 Tabitha Wanjiru1,2,3*
Tabitha Wanjiru1,2,3* Wallace Bulimo2
Wallace Bulimo2 Solomon Langat2Johnson Kinyua3Santos Yalwala1Gladys Kerich1Janet Ambale1Eric Garges1Robert Haynes1Gerald Kellar1John Eads1Fredrick Eyase1,2
Solomon Langat2Johnson Kinyua3Santos Yalwala1Gladys Kerich1Janet Ambale1Eric Garges1Robert Haynes1Gerald Kellar1John Eads1Fredrick Eyase1,2- 1Department of Emerging Infectious Diseases, Walter Reed Army Institute of Research-Africa, Nairobi, Kenya
- 2Centre for Virus Research, Kenya Medical Research Institute, Nairobi, Kenya
- 3Department of Biochemistry, Jomo Kenyatta University of Agriculture and Technology (JKUAT), Nairobi, Kenya
Introduction: Alphaviruses, members of the family Togaviridae, are globally distributed arthropod-borne viruses, including several medically significant pathogens such as Chikungunya virus and Sindbis virus. These viruses are primarily transmitted by mosquitoes and can cause a wide range of diseases in humans and animals, from febrile illnesses to neurological conditions. While many alphaviruses have been extensively studied, others, like Ndumu virus (NDUV), remain poorly understood. Surveillance of these viruses, is essential for monitoring their circulation within mosquito populations, enabling early detection and informed public health responses. This study sought to conduct comprehensive surveillance of pathogenic viruses in mosquito populations in North-eastern and coastal Kenya, during which NDUV was detected and characterized.
Methods: Adult mosquitoes were collected from Kwale, Malindi, Mombasa, and Isiolo counties using light traps. A total of 14,106 mosquitoes were grouped into 1,597 pools by species and geographic origin. Cell culture was conducted using Vero E6 cell line, followed by RNA extraction, library preparation, and sequencing with the Illumina MiSeq platform. Preliminary analysis was performed on the CZ-ID platform. Raw sequence reads underwent quality control with PrinseqLite v0.20.4 to remove adapters and filter low-quality reads. De Novo assembly was conducted using MEGAHIT v1.2.9, and final contigs were analyzed using BLASTn against the NCBI nucleotide (nt) database. Phylogenetic analysis was performed using the Maximum Likelihood method.
Results: Two pools from Malindi and one pool from Kwale showed cytopathic effects in four subsequent passages. NDUV was confirmed in pools with Culex annulioris, Mn. Africana and Culex univittatus species. Genomic analysis revealed lengths of 11,632, 11,646 and 11,646 nucleotides for the isolates. Phylogenetic analysis placed these isolates within the alphavirus clade, closely related to other NDUV strains.
Discussion: This detection underscores the importance of routine arbovirus surveillance to better understand the circulation and dynamics of neglected viruses like NDUV within endemic regions. Even though there exists serological evidence of exposure in humans, NDUV has not been directly linked to human or animal disease. The current data raises questions about its zoonotic potential and possible role as an emerging pathogen.
1 Introduction
Emerging and neglected arboviruses represent a persistent public health threat, particularly in regions with high mosquito biodiversity and limited surveillance. Arboviral diseases, including those caused by mosquito borne viruses, are common in tropical and subtropical regions where competent vector populations are abundant (1). Among these, alphaviruses are a genus of viruses within the Togaviridae family that include Chikungunya virus, Ross River virus, Mayaro virus, and Sindbis virus. These have a broad geographic distribution, ranging across the Americas, Africa, Asia, and the Pacific Islands (2). They replicate in both invertebrate hosts (mostly mosquitoes) and vertebrate hosts (e.g., birds, equids, amphibians, reptiles, rodents, swine, humans, and other primates). Aedes and Culex mosquitoes are the main vectors of alphaviruses (3). Some of these viruses are of great concern due to their ability to cause large outbreaks, particularly in areas with high mosquito populations (4). Alphavirus infections often result in clinical symptoms such as fever, rash, and arthralgia, but the severity of the disease can vary widely, with some cases presenting with more serious neurological or hemorrhagic outcomes.
NDUV, which was first isolated in 1959 from Mansonia uniformis mosquitoes in South Africa (5), has since been detected across several African countries, including Uganda and Kenya (6, 7). Recent findings have highlighted the broad host and vector range of NDUV, with detections in diverse mosquito species, such as Culex pipiens (8), Culex rubinotus, Aedes mcintoshi, Ae. Ochraceous, and Ae. tricholabis (6). In particular, studies in Kenya have revealed the virus’s presence in both rural and peri-urban mosquito populations, suggesting its widespread circulation. In addition to its detection in mosquitoes, NDUV has been identified in vertebrate hosts (9). It has been isolated from a domestic pig and bovine calf in Uganda and Kenya respectively (7, 10). In another study involving ticks, Ndumu virus (NDUV) was isolated from Rhipicephalus pulchellus species collected from both cattle and warthogs (9). Although serologic evidence for human infection has been demonstrated, there is no evidence supporting morbidity (10). However, its phylogenetic relationship with pathogenic alphaviruses highlights the potential for zoonotic transmission.
The role of Culex mosquitoes in NDUV transmission is particularly significant, as these mosquitoes are well-documented vectors for various arboviruses, including West Nile virus (WNV) and Japanese encephalitis virus (JEV). Studies have demonstrated the presence of NDUV in Culex pipiens, a widely distributed species known for its adaptability to urban and peri-urban environments (8), as well as in Culex rubinotus (6). The detection of Ndumu virus (NDUV) in Culex annulioris and Culex univittatus highlights the broad diversity of mosquito vectors potentially involved in its transmission. Culex annulioris, a species widely distributed across sub-Saharan Africa, is frequently associated with rural environments, including rice paddies, swamps, and irrigation systems (11). This species is recognized as a competent vector for several arboviruses, including Rift Valley fever virus (RVFV) and West Nile virus (WNV), due to its opportunistic feeding behaviour on both avian and mammalian hosts (12). Such feeding plasticity increases the likelihood of zoonotic spill over events, making Culex annulioris an important bridge vector in arbovirus transmission cycles. Similarly, Culex univittatus has been implicated in the transmission of WNV and Sindbis virus, with its widespread presence in urban, peri-urban, and rural habitats contributing to its epidemiological significance (13). This species exhibits a mixed feeding preference for birds and mammals, facilitating viral amplification within avian reservoirs while also posing a risk for human infections (14). Given their ability to act as bridge vectors, both Culex annulioris and Culex univittatus may play critical roles in the maintenance and transmission of NDUV in enzootic and epidemic settings.
