origin hypothesis for insect-associated viral sequences. These small, circular DNA viruses are increasingly recognized as abundant and diverse in insect populations worldwide (28) including those found in bats during metagenomic assays (24).
Choristoneura occidentalis granulovirus of the Baculoviridae family is generally considered an insect-specific pathogen, primarily infecting lepidopteran larvae. These viruses are exclusively pathogenic for arthropods, particularly insects, and have not been registered in bats previously (29). Low similarity (33.2%) and very small size (77 nt) of the obtained fragment of the hypothetical protein limit our analyses.
In this study, a contig (187 nt fragment of the polyprotein gene) 69.8% similar to Deformed wing virus was found. Deformed wing virus is a pathogen of insects belonging to the family Iflaviridae within the order Picornavirales. It was detected in bats in the USA (30) and Switzerland (31). It was supposed that this virus was introduced to bats and other mammals through the consumption of infected insects (32).
Interestingly, plant virus sequences were also identified, including those with moderate similarity (32.9%) by76 nt fragment of the coat protein-like protein gene to Grapevine leafroll-associated virus. This lower similarity percentage suggests either distant evolutionary relationships or potential dietary acquisition through insects that had previously fed on infected plant material, representing a multi-trophic chain of viral genetic material transfer.
The E-values for these matches ranged from highly significant for some sequences (particularly the insect virus matches) to moderately significant for others, providing varying levels of confidence in the taxonomic assignments. This variation underscores the importance of careful interpretation of metagenomic data, particularly when viruses cross taxonomic boundaries. Serological analyses for coronaviruses and influenza A virus in ELISA have shown negative results.
Discussion
More in-depth studies of the bat virome worldwide have begun in recent years, particularly after the pandemic, in which bats were suspected as a natural reservoir (32). This study is one of the few studies in Kazakhstan that has provided initial information on the bat virome in the Central Asian region. The virus families found in bats are important to studying possible natural anthropozoonotic foci. Finding contigs from viruses of Orthoherpesviridae, Adenoviridae, Circoviridae, Parvoviridae, and Retroviridae families in bats highlights their significance as major reservoirs of viral diversity. Bats are known to harbor many viruses because of their distinct ecological, physiological, and evolutionary characteristics, such as their capability to fly, their extended lifespans, and their tendency to roost in groups, which aids in the spread and persistence of viruses within their communities (33).
Orthoherpesviridae is a large family of DNA viruses that cause infections and some diseases in animals, including humans (34). Most bat herpesviruses are isolated from apparently healthy animals collected during captures as part of random surveillance programs. Orthoherpesviruses, typically latent in their hosts, underscore the complex interactions between bats and their viruses (35). Herpesviruses have evolved alongside their hosts, allowing for long-term persistence through latent infections and occasional shedding, which can facilitate cross-species transmission in specific circumstances (36). As noted earlier, the herpesvirus found in bats in Kazakhstan is a potentially novel species. Despite its similarity to Simplexvirus, the potential for bat herpesviruses to infect humans appears limited based on current evidence, though rare cases of cross-species transmission of herpesviruses have been documented in other mammalian systems (37). More complete genomic characterization of the bat herpesviruses identified in this study would help clarify their evolutionary relationships and assess their zoonotic potential. Perhaps in the future, dozens of new herpesviruses will be discovered in bats worldwide and in Kazakhstan, since their viral diversity is only beginning to be intensively studied.
The Adenoviridae family of viruses causes a wide range of diseases, mostly respiratory and gastrointestinal, in animals and humans (38). Adenovirus infections can be asymptomatic in hosts (39). Bat adenoviruses have been previously identified in multiple species across Europe, Asia, and the Americas, often showing host-specificity at the bat genus level (40). Adenoviruses are known to exhibit high host specificity but may also provide insight into host-virus co-evolution. In this research, the virus was detected in animals without any signs of disease, and its impact on the health of bats and other mammals needs to be elucidated in the future. The identification of sequences similar to Pipistrellus pipistrellus adenovirus in Myotis blythii is interesting as it suggests potential cross-genera transmission or the existence of conserved genomic regions across bat adenoviruses. The potential for zoonotic transmission of bat adenoviruses remains largely unexplored, though their usually strict host specificity makes cross-species jumps less likely than other viral families. Notably, a novel species of adenovirus from bats in Kazakhstan has been identified and genetically characterized before (41). The discovery of a new bat adenovirus in a local population is not unusual, as it is believed that there are hundreds of unknown and undiscovered adenovirus species in bats worldwide (40).
