 Kelsey H. Collins1,2
Kelsey H. Collins1,2 Walter Herzog1,2Graham Z. MacDonald1
Walter Herzog1,2Graham Z. MacDonald1 Raylene A. Reimer1,3
Raylene A. Reimer1,3 Jaqueline L. Rios1,2,4Ian C. Smith1Ronald F. Zernicke1,5,6
Jaqueline L. Rios1,2,4Ian C. Smith1Ronald F. Zernicke1,5,6 David A. Hart1,2,7,8*
David A. Hart1,2,7,8*- 1Human Performance Laboratory, Faculty of Kinesiology, University of Calgary, Calgary, AB, Canada
- 2McCaig Institute for Bone and Joint Health, University of Calgary, Calgary, AB, Canada
- 3Department of Biochemistry and Molecular Biology, University of Calgary, Calgary, AB, Canada
- 4CAPES Foundation, Brasilia, Brazil
- 5Departments of Orthopaedic Surgery and Biomedical Engineering, School of Kinesiology, University of Michigan, Ann Arbor, MI, United States
- 6Department of Surgery, Department of Physiology and Pharmacology, University of Calgary, Calgary, AB, Canada
- 7Department of Family Practice, The Centre for Hip Health and Mobility, University of British Columbia, Vancouver, BC, Canada
- 8Alberta Health Services Bone and Joint Health Strategic Clinical Network, Calgary, AB, Canada
Inflammation can arise in response to a variety of stimuli, including infectious agents, tissue injury, autoimmune diseases, and obesity. Some of these responses are acute and resolve, while others become chronic and exert a sustained impact on the host, systemically, or locally. Obesity is now recognized as a chronic low-grade, systemic inflammatory state that predisposes to other chronic conditions including metabolic syndrome (MetS). Although obesity has received considerable attention regarding its pathophysiological link to chronic cardiovascular conditions and type 2 diabetes, the musculoskeletal (MSK) complications (i.e., muscle, bone, tendon, and joints) that result from obesity-associated metabolic disturbances are less frequently interrogated. As musculoskeletal diseases can lead to the worsening of MetS, this underscores the imminent need to understand the cause and effect relations between the two, and the convergence between inflammatory pathways that contribute to MSK damage. Muscle mass is a key predictor of longevity in older adults, and obesity-induced sarcopenia is a significant risk factor for adverse health outcomes. Muscle is highly plastic, undergoes regular remodeling, and is responsible for the majority of total body glucose utilization, which when impaired leads to insulin resistance. Furthermore, impaired muscle integrity, defined as persistent muscle loss, intramuscular lipid accumulation, or connective tissue deposition, is a hallmark of metabolic dysfunction. In fact, many common inflammatory pathways have been implicated in the pathogenesis of the interrelated tissues of the musculoskeletal system (e.g., tendinopathy, osteoporosis, and osteoarthritis). Despite these similarities, these diseases are rarely evaluated in a comprehensive manner. The aim of this review is to summarize the common pathways that lead to musculoskeletal damage and disease that result from and contribute to MetS. We propose the overarching hypothesis that there is a central role for muscle damage with chronic exposure to an obesity-inducing diet. The inflammatory consequence of diet and muscle dysregulation can result in dysregulated tissue repair and an imbalance toward negative adaptation, resulting in regulatory failure and other musculoskeletal tissue damage. The commonalities support the conclusion that musculoskeletal pathology with MetS should be evaluated in a comprehensive and integrated manner to understand risk for other MSK-related conditions. Implications for conservative management strategies to regulate MetS are discussed, as are future research opportunities.
Methodology
The studies presented in this review were identified through PubMed Searches and the review of relevant papers in the area of diet-induced obesity, musculoskeletal health, musculoskeletal disease, and inflammation.
Introduction to Metabolic Syndrome and Obesity
Metabolic syndrome (MetS) is a cluster of conditions—visceral obesity, hypertension, dyslipidemia, and elevated fasting glucose—that increase an individual's risk for diabetes and cardiovascular complications (Alberti and Zimmet, 1998; Manuel et al., 2014). Human metabolism has evolved to efficiently convert chemical energy obtained through the consumption of food into thermal and chemical energy. Our body's metabolic pathways have developed to provide energy to tissues in times of physical threat and survival, or to efficiently conserve energy in times of food deprivation. Today, westernized societies have an abundance of food (food security) and many individuals have little need to perform physical activity. This combination has led to excessive nutrient storage, placing significant stress on our metabolic pathways, and leading to an increase in the prevalence of disease stemming from metabolic dysfunction (Miranda et al., 2005).
Concordant with the rise in MetS prevalence, there is also a global increase in the prevalence of musculoskeletal (MSK) diseases and disorders (Wearing et al., 2006). Recent evidence demonstrates that metabolic complications also increase the risk for the most prominent MSK diseases, such as sarcopenic obesity (muscle loss in obesity), osteoporosis, tendinopathy, and osteoarthritis, conditions which contribute significantly to disability and time lost from work. The resultant damage and pain associated with these conditions likely develops through low-level systemic inflammation (Hoy et al., 2014; Smith et al., 2014; Figure 1) in addition to loading due to obesity (Felson et al., 1988), and reduced ability to withstand loading due to sarcopenia.
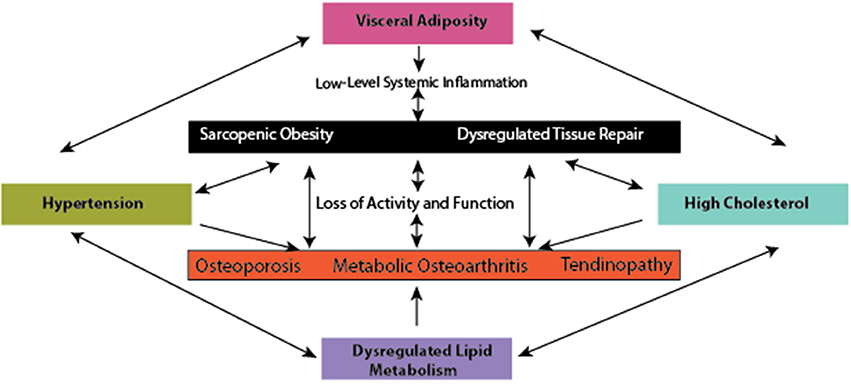
Figure 1. The interface between metabolic complications and musculoskeletal compromise.
MSK disease is of particular concern, for example, as osteoarthritis-related walking disability significantly increases risk for all-cause mortality and cardiovascular events, when controlling for other cofounders (Hawker et al., 2014). This suggests that MSK disability associated with MetS can contribute to the worsening of MetS through sedentary behavior. Although obesity has received considerable attention regarding chronic cardiovascular conditions and diseases, as well as diabetes, the MSK complications that result from obesity associated MetS are less frequently discussed and rarely evaluated comprehensively.
Muscle mass is a key predictor of longevity in older adults (Srikanthan and Karlamangla, 2014), and since muscle is highly plastic and undergoes regular remodeling, it is a vulnerable tissue in a chronic low-level inflammatory environment, such as that seen with metabolic dysfunction (Tidball, 2005; Fink et al., 2014; D'Souza et al., 2015; Collins et al., 2016a). For example, intramuscular lipid deposits increase with obesity and are also positively correlated with insulin resistance (Akhmedov and Berdeaux, 2013; Addison et al., 2014; Fellner et al., 2014), linking structural alterations with altered capacity for glucose homeostasis. Functionally, impaired muscle integrity, persistent atrophy, and lipid accumulation in muscle are risk factors for tendinopathy (Meyer and Ward, 2016), osteoporosis (Ormsbee et al., 2014), osteoarthritis (Lee et al., 2012), and integrity of a motion segment, such as the leg (Figure 2).
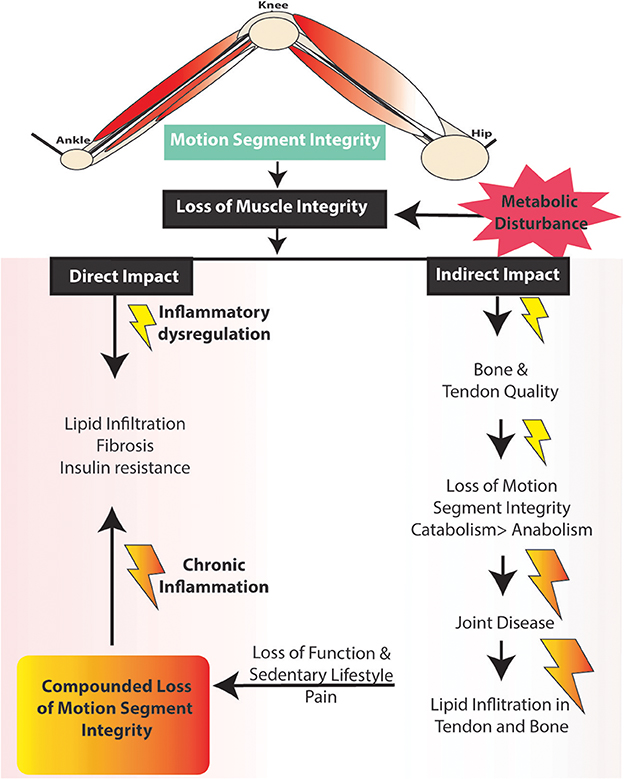
Figure 2. Potential impact of changes in muscle damage on lower limb motion segment integrity.
A Role for Metabolic Disturbance in Motion Segment Tissue Damage
A hypothesis has emerged proposing that metabolically mediated damage to MSK tissues may be one additional component of the MetS, as adipose-based inflammation links obesity, MetS, and MSK tissue damage (Hart and Scott, 2012; Zhuo et al., 2012). Increased visceral adiposity is linked to induction of increased levels of catabolic mediators (Fontana et al., 2007) and ultimately tissue damage (Figures 1 – 3). Additionally, the presence of hypertension may be linked to tissue damage through vasoconstriction and ultimately depriving tissues of appropriate nutrient exchange (McMaster et al., 2015). Moreover, high cholesterol is speculated to link dysregulated lipid metabolism and endothelial dysfunction and has been directly associated with tissue damage in tendons (Tilley et al., 2015), decreased bone mineral density (Makovey et al., 2009), and osteoarthritis damage (Farnaghi et al., 2017). In parallel, investigations into the impact of low-level systemic inflammation from metabolic disturbance on the onset of sarcopenic obesity tendinopathy, osteoporosis, and osteoarthritis have been conducted. Many of these authors report associations between low-level systemic inflammation from diet-induced obesity (DIO) with MSK disease outcomes (Table 1), however these diseases are seldom evaluated comprehensively to evaluate a common inflammatory pathway to disease induction and progression.
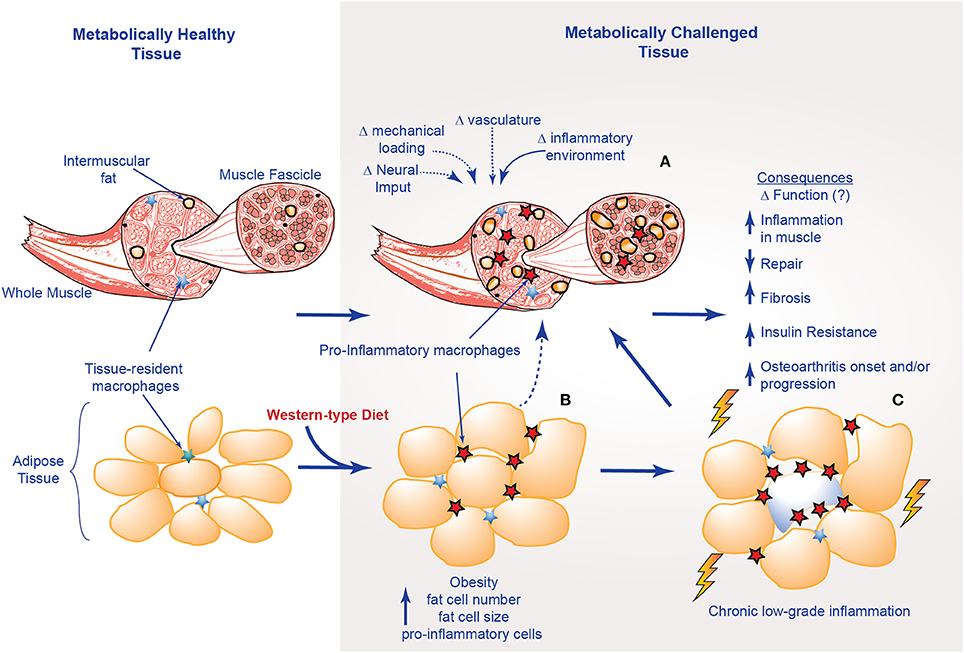
Figure 3. Structural and inflammatory changes in muscle with obesity; (A) factors that influence muscle structural integrity with metabolic challenge; (B) alterations in adipose tissue; (C) musculoskeletal consequences of chronic-low grade inflammation.
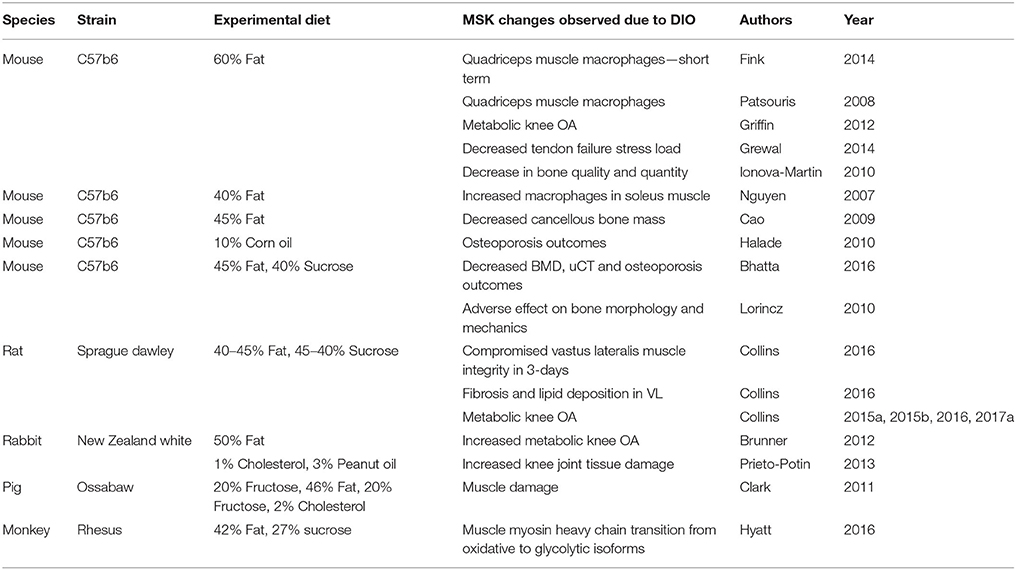
Table 1. Examples of MSK damage resulting from DIO.
As such, we suggest a hypothesis linking altered muscle integrity to direct and indirect consequences on the motion segment (Figures 2, 3). An obesogenic diet, resulting in over nutrition and development of a low-level systemic inflammation, acutely challenges associated tissues of the motion segment and can result in positive adaptation (i.e., through dynamic compensation that helps the tissue accommodate metabolic challenge), as well as negative adaptation (i.e., vulnerability to deleterious changes in tissue integrity). With these initial challenges, such tissue adaptive responses are balanced and help preserve the associated tissues and motion segment integrity. However, with chronic exposure to obesogenic diet and its inflammatory consequences, tissues demonstrate dysregulated repair, an imbalance toward negative adaptation resulting in regulatory failure, tissue damage (i.e., ectopic lipid storage and tissue fibrosis), and failure of motion segment integrity progressing to loss of function and ultimately, disease.
Aim and Scope of Review
The aim of this review is to summarize the links between induction of local and systemic inflammation, DIO, and a central role for muscle integrity in the inflammation-based pathogenesis of these MSK diseases. Early loss of muscle integrity can have both direct and indirect “ripple” effects downstream on tendons and bones, as well as the functioning of multi-tissue complex joints (e.g., the joint as an organ, Figure 2) (Frank et al., 2004; Loeser et al., 2012). As muscle plays a critical role in the mechanical and biological homeostasis of bones (through the muscle tendon unit indirectly and directly through the muscle/bone interface), tendons (directly through the muscle tendon unit), and joints (through loading and stabilization), we suggest that with the influence of dysregulated muscle loading, inflammation, and altered muscle integrity, failure of motion segment integrity is induced and exacerbated.
This review is focused on outcomes from studies using pre-clinical DIO models, as the gradual and progressive pathway toward MetS afforded by DIO provides critical insight into the short- and long-term pathophysiology, in addition to the phenotype of MSK diseases (Buettner et al., 2007; Nilsson et al., 2012). Evaluating diet-induced alterations allows for linking results across systems from food, through the gut and the associated microbiome, to early and late tissue-based changes, as well as the inclusion of potential epigenetic outcomes regarding temporal relations for the onset and progression of MSK disease.
Obesity and Impact on Muscle Integrity
Sarcopenic Obesity
Sarcopenic obesity, or low muscle mass and quality with increased fat mass, is not only associated with poor physical function (Zamboni et al., 2008), but also results in additional weight gain and an 8 to 11-fold increase in the risk for three or more additional physical disabilities (Baumgartner, 2000). Sarcopenic obesity was first defined clinically using two criteria: (a) an individual who is −2 standard deviations in muscle mass index (muscle mass/height2) compared to healthy, same-sex younger individuals; and (b) an individual who has a body fat percentage greater than the median body fat percentage for each sex (males: muscle mass index <7.26 kg/m2 with body fat percentage >27%, females: muscle mass index <5.45 kg/m2 body fat percentage >38%) (Baumgartner et al., 1998; Baumgartner, 2000). An alternative criteria for sarcopenic obesity involves falling below a linear regression-based threshold amount of lean mass given an amount of fat mass (Stenholm et al., 2008).
Sarcopenic obesity can predict disability and loss of activity in elderly adults (Baumgartner et al., 2004), and sarcopenic obesity is more closely associated with MetS than either obesity or sarcopenia alone. This suggests important roles for both fat accumulation and muscle loss in the etiology of MetS (Lim et al., 2010). Although the age-dependent declines in muscle structure and strength are well-documented, the mechanism by which MetS results in sarcopenic obesity remains to be clarified (Kob et al., 2014). As sarcopenic obesity results in disability, loss of activity, altered mechanical loading, and altered biological function in the muscle due to lipid deposition and its sequelae (Ormsbee et al., 2014), it is likely that sarcopenic obesity is central to the development of other musculoskeletal pathologies. In fact, data from our laboratory in a rat model revealed changes in the integrity of specific muscles as early as 3-days on a high-fat/high-sucrose (HFS) diet (Collins et al., 2016c), and correspond to long-term changes in systemic inflammation and gut microbiota (Collins et al., 2015a). These data support the notion that muscle may be among the first MSK tissues affected by DIO, and inflammation likely plays a substantial role in this loss of integrity.
