 Nandadeva Lokugamage1
Nandadeva Lokugamage1 Subhadip Choudhuri1
Subhadip Choudhuri1 Kempaiah Rayavara1
Kempaiah Rayavara1 Chien-Te Tseng1
Chien-Te Tseng1 Shinji Makino1
Shinji Makino1 Nisha Jain Garg1,2*
Nisha Jain Garg1,2*- 1Department of Microbiology and Immunology, University of Texas Medical Branch (UTMB), Galveston, TX, United States
- 2Institute for Human Infections and Immunity, UTMB, Galveston, TX, United States
Background: Nonsense-mediated mRNA decay (NMD) pathway recognizes the mRNAs of host and cytoplasmic pathogens harboring aberrant features and targets them for degradation. Poly(ADP-ribose) polymerases (PARPs) superfamily consists of 17 members, among which macrodomain and zinc finger PARPs function as regulators of RNA metabolism and transcription. In this study, we investigated whether crosstalk between NMD and PARPs regulates SARS-CoV-2 RNA stability and viral infection.
Methods: Transgenic mice (hACE2tg) expressing human angiotensin-converting enzyme 2, and human alveolar epithelial cells (Calu-3ACE2+, A549ACE2+), in which the expression of NMD factors and PARPs was modulated by molecular approaches were used for various studies.
Results: We found that NMD pathway targets endogenous and exogenous aberrant transcripts in human lung epithelial cells. Upon SARS-CoV-2 infection, the expression of NMD factors, up-framshift 1 and 2 (UPF1/UPF2) was decreased while PARP12 and PARP13 were significantly increased in Calu-3ACE2+ and A549ACE2+ cells and lung tissues of hACE2tg mice. Depletion of PARP12/PARP13 using target-specific (vs. scrambled) siRNAs significantly enhanced the stability of NMD targeted endogenous and exogenous aberrant transcripts and SARS-CoV-2 subgenomic S, E, M, and N mRNAs in A549ACE2+ cells, like what was noted in siUPF1/siUPF2-transfected lung epithelial cells. Conversely, overexpression of PARP12/PARP13 enhanced the NMD-dependent degradation of aberrant transcripts and SARS-CoV-2 subgenomic and genomic RNAs. Further, overexpression of PARP12/PARP13 had a dose-dependent effect in enhancing the anti-viral NMD activity and suppression of SARS-CoV-2 replication in infected cells.
Conclusion: We conclude that PARP12/PARP13 synergize with NMD pathway to regulate the viral mRNA stability and replication of SARS-CoV-2.
Introduction
Coronaviruses (CoVs) belong to the family Coronaviridae (classified as α, β, γ, and δ). While most human CoVs cause mild respiratory infections, severe acute respiratory syndrome CoV (SARS-CoV), Middle East respiratory syndrome CoV (MERS-CoV), and SARS-CoV-2 have caused serious respiratory illness and death. The clinical spectrum of SARS-CoV-2 exposure ranges from asymptomatic infection to life-threatening cardiopulmonary failure (1). Current vaccines have slowed SARS-CoV-2 spread worldwide, yet emerging variants continue to cause significant economic and public health burdens (2).
CoVs carry a positive-sense RNA genome (~30 kb) that is structurally polycistronic, containing multiple open reading frames (ORFs). Like other CoVs, SARS-CoV and SARS-CoV-2 particles are composed of a helical nucleocapsid that contains the viral genomic RNA and nucleocapsid (N) protein, enclosed within an envelope composed of spike (S), envelope (E), and membrane (M) viral proteins. Upon infection, the viral genomic RNA is released into the cytoplasm and undergoes translation of two large polyproteins from the two major ORFs (ORF1a/b) encoded in the 5′ two-thirds of the genome. The polyproteins are then processed by viral proteases to generate 15–16 nonstructural proteins, most of which are involved in viral RNA replication (3). Additionally, several subgenomic mRNAs that encode viral structural and accessory proteins are synthesized in CoV-infected cells (3). Although, our knowledge of CoV gene expression strategies has improved significantly in the last decade, our understanding of CoV–host interactions involved in the regulation of viral mRNA stability and viral gene expression remains limited.
The nonsense-mediated mRNA decay (NMD) pathway is important for maintaining host mRNA quality control. mRNAs with aberrant features, such as a premature termination codon (PTC), are bound by the exon junction complex and recognized by the NMD pathway, which leads to the decay of the target mRNA. The NMD pathway also targets PTC-free transcripts that contain long 3′ regions with GC-rich domains. For example, the NMD pathway targets the mRNAs of some viruses, including Semliki Forest virus (SFV) and Potato virus X (PVX) (4, 5). Like CoVs, SFV and PVX carry positive-sense genomic RNA and accumulate genomic and subgenomic mRNAs—both of which contain multiple ORFs—in infected cells. Because only the 5′ ORF in viral mRNAs is used for translation, the genomic RNA and most of the viral subgenomic mRNAs have a long 3′ region that is not translated, which can make them susceptible to NMD (3). Using the prototype β-CoV, murine hepatitis virus (MHV), we previously reported that the NMD pathway targets MHV genomic and subgenomic RNAs for degradation (6). Depletion of NMD factors (UPF1, UPF2, SMG5, and SMG6) by siRNAs enhanced MHV RNA stability and virus replication in mouse fibroblast cells (6). These results provided the first evidence for a novel role of NMD in CoV RNA decay.
The poly(ADP-ribose) polymerase (PARP) superfamily consists of 17 members characterized by the presence of a conserved PARP domain and the ability to catalyze ADP-ribosylation of various target proteins, including histones, enzymes, and transcription factors. PARP1, PARP2, and PARP3 can bind DNA, poly(ADP-ribosyl)ate themselves and their target proteins, and function in response to DNA damage. Macrodomain-containing PARP9, PARP14, and PARP15, and CCCH zinc finger (ZnF)-containing PARP7, PARP12, and PARP13 are emerging as regulators of RNA metabolism and transcription (7). PARP13 binds to target viral RNA (8), and it has been suggested that PARP13 may suppress the translation of bound RNAs by interfering with initiation (9) or activate mRNA decay by recruiting components of the 3′–5′ mRNA degradation machinery—a process distinct from, but functionally related to, the NMD pathway. Other studies have also indicated the antiviral activities of several PARPs (10), although whether PARPs interact with the NMD pathway to modulate viral RNA levels has not been elucidated.
In this study, we aimed to determine whether PARP12/PARP13 and the NMD pathway exert antiviral effects during SARS-CoV-2 infection. We hypothesized that, as RNA-binding proteins, PARP12/PARP13 may also interact with the NMD pathway to regulate viral RNA stability. To test this, we utilized molecular approaches to modulate the expression of NMD factors and PARP12/PARP13 in both in vitro and in vivo models of SARS-CoV-2 infection. We discuss the individual and synergistic roles of the NMD pathway and PARP12/PARP13 in controlling the RNA stability and replication of SARS-CoV-2.
Materials and methods
Ethics statement
Animal experiments were conducted following the National Institutes of Health guidelines for the housing and care of laboratory animals and in accordance with protocols approved by the Institutional Animal Care and Use Committee (#2002017A). All experiments involving infectious virus were conducted at UTMB in Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)-accredited animal biosafety level 3 and biosafety level 3 laboratories. All personnel handling infectious material received appropriate biosafety training. All reagents used in the study were of molecular grade with >99% purity.
Cell lines, mice, and SARS-CoV-2
African green monkey kidney epithelial Vero E6 (CRL-1586) cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA) and cultured in Eagle’s minimal essential medium (MEM) supplemented with 10% fetal bovine serum (FBS; Thermo Fisher, Waltham, MA). Human lung epithelial cells (Calu-3, HTB-55, ATCC) were grown in MEM supplemented with 20% FBS and subjected to clonal selection in 96-well plates. Individual clones were analyzed for ACE2 expression (11), and the Calu-3/2B4 clone expressing high levels of ACE2 was propagated and used in this study.
Human alveolar basal epithelial A549 cells (CCL-185, ATCC) were transduced with the lentiviral vector pLVX-Puro-hACE2 (NR-53522, ATCC) and subjected to clonal selection for hACE2 expression, as above. A stable clone (referred to as A549hACE2+) was propagated in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS for this study. SARS-CoV-2 (USA-WA1/2020, ATCC) original stock was passaged in Vero E6 cells at 37 °C in 5% CO2. The virus titer at passage 3 was determined, and virus aliquots at 1×107 50% tissue culture infectious doses (TCID50)/mL were stored at −80 °C.
C57BL/6 x C3H transgenic mice expressing human ACE2 were generated as previously described (12), and AC70 mice expressing high levels of hACE2 were used in this study. The hACE2tg mice (8-weeks old) were infected intranasally with SARS-CoV-2 (102 TCID50/mouse) in 60 μL of phosphate-buffered saline (PBS). Control mice were treated with PBS alone. Mice were euthanized using 2%–4% inhaled isoflurane followed by cervical dislocation. Lung tissues were obtained at 4–5 days post-infection.
Plasmids
The NS39 (NMD target) reporter is based on a nonsense-mutated human β-globin (NS39) transcript, which leads to a robust and highly reproducible reduction in mRNA abundance via NMD (13, 14). Specifically, the Renilla luciferase (rLuc) sequence coding for amino acids 1–311 was fused with the human β-globin gene with or without a nonsense mutation at position 39 to generate NS39 (NMD target) and wild-type (WT, NMD non-target) reporters, respectively. The two constructs were cloned into pCI-Neo (Promega) (13, 14), and recombinant plasmids were provided by Dr. Andreas E. Kulozik (University of Heidelberg) to Dr. Makino (6).
The cDNAs encoding human PARP12 and PARP13 were cloned into the pCAGGS plasmid (4,807 bp) under the control of a chimeric β-actin (chicken)–β-globulin (rabbit) promoter (CAG) and the human CMV-IE enhancer. Briefly, total RNA extracted from A549 cells was reverse transcribed using AccuScript high-fidelity reverse transcriptase (Agilent, Santa Clara, CA) and used as a template in PCR with high-fidelity DNA polymerase (New England Biolabs, Ipswich, MA) and target-specific oligonucleotides to amplify the full-length sequences coding for PARP12 (701 amino acids) and PARP13 (902 amino acids). The amplicons and pCAGGS were digested with Sacl/Xhol restriction enzymes and directionally ligated using a Gibson Assembly Cloning Kit (New England Biolabs, Ipswich, MA).
E. coli DH5α competent cells were transformed with the recombinant plasmids, and ampicillin-resistant bacterial clones were confirmed for accurate cloning of the insert by sequencing (GENEWIZ, Chelmsford, MA). Selected bacterial clones were grown in Luria broth containing 100 μg/mL ampicillin, and recombinant plasmids were purified by anion-exchange chromatography using a Plasmid Midi Kit (Qiagen, Germantown, MD).
Transfection and gene expression
Low-passage, healthy A549 cells (>90% viability) were used for all transfection studies. Opti-MEM reduced-serum medium was used to dilute shRNAi, plasmids, and transfection reagents, and cells were transfected in the absence of antibiotics to prevent cell death. In general, >80% cell viability was maintained throughout the experiments.
Briefly, ON-TARGETplus SMARTpool siRNAs designed for specific depletion of UPF1, UPF2, PARP12, and PARP13, as well as non-targeting control siRNA (siCtrl), were purchased from Horizon–PerkinElmer (Waltham, MA). A549 or A549hACE2+ cells were plated in 12-well plates (105 cells/well) in DMEM supplemented with 10% FBS. At approximately 70% confluency, cells were transfected in triplicate for 48 h with siRNAs (25–75 nM each) using TransIT-siQUEST transfection reagent (MIR2114, Mirus Bio, Madison, WI) or for 24 h with expression or reporter plasmids (0.02–1.0 μg each) using Lipofectamine 2000 (11668-019, Invitrogen, Waltham, MA). Depletion or overexpression of the targets was confirmed by RT-qPCR or Western blot analysis.
In some experiments, A549hACE2+ cells (105 cells/well, 12-well plates) were sequentially transfected in triplicate with siRNAs for 48 h and 1 μg of WT or NS39 reporter plasmids for 24 h. Cells were then infected with SARS-CoV-2 (multiplicity of infection [MOI]: 1.0) and incubated for 24 h at 37°C. Likewise, A549hACE2+ cells (105 cells/well, 12-well plates) transfected with PARP12/PARP13 expression plasmids (1 μg each) for 24 h were infected with SARS-CoV-2 (MOI: 1.0) for 24 h. The reporter or SARS-CoV-2 transcripts were monitored by RT-qPCR.
To monitor the synergy between PARP12/PARP13 and NMD factors, A549hACE2+ cells were seeded in 12-well plates (1 × 105 cells/well) and transfected in triplicate with siCtrl or UPF1- and UPF2-specific siRNAs (75 nM each) for 48 h and pCAGGS (1 μg) or PARP12 and PARP13 expression plasmids (0.02–1.0 μg each) for 24 h. Cells were then infected with SARS-CoV-2 (MOI: 0.1) for 24 h. Cells were analyzed in duplicate for SARS-CoV-2 subgenomic RNAs by RT-qPCR.