Despite the broad geographic and ecological distribution, the clinical impact of NDUV remains poorly understood. While it has not been directly associated with significant human morbidity, the limited availability of diagnostic tools and the potential for subclinical infections suggest that the virus’s public health impact may be underestimated. This study aimed to conduct comprehensive surveillance of mosquito populations in North-eastern and coastal Kenya. By identifying the mosquito species that may harbor NDUV, assessing the prevalence of the virus, and exploring its potential vertebrate hosts, this study contributes to a deeper understanding of the ecological and epidemiological characteristics of this alphavirus.
2 Materials and methods
2.1 Ethical approval
Ethical approval was obtained from the Kenya Medical Research Institute (KEMRI) Scientific and Ethics Review Unit (SERU) under protocol number KEMRI/SERU/CCR/4702 and WRAIR# 3101. Permission to conduct the study was granted by the National Council for Science, Technology, and Innovation (NACOSTI).
2.2 Study area
The study was carried out in four study areas, Kilifi County (Malindi) (3.2192°S, 40.1169°E), Kwale (4.1730°S, 39.4520°E), Mombasa (4.0435°S, 39.6682°E), and Isiolo county (0.3546°N, 37.5822°E) (Figure 1).
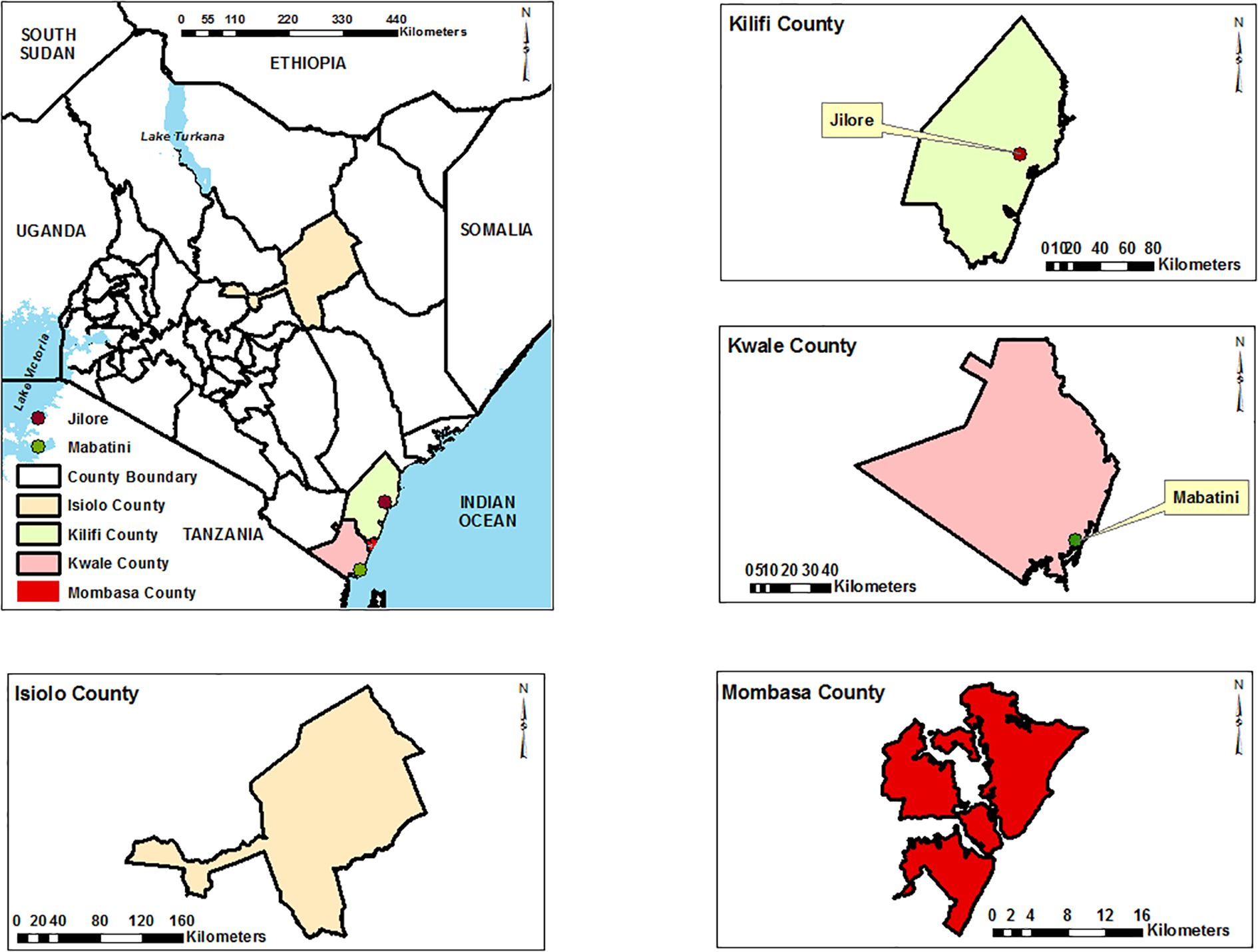
Figure 1. Map of coastal Kenya highlighting the mosquito sampling sites. Ndumu virus positive isolates were from samples collected in Jirole (Malindi) and Mabatini (Kwale). Base maps, boundaries and shape files of Kenyan map and administrative boundaries of the Counties were derived from GADM data version 4.1 (https://gadm.org) and the maps were generated using ArcGIS Version 10.2.2 (http://desktop.arcgis.com/en/arcmap) advanced license) courtesy of Samuel Owaka.
2.3 Entomological investigation
Sampling of adult mosquitoes was conducted between 18th June and 10th December 2023. Mosquitoes were collected using CDC miniature light traps (Model 512, John Hock Co., Gainesville, Florida, USA). Twelve (12) traps baited with carbonated dry ice (CO2) were deployed overnight (6pm-6am) in favorable habitat, including animal sheds and termite mounds. The samples were linked to the sites by geo-coding using a GPS. Captured mosquitoes were temporarily immobilized using triethylamine, preserved in liquid nitrogen, and transported to the laboratory at the Kenya Medical Research Institute in Kisumu, where they were stored at-80˚C until further processing. The mosquitoes were identified morphologically to species under dissecting microscope using taxonomy keys, including Edwards (1941) (15), Harbach (1988) (16) and Jupp (1986) (17). The identified mosquitoes were pooled in groups of 1 to 25 samples based on species, sex and collection site and stored at -80 degrees Celsius. In this study, mosquitoes were not pooled by abdominal status (i.e., unfed, blood-fed, gravid), this decision was based on the primary objective of virome detection and genomic characterization.
