 Yuxin Zhang1
Yuxin Zhang1 Mengzhu Li2Fangyu Lin1Rui Guo2Danhui Wu2Rui Shen2
Mengzhu Li2Fangyu Lin1Rui Guo2Danhui Wu2Rui Shen2 Vijay Harypursat2
Vijay Harypursat2 Yaokai Chen2*
Yaokai Chen2* Yemiao Chen3*
Yemiao Chen3*- 1Public Health Department, Chongqing Public Health Medical Center, Chongqing, China
- 2Department of Infectious Diseases, Chongqing Public Health Medical Center, Chongqing, China
- 3Biobank, Chongqing Public Health Medical Center, Chongqing, China
In the present era of highly active antiretroviral therapy (HAART), as a consequence of long-term antiretroviral medication consumption, extended patient survival, and persistent immune activation, people who live with HIV (PLWH) have become more susceptible to chronic diseases such as tumors, cardiovascular diseases, and diabetes mellitus when compared to the general population. Although the HIV-1 matrix protein p17 (p17) was initially thought to be a structural protein of HIV-1, recent studies have revealed its close association with elevated incidence of tumors, particularly lymphomas, in HIV-1-infected individuals. Experimental results indicate that p17 may promote the proliferation and clonogenicity of various cell types. Moreover, p17 and its variants are directly and indirectly linked to the occurrence and development of lymphomas. The sustained detection of p17 in clinical specimens—particularly lymph node biopsies and plasma samples from HIV/AIDS patients—points to its possible involvement in tumor microenvironment modulation. Here, we present a brief overview of the roles played by p17 in the regulation of cellular functions, promotion of lymphoma occurrence, and formation of the tumor microenvironment, as well as the potential molecular mechanisms which are fundamental to the functionality of p17. Additionally, we briefly outline other HIV-related tumors that p17 may likely be involved in. Investigation of p17 mechanisms and their interactions with oncogenic pathways is essential for understanding HIV-associated tumorigenesis. These findings may facilitate the development of novel therapeutic strategies for HIV-related malignancies
1 Introduction
The human immunodeficiency virus (HIV) is a virus that attacks the human immune system. The most severe stage of HIV infection results in acquired immunodeficiency syndrome (AIDS). Human Immunodeficiency Virus (HIV) mainly attacks and kills T-lymphocytes, especially CD4+ T-cells. These cells play a vital role in the immune system (1). Replication of HIV and its subsequent destruction of the immune system severely weakens immunity and leaves infected individulas vulnerable to a variety of opportunity infections and cancers (1). Since its first discovery in the 1980s, AIDS has claimed the lives of approximately 42.3 million people. According to UNAIDS, as of 2023, an estimated 39.9 million people were living with HIV globally, with approximately 1.6 million new infections each year (2). There is no cure for HIV infection. However, with universal access to HIV prevention, diagnosis, treatment, and care (including opportunistic infections), and widespread use of highly active HAART, HIV infection has become a chronic health condition. HIV still continues to pose a significant global health burden, with 30.7 million people living with HIV in 2023 receiving antiretroviral viral therapy and approximately 630,000 HIV-related deaths (3).
According to data provided by the American cancer society, Cancer Facts & Figures 2024, the burden of infection-related is ten times higher in HIV-infected individuals than in the general population (4). HIV-associated malignancies are generally divided into two categories: AIDS-defining cancers and AIDS-non-defining cancers (5). AIDS-defining cancers include Kaposi’s sarcoma, certain types of non-Hodgkin lymphomas such as Burkitt lymphoma and diffuse large B-cell lymphoma, and cervical cancer (5, 6). The development of these cancers is usually closely associated with immunosuppression due to HIV infection and is the hallmark lesion of HIV-infected individuals diagnosed with AIDS (7). Non-Hodgkin Lymphoma (NHL) is commonly found in HIV-infected individuals. According to data, the risk of developing non-Hodgkin lymphoma in HIV-infected individuals is about 12 times higher than that of the general population (6). In the era of combination antiretroviral therapy, lymphoma remains the most common cause of death in HIV-1 infected patients (8). This type of cancer is therefore classified as a defining cancer in HIV patients. In resource-poor areas, AIDS-defining cancers remain a major health threat among HIV-infected patients due to low ART coverage and limited medical resources (9).
In addition to ADCs, people with HIV are also more susceptible to developing non-AIDS-defining cancers (NADCs) than the general population (10). Non-AIDS-defining cancers (NADCs) are cancers that occur in HIV-infected individuals but are not used as criteria for an AIDS diagnosis. These cancers, which do not have defining characteristics but are also more common in HIV-infected individuals, include liver, lymphoma, and prostate cancers (10, 11). People living with HIV are approximately eight times more likely to be diagnosed with Hodgkin’s lymphoma compared to the general population (10), and HIV-infected individuals have an approximately five times higher risk of developing liver cancer compared to the general population (11). In addition to being associated with an increased risk of cancer, HIV infection is also associated with an increased risk of dying from cancer. HIV-infected individuals with multiple cancers are more likely to die from cancer than HIV-uninfected cancer patients (10, 11). As ART has become more widely available, HIV-infected people are living significantly longer, which gives them more time to be exposed to risk factors for cancer, such as long-term chronic inflammation, and immune activation status (12).
The etiology of these cancers appears to be multifactorial and may involve immunosuppression, frequent infections with oncogenic viruses, and high-risk behaviors including poor lifestyle choices (13, 14). The elevated risk of cancer in HIV-infected individuals is the result of synergistic effects of multiple mechanisms, including immunosuppression, chronic inflammation, and the direct pro-tumourigenic effects of viral proteins (15). Although HIV itself is not classified as a classical oncogenic virus (16), where HIV-encoded proteins play an important role in this process. Existing findings have been mainly in the HIV-1 envelope glycoprotein GP120, the regulatory protein Tat, and the auxiliary protein Nef which would have different abilities to promote tumor development (17–19). For example, HIV-1 envelope glycoprotein GP120 directly drives glioma cell proliferation and inhibits apoptosis by binding to the CXCR4/CCR5 receptor and activating the PI3K-AKT-mTOR pathway (20, 21), and promotes cancer progression by inducing chronic inflammation and immune-activated states (20–22); the regulatory protein Tat interferes with DNA double-strand break repair by inhibiting ATM kinase activity, leading to genomic and cell death (17–19). Tat interferes with DNA double-strand break repair by inhibiting ATM kinase activity, leading to genomic instability (21); and the auxiliary protein Nef enhances tumor cell survival and metastasis by down-regulating MHC-I molecules and activating the PAK2/NF-κB pathway (22, 23). Although part of the virus-host interaction network has been revealed, the dynamic regulatory mechanism of matrix protein P17 remains unclear. Compared to proteins such as GP120, P17 has unique pathological properties: lymphangiogenesis is essential to support lymphoma proliferation and survival, as well as tumor dissemination (24). HIV-1 matrix protein p17 is secreted by HIV-1-infected cells and accumulates and persists in lymph nodes of patients receiving highly active antiretroviral therapy in the absence of any HIV-1 replication activity (24). This review proposes the hypothesis that P17 may drive cancer progression by integrating immunosuppression with microenvironmental remodeling mechanisms.