Plaque assay
A549hACE2+ cells were sequentially transfected in triplicate with siCtrl or UPF1- and UPF2-specific siRNAs (75 nM each), pCAGGS (1 μg), or PARP12 and PARP13 expression plasmids (0.02–1.0 μg each), and then infected with SARS-CoV-2 (multiplicity of infection [MOI]: 0.1) for 24 h (as described above). Supernatants (spent medium) were serially diluted (10-fold dilutions, up to 10−6). Vero E6 cell monolayers were grown to 90% confluency in 6-well plates. Plates were inoculated in triplicate with 200 μL of diluted supernatants and incubated for 1 h with gentle rotation. The inoculum was then removed, plates were washed with 1× PBS, and monolayers were overlaid with 2 mL of MEM supplemented with 10% FBS containing 0.8% agarose and incubated for 48 h. After staining with 2 mL of 1× neutral red for 3–5 h, plaques were counted on plates with fewer than 300 plaques to avoid significant error due to overlapping.
Western blot
Cells (sample: buffer ratio, 1:10 v/v) were homogenized in RIPA buffer (9806, Cell Signaling, Danvers, MA), centrifuged at 10,000 × g, and the supernatants were collected as protein lysates. Protein lysates (5 μg) were electrophoresed on a 4%–20% Mini-PROTEAN TGX gel (Bio-Rad, Hercules, CA) and transferred to PVDF membranes using a Criterion Trans-Blot system (Bio-Rad). Membranes were blocked with 50 mM Tris-Cl (pH 7.5)/150 mM NaCl (TBS) containing 5% nonfat dry milk, washed with TBS–0.1% Tween-20 (TBST), and incubated overnight at 4 °C with antibodies against UPF1 (Ab133564, Abcam, Cambridge, MA), UPF2 (Ab153830, Abcam), PARP12 (Ab241967, Abcam), PARP13 (Ab154680, Abcam), or GAPDH (3683, Cell Signaling). All primary antibodies were used at a 1:1,000 dilution in TBST containing 1% BSA. Membranes were washed and incubated for 1 h with horseradish peroxidase (HRP)-conjugated anti-rabbit antibody (1:10,000 dilution; SouthernBiotech, Birmingham, AL). Color was developed with enhanced chemiluminescence detection reagent (GERPN2106, MilliporeSigma, Burlington, MA), and images were acquired and analyzed using an ImageQuant LAS4000 system (GE Healthcare).
RT-qPCR
Cells or tissues (sample: buffer ratio, 1:10 v/v) were homogenized in TRIzol reagent (15596-018, Invitrogen). Total RNA was isolated using the chloroform/isopropanol/ethanol method, treated with RNase-free DNase I (AM2222, Ambion, Austin, TX) to remove contaminating DNA, and analyzed for quality (OD260/280>1.8) and quantity (OD2601 = 40μg/mL) using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher). RNA (1 μg) was reverse transcribed using the iScript™ cDNA synthesis kit (1708841, Bio-Rad) and diluted to 100 μL with nuclease-free water. Real-time qPCR was performed on an iCycler thermal cycler (Bio-Rad) in a 20 μL reaction containing 50 ng cDNA, 10 μL iTaq Universal SYBR Green Supermix (1725120, Bio-Rad), and 500 nM oligonucleotide pairs (listed in Table 1).
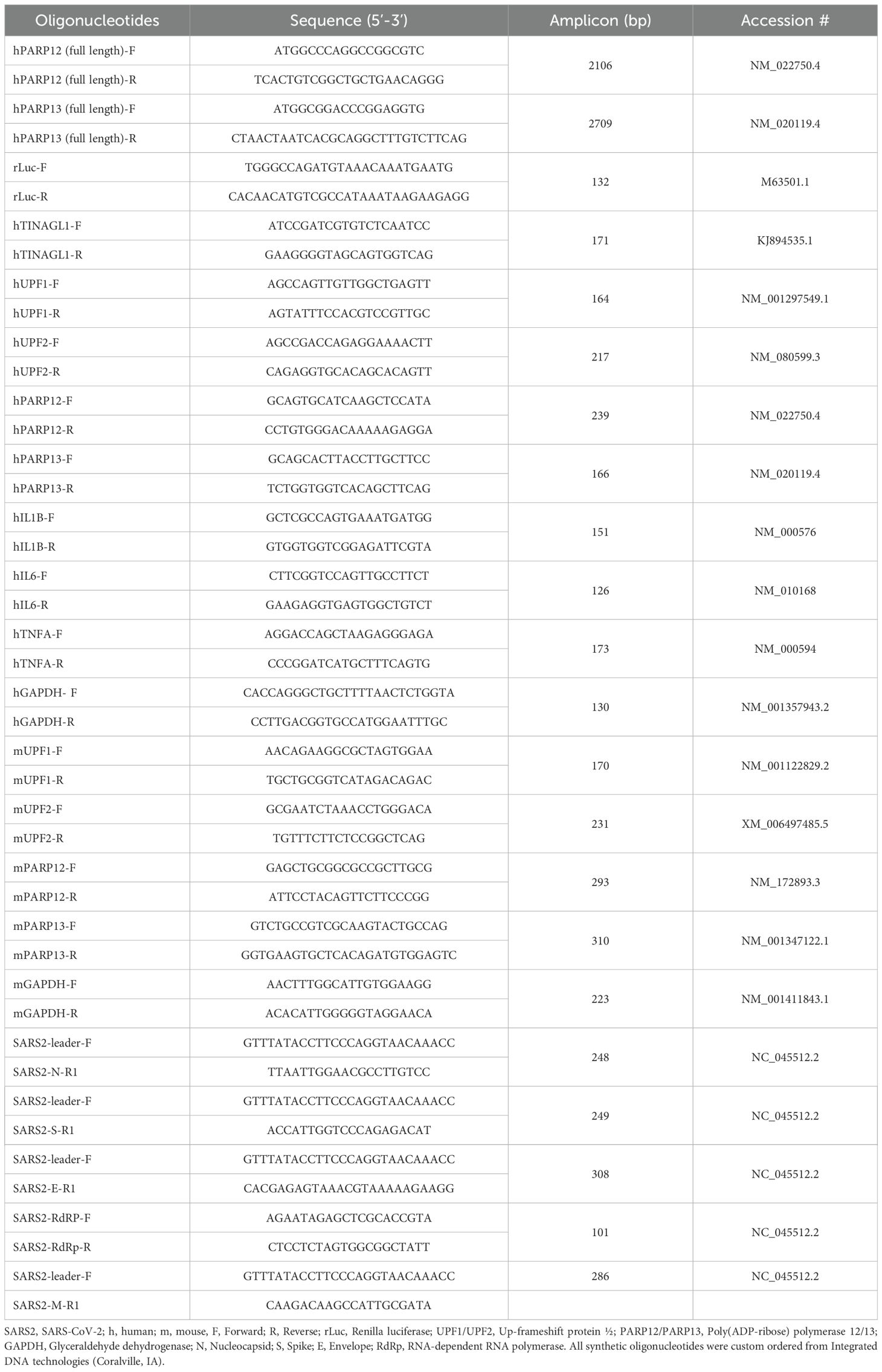
Table 1. Oligonucleotides used in this study.
Thermal cycling conditions were as follows: denaturation at 95°C for 15 s, followed by annealing/amplification at 60°C for 30 s for 40 cycles. Specific product amplification was confirmed by melt curve analysis. The PCR baseline-subtracted curve-fit model was applied for threshold cycle (Ct) determination, and Ct values of target mRNAs were normalized to the mean Ct values for housekeeping GAPDH mRNA. The relative change in target gene mRNA levels was calculated using the 2^–ΔΔCt method.
RNA-seq analysis
Confluent Calu-3/2B4 cells in 6-well plates were infected in triplicate with SARS-CoV-2 (MOI: 1.0) for 12, 24, and 48 h (control: mock infection). Total RNA was extracted as described above and analyzed for quality/quantity using a Bioanalyzer RNA 6000 Nano assay with an Agilent 2100 Bioanalyzer system (Santa Clara, CA). The poly(A)+ RNA, enriched from total RNA (1 μg) using oligo(dT)-attached magnetic beads, was incubated in 19.5 μL fragmentation buffer (Illumina, San Diego, CA) at 94°C for 8 min. A TruSeq RNA Sample Preparation Kit was used for first- and second-strand synthesis, adapter ligation, and library amplification, following the manufacturer’s instructions (Illumina).
Library quality was assessed on an Agilent DNA 1000 chip using the Agilent 2100 Bioanalyzer. A TruSeq SBS Kit v3 (Illumina) was used to perform paired-end 50-base sequencing on an Illumina HiSeq 1000, yielding approximately 100 million read pairs per sample.
RNA-seq reads were aligned to the human hg38 reference genome using STAR v2.4.0j software with the FeatureCounts function, and differential gene expression was analyzed using Bioconductor DESeq2 v.1.20.0. The Benjamini–Hochberg procedure was applied to calculate false discovery rate (FDR)–adjusted p-values of ≤0.01.
Immunohistochemistry
Five-micron, paraffin-embedded lung tissue-sections were deparaffinized in xylene, hydrated in graded alcohol, suspended in 0.01M sodium citrate buffer (pH6.0), and incubated in a boiling water bath for 10min to unmask the antigens. Slides were washed with 1X PBS and incubated for 10-min each with Bloxal blocking solution and 2.5% normal horse serum to block endogenous peroxidase activity and non-specific antibody binding. Next, slides were incubated for 12 h with primary antibodies against SMG1 (sc135563, Santa Cruz Biotechnology, Dallas, TX), UPF1 (sc393594, Santa Cruz), PARP12 (NBP1-56436, Novus Biologicals, Centennial, CO), and PARP13 (ab154680, Abcam). Primary antibodies were used at 1:50–1:200 dilutions in PBS–0.1% Tween-20. Slides were washed, incubated for 30 min with ImmPRESS Duet Double Staining Reagent (MP-7714) containing HRP–anti-rabbit and AP–anti-mouse IgG antibodies, sequentially stained for 10 min each with ImmPACT DAB EqV HRP (SK-4103) and ImmPACT Vector Red AP (SK-5105) substrates, and mounted with VectaMount AQ Aqueous Medium (H-5501). All immunohistochemistry reagents were obtained from Vector Laboratories (Newark, CA).
Slides were imaged using an Olympus BX-15 microscope (Center Valley, PA) equipped with a digital camera and Simple PCI software (v.6.0, Compix, Sewickley, PA). Each tissue section (n = 3 per group, at least two slides per tissue) was analyzed in nine microscopic fields. To evaluate the distribution of individual antigen expression, DAB- or AP-positive immunostained areas were scored as follows: (0) = < 10%, (1+) = 10-25%, (2+) = 25-50%, (3+) = 50-75%, and (4+) = > 75% of the scanned area. The intensity of staining was scored as (1) weak, (2+) moderate, and (3+) strong. Tissue single-antigen expression analysis was performed using a semi-quantitative combined scoring system. The combinative Quick IHC score was obtained from nine microscopic areas of each tissue on a single slide under 20X magnification and calculated by multiplying the score for the immunostained area by the score for staining intensity. Finally, an average score was calculated from each combinative score.
Statistical analysis
Mice were randomly distributed into treatment groups (n=5). In vitro studies were conducted with three biological replicates per treatment. All samples were analyzed in duplicate, and each experiment was performed twice using independent batches. Data were analyzed using GraphPad Prism software. Each biological sample is represented by a dot, and mean ± standard error of the mean (SEM) values are plotted as bar graphs.
The Kolmogorov–Smirnov test was performed to assess the normal distribution of data. Statistical significance between two groups was calculated using Student’s unpaired t-test with or without Welch’s correction or the nonparametric Mann–Whitney U-test. Significance among multiple groups was assessed using one-way ANOVA with Tukey’s post hoc test or the nonparametric Kruskal–Wallis/Dunn’s post hoc test. Statistical significance was accepted at p < 0.05.
Results
NMD pathway functions in human lung epithelial cells
UPF1 and UPF2 are essential for the normal functioning of NMD in the lungs. Western blot analysis showed that UPF1 and UPF2 were expressed in A549 cells. In comparison, UPF3—another NMD factor abundantly expressed in the reproductive organs of mice (15), was not detected in A549 lung epithelial cells. Transfection with siUPF1 and siUPF2 siRNAs at 50–75 nM (vs. siCtrl) resulted in 85%–95% depletion of UPF1 and UPF2, respectively (Figure 1a). No effects of the siRNAs were observed on GAPDH (background control) levels.
