 Yujuan Jiao
Yujuan Jiao Hongxin Wang1†
Hongxin Wang1† Yuqi Li
Yuqi Li Minghui Zhang
Minghui Zhang- 1Department of Pathology, Harbin Medical University, Harbin, China
- 2Department of Oncology, Chifeng City Hospital, Chifeng, China
Copper is an essential cofactor for all organisms. However, it can become toxic if its concentration rises above a specific level. This level is controlled by evolutionary conserved homeostatic mechanisms. Recently, a new type of cell death called cuproptosis has been found. The process represents a copper-dependent, regulated form of cell death that is distinct from all known death mechanisms and relies on mitochondrial respiration. The mechanism of cuproptosis involves the direct binding of copper to lipidated parts in the tricarboxylic acid (TCA) cycle. This results in the abnormal aggregation of lipoylated proteins and the destabilization of iron-sulfur cluster (Fe-S) proteins. These events induce proteotoxic stress, ultimately leading to cell death. Copper-induced cell death is controlled by proteolipid acylation, which is mediated by the mitochondrial iron-sulfur protein FDX1. Copper overload also inhibits the biosynthesis of iron-sulfur (Fe-S) clusters and impairs the activity of Fe-S enzymes. As a result, mitochondrial function is disrupted. Both copper-induced cell death and impaired copper homeostasis arise from the same mechanistic basis. The expression of copper import gene SLC31A1 (CTR1) and export genes ATP7A and ATP7B significantly influences cuproptosis. The tumor suppressor p53 may participate in this process by modulating glycolysis and mitochondrial metabolism. In contrast, glutathione (GSH) reduces copper ion cytotoxicity by binding copper to form a complex. The growth and spread of tumor cells is more dependent on copper than that of normal cells.The copper ionophore elesclomol (ES) kills cancer cells by transporting copper ions into them. This ES-Cu complex not only inhibits cancer cell proliferation but also activates an immune response. Moreover, when ES is combined with αPD-L1, it might increase the effectiveness of cancer treatment. This gives a new idea for treating cancer.
1 Introduction
Copper is an essential micronutrient that is critical for the maintenance of normal physiological functions. Recently, its complex roles in cancer biology have attracted broad research interest (Festa and Thiele, 2011; Garber, 2015; Guan et al., 2023). Intracellular copper levels are strictly controlled and must remain within a specific range. Even a slight increase can cause cytotoxicity or cell death, highlighting the critical role of copper homeostasis (Tian et al., 2023; Wang et al., 2023). Copper metabolism disorder, which involves either excessive copper accumulation or improper transport, is detrimental to humans (Scheiber et al., 2013). Excess copper generates reactive oxygen species (ROS) via the Fenton reaction. This, in turn, causes the oxidation and breakdown of proteins, lipids, and DNA, and contributes to the development of various human diseases (Gaetke and Chow, 2003; Roberts et al., 2009). The accumulation of copper can harm organisms by reducing the activity of the respiratory chain and producing ROS (Ruiz et al., 2021; Zischka and Einer, 2018). Significant accumulation of intracellular copper triggers a novel type of cell death known as “cuproptosis”. In 2022, Tsvetkov et al. first clearly proposed the concept of “cuproptosis,” marking a new phase in the research on copper-dependent cell death mechanisms (Tsvetkov et al., 2022). This process begins with copper binding directly to lipoylated components of the tricarboxylic acid (TCA) cycle. This interaction induces the aggregation of lipoylated proteins, resulting in proteotoxic stress and ultimately leading to cell death (Tsvetkov et al., 2022).
With growing research on copper metabolism and cuproptosis, a comprehensive understanding of the molecular mechanisms and therapeutic potential of copper and cuproptosis in cancer has become critically important. In this review, we first outline the fundamental regulatory mechanisms of intracellular copper homeostasis and metabolism. We then clarify the bidirectional positive feedback relationship between copper metabolism and cancer cell proliferation. Furthermore, we systematically summarize key molecular mechanisms underlying cuproptosis and evaluate the potential of cuproptosis-based cancer therapies, including their effects on the tumor immune microenvironment. Additionally, we explore potential copper-targeted therapeutic strategies, such as diagnostic screening using cuproptosis-related biomarkers and combined treatment approaches that couple cuproptosis with other forms of cell death for synergistic antitumor effects. Finally, in the context of conventional drugs,we discuss the current challenges and future directions of applying cuproptosis in cancer therapy.
2 Copper homeostasis and copper metabolism
2.1 Homeostatic mechanisms of copper
Copper is an essential trace element in animals and is widely distributed in tissues including muscle, bone, and liver (Festa and Thiele, 2011). Dietary copper is primarily absorbed through the gastrointestinal tract (Xiong et al., 2023), mainly in the small intestine. Copper uptake across the enterocyte apical membrane relies on copper transporter 1 (CTR1), which facilitates the entry of Cu+ (Nose et al., 2006). Alternatively, copper can be absorbed as Cu2+ via divalent metal transporter 1 (DMT1) and is subsequently reduced to Cu+ by cellular sulfhydryl groups (Sa et al., 2018; Gunshin et al., 1997). Copper is then exported across the basolateral membrane via the ATPase copper transporting alpha (ATP7A) protein (Wang et al., 2012). The targeted delivery of intracellular copper relies on multiple metallochaperone systems:(a) Cytochrome c oxidase copper chaperone COX17 (Cox17) transports copper to mitochondria and participates in cytochrome c oxidase assembly (Amaravadi et al., 1997); (b)The copper chaperone for superoxide dismutase (CCS) delivers copper to superoxide dismutase 1 (SOD1) in the cytoplasm and activates its antioxidant function (Culotta et al., 1997); (c) Antioxidant protein 1 (ATOX1) cooperates with P-type ATPases to deliver copper to the Golgi network for subsequent excretion or transmembrane transport (Lutsenko et al., 2007). Besides, other companion proteins send copper to specific places inside the cell (Banci et al., 2010). In the portal circulation, copper is transported to the liver primarily bound to serum proteins like albumin. Hepatocytes store excess copper using metallothioneins (MT1 and MT2) (Ge et al., 2022). Biliary copper excretion is mediated by ATPase copper transporting beta (ATP7B), a protein located on the bile duct membrane, and serves as a key pathway for maintaining systemic copper homeostasis (Roelofsen et al., 2000; Polishchuk et al., 2014). Intracellular free copper levels are tightly controlled and kept extremely low by the homeostatic system to prevent toxic reactions caused by copper accumulation (Ge et al., 2022; Lutsenko, 2010; Rae et al., 1999; Kim et al., 2008) (Figure 1).