2 p17 in the HIV-1 life cycle
p17 is encoded by the retroviral gene gag as a precursor, i.e., Pr55Gag (25). During or shortly after virus budding, Pr55Gag is cleaved by HIV-1 protease into mature proteins, including the matrix protein p17, the capsid protein p24, the nucleocapsid protein p7, protein p6, and two smaller spacer peptides p1 and p2. p17 protein is a 132 amino acid polypeptide containing five major α-helices and a highly basic platform of three β-strands (26). Crystal structural analysis indicates that p17 is trimeric in mature virions, which may contribute to virus assembly and Env glycoprotein incorporation into virions (27).
p17 was initially identified as the structural protein that forms a protective shell attached with its N-terminal membrane binding domain to the inner surface of the virion lipid (27, 28). An increasing number of studies have revealed that p17 [both the mature version and the precursor version (N-terminal of Pr55Gag)] performs multiple functions during the HIV-1 life cycle (29). During the HIV-1 replication cycle, p17 drives efficient viral proliferation through a multistage mechanism, and these functions simultaneously provide the molecular basis for carcinogenesis. During the early infection phase, p17 interacts with the host cell’s nuclear export machinery through its nuclear export signal (NES), facilitating the export of the viral preintegration complex (PIC) from the nucleus to the cytoplasm (30). This process ensures the efficient export of viral RNA from the nucleus by interacting with the host Crm1 protein to support the subsequent reverse transcription process (30, 31). Upon entering the late assembly stage, p17 also plays a key role in the accurate localization of viral RNA and viral assembly. p17 works in concert with other Pr55Gag regions (e.g. p24 and p7) to help accurately localize unspliced viral RNA to the assembly site, thereby facilitating the assembly of viral particles (28). In addition, p17 interacts with the HIV envelope protein Env, regulating its integration on the surface of viral particles and affecting viral infectivity (28, 31). Finally, loss of p17 function or mutation may result in impaired viral assembly and replication. In particular, mutations in the NES region of p17 may lead to the mislocalization of Pr55Gag and viral RNA, which can significantly affect viral replication and transmission (32, 33). Thus, p17 has diverse roles in the HIV-1 life cycle, including nuclear export, RNA localization, assembly, and integration of the envelope protein Env. The precise collaboration of these functions makes p17 an indispensable pleiotropic regulator of the HIV-1 replication cycle.
2.1 Secretion mechanism of P17
The secretion of the HIV-1 matrix protein p17 is accomplished primarily through multiple pathways. As a cleavage product of the viral Gag polyprotein, p17 is incorporated into mature viral particles during viral assembly and is naturally released into the extracellular environment with viral outgrowth (34). In addition, host cells can also secrete p17 via a non-classical pathway: p17 can be encapsidated in exosomes via intracellular multivesicular bodies (MVBs) and released the encapsidated p17 outside the cell through fusion with the cell membrane (35). In addition, the carbamylated modification of the N-terminus of p17 enables it to insert directly into the cell membrane and enter the extracellular space by membrane flipping or microvesicle shedding (36). Notably, the level of p17 secretion is regulated by a variety of factors, including the efficiency of cleavage of Gag precursors by HIV-1 protease and the regulation of the secretory process through pathways such as NF-κB when stimulated by inflammatory signals (37, 38). These fine-grained secretion mechanisms ensure that p17 can be efficiently distributed and fulfill its biological functions during infection.
Even though HAART is effective in inhibiting viral replication and reducing the level of viral RNA to undetectable levels, certain proteins of HIV-1, such as p24, p17, and gp120/gp41, are still able to persist in the patient’s lymph node germinal centers and can be detected even 5 to 13 months after treatment (19). The persistence may constantly stimulate B cells, causing them to be in a state of chronic activation (19). This chronic immune stimulation may disrupt the normal function of B cells, leading to abnormalities in their proliferation and differentiation (39, 40). In particular, in some specific HIV-associated lymphomas, B cells may be transformed into cancerous cells under continuous stimulation by viral proteins (19). Thus, the prolonged presence of P17 in immune cells, especially in immunologically active sites such as lymph nodes, may provide a sustained and favorable environment for cancer development (19).
3 p17 enhances cell proliferation and clonogenicity
The HIV-1 matrix protein p17 exhibits multifaceted regulatory functions in cellular activities through diverse molecular mechanisms. p17 through its interaction with human serum factors, significantly enhances lymphocyte proliferation and promotes HIV-1 viral replication (41). Additionally, when natural killer (NK) cells are exposed to p17, they display an enhanced proliferative capacity and increased secretion of pro-inflammatory cytokines, particularly interferon-gamma (IFN-γ) and tumor necrosis factor-alpha (TNF-α) (42, 43). These observations suggest that p17 serves as a crucial immunomodulator, potentially facilitating viral replication and dissemination via its effects on immune cell function.
While cell proliferation is essential for growth and tissue repair in organisms, HIV infection disrupts normal cellular proliferative processes, and compromises immune function. Studies in simian models have identified the regulatory role of p17 in B-cell growth, demonstrating the ability of p17 to enhance B-cell proliferation and clonogenicity via activation of proliferation signaling pathways and cell cycle progression (44). Notably, p17 variants (vp17s) with B-cell clone-forming activity have been identified in HIV-positive lymphoma patients, suggesting that they may be involved in lymphomagenesis (45). These variants are mainly dependent on the aberrant activation of the PAR1/EGFR/PI3K/Akt signaling pathway and on the insertion of an amino acid originating from the COOH-terminal region, which results in a characteristically unstable and misfolded protein conformation (46–48). This structural change allows vp17s to acquire a strong clonal B-cell growth-regulating ability, which directly promotes the formation of B-cell lymphomas. In terms of molecular mechanisms, misfolded vp17s trigger Gq protein-mediated EGFR transcriptional activation through specific binding to protease-activated receptor 1 (PAR1), which in turn activates the downstream PI3K/Akt signaling cascade, ultimately leading to dysfunction of cell cycle regulatory proteins (e.g., Cyclin D1, CDK1) and oncogenes (e.g., p53, Rb) (48, 49). Notably, the F1 peptide, which represents the core functional epitope of vp17s, is also capable of activating this signaling pathway, and the strength of regulation of the signaling pathway differs significantly between different vp17s variants (50). In addition, p17 inhibits the autophagy process in T cells by binding to OLA1 protein to over-activate GSK3β kinase, which in turn promotes the aberrant proliferation of HIV-infected T cells, which further highlights the pro-carcinogenic potential of the protein in multiple cell types (51). Overall, the role of p17 is not limited to direct immunomodulation, and its regulation through multiple signaling pathways, including cell cycle, proliferation, and autophagy, may play a key driving role in HIV-infected tumourigenesis.
Figure 1 reveals the mechanism of p17 and its variants in lymphocyte development and AIDS-associated lymphoma.1.Enhances lymphocyte proliferation and HIV-1 viral replication by interacting with human serum factors 2. Promotes NK cell proliferation and pro-inflammatory cytokine (IFN-γ, TNF-α) secretion 3. Activates proliferative signaling pathways and cell cycle progression and enhances B-cell clonogenicity 4. p17 variants (vp17s) associated with specific cellular receptor interactions 5.vp17s activate the EGFR/PI3K/Akt pathway via PAR1 receptor 6.Inhibition via OLA1-GSK3βaxis Autophagy and promotes proliferation of HIV-infected T cells Created with BioRender.com.