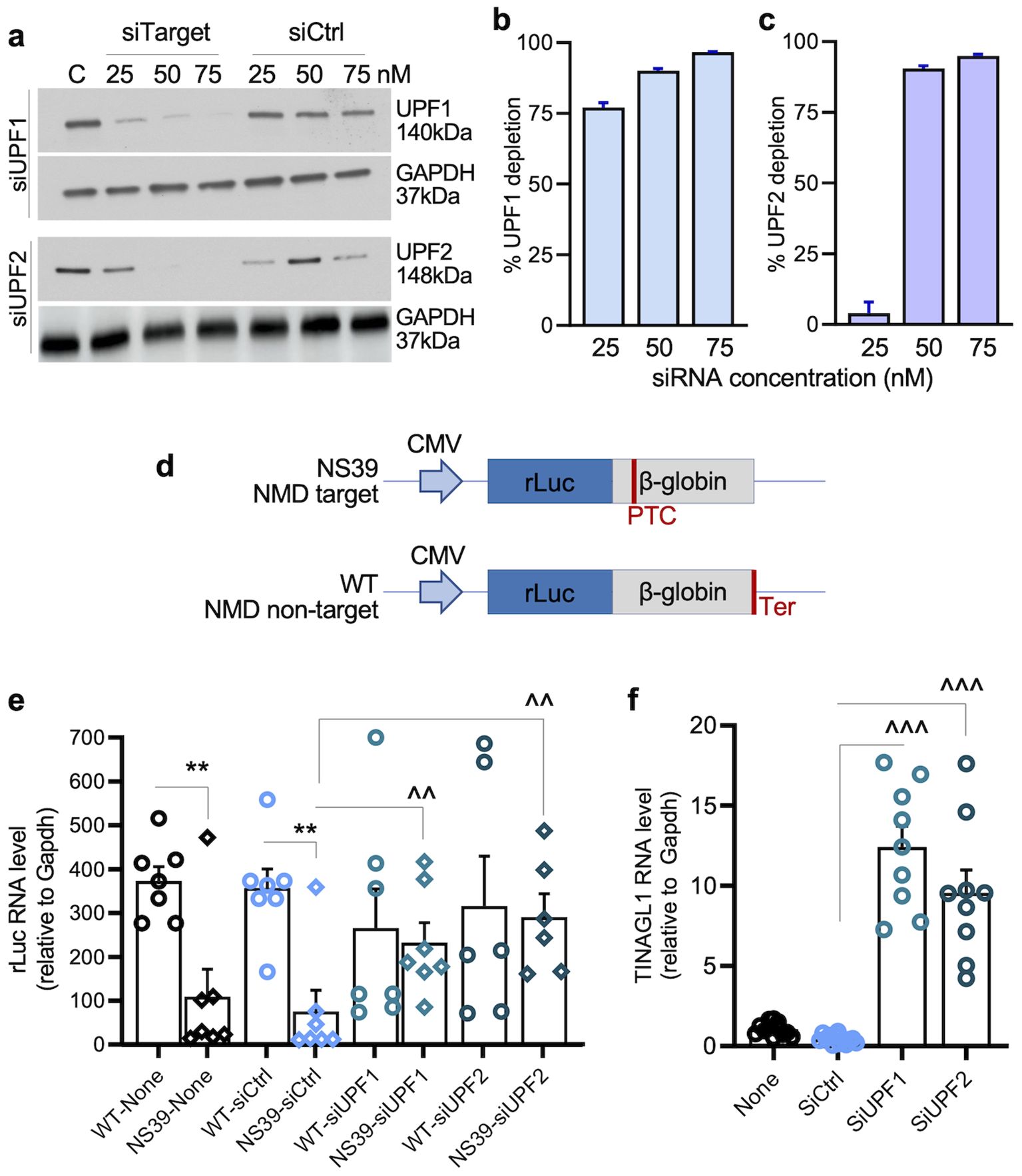
Figure 1. (a–c) NMD depletion in human epithelial cells. Human alveolar epithelial A549 cells were transfected with 25–75 nM of target-specific (siUPF1, siUPF2) or siCtrl siRNAs for 48 h. Representative western blots for UPF1 and UPF2 (control: GAPDH) are shown in panel a. Densitometry analysis of UPF1 and UPF2 bands (relative to GAPDH) was performed, and percent depletion of UPF1 and UPF2 in cells transfected with target-specific (vs. siCtrl) siRNAs was calculated for (b, c). (d) Schematic of NMD reporters. NS39 is an NMD target reporter consisting of an in-frame Renilla luciferase (rLuc) and β-globin fusion construct with a nonsense mutation at codon 39 of the β-globin open reading frame, resulting in the formation of a premature termination codon (PTC). Wild type (WT) is an NMD non-target reporter consisting of an rLuc/β-globin fusion construct with an accurate termination codon (Ter) and expresses full-length β-globin. The Renilla sequence is shown in blue and β-globin sequences in gray. The reporter constructs were cloned in the pCI-neo plasmid. (e) Effects of UPF1/UPF2 depletion on the exogenous NMD reporter. A549ACE2+ cells were sequentially transfected with 75 nM of siCtrl, siUPF1, or siUPF2 siRNAs for 48 h and 1 μg of WT or NS39 reporters for 24 h. The relative RNA levels of rLuc (normalized to Gapdh) were monitored by RT-qPCR. (f) UPF1/UPF2 depletion effects on the endogenous NMD reporter. A549ACE2+ cells transfected with siRNAs were analyzed for alternatively spliced TINAGL1 mRNA (endogenous NMD target) by RT-qPCR. In panels e–f, each biological sample (average of duplicate analysis) is plotted as a dot, and mean ± standard error of the mean (SEM) values derived from n = 6–8 biological samples per group are plotted as bars. Significance for e–f was calculated by one-way ANOVA with Tukey’s or Kruskal–Wallis/Dunn’s post hoc tests; p-values of ≤0.01 and ≤0.001 are plotted with 2 and 3 symbols, respectively (*WT vs. NS39; ^siCtrl vs. siUPF1 or siUPF2 in matched groups). Horizontal bars show the compared groups. p-values of ≤0.01,^^ and ≤0.001,^^^ are plotted (WT vs. NS39; ^siCtrl vs. siUPF1 or siUPF2).
To examine NMD function, UPF1- and UPF2-depleted cells were transfected with NS39 (NMD target) or WT (NMD non-target) reporters (Figure 1b). Both reporters express Renilla luciferase (rLuc) in fusion with β-globin; however, NS39 carries a nonsense mutation in β-globin that makes the RNA susceptible to degradation by the NMD pathway (13, 14). RT-qPCR analysis showed a 78.74%–86.47% lower accumulation of NS39 (vs. WT) reporter RNA in nontransfected and siCtrl-transfected cells, confirming the degradation of NS39 reporter mRNA by NMD in human epithelial cells (Figure 1c).
When transfected with siUPF1 or siUPF2, the accumulation of NS39 reporter RNA increased by 0.99–1.89-fold and 3.44–5.44-fold, respectively, compared to its level in controls (Figure 1c). No significant effects of UPF1 or UPF2 depletion were observed on WT (non-NMD target) reporter RNA levels (Figure 1c). Alternatively spliced mRNA for tubulointerstitial nephritis antigen-like 1 (TINAGL1), a known endogenous NMD target (16), also increased by 7.52–26.24-fold in UPF1- and UPF2-depleted (vs. control) cells (Figure 1d).
These results suggest that UPF1 and UPF2 are expressed and mediate NMD activity for degrading endogenous and exogenous aberrant transcripts in human lung epithelial cells. Consequently, UPF1/UPF2 depletion resulted in increased stability of NMD target transcripts in these cells.
Expression of NMD factors (UPF1/UPF2) in SARS-CoV-2 infection and their effects on SARS-CoV-2 transcripts
Because SARS-CoV-2 replicates in the cytoplasm and its genomic and subgenomic RNAs contain long 3′ regions, we hypothesized that SARS-CoV-2 RNAs might be NMD targets and that infection may alter the abundance of NMD factors. Indeed, mRNAs for UPF1 and UPF2 decreased by 82.41%–90.62% in lung tissues of hACE2tg mice infected with SARS-CoV-2 compared to mock-infected mice (Figures 2a, b). A549hACE2+ cells infected with SARS-CoV-2 for 24 h (vs. mock-infected) also exhibited 53.26% and 94.05% declines in mRNA levels for UPF1 and UPF2, respectively (Figures 2c, d).
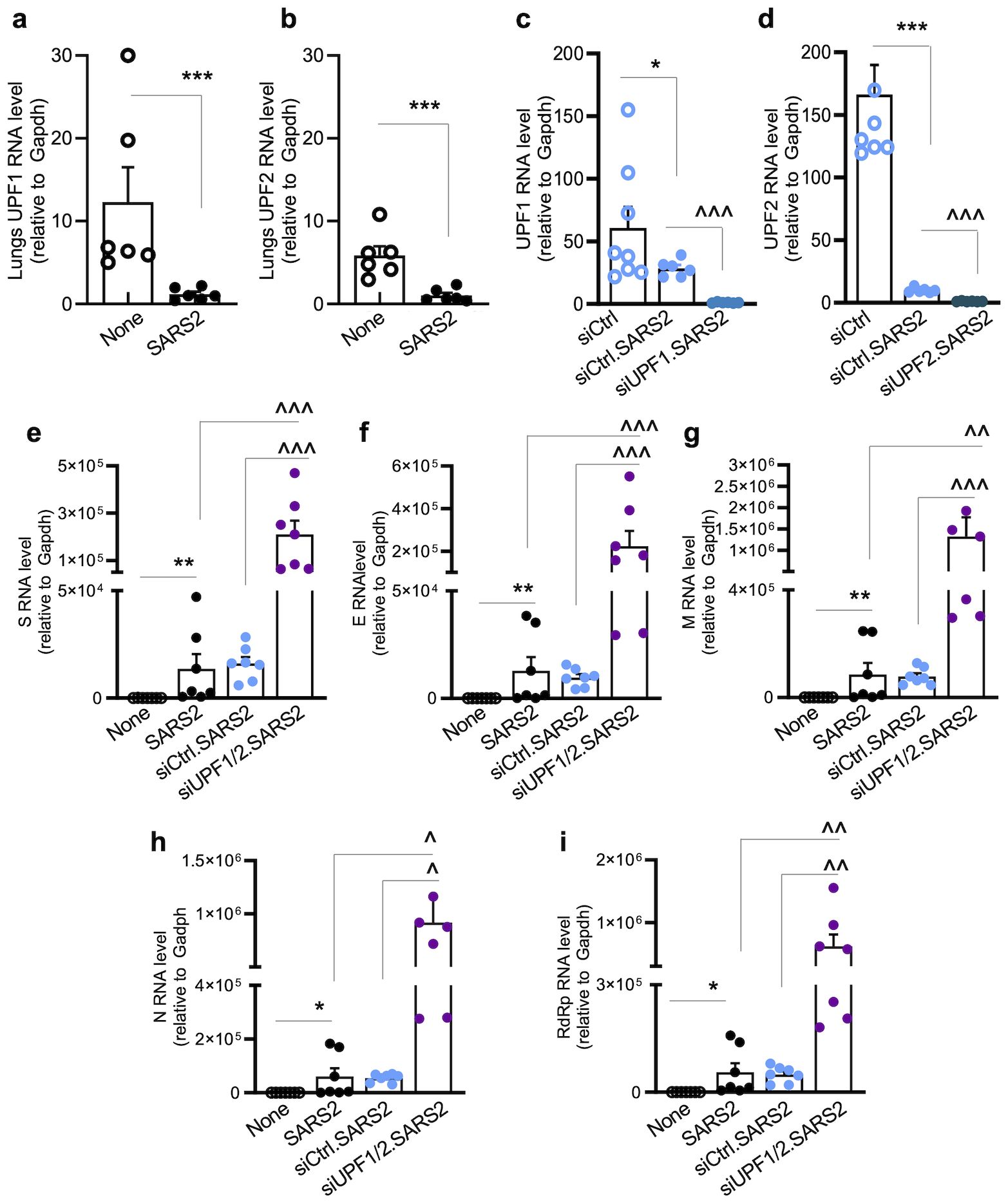
Figure 2. (a–d) SARS-CoV-2 suppresses UPF1/UPF2 expression. For in vivo studies (a, b), hACE2tg mice were intranasally challenged with 102 TCID50 of SARS-CoV-2, euthanized at 4–5 days post-infection, and lung tissues were harvested. For in vitro studies (c, d), A549ACE2+ cells were transfected with siCtrl, siUPF1, or siUPF2 siRNAs (75 nM each) and infected with SARS-CoV-2 (MOI: 1.0) for 24 h. Samples were analyzed in duplicate by RT-qPCR for mRNA levels of UPF1 and UPF2, and data were normalized to Gapdh. Controls: mock infected. Individual data points (average of duplicate values from each sample) and mean ± SEM values (n = 6–8 samples per group) are shown. (e–i) Effects of UPF1/UPF2 depletion on SARS-CoV-2 RNA. A549ACE2+ cells were nontransfected or transfected with siCtrl or siUPF1/siUPF2 siRNAs (75 nM each) for 48 h, and infected with SARS-CoV-2 (MOI: 1.0) for 24 h. RT-qPCR analysis of SARS-CoV-2 subgenomic RNAs (S, E, M, N) was performed using a common leader sequence and target-specific primers, and genomic RNA was amplified using RdRP-specific primers; relative levels of SARS-CoV-2 transcripts (normalized to Gapdh) were calculated. Individual data points from each biological sample (average of duplicate observations) and mean ± SEM values (n = 6–8 biological samples/group) are plotted. Student’s unpaired t-test with Mann–Whitney U-test (a, b) and one-way ANOVA with Tukey’s or Kruskal–Wallis/Dunn’s post hoc tests (c–i) were applied to calculate significance; p-values of ≤0.05, ≤0.01, and ≤0.001 are annotated with 1, 2, and 3 symbols, respectively (*control vs. infected; ^infected vs. siUPF1 and/or siUPF2-treated/infected). Horizontal bars show the compared groups. p-values of ≤0.05*,^, ≤0.01,^^, and ≤0.001,^^^ are plotted (control vs. infected; ^infected vs. siUPF1-treated/infected or siUPF2-treated/infected).