2.4 Mosquito sample preparation
One thousand five hundred ninety-six (1,596) mosquito pools were homogenized using a Mini-Beadruptor-16 (Biospec, Bartlesville, OK, USA) in 1000 µL of homogenization media (minimum essential media supplemented with 15% fetal Bovine Serum (FBS) (Gibco by Life Technologies, Grand Island, NY, USA), 2% L-glutamine (Sigma, Aldrich)and 2% antibiotic/antimycotic (Gibco by Life Technologies, Grand Island, NY, USA.) with zirconium beads (2.0 mm diameter) for 40 seconds. Subsequently, the homogenate was centrifuged at 14,000 rpm for 15 minutes at 4°C using a benchtop centrifuge (Eppendorf, USA). The supernatant was collected and inoculated into Vero cells while maintaining cold chain throughout the process.
2.5 Viral isolation
Virus isolation was performed in Vero (African green monkey kidney) cells (E6). Briefly, Vero cells were grown overnight at 37°C and 5% CO2 in minimum essential medium supplemented with 2% glutamine, 2% penicillin/streptomycin/amphotericin, 10% fetal bovine serum, and 7.5% NaHCO3 in 24-well plates (Corning, Incorporated). At 80% confluence, a 50 µL aliquot of the clarified supernatant from individual pools was inoculated into the wells. The plates were incubated for 1 hour in a humidified incubator at 37°C and 5% CO2 with gentle rocking of the plates every 15 minutes for virus adsorption. Following incubation, 1 mL of maintenance medium, comprising minimum essential medium supplemented with 2% glutamine, 2% penicillin/streptomycin/amphotericin, 2% fetal bovine serum, and 7.5% NaHCO3, was added. The plates were then cultured at 37°C and 5% CO2 and monitored daily for cytopathic effects (CPE) for 14 days. Cultures exhibiting CPE were harvested and further passaged by inoculating onto fresh monolayers of Vero cells (CCL 81™) in 25-cm3 cell culture flasks. After three successive passages, the supernatants of virus-infected Vero cell cultures exhibiting cytopathic effect of approximately 70% were harvested from the flasks for virus identification through next-generation sequencing.
2.6 Library preparation and next generation sequencing
Viral particles from CPE-positive cultures were recovered through 0.22µm filters (Millipore, Merck). From an aliquot of 140 μl of the supernatant, viral RNA was extracted using the QIAamp Viral RNA Mini Kit (Qiagen, Hilden, Germany) and eluted in one step with 60 μl of elution buffer. Paired-end libraries for high-throughput sequencing were prepared using Illumina Ribo-Zero Plus rRNA Depletion (Illumina, USA). The libraries were prepared following the manufacturer’s recommended protocol. The final library was denatured with NaOH and diluted further to 10pM before loading onto the Illumina MiSeq sequencing machine. Sequencing was performed using MiSeq Reagent V3 (Illumina, USA), in a 300-paired end cycle sequencing format.
2.7 Sequence analysis and virus identification
Initial analysis was performed using the CZ-ID platform, an integrated pipeline that offers quality control, de-hosting, duplicate removal, assembly, and viral identification capabilities. After this initial processing, the de-hosted sequence reads were retrieved for further analysis. To validate CZ-ID pipeline results, PrinseqLite v0.20.4 tool was used to filter low-quality reads and remove adapters on command line. De novo sequence assembly was conducted using MEGAHIT v1.2.9 (18) where Ndumu virus contigs were recovered and compared to CZ-ID pipeline results. This was followed by mapping the reads back to the generated contigs using BWA to produce SAM files. The subsequent generation of BAM files, along with sorting, indexing, and filtering of contigs that did not meet the specified read mapping thresholds, was handled using Samtools v1.20. Only contigs with an average depth of coverage of ≥10 and a length of ≥500 bp were retained for further analysis. These contigs were first compared against a local version of NCBI viral database using Diamond v2.0.4. To ensure specificity, putative viral contigs were further compared to the entire non-redundant protein database (nr), to exclude any non-viral contigs. A stringent e-value threshold of 1e-5 was employed throughout the homology searches to minimize false-positive hits.
2.8 Phylogenetic analysis of the identified RNA viruses
To describe the identified viruses in an evolutionary context, publicly available viruses belonging to these different groups, and more specifically those closely related to the viral strains obtained in the current study were downloaded and used as reference sequences in the reconstruction of phylogenetic trees. Closely related gene sequences were retrieved from NCBI viral database and used as reference sequences in reconstructing the phylogenetic relationship of the viral sequences. The combined set of sequences were aligned using MUSCLE software with default parameters (max iterations = 16) embedded in Molecular Evolutionary Genetics Analysis v.7.0 (MEGA7) (19) platform. The aligned sequences were edited using the Bioedit tool and maximum likelihood phylogenetic analysis carried out using IQ-TREE v1.6.12. The best model (GTR+G4 (General Time Reversible + Gamma)) and tree search was performed simultaneously based on 1000 bootstrap estimates and approximate likelihood ratio test (aLRT).