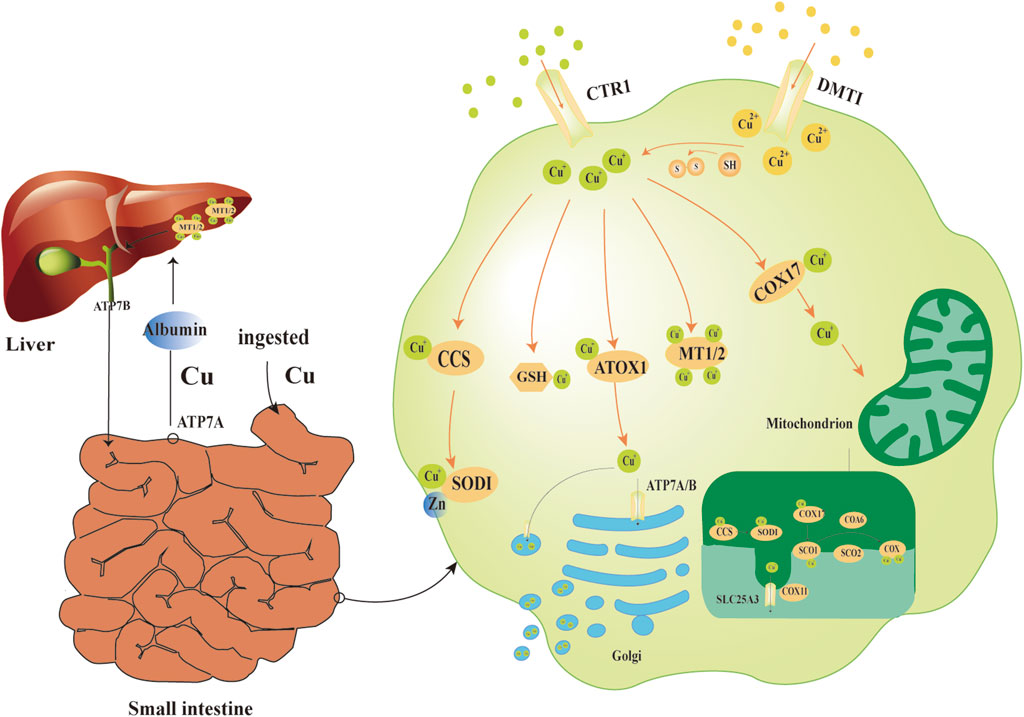
Figure 1. Physiological mechanisms of copper ion transport. The small intestine serves as the main site for copper absorption. Cu+ uptake across the enterocyte apical membrane relies primarily on CTR1, while Cu2+ uptake is mediated by DMT1. Within the cell, Cu2+ is reduced to Cu+ by -SH groups. The copper chaperone CCS delivers copper to SOD1 in the cytoplasm. Copper-binding enables SOD1 to form a stable dimer with zinc, which catalyzes the disproportionation of O2−. Cu+ can be captured by GSH and MT1/2, forming a dynamic intracellular copper pool. COX17 delivers Cu+ to mitochondrial SCO1 and COX11, a process essential for the assembly of COX. When copper levels are elevated, ATOX1 collaborates with ATP7A/B to transport copper ions to the Golgi apparatus for excretion. In the portal circulation, serum proteins (e.g., albumin) transport copper from the intestines to the liver. Excess copper is stored in hepatocytes through binding to MT1 and MT2, while ATP7B protein on the bile duct membrane facilitates hepatobiliary excretion to eliminate copper from the body.Abbreviations:CTR1 copper transporter 1,DMT1 divalent metal transporter 1, -SH sulfhydryl,CCS copper chaperone for superoxide dismutase, SOD1 superoxide dismutase 1, O2− superoxide anions,GSH glutathione, MT1/2 metallothioneins, COX17 cytochrome c oxidase copper chaperone COX17, SCO1 synthesis of cytochrome c oxidase 1, COX11 cytochrome c oxidase copper chaperone COX11,COX cytochrome c oxidase, ATOX1,antioxidant protein 1,ATP7A/B ATPase copper transporting alpha/beta.
Intracellular copper ions primarily bind to both copper-specific and non-copper-specific proteins for functional regulation and homeostasis maintenance. The binding mechanisms between these two types of proteins differ significantly. Copper proteins incorporate copper ions through highly specific and stable coordination—such as binding to histidine or cysteine residues—to enable enzymatic activity or support structural functions (Adman et al., 1991). In contrast, non-copper proteins (such as metallothionein) participate in copper metabolism through non-specific binding, buffering storage, and transport regulation (Kodama et al., 2012). These proteins play an auxiliary role in maintaining dynamic copper homeostasis within the cell. Additionally, copper chaperone proteins (e.g., ATOX1, CCS) are responsible for the targeted and precise delivery of copper ions (Lippard and Berg, 1994). Meanwhile, small molecules like glutathione help buffer intracellular copper levels and mitigate oxidative toxicity through reversible binding (Que et al., 2008; Solomon et al., 1996). Together, these mechanisms form a multi-layered and dynamically balanced copper homeostatic network, which protects cells from copper-induced toxicity while supporting essential biological processes that depend on copper.
2.2 Characteristics of copper metabolism
Copper is an essential trace element in living organisms and is widely present in diverse cells—from bacteria to humans (Kim et al., 2008). It serves as a cofactor for many key enzymes and plays a vital role in maintaining cellular homeostasis, a function well supported by scientific studies (Ge et al., 2022). Specifically, copper participates in regulating several fundamental cellular processes, such as mitochondrial respiration, antioxidant defense, and the biosynthesis of hormones, neurotransmitters, and pigments (Lippard and Berg, 1994; Que et al., 2008; Solomon et al., 1996). For example, copper acts as a key cofactor for mitochondrial cytochrome c oxidase and is essential for meeting the energy demands of rapidly proliferating cells (Lopez et al., 2019); It also serves as an essential cofactor for Cu/Zn SOD1, supporting cellular antioxidant defense mechanisms (Rae et al., 1999). It is noteworthy that despite its strong redox activity, copper rarely exists in a free form inside cells. Studies indicate that the intracellular concentration of free Cu2+ is extremely low under normal conditions, with an estimated upper limit of only 10–18 M (Lippard, 1999). This strict homeostatic regulation is essential for understanding copper’s role in pathological conditions.
Notably, dysregulation of copper metabolism can significantly impact physiological functions. Copper metabolism disorder, which involves either excessive copper accumulation or improper transport, is detrimental to humans (Scheiber et al., 2013). This phenomenon is particularly evident in tumor cells, which exhibit a higher demand for copper than normal cells (Denoyer et al., 2015). To support the energy and biosynthetic demands of rapid proliferation, copper levels are significantly elevated in tumor cells (Chen and Pervaiz, 2010). Clinical studies have revealed abnormally elevated copper levels in both tumor tissues and serum across multiple cancer types, such as thyroid (Baltaci et al., 2017), breast (Kuo et al., 2002; Feng et al., 2012), lung (Lossow et al., 2021; Jin et al., 2011), pancreatic (Lener et al., 2016), gallbladder (Basu et al., 2013), and colorectal cancers (Gupta et al., 1993). This elevation is strongly associated with poor patient prognosis.