Dysregulated Tissue Regeneration of Muscle—Primary Tissue Damaged by MetS
Muscle fiber damage happens on a daily basis and is generally considered to be a beneficial stimulus, leading to growth and adaptation through muscle regenerative processes (Karalaki et al., 2009). In muscle, monocyte and macrophage recruitment, as well as phagocytosis of necrotic material, occurs within the first 24 hours. Muscle is repaired through a series of tightly-controlled inflammatory processes (Akhmedov and Berdeaux, 2013). Specifically, muscle regeneration is a multistep process involving degeneration, regeneration, and remodeling, ultimately restoring structure and function (Laumonier and Menetrey, 2016).
The three most active cells in the regeneration of skeletal muscle are macrophages, satellite cells, and fibroblasts (Akhmedov and Berdeaux, 2013). The metabolic complications associated with obesity can result in an inappropriate temporal recruitment of these cells, which in turn leads to impaired angiogenesis and myocyte formation, while promoting the deposition of fibrotic and adipose tissue, ultimately leading to a reduction in structural integrity and functional capacity of a muscle (Karalaki et al., 2009).
With DIO, metabolic dysfunction and the presence of chronic low level inflammation can impair the “normal” inflammatory response and the regenerative capacities of skeletal muscles, resulting in a pseudo-injury (Collins et al., 2016c; Figure 3). For example, elevated levels of leptin, a satiety hormone now appreciated to have a role in low-level systemic inflammation, can impair angiogenesis, leading to tissue ischemia (Brown et al., 2015). IL-6 expression at the muscle level is a key mediator of macrophage infiltration and muscle repair (Zhang et al., 2013), while elevated expression of TGF-β promotes an increased fibrotic tissue deposition (Laumonier and Menetrey, 2016).
Efficient muscle regeneration can be attributed to satellite cells being readily available, and the cells' ability to re-establish residual pools to support multiple rounds of regeneration (Karalaki et al., 2009). Satellite cells are limited by the complex physiological environment in which they interact, an environment that can be significantly altered in individuals with obesity (D'Souza et al., 2015). A pathological host environment can limit a satellite cell's ability to be activated, proliferate, and differentiate into a muscle fiber (D'Souza et al., 2015; Meng et al., 2015). This was elegantly shown by Boldrin and colleagues through the transplantation of satellite cells from mdx mice, a genetic mouse model of muscular dystrophy, into a neutral environment (Boldrin et al., 2015). Despite the impairment of mdx satellite cells as a result of being in the pathogenic environment of an mdx mouse, following transplantation, mdx satellite cells were fully capable of being activated, and could proliferate and differentiate into a fully functional muscle fiber (Boldrin et al., 2015). Macrophages may also inhibit satellite cell activity, suggesting another mechanism by which low-level systemic inflammation may inhibit muscle repair (Tidball and Villalta, 2010).
Impairments in satellite cell activity have been reported to promote fibro/adipogenic progenitor cells (FAP) that normally aid in muscle regeneration, to differentiate into fibroblasts and/or adipocytes (Chapman et al., 2016). FAPs have also been demonstrated to be a source of intramuscular lipid deposition with rotator cuff/supraspinatus tendon injury (Liu et al., 2016). FAPs are thought to be vulnerable to reprogramming in the presence of low-level inflammation, which may result in increased lipid and fibrosis deposition, and may limit reversibility of fibrosis (Mann et al., 2011). Thus, chronic obesity and associated MetS, with inflammation and fatty infiltration of muscles, may lead to a compromise in the regeneration of muscle integrity.
In addition to the environmental challenges to the muscle regeneration posed by metabolic disturbance, skeletal muscle from individuals with obesity displays a greater number of glycolytic-fibers vs. oxidative-fibers when compared to a healthy individual (Pattanakuhar et al., 2017). Since oxidative-fibers generally contain a greater number of satellite cells relative to glycolytic fibers (Karalaki et al., 2009), individuals with obesity may also have fewer satellite cells. Based on this information, impairments in satellite cell function as a result of alterations in cell metabolism, a reduction in cell number, suppression of cell activation, depleted cell reserves, and impaired cell proliferation and differentiation may lead to impairments in muscle fiber regeneration (Akhmedov and Berdeaux, 2013).
Work from our laboratory has demonstrated that the oxidative soleus muscle is protected against HFS-induced damage over short-term and long-term exposures in a rat model (Collins et al., 2017b; Figure 4). By 3-days on HFS, dynamic increases in mRNA levels for superoxide dismutase (SOD2) in HFS animals implicate compensatory oxidative stress scavenging in the soleus muscle compared to control animals. By 2-weeks on HFS, increased mRNA levels for oxidative capacity [succinate dehydrogenase (SDH)] were detected compared to chow-fed controls, suggesting one adaptation strategy that the soleus muscle may employ with HFS metabolic challenge. Although the precise mechanism(s) by which the soleus is protected from metabolic disturbance-induced muscle damage remains to be clarified, it appears that increasing the oxidative capacity and the oxidative stress scavenging ability of muscles (i.e., with aerobic exercise) may be a beneficial strategy for mitigating obesity-induced muscle damage and its consequences.
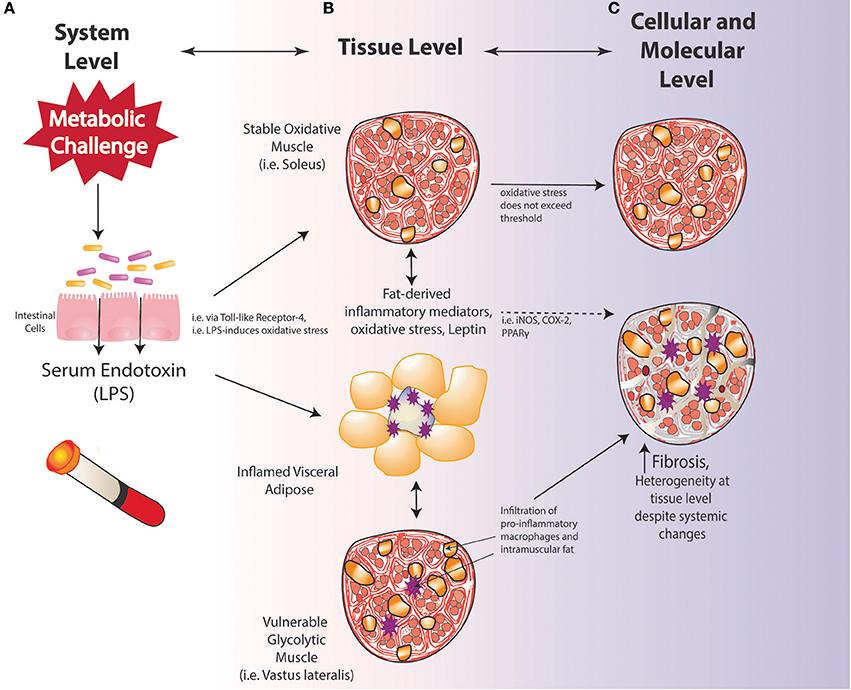
Figure 4. Vulnerability and protection of muscle with diet-induced obesity may be determined by oxidative capacity; (A) system level changes; (B) tissue-level changes; (C) cellular and molecular level alterations.
Inflammation related to obesity can also impair myocyte remodeling as a result of a reduction in protein synthesis due to elevated TNF-α levels (Brown et al., 2015). Furthermore, there is evidence for adipocyte-muscle cross talk in vitro, whereby adipocyte-derived inflammation can contribute to inflammation and atrophy in muscle cells subjected to a metabolically dysfunctional environment, possibly through IGF-1 (Pellegrinelli et al., 2015). Impaired protein synthesis can also prevent muscle from properly adapting to mechanical stimuli. Brown et al. (2015) demonstrated this inability, showing that following muscle damage, obese muscle displayed no adaptations, while lean mice displayed an increase in muscle wet weight and muscle fiber hypertrophy (Brown et al., 2015). Potential contributing factors to a reduction in protein synthesis may be elevated lipid metabolites resulting from impaired mitochondrial function contributing to elevated TNF-α levels, which are known to have inhibitory effects on IGF-1 (Akhmedov and Berdeaux, 2013). Decreases in IGF-1 result in the inhibition of the muscle growth signaling pathway (IGF-1 → P13K → Akt → mTOR), effectively blunting muscle protein synthesis (Brown et al., 2015). Furthermore, increased myostatin levels can also contribute to impaired growth in obese muscle. Myostatin is not only significantly up-regulated in obese skeletal muscle, but in adipose tissue as well, further inhibiting myogenesis, providing another avenue through which potential muscle-adipose cross talk may occur (Karalaki et al., 2009).
Functional Muscle Damage with Metabolic Derangement
Generally speaking, adults with obesity are reported to have significantly higher absolute strength in lower limb muscles, but lower strength when normalized to body mass (Tomlinson et al., 2016). When the upper limb muscles are evaluated, there are no statistical significant differences between individuals with obesity and normal-weight controls (Tomlinson et al., 2016). Potentially, the characterization of obesity by body mass, which is common in these studies, may be inappropriately representing body composition. We have shown, in a healthy and overweight population cohort, that body mass index (BMI) inappropriately estimates body composition in 30% of the population, with a specific disparity between BMI and body composition measurements in healthy females (Collins et al., 2017c). Also, there is a lack of data describing the effect of obesity on muscle integrity, and a lack of consistent protocols to assess muscle strength (Tomlinson et al., 2016). However, data from our lab (Collins et al., 2016a,c, 2017b) and others, in rodents (Ciapaite et al., 2015) and large mammals (Clark et al., 2011), have demonstrated deleterious alterations in muscle structural integrity with metabolic disturbance. Computational approaches modeling the gastrocnemius muscle have demonstrated that whole-muscle force is dependent on muscle integrity, specifically regarding reductions of muscle force due to intramuscular lipid (Rahemi et al., 2015). A zebrafish model of diet-induced obesity further demonstrated that obesity induces decreases in locomotor performance, isolated muscle isometric stress, work-loop power output, and muscle relaxation rates (Seebacher et al., 2017). Of note, these decrements in performance and function were not reversed with weight loss, generating interesting questions about the potential reversibility of impaired muscle function with obesity.
Additional sources of muscle damage and altered repair are advanced glycation end products (AGEs), which accumulate over time due to increased availability of glucose and hyperglycemia (Figure 5). Dietary AGEs can interfere with muscle healing and impair contractile function in a mouse model of obesity (Egawa et al., 2017). HFS diets can induce hyperglycemia in rodents, further linking diet-induced obesity to AGEs (Sumiyoshi et al., 2006). The receptor for AGEs on macrophages, called RAGE, is associated with a pro-inflammatory state, and RAGE/AGEs are reported to be involved in the onset and progression of metabolic disturbance, insulin resistance, and adipokine expression (Leuner et al., 2012; Hofmann et al., 2014). AGEs have been implicated in macrophage polarization toward M1 pro-inflammatory phenotypes, pro-inflammatory IL-6 secretion in adipose tissues, and initiating inflammatory cascades (Bopp et al., 2008; Frommhold et al., 2011; Nativel et al., 2013; Jin et al., 2015; Son et al., 2016) [i.e., NF-κB, p38 mitogen-activated protein kinase (MAPK)]. Reactive AGEs can also cross-link with collagen fibers, which subsequently can affect the fiber's mechanical and biological properties (Abate et al., 2013). Although AGE collagen cross-linking can be reversed (Asif et al., 2000), it is unclear whether glycation itself can be reversed. Of note, endurance exercise has been shown to attenuate AGEs in cardiac muscle of rats (Wright et al., 2014). With weight loss, reversal of metabolic dysfunction may not be fully achievable, and weight re-gain is common (Fothergill et al., 2016). As mentioned above, to what degree reversibility of skeletal muscle damage may be achieved in this context remains to be determined (Figures 2 – 4), and several factors could contribute to irreversibility (Mann et al., 2011). Likely, clarifying relations between the time of exposure to low-level systemic inflammation and muscle damage should be determined, as impaired muscle integrity has been observed following short-term exposure to a metabolic challenge, likely before “full” metabolic derangement has been achieved (Fink et al., 2014; Collins et al., 2016c). Also, some alterations in satellite cell function can be irreversible (Sacco and Puri, 2015). Generally, DNA methylation, or epigenetic changes, is an actively researched area being explored in the context of muscle (Carrió and Suelves, 2015), and DNA methylation is an important step in muscle cell differentiation (Brunk et al., 1996). However, it is likely that epigenetic changes, which are dynamic and induced by environmental changes, can induce cellular reprogramming, and thus could potentially limit the reversibility of muscle damage, even if the MetS-related inflammation is controlled (Carrió and Suelves, 2015). Investigations to clarify the role of methylation in physiological and pathophysiological muscle changes may provide valuable insight into the reversibility potential of muscle damage.
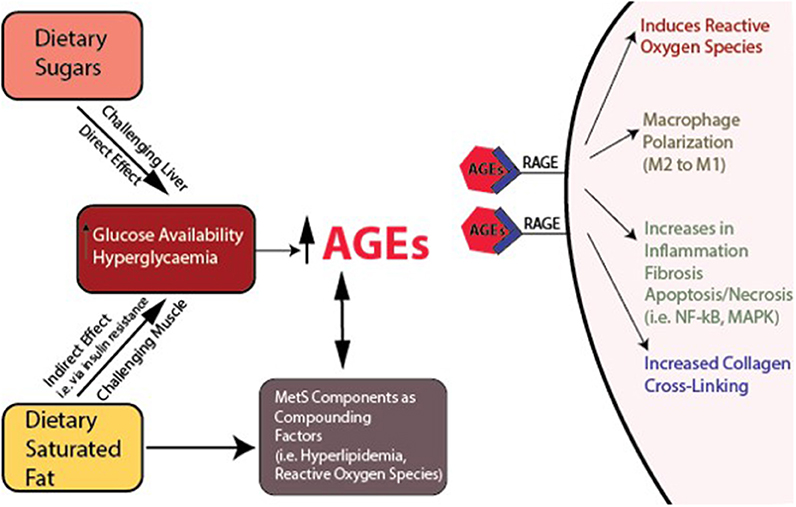
Figure 5. Interacting variables reinforcing metabolic dysfunction and sequelae. Links between dietary sugar, dietary saturated fat, increased hyperglycaemia, advanced glycation end products (AGEs), their receptors (RAGEs), and inflammation, macrophage polarization, and collagen cross-linking.
Dysregulated Homeostasis of Associated Motion Segment Tissues (Tendon, Bone, Cartilage, and Joint) with Obesity: Acute and Chronic
Muscle, tendon, cartilage, and bone tissues repair and regenerate over different timelines, and these different repair timelines may dictate vulnerability or resistance to damage with metabolic disturbance involving inflammatory processes. Both biological and mechanical stimuli are critical to homeostatic tissue regulatory mechanisms. Across tissues, remodeling rates slow with age. As discussed above, muscle tissue is likely the most vulnerable MSK tissue to perturbation with metabolic challenge. Repair of such tissues in the face of an active inflammatory environment is likely compromised, and resolution of repair is particularly challenging (Hart et al., 2004).
Altered muscle integrity can challenge the motion segment in a dynamic manner, until the associated tissues ultimately begin to fail (Figures 2, 3). Specifically, tendon healing is slower compared to muscle. In tendon, tenocytes migrate to the wound, initiating type III collagen synthesis. After several weeks to months, remodeling of tendinous tissue occurs such that collagen fibers are aligned in the direction of stress, and maturation occurs over the course of a year. Bone mechanotransduction, initiating the maintenance/remodeling processes (Scott et al., 2008), requires a series of events that can require almost 24-h to ultimately result in the synthesis of bone matrix proteins (Robling and Turner, 2009). Cartilage is considered to have limited intrinsic repair properties, although with the facilitation of autologous stem cells, biologics, and other transplants, some repair may be achievable (Chu et al., 2010). The impaired regenerative capacity of tendon, bone and cartilage likely indicate a lack of reversibility with altered integrity and subsequent compromise in function, underscoring a need for strategies aimed at primary prevention (before it occurs), secondary prevention (limiting the amount of damage as it occurs), tertiary prevention (prevention of progression of damage to disease), or reversal of damage.
Diet-Induced Obesity and Elements of the Dysregulated Inflammatory Response
Inflammatory Mediator Alterations in Obesity
Cytokines and Chemokines
Activation of the innate immune system is critical in the pathogenesis of type 2 diabetes and tissue damage. Some of the key signaling pathways involved in this process are nuclear factor –κB (NF-κB), c-Jun N-terminal Kinase (JNK), and the NLRP3 inflammasome, all resulting in transcription of pro-inflammatory cytokines (Lackey and Olefsky, 2015). NF-κB can be activated by elevated levels of pro-inflammatory cytokines such as TNF-α, which is increased in the adipose tissue of obese and diabetic animals. Neutralizing TNF-α has been shown to reduce insulin resistance (Hotamisligil et al., 1993). Additionally, JNK activity increases in tissues that are sensitive to insulin, is activated by ER stress, and directly inhibits insulin signaling (Gual et al., 2005; Lackey and Olefsky, 2015). The inflammasome is a protein complex that matures and secretes inflammatory cytokines, such as IL-1β and IL-18. Similar to NF-κB, the inflammasome can be activated by pro-inflammatory cytokines, lipopolysaccharide (LPS), and some forms of low density lipoproteins (LDLs). LPS, low density lipoproteins, and the AGE-product of LDLs can signal through Toll-like Receptor-4 (TLR-4) (Hodgkinson et al., 2008), activating IL-1β and IL-18 signaling.