2.9 Species confirmation of mosquito pools
COI gene amplification and sequencing were conducted to confirm the morphological identification of mosquito species in each pool. DNA extraction was performed on the bulk pools of the crude homogenates using a QIAamp DNA extraction kit (Qiagen, Germany), according to the manufacturer’s instructions. Two extraction blanks were included during the extraction process and subsequently used during PCR and sequencing. The extracted DNA was quantified using a Qubit fluorometer 2.0. Amplification of the COI gene was then carried out on <1µg of extracted DNA, using the universal pair of primers for metazoan invertebrates LCO1490/HCO2198 (20). These primers amplify an approximately 710-bp region of the COI gene of arthropod vectors. COI amplicons were generated from a 25µL PCR containing 12.5 µl AmpliTaq Gold 360 master mix (Applied Biosystems, USA), 9.5µl DNase/RNase-free water, and 0.5µl each of the forward and reverse primers at 25µM. The PCR cycling conditions were set as follows; initial denaturation at 95°C for 10 min, 35 cycles of 95°C for 30 s, 49°C for 30 s, 72°C for 30 s, and a final extension of 72°C for 10 min. The COI amplicons were first purified using AMPure XP beads (Beckman Coulter, USA). The purified products were quantified using a Qubit dsDNA HS assay kit (Invitrogen, USA) with a Qubit fluorometer 2.0. Based on the concentration of the quantified products, the volume of PCR products that yielded 200 fmol was determined and used as starting material for MinION library preparation. Library preparation was carried out using a ligation sequencing kit (SQK-LSK114), following the manufacturers protocol with the exclusion of the DNA fragmentation step. Briefly, 200 fmol of the purified products were end repaired using a NEBNext Ultra II end repair and dA-tailing module (New England Biolabs [NEB], UK). The end-repaired DNA for each sample was individually barcoded using Native Barcoding Expansion 1-12 (EXP-NBD114), which was achieved with the use of NEB Blunt/TA ligase master mix (NEB, UK). An equal amount from each of the 200-fmol barcoded libraries was combined into a single pool, which was then purified with AMPure XP beads (Beckman Coulter, USA). Adapter ligation of the purified library was done with NEBNext quick ligation module (NEB, UK) and the libraries were further purified using AMPure XP beads, with a final wash of the beads being carried out using short fragment buffer (SFB) provided with the SQK-LSK114 kit. The final library was loaded onto the flow cell (FLO-MIN106D) and sequenced using the workflows provided in the MinKNOW software. Raw reads were processed in real-time using the EPI2ME platform (Run on cloud), which performed base-calling with Guppy (v4.2.2), quality filtering (minimum Q-score 7), and taxonomic classification using the ‘What’s in my Pot’ (WIMP) workflow. Taxonomic assignments were validated by BLAST searches against the NCBI nucleotide database and cross-checked with the Barcode of Life Data System (BOLD) for species confirmation. Only sequences with ≥98% identity and ≥90% query coverage were considered reliable identifications.
2.10 Genetic distance analysis
To assess the genetic distance between different strains of Ndumu Virus and alphaviruses, we used the BioPython software v1.85 to calculate pairwise genetic distances based on the nucleotide sequences. The distances were computed using the Kimura 2-parameter model, p-distance. A pairwise distance matrix was constructed from the calculated genetic distances. The matrix was visualized as a heatmap using Seaborn (v0.11.0) in Python.
3 Results
3.1 Ndumu virus isolation on Vero cells
A total of one thousand five hundred ninety-six (1,596) mosquito pools were processed for cell culture, three pools originating from Malindi and Kwale exhibited cytopathic effects (CPE) on day 6 post inoculation (Table 1). Ndumu virus was confirmed in Culex annulioris, Mn. africana and Culex univittatus mosquito species. The remaining 1,593 pools did not show any CPE across four blind passages and were considered negative using this approach (Supplementary Material S1).
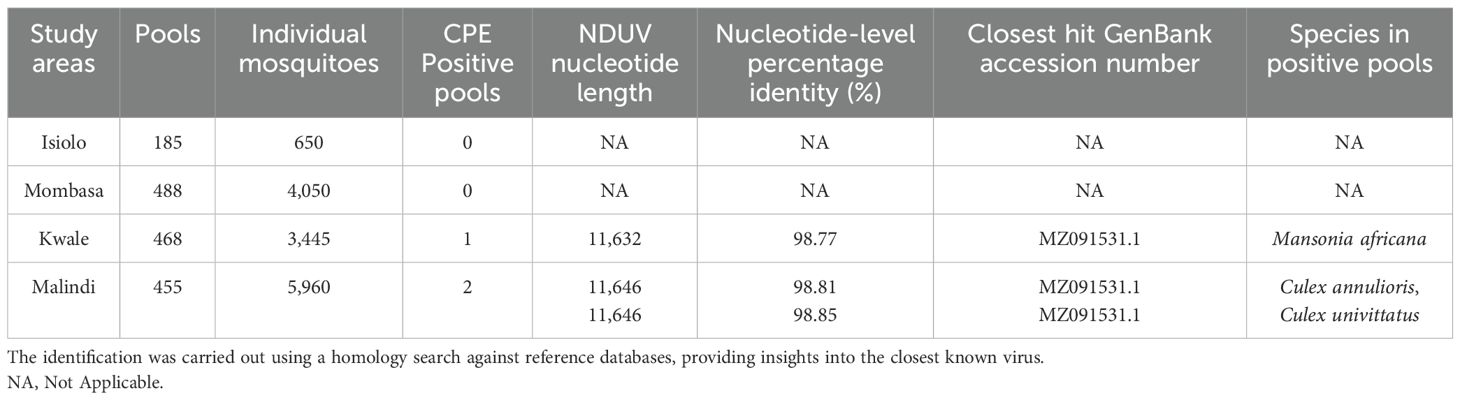
Table 1. Viruses identified in the study and their identification details based on sequence analysis.
3.2 Phylogenetic relationships of identified NDUV sequences
Phylogenetic analysis was conducted to characterize the Ndumu virus (NDUV) strains isolated from mosquito pools in this study and to determine their evolutionary relationships with other alphaviruses. The three Kenyan NDUV isolates formed a well-supported monophyletic clade closely related to a strain previously isolated from domestic pigs in Uganda, suggesting regional circulation and evolutionary divergence. The broader analysis confirmed that the NDUV strains clustered distinctly from Middelburg virus and other members of the Semliki Forest antigenic complex, while remaining within the Old World alphavirus clade (Figure 2).

Figure 2. Maximum likelihood phylogenetic tree constructed using nucleotide sequences of Ndumu virus (NDUV) isolates obtained in this study with Alphavirus sequences retrieved from GenBank. Bootstrap support values (1,000 replicates) are indicated at the branch nodes. Isolates detected in this study are highlighted in blue.
3.3 Metabarcoding analysis
The COI sequence analysis provided a molecular identity for the mosquito specimens. These results were then compared to the species identity provided using morphologically based taxonomic keys (Table 2). The results obtained showed that all the species that were collected in this study had concurring morphological and molecular identities.