To evaluate whether NMD targets SARS-CoV-2 RNAs, A549hACE2+ cells were transfected with siCtrl or siUPF1/siUPF2 (75 nM each) siRNAs before infection (MOI: 1.0), and viral subgenomic RNAs were analyzed at 24 h. Transfection with siUPF1 and siUPF2 before SARS-CoV-2 infection led to almost complete depletion of UPF1 and UPF2 RNA, respectively (Figures 2c, d). We detected the accumulation of SARS-CoV-2 S, E, M, and N subgenomic RNAs in nontransfected or siCtrl-transfected A549hACE2+ cells at 24 h (Figures 2e–h).
Importantly, 12.1–14.4-fold, 16.4–22.1-fold, 14.4–15.8-fold, and 14.2–15.8-fold further increases in S, E, M, and N subgenomic RNAs, respectively, were observed in UPF1/UPF2-depleted (vs. control) cells infected with SARS-CoV-2 (Figures 2e–h). A similar pattern of increase in genomic RNA for RdRP was also noted in UPF1/UPF2-depleted (vs. siCtrl) cells infected with SARS-CoV-2 (Figure 2i).
Together, these results show that NMD effectors are suppressed in lung epithelial cells and mouse lung tissue during SARS-CoV-2 infection. Our findings using UPF1/UPF2-depleted human lung epithelial cells demonstrate that SARS-CoV-2 subgenomic and genomic RNAs are targeted for degradation by the NMD pathway.
Expression and effects of PARP12/PARP13 in SARS-CoV-2 infection
PARP12 and PARP13 are known to bind viral RNA and suggested to suppress viral replication by interfering with translation initiation (9) or by activating mRNA decay via the 3’-5’ mRNA degradation machinery (17). PARP13/TRIM25 complex also binds RNA to regulate gene expression, including genes involved in priming anti-viral immunity (18). In this study, we focused on understanding how NMD-PARPs interactions may occur in the context of SARS-CoV-2 infection.
We first determined whether PARP expression levels changed in response to SARS-CoV-2 infection. To do this, we extracted PARP-related data from global gene expression profiling of Calu3/2B4hACE2+ lung epithelial cells infected with SARS-CoV-2 (MOI: 1.0) for 12, 24, and 48 h. Of the 16 PARPs identified in the RNA-seq dataset, expression levels for PARP1–3 were slightly decreased, while those for PARP4–8, PARP11, PARP15, and PARP16 were slightly increased during 24–48 h post-infection (Figure 3a). The relative mRNA levels encoding PARP9–10 and PARP12–14 increased by 0.51–2.85-fold in SARS-CoV-2-infected cells at 24–48 h (Figure 3a). Furthermore, mRNAs encoding PARP12 and PARP13 were increased by 2.27–3.0-fold in the lungs of SARS-CoV-2-infected (vs. mock-infected) hACE2tg mice (Figures 3b, c).
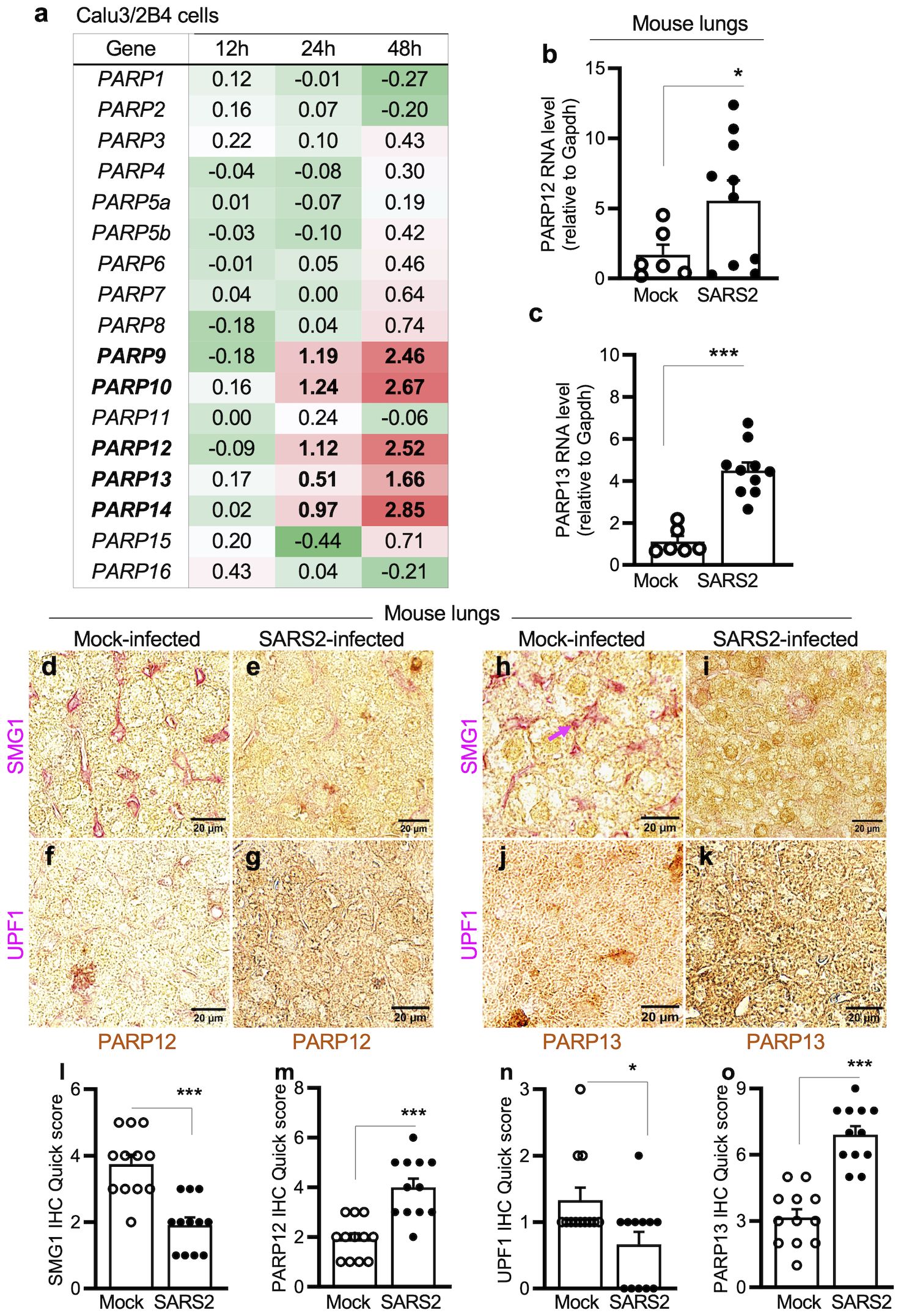
Figure 3. (a–c) Expression of poly(ADP-ribose) polymerases in response to SARS-CoV-2 infection. For in vitro studies (a), human lung epithelial Calu-3/2B4 cells (ACE2+ clone) were infected in triplicate with SARS-CoV-2 (MOI: 1.0) for 12, 24, and 48 h and subjected to RNA-seq analysis. Averages of log2 fold changes in mRNAs for PARP1–PARP16 in infected (vs. mock-infected) cells after normalizing to housekeeping controls are shown (red: upregulation; green: downregulation). Significance was calculated by Student’s unpaired t-test with Mann–Whitney U-test; bolded values show significance (p ≤ 0.01) between control vs. infected samples. For in vivo studies (b, c), hACE2tg mice were intranasally challenged with 102TCID50 SARS-CoV-2 (controls: mock infected) for 4–5 days. RT-qPCR for PARP12- and PARP13-encoding mRNAs (normalized to Gapdh) in lung tissues was performed. Individual data points from each mouse (average of duplicate observations) and mean ± SEM values (n=6–10 mice/group) are plotted. (d–o) Immunohistochemistry for PARPs and NMD factors in lungs of mice infected with SARS-CoV-2. The hACE2tg mice were intranasally challenged with 102TCID50 SARS-CoV-2 (controls: mock infected) for 4–5 days. Paraffin-embedded 5-μm lung tissue sections were subjected to dual immunohistochemistry. Representative images identifying the expression of PARP12 (d–g) and PARP13 (h–k) (brown) and NMD factors SMG1 (d,e, h, i) and UPF1 (f, g, j, k) (magenta) are shown (scale bar: 20 μm), and quick IHC scores for each antigen are plotted in l–o. Data points for each tissue section (average of scanning in 9 microscopic fields) and mean ± SEM values (n ≥ 6 mice/group, 3 tissue sections/mouse) are shown. Significance comparing control and infected groups was calculated by Student’s unpaired t-test with Mann–Whitney U-test. In all bar graphs, p-values are plotted as *p≤0.05, **p≤0.01, ***p≤0.001. Horizontal bars show the compared groups. In bar graphs, p-values are plotted as p≤0.05*, p≤0.01, p≤0.001*. Horizontal bars show the compared groups.
Immunohistochemical staining provided additional evidence of changes in PARPs (and NMD effectors) in response to SARS-CoV-2 infection. PARP12 and PARP13 levels increased by 106.45%–127.27%, whereas NMD effectors SMG1 and UPF1 decreased by 35.95%–52.0% in lung tissues of SARS-CoV-2-infected (vs. mock-infected) mice (Figures 3d–o).
To determine whether PARP12/PARP13 exert antiviral effects, A549hACE2+ cells were transfected with siPARP12 and siPARP13 siRNAs and subsequently infected with SARS-CoV-2. Western blot analysis showed that siPARP12 and siPARP13 at 50–75 nM (vs. siCtrl) resulted in 50%–84% and 95%–99% depletion of PARP12 and PARP13, respectively (Figures 4a–c). No effects of PARP12 or PARP13 depletion were observed on GAPDH levels.