Tumor cells adapt their copper metabolism in multiple ways to support malignant proliferation. First, many cancer cells exhibit high expression of CTR1, leading to significantly enhanced copper uptake (Zimnicka et al., 2014). Second, cancer cells modify the expression and localization of copper chaperone proteins to preferentially deliver copper ions to enzymes involved in proliferation and angiogenesis, thereby promotes these key processes (Suwara and Hartman, 2025). Moreover, increased intracellular copper levels activate multiple signaling pathways, such as mitogen-activated protein kinase/extracellular signal-regulated kinase (MEK/ERK) and B-rapidly accelerated fibrosarcoma (BRAF),which stimulate cell proliferation, promote angiogenesis via vascular endothelial growth factor (VEGF) activation (Pan et al., 2002), and enhance tumor invasion and metastasis by inducing epithelial-mesenchymal transition (EMT) (Zhang et al., 2025). However, excessive copper accumulation acts as a “double-edged sword” in tumors. On one hand, copper promotes tumor growth; on the other hand, excess copper induces high levels of ROS through the Fenton reaction, leading to severe oxidative damage in proteins, lipids, and DNA (Banci et al., 2010; Basu et al., 2013). Furthermore, high copper levels can directly or indirectly modulate key apoptotic pathway proteins, leading to programmed cell death (Guo et al., 2025).
Disrupted copper metabolism also significantly influences immune regulation within the tumor microenvironment (TME). TME is a complex ecosystem composed of tumor cells, extracellular matrix (ECM), innate and adaptive immune cells, inflammatory cells, tumor-associated macrophages (TAMs), cancer-associated fibroblasts (CAFs), tumor-associated neutrophils, myeloid-derived suppressor cells (MDSCs), endothelial cells, microvascular networks, as well as various cytokines and chemokines (Wu et al., 2022). These components interact dynamically to form an immunosuppressive microenvironment (Yang et al., 2021; Mo et al., 2021). Interestingly, studies indicate that elevated copper levels in the TME are positively correlated with increased expression of the programmed death-ligand 1(PD-L1) (Cobine and Brady, 2022). PD-L1 binds to programmed cell death protein 1(PD-1) on T cells and suppresses downstream signaling pathways such as rat sarcoma/mitogen-activated protein kinase (Ras/MAPK) and phosphatidylinositol-3-kinase/protein kinase B (PI3K/AKT), leading to T cell apoptosis and exhaustion, thereby facilitating tumor immune escape (Kciuk et al., 2023).
In this complex microenvironment, the functional state of macrophages plays a particularly critical role. Macrophages can be categorized into three main types: the unactivated M0 type, the classically activated pro-inflammatory M1 type, and the alternatively activated anti-inflammatory M2 type (Chaintreuil et al., 2023). M1 macrophages inhibit tumor growth by secreting cytokines like tumor necrosis factor-alpha (TNF-α), while M2 macrophages promote tumor progression and angiogenesis through factors such as interleukin-6(IL-6), interleukin-10(IL-10), and VEGF (54). Studies indicate that excess copper markedly suppresses macrophage function and promotes polarization toward the M2 phenotype, thereby enhancing immunosuppression (Zhao and Zhao, 2019; Tang et al., 2023). Moreover, an effective anti-tumor response by immune cells relies on normal mitochondrial respiratory function (Chang et al., 2022). However, copper overload disrupts mitochondrial function, leading to impaired energy metabolism and increased oxidative stress (Roberts et al., 2008; Sokol et al., 1994; Gu et al., 2000). Consequently, immune cells lose their capacity to effectively recognize and attack tumor cells, resulting in a weakened anti-tumor immune response (Chang et al., 2022).
In summary, cancer cells display distinct copper metabolic characteristics, primarily marked by increased intracellular copper levels and a strong reliance on copper-dependent oncogenic mechanisms (Yang et al., 2023). The link between copper metabolism and tumor development has been widely investigated, with substantial evidence supporting copper’s key role in tumor initiation and progression (Ishida et al., 2013). At the metabolic level, copper promotes metabolic reprogramming in tumor cells by regulating oxidative phosphorylation and energy metabolism, thereby supporting their rapid proliferation. At the molecular level, it elevates cellular oxidative stress and stimulates angiogenesis, thereby accelerating tumor progression. Concurrently, dysregulated copper metabolism is also a consequence of the malignant phenotype in cancer cells. Tumor cells often upregulate the CTR1 to meet their elevated copper demand (Zimnicka et al., 2014). Additionally, hypoxia-inducible factor-1α(HIF-1α) indirectly stabilizes and activates the transcription of multiple copper metabolism-related genes, such as those regulating CTR1 (Su et al., 2022). This mechanism further enhances copper accumulation in tumor cells and establishes a positive feedback loop that continuously drives malignant progression.
3 The onset and development of cuproptosis
3.1 Cuproptosis
Recently, a novel form of copper-dependent cell death, reliant on mitochondrial respiration, has been identified and termed “cuproptosis” (Tsvetkov et al., 2022). It describes how copper affects cells through signaling pathways and both enzymatic and non-enzymatic activities (Ge et al., 2022). Cuproptosis can be controlled by using genes or drugs to target proteins related to copper homeostasis. During this process, copper enhances mitochondria-dependent energy metabolism and induces ROS accumulation, resulting in cytotoxicity—a mechanism now referred to as “cuproptosis” (Ge et al., 2022). Cell death involves complex signaling pathways and molecular mechanisms (Tang et al., 2019), which lead to changes in proteins and lipids (Carneiro and El-Deiry, 2020). The main types of cell death include necrosis (Weinlich et al., 2017), pyroptosis (Bergsbaken et al., 2009), ferroptosis (Dixon et al., 2012) and other forms. Previous studies have shown that elesclomol (ES) induces apoptosis through the ROS-dependent mechanism (Hasinoff et al., 2014; Kirshner et al., 2008). However, the cell death caused by ES does not involve the breakdown of the caspase-3, a marker of apoptosis,and not activate caspase-3 (Elmore, 2007). The killing potential of ES was not affected when cells were knocked out of apoptosis effector or treated with other inhibitors of known cell death mechanisms, suggesting that the cell death pathway induced by copper ionophores is different from known cell death pathways (Tsvetkov et al., 2022).
Studies have demonstrated that cuproptosis is closely linked to mitochondrial protein lipoylation. Protein lipoylation is a key post-translational modification involving the covalent attachment of an acyl group (e.g., acetyl, propionyl, succinyl, etc.) to specific amino acid residues, most commonly the ε-amino group of lysine (Resh, 2016). The modification is known to occur only in four key metabolic enzymes: dihydrolipoamide branched-chain transacylase E2 (DBT), dihydrolipoamide-S-succinyltransferase (DLST),dihydrolipoamide-S-acetyltransferase (DLAT) and glycine cleavage system H protein (GCSH) (Solmonson and DeBerardinis, 2018; Rowland et al., 2018). All these enzymes play essential roles in directing carbon sources into the tricarboxylic acid cycle (TCA cycle): DBT, as part of the branched-chain ketoacid dehydrogenase complex, catalyzes the conversion of branched-chain α-ketoacids to acyl-CoA through lipoylation (Lin et al., 2024); DLST promotes the transformation of α-ketoglutarate to succinyl-CoA within the α-ketoglutarate dehydrogenase complex (Lin et al., 2024); DLAT, a core component of the pyruvate dehydrogenase complex, converts pyruvate to acetyl-CoA (La et al., 2022); GCSH serves as a lipoyl carrier protein and enhances enzymatic activity by modifying DLST and DLAT. Together, they regulate the generation of TCA cycle intermediate metabolites (Mukha et al., 2022). These lipoylation modifications are crucial for sustaining normal enzyme function (Rowland et al., 2018), and ferredoxin 1(FDX1) is considered a key upstream regulator of this process.