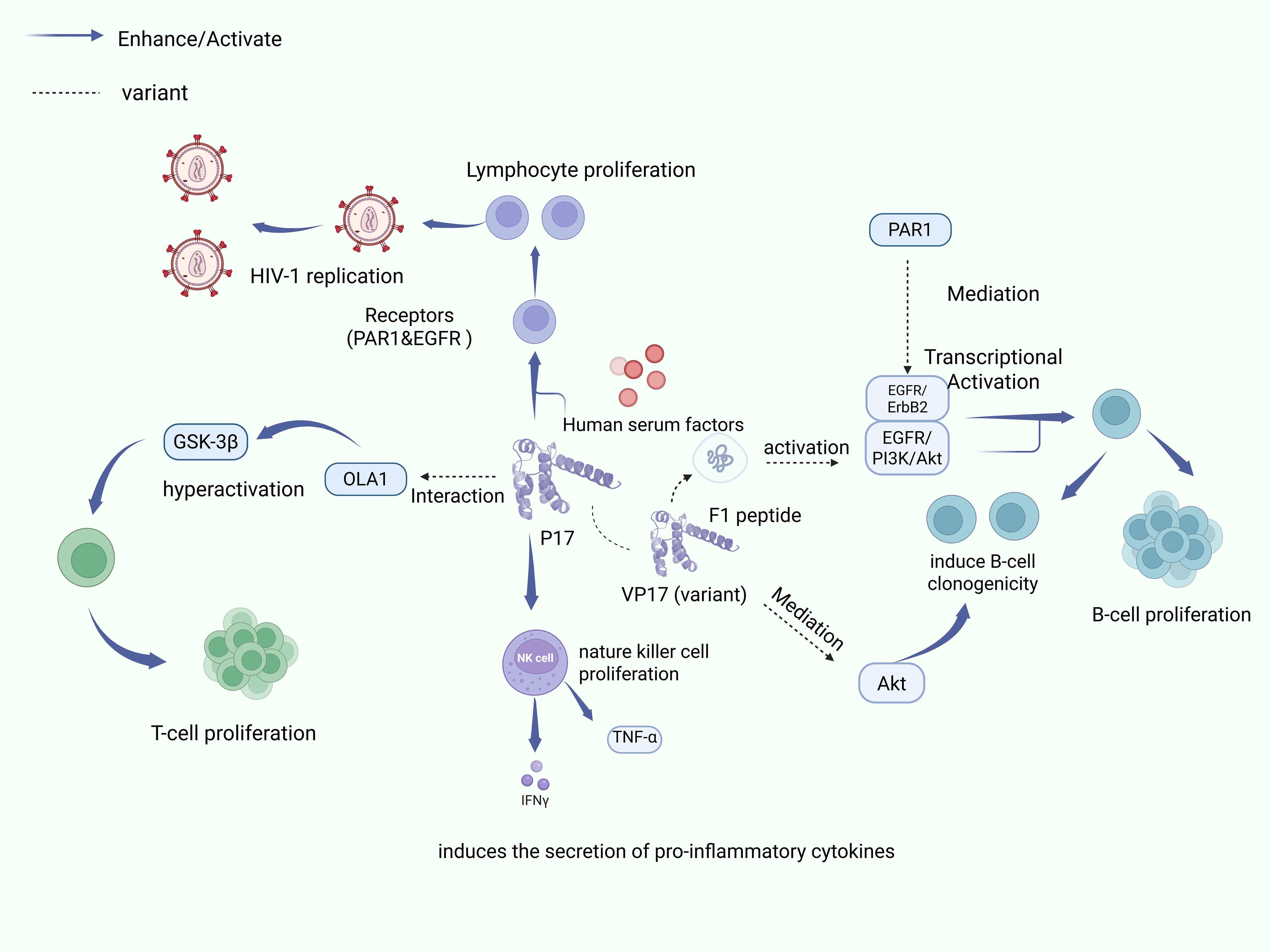
Figure 1. p17 enhances cell proliferation and clonogenicity.
4 p17 and its variants induce lymphogenesis
Lymphocytes are fundamental components of the immune system, and orchestrate the regulation and maintenance of immune responses (52). The process of lymphocyte genesis encompasses both production and differentiation of these crucial immune cells (53). In the context of HIV infection, p17 and its variants emerge as significant factors that influence lymphocyte genesis, with certain variants associated with increased lymphomagenesis risk via their effects on tumor cell proliferation, transformation, and signaling pathway activation.
4.1 p17 activates signaling pathways and transcription factors in lymphocyte development
The HIV-1 matrix protein p17 is able to regulate the developmental and proliferative processes of lymphocytes through a variety of mechanisms. Studies have shown that the p17 protein can act through the activation of key transcription factors required for lymphocyte proliferation (54). c-Myc can co-promote B-cell growth by up-regulating cellular metabolism and enhancing cell survival in hypoxic environments (54). p17 also induces phosphorylation modification and activation of the transcriptional activities of CREB and c-Myc through up-regulation of PKA and MEK/ERK signaling pathways, thereby contributing to lymphocyte development and lymphoma progression. Phosphorylation modifications and activates their transcriptional activities, thereby driving lymphocyte developmental processes and thus promoting lymphoma progression (55).
In a transgenic mouse model carrying HIV-1 antigen, researchers observed that the p17 protein showed a high expression state outside the cells in the lymphoma model. This high expression state of p17 was able to significantly stimulate activated B cells while upregulating the expression level of RAG1 recombinase, and this regulatory effect of multiple mechanisms may lead to increased genomic instability and promote malignant transformation of cells (19, 54). Notably, different HIV-1 p17 variants exhibit differential regulation of PTEN activation and B cell growth. Specifically: first, the R76G mutant significantly enhanced B-cell clone formation through specific activation of the PTEN/PI3K/Akt signaling pathway (50); second, the S75X variant originating from the Ugandan strain was able to activate the PI3K/Akt pathway through its variant-unique molecular mechanism, which further promoted aberrant B-cell proliferation and malignant transformation (50, 55). These findings not only reveal the pleiotropic role of p17 variants in lymphoma development but also lay the molecular foundation for the development of targeted therapeutic strategies. Together, these findings reveal the pleiotropic mechanisms of p17 in lymphoma development, but further exploration is needed to determine the timeliness of p17 exposure and the feasibility of targeting oncogenic variants for treatment.
4.2 p17 variants in HIV-associated lymphoma development
Lymphangiogenesis plays a key role in both the growth and metastasis of AIDS-related lymphomas. A common feature of lymphocytic malignancies is the presence of chromosomal translocations between antigen receptor genes and proto-oncogenes (54). However, differences in mutations in p17 can have different effects. For example, mutations in the p17 n-terminal structure may impair viral infectivity, fusion, and envelope binding. Differently, c-terminal mutations may enhance pathogenicity (19). Another study found that c-terminal insertion mutations occur frequently in p17, and these insertion mutations are more common in plasma viral particles from patients with non-Hodgkin’s lymphoma (NHL) (49).
In addition, studies on recombinant p17 and variant p17- lyrm derived from splenic marginal zone lymphomas have shown their effects on intracellular signaling (56). In addition, p17 affects the immune response by inducing the migration of immature plasma cell-like dendritic cells to lymph nodes and disrupting the coordination between the innate and adaptive immune systems (57). These studies suggest a strong association between p17 polymorphisms and lymphoma development. Some studies have shown a higher prevalence of vp17s mutations in HIV-associated lymphomas compared to non-HIV lymphomas. For example, COOH-terminal amino acid insertions generate unstable vp17s variants, altering protein conformation and reducing immune recognition. These mutations may also damage HIV-1 reverse transcriptase efficiency (58), and then enhancing viral infectivity, replication capacity, and lymphoma development (59).