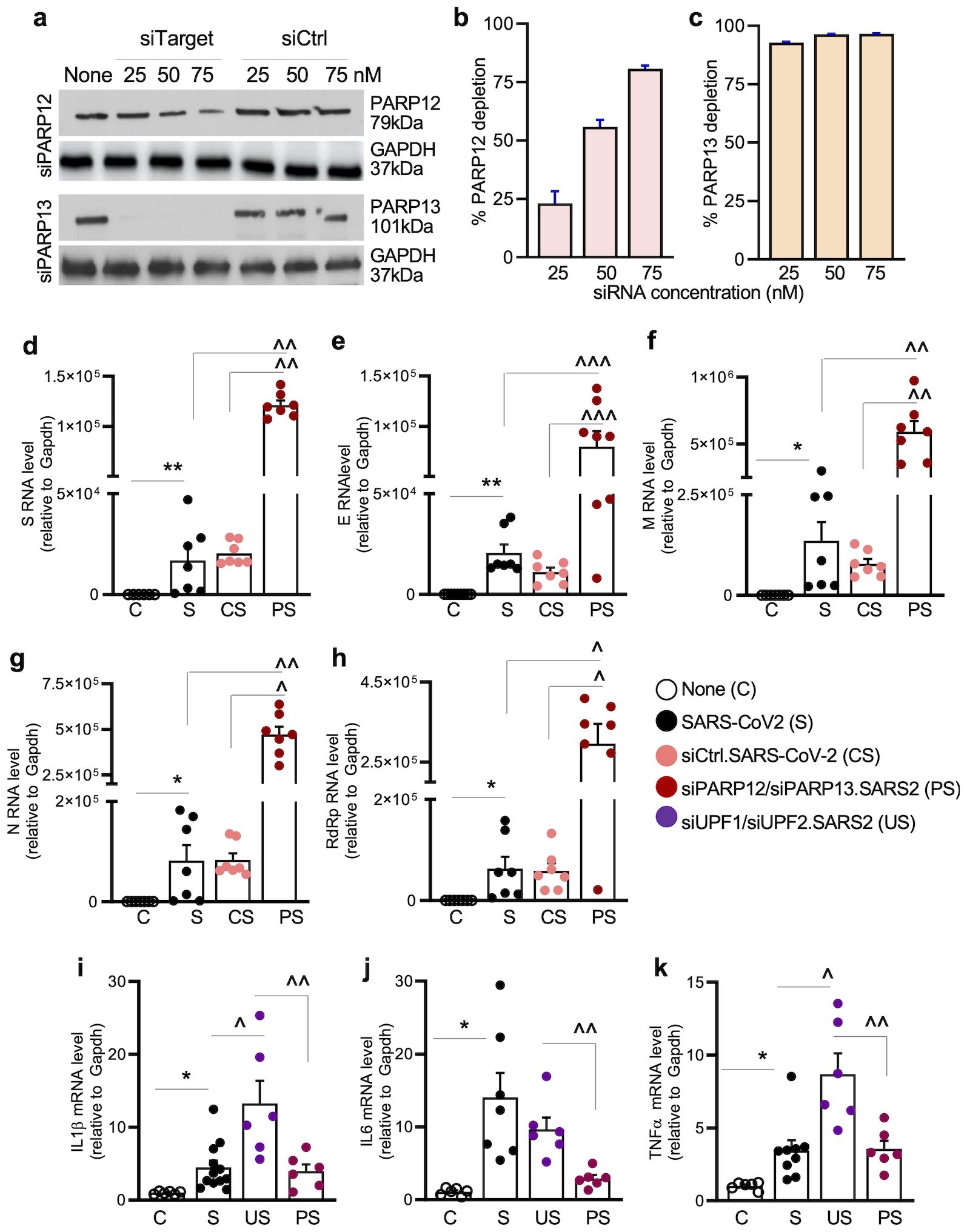
Figure 4. (a–c) PARP12/PARP13 depletion. A549ACE2+ cells were transfected with 25-75nM of control (siCtrl) or target-specific (siPARP12, siPARP13) siRNAs for 48 h. Representative western blots for PARP12 and PARP13 (control: GAPDH) are shown in (a). Densitometry analysis of PARP12 and PARP13 bands (relative to GAPDH) was performed, and percent depletion of PARP12 and PARP13 in cells transfected with target-specific (vs. siCtrl) siRNAs was calculated for (b, c). (d–k) Effects of PARP12/PARP13 depletion on SARS-CoV-2 RNA stability and cytokine response. A549ACE2+ cells were not transfected or were transfected with siRNAs (75nM each) for 48h, and mock infected or infected with SARS-CoV-2 (MOI: 1.0) for 24 h. RT-qPCR was performed to examine the levels of SARS-CoV-2 subgenomic RNAs (S, E, M, N) and genomic RdRP RNA (d–h), and host cytokine gene expression (IL1β, IL6, and TNFα (i–k). Data were normalized to Gapdh. Individual data points from each biological sample (average of duplicate observations) and mean ± SEM values (n=6–8 biological samples/group) are plotted. Significance was calculated by one-way ANOVA with Tukey’s or Kruskal–Wallis/Dunn’s post hoc tests; p-values of ≤0.05, ≤0.01, and ≤0.001 are annotated with 1, 2, and 3 symbols, respectively (*control vs. infected; ^infected vs. siPARP12 and/or siPARP13-treated/infected). Horizontal bars show the compared groups. p-values of ≤0.05*,^, ≤0.01,^^, and ≤0.001,^^^ are plotted (control vs. infected; ^infected vs. siPARP12-treated/infected or siPARP13-treated/infected).
RT-qPCR analysis of A549hACE2+ cells transfected with siPARP12/siPARP13 (75 nM each) or siCtrl siRNA followed by SARS-CoV-2 infection (MOI: 1.0) was conducted to evaluate the effects of PARP12/PARP13 on viral RNA. The siCtrl transfection did not affect the amounts of S, E, M, and N subgenomic RNAs after SARS-CoV-2 infection for 24 h (Figures 4e–h), whereas 6.5–7.8-fold, 6.1–8.2-fold, 5.8–6.5-fold, and 6.8–7.6-fold increases in S, E, M, and N subgenomic RNA levels, respectively, were observed in PARP12/PARP13-depleted (vs. control) cells infected with SARS-CoV-2 (Figures 4e–h). A similar increase in RdRP-encoding genomic RNA was also observed in PARP12/PARP13-depleted (vs. siCtrl) cells infected with SARS-CoV-2 (Figure 4h).
Moreover, a significant increase in the expression of proinflammatory cytokines (IL1β, IL6, and TNFα) was observed in SARS-CoV-2-infected (vs. mock-infected) cells (Figures 4i–k). A further >2-fold increase in IL1β and TNFα gene expression was observed in UPF1/UPF2-depleted (vs. control) cells but not in PARP12/PARP13-depleted (vs. control) cells infected with SARS-CoV-2.To further verify the effects of PARP12/PARP13 on SARS-CoV-2, A549hACE2+ cells were transfected with 1 μg of expression plasmids (pCAGGS.PARP12 and/or pCAGGS.PARP13) prior to SARS-CoV-2 infection (MOI: 1.0). RT-qPCR analysis showed a 587–663-fold increase in PARP12 and PARP13 expression levels in cells transfected with recombinant (vs. empty pCAGGS) plasmids (Figures 5a, b). Upon infection with SARS-CoV-2 (MOI: 1.0) for 24 h, PARP12/PARP13-overexpressing A549hACE2+ cells exhibited 55.8%–85.1%, 69.5%–86.3%, 87.5%–95.3%, and 66.6%–84.2% declines in subgenomic RNAs encoding S, E, M, and N, respectively, compared to nontransfected or pCAGGS-transfected cells infected with SARS-CoV-2 (Figures 5c–f). A similar decline in RdRP-encoding genomic RNA was observed in PARP12/PARP13-overexpressing (vs. control) cells infected with SARS-CoV-2 (Figure 5g). SARS-CoV-2 transcripts were not detectable in mock-infected cells.
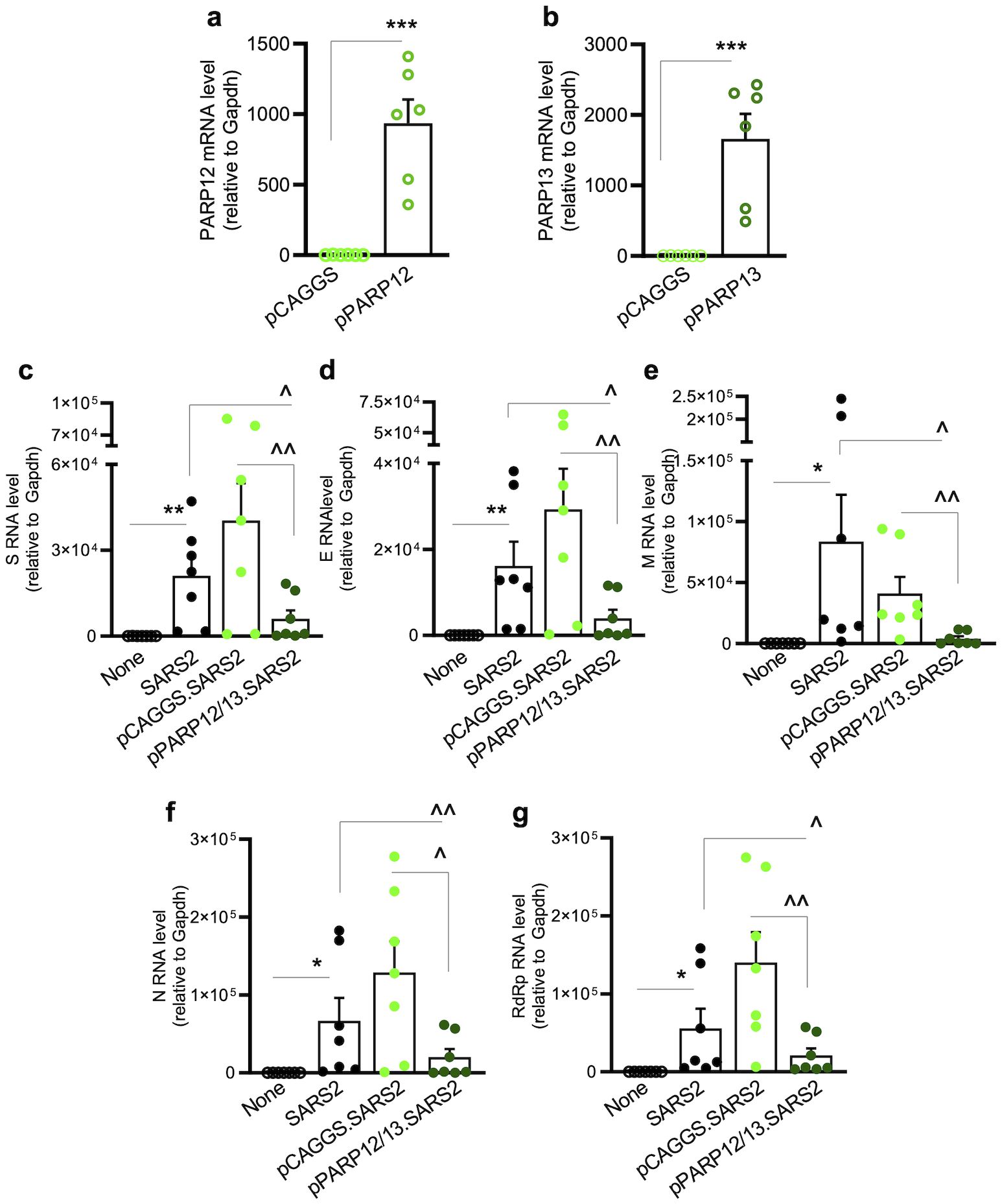
Figure 5. PARP12/PARP13 overexpression decreases SARS-CoV-2 RNA levels. A549ACE2+ cells were transfected with pCAGGS, pCAGGS-PARP12 and/or pCAGGS-PARP13 expression plasmids (1μg each) for 48h, mock infected or infected with SARS-CoV-2 (MOI: 1.0) for 24 h, and analyzed by RT-qPCR. (a, b) PARP12 and PARP13 transcript levels. (c–f) SARS-CoV-2 subgenomic RNAs (S, E, M, N) were amplified with a common leader sequence and target-specific primers, and genomic RNA was amplified using RdRp-specific primers. Target-specific Ct values were normalized to Gapdh Ct values, and relative RNA levels were calculated. Individual data-points from each biological sample (average of duplicate observations) and mean ± SEM values (n=5–8 biological samples per group) are plotted. Significance was calculated by Student’s unpaired t-test with Mann-Whitney U-test (a, b) and one-way ANOVA with Tukey’s or Kruskal-Wallis/Dunn’s post-hoc tests (c–h), and p-values of ≤0.05, ≤0.01, and ≤0.001 are shown with 1, 2, and 3 symbols, respectively (*control vs. infected; ^infected vs. siPARP12 and/or siPARP13-treated/infected). Horizontal bars show the compared groups. p-values of ≤0.05*,^, ≤0.01,^^, and ≤0.001,^^^ are plotted (control vs. infected; ^infected vs. siPARP12-treated/infected or siPARP13-treated/infected).
Together, the results presented in Figures 3, 4, 5 suggest that (a) PARP12 and PARP13 were markedly increased in lung epithelial cells and lung tissue of hACE2tg mice in response to SARS-CoV-2 infection; (b) our findings provide additional evidence for the inhibitory effects of SARS-CoV-2 on NMD effectors in mice; and (c) a significant increase in viral transcripts upon PARP12/PARP13 depletion, and an extensive decline in SARS-CoV-2 transcripts upon PARP12/PARP13 overexpression, suggest that PARP12 and PARP13 constitute part of the host defense mechanism against viral infection. Moreover, an increase in antiviral immune response following depletion of UPF1/UPF2—but not PARP12/PARP13—further supports the conclusion that downregulation of UPF1/UPF2 and upregulation of PARP12/PARP13 by SARS-CoV-2 occur as part of a host defense mechanism to regulate the antiviral state.
PARP12/PARP13 enhance NMD-mediated degradation of SARS-CoV-2 RNA and control viral replication
Considering that PARP12/PARP13 bind viral RNA as well as exonucleases of the NMD pathway (17), and based on our findings in the above figures (Figures 3-5), we aimed to test whether PARP12/PARP13 promote the degradation of SARS-CoV-2 RNA in the cytosol via the NMD pathway.
We first examined the effects of PARP12/PARP13 on NMD reporters. A549hACE2+ cells were transfected with siCtrl, siPARP12, or siPARP13 siRNAs for 48 h and then transfected with NS39 and WT reporters, which encode an rLuc ORF in-frame fused with β-globin. Because of a nonsense mutation in β-globin, NS39 transcripts are susceptible to the NMD pathway. RT-qPCR analysis showed 88.93%–90.01% lower accumulation of NS39 reporter RNA than WT reporter RNA in nontransfected and siCtrl-transfected cells (Figure 6a). The amount of NS39 reporter RNA increased by 3.08–3.98-fold and 1.52–2.09-fold, respectively, in PARP12- and PARP13-depleted (vs. control) A549hACE2+ cells (Figure 6a). No significant effects of PARP12 or PARP13 depletion were noted on WT reporter RNA levels.