FDX1 is a small iron-sulfur cluster protein that transfers electrons from Nicotinamide Adenine Dinucleotide Phosphate (NADPH) to the mitochondrial cytochrome P450 system via ferredoxin reductase (FDXR). It participates in key biological processes including steroid synthesis, cholesterol metabolism, and bile acid production (Sheftel et al., 2010). In lung adenocarcinoma, downregulation of FDX1 leads to significant alterations in metabolites associated with glucose metabolism, fatty acid oxidation, and amino acid metabolism (Zhang et al., 2021); In gliomas, cellular myelocytomatosis oncogene (c-MYC) upregulates FDX1, thereby inhibiting Microtubule-associated protein 1A/1B-light chain 3 (LC3) mediated mitochondrial autophagy (Guowei et al., 2023). Additionally, FDX1 contributes to the regulation of apoptosis and autophagy in polycystic ovary syndrome (Xing et al., 2023). Notably, FDX1 plays a critical role in cuproptosis: when excess copper ions (particularly in the presence of copper ionophores) enter mitochondria, FDX1 reduces Cu2+ to the more reactive Cu+. The reduced Cu+ directly binds to lipoylated proteins (e.g., DLAT) in the TCA cycle, causing abnormal oligomerization and aggregation, which ultimately leads to serious proteotoxic stress. Simultaneously, this process causes destabilization and functional impairment of iron-sulfur (Fe-S) cluster proteins, ultimately resulting in irreversible cell death.
3.2 Selection and research on elosclomol
Cuproptosis strongly depends on copper ionophores—small molecules that bind extracellular copper ions, transport them into cells (especially mitochondria), and thereby induce severe cell death (Hunsaker and Franz, 2019; Oliveri, 2020; Guo et al., 2023). Copper ionophores do not depend on intracellular small molecule chaperones; instead, they directly promote toxic effects by inducing abnormal copper accumulation (Tsvetkov et al., 2022). Additionally, naturally produced copper ionophores have shown strong antimicrobial effects (Raffa et al., 2021; Patteson et al., 2021). So far, several types of copper ionophores have been created, such as disulfide (DSF), 8-hydroxyquinoline (8-HQ), pyrithione, and ES (Kahlson and Dixon, 2022). Among these ionophores, ES is special. It can direct the transport of copper from outside the cell into the mitochondria, which causes copper to build up in the cell. Within the mitochondria, Cu2+ is changed to Cu+, leading to the production of ROS (Nagai et al., 2012).
Studies on ES further revealed that cell death induced by copper ionophores primarily relies on the continuous accumulation of intracellular copper (Tsvetkov et al., 2022). ES binds to extracellular Cu2+ through its nitrogen and sulfur atoms, which act as the primary binding sites (Helsel and Franz, 2015). Specifically, upon Cu2+ binding, ES undergoes deprotonation at its N1 and N2 atoms, resulting in the formation of a neutral ES-Cu2+ complex (Yadav et al., 2013). After entering the cell, the ES-Cu2+ complex is transported to the mitochondria. Within the mitochondria, Cu2+ is reduced to Cu+ by the action of FDX1. Subsequently, Cu+ is released from the complex, while ES continues to be exported, thereby facilitating the repeated shuttling of the ES-Cu2+ complex. This cycle causes copper to build up continuously inside the cell. At the same time, ROS levels keep rising steadily. Eventually, the cell’s antioxidant defenses become overwhelmed, triggering oxidative stress (Nagai et al., 2012). Compared with normal cells, cancer cells not only have higher basic levels of ROS and lower antioxidant abilities, but they also show higher oxidative stress levels and rely more on mitochondrial metabolism (Gupte and Mumper, 2009; Trachootham et al., 2009). These characteristics render cancer cells highly sensitive to ES-induced copper toxicity, with their sensitivity being significantly higher than that of healthy cells (Zheng et al., 2022).
However, ES is quickly removed and broken down in the body. This makes it harder for copper ions to enter cancer cells. Despite this, ES has been found to be effective at transporting copper (Nagai et al., 2012), And this high effectiveness might be the reason why we see strong anti-cancer activity in many different types of tumors, such as lung cancer, melanoma, and sarcoma (Berkenblit et al., 2007). In a study on breast cancer, it was reported that ES caused cell death through apoptosis (Qu et al., 2010). Recently, ES has also been used to treat Menkes disease, which is a neurological disorder that happens because of copper deficiency (Guthrie et al., 2020). Based on the fundamental differences in copper metabolism between cancer and normal cells—particularly the elevated copper levels and copper-dependent pro-oncogenic mechanisms in malignancies—ES has emerged as a promising multifunctional drug candidate. It effectively kills cancer cells while also protecting neuronal cells (Gao et al., 2021). Normal cells possess robust copper homeostatic regulation (Ge et al., 2022), intact mitochondrial function, and a potent antioxidant defense system that maintains ROS at low levels (Nagai et al., 2012). As a result, ES-Cu complexes are rarely activated in normal cells, minimizing oxidative stress and preventing cellular damage. So, boosting the level of ES in tumor tissues helps get more copper ions into cancer cells, which leads to the death of those cells (Nagai et al., 2012).
4 Relevant mechanisms affecting the occurrence of cuproptosis
4.1 Copper overload disrupts the synthesis of Fe-S cluster proteins
Fe-S clusters are cofactors made up of iron and inorganic sulfur, which play a crucial role in a variety of biological processes, including mitochondrial energy production, amino acid synthesis, cofactor synthesis, and tRNA modification (Lill, 2009). The synthesis of Fe-S clusters involves four steps: 1) Nitrogen fixation 1 homolog (NFS1) catalyzes the conversion of cysteine to alanine, producing persulfide intermediates in the process; 2) The persulfide intermediate binds to iron, forming a [2Fe-2S] cluster on the scaffold protein of the iron-sulfur cluster assembly enzyme (ISCU); 3) The [2Fe-2S] cluster is released from ISCU with the assistance of a partner protein. Some of these clusters are directly inserted into [2Fe-2S] type iron-sulfur proteins, while others go on to form [4Fe-4S] clusters with the cooperation of iron-sulfur cluster assembly 1(ISCA1) and iron-sulfur cluster assembly 2(ISCA2); 4) The ultimately generated [4Fe-4S] clusters are transferred to the [4Fe-4S] type lipoprotein Fe-S protein for maturation (Gourdoupis et al., 2018; Lill and Freibert, 2020; Weiler et al., 2020) (Figure 2).