TLR-4 recognition of saturated fatty acids is necessary to enable NF-κB signaling and induce expression of pro-inflammatory cytokines (TNF-α, IL-6, and MCP-1) (Jialal et al., 2014; Lackey and Olefsky, 2015). TLRs sense pathogen-associated molecular patterns and damage-associated molecular patterns (PAMPs), and regulate the inflammatory responses to mitigate tissue repair (Lee et al., 2013). TLR-4 gene expression and protein content are increased in muscle from patients with obesity and diabetes, potentially contributing to insulin resistance, as well as compromised muscle integrity observed with metabolic disturbance (Reyna et al., 2008). In the context of OA, TLRs can modulate the catabolic pathways and maintain joint homeostasis (Houard et al., 2013). Of interest, the lubricating molecule proteoglycan-4 (PRG-4) has been shown to modulate the inflammatory response through competitive inhibition of TLR-4 receptors, suggesting that as PRG-4 concentration is reduced with OA severity, inflammatory modulation may also be reduced, contributing to the progression of OA (Iqbal et al., 2016). In tendinopathy, there may not be a clear role for TLRs, as catabolic processes in Achilles tendinopathy seem to occur independently of TLR4-induced gene expression from IL-1β or TNF-α (de Mos et al., 2009). In bone, LPS-induced activation of TLR-4 in neutrophils is reported to upregulate the catabolic RANKL osteoclast cascade, linking TLR based inflammation to increased bone resorption (Chakravarti et al., 2009).
Appropriate levels of pro-inflammatory cytokines are necessary for tissue homeostasis. However, exogenous exposure to pro-inflammatory cytokines, or endogenous high levels of pro-inflammatory cytokines, are associated with damage across all musculoskeletal tissues. In muscle, TNF-α, IL-1β, and IL-6 activate transcription of MuRF-1 and MAFBx/atrogin-1, the key muscle atrophy pathway, through IGF/Akt-1 (Akhmedov and Berdeaux, 2013). For example, IL-6 and TNF-α inhibit bone-forming osteoblast cells, and NF-κb, RANKL, TNF-α, IL-6, M-CSF, and MCP-1 can contribute to osteoclast recruitment, maturation, and inhibit osteoclast apoptosis (Roy et al., 2016). In tendon, pro-inflammatory cytokines (IL-1β, TNF-α, IL-6) can disrupt tissue homeostasis, by inducing extracellular matrix degradation, induce other pro-inflammatory cytokines, which result in necrotic and apoptotic cell changes, and affect collagen and elastin expression (Schulze-Tanzil et al., 2011). Cytokine effects in tendon are modulated by mechanical loading in this complex mechano-biological environment (Killian et al., 2012).
The knee joint is a complex organ system with many resident inflammatory cells, particularly in the synovium and infrapatellar fat pad. These tissues secrete pro-inflammatory cytokines, like IL1-α and IL-1β (Sanchez-Adams et al., 2014). In humans, systemic and synovial fluid inflammatory profiles can differentiate between patients based on OA severity, suggesting that cytokine levels may help define OA pathogenesis (Heard et al., 2013). In addition to the infrapatellar fat pad, there is also a synovial adipose depot, and these two depots differ significantly from each other in lean individuals with OA (Harasymowicz et al., 2017). Moreover, infrapatellar fat pads and synovium adipose depots differed in adipocyte size, fibrosis, and macrophage infiltration in OA patients with obesity compared to lean OA patients (Harasymowicz et al., 2017). High glucose concentrations can contribute to increased chondrocyte responsiveness to cytokines, increased levels of reactive oxygen species, leading to an overproduction of IL-6 and PGE2 (Laiguillon et al., 2015). These cytokines can also contribute to mitochondrial dysfunction within the joint, and, in turn, mitochondrial dysfunction can amplify chondrocyte responsiveness to cytokines (Vaamonde-García et al., 2012).
Unpublished data from the author's laboratories suggest that intramuscular lipid deposits and fibrotic material in the vastus lateralis muscle of the knee are associated with the presence of metabolic-induced knee joint damage after a 12-week obesity induction period (Figure 6). Based on the inflammatory mediator profile of joints undergoing damage (Collins et al., 2015a,b, 2016b), inflammation is likely playing a significant role in this process and in the relations between muscle damage and joint damage. Ongoing efforts will probe the role of associated inflammation on muscle integrity in the onset and progression of such metabolic joint damage and whether it leads to overt OA.
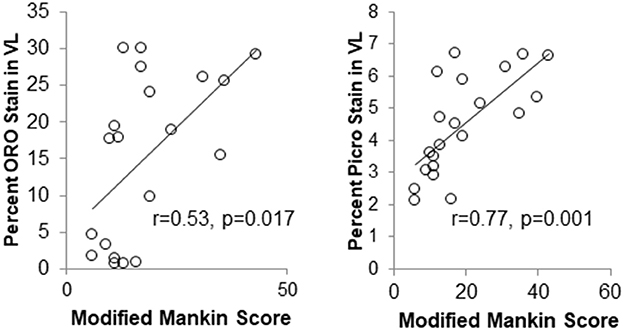
Figure 6. Markers of muscle integrity are associated with metabolic OA severity after 12-weeks of high-fat high-sucrose diet-induced obesity. (ORO, Oil Red O stain for intramuscular fat; Picro, Picrosirius stain for collagen).
Given the systemic-to-local hypothesis in metabolic OA (Zhuo et al., 2012; Collins et al., 2016b; Berenbaum et al., 2017), we have evaluated hip, knee, and shoulder joints using the HFS rat model system for 12-weeks (Collins et al., 2017a). A HFS diet, in the absence of trauma, resulted in significant increases in joint damage/OA-like changes in the shoulder and knee joints of rats after a standard 12-week obesity induction period. The hip joint, however, was not significantly affected by DIO, which is consistent with findings from human epidemiological studies. Total joint damage, assessed by adding the individual Modified Mankin Scores across all three joints, was increased in DIO animals compared to chow-fed animals, and was associated with the percentage of body fat. Positive significant predictive relations for total joint score were found between body fat and two serum mediators (IL-1α and VEGF). These data suggest that systemic inflammatory alterations from DIO in this model system may result in a higher incidence of knee, shoulder, and multi-joint OA-like/joint damage with the HFS diet over the long term (Collins et al., 2017a). Due to the preliminary nature of these studies, longitudinal experiments with multiple time points are needed to validate these proposed relations. If these relations are supported in future studies, then not all joints are affected equally by obesity-associated inflammation in MetS. Studying these relations are an ongoing direction of current research.
Adipokines
Adipocytes release adipokines such as leptin, adiponectin, visfatin, and resistin as a signaling mechanism in addition to passively storing energy, which can cause and exacerbate chronic low-level systemic inflammation (Gomez et al., 2009). Adipokines have been shown to induce pro-inflammatory mediators in activated CD4+ T cells from osteoarthritis patients, demonstrating that systemic mediators may play a role in osteoarthritis (Scotece et al., 2017). After interaction with such activated CD4+ T cells, chondrocytes demonstrate increased expression of MMP-13 and decreased expression of collagen-2 and aggrecan.
Leptin is a satiety hormone that increases in a near-linear fashion to body fat (Friedman and Halaas, 1998). It inhibits appetite and regulates body weight, energy expenditure, and maintains glucose homeostasis. However, with metabolic disturbance, individuals become leptin resistant. Leptin is a class-1 cytokine secreted from adipose tissue, and high concentrations of leptin are associated with musculoskeletal tissue damage (Zabeau et al., 2003; Collins et al., 2015a,b, 2016b,c). Leptin activates downstream pro-inflammatory pathways (IL-2, IFNγ) and inhibits the anti-inflammatory pathway (IL-4) (Lechler et al., 1998). Aside from being produced by adipose tissue, leptin levels can be increased by TNF-α, IL-1, and LPS, creating a positive-feedback loop with low-level chronic inflammation (Grunfeld et al., 1996).
Leptin signaling is critical to conventional muscle maintenance (Akhmedov and Berdeaux, 2013), and basal levels of satellite cells are reduced in animals with impaired leptin signaling (Peterson et al., 2008). However, hyperphysiological levels of leptin also stimulate the proliferation and activation of macrophages, which may be a mechanism by which leptin concentration influences tissue damage (Santos-Alvarez et al., 1999). Leptin may also positively upregulate myostatin (Rodríguez et al., 2015), a member of the TGF-β superfamily that negatively regulates muscle mass and growth, and a molecule which is up-regulated in muscle from individuals with metabolic disturbance (Hittel et al., 2009). Upregulation of follistatin has been an effective treatment for muscle degenerative diseases by increasing muscle growth due to its ability to inhibit myostatin, a negative regulator of muscle mass. Follistatin also alleviates synovitis and mitigates OA-like changes from inflammatory arthritis in mice (Yamada et al., 2014). As muscle has a role in OA pathogenesis, protecting both muscles and joints with follistatin represents an attractive therapeutic opportunity for MetS-induced musculoskeletal damage.
Leptin also may be involved in mediating the pathogenesis of osteoarthritis in humans (Fowler-Brown et al., 2014) and other animals (Griffin et al., 2009, 2010, 2012; Collins et al., 2015a,b, 2016b). Leptin is found in synovial fluid of humans (Lübbeke et al., 2013) and other animals (Collins et al., 2015a,b, 2016b). Unpublished mRNA data (Table 2); from our laboratory indicate increased mRNA expression levels for leptin in the fat pad and synovium of rats with DIO compared to those on a chow diet, after a 12-week obesity induction period.
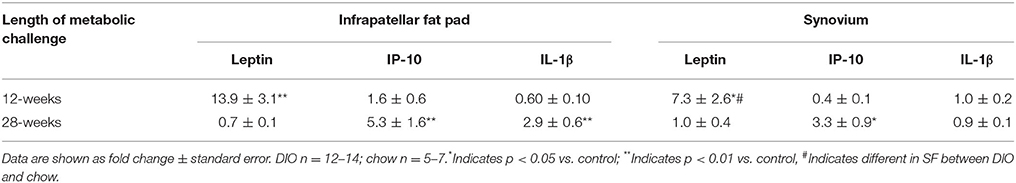
Table 2. Differential mRNA levels for leptin, IP-10 and IL-1β with 12- and 28-weeks of diet-induced obesity.
Additionally, leptin may be involved in mediating the pathogenesis of osteoarthritis in humans (Fowler-Brown et al., 2014) and other animals (Griffin et al., 2009, 2010, 2012; Collins et al., 2015a,b, 2016b). Leptin is found in synovial fluid of humans (Lübbeke et al., 2013) and other animals (Collins et al., 2015a,b, 2016b). Increased mRNA levels for leptin were accompanied by increased leptin in the serum and synovial fluid (Collins et al., 2016b). After 28-weeks of DIO, the fat pad and synovium demonstrated disparate up-regulation of IL-1β, although IL-1β was detected in the synovial fluid of these animals (Collins et al., 2015a). These findings suggest that the fat pad and synovium contribute to increased pro-inflammatory synovial fluid profiles (Collins et al., 2016b). However, cartilage explants are not substantially damaged when exposed to physiological levels of leptin (Griffin et al., 2010), despite reports of associations between leptin and MMPs (Koskinen et al., 2011), calling into question the direct involvement of leptin in eliciting cartilage damage.
In the context of bone, leptin influences the formtion and resorption of mineralized tissue by increasing the activity of osteoclasts through RANKL (Ducy et al., 2000). However, there are also reports that leptin supports bone growth and bone MSC differentiation into osteoblasts (Thomas et al., 1999), so the precise mechanism for leptin's effects on bone is unclear, but likely is dependent on exposure and dose (Kawai et al., 2009). Low-level systemic inflammation and leptin can negatively influence tendon structural integrity (Abate, 2014; Abate et al., 2016), can result in accelerated heterotopic ossification in tendon tissues (Jiang et al., 2017) and may be associated with increases in tendon ruptures (Ji et al., 2010). Taken together, increases in systemic leptin—or mediators downstream of leptin—appear to have deleterious effects on all major musculoskeletal tissues. These data support the notion that catabolic activity as a result of increased leptin is an important pathway to clarify in the context of global musculoskeletal integrity (Griffin et al., 2009). More details regarding the interface among leptin signaling, inflammation, metabolism, and musculoskeletal disorders are detailed elsewhere (Abella et al., 2017b).
Resistin is an adipokine involved in insulin resistance, inflammation, and energy homeostasis. Serum resistin levels are reported to be derived from visceral adipose tissue (Milan et al., 2002). There is conflicting evidence as to whether resistin is associated with bone mineral density (Mohiti-Ardekani et al., 2014). Within the joint, synovial fluid levels of resistin are associated with inflammatory and catabolic molecules in the joints of human osteoarthritis patients (Koskinen et al., 2014). However, resistin and visfatin, demonstrated positive predictive relations with recovery from upper extremity soft tissue disorders such as tendinopathy, and are thought to be related to anti-inflammatory response mechanisms (Rechardt et al., 2014).
Adiponectin, another adipokine, is derived from visceral fat, and is thought to serve a protective role on cardiovascular health and glucose homeostasis (Milan et al., 2002). As body fat decreases, adiponectin levels generally increase, and adiponectin may modulate adipose tissue regulation via NF-κB (Ajuwon and Spurlock, 2005). Specifically, adiponectin treatment in obese mice increases bacterial clearance and hematopoietic progenitor proliferation in bone marrow (Masamoto et al., 2016). Adiponectin was shown to be significantly correlated with bone mineral density in a group of osteoporotic and healthy patients (Mohiti-Ardekani et al., 2014). Adiponectin has also been proposed as a systemic biomarker of OA. Plasma adiponectin was significantly higher in a population of OA patients and was also higher in women with erosive hand OA compared to patients with non-erosive OA (Filková et al., 2009). In a separate cohort of hand OA patients, the individuals with the highest levels of adiponectin demonstrated a decreased risk for hand OA progression (Yusuf et al., 2011). However, the study populations were not the same in these two reports (i.e., total numbers, males and females vs. females alone, and use of European populations potentially differing genetically) and therefore, additional research needs to be performed to better evaluate this relationship. Additionally, in a study evaluating 76 males in Thailand with knee OA revealed that levels of adiponectin in synovial fluid correlated with disease severity (Honsawek and Chayanupatkul, 2010). In muscle, adiponectin has been shown to increase fatty acid oxidation and glucose uptake, and to attenuate local inflammation (Nigro et al., 2015). Adiponectin has been proposed as a treatment for diabetic tendinopathy (Rothan et al., 2013), suggesting it may be protective and have favorable anti-inflammatory effects across MSK tissues.
Progranulin is a more recently identified adipokine that may also have anti-inflammatory characteristics. mRNA levels for progranulin are increased in cartilage, synovium, and infrapatellar fat pads from OA patients, and mRNA levels are increased in response to pro-inflammatory stimulation (Abella et al., 2016). Specifically, progranulin has been shown to counteract pro-inflammatory molecule expression (i.e., NOS, MMP-13) induced by IL-1β and the LPS-TLR-4 axis (Abella et al., 2016). Attstrin, a progranulin-derived peptide, is a promising therapeutic candidate for osteoarthritis (Abella et al., 2017a) given that progranulin can counteract IL-1 driven inflammation through TNFR1 in human chondrocytes, and intraarticular injection of progranulin-derived attstrin prevented OA-progression in a surgical model of murine OA (Xia et al., 2015).
Obesity and Involvement of Cells of the Inflammatory System
Macrophages
Generally speaking, macrophages are dichotomized into M1 (pro-inflammatory) and M2 (anti-inflammatory) phenotypes based on their activity during tissue repair processes (Novak and Koh, 2013). However, it is understood that this dichotomy represents an oversimplification based on in vitro data which may not accurately represent in vivo states (Martinez and Gordon, 2014). Macrophages are described to demonstrate a high degree of functional plasticity and their phenotypes can change based on environmental stimuli (Stout and Suttles, 2004). It is likely useful to consider macrophage activation as a spectrum rather than a binary categorization (Mosser and Edwards, 2008), but for the purposes of this review, and concordant with the current musculoskeletal literature, macrophage activity and these relations are generalized using the M1/M2 paradigm.
Macrophages are derived from monocyte precursor cells. Tissues have resident macrophages, which are responsible for general tissue maintenance. These macrophages are described as alternatively activated, or M2 macrophages, and are induced by TGF-β, IL-4, and IL-13 (Gordon and Martinez, 2010). M1 macrophages (classically activated macrophages) are key phagocytes within tissues, and are induced through IFN-γ activation and LPS-induced TLR signaling or from detection of pathogen-associated molecular patterns (Lampiasi et al., 2016). With obesity, stress, loading, or tissue and niche specific control mechanisms, M2-type macrophages may experience a phenotypic shift. This shift may be influenced by exposure to certain cytokines through their general signaling mechanisms or in the presence of other conditions, including IL-6 (Braune et al., 2017) or TNF-α (Wu et al., 2015). AGEs can also play a role in M2 to M1 polarization (Jin et al., 2015) (Figure 5). Typically, an imbalance in the ratio of M1:M2 macrophages is considered maladaptive, creating an imbalance toward tissue degradation and an absence of adequate repair. MAPK is a key pathway in macrophage-mediated inflammatory responses and may play a significant role in diseases mediated by macrophages (Yang et al., 2014).
Within 24 hours following muscle damage, thousands of macrophages have infiltrated the damaged tissue (Tidball, 2005; Grounds, 2011). Pro-inflammatory M1-macrophages first perform cell lysis, removing necrotic muscle tissue debris (Laumonier and Menetrey, 2016). Anti-inflammatory M2-macrophages infiltrate the damaged site once M1-macrophages have removed necrotic debris (~48 hours following injury), helping resolve the inflammatory response while promoting myogenesis (Akhmedov and Berdeaux, 2013). Through the secretion of a number of growth factors, macrophages promote angiogenesis (FGF, TGF-β), synthesis of ECM proteins (TGF-β), and activation of satellite cells, all promoting myogenesis (Grounds, 2011).
It is possible that an increased presence of M1 macrophage cells in a tissue is an early sign of disrupted tissue homeostasis and repair. With diet-induced obesity, M1-type macrophages are present in the quadriceps muscle of mice (Fink et al., 2014), rats (Collins et al., 2016c), and humans (Fink et al., 2014; Khan et al., 2015). In bone, osteoclasts are considered M1-type macrophages. With a high-fat diet, bone loss is accelerated in young mice due to increased osteoclastogenesis (Shu et al., 2015), and as noted, pro-inflammatory cytokines increase osteoclast activity in humans with obesity through activation of the NF-κB/RANKL pathway (Cao, 2011). In the context of the joint as an organ (Frank et al., 2004), the infrapatellar fat pad does not demonstrate inflammation or M1 polarization prior to knee OA in mice (Barboza et al., 2017). Synovial membrane samples from OA patients reveal markers for M1 macrophages (IL-1α, IL-1β, and TNF-α) and decreased levels of the M2 marker IL-1RA (Smith et al., 1997). As such, it is challenging to determine if levels of M1/M2 macrophages are a cause or consequence of disease. In rabbits, intra-articular injections of the pro-inflammatory mediators IL-1 and TNF-α stimulate M1 activation and degrade cartilage, suggesting macrophage polarization may occur in synovial cells (Pettipher et al., 1986; Henderson and Pettipher, 1989). Conditional macrophage depletion in obese mice with a knee injury does not mitigate OA severity (Wu et al., 2017b). Moreover, in mice where macrophages were depleted, there was an increase in neutrophils and CD3+ T cells compared to control animals (Wu et al., 2017b). These data suggest that a potential redundancy may exist between macrophages and other inflammatory cells. Macrophages may modulate inflammatory homeostasis within the joint OA and obesity, and that a basal level of macrophages are likely needed to maintain joint health (Wu et al., 2017b). While this study is not conclusive, it demonstrates that the relations among macrophage polarization and presence in obesity and OA are complex. It is important to note that what constitutes a healthy or maladaptive balance in macrophage phenotype in one tissue or disease may differ from another tissue or disease process. As such, these findings are likely context- and tissue-dependent. More studies are needed to better understand whether synovial macrophage polarization, mitigation/modulation, or ablation is a promising therapeutic target in OA pathogenesis (Sun et al., 2016).