Figure 2 shows the P17 acts in lymphocytes through several mechanisms: (1) P17 regulates PKA and MEK/ERK signaling pathways, leading to CREB and c-Myc activation, which in turn promotes lymphocyte development; (2) the R76G mutation in P17 induces activation of the PTEN/PI3K/Akt pathway, boosting B-cell cloning; and (3) the S75X variant of P17 stimulates B cell proliferation and transformation through activation of the PI3K/Akt pathway. Created with BioRender.com.
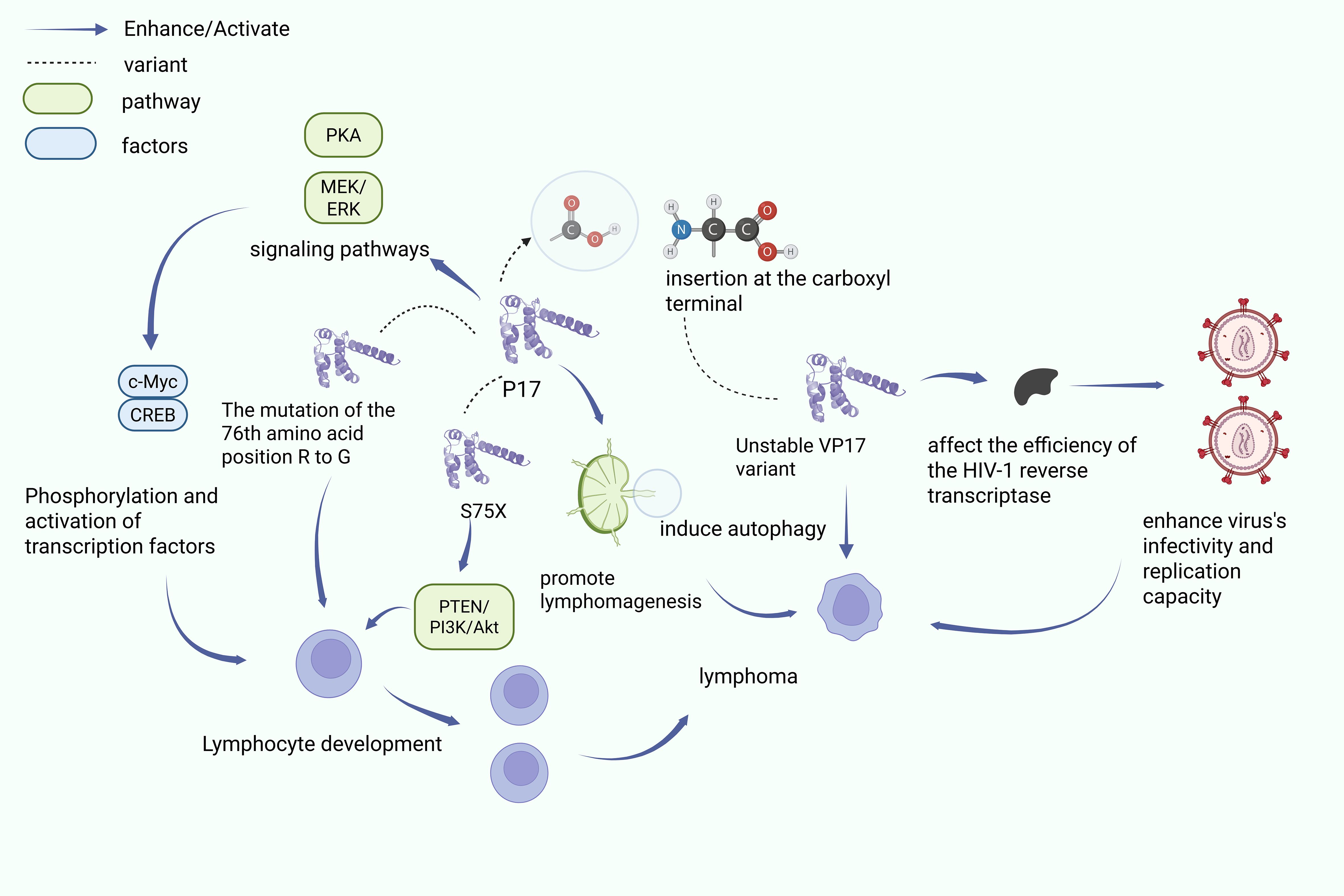
Figure 2. p17 and its variants induce lymphogenesis.
5 p17 modulation of the tumor microenvironment
In HIV-1 patients, infected cells release significant amounts of p17 independent of viral particles (45). Notably, even during highly active antiretroviral therapy (HAART) when viral replication is suppressed, p17 remains detectable in germinal centers of lymph nodes (59), plasma (60), and hepatic endothelial cells (61). This persistent presence suggests a complex role in the modulation of the local microenvironment through multiple mechanisms.
Some studies show that the regulatory mechanisms of p17 on the vascular system are diverse: Firstly, p17 can stimulate angiogenesis through CXCR1/CXCR2 chemokine receptors (32, 61); On the other hand, its binding to endothelin-1 (ET-1) and its (24) type receptor (ETB) can also promote lymphatic angiogenesis. When induced ET-1/ETB activation, p17 can also trigger PI3K/Akt and ERK1/2, two key signal transduction pathways, which provide ideal microenvironmental conditions for promoting lymphangiogenesis and tumor metastasis (62, 63). In addition, p17 can stimulate brain endothelial cell angiogenesis through EGFR1, which increases the likelihood of comorbidities in HIV patients (62). P17 also activates epidermal growth factor receptors on brain endothelial cells, stimulating downstream signaling pathways responsible for initiating angiogenesis (62). Additionally, p17 influences adipocyte development and function via PPARγ pathway activation, promoting differentiation and proliferation. In the tumor microenvironment, there are complex interactions between angiogenesis, lymphangiogenesis, and lipidogenesis, and these processes contribute to tumor development to varying degrees (63). The p17 protein also interacts with CXCR 2 and Syndecan-2 to activate hepatic stellate cells (64), and can regulate the secretion of von Willy brand factors in endothelial cells, affecting the coagulation process (65).
The ability of p17 to bind heparan sulfate proteoglycans (HSPGs) in lymphoid tissues may lead to elevated local concentrations, potentially sequestering additional viral proteins, and facilitating tumorigenesis. Recent studies confirming high p17 expression in lymph node tissues of AIDS patients provide strong evidence for its role in creation of a tumor-supportive microenvironment (66, 67).
Figure 3 depicts the comprehensive influence of p17 on the tumor microenvironment through multiple mechanisms, i.e., (1) induction of pro-inflammatory phenotypes in monocytes via STAT1 activation and inflammatory gene upregulation; (2) promotion of angiogenesis through CXCR1/CXCR2 activation in tumor cells and brain endothelial cells via the EGFR1 pathway; (3) enhancement of lymphangiogenesis through the endothelin-1/endothelin B receptor axis; (4) regulation of hepatic stellate cell activation via CXCR2/Syndecan-2 interaction; (5) modulation of adipocyte development through PPARγ pathway activation; and (6) influence on coagulation through von Willebrand factor secretion in endothelial cells. Created with BioRender.com.