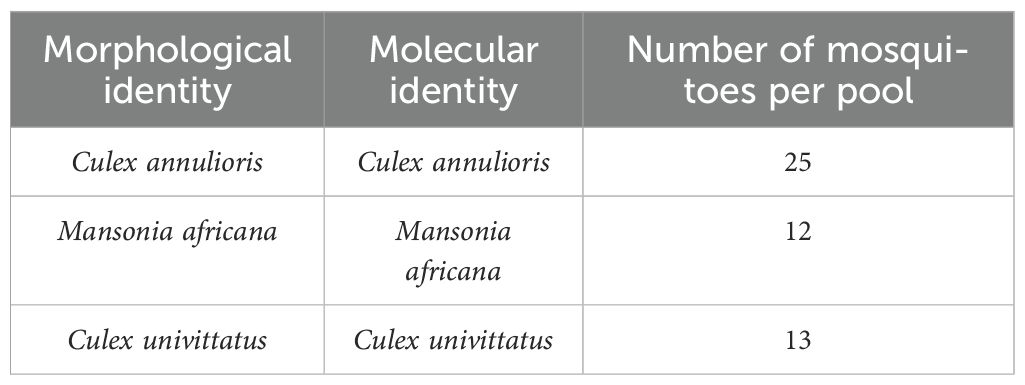
Table 2. Morphological and COI molecular identification of mosquito species in which Ndumu virus was isolated and corresponding pool sizes.
3.4 Genetic distance
To determine the degree of similarity and divergence, a genetic heat map was generated to visually represent pairwise genetic distances between the isolates and reference sequences (Figure 3).
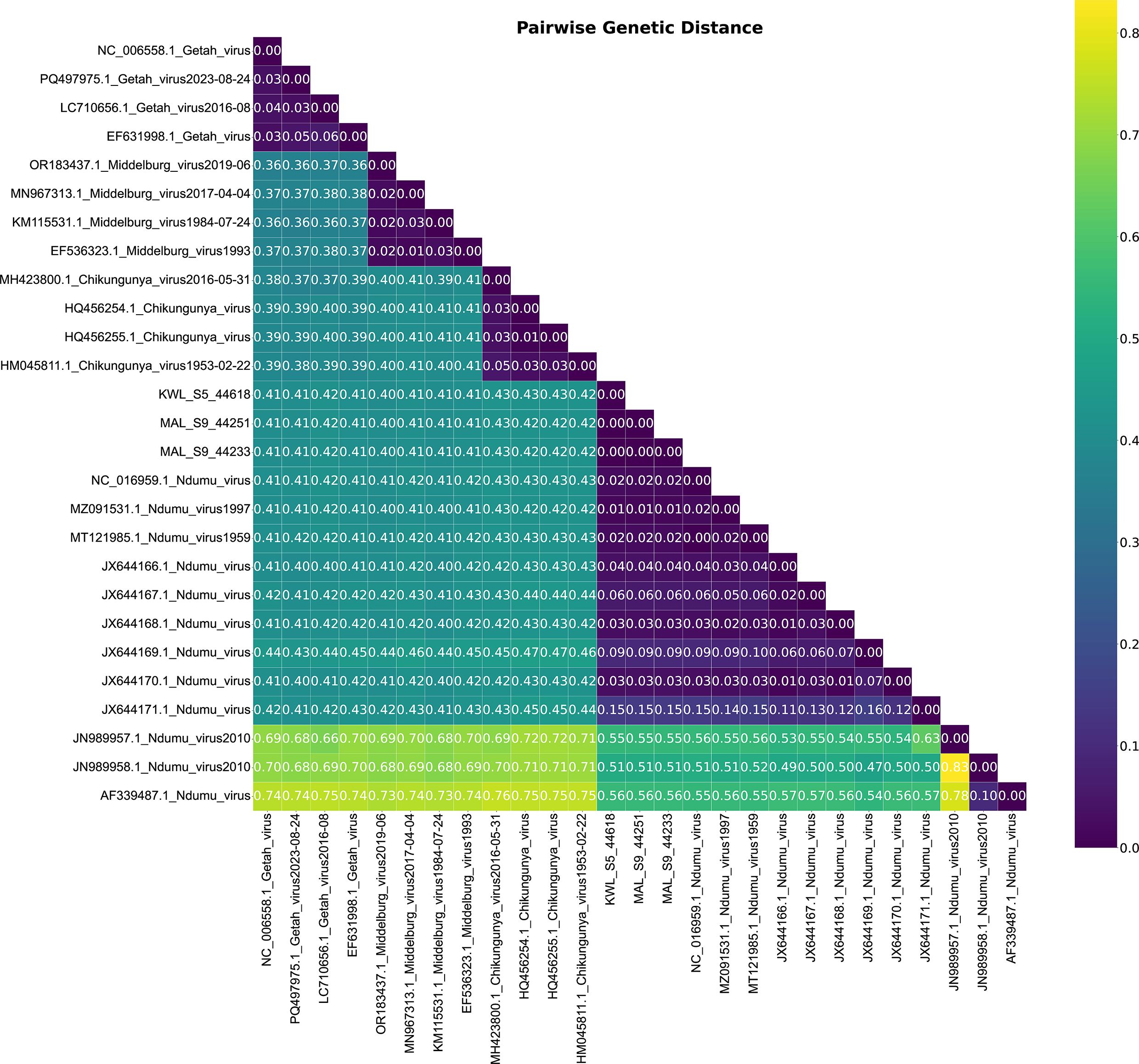
Figure 3. Pairwise genetic distance heatmap of Ndumu Virus and Alphavirus strains. The analysis highlights the genetic relationships between the strains, providing insights into their evolutionary distance.
Heatmap analysis based on pairwise p-distances of full genome sequences revealed a clear clustering pattern among Ndumu virus (NDUV) isolates and distinct separation from other alphaviruses. The intra-group distances among the NDUV isolates ranged from 0.01 to 0.15, indicating high sequence similarity, with most isolates showing minimal divergence (0.01–0.06), except for one JX644171.1 notably divergent isolate with a distance of 0.15. In contrast, distances between the NDUV isolates and other alphaviruses such as Middelburg virus, Getah virus, and Chikungunya virus (CHIKV) were substantially higher, ranging from 0.400 to 0.430. Specifically, Middelburg virus showed distances of 0.400–0.410, while Getah virus and CHIKV ranged between 0.410–0.430. Consistent with other alphaviruses, the genome organization of NDUV retained the canonical arrangement of non-structural and structural proteins, with conserved functional domains, further supporting the biological stability of NDUV despite its genetic distinctiveness (Figure 4).