Specific Cell Involvement in Metabolic Overload with Obesity
Many cells (e.g., adipocytes, hepatocytes, and myocytes) contribute to the conversion of food into chemical energy and can be placed under significant metabolic stress when exposed to a typical western-style diet. Significant increases in the postprandial flux of metabolic substrates results in drastic nutrient spikes characterized by elevated glucose, free fatty acid (FFA), and triglyceride (TG) levels (Weiss et al., 2013). Chronic nutrient overload places metabolic pathways under significant stress, overwhelming subcutaneous adipose depots, leading to adipocyte mitochondrial dysfunction and impaired insulin sensitivity (Miranda et al., 2005). Normally, insulin inhibits lipolysis and promotes glucose transport. However, during chronic metabolic overload, mitochondrial dysfunction can disrupt adipocyte insulin signaling pathways, leading to impaired glucose transport into adipocytes and the inability to suppress cell lipolysis, further increasing blood glucose and free fatty acid (FFA) levels in circulation (Miranda et al., 2005). As a result of increased circulating blood glucose and FFA levels, hepatocytes and myocytes are placed under great metabolic stress.
Mitochondrial Dysfunction and Adipose Adaptations with Metabolic Challenge
With the onset of metabolic challenge, the imbalance of redox states and mitochondrial dysfunction may be instrumental to the development of insulin resistance and MetS (Long et al., 2015). With nutrient overload, mitochondrial dysfunction in adipose tissue will give rise to an increase in inflammatory mediators (adipokines) resulting in tissue remodeling, and potentially cell death (Trayhurn, 2005; Vernochet et al., 2014). Adipokine dysregulation can result in the fibrosis, or inadequate healing response, of adipose tissue (Kwon et al., 2012), as well as liver (Chiang et al., 2011). As adipose depots become inflamed and fibrotic, they become limited in their energy storing and endocrine function, resulting in the deposition of ectopic lipid in other tissues coupled with the presences of insulin resistance (i.e., muscle and liver) (Sun et al., 2013). Altered mitochondrial function, ectopic lipid formation, and incomplete or inadequate healing responses are hallmarks of MSK diseases, and are likely initiated by low-level systemic inflammation.
Mechanisms of Tissue Metabolic Dysfunction with Obesity
Once subcutaneous adipose depots become overwhelmed, lipid spill-over leads to ectopic lipid accumulation in visceral adipose tissue depots and other insulin sensitive tissues, such as muscle (Miranda et al., 2005). In muscle, the nutrient overload placed on myocytes, similar to adipocytes, can lead to metabolically dysfunctional cells, further contributing to the already dysregulated inflammatory environment. The complexity and interconnectedness between these processes and mitochondrial dysfunction are illustrated in Figure 7.
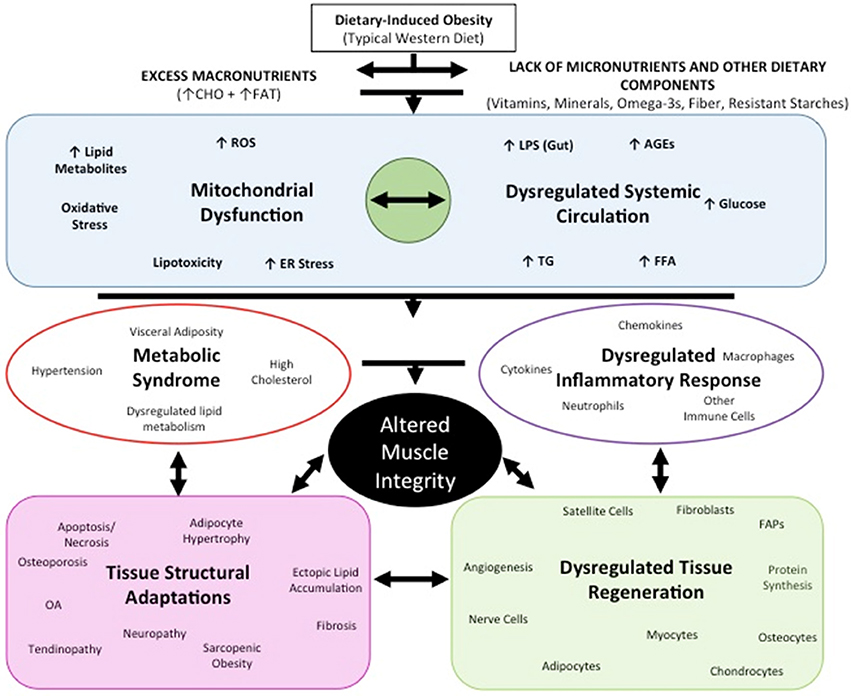
Figure 7. Overview of tissue adaptations and processes with diet-induced obesity.
Dietary Considerations, Nutrient Content, and Obesity Phenotypes
While discussing inflammation from DIO, it is important to consider the composition of the experimental diets, as the dietary profile and energy density of a given diet likely will impact the resultant inflammatory profile in each preclinical model (reviewed in Hariri and Thibault, 2010). For example, diets rich in fat elicit different types of glucose intolerance compared to diets rich in sugar, whereby dietary fat largely compromises muscular glucose tolerance, and sugar challenges liver lipogenic and gluconeogenic enzyme activities (Sumiyoshi et al., 2006). As such, different dietary composition may yield different findings or differences in the severity of disease outcomes.
Fat and sugar dietary constituents can contribute to different animal phenotypes, whereby animals on a high sugar diet do not necessarily gain weight, but demonstrate a conversion of lean body mass to fat body mass (Sumiyoshi et al., 2006). Diets high in saturated fat, however, generally result in increased body fat and body mass, although the type of fat employed (i.e., omega-3 fats) can also have anti-inflammatory effects, and can modulate body fat and body mass (Schemmel et al., 1969; Sumiyoshi et al., 2006; Hariri and Thibault, 2010; Wu et al., 2014, 2017a). To add to the complexity of this matter, many animal species, like humans, demonstrate obesity prone and obesity resistant phenotypes (Schemmel et al., 1969). Arguably, skeletal muscle oxidative metabolism (Shahrokhi et al., 1993) and genetic differences in fat oxidation (Ji and Friedman, 2007; Marrades et al., 2007) may affect whether an animal is prone or resistant to obesity. This feature allows for the experimental evaluation of animals that are exposed to the same amounts of an obesogenic diet, but develop disparate increases in body mass. With MSK disease, this is an important consideration, as tissue damage can be evaluated between animals with similar mass, but increased body fat and low-level inflammation (Hariri and Thibault, 2010). Epidemiological studies reveal MetS can also occur in individuals with typical body weight or that are underweight (e.g., HIV-infected patients), with increases in body fat and metabolic derangement, which underscores the relevance of evaluating obesity resistant animals as a surrogate for this phenomenon to understand MSK diseases in the context of MetS (Drelichowska et al., 2015).
Nutrient Sensing, Mitochondrial Dysfunction, and Metabolic Dysfunction
Mitochondria are the primary cellular organelles that generate adenosine triphosphate (ATP). Mitochondrial dysfunction is attributed to excessive nutrient processing, resulting in the uncoupling of oxidative phosphorylation, effectively increasing reactive oxygen species (ROS) generation and decreasing ATP production (Weiss et al., 2013). The free radical theory states that an imbalance between the generation of ROS and antioxidants produced by peroxisomes can result in ROS stealing electrons from other cellular sources, resulting in cellular damage (Ashok and Ali, 1999). Under nutrient overload, as seen in obesity, peroxisomal numbers and/or activity can be impaired as a result of micronutrient deficiencies and/or excessive inflammatory cytokine (e.g., TNF-α) accumulation resulting from elevated oxidative stress and the accumulation of metabolic by-products.
When exposed to nutrient overload, nutrient sensors [i.e., AMP-activated protein kinase (AMPK), sirtuins (SIRT), and mechanistic target of rapamycin, (mTOR)] become impaired, impair mitochondrial function, and can contribute to systemic inflammation, adiposity, and lipotoxicity. AMPK activity also plays a role in bone homeostasis, as an essential mediator of fat and glucose metabolism on bone remodeling (Jeyabalan et al., 2012; reviewed in Finkel, 2015). In skeletal muscle, leptin has been shown to activate AMPK (Salminen and Kaarniranta, 2012), inhibiting FA synthesis, increasing FA oxidation and glucose uptake (Minokoshi et al., 2002). Moreover, limiting nutrient supply and the associated increase in SIRT-1 activity are hypothesized to enhance muscle cell proliferation, while nutrient overload, or age-related loss of SIRT-1, is expected to provide an unfavorable environment for cell proliferation (Akhmedov and Berdeaux, 2013). Likely, reduced cell proliferation occurs, in part, through age-related loss of SIRT, potentially as a consequence of reduced AMPK activity, which may result in loss or dysfunction of mitochondrial activity (Wang et al., 2015; Berenbaum et al., 2017).
In chondrocytes from human OA patients, mitochondrial activity is deficient, leading to a catabolic cascade of responses. Activating AMPK/SIRT-1, mitochondrial master regulator PGC-1a, and FOXO3A mediates chondroprotection, suggesting these pathways may also be critical to maintaining mitochondrial function in chondrocytes, which is critical to preserving cartilage tissue structure (Zhao et al., 2014; Wang et al., 2015; Berenbaum et al., 2017). Mitigating age-related changes in oxidative stress in cartilage (i.e., through SIRT-3) protects against OA-progression and osteoclastogenesis in bone, suggesting that improving the resistance of cartilage to oxidative stress may be one therapeutic target for OA, or for maintaining bone homeostasis (Fu et al., 2016; Huh et al., 2016).
Reactive Oxygen Species (ROS) and Oxidative Stress Alterations with Obesity
As a by-product of mitochondrial metabolism and homeostasis, ROS are produced and are highly regulated. With cell damage, mitochondrial dysfunction, or oxidative stress, ROS levels accumulate and are associated with the onset of metabolic dysfunction (Serra et al., 2013). ROS accumulation can lead to: lipid peroxidation and disruption of the cellular membrane; ER stress, resulting in protein mis-folding and unfolding, and a decrease in protein synthesis, ultimately rendering cells incapable of clearing misfolded proteins; and activation of Caspase-3 and cell apoptosis (Weiss et al., 2013). Furthermore, oxidative stress in tissues is reported to be an important pathogenic mechanism of MetS onset (Furukawa et al., 2004).
Increased fat mass has been linked to increased systemic markers of oxidative stress in humans and mice (Figure 7) (Furukawa et al., 2004). For example, increased peroxide and reduced endothelial nitric oxide synthase have been associated with cancellous bone loss in an obesity model (Ohnishi et al., 2009). In bone, reactive oxygen species and oxidative stress are critical to osteoclast differentiation. As such, ROS may contribute to osteoporosis and bone catabolism, through activation of RANKL—or receptor activator of NF-κB ligand- influencing osteoclast activity, as well as other pathways downstream such as NF-κB (Callaway and Jiang, 2015). With increased systemic inflammation associated with obesity, bone marrow macrophages and their progenitors can be increased (Singer et al., 2014), with related stimulation of osteoclastogenesis and reduced osteoblast development (Kyung et al., 2009; Halade et al., 2011). Yue and co-workers also reported that leptin produced from obese adipose tissue can bind to leptin receptors on mesenchymal stem cells promoting differentiation to adipocytes and inhibiting osteoblast formation (Yue et al., 2016). The central effects of leptin can also promote cancellous bone loss via the sympathetic nervous system (Ducy et al., 2000; Takeda et al., 2002).
In skeletal muscle, although low-levels of ROS are necessary for force production, high levels of ROS can result in great susceptibility to fatigue and contractile dysfunction (Powers et al., 2011) and are associated with reduced muscle repair capacity (Kozakowska et al., 2015). There may be a link between dysfunctional repair and overuse tendinopathies through ROS production, but this idea remains to be tested experimentally (Bestwick and Maffulli, 2004; Longo et al., 2008). Although the signaling pathway between ROS and osteoarthritis is not clear, there are examples of antioxidant therapy (e.g., dietary polyphenols and hyaluronic acid) that are useful in human studies (reviewed in Lepetsos and Papavassiliou, 2016).
Lipotoxicity and Insulin Resistance with Obesity
Accumulation of lipids and lipid by-products can result in dysfunction in myocyte metabolic pathways. Lipid concentration is considered a strong indicator of the sensitivity of myocytes insulin-mediated pathways, as well as adipocyte and hepatocyte insulin sensitivity, significantly affecting substrate metabolism and circulating metabolite concentrations (Weiss et al., 2013). The adverse metabolic effects associated with ectopic lipid storage are supported by lipodystrophy studies, showing that muscle lipid accumulation can result in severe insulin resistance and diabetes (Weiss et al., 2013). In addition to the liver, primarily utilized for short term energy storage when circulating nutrient levels are elevated, skeletal muscle is a primary site of glucose uptake and utilization (Yu et al., 2012). Skeletal muscle insulin resistance largely contributes to whole body glucose levels and the presence of a pro-inflammatory environment, as the body is comprised of 40–50% muscle by weight (DeFronzo and Tripathy, 2009; Weiss et al., 2013). In response to low-level inflammation from DIO, extracellular matrix remodeling may contribute to the onset of insulin resistance, thereby inducing collagen synthesis and muscle fibrosis, and contributing to decreased insulin signaling (Williams et al., 2015).
Insulin resistance can also impact tissues of the joint organ system, because insulin resistance has been linked to chondrocyte dysfunction, and insulin appears to have a protective role for synoviocytes, whereby insulin blunts TNF-induced matrix metalloproteinase release (Hamada et al., 2015). However, patients with diabetes exhibit increased synovial levels of TNF-α and macrophages, suggesting that insulin resistance may impair the protective effect of insulin in the joint (Hamada et al., 2016), and greater insulin resistance is related to lower femoral neck strength (Srikanthan et al., 2014).
Accumulation of Metabolic By-Products and Toxic Lipid Metabolites in Obesity
Elevated levels of oxidative stress can result in incomplete substrate oxidation. These impairments can lead to the excessive accumulation of toxic lipid metabolites (e.g., diacylglycerol, fatty acetyl CoA, and ceramides) and ROS, all natural by-products of cellular metabolism (Broussard and Devkota, 2016). Fatty acid trafficking in muscle may be one of the key factors involved in the onset of insulin resistance, by changing the availability of substrates involved in formation and clearance of harmful lipid intermediates (diacylglycerides and ceramide) (Mittendorfer, 2011). The balance between synthesis and breakdown of these intermediates may influence the balance between the formation and breakdown of FA and TGs, resulting in the storage and development of lipid depots in non-adipose tissues (Mittendorfer, 2011) from alterations in clearance mechanisms.
Hyperglycemia and Advanced Glycation End Products with Obesity
Advanced glycation end products (AGEs) have a pronounced effect on many proteins, particularly collagen. Generally, AGEs accumulate in tissues and form cross-links with targeted proteins, alter cell structure, and interact with receptors that induce oxidative stress and inflammation, resulting in tissue damage (Sanguineti et al., 2014), Figure 5. Specifically, AGEs can alter the physiological failure behavior of some tissues, including tendons (Fessel et al., 2014), by increasing lateral spacing, accumulating cross-links between collagen fibrils, and reducing mechanical properties of tissues by reducing sliding behavior (Gautieri et al., 2017). Moreover, AGEs affect growth and modulate the physiological processes of OA (Franke et al., 2009), and damage-associated molecular patterns (DAMPS) can bind to RAGE and contribute to the pathogenesis of OA (Rosenberg et al., 2017). AGEs can induce muscle atrophy in a RAGE-mediated AMPK down-regulated manner (Chiu et al., 2016) and may affect skeletal muscle growth and contractile function in mice fed a diet high in AGEs (Egawa et al., 2017). In bone, AGEs are associated with decreased bone mineral density and impaired bone quality, eliciting oxidative stress and inflammatory responses in bone cells, while altering material properties of bone collagen fibers via cross-linking (Yamamoto and Sugimoto, 2016).
Obesity, Bacterial Lipopolysaccharide (LPS), and Gut Microbiota
It is well-established that the gut microbiota is altered with DIO in a manner that promotes a pro-inflammatory environment (Cani et al., 2009; Cândido et al., 2017). Through changes in tight junction proteins and intestinal barrier integrity, components of gram-negative bacteria may leak into the systemic circulation, resulting in increased systemic LPS concentrations in obese animals including humans (Figure 8) (Cani et al., 2008). Systemic LPS, inflammation, and metabolic disturbance resulting from altered gut microbiota and a leaky gut are believed to be key initiating factors leading to insulin resistance (Cani et al., 2007). In the context of musculoskeletal disease, an impaired gut barrier function has been implicated in rheumatoid arthritis and OA, and may present a viable therapeutic approach for disease management (Scher and Abramson, 2011). However, to date, cause and effect relations between rheumatoid arthritis and the gut microbiota are still being explored (Bravo-Blas et al., 2016).
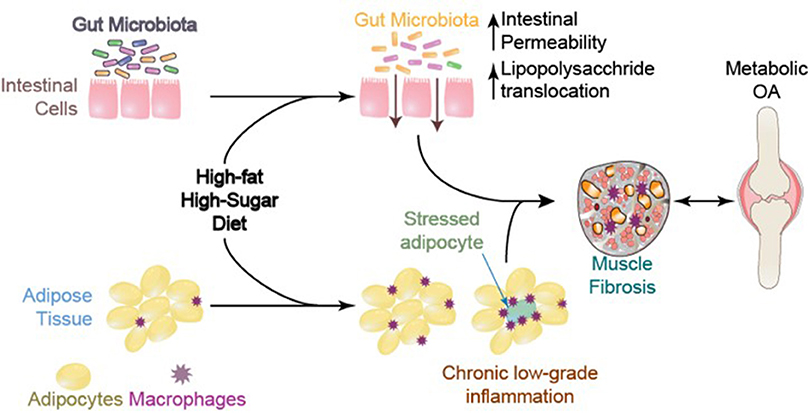
Figure 8. Systemic mediators and compromised muscle integrity are associated with Metabolic OA onset and progression, adapted from Collins et al. (2015a).