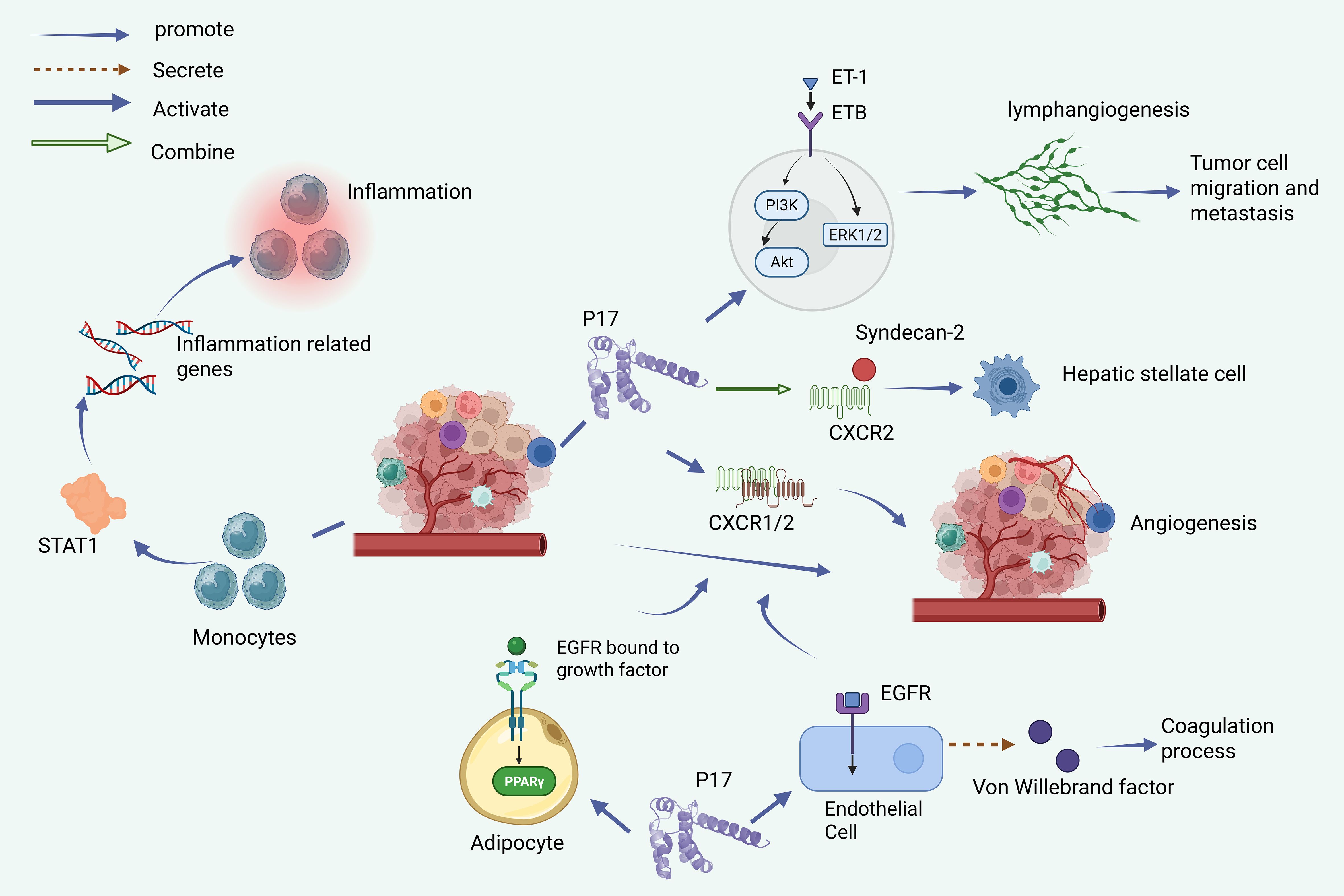
Figure 3. p17 modulates the tumor micro-environment.
6 Effects of p17 on other comorbid cancers
Table 1 shows the most relevant oncogenic mechanisms of p17. In addition, studies have suggested that p17 has a broad indirect mechanism of action in many cancer processes. First, p17 is able to penetrate the blood-brain barrier and induce neurotoxicity, which may be associated with the promotion of CNS neoplasia (61). In addition, p17 variants are associated with neurological complications (72). In studies of triple-negative breast cancer subtype MDA-MB-231, p17 was found to stimulate cancer cell proliferation and metastasis by binding the CXCR2 receptor and activating the ERK1/2 signaling pathway (70). Based on the analysis of existing studies, future research can focus on how p17 and its structural changes interact with cell membrane receptors such as CXCR2 and other g protein-coupled receptors, and then affect the proliferation and metastasis of cancer cells at a more diverse molecular level. This research field and direction will lay the foundation for the treatment of a variety of tumors in hiv patients. In addition, p17 was found to be able to interfere with lipid metabolism in hepatocytes by affecting the STAT pathway, suggesting that p17 may be involved in liver dysfunction in Hiv-Infected patients (71). In general, these lines of evidence suggest that p17 and its variants potentially influence the progression of multiple cancer types in people living with HIV through various mechanisms. Future studies could deeply analyze the mechanism of action of p17, which helps to overcome the current limitations of tumor development in clinical treatment of people living with HIV.
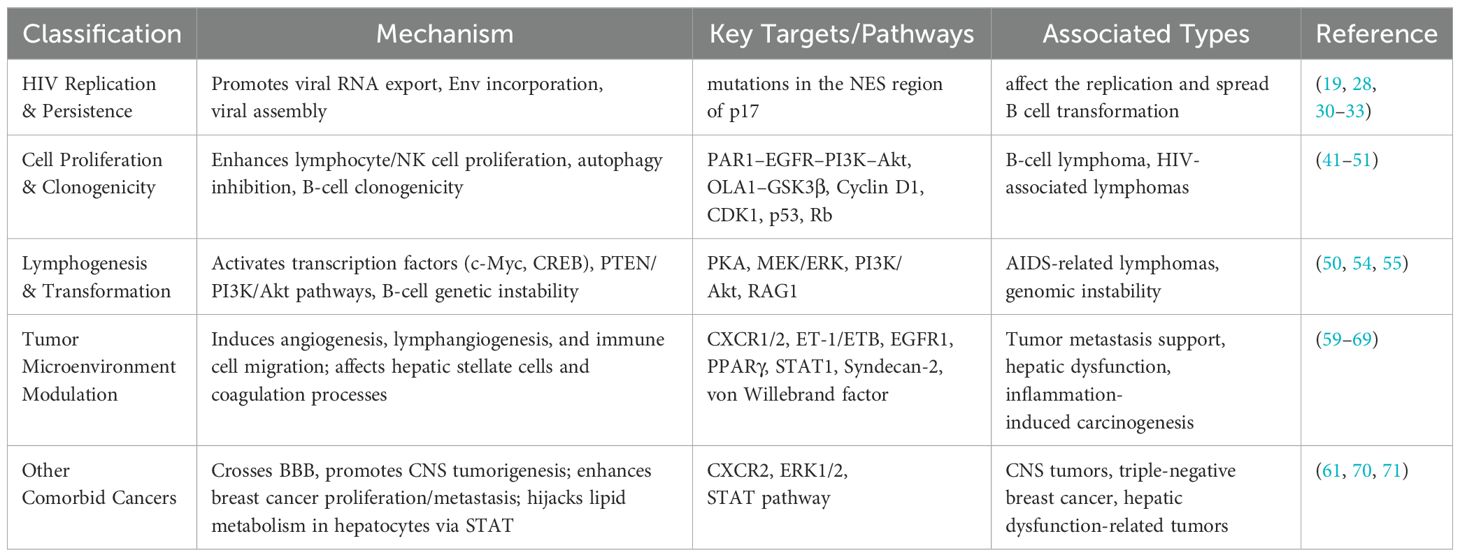
Table 1. The most relevant oncogenic mechanisms of p17.