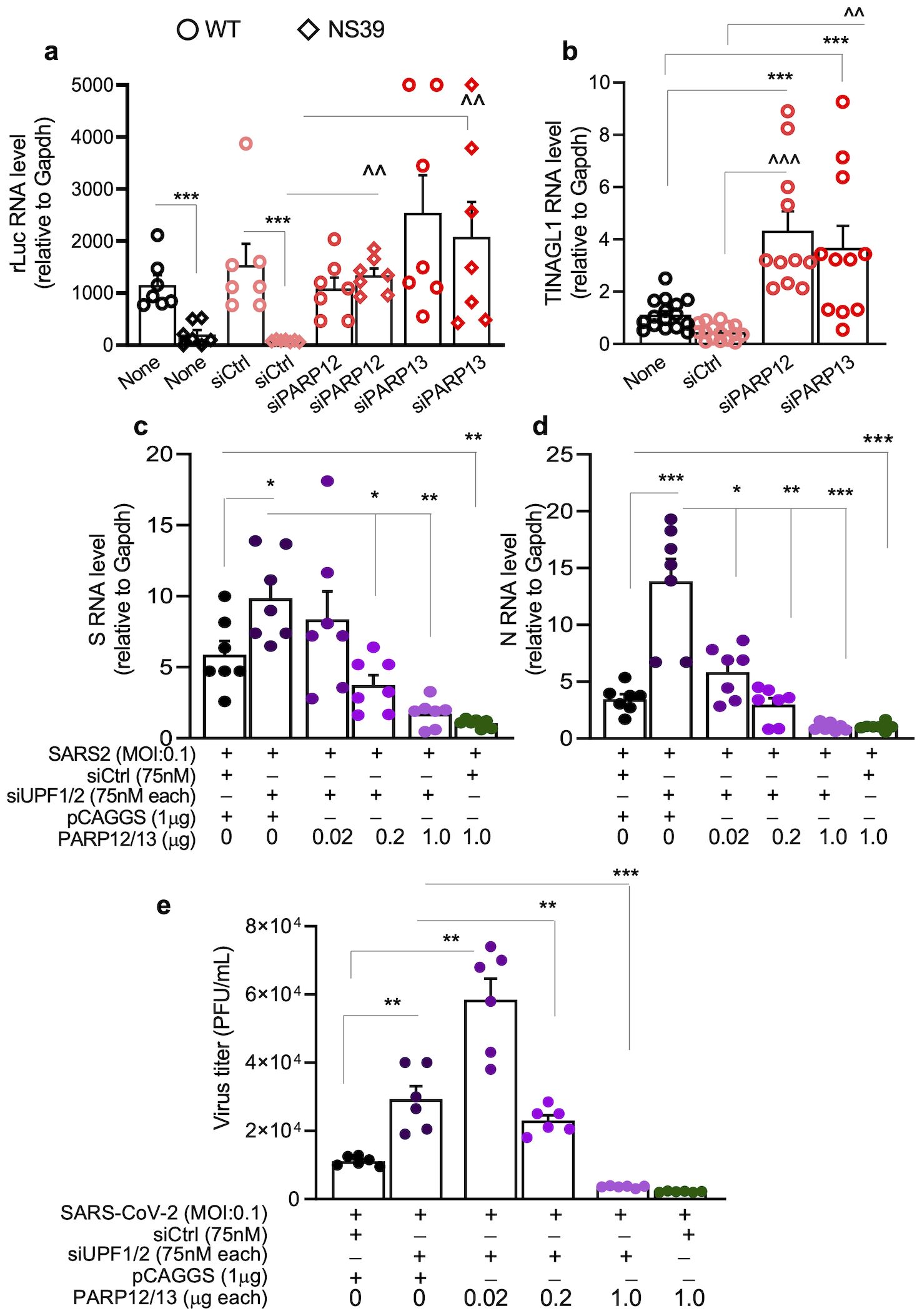
Figure 6. PARP12/PARP13-NMD regulation of SARS-CoV-2 RNA decay. (a, b) PARP12/PARP13 depletion enhances the stability of NMD target reporters. A549hACE2+ cells were transfected with siCtrl, siPARP12, or siPARP13 siRNAs (75nM each) for 48h and WT (NMD non-target) or NS39 (NMD target) reporter (1µg each) for 24h. Cells were analyzed by RT-qPCR for (a) rLuc encoded by WT and NS39 reporters and (b) TINAGL1 (endogenous NMD target) levels. Each biological sample (average of duplicate analysis) is plotted as a dot, and mean ± SEM values derived from n=6–8 biological samples per group are plotted as bars. Significance was calculated by one-way ANOVA with Tukey’s or Kruskal–Wallis/Dunn’s post hoc tests; p-values of 0.05, ≤0.01, and ≤0.001 are plotted with 1, 2, and 3 symbols, respectively (*WT vs. NS39; ^siCtrl vs. siPARP12 or siPARP13 in NS39-transfected groups). Horizontal bars show the compared groups. (c, d) Effects of PARP12/PARP13 and UPF1/UPF2 on SARS-CoV-2 transcripts. A549ACE2+ cells were transfected with siCtrl or siUPF1 and siUPF2 siRNAs (75nM each) for 48h, and 0.02, 0.2, or 1.0-µg of PAPR12 and PARP13 encoding pCAGGS plasmids (control: 1μg pCAGGS) for 24h. Cells were then infected with SARS-CoV-2 (MOI: 0.1) for 24h, and analyzed by RT-qPCR for S and N subgenomic RNAs using leader sequence and target-specific reverse primers. For panels a–d, target-specific Ct values were normalized to Gapdh Ct values, and relative RNA levels of the targets were calculated. Individual data points from each biological sample (average of duplicate observations) and mean ± SEM values (n = 6–8 biological samples per group) are plotted. (e) PARP12/PARP13 and UPF1/UPF2 regulation of SARS-CoV-2 replication. A549ACE2+ cells were sequentially transfected with siCtrl or siUPF1 and siUPF2 siRNAs (75nM each, 48h) and PAPR12- and PARP13-encoding pCAGGS plasmids (0.02-1.0-µg each, 24h) and then infected with SARS-CoV-2 (MOI: 0.1, 24h). Culture supernatants (10-fold dilutions up to 10-6) were analyzed in triplicate by plaque assay, as described in Methods. Significance for c–e was calculated by one-way ANOVA with Tukey’s or Kruskal–Wallis/Dunn’s post hoc tests; p-values are plotted as *p≤0.05, **p≤0.01, ***p≤0.001. Horizontal bars show the compared groups. For (a, b) p-values of ≤0.05*,^, ≤0.01,^^, and ≤0.001,^^^ are plotted (WT vs. NS39; ^siCtrl vs. siPARP12 or siPARP13 in NS39-transfected groups). For (c–e), p-values are plotted as ≤0.05*, ≤0.01, ≤0.001* and horizontal bars show the compared groups.
Alternatively spliced mRNA for TINAGL1 (an endogenous NMD target) also increased by 3.37–9.76-fold and 2.62–7.92-fold, respectively, in PARP12- and PARP13-depleted (vs. control) cells (Figure 6b). These results suggest that PARP12 and PARP13 signal NMD-mediated degradation of endogenous and exogenous aberrant transcripts in human lung epithelial cells.
To further evaluate PARP12/PARP13 synergy with NMD in modulating SARS-CoV-2 RNA stability, A549hACE2+ cells were sequentially transfected with siUPF1/UPF2 siRNAs (75 nM each) and PARP12/PARP13 expression plasmids (0.02–1.0 μg each) and then infected with SARS-CoV-2 (MOI: 0.1) for 24 h. Cells transfected with siCtrl and pCAGGS served as negative controls, while cells depleted of UPF1/UPF2 or overexpressing PARP12/PARP13 served as positive controls. No SARS-CoV-2 RNA was detected in cells transfected with siCtrl, UPF1/UPF2 siRNAs, or empty or PARP12/13 expression plasmids prior to infection.
After SARS-CoV-2 infection, cells transfected with siUPF1/siUPF2 (vs. siCtrl or pCAGGS) exhibited a 68.50%–338.54% increase in subgenomic RNAs encoding S and N viral proteins (Figures 6c, d). Overexpression of PARP12/PARP13 with UPF1/UPF2 depletion had a dose-dependent effect: transfection of 0.02 μg, 0.2 μg, and 1.0 μg of each PARP12 and PARP13 expression plasmid suppressed the accumulation of subgenomic S RNA by 16.52%, 65.91%, and 83.22%, respectively, and of N RNA by 62.32%, 80.64%, and 92.98%, respectively, compared to cells transfected with siUPF1/siUPF2 alone (Figures 6c, d). SARS-CoV-2 transcripts were also decreased by 89.80%–93.02% in infected cells transfected with PARP12/PARP13 expression plasmids alone (vs. siCtrl/pCAGGS), consistent with the results shown in Figure 5.
These findings suggest that PARP12 and PARP13 synergize with the NMD pathway to enhance the degradation of aberrant host transcripts and SARS-CoV-2 RNAs in human lung epithelial cells.
To confirm that PARP12/PARP13 crosstalk with NMD to regulate SARS-CoV-2 replication, A549hACE2+ cells transfected with siUPF1/UPF2 siRNAs (75 nM each) and PARP12/PARP13 expression plasmids (0.02–1.0 μg each) were infected with SARS-CoV-2 (MOI: 0.1) for 24 h. Supernatants were titrated by plaque assay to evaluate the effects of UPF1/UPF2 and PARP12/PARP13 on viral replication. These data showed a 163% increase in infectious SARS-CoV-2 titers when supernatants from UPF1/UPF2-depleted (vs. siCtrl/pCAGGS-transfected) cells were used.
Overexpression of PARP12/PARP13 with UPF1/UPF2 depletion had a dose-dependent effect such that 0.02 μg each of PARP12 and PARP13 expression plasmids had a nonsignificant effect on siUPF1/siUPF2-dependent viral replication, whereas co-transfection of 0.2 μg (each) and 1 μg (each) of PARP12 and PARP13 expression plasmids suppressed the siUPF1/siUPF2-dependent increase in viral replication by 60.68% and 93.84%, respectively. These results suggest that PARP12/PARP13 crosstalk with UPF1/UPF2 to regulate NMD activity and control SARS-CoV-2 replication.
Discussion
NMD is a translation-dependent process that targets truncated transcripts through a set of highly conserved factors from yeast to humans (19). Briefly, NMD-associated effectors—including Ser/Thr protein kinase (SMG1), RNA helicase and ATPase (UPF1), and Regulator of Nonsense Transcripts 2 (UPF2)—recognize truncated open reading frames on the exon junction complex. Subsequent phosphorylation of UPF1 triggers the recruitment and/or signaling of (a) SMG6 endonucleolytic activity; (b) SMG5/SMG7 and SMG5/proline-rich nuclear receptor coactivator 2 (PNRC2)-mediated 5′ decapping and 3′ deadenylation, respectively, of cleaved mRNA products; and (c) exosome- and 5′–3′ exoribonuclease (XRN1)-mediated exonucleolytic decay of mRNA fragments (19, 20). Recent system-wide studies have found that NMD participates in the surveillance and degradation of positive-sense RNA viruses that undergo RNA synthesis and viral assembly in the host cytoplasm, and that these viruses have evolved mechanisms to disrupt antiviral NMD activity (21, 22). For example, knockdown of NMD factors (UPF1, SMG5, or SMG7) increased RNA synthesis and replication of SFV and Sindbis alphaviruses in HeLa cells (4). Viral proteins of Zika virus (ZIKV), West Nile virus, and hepatitis C virus have been suggested to degrade UPF1 (23, 24). The 3’ untranslated regions (UTR) of Rous sarcoma virus and Turnip crinkle virus (25, 26), as well as two proteins of human T-cell lymphotropic virus (27, 28), were implicated in protecting viral mRNA from NMD machinery through unknown mechanisms.
Our team previously demonstrated that MHV RNA, a mammalian coronavirus, is an NMD target, and depletion of NMD factors (UPF1, UPF2, SMG5, or SMG6) facilitated more efficient MHV replication in cultured cells (6). The observations from this study provide further evidence that NMD exerts an inhibitory effect on SARS-CoV-2. This is supported by the findings that SARS-CoV-2 infection suppressed the expression of UPF1, UPF2, and SMG1 in lung epithelial cells and lung tissues of infected mice. Additionally, UPF1/UPF2 depletion enhanced the abundance of subgenomic mRNAs for structural proteins (S, E, M, N) and the genomic RNA of SARS-CoV-2, thereby facilitating more efficient replication of the virus in human lung epithelial cells.
How coronaviruses regulate the NMD pathway is not yet fully understood. We previously found that the nucleocapsid (N) protein of MHV suppresses the NMD pathway (6) and others have reported that the SARS-CoV-2 N protein interacts with NMD factors (29). Recently, the SARS-CoV-2 N protein was shown to directly bind UPF2 and inhibit the helicase activity of UPF1, thereby disrupting the formation of the UPF1/UPF2 complex and inhibiting the NMD pathway (30, 31). These studies provide insight into viral N protein interactions with UPF1/UPF2 and highlight the need for further research to delineate how SARS-CoV-2 inhibits the NMD pathway and how NMD targets viral RNA.