Circoviridae viruses, which have been identified in bat species, are generally small DNA viruses with circular genomes (24). Although subclinical infections are common with circoviruses, they are thought to be associated with infectious anemia in chickens, pigeon circovirus disease, and multisystem post-weaning wasting syndrome in pigs (42). Contigs of this virus have previously been detected in bats and appear to be common there (43, 44). The circovirus identified in this study was 72% similar to Myotis capaccinii feces-associated cyclovirus 2, indicating its significant genetic distance from the described species of this family. Its discovery so far has only scientific significance in the study of the diversity of bat circoviruses. A role in pathogenicity remains to be determined.
Retroviridae. Although retroviruses such as human immunodeficiency virus (HIV) are among the most important families of viruses that have been transmitted from animals to humans, it remains to be seen whether bat retroviruses are capable of infecting and causing disease in humans (45). Although primarily associated with avian and porcine hosts, their detection in bats may indicate a broader host range and ecological niche than previously thought. Similarly, the discovery of retroviral sequences such as the Myotis daubentonii endogenous retrovirus, highlights the potential for viral integration into host genomes, a phenomenon that has likely played a role in the evolutionary dynamics of bat immunity (5). Retroviridae detected in Myotis blythii from Kazakhstan adds to the growing evidence that retroviruses are widespread in bat populations globally. While retroviruses such as HIV have demonstrated significant zoonotic potential, the risk posed by bat retroviruses remains largely theoretical at present. Continued surveillance and characterization of bat retroviruses, particularly focusing on receptor usage and potential barriers to cross-species transmission, will be important for risk assessment.
Bat parvoviruses are represented by avian Anser anser dependoparvovirus and human Adeno-associated virus 2. The identification of parvoviruses in bat contigs aligns with earlier research, which indicates that these virus families often infect mammals, including bats, typically without leading to apparent disease. Parvoviruses are often associated with gastrointestinal or systemic infections in mammals but remain understudied in their bat hosts (45). The discovery of a bat parvovirus similar to avian and human parvoviruses in this research is an interesting case, but perhaps this family has not yet been studied enough in local bats. In addition to the above-mentioned Orthoherpesviridae and Adenoviridae viral families, representatives of the Circoviridae and Parvoviridae families may also be potentially novel species of mammalian viruses. Their similarity to known viruses varied from 71.52% to 100%, but further studies are needed to obtain their complete genomes. This also applies to the discovered insect viruses.
Our study has some limitations. Short viral sequences (180–627 nt) were identified in the metagenomic sequencing, and further investigations, including virus propagation, are necessary to obtain complete genomes.
A significant presence of insect viruses or their close relatives of supposedly dietary origin is revealed in bats. These include a diverse range of viral types, such as densoviruses, circular viruses, baculoviruses, and iflaviruses. The prevalence of these viruses in bats varies, with some viral groups showing notable presence in certain bat populations. The evolutionary links observed between certain insect and bat viruses highlight the interconnectedness of viromes across different taxa.
The novelty of the data obtained on the presence of insect viruses in bats is relative, expanding our understanding of the diversity of their virome, the components of which, as it turns out, are representatives of viruses from different kingdoms.
Further research is essential to fully elucidate the nature of these interactions, including determining the full host range of these viruses, their ability to replicate in bat cells, and their potential impact on bat health and the broader ecosystem. Comprehensive studies utilizing metagenomics, targeted viral detection assays, and investigations into viral replication and persistence in bats are needed to gain a deeper understanding of this complex and evolving field. Bats, as predators of insects, might also play a role in the ecology and dissemination of insect viruses within insect populations through predation and fecal shedding of viral particles (27).
It may seem that the various virus families discovered in this study are more similar to human, insect, and plant viruses than to related bat viruses that were previously collected from neighboring regions such as China, Russia, and others. We think that bats have been very little studied virologically to date and that several novel species can be found even within a single population, which is often the case. Therefore, we believe that with the accumulation of large amounts of information from different countries, conclusions can be made about the global distribution of viral families.
The findings highlight bats’ critical role as reservoirs for various viral families. This discovery holds significant implications for comprehending viral ecology, the risk of zoonotic transmission, and the co-evolutionary dynamics between hosts and viruses. Future studies should focus on characterizing these viruses’ functions, assessing their capability to infect other species, and examining the ecological influences on viral diversity in bats. Moreover, conducting genomic surveillance of bats in various geographic areas and habitats will enhance our understanding of their contribution to global viral evolution and emergence.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: NCBI GenBank, accession ERS24838366–ERS24838369, PP410068, OP161118, PP410066, KY370032, MW046577, MN851303, MF615314, JQ951958, JX020762, HQ442266, JX185662, DQ333351, JX185667, AF375296, JX185429, KF963252, KM598385.