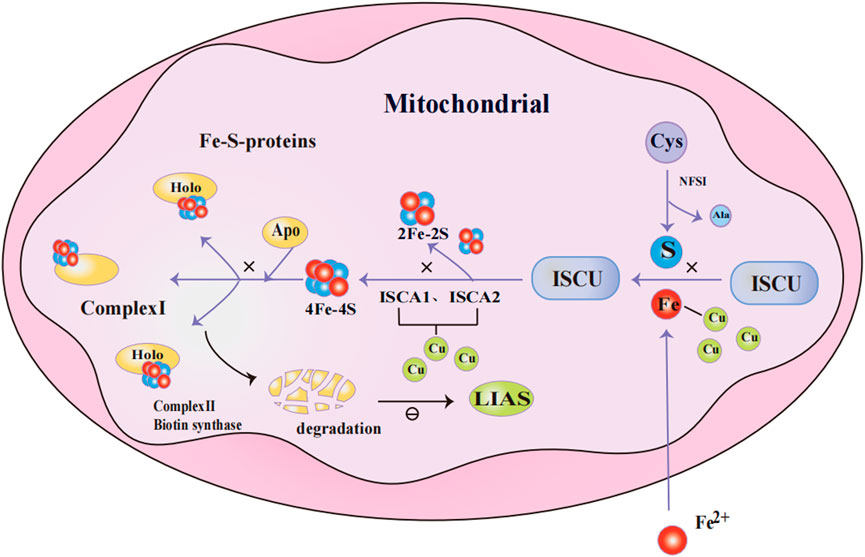
Figure 2. Mechanism of Fe-S cluster protein production in mitochondria. NFS1 catalyzes the conversion of Cys to Ala and persulfide; persulfide binds to Fe2+ and forms [2Fe-2S] clusters on the ISCU; [2Fe-2S] clusters are released from ISCU with the assistance of chaperone proteins, while others further assemble into [4Fe-4S] clusters through the collaboration of ISCA1 and ISCA2. The [4Fe-4S] cluster is delivered to [4Fe-4S]-type apolipoprotein to facilitate their maturation into functional Fe-S proteins. When excess Cu+ accumulates in mitochondria, it competes with Fe2+ for metal-binding sites in Fe-S cluster assembly proteins. Additionally, Cu+ binds to ISCA1 and ISCA2, disrupting the formation of Fe-S clusters. When Fe-S clusters are deficient, Fe-S protein precursors (e.g., apo-proteins) that fail to assemble correctly and acquire cofactors are marked for degradation. This results in reduced LIAS levels, which subsequently inhibits protein lipoylation.Abbreviations: NFS1 nitrogen fixation 1 homolog,Cys Cysteine,Ala Alanine, ISCU iron-sulfur cluster assembly enzyme, ISCA1/2 iron-sulfur cluster assembly 1/2,Fe-S cluster iron-sulfur cluster, LIAS lipoic acid synthetase.
Excessive copper can compete with iron for metal-binding sites in Fe-S cluster assembly proteins, thereby hindering the biosynthesis of Fe-S clusters in Escherichia coli under both aerobic and anaerobic conditions (Tan et al., 2014). In addition, copper has been shown to impair the assembly of [4Fe-4S] clusters in vitro (Brancaccio et al., 2017). In vivo, too much copper inhibits Fe-S clusters from forming, which reduces Fe-S enzyme activity and harms mitochondrial function (Lill and Freibert, 2020). Mechanistic studies show that human ISCA1, ISCA2, and ISCU bind copper strongly, disrupting the assembly of iron-sulfur clusters (Du et al., 2023). Copper overload reduces overall Fe-S cluster production in cells, leading to functional loss of Fe-S proteins and disruption of mitochondrial homeostasis. In contrast, non-Fe-S proteins remain largely unaffected, indicating the specificity of this process. In human cells, a shortage of Fe-S clusters causes Fe-S apoproteins to fail to mature properly (Gao et al., 2021). These immature proteins are recognized as misfolded or unstable and are targeted for degradation via mechanisms such as the ubiquitin-proteasome system (Maio and Rouault, 2020). This process prevents toxic protein aggregation and helps maintain intracellular stability. When Fe-S cluster biosynthesis is disrupted, lipoic acid synthetase (LIAS) levels drop, inhibiting protein lipidation (Pan et al., 2018; Wang et al., 2017). Collectively, these findings indicate that Fe-S cluster assembly and the functionality of their downstream proteins represent a key molecular target of copper toxicity.
4.2 FDX1 and protein palmitoylation are important elements in cuproptosis
At the subcellular level, mitochondria are highly sensitive to copper-induced cytotoxicity (Lucena-Valera et al., 2023). Cells that rely on mitochondrial respiration are more sensitive to copper ionophores. When mitochondrial function inhibitors and mitochondrial antioxidants were used, they significantly impacted the cells’ sensitivity to copper ionophores. However, mitochondrial oxidative phosphorylation decoupling agents had no effect on copper toxicity, indicating that mitochondrial respiration is crucial in the process of cuproptosis. For example, in ES-sensitive human lung adenocarcinoma (ABC-1) cells, time-dependent dysregulation was observed in multiple TCA cycle-related metabolites, indicating copper’s direct disruption of energy metabolism (Tsvetkov et al., 2022).
FDX1 serves as a key regulator in this process, encoding components of the sulfolipid metabolic pathway and directly modulating protein lipoylation (Rowland et al., 2018).
During lipoyl synthesis, LIAS forms transient covalent bonds with lipoyl carrier proteins and interacts with enzymes including GCSH and lipoyltransferase (LIPT1) (Gourdoupis et al., 2018; Wang et al., 2017; Seidel et al., 2013; Choi et al., 2019). It’s been shown that LIAS function relies on the normal lipidation of Fe-S cluster proteins. When Fe-S cluster biosynthesis is disrupted, LIAS levels drop, inhibiting protein lipoylation (Pan et al., 2018; Wang et al., 2017). FDX1 functions upstream of LIAS and regulates lipoylation by promoting Fe-S cluster assembly and maintaining LIAS stability (Gourdoupis et al., 2018). It directly binds to LIAS and supplies electronic support for S-adenosylmethionine (SAM)-dependent lipoylation reactions. In the presence of ES, large amounts of Cu2+ enter mitochondria. FDX1 is a direct target of the ES-Cu2+ complex, which binds to and inhibits FDX1 function, resulting in mitochondrial dysfunction (Ni et al., 2019). Additionally, FDX1 acts as a mitochondrial reductase that reduces Cu2+ in the ES-Cu2+ complex to Cu+ (Zheng et al., 2022). The resulting Cu+ induces destabilization and loss of Fe-S cluster proteins, which disrupts LIAS synthesis and inhibits protein lipoylation, ultimately blocking the TCA cycle; binding of lipoylated proteins to Cu+ triggers protein oligomerization, resulting in proteotoxic stress and ultimately inducing cell death (Tsvetkov et al., 2022) (Figure 3).