The human genome encodes 17 PARPs, categorized into four subfamilies based on the presence or absence of structural motifs beyond the PARP domain. Signature motifs other than the PARP domain have not been identified in unclassified PARPs (PARP4, PARP6, PARP8, PARP10, PARP11, and PARP16). PARP1–3 bind DNA and play roles in DNA damage control (32). PARP9, PARP14, and PARP15 contain macrodomains that enable interaction with ADP-ribosylated proteins and have been shown to signal the transcription of interferon-stimulated genes and modulate the innate immune response (33, 34). PARP5a/b contain protein-binding ankyrin repeats and were initially identified as regulators of telomere elongation (35). Finally, PARP7, PARP12, and PARP13 contain one or more CCCH ZnF motifs for RNA binding and regulate RNA metabolism and translation (36). Overall, due to their distinct functional domains and enzymatic activities, PARPs have been implicated in diverse biological processes in health and disease and have also been attributed antiviral roles (10). For instance, PARP13 was found to inhibit retrovirus replication (37), and PARP12 and PARP13 have been shown to restrict replication of a broad range of cytoplasmic viral species (38–41). Published reports (42) and our data demonstrate that SARS-CoV-2 induces the expression of PARP9–10 and PARP12–14 in infected lung epithelial cells and lung tissues of infected mice. RNA-seq analyses of human bronchial cells, bronchoalveolar lavage fluid, and lung biopsies from ferrets, adenovirus–hACE2-expressing mice, and COVID-19 patients also revealed significant increases in PARP9, PARP12, and PARP14 across all tissues, and PARP13 specifically in lung tissues of SARS-CoV-2-infected hosts (43, 44). Furthermore, our data show that PARP12 and PARP13 restrict the accumulation of viral transcripts and the replication of SARS-CoV-2 in infected cells. An in silico drug screening study suggested that a specific PARP1 inhibitor, CVL218, may also offer therapeutic efficacy against SARS-CoV-2 (45), although neither published literature nor transcriptomic studies, nor our results, have shown PARP1 activation by SARS-CoV-2 infection in vitro or in vivo. Further studies employing chemical and genetic modulation of PARP1 are needed to verify its role in SARS-CoV-2 infection.
Overall, our findings provide strong evidence for the role of PARP12/PARP13 in inhibiting SARS-CoV-2 replication and support the development of PARP12/PARP13 agonists as potential broad-range antivirals.
Despite several studies, a unifying mechanism by which PARPs regulate the replication of RNA viruses has not yet been proposed. Initially, PARP13 was suggested to enhance retinoic acid-inducible gene I and interferon regulatory factor 3 signaling (46) or to degrade interferon mRNAs (8), thereby shaping anti-viral innate immunity. Subsequently, PARP13 was found to bind the 3′ UTR of MHV, the 5′ UTRs of human immunodeficiency virus, and the middle of the polycistronic transcript of Ebola virus (39, 47, 48). The specific viral motifs recognized by PARP13 were not identified in these studies, although it was suggested that CpG motifs in viral genomes are key to PARP13 binding (49) and its antiviral effects.
Our study shows that SARS-CoV-2, which harbors a CpG-suppressive genome (50) is also susceptible to PARP13-mediated decay. Others have documented that full-length PARP13 is more potent in inhibiting human immunodeficiency virus and influenza A virus than its short ZAP13 isoform, which lacks the C-terminal PARP domain (51, 52). Indeed, PARP13 binding to viral RNA has been shown to facilitate the recruitment of poly(A)-specific ribonuclease, RNA exosome enzymes, and decapping enzymes or RNA helicases (48, 53, 54) all of which are downstream effectors of the NMD pathway. However, confirmatory evidence for the role of full-length or short-length PARP13 in regulating NMD activity in the presence or absence of viral infection has not yet been provided in the published literature.
Our results demonstrate that PARP13 expression correlates directly with NMD activity: depletion of PARP13 enhanced the stability of both endogenous and exogenous NMD target mRNAs, whereas PARP13 overexpression had a dose-dependent effect on the degradation of NMD target mRNAs and SARS-CoV-2 transcripts. Moreover, PARP13 overexpression enhanced the NMD-mediated control of SARS-CoV-2 replication in a dose-dependent manner. Based on published studies and our findings, we propose that PARP13 binding to cytoplasmic viral RNAs facilitates the recruitment and activation of the RNA decay machinery, leading to the degradation of viral RNAs and inhibition of viral replication.
Unlike PARP13, PARP12 contains a catalytically active PARP domain capable of mono-ADP-ribosylating target proteins. The observation of increased PARP12 expression in cells that cleared Venezuelan equine encephalitis virus (VEEV) replication compared to those with persistent VEEV infection led to several studies showing that PARP12 restricted the replication of encephalomyocarditis virus, vesicular stomatitis virus, and Sindbis virus, among others, in various cell types (40, 55). It was hypothesized that the PARP12 ZnF motif might be essential for inhibition of viral replication. Indeed, PARP12 has been shown to translocate from the Golgi bodies to stress granules under stress stimuli, including infection with RNA viruses, and this translocation activity was abrogated upon mutation of the ZnF domains (56). Whether the PARP12 ZnF motifs bind viral RNA in stress granules, and whether such binding is required for viral RNA degradation, has not yet been established. Other studies have shown that PARP12 inhibits ZIKV replication via ADP-ribosylation and proteasomal degradation of the viral NS1 and NS3 proteins; this activity did not require the ZnF domains (57). Our findings provide initial evidence that PARP12, like PARP13, influences NMD activity and enhances the degradation of NMD-targeted aberrant transcripts and SARS-CoV-2 RNAs. Thus, PARP12 overexpression restricted SARS-CoV-2 replication in infected lung epithelial cells. Further studies will be needed to delineate the specific roles of the PARP12 ZnF and PARP domains in shaping the antiviral activity of NMD factors.
Our findings of increased antiviral immune response following UPF1/UPF2 depletion allow us to propose that UPF1/UPF2 and PARP12/PARP13 may also influence host defense mechanisms to establish an antiviral state. Indeed, PARP12/PARP13 bind exonucleases of the NMD pathway (17), and PARP13 (along with TRIM25) binds RNA to regulate gene expression, including genes that initiate antiviral immunity (18). Several other PARPs upregulated in response to SARS-CoV-2 and model coronavirus infections also exert antiviral activity (10). SARS-CoV-2 was shown to dysregulate the PARP antiviral response by disturbing NAD biosynthetic pathways that provide substrates for PARP activation. This disruption could be rescued by pharmacological activation of NAD synthesis (58), indicating that noncanonical PARP activities are limited by NAD availability and that pharmacological interventions enhancing NAD levels should be considered to improve innate immunity against coronaviruses.
PARP12 and PARP13 themselves function as interferon-stimulated genes. PARP12, in particular, acts as a strong inhibitor of ZIKV replication through ADP-ribosylation of NS1 and NS3, triggering their proteasome-mediated degradation and reducing their availability for viral replication (57). We thus postulate that while inhibition of the NMD pathway favors SARS-CoV-2 RNA stability and viral replication, a decline in UPF1 and UPF2—together with upregulation of PARP12/PARP13—may benefit the host by promoting a balanced antiviral immune response.
In summary, this study investigated the role of the NMD pathway and PARP12/PARP13 in SARS-CoV-2 infection. We demonstrate that (a) UPF1/UPF2-mediated NMD degrades endogenous and exogenous aberrant transcripts in human lung epithelial cells; (b) NMD activity targets SARS-CoV-2 RNA for degradation; and (c) SARS-CoV-2 suppresses the expression of NMD effectors in lung epithelial cells and lung tissue of mice, thereby facilitating viral replication. We also found that (d) PARP12/PARP13 were increased in lung epithelial cells and lung tissue of SARS-CoV-2-infected mice, and (e) PARP12/PARP13 had antiviral effects, as indicated by a direct correlation between PARP12/PARP13 expression and inhibition of RNA stability and replication of SARS-CoV-2 in lung epithelial cells. Furthermore, (f) PARP12/PARP13 augmented NMD activity in degrading endogenous and exogenous aberrant transcripts as well as SARS-CoV-2 RNA in human lung epithelial cells. These results support the proposition that PARP12/PARP13 agonists may serve as adjuvants to enhance the efficacy of antiviral therapies in controlling SARS-CoV-2 infection and COVID-19 pathogenesis.
Data availability statement
The data presented in the study are deposited ni the Gene Expression Omnibus repository, accession number GSE255647.
Ethics statement
The animal study was approved by Institutional Animal Care and Use Committee. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
NL: Methodology, Formal Analysis, Data curation, Writing – review & editing, Investigation. SC: Data curation, Writing – review & editing, Formal Analysis, Investigation. KR: Formal Analysis, Investigation, Writing – review & editing. C-TT: Resources, Writing – review & editing, Supervision. SM: Resources, Writing – review & editing. NG: Project administration, Conceptualization, Writing – review & editing, Resources, Writing – original draft, Software, Funding acquisition, Supervision.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported in part by a grant from the Institute for Human Infections and Immunity at University of Texas Medical Branch (UTMB), Galveston to NJG and Public Health Service grant R01AI146081 to SM from the National Institutes of Health. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
COVID-19, Coronavirus disease of 2019; hACE2, human angiotensin-converting enzyme 2 receptor; NMD, nonsense-mediated mRNA decay; NS39, NMD target construct; PARP, poly(ADP)ribose polymerase; PTC, premature termination codon; SARS-CoV-2, Severe acute respiratory syndrome coronavirus 2; UPF1/UPF2, up-framshift 1 or 2; UTR, untranslated region. S, spike; E, envelope; M, membrane; N, nucleocapsid; RdRP, RNA-d-RNA polymerase are SARS-CoV-2 proteins.