We were among the first to identify a significant linkage between constituents of the gut microbiota (Methanobrevibacter and Lactobacillis spp.) and musculoskeletal changes in metabolic OA/joint damage severity (Collins et al., 2015a) and the onset of compromised muscle integrity (Collins et al., 2016c). These findings link systemic (serum) and local (synovial fluid) to LPS and musculoskeletal changes with diet-induced obesity (Huang et al., 2016) (Figure 9). Using a fecal transplant intervention, lipid profiles can be transferred from donors to germ-free hosts, and altered muscle integrity can be recapitulated, suggesting that the gut microbiota may have a direct effect on muscle development and integrity (Yan et al., 2016). The gut-OA pathophysiological link indicates that dietary and microbiota-based interventions may be important therapeutic opportunities that should be evaluated in the context of MSK health (Collins et al., 2015a; Huang et al., 2016; Berenbaum et al., 2017). There is a need for studies detailing the causal mechanisms of interactions among the gut-microbiota, LPS, and MSK changes. However, given the short-term changes in gut microbiota composition, as well as the decrease in variance in relative abundance across species, studies promoting gut microbiota diversity could be useful in this context. One such candidate is evaluating the impact of prebiotic fiber, which may positively modulate the gut microbiota to promote improved host interactions (Bomhof et al., 2014; Paul et al., 2016; Nicolucci et al., 2017; Parnell et al., 2017).
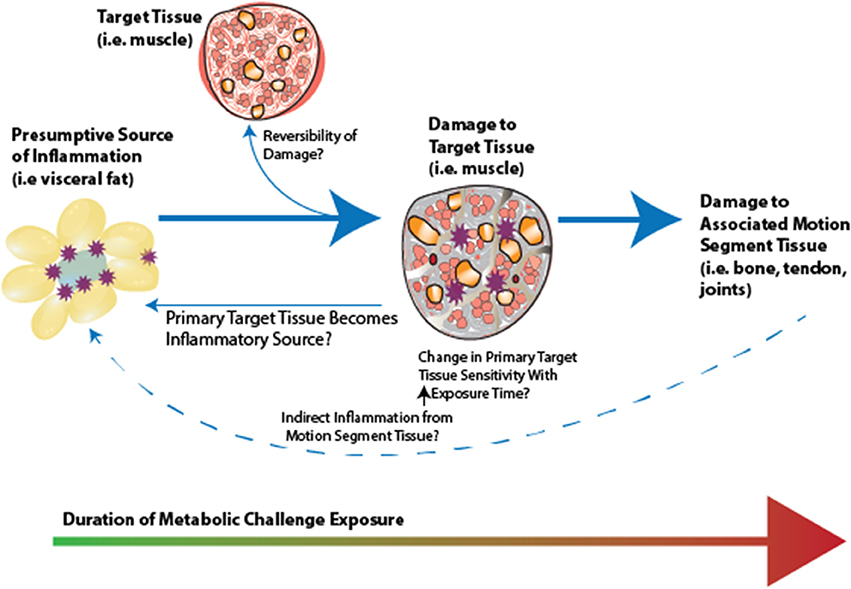
Figure 9. Conceptual framework for inflammatory initiation by a presumptive inflammatory source (i.e., visceral fat, gut microbiota), initiation of adaptation, and subsequent damage in primary target tissue. There is likely a “ripple effect” to the motion segment tissues. Whether associated motion segment tissues becomie inflammatory sources, affecting a presumptive inflammatory source, as well as whether damage is reversible in these tissues, represent interesting open research questions in this area.
Implications for Conservative Care Strategies Related to MetS and Inflammation
Information regarding pharmacologic management for obesity and metabolic syndrome can be found elsewhere (Apovian et al., 2015), and promising therapeutic targets are discussed throughout this review (and highlighted in Table 3).

Table 3. Remaining questions, research agenda, and evidence-supported candidate pathways, targets, and conservative care opportunities.
The inflammatory interface between damage to MSK components and MetS should be a critical consideration, as protecting and preserving MSK structural integrity could be a key approach to conservative management of MetS. Presently, clinical guidelines indicate that weight loss and exercise are good evidence-based conservative approaches for MSK conditions (McAlindon et al., 2014). In the context of osteoarthritis specifically, the combined effect of 18-months of dietary modulation and a combination program involving both aerobic and strength training-based exercise was found to induce weight loss, reduce knee compressive forces, reduce serum IL-6 levels, decrease infrapatellar fat pad size, decrease pain, and increase function in overweight or obese adults with knee OA (Messier et al., 2013; Beavers et al., 2015; Pogacnik Murillo et al., 2017). As such, careful characterization of exercise and dietary interventions in human and other animal models is needed to implement these strategies.
Although there are several candidates for dietary intervention, prebiotic fiber and probiotic supplementation targeting the microbiome (Nicolucci et al., 2017; Parnell et al., 2017), and decreased intake of dietary sugar (Te Morenga et al., 2013) are three safe, readily available, and translatable dietary interventions to protect against MSK damage due to metabolic disturbance that warrant further investigation. As the onset of MSK damage with metabolic disturbance does not have a readily identified starting point, studies are needed to evaluate the efficacy and utility of these dietary modulations in the patient population of interest (e.g., in prevention vs. reversibility and acute vs. chronic), as well as unintended consequences of these modifications. It would be critical to monitor muscle, tendon, bone, and cartilage tissues simultaneously while evaluating these dietary interventions, to facilitate an understanding of the benefits and interconnectedness of these MSK tissue changes and diseases.
Opportunities and Future Research Directions
There are several gaps in the current understanding of the effects of low-level systemic inflammation vs. metabolic disturbance on musculoskeletal health. Many of these gaps are summarized in Table 3. Whether muscle is a primary target tissue in the motion segment, and whether muscular changes directly result in subsequent changes in bone, tendon, and joints, remains to be assessed and clarified. The studies presented here underscore the need for evaluating multiple musculoskeletal tissues and diseases in concert with MetS (as well as defining which elements of the MetS are responsible for which consequences of obesity). This approach will facilitate an understanding of how damage in each tissue develops with respect to the other tissues comprising a motion segment, how the different tissues may contribute to homeostasis or damage of the motion segment, and what role that MetS/inflammation plays in manifesting the risks of tissue damage. At present, it is unclear if: MetS-related inflammation increases in severity with chronicity; systemic inflammation is constant with continued metabolic disturbance; or MetS-related inflammation is dynamic and fluctuating. It may be useful to evaluate tissues as “sources” or “targets” of low-level systemic inflammation (Figure 9). This conceptual framework is demonstrated in the data from our previous studies, which suggest a systemic-to-local time-course shift from serum to synovial fluid, and serum to muscle changes (Collins et al., 2016a,b). Briefly, metabolic dysregulation initiates an inflammatory cascade, whereby an early perturbation in a presumptive inflammatory source (i.e., visceral fat and gut microbiota) results in target tissue (i.e., muscle) adaptation and subsequent damage. Although the source of the inflammation is unclear, as are the specific pathways involved, target tissue inflammation and damage may affect subsequent associated motion segment tissues (i.e., bone, tendon, and joints), and these relations are affected by many factors (i.e., the dynamic make-up/profile of the inflammatory components, time of exposure, and severity of the metabolic disturbance).
Furthermore, adaptation and impact of sources of inflammation on target tissues are important knowledge gaps that need to be addressed. It would be of value to determine if metabolic dysfunction and regulation are integrated and interdependent, or if source tissues and target tissues of the motion segment are regulated and affected differently and independently, at least in a first approximation. Identifying the potential for reversibility of tissue damage with diet-induced obesity is another critical area of research that should be addressed. The mechanism(s) by which target tissue or motion segment tissue changes may contribute to MetS and inflammation are incompletely understood at this time. Time-course studies are needed to answer these questions.
Discussion of the impact of metabolic disturbance on neuroregulatory systems, and a role for neuroinflammation-mediated influence on loss of musculoskeletal integrity was limited in this review, but this may be a potentially important area for future research. Nearly all of the tissues of a motion segment discussed are innervated except for adult cartilage, and thus, neuro-regulation and how it is affected primarily and secondarily by MetS associated inflammation, may be critical in understanding the underlying mechanistic considerations. In particular, how these changes in neuroregulation and neuroinflammation contribute to a loss of muscle integrity is an interesting area for future research.
Summary
Studies presented in this review implicate several pathways that may be critical for the onset and progression of systemic inflammation due to metabolic disturbance and musculoskeletal damage. Some of the pathways that could be involved are mitogen-activated protein kinases (MAPK, p38 MAPK, JNK), myeloid differentiation primary response gene 88 (MyD88), NFκB, and the NLRP3 inflammasome. Likely, some or all of these pathways may be activated in parallel creating a potentially positive-feedback based redundant system. Investigations targeting specific pathways in the context of metabolic disturbance and MSK disease will provide much needed mechanistic insights to better understand the consequences of diet-induced obesity.
A motion segment, such as a leg is comprised of a number of interdependent components (i.e., muscles, tendons, bones, and articular joints) that rely on the integrated biological and mechanical integrity of all tissues for optimal function. Thus, the direct and indirect impact of dysregulation via MetS and associated inflammation on any one element may have a “ripple” effect that may be compounded over time. There are many important and relevant gaps in this research area, and addressing them will provide valuable insights into the relations among the motion segment, common inflammatory pathways, and resulting MSK disease.
Author Contributions
KC was responsible for conception and design, collection, and assembly of data analysis and interpretation of the data, drafting of the article, critical revision of the article for important intellectual content and final approval of the article. WH, GM, RR, JR, IS, and RZ were responsible for conception and design, collection and assembly of data analysis, interpretation of the data, critical revision of the article for important intellectual content and final approval of the article. DH was responsible for conception and design, analysis and interpretation of the data, drafting of the article, critical revision of the article for important intellectual content and final approval of the article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Canadian Institutes of Health Research # RT736475 and MOP 115076, the Canada Research Chair Programme, the Alberta Innovates Health Solutions Osteoarthritis Team Grant, Alberta Innovates Health Solutions, Alberta Health Services Strategic Clinical Network Program, Canadian Institutes of Health Research Banting and Best Canada Graduate Scholarship, and the Killam Foundation. Due to space and scope limitations, we apologize to authors in this area whose work we were unable to include in the present review.
References
Abate, M. (2014). How obesity modifies tendons (implications for athletic activities). Muscles Ligaments Tendons J. 4, 298–302.
Abate, M., Salini, V., and Andia, I. (2016). How Obesity affects tendons? Adv. Exp. Med. Biol. 920, 167–177. doi: 10.1007/978-3-319-33943-6_15
Abate, M., Schiavone, C., Salini, V., and Andia, I. (2013). Occurrence of tendon pathologies in metabolic disorders. Rheumatology 52, 599–608. doi: 10.1093/rheumatology/kes395
Abella, V., Pino, J., Scotece, M., Conde, J., Lago, F., Gonzalez-Gay, M. A., et al. (2017a). Progranulin as a biomarker and potential therapeutic agent. Drug Discov. Today 22, 1557–1564. doi: 10.1016/J.DRUDIS.2017.06.006
Abella, V., Scotece, M., Conde, J., López, V., Pirozzi, C., Pino, J., et al. (2016). The novel adipokine progranulin counteracts IL-1 and TLR4-driven inflammatory response in human and murine chondrocytes via TNFR1. Sci. Rep. 6:20356. doi: 10.1038/srep20356
Abella, V., Scotece, M., Conde, J., Pino, J., Gonzalez-Gay, M. A., Gómez-Reino, J. J., et al. (2017b). Leptin in the interplay of inflammation, metabolism and immune system disorders. Nat. Rev. Rheumatol. 13, 100–109. doi: 10.1038/nrrheum.2016.209
Addison, O., Drummond, M. J., LaStayo, P. C., Dibble, L. E., Wende, A. R., McClain, D. A., et al. (2014). Intramuscular fat and inflammation differ in older adults: the impact of frailty and inactivity. J. Nutr. Health Aging 18, 532–538. doi: 10.1007/s12603-014-0019-1
Ajuwon, K. M., and Spurlock, M. E. (2005). Adiponectin inhibits LPS-induced NF- B activation and IL-6 production and increases PPAR 2 expression in adipocytes. AJP Regul. Integr. Comp. Physiol. 288, R1220–R1225. doi: 10.1152/ajpregu.00397.2004
Akhmedov, D., and Berdeaux, R. (2013). The effects of obesity on skeletal muscle regeneration. Front. Physiol. 4:371. doi: 10.3389/fphys.2013.00371
Alberti, K. G., and Zimmet, P. Z. (1998). Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet. Med. 15, 539–553. doi: 10.1002/(SICI)1096-9136(199807)15:7<539::AID-DIA668>3.0.CO;2-S
Apovian, C. M., Aronne, L. J., Bessesen, D. H., McDonnell, M. E., Murad, M. H., Pagotto, U., et al. (2015). Pharmacological management of obesity: an endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 100, 342–362. doi: 10.1210/jc.2014-3415
Ashok, B. T., and Ali, R. (1999). The aging paradox: free radical theory of aging. Exp. Gerontol. 34, 293–303. doi: 10.1016/S0531-5565(99)00005-4
Asif, M., Egan, J., Vasan, S., Jyothirmayi, G. N., Masurekar, M. R., Lopez, S., et al. (2000). An advanced glycation endproduct cross-link breaker can reverse age-related increases in myocardial stiffness. Proc. Natl. Acad. Sci. U.S.A. 97, 2809–2813. doi: 10.1073/pnas.040558497
Barboza, E., Hudson, J., Chang, W.-P., Kovats, S., Towner, R. A., Silasi-Mansat, R., et al. (2017). Pro-fibrotic infrapatellar fat pad remodeling without M1-macrophage polarization precedes knee osteoarthritis in diet-induced obese mice. Arthritis Rheumatol. 69, 1221–1232. doi: 10.1002/art.40056
Baumgartner, R. N. (2000). Body composition in healthy aging. Ann. N. Y. Acad. Sci. 904, 437–448. doi: 10.1111/j.1749-6632.2000.tb06498.x
Baumgartner, R. N., Koehler, K. M., Gallagher, D., Romero, L., Heymsfield, S. B., Ross, R. R., et al. (1998). Epidemiology of sarcopenia among the elderly in New Mexico. Am. J. Epidemiol. 147, 755–763.