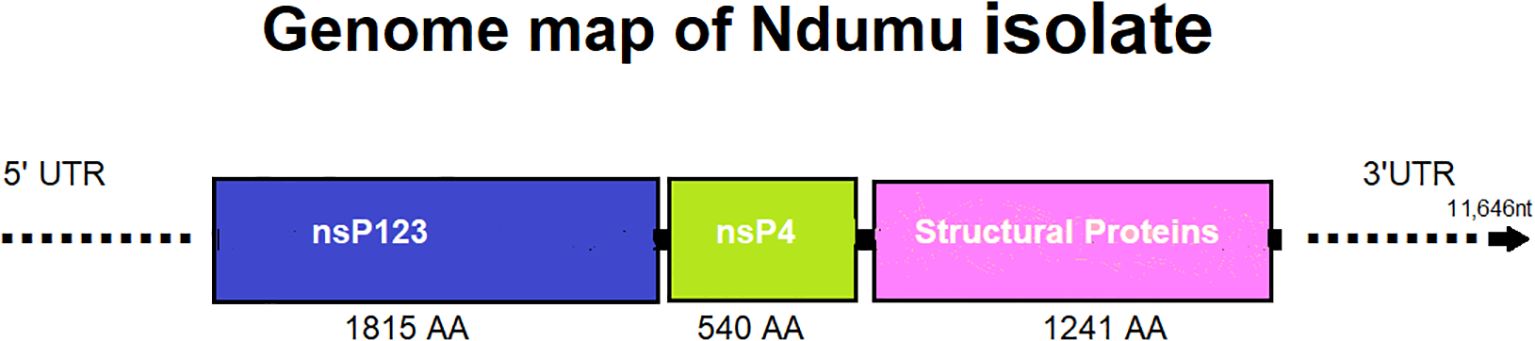
Figure 4. Genome organization of a representative Ndumu virus isolate (11,646 nt length), illustrating the typical structure observed across all detected isolates. The genome includes regions encoding non-structural proteins (nsP1–nsP3, nsP4) and the structural protein region.
4 Discussion
This study confirms the circulation of Ndumu virus (NDUV) in mosquito vectors in coastal Kenya, expanding its known geographic range and underscoring its potential public health relevance. Detection of NDUV in Culex annulioris, Mansonia africana, and Culex univittatus suggests a broader vector range than previously recognized, raising important questions regarding the roles these mosquito species may play in the maintenance and transmission of the virus. The confirmation of NDUV in three mosquito pools exhibiting cytopathic effects (CPE) in Vero cells provides evidence of active viral replication and suggests the potential for vertebrate infectivity. In contrast, the absence of CPE in the remaining 1,593 pools may reflect the absence of virus, the presence of non-cytolytic viruses, or viruses present at low titers undetectable by cell culture alone. These findings highlight the limitations of CPE-based surveillance methods and underscore the importance of integrating complementary molecular approaches such as untargeted metagenomic sequencing to improve sensitivity and comprehensiveness in arbovirus surveillance.
Our detection of NDUV in multiple mosquito species with diverse feeding behaviors, including Culex pipiens, Aedes aegypti, Anopheles funestus, and Eretmapodites chrysogaster, suggests a broad ecological adaptability of the virus within vector populations. Several of these species are known to feed on both humans and domestic animals, raising the possibility of cross-species transmission and involvement of vertebrate hosts in the virus’s life cycle. Notably, NDUV was previously isolated from domestic pigs in Uganda (7), and experimental infections in mice and non-human primates have shown variable pathogenicity and viremia (21), supporting the potential for vertebrate involvement including goats, cattle and warthogs (9, 22). Additionally, prior documentation of vertical transmission in mosquitoes (8) may explain the sustained circulation of the virus even in the absence of vertebrate involvement. Together, our findings point to a complex ecological system in which NDUV may be maintained through both horizontal (vector-to-host) and vertical (mosquito-to-offspring) transmission, warranting further studies to characterize the role of domestic and wild vertebrates in the natural transmission cycle.
Phylogenetic analysis revealed that the three Kenyan Ndumu virus (NDUV) isolates form a well-supported monophyletic clade, clustering closely with an NDUV strain previously isolated from domestic pigs in Uganda (7), suggesting regional circulation and possible host adaptation. While our sequences clearly diverge from other alphaviruses, they remain within the Old World alphavirus lineage. Interestingly, both our study and previous work by (3) in South Africa show NDUV forming a sister clade to Middelburg virus, reinforcing the notion of shared ancestry within the Semliki Forest antigenic complex. Contrastingly, work by (6) suggested a potential branching of NDUV from Chikungunya virus; however, our broader phylogenetic reconstruction, including full genome alignments and robust bootstrap support, places NDUV distinctly from both Chikungunya and Middelburg viruses. Collectively, these findings support the genetic distinctiveness of Kenyan NDUV strains and highlight their evolutionary divergence, possibly shaped by geographic, ecological, or host-associated pressures.
COI-based metabarcoding provided reliable molecular confirmation of mosquito species, supplementing morphological identification and ensuring accurate vector-virus associations. Although this approach can also detect non-mosquito COI sequences from bloodmeals (23), no vertebrate sequences were recovered in our study. This is due to our sample preparation strategy, which prioritized whole mosquitoes for viral detection rather than dissected, blood-fed individuals.
The heatmap analysis based on pairwise distances provides critical insight into the genetic diversity and evolutionary relationships of the Ndumu virus (NDUV) isolates. The low intra-species distances (0.01–0.06) among most isolates suggest recent common ancestry and indicate that these viruses may be circulating within a relatively localized ecological niche or transmission cycle. The one isolate with a distance of 0.15, while still within species-level variation, may represent a more divergent lineage or reflecting natural evolutionary drift. Importantly, the consistently high divergence observed between the NDUV isolates and other alphaviruses, particularly Chikungunya virus (0.410–0.430) and Getah virus (0.410–0.420), underscores the clear phylogenetic distinction between NDUV and members of the Semliki Forest antigenic complex (24). Middelburg virus, which exhibited slightly lower divergence (0.400–0.410), may share an older common ancestor with NDUV, supporting previous phylogenetic observations that place these two viruses within a broader clade of Old World alphaviruses (2, 25).