7 Conclusion
Existing research still has important limitations in its understanding of the carcinogenic effects of p17: for example, animal simulations in the HIV state are still lacking, and most of the current evidence comes from in vitro experiments. Moreover, the functional heterogeneity of p17 variant and its interaction with host genetic background have not been systematically resolved. In addition, the synergistic carcinogenic effect of p17 with other key viral proteins (such as Tat and Nef) still needs to be further studied. The study of the P17 mechanism and its multiple oncogenic pathways is a very important part of research into HIV-associated tumours. The study of P17’s relevance to HIV tumors could in the future lead to precise oncological treatments for HIV-infected patients, targeting specific receptors or specific immunomodulation.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author/s.
Author contributions
YZ: Investigation, Resources, Supervision, Writing – original draft, Writing – review & editing. ML: Resources, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. FL: Investigation, Software, Writing – original draft. RG: Visualization, Writing – review & editing. DW: Validation, Visualization, Writing – review & editing. RS: Writing – review & editing. VH: Writing – review & editing. YKC: Funding acquisition, Writing – original draft, Writing – review & editing. YeC: Funding acquisition, Resources, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by grants from the Joint Project of Chongqing Health Commission and Chongqing Science & Technology Bureau (2024MSXM178), the Chongqing Technology Innovation and Application Development Special Project (CSTB2022TIAD-KPX0158), the Open Project of Jiangsu Provincial Science and Technology Resources (Clinical Resources) Coordination Service Platform (TC2022B009).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Zaongo SD, Ouyang J, Chen Y, Jiao YM, Wu H, and Chen Y. HIV infection predisposes to increased chances of HBV infection: current understanding of the mechanisms favoring HBV infection at each clinical stage of HIV infection. Front Immunol. (2022) 13:853346. doi: 10.3389/fimmu.2022.853346
2. UNAIDS. Global HIV & AIDS statistics — Fact sheet (2024). Available online at: https://www.unaids.org/sites/default/files/media_asset/UNAIDS_FactSheet_en.pdf (Accessed January 9, 2025).
3. WHO. HIV statistics, globally and by WHO region (2024). Available online at: https://cdn.who.int/media/docs/default-source/hq-hiv-hepatitis-and-stis-library/hiv-epi-fact-sheet-march-2025.pdf?sfvrsn=61d39578_12 (Accessed 1 February, 2025).
4. Siegel RL, Giaquinto AN, and Jemal A. Cancer statistics, 2024. CA A Cancer J Clinicians. (2024) 74:12–49. doi: 10.3322/caac.21820
5. Shmakova A, Germini D, and Vassetzky Y. HIV-1, HAART and cancer: A complex relationship. Int J Cancer. (2020) 146:2666–79. doi: 10.1002/ijc.32730
6. Yarchoan R and Uldrick TS. HIV-associated cancers and related diseases. N Engl J Med. (2018) 378:1029–41. doi: 10.1056/NEJMra1615896
7. Cesarman E, Damania B, Krown SE, Martin J, Bower M, and Whitby D. Kaposi sarcoma. Nat Rev Dis Primers. (2019) 5:9. doi: 10.1038/s41572-019-0060-9
8. Levine AM. AIDS-related Malignancies: the emerging epidemic. J Natl Cancer Inst. (1993) 85:1382–97. doi: 10.1093/jnci/85.17.1382
9. Montaño MA, Chagomerana MB, Borok M, Painschab M, Uldrick TS, and Bender Ignacio RA. Impact of antiretroviral therapy on cancer treatment outcomes among people living with HIV in low- and middle-income countries: a systematic review. Curr HIV/AIDS Rep. (2021) 18:105–16. doi: 10.1007/s11904-021-00542-5
11. Pinzone MR, Fiorica F, Di Rosa M, Malaguarnera G, Malaguarnera L, Cacopardo B, et al. Non-AIDS-defining cancers among HIV-infected people. Eur Rev Med Pharmacol Sci. (2012) 16(10):1377–88.
12. Omar A, Marques N, and Crawford N. Cancer and HIV: the molecular mechanisms of the deadly duo. Cancers (Basel). (2024) 16:546. doi: 10.3390/cancers16030546
13. Casper C. The increasing burden of HIV-associated Malignancies in resource-limited regions. Annu Rev Med. (2011) 62:157–70. doi: 10.1146/annurev-med-050409-103711
14. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. A review of human carcinogens: Part B: Biological agents. Vol. 100B. IARC Monogr Eval Carcinog Risks Hum. Lyon, France: World Health Organization. (2012). pp. 1–441.
15. Grulich AE, van Leeuwen MT, Falster MO, and Vajdic CM. Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: a meta-analysis. Lancet. (2007) 370:59–67. doi: 10.1016/S0140-6736(07)61050-2
16. Valentín-Guillama G, López S, Kucheryavykh YV, Chorna NE, Pérez J, Ortiz-Rivera J, et al. HIV-1 envelope protein gp120 promotes proliferation and the activation of glycolysis in glioma cell. Cancers. (2018) 10:301. doi: 10.3390/cancers10090301
17. Hodgson DM, Yirmiya R, Chiappelli F, and Taylor AN. Intracerebral HIV glycoprotein (Gp120) enhances tumor metastasis via centrally released interleukin-1. Brain Res. (1998) 781:244–51. doi: 10.1016/s0006-8993(97)01243-2
18. Benlarbi M, Richard J, Bourassa C, Tolbert WD, Chartrand-Lefebvre C, Gendron-Lepage G, et al. Plasma human immunodeficiency virus 1 soluble glycoprotein 120 association with correlates of immune dysfunction and inflammation in antiretroviral therapy-treated individuals with undetectable viremia. J Infect Dis. (2024) 229:763–74. doi: 10.1093/infdis/jiad503
19. Carroll VA, Lafferty MK, Marchionni L, Bryant JL, Gallo RC, and Garzino-Demo A. Expression of HIV-1 matrix protein P17 and association with B-cell lymphoma in HIV-1 transgenic mice. Proc Natl Acad Sci United States America. (2016) 113:13168–73. doi: 10.1073/pnas.1615258113
20. Kast RE. Feedback between glial tumor necrosis factor-alpha and gp120 from HIV-infected cells helps maintain infection and destroy neurons. Neuroimmunomodulation. (2002) 10:85–92. doi: 10.1159/000065184
21. Srivastava DK, Tendler CL, Milani D, English MA, Licht JD, and Wilson SH. The HIV-1 transactivator protein Tat is a potent inducer of the human DNA repair enzyme beta-polymerase. AIDS (London England). (2001) 15:433–40. doi: 10.1097/00002030-200103090-00001
22. Arhel NJ and Kirchhoff F. Implications of Nef: host cell interactions in viral persistence and progression to AIDS. Curr Top Microbiol Immunol. (2009) 339:147–75. doi: 10.1007/978-3-642-02175-6_8
23. Fenard D, Yonemoto W, de Noronha C, Cavrois M, Williams SA, and Greene WC. Nef is physically recruited into the immunological synapse and potentiates T cell activation early after TCR engagement. J Immunol. (2005) 175:6050–7. doi: 10.4049/jimmunol.175.9.6050
24. Caccuri F, Rueckert C, Giagulli C, Schulze K, Basta D, Zicari S, et al. HIV-1 matrix protein P17 promotes lymphangiogenesis and activates the endothelin-1/endothelin B receptor axis. Arteriosclerosis thrombosis Vasc Biol. (2014) 34:846–56. doi: 10.1161/atvbaha.113.302478