Baumgartner, R. N., Wayne, S. J., Waters, D. L., Janssen, I., Gallagher, D., and Morley, J. E. (2004). Sarcopenic obesity predicts instrumental activities of daily living disability in the elderly. Obes. Res. 12, 1995–2004. doi: 10.1038/oby.2004.250
Beavers, K. M., Beavers, D. P., Newman, J. J., Anderson, A. M., Loeser, R. F., Nicklas, B. J., et al. (2015). Effects of total and regional fat loss on plasma CRP and IL-6 in overweight and obese, older adults with knee osteoarthritis. Osteoarthr. Cartil. 23, 249–256. doi: 10.1016/j.joca.2014.11.005
Berenbaum, F., Griffin, T. M., and Liu-Bryan, R. (2017). metabolic regulation of inflammation in osteoarthritis. Arthritis Rheumatol. 69, 9–21. doi: 10.1002/art.39842
Bestwick, C. S., and Maffulli, N. (2004). Reactive oxygen species and tendinopathy: do they matter? Br. J. Sports Med. 38, 672–674. doi: 10.1136/bjsm.2004.012351
Boldrin, L., Zammit, P. S., and Morgan, J. E. (2015). Satellite cells from dystrophic muscle retain regenerative capacity. Stem Cell Res. 14, 20–29. doi: 10.1016/j.scr.2014.10.007
Bomhof, M. R., Saha, D. C., Reid, D. T., Paul, H. A., and Reimer, R. A. (2014). Combined effects of oligofructose and Bifidobacterium animalis on gut microbiota and glycemia in obese rats. Obesity 22, 763–771. doi: 10.1002/oby.20632
Bopp, C., Bierhaus, A., Hofer, S., Bouchon, A., Nawroth, P. P., Martin, E., et al. (2008). Bench-to-bedside review: the inflammation-perpetuating pattern-recognition receptor RAGE as a therapeutic target in sepsis. Crit. Care 12, 201. doi: 10.1186/cc6164
Braune, J., Weyer, U., Hobusch, C., Mauer, J., Brüning, J. C., Bechmann, I., et al. (2017). IL-6 regulates M2 polarization and local proliferation of adipose tissue macrophages in obesity. J. Immunol. 198, 2927–2934. doi: 10.4049/jimmunol.1600476
Bravo-Blas, A., Wessel, H., and Milling, S. (2016). Microbiota and arthritis. Curr. Opin. Rheumatol. 28, 1. doi: 10.1097/BOR.0000000000000261
Broussard, J. L., and Devkota, S. (2016). The changing microbial landscape of Western society: diet, dwellings and discordance. Mol. Metab. 5, 737–742. doi: 10.1016/j.molmet.2016.07.007
Brown, L. A., Lee, D. E., Patton, J. F., Perry, R. A., Brown, J. L., Baum, J. I., et al. (2015). Diet-induced obesity alters anabolic signalling in mice at the onset of skeletal muscle regeneration. Acta Physiol. 215, 46–57. doi: 10.1111/apha.12537
Brunk, B. P., Goldhamer, D. J., and Emerson, C. P. Jr. (1996). Regulated demethylation of the myoD Distal enhancer during skeletal myogenesis. Dev. Biol. 177, 490–503. doi: 10.1006/dbio.1996.0180
Buettner, R., Schölmerich, J., and Bollheimer, L. C. (2007). High-fat diets: modeling the metabolic disorders of human obesity in rodents. Obesity 15, 798–808. doi: 10.1038/oby.2007.608
Callaway, D. A., and Jiang, J. X. (2015). Reactive oxygen species and oxidative stress in osteoclastogenesis, skeletal aging and bone diseases. J. Bone Miner. Metab. 33, 359–370. doi: 10.1007/s00774-015-0656-4
Cândido, F. G., Valente, F. X., Grześkowiak, Ł. M., Moreira, A. P. B., Rocha, D. M. U. P., and Alfenas, R. C. G. (2017). Impact of dietary fat on gut microbiota and low-grade systemic inflammation: mechanisms and clinical implications on obesity. Int. J. Food Sci. Nutr. 69, 125–143. doi: 10.1080/09637486.2017.1343286
Cani, P. D., Amar, J., Iglesias, M. A., Poggi, M., Knauf, C., Bastelica, D., et al. (2007). Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56, 1761–1772. doi: 10.2337/db06-1491
Cani, P. D., Bibiloni, R., Knauf, C., Waget, A., Neyrinck, A. M., Delzenne, N. M., et al. (2008). Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 57, 1470–1481. doi: 10.2337/db07-1403
Cani, P. D., Possemiers, S., Van de Wiele, T., Guiot, Y., Everard, A., Rottier, O., et al. (2009). Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 58, 1091–1103. doi: 10.1136/gut.2008.165886
Cao, J. J. (2011). Effects of obesity on bone metabolism. J. Orthop. Surg. Res. 6:30. doi: 10.1186/1749-799X-6-30
Carrió, E., and Suelves, M. (2015). DNA methylation dynamics in muscle development and disease. Front. Aging Neurosci. 7:19. doi: 10.3389/fnagi.2015.00019
Chakravarti, A., Raquil, M.-A., Tessier, P., and Poubelle, P. E. (2009). Surface RANKL of Toll-like receptor 4–stimulated human neutrophils activates osteoclastic bone resorption. Blood 114, 1633–1644. doi: 10.1182/blood-2008
Chapman, M. A., Meza, R., and Lieber, R. L. (2016). Skeletal muscle fibroblasts in health and disease. Differentiation 92, 108–115. doi: 10.1016/j.diff.2016.05.007
Chiang, D. J., Pritchard, M. T., and Nagy, L. E. (2011). Obesity, diabetes mellitus, and liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G697–G702. doi: 10.1152/ajpgi.00426.2010
Chiu, C.-Y., Yang, R.-S., Sheu, M.-L., Chan, D.-C., Yang, T.-H., Tsai, K.-S., et al. (2016). Advanced glycation end-products induce skeletal muscle atrophy and dysfunction in diabetic mice via a RAGE-mediated, AMPK-down-regulated, Akt pathway. J. Pathol. 238, 470–482. doi: 10.1002/path.4674
Chu, C. R., Szczodry, M., and Bruno, S. (2010). Animal models for cartilage regeneration and repair. Tissue Eng. Part B Rev. 16, 105–115. doi: 10.1089/ten.TEB.2009.0452
Ciapaite, J., van den Berg, S. A., Houten, S. M., Nicolay, K., van Dijk, K. W., and Jeneson, J. A. (2015). Fiber-type-specific sensitivities and phenotypic adaptations to dietary fat overload differentially impact fast- versus slow-twitch muscle contractile function in C57BL/6J mice. J. Nutr. Biochem. 26, 155–164. doi: 10.1016/j.jnutbio.2014.09.014
Clark, B. A., Alloosh, M., Wenzel, J. W., Sturek, M., and Kostrominova, T. Y. (2011). Effect of diet-induced obesity and metabolic syndrome on skeletal muscles of Ossabaw miniature swine. Am. J. Physiol. Endocrinol. Metab. 300, E848–E857. doi: 10.1152/ajpendo.00534.2010
Collins, K. H., Hart, D. A., Reimer, R. A., Seerattan, R. A., Banker, C. W., Sibole, S. C., et al. (2016a). High-fat high-sucrose diet leads to dynamic structural and inflammatory alterations in the rat vastus lateralis muscle. J. Orthop. Res. 34, 2069–2078. doi: 10.1002/jor.23230
Collins, K. H., Hart, D. A., Reimer, R. A., Seerattan, R. A., and Herzog, W. (2016b). Response to diet-induced obesity produces time-dependent induction and progression of metabolic osteoarthritis in rat knees. J. Orthop. Res. 34, 1010–1018. doi: 10.1002/jor.23103
Collins, K. H., Hart, D. A., Seerattan, R. A., Reimer, R. A., and Herzog, W. (2017a). High-fat/high-sucrose diet-induced obesity results in joint-specific development of osteoarthritis-like degeneration in a rat model. Bone Jt. Res. doi: 10.1302/2046-3758.73.BJR-2017-0201.R2. [Epub ahead of print].
Collins, K. H., Hart, D., Smith, I., Issler, A., Reimer, R., Seerattan, R., et al. (2017b). Acute and chronic changes in rat soleus muscle after high-fat high-sucrose diet. Physiol. Rep. 5:e13270. doi: 10.14814/phy2.13270
Collins, K. H., Paul, H. A., Hart, D. A., Reimer, R. A., Smith, I. C., Rios, J. L., et al. (2016c). A high-fat high-sucrose diet rapidly alters muscle integrity, inflammation and gut microbiota in male rats. Sci. Rep. 6:37278. doi: 10.1038/srep37278
Collins, K. H., Paul, H. A., Reimer, R. A., Seerattan, R.-A., Hart, D. A., and Herzog, W. (2015a). Relationship between inflammation, the gut microbiota, and metabolic osteoarthritis development: studies in a rat model. Osteoarthr. Cartil. 23, 1989–1998. doi: 10.1016/j.joca.2015.03.014
Collins, K. H., Reimer, R. A., Seerattan, R.-A. A., Leonard, T. R., and Herzog, W. (2015b). Using diet-induced obesity to understand a metabolic subtype of osteoarthritis in rats. Osteoarthr. Cartil. 23, 957–965. doi: 10.1016/j.joca.2015.01.015
Collins, K. H., Sharif, B., Sanmartin, C., Reimer, R. A., Herzog, W., Chin, R., et al. (2017c). Association of body mass index (BMI) and percent body fat among BMI-defined non-obese middle-aged individuals: insights from a population-based Canadian sample. Can. J. Public Heal. 107, 520. doi: 10.17269/cjph.107.5652
D'Souza, D. M., Trajcevski, K. E., Al Sajee, D., Wang, D. C., Thomas, M., Anderson, J. E., et al. (2015). Diet induced obesity impairs muscle satellite cell activation and muscle repair through alterations in hepatocyte growth factor signaling. Physiol. Rep. 3:e12506. doi: 10.14814/phy2.12506
de Mos, M., Joosten, L. A. B., Oppers-Walgreen, B., van Schie, J. T. M., Jahr, H., van Osch, G. J. V. M., et al. (2009). Tendon degeneration is not mediated by regulation of Toll-like receptors 2 and 4 in human tenocytes. J. Orthop. Res. 27, 1043–1047. doi: 10.1002/jor.20834
DeFronzo, R. A., and Tripathy, D. (2009). Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 32, S157–S163. doi: 10.2337/dc09-S302
Drelichowska, J., Kwiatkowska, W., Knysz, B., and Witkiewicz, W. (2015). Metabolic syndrome in HIV-positive patients. HIV AIDS Rev. 14, 35–41. doi: 10.1016/j.hivar.2014.09.002
Ducy, P., Amling, M., Takeda, S., Priemel, M., Schilling, A. F., Beil, F. T., et al. (2000). Leptin inhibits bone formation through a hypothalamic relay: a central control of bone mass. Cell 100, 197–207. doi: 10.1016/S0092-8674(00)81558-5
Egawa, T., Tsuda, S., Goto, A., Ohno, Y., Yokoyama, S., Goto, K., et al. (2017). Potential involvement of dietary advanced glycation end products in impairment of skeletal muscle growth and muscle contractile function in mice. Br. J. Nutr. 117, 21–29. doi: 10.1017/S0007114516004591
Farnaghi, S., Prasadam, I., Cai, G., Friis, T., Du, Z., Crawford, R., et al. (2017). Protective effects of mitochondria-targeted antioxidants and statins on cholesterol-induced osteoarthritis. FASEB J. 31, 356–367. doi: 10.1096/fj.201600600R
Fellner, C., Schick, F., Kob, R., Hechtl, C., Vorbuchner, M., Büttner, R., et al. (2014). Diet-induced and age-related changes in the quadriceps muscle: MRI and MRS in a rat model of sarcopenia. Gerontology 60, 530–538. doi: 10.1159/000360289
Felson, D. T., Anderson, J. J., Naimark, A., Walker, A. M., and Meenan, R. F. (1988). Obesity and knee osteoarthritis. The Framingham Study. Ann. Intern. Med. 109, 18–24.
Fessel, G., Li, Y., Diederich, V., Guizar-Sicairos, M., Schneider, P., Sell, D. R., et al. (2014). Advanced glycation end-products reduce collagen molecular sliding to affect collagen fibril damage mechanisms but not stiffness. PLoS ONE 9:e110948. doi: 10.1371/journal.pone.0110948
Filková, M., Lisková, M., Hulejová, H., Haluzík, M., Gatterová, J., Pavelková, A., et al. (2009). Increased serum adiponectin levels in female patients with erosive compared with non-erosive osteoarthritis. Ann. Rheum. Dis. 68, 295–296. doi: 10.1136/ard.2008.095737
Fink, L. N., Costford, S. R., Lee, Y. S., Jensen, T. E., Bilan, P. J., Oberbach, A., et al. (2014). Pro-inflammatory macrophages increase in skeletal muscle of high fat-fed mice and correlate with metabolic risk markers in humans. Obesity 22, 747–757. doi: 10.1002/oby.20615
Fontana, L., Eagon, J. C., Trujillo, M. E., Scherer, P. E., and Klein, S. (2007). Visceral fat adipokine secretion is associated with systemic inflammation in obese humans. Diabetes 56, 1010–1013. doi: 10.2337/db06-1656
Fothergill, E., Guo, J., Howard, L., Kerns, J. C., Knuth, N. D., Brychta, R., et al. (2016). Persistent metabolic adaptation 6 years after “The Biggest Loser” competition. Obesity 24, 1612–1619. doi: 10.1002/oby.21538
Fowler-Brown, A., Kim, D. H., Shi, L., Marcantonio, E., Wee, C. C., Shmerling, R. H., et al. (2014). The mediating effect of leptin on the relationship between body weight and knee osteoarthritis in older adults. Arthritis Rheumatol. 67, 169–175. doi: 10.1002/art.38913
Frank, C. B., Shrive, N. G., Boorman, R. S., Lo, I. K. Y., and Hart, D. A. (2004). New perspectives on bioengineering of joint tissues: joint adaptation creates a moving target for engineering replacement tissues. Ann. Biomed. Eng. 32, 458–465. doi: 10.1023/B:ABME.0000017548.85451.b7
Franke, S., Sommer, M., Rüster, C., Bondeva, T., Marticke, J., Hofmann, G., et al. (2009). Advanced glycation end products induce cell cycle arrest and proinflammatory changes in osteoarthritic fibroblast-like synovial cells. Arthritis Res. Ther. 11, R136. doi: 10.1186/ar2807
Friedman, J. M., and Halaas, J. L. (1998). Leptin and the regulation of body weight in mammals. Nature 395, 763–770. doi: 10.1038/27376
Frommhold, D., Kamphues, A., Dannenberg, S., Buschmann, K., Zablotskaya, V., Tschada, R., et al. (2011). RAGE and ICAM-1 differentially control leukocyte recruitment during acute inflammation in a stimulus-dependent manner. BMC Immunol. 12:56. doi: 10.1186/1471-2172-12-56
Fu, Y., Kinter, M., Hudson, J., Humphries, K. M., Lane, R. S., White, J. R., et al. (2016). Aging promotes sirtuin 3-dependent cartilage superoxide dismutase 2 acetylation and osteoarthritis. Arthritis Rheumatol. 68, 1887–1898. doi: 10.1002/art.39618
Furukawa, S., Fujita, T., Shimabukuro, M., Iwaki, M., Yamada, Y., Nakajima, Y., et al. (2004). Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Invest. 114, 1752–1761. doi: 10.1172/JCI21625
Gautieri, A., Passini, F. S., Silván, U., Guizar-Sicairos, M., Carimati, G., Volpi, P., et al. (2017). Advanced glycation end-products: mechanics of aged collagen from molecule to tissue. Matrix Biol. 15, 95–108. doi: 10.1016/j.matbio.2016.09.001
Gomez, R., Lago, F., Gomez-Reino, J., Dieguez, C., and Gualillo, O. (2009). Adipokines in the skeleton: influence on cartilage function and joint degenerative diseases. J. Mol. Endocrinol. 43, 11–18. doi: 10.1677/JME-08-0131
Gordon, S., and Martinez, F. O. (2010). Alternative activation of macrophages: mechanism and functions. Immunity 32, 593–604. doi: 10.1016/j.immuni.2010.05.007
Griffin, T. M., Fermor, B., Huebner, J. L., Kraus, V. B., Rodriguiz, R. M., Wetsel, W. C., et al. (2010). Diet-induced obesity differentially regulates behavioral, biomechanical, and molecular risk factors for osteoarthritis in mice. Arthritis Res. Ther. 12, R130. doi: 10.1186/ar3068
Griffin, T. M., Huebner, J. L., Kraus, V. B., and Guilak, F. (2009). Extreme obesity due to impaired leptin signaling in mice does not cause knee osteoarthritis. Arthritis Rheum. 60, 2935–2944. doi: 10.1002/art.24854
Griffin, T. M., Huebner, J. L., Kraus, V. B., Yan, Z., and Guilak, F. (2012). Induction of osteoarthritis and metabolic inflammation by a very high-fat diet in mice: effects of short-term exercise. Arthritis Rheum. 64, 443–453. doi: 10.1002/art.33332
Grounds, M. D. (2011). “Regeneration of Muscle,” in eLS (Chichester: John Wiley & Sons, Ltd). doi: 10.1002/9780470015902.a0001106.pub2
Grunfeld, C., Zhao, C., Fuller, J., Pollack, A., Moser, A., Friedman, J., et al. (1996). Endotoxin and cytokines induce expression of leptin, the ob gene product, in hamsters. J. Clin. Invest. 97, 2152–2157. doi: 10.1172/JCI118653
Gual, P., Le Marchand-Brustel, Y., and Tanti, J.-F. (2005). Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie 87, 99–109. doi: 10.1016/j.biochi.2004.10.019
Halade, G. V., El Jamali, A., Williams, P. J., Fajardo, R. J., and Fernandes, G. (2011). Obesity-mediated inflammatory microenvironment stimulates osteoclastogenesis and bone loss in mice. Exp. Gerontol. 46, 43–52. doi: 10.1016/j.exger.2010.09.014
Hamada, D., Maynard, R., Schott, E., Drinkwater, C. J., Ketz, J. P., Kates, S. L., et al. (2016). Suppressive effects of insulin on tumor necrosis factor-dependent early osteoarthritic changes associated with obesity and type 2 diabetes mellitus. Arthritis Rheumatol. 68, 1392–1402. doi: 10.1002/art.39561
Hamada, D., Maynard, R., Schott, E., Drinkwater, C. J., Ketz, J. P., Kates, S. L., et al. (2015). Insulin Suppresses TNF-dependent Early Osteoarthritic Changes Associated with Obesity and Type 2 Diabetes. Arthritis Rheumatol.
Harasymowicz, N. S., Clement, N. D., Azfer, A., Burnett, R., Salter, D. M., and Simpson, A. H. W. R. (2017). Regional differences between perisynovial and infrapatellar adipose tissue depots and their response to class II and III obesity in patients with OA. Arthritis Rheumatol. 69, 1396–1406. doi: 10.1002/art.40102
Hariri, N., and Thibault, L. (2010). High-fat diet-induced obesity in animal models. Nutr. Res. Rev. 23, 270–299. doi: 10.1017/S0954422410000168
Hart, D. A., Kydd, A. S., Frank, C. B., and Hildebrand, K. A. (2004). Tissue repair in rheumatoid arthritis: challenges and opportunities in the face of a systemic inflammatory disease. Best Pract. Res. Clin. Rheumatol. 18, 187–202. doi: 10.1016/j.berh.2004.02.007
Hart, D. A., and Scott, A. (2012). Getting the dose right when prescribing exercise for connective tissue conditions: the Yin and the Yang of tissue homeostasis. Br. J. Sports Med. 46, 696–698. doi: 10.1136/bjsports-2011-090083
Hawker, G. A., Croxford, R., Bierman, A. S., Harvey, P. J., Ravi, B., Stanaitis, I., et al. (2014). All-cause mortality and serious cardiovascular events in people with hip and knee osteoarthritis: a population based cohort study. PLoS ONE 9:e91286. doi: 10.1371/journal.pone.0091286
Heard, B. J., Fritzler, M. J., Wiley, J. P., McAllister, J., Martin, L., El-Gabalawy, H., et al. (2013). Intraarticular and systemic inflammatory profiles may identify patients with osteoarthritis. J. Rheumatol. 40, 1379–1387. doi: 10.3899/jrheum.121204
Henderson, B., and Pettipher, E. R. (1989). Arthritogenic actions of recombinant IL-1 and tumour necrosis factor alpha in the rabbit: evidence for synergistic interactions between cytokines in vivo. Clin. Exp. Immunol. 75, 306–310.