The genome organization of the Ndumu virus (NDUV) isolates in this study follows the canonical structure observed in alphaviruses, comprising nonstructural proteins (nsPs) followed by structural proteins. Specifically, the viral genome encodes nsp1, nsp2, and nsp3 as part of a large replicase polyprotein (P123), with nsp4 expressed via a read through mechanism from an opal stop codon (26). Functionally, nsp1 is responsible for capping viral RNAs and anchoring the replication complex to intracellular membranes (27). Protein nsp2 has helicase and protease activity and is crucial for polyprotein processing and host shutoff (28), nsp3, although less well understood, is thought to contribute to replication complex formation and host interaction, often containing hypervariable domains associated with host range (29). The nsp4 encodes the RNA-dependent RNA polymerase (RdRp), essential for viral genome replication and subgenomic RNA synthesis (30). The structural polyprotein encodes the capsid (C), E3, E2, 6K, and E1 proteins, which are post-translationally processed to form the viral nucleocapsid and envelope glycoproteins (26). These proteins mediate virion assembly, budding, and host cell entry through receptor binding and membrane fusion (31). The retention of this gene arrangement and functionality among our NDUV isolates confirms their classification within the Alphavirus genus and may underscores the observed cytopathic effects in mammalian cells. However, the 5′ and 3′ untranslated regions (UTRs) we obtained appear distinct, the divergence observed in NDUV UTRs may indicate virus-specific regulatory mechanisms or adaptations to distinct host/vector environments. These differences could potentially influence replication efficiency, immune evasion, or tissue tropism. However, their exact functional consequences remain unclear calling for further studies.
Despite its widespread distribution in Africa and serological evidence of human exposure, no human illnesses have been conclusively attributed to NDUV infection (21). However, the assumption that NDUV does not cause disease in humans remains unverified. Given the detection of NDUV in multiple mosquito species with varying host preferences, a targeted search for human infections is warranted.
Conclusion
This study provides strong genomic, phylogenetic, and functional evidence that Ndumu virus is an emerging alphavirus with the ability to infect both mosquitoes and mammalian cells. While no human cases have been reported, its phylogenetic proximity to other pathogenic alphaviruses and its ability to cause CPE suggest a potential public health concern. Further investigations, including serological studies, in vivo infection models, and vector competence assays, are needed to assess its zoonotic risk and transmission dynamics.
Recommendations
● Perform seroprevalence studies in humans and animals to check for unnoticed infections.
● Conduct in vivo studies in mammalian models (e.g., mice) to evaluate virulence.
● Screen vertebrate hosts (e.g., birds, pigs, rodents) to identify possible reservoirs.
● Retrospectively screen febrile illness samples for Ndumu virus presence.
● Enhance mosquito surveillance programs in endemic and neighboring regions.
● Investigate coinfections with other arboviruses (e.g., West Nile virus, Dengue, Chikungunya) to assess epidemiological implications.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: GenBank, accession PV472102, PV472103, and PV472104.
Ethics statement
Ethical approval was obtained from the Kenya Medical Research Institute (KEMRI) Scientific and Ethics Review Unit (SERU) under protocol number KEMRI/SERU/CCR/4702 and WRAIR# 3101. Permission to conduct the study was granted by the Nat ional Council for Science, Technology, and Innovation (NACOSTI).
Author contributions
TW: Data curation, Conceptualization, Methodology, Formal Analysis, Visualization, Writing – review & editing, Writing – original draft, Software, Investigation. WB: Writing – original draft, Methodology, Writing – review & editing, Supervision. SL: Methodology, Software, Writing – review & editing, Conceptualization, Writing – original draft, Visualization, Formal Analysis, Data curation. JK: Writing – review & editing, Writing – original draft. SY: Writing – original draft, Writing – review & editing, Data curation. GlK: Writing – original draft, Formal Analysis, Writing – review & editing. JA: Formal Analysis, Writing – review & editing, Writing – original draft. EG: Resources, Project administration, Writing – original draft, Writing – review & editing. RH: Project administration, Validation, Writing – review & editing, Writing – original draft, Resources. GeK: Validation, Resources, Writing – review & editing, Writing – original draft, Project administration. JE: Writing – original draft, Resources, Writing – review & editing, Project administration, Validation. FE: Writing – original draft, Validation, Funding acquisition, Project administration, Supervision, Resources, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was funded by the Armed Forces Health Surveillance Branch (AFHSB) and its Global Emerging Infections Surveillance (GEIS) Section, FY2022 ProMIS ID: P0116_22_KY and FY2023 ProMIS ID P0094_23_KY.
Acknowledgments
We thank Hellen Koka, Victor Ofula, Dr. Samson Konongoi, Francis Mulwa, Dr. Edith Chepkorir, Bryson Kimemia and Jane Thiiru for their expert contribution in cell culture and data analysis.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
This Material has been reviewed by the Walter Reed Army Institute of Research. There is no objection to its presentation and/or publication. The opinions or assertions contained herein are the private views of the author, and are not to be construed as official, or as reflecting true views of the Department of the Army or the Department of Defense.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fviro.2025.1617756/full#supplementary-material
References
1. Powell JR and Tabachnick WJ. History of domestication and spread of Aedes aEgypti–a review. Mem Inst Oswaldo Cruz. (2013) 108:11–7. doi: 10.1590/0074-0276130395