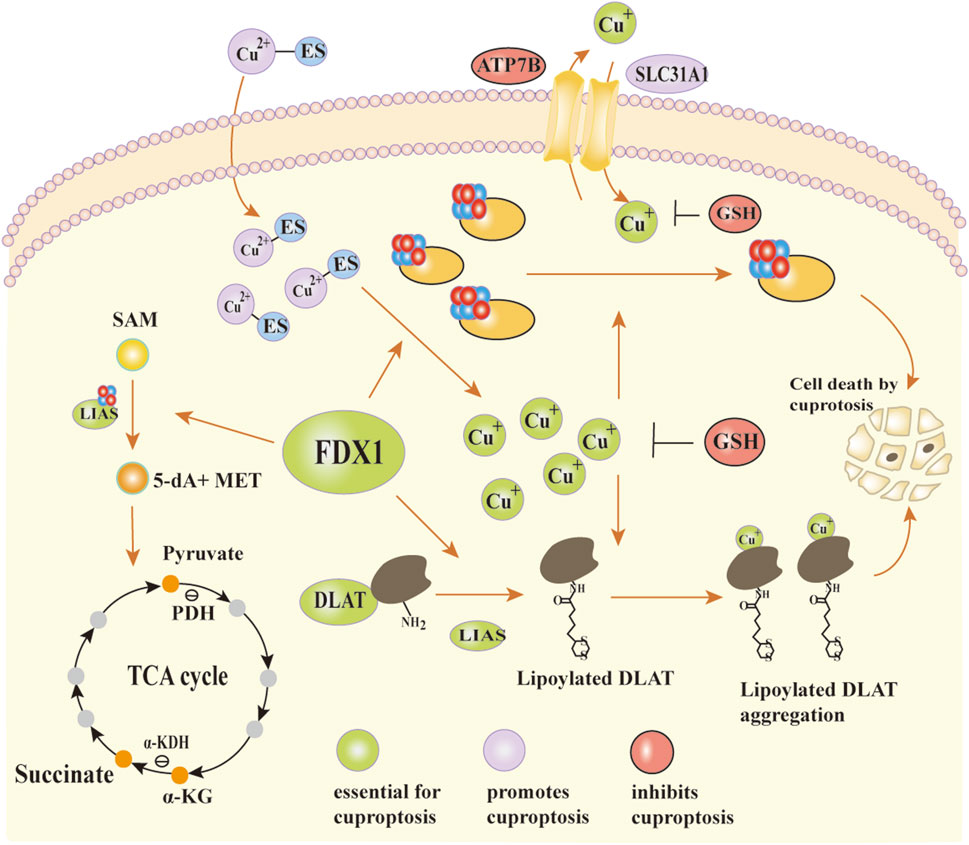
Figure 3. The mechanism of action of FDX1 and the occurrence of copper death. In the synergistic interaction between LIAS and Fe-S clusters, SAM is catalytically converted to 5′-dA and MET. MET provides carbon skeletons and energy substrates for the TCA cycle through subsequent metabolic processes. FDX1 plays a critical role in maintaining Fe-S cluster stability and supporting LIAS function. It not only assists in Fe-S cluster assembly but also directly binds to LIAS, providing electronic support for protein lipoylation reactions. Cu+ can enter mitochondria through SLC31A1 and is exported via an ATP7B-dependent transport mechanism. In the presence of the copper ionophore ES, excess Cu2+ is transported into mitochondria, forming a Cu2+-ES complex that binds to FDX1. This interaction partially inhibits FDX1 function and contributes to mitochondrial dysfunction. Meanwhile, FDX1 reduces Cu2+ to Cu+. Cu+ disrupts Fe-S cluster biosynthesis through two main mechanisms: it competes with Fe2+ for metal-binding sites in Fe-S assembly proteins, and it binds to lipoylated proteins, inducing abnormal oligomerization and proteotoxic stress. These effects ultimately lead to cell death.Abbreviations:LIAS lipoic acid synthetase, Fe-S clusters iron-sulfur cluster, SAM S-adenosylmethionine, 5′-dA 5′-deoxyadenosine, MET methionine, TCA cycle tricarboxylic acid cycle, FDX1 ferredoxin1, SLC31A1 solute carrier family 31, ATP7B ATPase copper transporting beta.
Genetic studies demonstrated that deletion of FDX1 or LIAS inhibits copper-induced cell death. Furthermore, FDX1 knockout enhanced cellular resistance to the copper ionophore ES, but did not significantly affect apoptosis inducers or ferroptosis agents (Tsvetkov et al., 2022). The cytotoxic activity of most copper ion-sensitive compounds (e.g., ES) was markedly reduced in cells cultured under glycolytic conditions, indicating a close link between copper metabolism and mitochondrial-mediated protein lipoylation processes (Tsvetkov et al., 2022). Studies have found that knockdown of FDX1 or lipoylation-related enzymes effectively protects cells from copper-induced toxicity (Tsvetkov et al., 2022). Metabolite analysis showed that FDX1 deficiency impairs protein lipoylation by inhibiting pyruvate dehydrogenase (PDH) and α-ketoglutarate dehydrogenase (KGDH) activity in the TCA cycle. This disruption leads to depletion of succinate and accumulation of both pyruvate and α-ketoglutarate (Ni et al., 2019). Knockdown of FDX1 not only completely blocked protein lipoylation but also significantly suppressed cellular respiration, an effect consistent with LIAS deficiency. Additionally, FDX1 knockdown resulted in elevated levels of SAM, an essential substrate for LIAS in the lipoic acid synthesis pathway. These findings suggest that FDX1 functions as an upstream regulator of protein lipoylation by controlling the function of LIAS (Tsvetkov et al., 2022). Lipoylated proteins are essential for copper binding (Tsvetkov et al., 2022).
When copper binds to lipoylated proteins, it causes the proteins to lose their function. Treatment with a copper ionophore can partly fix these functional defects. Additionally, copper binding to lipoylated proteins in the TCA cycle markedly enhances their toxicity. Native gel electrophoresis showed that DLAT’s oligomerization depends on its lipoylation state. Mass spectrometry analysis revealed that copper ionophore treatment causes Fe-S cluster proteins to degrade in an FDX1-dependent way and triggers protein toxicity stress (Tsvetkov et al., 2022). These findings align with previous observations in bacteria and yeast, where copper was found to destabilize proteins containing Fe-S clusters (Brancaccio et al., 2017; Macomber and Imlay, 2009; Chillappagari et al., 2010).
4.3 There’s a link between copper uptake and export genes and copper-caused cell death
Copper ionophores disrupt the balance of intracellular copper levels, affecting regulatory mechanisms like the copper import gene solute carrier family 31(SLC31A1) and the copper export genes ATP7A and ATP7B. These genes are closely associated with mutations in the copper metabolism-related diseases Menkes disease and Wilson’s disease, respectively (Lutsenko, 2010; Nevitt et al., 2012). CTR1 serves as the primary channel for copper uptake into cells (Lee et al., 2002). ATP7A functions as a steady-state copper exporter, regulating intracellular copper levels to shield cells from copper toxicity (Aubert et al., 2020). ATP7A is highly expressed in enterocytes and plays a crucial role in both copper uptake and excretion processes (Du et al., 2023). Furthermore, ATP7A has the capability to supply copper to various copper-dependent enzymes, thereby influencing numerous facets of tumorigenesis, such as cell proliferation, metastasis, and angiogenesis (Denoyer et al., 2015). Research has demonstrated that the expression of ATP7A in cancerous tissues is much higher than in the adjacent tissues (Li et al., 2019).