References
1. Younger DS. Coronavirus 2019: clinical and neuropathological aspects. Curr Opin Rheumatol. (2021) 33:49–57. doi: 10.1097/BOR.0000000000000769
2. Watson OJ, Barnsley G, Toor J, Hogan AB, Winskill P, and Ghani AC. Global impact of the first year of COVID-19 vaccination: a mathematical modelling study. Lancet Infect Dis. (2022) 22:1293–302. doi: 10.1016/S1473-3099(22)00320-6
3. Kim D, Lee JY, Yang JS, Kim JW, Kim VN, and Chang H. The architecture of SARS-coV-2 transcriptome. Cell. (2020) 181:914–921 e10. doi: 10.1016/j.cell.2020.04.011
4. Balistreri G, Horvath P, Schweingruber C, Zund D, McInerney G, Merits A, et al. The host nonsense-mediated mRNA decay pathway restricts Mammalian RNA virus replication. Cell Host Microbe. (2014) 16:403–11. doi: 10.1016/j.chom.2014.08.007
5. Garcia D, Garcia S, and Voinnet O. Nonsense-mediated decay serves as a general viral restriction mechanism in plants. Cell Host Microbe. (2014) 16:391–402. doi: 10.1016/j.chom.2014.08.001
6. Wada M, Lokugamage KG, Nakagawa K, Narayanan K, and Makino S. Interplay between coronavirus, a cytoplasmic RNA virus, and nonsense-mediated mRNA decay pathway. Proc Natl Acad Sci U.S.A. (2018) 115:E10157–66. doi: 10.1073/pnas.1811675115
7. Ke Y, Zhang J, Lv X, Zeng X, and Ba X. Novel insights into PARPs in gene expression: regulation of RNA metabolism. Cell Mol Life Sci. (2019) 76:3283–99. doi: 10.1007/s00018-019-03120-6
8. Schwerk J, Soveg FW, Ryan AP, Thomas KR, Hatfield LD, Ozarkar S, et al. RNA-binding protein isoforms ZAP-S and ZAP-L have distinct antiviral and immune resolution functions. Nat Immunol. (2019) 20:1610–20. doi: 10.1038/s41590-019-0527-6
9. Zhu Y, Wang X, Goff SP, and Gao G. Translational repression precedes and is required for ZAP-mediated mRNA decay. EMBO J. (2012) 31:4236–46. doi: 10.1038/emboj.2012.271
10. Malgras M, Garcia M, Jousselin C, Bodet C, and Leveque N. The antiviral activities of poly-ADP-ribose polymerases. Viruses. (2021) 13:582–95. doi: 10.3390/v13040582
11. Yoshikawa T, Hill TE, Yoshikawa N, Popov VL, Galindo CL, Garner HR, et al. Dynamic innate immune responses of human bronchial epithelial cells to severe acute respiratory syndrome-associated coronavirus infection. PloS One. (2010) 5:e8729. doi: 10.1371/journal.pone.0008729
12. Tseng CT, Huang C, Newman P, Wang N, Narayanan K, Watts DM, et al. Severe acute respiratory syndrome coronavirus infection of mice transgenic for the human Angiotensin-converting enzyme 2 virus receptor. J Virol. (2007) 81:1162–73. doi: 10.1128/JVI.01702-06
13. Thermann R, Neu-Yilik G, Deters A, Frede U, Wehr K, Hagemeier C, et al. Binary specification of nonsense codons by splicing and cytoplasmic translation. EMBO J. (1998) 17:3484–94. doi: 10.1093/emboj/17.12.3484
14. Boelz S, Neu-Yilik G, Gehring NH, Hentze MW, and Kulozik AE. A chemiluminescence-based reporter system to monitor nonsense-mediated mRNA decay. Biochem Biophys Res Commun. (2006) 349:186–91. doi: 10.1016/j.bbrc.2006.08.017
15. Shum EY, Jones SH, Shao A, Chousal JN, Krause MD, Chan WK, et al. The antagonistic gene paralogs upf3a and upf3b govern nonsense-mediated RNA decay. Cell. (2016) 165:382–95. doi: 10.1016/j.cell.2016.02.046
16. Colombo M, Karousis ED, Bourquin J, Bruggmann R, and Muhlemann O. Transcriptome-wide identification of NMD-targeted human mRNAs reveals extensive redundancy between SMG6- and SMG7-mediated degradation pathways. RNA. (2017) 23:189–201. doi: 10.1261/rna.059055.116
17. Todorova T, Bock FJ, and Chang P. PARP13 regulates cellular mRNA post-transcriptionally and functions as a pro-apoptotic factor by destabilizing TRAILR4 transcript. Nat Commun. (2014) 5:5362. doi: 10.1038/ncomms6362
18. Busa VF, Ando Y, Aigner S, Yee BA, Yeo GW, and Leung AKL. Transcriptome regulation by PARP13 in basal and antiviral states in human cells. iScience. (2024) 27:109251. doi: 10.1016/j.isci.2024.109251
19. Mailliot J, Vivoli-Vega M, and Schaffitzel C. No-nonsense: insights into the functional interplay of nonsense-mediated mRNA decay factors. Biochem J. (2022) 479:973–93. doi: 10.1042/BCJ20210556
20. Kurosaki T, Popp MW, and Maquat LE. Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat Rev Mol Cell Biol. (2019) 20:406–20. doi: 10.1038/s41580-019-0126-2
21. Kebaara BW and Atkin AL. Long 3'-UTRs target wild-type mRNAs for nonsense-mediated mRNA decay in Saccharomyces cerevisiae. Nucleic Acids Res. (2009) 37:2771–8. doi: 10.1093/nar/gkp146
22. Imamachi N, Salam KA, Suzuki Y, and Akimitsu N. A GC-rich sequence feature in the 3' UTR directs UPF1-dependent mRNA decay in mammalian cells. Genome Res. (2017) 27:407–18. doi: 10.1101/gr.206060.116
23. Fontaine KA, Leon KE, Khalid MM, Tomar S, Jimenez-Morales D, Dunlap M, et al. The cellular NMD pathway restricts zika virus infection and is targeted by the viral capsid protein. mBio. (2018) 9:e02126-18. doi: 10.1128/mBio.02126-18
24. Ramage HR, Kumar GR, Verschueren E, Johnson JR, Von Dollen J, Johnson T, et al. A combined proteomics/genomics approach links hepatitis C virus infection with nonsense-mediated mRNA decay. Mol Cell. (2015) 57:329–40. doi: 10.1016/j.molcel.2014.12.028
25. Withers JB and Beemon KL. Structural features in the Rous sarcoma virus RNA stability element are necessary for sensing the correct termination codon. Retrovirology. (2010) 7:65. doi: 10.1186/1742-4690-7-65
26. May JP, Yuan X, Sawicki E, and Simon AE. RNA virus evasion of nonsense-mediated decay. PloS Pathog. (2018) 14:e1007459. doi: 10.1371/journal.ppat.1007459
27. Fiorini F, Robin JP, Kanaan J, Borowiak M, Croquette V, Le Hir H, et al. HTLV-1 Tax plugs and freezes UPF1 helicase leading to nonsense-mediated mRNA decay inhibition. Nat Commun. (2018) 9:431. doi: 10.1038/s41467-017-02793-6
28. Nakano K, Ando T, Yamagishi M, Yokoyama K, Ishida T, Ohsugi T, et al. Viral interference with host mRNA surveillance, the nonsense-mediated mRNA decay (NMD) pathway, through a new function of HTLV-1 Rex: implications for retroviral replication. Microbes Infect. (2013) 15:491–505. doi: 10.1016/j.micinf.2013.03.006
29. Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. (2020) 583:459–68. doi: 10.1038/s41586-020-2286-9
30. Nuccetelli V, Mghezzi-Habellah M, Deymier S, Roisin A, Gérard-Baraggia F, Rocchi C, et al. The SARS-CoV-2 nucleocapsid protein interferes with the full enzymatic activation of UPF1 and its interaction with UPF2. Nucleic Acids Res. (2025) 53:gkaf010. doi: 10.1093/nar/gkaf010
31. Mallick M, Boehm V, Xue G, Blackstone M, Gehring Niels H, and Chakrabarti S. Modulation of UPF1 catalytic activity upon interaction of SARS-CoV-2 Nucleocapsid protein with factors involved in nonsense mediated-mRNA decay. Nucleic Acids Res. (2024) 52:13325–39. doi: 10.1093/nar/gkae829
32. Huang D and Kraus WL. The expanding universe of PARP1-mediated molecular and therapeutic mechanisms. Mol Cell. (2022) 82:2315–34. doi: 10.1016/j.molcel.2022.02.021
33. Iwata H, Goettsch C, Sharma A, Ricchiuto P, Goh WW, Halu A, et al. PARP9 and PARP14 cross-regulate macrophage activation via STAT1 ADP-ribosylation. Nat Commun. (2016) 7:12849. doi: 10.1038/ncomms12849
34. Higashi H, Maejima T, Lee LH, Yamazaki Y, Hottiger MO, Singh SA, et al. A study into the ADP-ribosylome of IFN-gamma-stimulated THP-1 human macrophage-like cells identifies ARTD8/PARP14 and ARTD9/PARP9 ADP-ribosylation. J Proteome Res. (2019) 18:1607–22. doi: 10.1021/acs.jproteome.8b00895
35. Zimmerlin L and Zambidis ET. Pleiotropic roles of tankyrase/PARP proteins in the establishment and maintenance of human naive pluripotency. Exp Cell Res. (2020) 390:111935. doi: 10.1016/j.yexcr.2020.111935
36. Brady PN, Goel A, and Johnson MA. Poly(ADP-ribose) polymerases in host-pathogen interactions, inflammation, and immunity. Microbiol Mol Biol Rev. (2019) 83:e00038-18. doi: 10.1128/MMBR.00038-18
37. Gao G, Guo X, and Goff SP. Inhibition of retroviral RNA production by ZAP, a CCCH-type zinc finger protein. Science. (2002) 297:1703–6. doi: 10.1126/science.1074276
38. Bick MJ, Carroll JW, Gao G, Goff SP, Rice CM, and MacDonald MR. Expression of the zinc-finger antiviral protein inhibits alphavirus replication. J Virol. (2003) 77:11555–62. doi: 10.1128/jvi.77.21.11555-11562.2003
39. Muller S, Moller P, Bick MJ, Wurr S, Becker S, Gunther S, et al. Inhibition of filovirus replication by the zinc finger antiviral protein. J Virol. (2007) 81:2391–400. doi: 10.1128/JVI.01601-06
40. Atasheva S, Akhrymuk M, Frolova EI, and Frolov I. New PARP gene with an anti-alphavirus function. J Virol. (2012) 86:8147–60. doi: 10.1128/JVI.00733-12
41. Chiu HP, Chiu H, Yang CF, Lee YL, Chiu FL, Kuo HC, et al. Inhibition of Japanese encephalitis virus infection by the host zinc-finger antiviral protein. PloS Pathog. (2018) 14:e1007166. doi: 10.1371/journal.ppat.1007166
42. Grunewald ME, Shaban MG, Mackin SR, Fehr AR, and Perlman S. Murine coronavirus infection activates the aryl hydrocarbon receptor in an indoleamine 2,3-dioxygenase-independent manner, contributing to cytokine modulation and proviral TCDD-inducible-PARP expression. J Virol. (2020) 94:e0176. doi: 10.1128/JVI.01743-19
43. Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Moller R, et al. Imbalanced host response to SARS-coV-2 drives development of COVID-19. Cell. (2020) 181:1036–1045 e9. doi: 10.1016/j.cell.2020.04.026
44. Lieberman NAP, Peddu V, Xie H, Shrestha L, Huang ML, Mears MC, et al. In vivo antiviral host transcriptional response to SARS-CoV-2 by viral load, sex, and age. PloS Biol. (2020) 18:e3000849. doi: 10.1371/journal.pbio.3000849
45. Ge Y, Tian T, Huang S, Wan F, Li J, Li S, et al. An integrative drug repositioning framework discovered a potential therapeutic agent targeting COVID-19. Signal Transduction Targeted Ther. (2021) 6:165. doi: 10.1038/s41392-021-00568-6
46. Hayakawa S, Shiratori S, Yamato H, Kameyama T, Kitatsuji C, Kashigi F, et al. ZAPS is a potent stimulator of signaling mediated by the RNA helicase RIG-I during antiviral responses. Nat Immunol. (2011) 12:37–44. doi: 10.1038/ni.1963
47. Mao R, Nie H, Cai D, Zhang J, Liu H, Yan R, et al. Inhibition of hepatitis B virus replication by the host zinc finger antiviral protein. PloS Pathog. (2013) 9:e1003494. doi: 10.1371/journal.ppat.1003494
48. Zhu Y, Chen G, Lv F, Wang X, Ji X, Xu Y, et al. Zinc-finger antiviral protein inhibits HIV-1 infection by selectively targeting multiply spliced viral mRNAs for degradation. Proc Natl Acad Sci U.S.A. (2011) 108:15834–9. doi: 10.1073/pnas.1101676108
49. Kmiec D, Nchioua R, Sherrill-Mix S, Sturzel CM, Heusinger E, Braun E, et al. CpG frequency in the 5' Third of the env gene determines sensitivity of primary HIV-1 strains to the zinc-finger antiviral protein. mBio. (2020) 11:e02903. doi: 10.1128/mBio.02903-19
50. Matyasek R and Kovarik A. Mutation patterns of human SARS-coV-2 and bat raTG13 coronavirus genomes are strongly biased towards C>U transitions, indicating rapid evolution in their hosts. Genes (Basel). (2020) 11:761. doi: 10.3390/genes11070761
51. Liu CH, Zhou L, Chen G, and Krug RM. Battle between influenza A virus and a newly identified antiviral activity of the PARP-containing ZAPL protein. Proc Natl Acad Sci U.S.A. (2015) 112:14048–53. doi: 10.1073/pnas.1509745112
52. Ficarelli M, Wilson H, Pedro Galao R, Mazzon M, Antzin-Anduetza I, Marsh M, et al. KHNYN is essential for the zinc finger antiviral protein (ZAP) to restrict HIV-1 containing clustered CpG dinucleotides. Elife. (2019) 8:e46767. doi: 10.7554/eLife.46767
53. Chen G, Guo X, Lv F, Xu Y, and Gao G. p72 DEAD box RNA helicase is required for optimal function of the zinc-finger antiviral protein. Proc Natl Acad Sci U.S.A. (2008) 105:4352–7. doi: 10.1073/pnas.0712276105
54. Luo X, Wang X, Gao Y, Zhu J, Liu S, Gao G, et al. Molecular mechanism of RNA recognition by zinc-finger antiviral protein. Cell Rep. (2020) 30:46–52 e4. doi: 10.1016/j.celrep.2019.11.116
55. Atasheva S, Frolova EI, and Frolov I. Interferon-stimulated poly(ADP-Ribose) polymerases are potent inhibitors of cellular translation and virus replication. J Virol. (2014) 88:2116–30. doi: 10.1128/JVI.03443-13
56. Welsby I, Hutin D, Gueydan C, Kruys V, Rongvaux A, and Leo O. PARP12, an interferon-stimulated gene involved in the control of protein translation and inflammation. J Biol Chem. (2014) 289:26642–57. doi: 10.1074/jbc.M114.589515
57. Li L, Zhao H, Liu P, Li C, Quanquin N, Ji X, et al. PARP12 suppresses Zika virus infection through PARP-dependent degradation of NS1 and NS3 viral proteins. Sci Signal. (2018) 11:eaa59332. doi: 10.1126/scisignal.aas9332
Keywords: SARS-CoV-2, poly(ADP)ribose polymerase, nonsense mediated RNA decay, antiviral response, COVID-19
Citation: Lokugamage N, Choudhuri S, Rayavara K, Tseng C-T, Makino S and Garg NJ (2025) Effects of the cross-talk between PARP12/PARP13 and nonsense mediated RNA decay pathway on RNA stability and replication of SARS-CoV-2. Front. Virol. 5:1691166. doi: 10.3389/fviro.2025.1691166
Received: 22 August 2025; Accepted: 27 October 2025;
Published: 20 November 2025.
Edited by:
Jayasri Das Sarma, Indian Institute of Science Education and Research Kolkata, IndiaReviewed by:
Rahul Basu, University of Texas at San Antonio, United StatesLucky Sarkar, Lerner Research Institute, United States
Copyright © 2025 Lokugamage, Choudhuri, Rayavara, Tseng, Makino and Garg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nisha Jain Garg, bmlnYXJnQHV0bWIuZWR1