Ethics statement
The animal study was approved by Research and Production Center for Microbiology and Virology Local Ethics Commission. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
KK: Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Writing – original draft, Writing – review & editing. TS: Formal analysis, Methodology, Writing – original draft, Writing – review & editing. AK: Investigation, Writing – original draft, Writing – review & editing. YKa: Resources, Writing – original draft, Writing – review & editing. YKh: Resources, Writing – original draft, Writing – review & editing. SN: Resources, Writing – original draft, Writing – review & editing. SG: Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by a grant AP19678255 from the Ministry of Science and Higher Education of the Republic of Kazakhstan.
Acknowledgments
The authors are grateful to Andrey Gavrilov for his help in sampling from bats.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2025.1582410/full#supplementary-material
References
1. Simmons NB and Cirranello AL. Bat Species of the World: A taxonomic and geographic database. Version 1.7 (2025) (Accessed March 30, 2025).
2. Luis AD, Hayman DT, O’Shea TJ, Cryan PM, Gilbert AT, Pulliam JR, et al. A comparison of bats and rodents as reservoirs of zoonotic viruses: are bats special? Proc Biol Sci. (2013) 280:20122753. doi: 10.1098/rspb.2012.2753
3. Wang Y, Xu P, Han Y, Zhao W, Zhao L, Li R, et al. Unveiling bat-borne viruses: a comprehensive classification and analysis of virome evolution. Microbiome. (2024) 12:235. doi: 10.1186/s40168-024-01955-1
4. He B, Li Z, Yang F, Zheng J, Feng Y, Guo H, et al. Virome profiling of bats from Myanmar by metagenomic analysis of tissue samples reveals more novel mammalian viruses. PloS One. (2013) 8:e61950. doi: 10.1371/journal.pone.0061950
5. Kohl C, Brinkmann A, Radonić A, Dabrowski PW, Mühldorfer K, Nitsche A, et al. The virome of German bats: comparing virus discovery approaches. Sci Rep. (2021) 11:7430. doi: 10.1038/s41598-021-86435-4
6. Cui J, Li F, and Shi ZL. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. (2019) 17:181–92. doi: 10.1038/s41579-018-0118-9
7. Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, and Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. New Engl J Med. (2012) 367:1814–20. doi: 10.1056/NEJMoa1211721
8. Lecuit M and Eloit M. The human virome: new tools and concepts. Trends Microbiol. (2013) 21:510–5. doi: 10.1016/j.tim.2013.07.001
9. Radford AD, Chapman D, Dixon L, Chantrey J, Darby AC, and Hall N. Application of next-generation sequencing technologies in virology. J Gen Virol. (2012) 93:1853–68. doi: 10.1099/vir.0.043182-0
10. Drexler JF, Geipel A, König A, Corman VM, van Riel D, Leijten LM, et al. Bats carry pathogenic hepadnaviruses antigenically related to hepatitis B virus and capable of infecting human hepatocytes. Proc Natl Acad Sci United States America. (2013) 110:16151–6. doi: 10.1073/pnas.1308049110
11. He B, Yang F, Yang W, Zhang Y, Feng Y, Zhou J, et al. Characterization of a novel G3P[3] rotavirus isolated from a lesser horseshoe bat: a distant relative of feline/canine rotaviruses. J Virol. (2013) 87:12357–66. doi: 10.1128/JVI.02013-13
12. He B, Zhang Y, Xu L, Yang W, Yang F, Feng Y, et al. Identification of diverse alphacoronaviruses and genomic characterization of a novel severe acute respiratory syndrome-like coronavirus from bats in China. J Virol. (2014) 88:7070–82. doi: 10.1128/JVI.00631-14
13. Brinkmann A, Kohl C, Radonić A, Dabrowski PW, Mühldorfer K, Nitsche A, et al. First detection of bat-borne Issyk-Kul virus in Europe. Sci Rep. (2020) 10:22384. doi: 10.1038/s41598-020-79468-8
14. Salmier A, Tirera S, de Thoisy B, Franc A, Darcissac E, Donato D, et al. Virome analysis of two sympatric bat species (Desmodus rotundus and Molossus molossus) in French Guiana. PloS One. (2017) 12:e0186943. doi: 10.1371/journal.pone.0186943
15. Drexler JF, Corman VM, Müller MA, Maganga GD, Vallo P, Binger T, et al. Bats host major mammalian paramyxoviruses. Nat Commun. (2012) 3:796. doi: 10.1038/ncomms1796
16. Bekenov AB, Grachev Yu. A, Mazin VN, and Shubin VI. Mammals. In: Gvozdev EV, editor. Book of the genetic resources of the fauna of the kazakh SSR. Academy of Sciences of the Kazakh SSR; Institute of Zoology, Alma-Ata (1989). p. 215. Available at: https://kazneb.kz/ru/bookView/view?brId=1030531&simple=true. Part 1. (Accessed May 5, 2025).