A shared mechanism underlies both copper-induced cell death and dysregulation of copper homeostasis. In human embryonic kidney 293 cells expressing the SV40 large T antigen (HEK293T) and ABC1 cells, when SLC31A1 is overexpressed, the cells’ sensitivity to physiological copper concentrations rises significantly. Elevated copper concentrations result in a reduction in the total quantity of proteins related to mitochondrial respiration, a decrease in protein lipidation, a decline in Fe-S cluster proteins, and an upregulation of heat shock protein 70(HSP70) levels (Tsvetkov et al., 2022). However, inhibitors of ferroptosis, necrosis, and apoptosis have proven ineffective in preventing copper-induced cell death. Conversely, copper chelators with bathocuproine disulfonic acid (BCS) and tetrathiomolybdate (TTM), as well as genetic knockout (KO) of FDX1 and LIAS, have all shown the ability to partially rescue cells from copper-induced death (Tsvetkov et al., 2022). Loss-of-function experiments have further shown that silencing ATP7A can markedly inhibit the growth and metastasis of breast and lung cancer cells (Shanbhag et al., 2019). These findings underscore the pivotal role of copper homeostasis mechanisms in cell survival, tumorigenesis, and sensitivity to copper toxicity. They also suggest that genes involved in copper homeostasis (e.g., SLC31A1, ATP7A) may represent potential therapeutic targets.
4.4 p53 may enhance or promote copper-induced cell death
Tumor Protein p53 (TP53) is the gene most frequently mutated in human cancers, and the majority of these cancer-related protein 53 (p53) mutations are missense mutations, typically found in its DNA-binding domains. Many of these mutant p53 (mtp53) proteins promote cancer development by gaining new, harmful functions, known as “gain-of-function” (GOF) activity (Mantovani et al., 2019; Zhang et al., 2020). Studies have demonstrated that various mtp53 proteins can play distinct roles in regulating glycolysis and mitochondrial metabolism (Eriksson et al., 2017). The p53 protein, encoded by the TP53 gene, is a key transcription factor activated by diverse cellular stress signals—including DNA damage (Fagherazzi et al., 2025), hypoxia (Simon-Molas et al., 2021), and oncogene activation—to execute central regulatory functions (Gong et al., 2016). Activated p53 transcriptionally regulates downstream target genes (e.g., the cell cycle inhibitor p21)to arrest the cell cycle at the G1/S checkpoint, providing time for DNA repair. If the damage is irreparable, p53 initiates apoptosis to eliminate abnormal cells (Liu et al., 2024; Baliakas and Soussi, 2025). Additionally, p53 plays a broad role in regulating cellular metabolism, influencing key processes such as glycolysis, oxidative phosphorylation, and antioxidant responses (Sanford et al., 2023). It also induces cellular senescence and irreversible cell cycle arrest, thereby suppressing tumor progression (Liu et al., 2024). p53 also plays an important role in maintaining intracellular copper homeostasis through two primary mechanisms:
On one hand, p53 inhibits the copper chaperone ATOX1 to reduce free intracellular copper availability and prevents excess copper accumulation (Tsymbal et al., 2023). It also indirectly senses copper levels by modulating glutathione (GSH) concentrations (Xiong et al., 2023). On the other hand, under pathological or experimental conditions with abnormally high copper, p53 eliminates copper-damaged cells to protect the organism (Xiong et al., 2023).
Possible mechanisms of action of p53 in copper-induced cell death, also known as “cuproptosis,” include:
1. Promote cuproptosis sensitivity: p53 may enhance cells’ sensitivity to cuproptosis by suppressing glycolysis and facilitating the shift towards mitochondrial metabolism. Firstly, p53 reduces glucose uptake by suppressing the expression and activity of glucose transporters such as glucose transporter 1 (GLUT1) and glucose transporter 4 (GLUT4) (Kawauchi et al., 2008; Schwartzenberg-Bar-Yoseph et al., 2004). Secondly,p53 suppresses glycolysis by inhibiting several key catalytic steps within the glycolytic pathway. For example, p53 inhibits the conversion of glucose to glucose-6-phosphate by promoting the degradation of Hexokinase 2(HK2) mRNA (Wang et al., 2014). In the final step of glycolysis, pyruvate is converted to lactate by lactate dehydrogenase (LDHA/B). p53 represses LDHA expression either by directly binding to its promoter or by promoting degradation of the LDHA transcription factor HIF-1α (Zhou et al., 2019; Semenza, 2010). This can result in the accumulation of intracellular pyruvate, which subsequently enters the TCA cycle and supports oxidative phosphorylation.
2. Inhibit glucose metabolism: p53 suppresses glucose metabolism through multiple mechanisms. Glucose serves as the primary source of ATP in mammalian cells. When glucose is restricted, intracellular ATP levels decrease, leading to an increase in the AMP/ATP ratio. This activates the AMPK signaling pathway (Herzig and Shaw, 2018). The activation of AMPK then induces the phosphorylation and activation of p53, creating a feedback loop between p53 and glucose metabolism. This loop further promotes the shift from glycolysis to oxidative phosphorylation (Jones et al., 2005);
3. Enhance oxidative phosphorylation: p53 enhances mitochondrial oxidative phosphorylation and facilitates cuproptosis by promoting cytochrome c oxidase (COX) complex assembly through induction of synthesis of cytochrome c oxidase 2(SCO2) expression, and by assisting in the proper assembly of respiratory complex I via the target gene AIF-encoded mitochondrial membrane proteins (Stambolsky et al., 2006).
4. Maintain mitochondrial function: p53 enhances the TCA cycle and oxidative phosphorylation through multiple mechanisms. P53 promotes the conversion of pyruvate to acetyl-CoA by activating the PDH (Zhang et al., 2011; Contractor and Harris, 2012), a key process required for cuproptosis within the TCA cycle. P53 boosts acetyl-CoA production by enhancing fatty acid oxidation and suppressing lipid synthesis (Liu et al., 2014; Assaily et al., 2011; Moon et al., 2019), thereby facilitating the initiation of the TCA cycle. Studies have shown that p53 activity is essential for maintaining mitochondrial integrity, cytochrome c oxidase activity, and respiratory function. Deletion or mutation of p53 leads to a significant decline in mitochondrial function (Matoba et al., 2006; Saleem et al., 2009; Zhou et al., 2003);
5. Modulate GSH biogenesis: p53 may play a dual role in copper-induced cell death by differentially regulating the biosynthesis of GSH(12). GSH is a crucial copper-binding ligand that swiftly binds to Cu+ to form a stable Cu+-GSH complex (Tsvetkov et al., 2022). Based on its ability to bind copper ions, GSH safeguards cells from copper-mediated cytotoxicity by preventing harmful interactions between copper and proteins (Sa et al., 2018). However, the depletion of intracellular GSH, results in copper-dependent cell death. This phenomenon can be mitigated by the use of exogenous copper chelators, such as TTM.
In summary, a necessary condition for the occurrence of copper-induced cell death is that the concentration of copper ions exceeds normal levels. When copper overload happens, copper competes with iron for the metal-binding sites in Fe-S cluster assembly proteins. This competition interferes with the formation of Fe-S cluster proteins. This leads to a decrease in LIAS levels and inhibits the acylation of proteins. However, in the presence of FDX1, LIAS can be stably generated and directly binds to FDX1. This interaction promotes the lipoylation of proteins. The lipoylated proteins then bind to Cu+, which initiates protein oligomerization. This oligomerization leads to protein toxic stress, ultimately inducing cell death. Overexpression of genes involved in copper homeostasis can enhance a cell’s sensitivity to copper concentrations. Additionally, p53 increases cellular sensitivity to cuproptosis by suppressing glycolysis and maintaining normal mitochondrial function.