2. Braack L, Gouveia De Almeida AP, Cornel AJ, Swanepoel R, and De Jager C. Mosquito-borne arboviruses of African origin: Review of key viruses and vectors. Parasites Vectors. (2018) 11. doi: 10.1186/s13071-017-2559-9
3. Guarido MM, Fourie I, Meno K, Mendes A, Riddin MA, Macintyre C, et al. Regions of South Africa, 2014 to 2018. Viruses. (2023) 15:414. doi: 10.3390/v15020414
4. Weaver SC and Barrett ADT. Transmission cycles, host range, evolution and emergence of arboviral disease. Nat Rev Microbiol. (2004) 2:789–801. doi: 10.1038/nrmicro1006
5. Kokernot RH, McIntoshi BM, and Worth CB. Ndumu virus, a hitherto unknown agent, isolated from culicine mosquitoes collected in northern Natal, Union of South Africa. Am J Trop Med Hyg. (1961) 10:383–6. doi: 10.4269/ajtmh.1961.10.383
6. Ochieng C, Lutomiah J, Makio A, Koka H, Chepkorir E, Yalwala S, et al. Mosquito-borne arbovirus surveillance at selected sites in diverse ecological zones of Kenya; 2007 - 2012. Virol J. (2013) 10:1–10. doi: 10.1186/1743-422X-10-140
7. Masembe C, Michuki G, Onyango M, Rumberia C, Norling M, Bishop RP, et al. Viral metagenomics demonstrates that domestic pigs are a potential reservoir for Ndumu virus. Virol J. (2012) 9:1. doi: 10.1186/1743-422X-9-218
8. Lutomiah J, Ongus J, Linthicum KJ, and Sang R. Natural vertical transmission of ndumu virus in culex pipiens (Diptera: Culicidae) mosquitoes collected as larvae. J Med Entomol. (2014) 51:1091–5. doi: 10.1603/ME14064
9. Lwande OW, Lutomiah J, Obanda V, Gakuya F, Mutisya J, Mulwa F, et al. Isolation of tick and mosquito-borne arboviruses from ticks sampled from livestock and wild animal hosts in Ijara District, Kenya. Vector-Borne Zoonotic Dis. (2013) 13:637–42. doi: 10.1089/vbz.2012.1190
10. Moolla N, Viljoen N, Patharoo V, Grobbelaar A, Ismail A, and Weyer J. Near-complete genome sequence of Ndumu virus from Garissa, Kenya, 1997. Microbiol Resour Announc. (2021) 10:550–1. doi: 10.1128/MRA.00551-21
11. Muturi EJ, Shililu JI, Gu W, Jacob BG, Githure JI, and Novak RJ. Larval habitat dynamics and diversity of Culex mosquitoes in rice agro-ecosystem in Mwea, Kenya. Am J Trop Med Hyg. (2007) 76:95–102. doi: 10.4269/ajtmh.2007.76.95
12. Turell MJ, Linthicum KJ, Patrican LA, Glyn Davies F, Kairo A, and Bailey CL. Vector competence of selected african mosquito (Diptera: Culicidae) species for rift valley fever virus. J Med Entomol. (2008) 45:102–8. doi: 10.1093/jmedent/45.1.102
13. Mackenzie. Investigating species co-occurrence patterns when species are detected. J Anim Ecol. (2004) 73(3):546–55. doi: 10.1111/j.0021-8790.2004.00828.x
14. Hubálek Z and Halouzka J. West Nile fever - A reemerging mosquito-borne viral disease in Europe. Emerg Infect Dis. (1999) 5:643–50.
15. Edwards. Mosquitoes of the Ethiopian region III. Ann Entomol Soc Am. (1941) 35:475–5. doi: 10.1093/aesa/35.4.475a
16. Harbach RE. The Culicidae (Diptera): a review of taxonomy, classification and phylogeny. Contrib. Am. Entomol. Inst. (1988) 24(1):1–240.
18. Li D, Liu CM, Luo R, Sadakane K, and Lam TW. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. (2015) 31:1674–6. doi: 10.1093/bioinformatics/btv033
19. Kumar S, Stecher G, and Tamura K. MEGA7: Molecular evolutionary genetics analysis version 7.0. molecular biology and evolution. Mol Biol Evol. (2016) 33(7):1870–4. doi: 10.1093/molbev/msw054
20. Lutz R, F O, B M, H W, and Vrijenhoek R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol. (2022) 16:1–23.
21. Jupp PG, McIntosh BM, and Anderson D. Culex (Eumelanomyia) rubinotus theobald as a vector of Banzi, Germiston and Witwatersrand viruses. J Med Ent. (1976) 12(6):647–51.
22. Lutomiah J, Omondi D, Masiga D, Mutai C, Mireji PO, Ongus J, et al. Blood meal analysis and virus detection in blood-fed mosquitoes collected during the 2006–2007 rift valley fever outbreak in Kenya. Vector-Borne Zoonotic Dis. (2014) 14:656–64. doi: 10.1089/vbz.2013.1564
23. Borland EM and Kading RC. Modernizing the toolkit for arthropod bloodmeal identification. Insects. (2021) 12:1–27. doi: 10.3390/insects12010037
25. van Niekerk AJ. Towards inclusive growth in Africa. Dev South Afr. (2020) 37:519–33. doi: 10.1080/0376835X.2020.1736004
26. Strauss JH and Strauss EG. The alphaviruses: Gene expression, replication, and evolution. Microbiol Rev. (1994) 58:491–562. doi: 10.1128/mr.58.3.491-562.1994
27. Peränen J, Laakkonen P, Hyvönen M, and Kääriäinen L. The alphavirus replicase protein nsP1 is membrane-associated and has affinity to endocytic organelles. Virology. (1995) 208:610–20. doi: 10.1006/viro.1995.1192
28. Rupp JC, Sokoloski KJ, Gebhart NN, and Hardy RW. Alphavirus RNA synthesis and non-structural protein functions. J Gen Virol. (2015) 96:2483–500. doi: 10.1099/jgv.0.000249
29. Fros JJ and Pijlman GP. Alphavirus infection: host cell shut-off and inhibition of antiviral responses. Viruses. (2016) 8:166. https://pmc.ncbi.nlm.nih.gov/articles/PMC4926186/.
30. A.lemm J, Rümenapf T, Strauss EG, Strauss JH, and M.rice C. Polypeptide requirements for assembly of functional Sindbis virus replication complexes: A model for the temporal regulation of minus- and plus-strand RNA synthesis. EMBO J. (1994) 13:2925–34. doi: 10.1002/j.1460-2075.1994.tb06587.x?download=true
Keywords: Ndumu virus (NDUV), emerging arboviruses, zoonotic potential, mosquito borne viruses, next generation sequencing (NGS)
Citation: Wanjiru T, Bulimo W, Langat S, Kinyua J, Yalwala S, Kerich G, Ambale J, Garges E, Haynes R, Kellar G, Eads J and Eyase F (2025) Detection and genomic characterization of Ndumu virus in Coastal Kenya from Culex and Mansonia africana mosquitoes. Front. Virol. 5:1617756. doi: 10.3389/fviro.2025.1617756
Received: 24 April 2025; Accepted: 19 June 2025;
Published: 08 July 2025.
Edited by:
Shengzhang Dong, Johns Hopkins University, United StatesReviewed by:
Edwin Ogola, National Institute of Allergy and Infectious Diseases (NIH), United StatesJuan Camilo Hernandez-Valencia, University of South Bohemia in České Budějovice, Czechia
Copyright © 2025 Wanjiru, Bulimo, Langat, Kinyua, Yalwala, Kerich, Ambale, Garges, Haynes, Kellar, Eads and Eyase. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tabitha Wanjiru, d2FuamlydXRhYnNAZ21haWwuY29t