25. Ono A, Orenstein JM, and Freed EO. Role of the gag matrix domain in targeting human immunodeficiency virus type 1 assembly. J Virol. (2000) 74:2855–66. doi: 10.1128/jvi.74.6.2855-2866.2000
26. Klingler J, Anton H, Réal E, Zeiger M, Moog C, Mély Y, et al. How HIV-1 gag manipulates its host cell proteins: A focus on interactors of the nucleocapsid domain. Viruses. (2020) 12:888. doi: 10.3390/v12080888
27. Verli H, Calazans A, Brindeiro R, Tanuri A, and Guimarães JA. Molecular dynamics analysis of HIV-1 matrix protein: clarifying differences between crystallographic and solution structures. J Mol Graphics modelling. (2007) 26:62–8. doi: 10.1016/j.jmgm.2006.09.009
28. Marie V and Gordon ML. The HIV-1 gag protein displays extensive functional and structural roles in virus replication and infectivity. Int J Mol Sci. (2022) 23:7569. doi: 10.3390/ijms23147569
29. Caccuri F, Marsico S, Fiorentini S, Caruso A, and Giagulli C. HIV-1 Matrix Protein p17 and its Receptors. Curr Drug Targets. (2016) 17:23–32. doi: 10.2174/1389450116666150825110840
30. Dupont S, Sharova N, DéHoratius C, Virbasius CM, Zhu X, Bukrinskaya AG, et al. A novel nuclear export activity in HIV-1 matrix protein required for viral replication. Nature. (1999) 402(6762):681–5. doi: 10.1038/45272
31. Chiu HC, Huang WR, Wang YY, Li JY, Liao TL, Nielsen BL, et al. Heterogeneous nuclear ribonucleoprotein A1 and lamin A/C modulate nucleocytoplasmic shuttling of avian reovirus p17. J Virol. (2019) 93(20):e00851–19. doi: 10.1128/JVI.00851-19
32. Ohori Y, Okazaki H, Watanabe S, Tochio N, Arai M, Kigawa T, et al. Flexible and rigid structures in HIV-1 p17 matrix protein monitored by relaxation and amide proton exchange with NMR. Biochim Biophys Acta. (2014) 1844(3):520–6. doi: 10.1016/j.bbapap.2013.12.010
33. Dong X, Li H, Derdowski A, Ding L, Burnett A, Chen X, et al. Ap-3 directs the intracellular trafficking of HIV-1 Gag and plays a key role in particle assembly. Cell. (2005) 120:663–74. doi: 10.1016/j.cell.2004.12.023
34. Freed EO. HIV-1 assembly, release and maturation. Nat Rev Microbiol. (2015) 13:484–96. doi: 10.1038/nrmicro3490
35. Perlman M and Resh MD. Identification of an intracellular trafficking and assembly pathway for HIV-1 gag. Traffic. (2006) 7:731–45. doi: 10.1111/j.1398-9219.2006.00428.x
36. Wu Z, Alexandratos J, Ericksen B, Lubkowski J, Gallo RC, and Lu W. Total chemical synthesis of N-myristoylated HIV-1 matrix protein p17: structural and mechanistic implications of p17 myristoylation. Proc Natl Acad Sci U S A. (2004) 101:11587–92. doi: 10.1073/pnas.0404649101
37. Sudo S, Haraguchi H, Hirai Y, Gatanaga H, Sakuragi J, Momose F, et al. Efavirenz enhances HIV-1 gag processing at the plasma membrane through Gag-Pol dimerization. J Virol. (2013) 87(6):3348–60. doi: 10.1128/JVI.02306-12
38. Henrick BM, Yao XD, Rosenthal KL, and INFANT study team. HIV-1 structural proteins serve as PAMPs for TLR2 heterodimers significantly increasing infection and innate immune activation. Front Immunol. (2015) 6:426. doi: 10.3389/fimmu.2015.00426
39. Shen X and Tomaras GD. Alterations of the B-cell response by HIV-1 replication. Curr HIV/AIDS Rep. (2011) 8:23–30. doi: 10.1007/s11904-010-0064-2
40. Swingler S, Zhou J, Swingler C, Dauphin A, Greenough T, Jolicoeur P, et al. Evidence for a pathogenic determinant in HIV-1 Nef involved in B cell dysfunction in HIV/AIDS. Cell Host Microbe. (2008) 4(1):63–76. doi: 10.1016/j.chom.2008.05.015
41. De Francesco MA, Caruso A, Fallacara F, Canaris AD, Dima F, Poiesi C, et al. Hiv P17 enhances lymphocyte proliferation and HIV-1 replication after binding to a human serum factor. AIDS (London England). (1998) 12:245–52. doi: 10.1097/00002030-199803000-00001
42. Vitale M, Caruso A, De Francesco MA, Rodella L, Bozzo L, Garrafa E, et al. HIV-1 matrix protein P17 enhances the proliferative activity of natural killer cells and increases their ability to secrete proinflammatory cytokines. Br J haematology. (2003) 120:337–43. doi: 10.1046/j.1365-2141.2003.04053.x
43. Caccuri F, D’Ursi P, Uggeri M, Bugatti A, Mazzuca P, Zani A, et al. Evolution toward beta common chain receptor usage links the matrix proteins of HIV-1 and its ancestors to human erythropoietin. Proc Natl Acad Sci United States America. (2021) 118:e2021366118. doi: 10.1073/pnas.2021366118
44. Fiorentini S, Riboldi E, Facchetti F, Avolio M, Fabbri M, Tosti G, et al. HIV-1 matrix protein P17 induces human plasmacytoid dendritic cells to acquire a migratory immature cell phenotype. Proc Natl Acad Sci United States America. (2008) 105:3867–72. doi: 10.1073/pnas.0800370105
45. Caccuri F, Messali S, Zani A, Campisi G, Giovanetti M, Zanussi S, et al. HIV-1 mutants expressing B cell clonogenic matrix protein P17 variants are increasing their prevalence worldwide. Proc Natl Acad Sci United States America. (2022) 119:e2122050119. doi: 10.1073/pnas.2122050119
46. He W, Mazzuca P, Yuan W, Varney K, Bugatti A, Cagnotto A, et al. Identification of amino acid residues critical for the B cell growth-promoting activity of HIV-1 matrix protein P17 variants. Biochim Biophys Acta Gen Subj. (2019) 1863:13–24. doi: 10.1016/j.bbagen.2018.09.016
47. Giagulli C, Caccuri F, Zorzan S, Bugatti A, Zani A, Filippini F, et al. B-cell clonogenic activity of HIV-1 P17 variants is driven by PAR1-mediated EGF transactivation. Cancer Gene Ther. (2021) 28:649–66. doi: 10.1038/s41417-020-00246-9
48. Dolcetti R, Giagulli C, He W, Selleri M, Caccuri F, Eyzaguirre LM, et al. Role of HIV-1 matrix protein P17 variants in lymphoma pathogenesis. Proc Natl Acad Sci United States America. (2015) 112:14331–6. doi: 10.1073/pnas.1514748112
49. Giagulli C, D’Ursi P, He W, Zorzan S, Caccuri F, Varney K, et al. A single amino acid substitution confers B-cell clonogenic activity to the HIV-1 matrix protein P17. Sci Rep. (2017) 7:6555. doi: 10.1038/s41598-017-06848-y
50. Giagulli C, Marsico S, Magiera AK, Bruno R, Caccuri F, Barone I, et al. Opposite effects of HIV-1 P17 variants on PTEN activation and cell growth in B cells. PloS One. (2011) 6:e17831. doi: 10.1371/journal.pone.0017831
51. Lu J, Jia J, Zhang J, and Liu X. HIV P17 Enhances T Cell Proliferation by Suppressing Autophagy through the P17-Ola1-Gsk3β Axis under Nutrient Starvation. J Med Virol. (2021) 93:3607–20. doi: 10.1002/jmv.26423
52. Morimoto K and Nakajima K. Role of the immune system in the development of the central nervous system. Front Neurosci. (2019) 13:916. doi: 10.3389/fnins.2019.00916
53. Moro-García MA, Mayo JC, Sainz RM, and Alonso-Arias R. Influence of inflammation in the process of T lymphocyte differentiation: proliferative, metabolic, and oxidative changes. Front Immunol. (2018) 9:339. doi: 10.3389/fimmu.2018.00339
54. Li S, Bozzo L, Wu Z, Lu W, and Romerio F. The HIV-1 matrix protein P17 activates the transcription factors C-Myc and CREB in human B cells. New microbiologica. (2010) 33:13–24.