17. Gvozdev EV and Strautman EI. Nasekomoyadnye i dvukrylye. In: Mlekopitayushchie Kazakhstana [Insectivora and chiroptera. Mammals of Kazakhstan, vol. IV. Nauka Kazakhskoy SSR., Alma-Ata (1985). p. 280.
18. Kandeil A, Gomaa MR, Shehata MM, El Taweel AN, Mahmoud SH, Bagato O, et al. Isolation and characterization of a distinct influenza A virus from Egyptian bats. J Virol. (2019) 93:e01059-18. doi: 10.1128/JVI.01059-18
19. Karamendin K, Kydyrmanov A, and Fereidouni S. Has avian influenza virus H9 originated from a bat source? Front Vet Sci. (2024) 10:1332886. doi: 10.3389/fvets.2023.1332886
20. Sabyrzhan T, Kydyrmanov A, Nuralibekov S, Khan E, Kassymbekov Y, Kumar M, et al. A new herpesvirus discovered during the study of viral diversity in bats of Kazakhstan. Microbiol Virol. (2025) 1:168–79. doi: 10.53729/MV-AS.2025.01.09
21. Razafindratsimandresy R, Jeanmaire EM, Counor D, Vasconcelos PF, Sall AA, and Reynes JM. Partial molecular characterization of alphaherpesviruses isolated from tropical bats. J Gen Virol. (2009) 90:44–7. doi: 10.1099/vir.0.006825-0
22. Buigues J, Viñals A, Martínez-Recio R, Monrós JS, Sanjuán R, and Cuevas JM. Full-genome sequencing of dozens of new DNA viruses found in Spanish bat feces. Microbiol Spectr. (2024) 12:e0067524. doi: 10.1128/spectrum.00675-24
23. Wu Z, Lu L, Du J, Yang L, Ren X, Liu B, et al. Comparative analysis of rodent and small mammal viromes to better understand the wildlife origin of emerging infectious diseases. Microbiome. (2018) 6:178. doi: 10.1186/s40168-018-0554-9
24. Vidovszky MZ, Kapitány S, Gellért Á, Harrach B, Görföl T, Boldogh SA, et al. Detection and genetic characterization of circoviruses in more than 80 bat species from eight countries on four continents. Vet Res Commun. (2023) 47:1561–73. doi: 10.1007/s11259-023-10111-3
25. Wu Z, Ren X, Yang L, Hu Y, Yang J, He G, et al. Virome analysis for identification of novel mammalian viruses in bat species from Chinese provinces. J Virol. (2012) 86:10999–1012. doi: 10.1128/JVI.01394-12
26. Cui J, Tachedjian G, Tachedjian M, Holmes EC, Zhang S, and Wang LF. Identification of diverse groups of endogenous gammaretroviruses in mega- and microbats. J Gen Virol. (2012) 93:2037–45. doi: 10.1099/vir.0.043760-0
27. Ge X, Li Y, Yang X, Zhang H, Zhou P, Zhang Y, et al. Metagenomic analysis of viruses from bat fecal samples reveals many novel viruses in insectivorous bats in China. J Virol. (2012) 86(8):4620–30. doi: 10.1128/jvi.06671-11
28. Rosario K, Dayaram A, Marinov M, Ware J, Kraberger S, Stainton D, et al. Diverse circular ssDNA viruses discovered in dragonflies (Odonata: Epiprocta). J Gen Virol. (2012) 93:2668–81. doi: 10.1099/vir.0.045948-0
29. Ikeda M, Hamajima R, and Kobayashi M. Baculoviruses: diversity, evolution and manipulation of insects. Entomol Sci. (2015) 18:1–20. doi: 10.1111/ens.12105
30. Juergens KB, Huckabee J, and Greninger AL. Two novel iflaviruses discovered in bat samples in washington state. Viruses. (2022) 14:994. doi: 10.3390/v14050994
31. Hardmeier I, Aeberhard N, Qi W, Schoenbaechler K, Kraettli H, Hatt J-M, et al. Metagenomic analysis of fecal and tissue samples from 18 endemic bat species in Switzerland revealed a diverse virus composition including potentially zoonotic viruses. PloS One. (2021) 16:e0252534. doi: 10.1371/journal.pone.0252534
32. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. (2020) 579:270–3. doi: 10.1038/s41586-020-2012-7
33. Irving AT, Ahn M, Goh G, Anderson DE, and Wang LF. Lessons from the host defences of bats, a unique viral reservoir. Nature. (2021) 589:363–70. doi: 10.1038/s41586-020-03128-0
34. VSasaki M, Setiyono A, Handharyani E, Kobayashi S, Rahmadani I, Taha S, et al. Isolation and characterization of a novel alphaherpesvirus in fruit bats. J Virol. (2014) 88:9819–29. doi: 10.1128/JVI.01277-14
35. Gatherer D, Depledge DP, Hartley CA, Szpara ML, Vaz PK, Benkő M, et al. ICTV virus taxonomy profile: herpesviridae 2021. J Gen Virol. (2021) 102:1673. doi: 10.1099/jgv.0.001673
36. Wibbelt G, Kurth A, Yasmum N, Bannert M, Nagel S, Nitsche A, et al. Discovery of herpesviruses in bats. J Gen Virol. (2007) 88:2651–5. doi: 10.1099/vir.0.83045-0
37. Russell GC, Stewart JP, and Haig DM. Malignant catarrhal fever: a review. Vet J. (2009) 179:324–35. doi: 10.1016/j.tvjl.2007.11.007
38. Benkő M, Aoki K, Arnberg N, Davison AJ, Echavarría M, Hess M, et al. ICTV virus taxonomy profile: adenoviridae 2022. J Gen Virol. (2022) 103:1721. doi: 10.1099/jgv.0.001721
39. Harrach B, Tarján ZL, and Benkő M. Adenoviruses across the animal kingdom: a walk in the zoo. FEBS Lett. (2019) 593:3660–73. doi: 10.1002/1873-3468.13687
40. Vidovszky MZ, Kohl C, Boldogh S, Gorfol T, Wibbelt G, Kurth A, et al. Random sampling of the Central European bat fauna reveals the existence of numerous hitherto unknown adenoviruses. Acta Vet Hung. (2015) 63:508–25. doi: 10.1556/004.2015.047
41. Karamendin K, Kydyrmanov A, Sabyrzhan T, Nuralibekov S, Kasymbekov Y, and Khan Y. Detection and phylogenetic characterization of a novel adenovirus found in lesser mouse-eared bat (Myotis blythii) in south Kazakhstan. Viruses. (2023) 15:1139. doi: 10.3390/v15051139
42. Meng X-J. Circoviridae. In: Knipe DM and Howley PM, editors. Fields virology. Wolters Kluwer/Lippincott Williams & Wilkins Health, Philadelphia, PA (2013). p. 2664.
43. Wang LF and Anderson DE. Viruses in bats and potential spillover to animals and humans. Curr Opin Virol. (2019) 34:79–89. doi: 10.1016/j.coviro.2018.12.007
44. Dhandapani G, Yoon SW, Noh JY, Jang SS, Kim MC, Lim HA, et al. Detection of bat-associated circoviruses in Korean bats. Arch Virol. (2021) 166:3013–21. doi: 10.1007/s00705-021-05202-y
Keywords: bat, virus, metagenome, virome, Kazakhstan, next generation sequencing
Citation: Karamendin K, Sabyrzhan T, Kydyrmanov A, Kasymbekov Y, Khan Y, Nuralibekov S and Goodman SJ (2025) Virological surveillance of bats in Southern Kazakhstan. Front. Virol. 5:1582410. doi: 10.3389/fviro.2025.1582410
Received: 24 February 2025; Accepted: 23 April 2025;
Published: 23 June 2025.
Edited by:
Claudia Alteri, University of Milan, ItalyReviewed by:
George William Carnell, University of Cambridge, United KingdomLucia Moreira, Universidad de la República, Uruguay
Renata Budaszewski, Universidade Estadual do Rio Grande do Sul, Brazil
Copyright © 2025 Karamendin, Sabyrzhan, Kydyrmanov, Kasymbekov, Khan, Nuralibekov and Goodman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kobey Karamendin, a29iZXkua2FyYW1lbmRpbkBnbWFpbC5jb20=