Hittel, D. S., Berggren, J. R., Shearer, J., Boyle, K., and Houmard, J. A. (2009). Increased secretion and expression of myostatin in skeletal muscle from extremely obese women. Diabetes 58, 30–38. doi: 10.2337/db08-0943
Hodgkinson, C. P., Laxton, R. C., Patel, K., and Ye, S. (2008). Advanced glycation end-product of low density lipoprotein activates the toll-like 4 receptor pathway implications for diabetic atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 28, 2275–2281. doi: 10.1161/ATVBAHA.108.175992
Hofmann, B., Yakobus, Y., Indrasari, M., Nass, N., Santos, A. N., Kraus, F. B., et al. (2014). RAGE influences the development of aortic valve stenosis in mice on a high fat diet. Exp. Gerontol. 59, 13–20. doi: 10.1016/j.exger.2014.05.001
Honsawek, S., and Chayanupatkul, M. (2010). Correlation of plasma and synovial fluid adiponectin with knee osteoarthritis severity. Arch. Med. Res. 41, 593–598. doi: 10.1016/j.arcmed.2010.11.007
Hotamisligil, G., Shargill, N., and Spiegelman, B. (1993). Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 259, 87–91. doi: 10.1126/science.7678183
Houard, X., Goldring, M. B., and Berenbaum, F. (2013). Homeostatic mechanisms in articular cartilage and role of inflammation in osteoarthritis. Curr. Rheumatol. Rep. 15, 375. doi: 10.1007/s11926-013-0375-6
Hoy, D., Geere, J.-A., Davatchi, F., Meggitt, B., and Barrero, L. H. (2014). A time for action: opportunities for preventing the growing burden and disability from musculoskeletal conditions in low- and middle-income countries. Best Pract. Res. Clin. Rheumatol. 28, 377–393. doi: 10.1016/j.berh.2014.07.006
Huang, Z. Y., Stabler, T., Pei, F. X., and Kraus, V. B. (2016). Both systemic and local lipopolysaccharide (LPS) burden are associated with knee OA severity and inflammation. Osteoarthr. Cartil. 24, 1769–1775. doi: 10.1016/j.joca.2016.05.008
Huh, J.-E., Shin, J. H., Jang, E. S., Park, S. J., Park, D. R., Ko, R., et al. (2016). Sirtuin 3 (SIRT3) maintains bone homeostasis by regulating AMPK-PGC-1β axis in mice. Sci. Rep. 6:22511. doi: 10.1038/srep22511
Iqbal, S. M., Leonard, C. C., Regmi, S., De Rantere, D., Tailor, P., Ren, G., et al. (2016). Lubricin/proteoglycan 4 binds to and regulates the activity of toll-like receptors in vitro. Sci. Rep. 6:18910. doi: 10.1038/srep18910
Jeyabalan, J., Shah, M., Viollet, B., and Chenu, C. (2012). AMP-activated protein kinase pathway and bone metabolism. J. Endocrinol. 212, 277–290. doi: 10.1530/JOE-11-0306
Ji, H., and Friedman, M. I. (2007). Reduced capacity for fatty acid oxidation in rats with inherited susceptibility to diet-induced obesity. Metabolism 56, 1124–1130. doi: 10.1016/j.metabol.2007.04.006
Ji, J., wang, Z., Shi, D., Gao, X., and Jiang, Q. (2010). Pathologic changes of Achilles tendon in leptin-deficient mice. Rheumatol. Int. 30, 489–493. doi: 10.1007/s00296-009-1001-9
Jialal, I., Kaur, H., and Devaraj, S. (2014). Toll-like receptor status in obesity and metabolic syndrome: a translational perspective. J. Clin. Endocrinol. Metab. 99, 39–48. doi: 10.1210/jc.2013-3092
Jiang, H., Chen, Y., Chen, G., Tian, X., Tang, J., Luo, L., et al. (2017). Leptin accelerates the pathogenesis of heterotopic ossification in rat tendon tissues via mTORC1 signaling. J. Cell. Physiol. 233, 1017–1028. doi: 10.1002/jcp.25955
Jin, X., Yao, T., Zhou, Z., Zhu, J., Zhang, S., Hu, W., et al. (2015). Advanced glycation end products enhance macrophages polarization into M1 phenotype through activating RAGE/NF-κB pathway. Biomed Res. Int. 2015:732450. doi: 10.1155/2015/732450
Karalaki, M., Fili, S., Philippou, A., and Koutsilieris, M. (2009). Muscle regeneration: cellular and molecular events. In Vivo 23, 779–796.
Kawai, M., Devlin, M. J., and Rosen, C. J. (2009). Fat targets for skeletal health. Nat. Rev. Rheumatol. 5, 365–372. doi: 10.1038/nrrheum.2009.102
Khan, I. M., Perrard, X.-Y., Brunner, G., Lui, H., Sparks, L. M., Smith, S. R., et al. (2015). Intermuscular and perimuscular fat expansion in obesity correlates with skeletal muscle T cell and macrophage infiltration and insulin resistance. Int. J. Obes. 39, 1607–1618. doi: 10.1038/ijo.2015.104
Killian, M. L., Cavinatto, L., Galatz, L. M., and Thomopoulos, S. (2012). The role of mechanobiology in tendon healing. J. Shoulder Elb. Surg. 21, 228–237. doi: 10.1016/j.jse.2011.11.002
Kob, R., Bollheimer, L. C., Bertsch, T., Fellner, C., Djukic, M., Sieber, C. C., et al. (2014). Sarcopenic obesity: molecular clues to a better understanding of its pathogenesis? Biogerontology 16, 15–29. doi: 10.1007/s10522-014-9539-7
Koskinen, A., Vuolteenaho, K., Moilanen, T., and Moilanen, E. (2014). Resistin as a factor in osteoarthritis: synovial fluid resistin concentrations correlate positively with interleukin 6 and matrix metalloproteinases MMP-1 and MMP-3. Scand. J. Rheumatol. 43, 249–253. doi: 10.3109/03009742.2013.853096
Koskinen, A., Vuolteenaho, K., Nieminen, R., Moilanen, T., and Moilanen, E. (2011). Leptin enhances MMP-1, MMP-3 and MMP-13 production in human osteoarthritic cartilage and correlates with MMP-1 and MMP-3 in synovial fluid from oa patients. Clin. Exp. Rheumatol. 29, 57–64.
Kozakowska, M., Pietraszek-Gremplewicz, K., Jozkowicz, A., and Dulak, J. (2015). The role of oxidative stress in skeletal muscle injury and regeneration: focus on antioxidant enzymes. J. Muscle Res. Cell Motil. 36, 377–393. doi: 10.1007/s10974-015-9438-9
Kwon, E.-Y., Shin, S.-K., Cho, Y.-Y., Jung, U., Kim, E., Park, T., et al. (2012). Time-course microarrays reveal early activation of the immune transcriptome and adipokine dysregulation leads to fibrosis in visceral adipose depots during diet-induced obesity. BMC Genomics 13:450. doi: 10.1186/1471-2164-13-450
Kyung, T.-W., Lee, J.-E., Van Phan, T., Yu, R., and Choi, H.-S. (2009). Osteoclastogenesis by bone marrow-derived macrophages is enhanced in obese mice. J. Nutr. 139, 502–506. doi: 10.3945/jn.108.100032
Lackey, D. E., and Olefsky, J. M. (2015). Regulation of metabolism by the innate immune system. Nat. Rev. Endocrinol. 12, 15–28. doi: 10.1038/nrendo.2015.189
Laiguillon, M.-C., Courties, A., Houard, X., Auclair, M., Sautet, A., Capeau, J., et al. (2015). Characterization of diabetic osteoarthritic cartilage and role of high glucose environment on chondrocyte activation: toward pathophysiological delineation of diabetes mellitus-related osteoarthritis. Osteoarthr. Cartil. 23, 1513–1522. doi: 10.1016/j.joca.2015.04.026
Lampiasi, N., Russo, R., and Zito, F. (2016). The alternative faces of macrophage generate osteoclasts. Biomed Res. Int. 2016, 1–9. doi: 10.1155/2016/9089610
Laumonier, T., and Menetrey, J. (2016). Muscle injuries and strategies for improving their repair. J. Exp. Orthop. 3, 15. doi: 10.1186/s40634-016-0051-7
Lechler, R. I., Lord, G. M., Matarese, G., Howard, J. K., Baker, R. J., and Bloom, S. R. (1998). Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature 394, 897–901. doi: 10.1038/29795
Lee, H., Lee, S., Cho, I.-H., and Lee, S. J. (2013). Toll-like receptors: sensor molecules for detecting damage to the nervous system. Curr. Protein Pept. Sci. 14, 33–42. doi: 10.2174/1389203711314010006
Lee, S., Kim, T.-N., and Kim, S.-H. (2012). Sarcopenic obesity is more closely associated with knee osteoarthritis than is nonsarcopenic obesity: a cross-sectional study. Arthritis Rheumatol. 64, 3947–3954. doi: 10.1002/art.37696
Lepetsos, P., and Papavassiliou, A. G. (2016). ROS/oxidative stress signaling in osteoarthritis. Biochim. Biophys. Acta Mol. Basis Dis. 1862, 576–591. doi: 10.1016/j.bbadis.2016.01.003
Leuner, B., Max, M., Thamm, K., Kausler, C., Yakobus, Y., Bierhaus, A., et al. (2012). RAGE influences obesity in mice. Effects of the presence of RAGE on weight gain, AGE accumulation, and insulin levels in mice on a high fat diet. Z. Gerontol. Geriatr. 45, 102–108. doi: 10.1007/s00391-011-0279-x
Lim, S., Kim, J. H., Yoon, J. W., Kang, S. M., Choi, S. H., Park, Y. J., et al. (2010). Sarcopenic obesity: prevalence and association with metabolic syndrome in the Korean Longitudinal Study on Health and Aging (KLoSHA). Diabetes Care 33, 1652–1654. doi: 10.2337/dc10-0107
Liu, X., Ning, A. Y., Chang, N. C., Kim, H., Nissenson, R., Wang, L., et al. (2016). Investigating the cellular origin of rotator cuff muscle fatty infiltration and fibrosis after injury. Muscles Ligaments Tendons J. 6, 6–15. doi: 10.11138/mltj/2016.6.1.006
Loeser, R. F., Goldring, S. R., Scanzello, C. R., and Goldring, M. B. (2012). Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 64, 1697–1707. doi: 10.1002/art.34453
Long, Z., Zhang, X., Sun, Q., Liu, Y., Liao, N., Wu, H., et al. (2015). Evolution of metabolic disorder in rats fed high sucrose or high fat diet: Focus on redox state and mitochondrial function. Gen. Comp. Endocrinol. 242, 92–100. doi: 10.1016/j.ygcen.2015.10.012
Longo, U. G., Olivia, F., Denaro, V., Maffulli, N., and Maffulli, N. (2008). Oxygen species and overuse tendinopathy in athletes. Disabil. Rehabil. 30, 1563–1571. doi: 10.1080/09638280701785643
Lübbeke, A., Finckh, A., Puskas, G. J., Suva, D., Lädermann, A., Bas, S., et al. (2013). Do synovial leptin levels correlate with pain in end stage arthritis? Int. Orthop. 37, 2071–2079. doi: 10.1007/s00264-013-1982-6
Makovey, J., Chen, J. S., Hayward, C., Williams, F. M. K., and Sambrook, P. N. (2009). Association between serum cholesterol and bone mineral density. Bone 44, 208–213. doi: 10.1016/j.bone.2008.09.020
Mann, C. J., Perdiguero, E., Kharraz, Y., Aguilar, S., Pessina, P., Serrano, A. L., et al. (2011). Aberrant repair and fibrosis development in skeletal muscle. Skelet. Muscle 1:21. doi: 10.1186/2044-5040-1-21
Manuel, D. G., Tuna, M., Hennessy, D., Bennett, C., Okhmatovskaia, A., Fines, P., et al. (2014). Projections of preventable risks for cardiovascular disease in Canada to 2021: a microsimulation modelling approach. C. Open 2, E94–E101. doi: 10.9778/cmajo.2012-0015
Marrades, M. P., Martínez, J. A., and Moreno-Aliaga, M. J. (2007). Differences in short-term metabolic responses to a lipid load in lean (resistant) vs obese (susceptible) young male subjects with habitual high-fat consumption. Eur. J. Clin. Nutr. 61, 166–174. doi: 10.1038/sj.ejcn.1602500
Martinez, F. O., and Gordon, S. (2014). The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 6, 1–13. doi: 10.12703/P6-13
Masamoto, Y., Arai, S., Sato, T., Yoshimi, A., Kubota, N., Takamoto, I., et al. (2016). Adiponectin enhances antibacterial activity of hematopoietic cells by suppressing bone marrow inflammation. Immunity 44, 1422–1433. doi: 10.1016/j.immuni.2016.05.010
McAlindon, T. E., Bannuru, R. R., Sullivan, M. C., Arden, N. K., Berenbaum, F., Bierma-Zeinstra, S. M., et al. (2014). OARSI guidelines for the non-surgical management of knee osteoarthritis. Osteoarthr. Cartil. 22, 363–388. doi: 10.1016/j.joca.2014.01.003
McMaster, W. G., Kirabo, A., Madhur, M. S., and Harrison, D. G. (2015). Inflammation, immunity, and hypertensive end-organ damage. Circ. Res. 116, 1022–1033. doi: 10.1161/CIRCRESAHA.116.303697
Meng, J., Bencze, M., Asfahani, R., Muntoni, F., and Morgan, J. E. (2015). The effect of the muscle environment on the regenerative capacity of human skeletal muscle stem cells. Skelet. Muscle 5, 11. doi: 10.1186/s13395-015-0036-8
Messier, S. P., Mihalko, S. L., Legault, C., Miller, G. D., Nicklas, B. J., DeVita, P., et al. (2013). Effects of intensive diet and exercise on knee joint loads, inflammation, and clinical outcomes among overweight and obese adults with knee osteoarthritis: the IDEA randomized clinical trial. JAMA 310, 1263–1273. doi: 10.1001/jama.2013.277669
Meyer, G. A., and Ward, S. R. (2016). Developmental biology and regenerative medicine: addressing the vexing problem of persistent muscle atrophy in the chronically torn human rotator cuff. Phys. Ther. 96, 722–733. doi: 10.2522/ptj.20150029
Milan, G., Granzotto, M., Scarda, A., Calcagno, A., Pagano, C., Federspil, G., et al. (2002). Resistin and adiponectin expression in visceral fat of obese rats: effect of weight loss. Obes. Res. 10, 1095–1103. doi: 10.1038/oby.2002.149
Minokoshi, Y., Kim, Y.-B., Peroni, O. D., Fryer, L. G. D., Müller, C., Carling, D., et al. (2002). Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 415, 339–343. doi: 10.1038/415339a
Miranda, P. J., DeFronzo, R. A., Califf, R. M., and Guyton, J. R. (2005). Metabolic syndrome: definition, pathophysiology, and mechanisms. Am. Heart J. 149, 33–45. doi: 10.1016/j.ahj.2004.07.013
Mittendorfer, B. (2011). Origins of metabolic complications in obesity: adipose tissue and free fatty acid trafficking. Curr. Opin. Clin. Nutr. Metab. Care 14, 535–541. doi: 10.1097/MCO.0b013e32834ad8b6
Mohiti-Ardekani, J., Soleymani-Salehabadi, H., Owlia, M. B., and Mohiti, A. (2014). Relationships between serum adipocyte hormones (adiponectin, leptin, resistin), bone mineral density and bone metabolic markers in osteoporosis patients. J. Bone Miner. Metab. 32, 400–404. doi: 10.1007/s00774-013-0511-4
Mosser, D. M., and Edwards, J. P. (2008). Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969. doi: 10.1038/nri2448
Nativel, B., Marimoutou, M., Thon-Hon, V. G., Gunasekaran, M. K., Andries, J., Stanislas, G., et al. (2013). Soluble HMGB1 is a novel adipokine stimulating IL-6 secretion through RAGE receptor in SW872 preadipocyte cell line: contribution to chronic inflammation in fat tissue. PLoS ONE 8:e76039. doi: 10.1371/journal.pone.0076039
Nicolucci, A. C., Hume, M. P., Martínez, I., Mayengbam, S., Walter, J., and Reimer, R. A. (2017). Prebiotics reduce body fat and alter intestinal microbiota in children who are overweight or with obesity. Gastroenterology 153, 711–722. doi: 10.1053/j.gastro.2017.05.055
Nigro, E., Scudiero, O., Monaco, M. L., Palmieri, A., Mazzarella, G., Costagliola, C., et al. (2015). New insight into adiponectin role in obesity and obesity-related diseases. Biomed Res. Int. 2014:658913. doi: 10.1155/2014/658913
Nilsson, C., Raun, K., Yan, F., Larsen, M. O., and Tang-Christensen, M. (2012). Laboratory animals as surrogate models of human obesity. Acta Pharmacol. Sin. 33, 173–181. doi: 10.1038/aps.2011.203
Novak, M. L., and Koh, T. J. (2013). Macrophage phenotypes during tissue repair. J. Leukoc. Biol. 93, 875–881. doi: 10.1189/jlb.1012512
Ohnishi, T., Bandow, K., Kakimoto, K., Machigashira, M., Matsuyama, T., and Matsuguchi, T. (2009). Oxidative stress causes alveolar bone loss in metabolic syndrome model mice with type 2 diabetes. J. Periodontal Res. 44, 43–51. doi: 10.1111/j.1600-0765.2007.01060.x
Ormsbee, M. J., Prado, C. M., Ilich, J. Z., Purcell, S., Siervo, M., Folsom, A., et al. (2014). Osteosarcopenic obesity: the role of bone, muscle, and fat on health. J. Cachexia. Sarcopenia Muscle 5, 183–192. doi: 10.1007/s13539-014-0146-x
Parnell, J. A., Klancic, T., and Reimer, R. A. (2017). Oligofructose decreases serum lipopolysaccharide and plasminogen activator inhibitor-1 in adults with overweight/obesity. Obesity 25, 510–513. doi: 10.1002/oby.21763
Pattanakuhar, S., Pongchaidecha, A., Chattipakorn, N., and Chattipakorn, S. C. (2017). The effect of exercise on skeletal muscle fibre type distribution in obesity: from cellular levels to clinical application. Obes. Res. Clin. Pract. 11, 112–132. doi: 10.1016/J.ORCP.2016.09.012
Paul, H. A., Bomhof, M. R., Vogel, H. J., Reimer, R. A., and Slupsky, C. M. (2016). Diet-induced changes in maternal gut microbiota and metabolomic profiles influence programming of offspring obesity risk in rats. Sci. Rep. 6:20683. doi: 10.1038/srep20683
Pellegrinelli, V., Rouault, C., Rodriguez-Cuenca, S., Albert, V., Edom-Vovard, F., Vidal-Puig, A., et al. (2015). Human adipocytes induce inflammation and atrophy in muscle cells during obesity. Diabetes 64, 3121–3134. doi: 10.2337/db14-0796
Peterson, J. M., Bryner, R. W., and Alway, S. E. (2008). Satellite cell proliferation is reduced in muscles of obese Zucker rats but restored with loading. AJP Cell Physiol. 295, C521–C528. doi: 10.1152/ajpcell.00073.2008
Pettipher, E. R., Higgs, G. A., and Henderson, B. (1986). Interleukin 1 induces leukocyte infiltration and cartilage proteoglycan degradation in the synovial joint. Proc. Natl. Acad. Sci. U.S.A. 83, 8749–8753.