55. Martorelli D, Muraro E, Mastorci K, Dal Col J, Faè DA, Furlan C, et al. A natural HIV P17 protein variant up-regulates the LMP-1 EBV oncoprotein and promotes the growth of EBV-infected B-lymphocytes: implications for EBV-driven lymphomagenesis in the HIV setting. Int J Cancer. (2015) 137:1374–85. doi: 10.1002/ijc.29494
56. Selleri M, Dolcetti R, Caccuri F, Giombini E, Rozera G, Abbate I, et al. In-depth analysis of compartmentalization of HIV-1 matrix protein P17 in PBMC and plasma. New microbiologica. (2017) 40:58–61.
57. Mazzuca P, Marsico S, Schulze K, Mitola S, Pils MC, Giagulli C, et al. Role of autophagy in HIV-1 matrix protein P17-driven lymphangiogenesis. J Virol. (2017) 91:10-1128. doi: 10.1128/jvi.00801-17
58. Caccuri F, Muraro E, Gloghini A, Turriziani O, Riminucci M, Giagulli C, et al. Lymphomagenic properties of a HIV P17 variant derived from a splenic marginal zone lymphoma occurred in a HIV-infected patient. Hematological Oncol. (2019) 37:176–84. doi: 10.1002/hon.2562
59. Zani A, Messali S, Bugatti A, Uggeri M, Rondina A, Sclavi L, et al. Molecular mechanisms behind the generation of pro-oncogenic HIV-1 matrix protein P17 variants. J Gen Virol. (2024) 105:001982. doi: 10.1099/jgv.0.001982
60. Fiorentini S, Marini E, Caracciolo S, and Caruso A. Functions of the HIV-1 matrix protein P17. New microbiologica. (2006) 29:1–10.
61. Caccuri F, Neves V, Gano L, Correia JDG, Oliveira MC, Mazzuca P, et al. The HIV-1 matrix protein P17 does cross the blood-brain barrier. J Virol. (2022) 96:e0120021. doi: 10.1128/jvi.01200-21
62. Liu D, Zeinolabediny Y, Caccuri F, Ferris G, Fang WH, Weston R, et al. P17 from HIV induces brain endothelial cell angiogenesis through EGFR-1-mediated cell signalling activation. Lab investigation; A J Tech Methods Pathol. (2019) 99:180–90. doi: 10.1038/s41374-018-0147-z
63. Basta D, Latinovic O, Lafferty MK, Sun L, Bryant J, Lu W, et al. Angiogenic, lymphangiogenic and adipogenic effects of HIV-1 matrix protein P17. Pathog Dis. (2015) 73:ftv062. doi: 10.1093/femspd/ftv062
64. Renga B, Francisci D, Schiaroli E, Carino A, Cipriani S, D’Amore C, et al. The HIV matrix protein P17 promotes the activation of human hepatic stellate cells through interactions with CXCR2 and syndecan-2. PloS One. (2014) 9:e94798. doi: 10.1371/journal.pone.0094798
65. Bugatti A, Marsico S, Mazzuca P, Schulze K, Ebensen T, Giagulli C, et al. Role of autophagy in von willebrand factor secretion by endothelial cells and in the in vivo thrombin-antithrombin complex formation promoted by the HIV-1 matrix protein P17. Int J Mol Sci. (2020) 21:2022. doi: 10.3390/ijms21062022
66. Giagulli C, Magiera AK, Bugatti A, Caccuri F, Marsico S, Rusnati M, et al. HIV-1 matrix protein P17 binds to the IL-8 receptor CXCR1 and shows IL-8-like chemokine activity on monocytes through Rho/ROCK activation. Blood. (2012) 119:2274–83. doi: 10.1182/blood-2011-06-364083
67. Feng Y, Wang Z, Zeng D, Song S, Yang Y, Wang A, et al. High expression of HIV-1 matrix protein P17 in both lymphoma and lymph node tissues of aids patients. Pathology Res Pract. (2022) 237:154061. doi: 10.1016/j.prp.2022.154061
68. Caccuri F, Giagulli C, Bugatti A, Benetti A, Alessandri G, Ribatti D, et al. HIV-1 matrix protein P17 promotes angiogenesis via chemokine receptors CXCR1 and CXCR2. Proc Natl Acad Sci United States America. (2012) 109:14580–5. doi: 10.1073/pnas.1206605109
69. Renga B, Francisci D, D’Amore C, Schiaroli E, Mencarelli A, Cipriani S, et al. The HIV matrix protein P17 subverts nuclear receptors expression and induces a stat1-dependent proinflammatory phenotype in monocytes. PloS One. (2012) 7:e35924. doi: 10.1371/journal.pone.0035924
70. Caccuri F, Giordano F, Barone I, Mazzuca P, Giagulli C, Andò S, et al. HIV-1 matrix protein P17 and its variants promote human triple negative breast cancer cell aggressiveness. Infect Agents Cancer. (2017) 12:49. doi: 10.1186/s13027-017-0160-7
71. Renga B, Francisci D, Carino A, Marchianò S, Cipriani S, Chiara Monti M, et al. The Hiv matrix protein P17 induces hepatic lipid accumulation via modulation of nuclear receptor transcriptoma. Sci Rep. (2015) 5:15403. doi: 10.1038/srep15403
Keywords: human immunodeficiency virus (HIV), matrix protein p17, lymphoma, cell proliferation, tumor microenvironment
Citation: Zhang Y, Li M, Lin F, Guo R, Wu D, Shen R, Harypursat V, Chen Y and Chen Y (2025) HIV-1 matrix protein p17: a key factor in HIV-associated cancers. Front. Virol. 5:1584507. doi: 10.3389/fviro.2025.1584507
Received: 01 March 2025; Accepted: 23 April 2025;
Published: 15 May 2025.
Edited by:
Moises Leon Juarez, Instituto Nacional de Perinatología (INPER), MexicoReviewed by:
Ayantika Sen, Stanford University, United StatesVictor Javier Cruz Holguin, National Polytechnic Institute of Mexico (CINVESTAV), Mexico
Copyright © 2025 Zhang, Li, Lin, Guo, Wu, Shen, Harypursat, Chen and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yaokai Chen, eWFva2FpY2hlbkBob3RtYWlsLmNvbQ==; Yemiao Chen, eWVtaWFvY2hlbkAxMjYuY29t