Pogacnik Murillo, A. L., Eckstein, F., Wirth, W., Beavers, D., Loeser, R. F., Nicklas, B. J., et al. (2017). Impact of Diet and/or Exercise Intervention on Infrapatellar Fat Pad Morphology: Secondary Analysis from the Intensive Diet and Exercise for Arthritis (IDEA) Trial. Cells Tissues Organs 203, 258–266. doi: 10.1159/000449407
Powers, S. K., Ji, L. L., Kavazis, A. N., and Jackson, M. J. (2011). Reactive oxygen species: impact on skeletal muscle. Compr. Physiol. 1, 941–969. doi: 10.1002/cphy.c100054
Rahemi, H., Nigam, N., and Wakeling, J. M. (2015). The effect of intramuscular fat on skeletal muscle mechanics: implications for the elderly and obese. J. R. Soc. Interface 12:20150365. doi: 10.1098/rsif.2015.0365
Rechardt, M., Viikari-Juntura, E., and Shiri, R. (2014). Adipokines as predictors of recovery from upper extremity soft tissue disorders. Rheumatology 53, 2238–2242. doi: 10.1093/rheumatology/keu272
Reyna, S. M., Ghosh, S., Tantiwong, P., Meka, C. S. R., Eagan, P., Jenkinson, C. P., et al. (2008). Elevated toll-like receptor 4 expression and signaling in muscle from insulin-resistant subjects. Diabetes 57, 2595–2602. doi: 10.2337/db08-0038
Robling, A. G., and Turner, C. H. (2009). Mechanical signaling for bone modeling and remodeling. Crit. Rev. Eukaryot. Gene Exp. 19, 319–338.
Rodríguez, A., Becerril, S., Méndez-Giménez, L., Ramírez, B., Sáinz, N., Catalán, V., et al. (2015). Leptin administration activates irisin-induced myogenesis via nitric oxide-dependent mechanisms, but reduces its effect on subcutaneous fat browning in mice. Int. J. Obes. 39, 397–407. doi: 10.1038/ijo.2014.166
Rosenberg, J. H., Rai, V., Dilisio, M. F., and Agrawal, D. K. (2017). Damage-associated molecular patterns in the pathogenesis of osteoarthritis: potentially novel therapeutic targets. Mol. Cell. Biochem. 434, 171–179. doi: 10.1007/s11010-017-3047-4
Rothan, H. A., Suhaeb, A. M., and Kamarul, T. (2013). Recombinant human adiponectin as a potential protein for treating diabetic tendinopathy promotes tenocyte progenitor cells proliferation and tenogenic differentiation in vitro. Int. J. Med. Sci. 10, 1899–1906. doi: 10.7150/ijms.6774
Roy, B., Curtis, M. E., Fears, L. S., Nahashon, S. N., and Fentress, H. M. (2016). Molecular mechanisms of obesity-induced osteoporosis and muscle atrophy. Front. Physiol. 7:439. doi: 10.3389/fphys.2016.00439
Sacco, A., and Puri, P. L. (2015). Regulation of muscle satellite cell function in tissue homeostasis and aging. Cell Stem Cell 16, 585–587. doi: 10.1016/j.stem.2015.05.007
Salminen, A., and Kaarniranta, K. (2012). AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res. Rev. 11, 230–241. doi: 10.1016/j.arr.2011.12.005
Sanchez-Adams, J., Leddy, H. A., McNulty, A. L., O'Conor, C. J., Guilak, F., O'Conor, C. J., et al. (2014). The mechanobiology of articular cartilage: bearing the burden of osteoarthritis. Curr. Rheumatol. Rep. 16, 451. doi: 10.1007/s11926-014-0451-6
Sanguineti, R., Puddu, A., Mach, F., Montecucco, F., and Viviani, G. L. (2014). Advanced glycation end products play adverse proinflammatory activities in osteoporosis. Mediators Inflamm. 2014:975872. doi: 10.1155/2014/975872
Santos-Alvarez, J., Goberna, R., and Sánchez-Margalet, V. (1999). Human leptin stimulates proliferation and activation of human circulating monocytes. Cell. Immunol. 194, 6–11. doi: 10.1006/cimm.1999.1490
Schemmel, R., Mickelsen, O., and Tolgay, Z. (1969). Dietary obesity in rats: influence of diet, weight, age, and sex on body composition. Am. J. Physiol. 216, 373–379.
Scher, J. U., and Abramson, S. B. (2011). The microbiome and rheumatoid arthritis. Nat. Rev. Rheumatol. 7, 569–578. doi: 10.1038/nrrheum.2011.121
Schulze-Tanzil, G., Al-Sadi, O., Wiegand, E., Ertel, W., Busch, C., Kohl, B., et al. (2011). The role of pro-inflammatory and immunoregulatory cytokines in tendon healing and rupture: new insights. Scand. J. Med. Sci. Sports 21, 337–351. doi: 10.1111/j.1600-0838.2010.01265.x
Scotece, M., Pérez, T., Conde, J., Abella, V., López, V., Pino, J., et al. (2017). Adipokines induce pro-inflammatory factors in activated Cd4+ T cells from osteoarthritis patient. J. Orthop. Res. 35, 1299–1303. doi: 10.1002/jor.23377
Scott, A., Khan, K. M., Duronio, V., and Hart, D. A. (2008). Mechanotransduction in human bone: in vitro cellular physiology that underpins bone changes with exercise. Sports Med. 38, 139–160.
Seebacher, F., Tallis, J., McShea, K., and James, R. S. (2017). Obesity-induced decreases in muscle performance are not reversed by weight loss. Int. J. Obes. 41, 1271–1278. doi: 10.1038/ijo.2017.81
Serra, D., Mera, P., Malandrino, M. I., Mir, J. F., and Herrero, L. (2013). Mitochondrial fatty acid oxidation in obesity. Antioxid. Redox Signal. 19, 269–284. doi: 10.1089/ars.2012.4875
Shahrokhi, A., Pagliassotti, M. J., Shahrokhi, K. A., and Hill, J. O. (1993). Skeletal muscle glucose metabolism on obesity-prone and obesity-resistant rats. Am. J. Physiol. 264, R1224–R1228.
Shu, L., Beier, E., Sheu, T., Zhang, H., Zuscik, M. J., Puzas, E. J., et al. (2015). High-fat diet causes bone loss in young mice by promoting osteoclastogenesis through alteration of the bone marrow environment. Calcif. Tissue Int. 96, 313–323. doi: 10.1007/s00223-015-9954-z
Singer, K., DelProposto, J., Lee Morris, D., Zamarron, B., Mergian, T., Maley, N., et al. (2014). Diet-induced obesity promotes myelopoiesis in hematopoietic stem cells. Mol. Metab. 3, 664–675. doi: 10.1016/j.molmet.2014.06.005
Smith, E. U. R., Hoy, D. G., Cross, M. J., Sanchez-Riera, L., Blyth, F., Buchbinder, R., et al. (2014). Burden of disability due to musculoskeletal (MSK) disorders. Best Pract. Res. Clin. Rheumatol. 28, 353–366. doi: 10.1016/j.berh.2014.08.002
Smith, M. D., Triantafillou, S., Parker, A., Youssef, P. P., and Coleman, M. (1997). Synovial membrane inflammation and cytokine production in patients with early osteoarthritis. J. Rheumatol. 24, 365–371.
Son, K. H., Son, M., Ahn, H., Oh, S., Yum, Y., Choi, C. H., et al. (2016). Age-related accumulation of advanced glycation end-products-albumin, S100β, and the expressions of advanced glycation end product receptor differ in visceral and subcutaneous fat. Biochem. Biophys. Res. Commun. 477, 271–276. doi: 10.1016/j.bbrc.2016.06.056
Srikanthan, P., Crandall, C. J., Miller-Martinez, D., Seeman, T. E., Greendale, G. A., Binkley, N., et al. (2014). Insulin resistance and bone strength: findings from the study of midlife in the United States. J. Bone Miner. Res. 29, 796–803. doi: 10.1002/jbmr.2083
Srikanthan, P., and Karlamangla, A. S. (2014). Muscle mass index as a predictor of longevity in older adults. Am. J. Med. 127, 547–553. doi: 10.1016/j.amjmed.2014.02.007
Stenholm, S., Harris, T. B., Rantanen, T., Visser, M., Kritchevsky, S. B., and Ferrucci, L. (2008). Sarcopenic obesity: definition, cause and consequences. Curr. Opin. Clin. Nutr. Metab. Care 11, 693–700. doi: 10.1097/MCO.0b013e328312c37d
Stout, R. D., and Suttles, J. (2004). Functional plasticity of macrophages: reversible adaptation to changing microenvironments. J. Leukoc. Biol. 76, 509–513. doi: 10.1189/jlb.0504272
Sumiyoshi, M., Sakanaka, M., and Kimura, Y. (2006). Chronic intake of high-fat and high-sucrose diets differentially affects glucose intolerance in mice. J. Nutr. 136, 582–587. doi: 10.1093/jn/136.3.582
Sun, A. R., Friis, T., Sekar, S., Crawford, R., Xiao, Y., and Prasadam, I. (2016). Is synovial macrophage activation the inflammatory link between obesity and osteoarthritis? Curr. Rheumatol. Rep. 18, 57. doi: 10.1007/s11926-016-0605-9
Sun, K., Tordjman, J., Clément, K., and Scherer, P. E. (2013). Fibrosis and adipose tissue dysfunction. Cell Metab. 18, 470–477. doi: 10.1016/j.cmet.2013.06.016
Takeda, S., Elefteriou, F., Levasseur, R., Liu, X., Zhao, L., Parker, K. L., et al. (2002). Leptin regulates bone formation via the sympathetic nervous system. Cell 111, 305–317. doi: 10.1016/S0092-8674(02)01049-8
Te Morenga, L., Mallard, S., and Mann, J. (2013). Dietary sugars and body weight: systematic review and meta-analyses of randomised controlled trials and cohort studies. BMJ 346:e7492. doi: 10.1136/bmj.e7492
Thomas, T., Gori, F., Khosla, S., Jensen, M. D., Burguera, B., and Riggs, B. L. (1999). Leptin acts on human marrow stromal cells to enhance differentiation to osteoblasts and to inhibit differentiation to adipocytes 1. Endocrinology 140, 1630–1638. doi: 10.1210/endo.140.4.6637
Tidball, J. G. (2005). Inflammatory processes in muscle injury and repair. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288, R345–R353. doi: 10.1152/ajpregu.00454.2004
Tidball, J. G., and Villalta, S. A. (2010). Regulatory interactions between muscle and the immune system during muscle regeneration. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298, R1173–R1187. doi: 10.1152/ajpregu.00735.2009
Tilley, B. J., Cook, J. L., Docking, S. I., and Gaida, J. E. (2015). Is higher serum cholesterol associated with altered tendon structure or tendon pain? A systematic review. Br. J. Sports Med. 49, 1504–1509. doi: 10.1136/bjsports-2015-095100
Tomlinson, D. J., Erskine, R. M., Morse, C. I., Winwood, K., and Onambélé-Pearson, G. (2016). The impact of obesity on skeletal muscle strength and structure through adolescence to old age. Biogerontology 17, 467–483. doi: 10.1007/s10522-015-9626-4
Trayhurn, P. (2005). Adipose tissue in obesity—an inflammatory issue. Endocrinology 146, 1003–1005. doi: 10.1210/en.2004-1597
Vaamonde-García, C., Riveiro-Naveira, R. R., Valcárcel-Ares, M. N., Hermida-Carballo, L., Blanco, F. J., and López-Armada, M. J. (2012). Mitochondrial dysfunction increases inflammatory responsiveness to cytokines in normal human chondrocytes. Arthritis Rheum. 64, 2927–2936. doi: 10.1002/art.34508
Vernochet, C., Damilano, F., Mourier, A., Bezy, O., Mori, M. A., Smyth, G., et al. (2014). Adipose tissue mitochondrial dysfunction triggers a lipodystrophic syndrome with insulin resistance, hepatosteatosis, and cardiovascular complications. FASEB J. 28, 4408–4419. doi: 10.1096/fj.14-253971
Wang, Y., Zhao, X., Lotz, M., Terkeltaub, R., and Liu-Bryan, R. (2015). Mitochondrial biogenesis is impaired in osteoarthritis chondrocytes but reversible via peroxisome proliferator-activated receptor γ coactivator 1α. Arthritis Rheumatol. 67, 2141–2153. doi: 10.1002/art.39182
Wearing, S. C., Hennig, E. M., Byrne, N. M., Steele, J. R., and Hills, A. P. (2006). Musculoskeletal disorders associated with obesity: a biomechanical perspective. Obes. Rev. 7, 239–250. doi: 10.1111/j.1467-789X.2006.00251.x
Weiss, R., Bremer, A. A., and Lustig, R. H. (2013). What is metabolic syndrome, and why are children getting it? Ann. N. Y. Acad. Sci. 1281, 123–140. doi: 10.1111/nyas.12030
Williams, A. S., Kang, L., and Wasserman, D. H. (2015). The extracellular matrix and insulin resistance. Trends Endocrinol. Metab. 26, 357–366. doi: 10.1016/j.tem.2015.05.006
Wright, K. J., Thomas, M. M., Betik, A. C., Belke, D., and Hepple, R. T. (2014). Exercise training initiated in late middle age attenuates cardiac fibrosis and advanced glycation end-product accumulation in senescent rats. Exp. Gerontol. 50, 9–18. doi: 10.1016/j.exger.2013.11.006
Wu, C.-L., Jain, D., McNeill, J. N., Little, D., Anderson, J. A., Huebner, J. L., et al. (2014). Dietary fatty acid content regulates wound repair and the pathogenesis of osteoarthritis following joint injury. Ann. Rheum. Dis. 74, 2076–2083. doi: 10.1136/annrheumdis-2014-205601
Wu, C.-L., Kimmerling, K. A., Little, D., and Guilak, F. (2017a). Serum and synovial fluid lipidomic profiles predict obesity-associated osteoarthritis, synovitis, and wound repair. Sci. Rep. 7:44315. doi: 10.1038/srep44315
Wu, C.-L., McNeill, J., Goon, K., Little, D., Kimmerling, K., Huebner, J., et al. (2017b). Conditional macrophage depletion increases inflammation and does not inhibit the development of osteoarthritis in obese MaFIA mice. Arthritis Rheumatol. 69, 1772–1783. doi: 10.1002/art.40161
Wu, X., Xu, W., Feng, X., He, Y., Liu, X., Gao, Y., et al. (2015). TNF-a mediated inflammatory macrophage polarization contributes to the pathogenesis of steroid-induced osteonecrosis in mice. Int. J. Immunopathol. Pharmacol. 28, 351–361. doi: 10.1177/0394632015593228
Xia, Q., Zhu, S., Wu, Y., Wang, J., Cai, Y., Chen, P., et al. (2015). Intra-articular transplantation of atsttrin-transduced mesenchymal stem cells ameliorate osteoarthritis development. Stem Cells Transl. Med. 4, 523–531. doi: 10.5966/sctm.2014-0200
Yamada, J., Tsuji, K., Miyatake, K., Matsukura, Y., Abula, K., Inoue, M., et al. (2014). Follistatin alleviates synovitis and articular cartilage degeneration induced by carrageenan. Int. J. Inflam. 2014, 1–9. doi: 10.1155/2014/959271
Yamamoto, M., and Sugimoto, T. (2016). Advanced glycation end products, diabetes, and bone strength. Curr. Osteoporos. Rep. 14, 320–326. doi: 10.1007/s11914-016-0332-1
Yan, H., Diao, H., Xiao, Y., Li, W., Yu, B., He, J., et al. (2016). Gut microbiota can transfer fiber characteristics and lipid metabolic profiles of skeletal muscle from pigs to germ-free mice. Sci. Rep. 6:31786. doi: 10.1038/srep31786
Yang, Y., Kim, S. C., Yu, T., Yi, Y.-S., Rhee, M. H., Sung, G.-H., et al. (2014). Functional roles of p38 mitogen-activated protein kinase in macrophage-mediated inflammatory responses. Mediators Inflamm. 2014:352371. doi: 10.1155/2014/352371
Yu, H., Zhou, D., Jia, W., and Guo, Z. (2012). Locating the source of hyperglycemia: liver versus muscle. J. Diabetes 4, 30–36. doi: 10.1111/j.1753-0407.2011.00170.x
Yue, R., Zhou, B. O., Shimada, I. S., Zhao, Z., and Morrison, S. J. (2016). Leptin receptor promotes adipogenesis and reduces osteogenesis by regulating mesenchymal stromal cells in adult bone marrow. Cell Stem Cell 18, 782–796. doi: 10.1016/j.stem.2016.02.015
Yusuf, E., Ioan-Facsinay, A., Bijsterbosch, J., Klein-Wieringa, I., Kwekkeboom, J., Slagboom, P. E., et al. (2011). Association between leptin, adiponectin and resistin and long-term progression of hand osteoarthritis. Ann. Rheum. Dis. 70, 1282–1284. doi: 10.1136/ard.2010.146282
Zabeau, L., Lavens, D., Peelman, F., Eyckerman, S., Vandekerckhove, J., and Tavernier, J. (2003). The ins and outs of leptin receptor activation. FEBS Lett. 546, 45–50. doi: 10.1016/S0014-5793(03)00440-X
Zamboni, M., Mazzali, G., Fantin, F., Rossi, A., and Di Francesco, V. (2008). Sarcopenic obesity: a new category of obesity in the elderly. Nutr. Metab. Cardiovasc. Dis. 18, 388–395. doi: 10.1016/j.numecd.2007.10.002
Zhang, C., Li, Y., Wu, Y., Wang, L., Wang, X., and Du, J. (2013). Interleukin-6/signal transducer and activator of transcription 3 (STAT3) pathway is essential for macrophage infiltration and myoblast proliferation during muscle regeneration. J. Biol. Chem. 288, 1489–1499. doi: 10.1074/jbc.M112.419788
Zhao, X., Petursson, F., Viollet, B., Lotz, M., Terkeltaub, R., and Liu-Bryan, R. (2014). Peroxisome proliferator-activated receptor γ Coactivator 1α and FoxO3A mediate chondroprotection by AMP-activated protein kinase. Arthritis Rheumatol. 66, 3073–3082. doi: 10.1002/art.38791
Keywords: joint diseases, muscle, bone, tendon, NFkB, MAPK
Citation: Collins KH, Herzog W, MacDonald GZ, Reimer RA, Rios JL, Smith IC, Zernicke RF and Hart DA (2018) Obesity, Metabolic Syndrome, and Musculoskeletal Disease: Common Inflammatory Pathways Suggest a Central Role for Loss of Muscle Integrity. Front. Physiol. 9:112. doi: 10.3389/fphys.2018.00112
Received: 30 October 2017; Accepted: 05 February 2018;
Published: 23 February 2018.
Edited by:
Alexandrina Ferreira Mendes, University of Coimbra, PortugalReviewed by:
Oreste Gualillo, Servicio Gallego de Salud, SpainFrancis Berenbaum, Université Pierre et Marie Curie, France
Tom Appleton, University of Western Ontario, Canada
Copyright © 2018 Collins, Herzog, MacDonald, Reimer, Rios, Smith, Zernicke and Hart. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David A. Hart, aGFydGRAdWNhbGdhcnkuY2E=