5 The role of cuproptosis in tumor cells
CTR1, a copper transport protein, plays a key role in maintaining intracellular copper homeostasis (Zhou et al., 2023). CTR1-mediated intracellular copper overload can induce cuproptosis in tumor cells. At the same time, it also causes an upregulation of expression levels of PD-L1 on the surface of cancer cells (Cobine and Brady, 2022; Zhang et al., 2023). This mechanism provides an important basis for boosting the antitumor effects of anti-programmed death-ligand 1 monoclonal antibody (αPD-L1) antibodies (Chen et al., 2022). Since cuproptosis relies on mitochondrial respiration, metabolically active tumors may be more vulnerable to this form of cell death. However, tumor cells can also develop resistance to cuproptosis through various mechanisms. For example, high intracellular levels of GSH can chelate copper ions, thereby inactivating them (Wang et al., 2025). Alternatively, activation of the HIF-1α signaling pathway in the hypoxic tumor microenvironment can promote resistance to cuproptosis. This occurs through two mechanisms: upregulation of metallothionein MT2A to bind copper ions, and degradation of the key protein DLAT (Yang et al., 2025). Therefore, targeting cuproptosis has emerged as a promising anti-tumor strategy.
In advanced lung adenocarcinoma, elevated copper levels activate spliced x-box-binding protein 1 (XBP1s)-mediated super-enhancers (SEs), which promote mahogunin ring finger 1 (MGRN1) transcription. This subsequently enhances ubiquitination and degradation of LIPT1 (Gu et al., 2025). This mechanism not only drives glycolytic metabolic reprogramming but also significantly suppresses cuproptosis and promotes tumor progression. In melanoma, a study utilized endogenous copper ions and click chemistry cascade reactions to successfully amplify the cuproptosis effect, effectively inhibiting tumor growth (Liu et al., 2025). In osteosarcoma, cuproptosis induces PD-L1 expression. To exploit this mechanism, a study developed a tetrahedral framework nucleic acid (tFNA) delivery system carrying both a PD-L1 monoclonal antibody and the cuproptosis inducer ESCu. This system simultaneously blocked PD-L1 and induced cuproptosis, significantly enhancing chimeric antigen receptor T (CAR-T) cell infiltration and antitumor efficacy within the tumor microenvironment (Huang et al., 2025). Moreover, in solid tumors—such as clear cell renal cell carcinoma and papillary thyroid carcinoma—hypoxia suppresses cuproptosis by activating HIF-1α, modulating the pyruvate dehydrogenase kinase 1/3(PDK1/3)-DLAT axis, and upregulating metallothionein MT2A expression. Studies indicate that combining HIF-1α inhibitors can effectively reverse this resistance and enhance cuproptosis-induced therapeutic responses (Yang et al., 2025).
In addition to the mechanisms mentioned above, the copper ionophore ES has demonstrated significant anti-tumor potential, and its effects are closely linked to cuproptosis. The antitumor activity of ES primarily stems from its capacity to disrupt the cytoskeleton of tumor cells (Gehrmann, 2006). In colorectal cancer (CRC), studies have shown that the death of colorectal cancer cells induced by ES depends on the buildup of ROS when copper is added (Gao et al., 2021). Its capacity to promote the generation of ROS is primarily due to its targeted effect on the mitochondrial electron transport (Blackman et al., 2012). Even without the addition of exogenous copper, ES can slow down the growth of CRC cells. Moreover, in both in vitro and in vivo experiments, ES reduced the expression of ATP7A. ATP7A expression is higher in CRC tissues than in normal tissues. CRC cells enhance tumor growth and development by increasing ATP7A expression. This further indicates that ES has great therapeutic potential (Gao et al., 2021).
Kirsten ratsarcoma viral oncogene homolog (KRAS) mutations have also been found to be common in lung, pancreatic, colorectal, and other types of tumors (Simanshu et al., 2017). The oncogenic activity of KRAS boosts the requirement for copper bioavailability (Shanbhag et al., 2019), stabilizes ATP7A, and promotes its expression on the cell surface. When copper levels are high, CTR1 is taken into the cell and broken down, while ATP7A is moved to the cell surface to remove the excess copper ions. KRAS mutations remodel the copper homeostasis network in intestinal epithelial cells through transcriptional and post-transcriptional mechanisms. This reprogramming makes mutant cells dependent on high copper levels and increases their sensitivity to copper chelators. In KRAS mutant models, macrophages promote copper uptake via a non-classical pathway to supply bioavailable copper for tumor growth, while ATP7A alleviates copper toxicity through efflux mechanisms. Notably, inhibiting macrophage activity reduces copper uptake, suppresses copper-dependent signaling, and delays tumor progression (Aubert et al., 2020).
Since the gastrointestinal tract is where the body mainly absorbs copper, the antitumor effects of ES could be closely linked to problems with copper metabolism (Gao et al., 2021). Studies have found that the stomach acid-reducing drug omeprazole can stop the overproduction of melanin by preventing too much copper from causing ATP7A to appear on the cell surface (Matsui et al., 2015). Furthermore, ES stops cisplatin-resistant lung cancer cells from growing by boosting the levels of ROS (Wangpaichitr et al., 2009). Paclitaxel is another medication that makes tumor cells more vulnerable by damaging their cytoskeleton. When ES is used together with paclitaxel, it has been shown to be just as well-tolerated as paclitaxel on its own (Berkenblit et al., 2007).
Therefore, by increasing the level of ES in tumor tissues can help transport more copper ions into cancer cells, inducing cancer cell death (Nagai et al., 2012). Boda Guo et al. designed an amphiphilic biodegradable polymer (PHPM) characterized by the presence of ROS-sensitive thioketone bonds and dangling paired carboxylic acids on the backbone, followed by PHPM encapsulation of ES and Cu into NP@ESCu (Guo et al., 2023). Studies have indicated that NP@ESCu remarkably enhances the transport of intracellular copper. It also causes serious mitochondrial damage in cancer cells through the upregulation of ROS. This process results in the shrinking of mitochondria, an increase in membrane density, and a decrease or complete loss of mitochondrial crests. These changes ultimately lead to a type of cell death known as cuproptosis (Guo et al., 2023).
Beyond directly inducing tumor cell death, NP@ESCu also enhances antitumor efficacy through multiple immunomodulatory mechanisms:NP@ESCu can cause dendritic cells (DCs) to mature and encourage cluster of differentiation 8-positive T (CD8+ T) cells to infiltrate tumor tissues. Additionally, NP@ESCu spurs the change of TAMs from the immunosuppressive M2 phenotype to the immunostimulatory M1 phenotype; NP@ESCu can reprogram the TME. It activates systemic anti-tumor immune responses and induces long-term immune memory (Guo et al., 2023) (Figure 4). Studies have indicated that the characteristics of the TME can influence the therapeutic effectiveness of αPD-L1. In fact, they may even restrict its efficacy (Wang et al., 2021). Therefore, the combination of NP@ESCu and αPD-L1 is likely to further boost the anti-tumor effect through a synergistic action. This offers a more effective treatment approach.
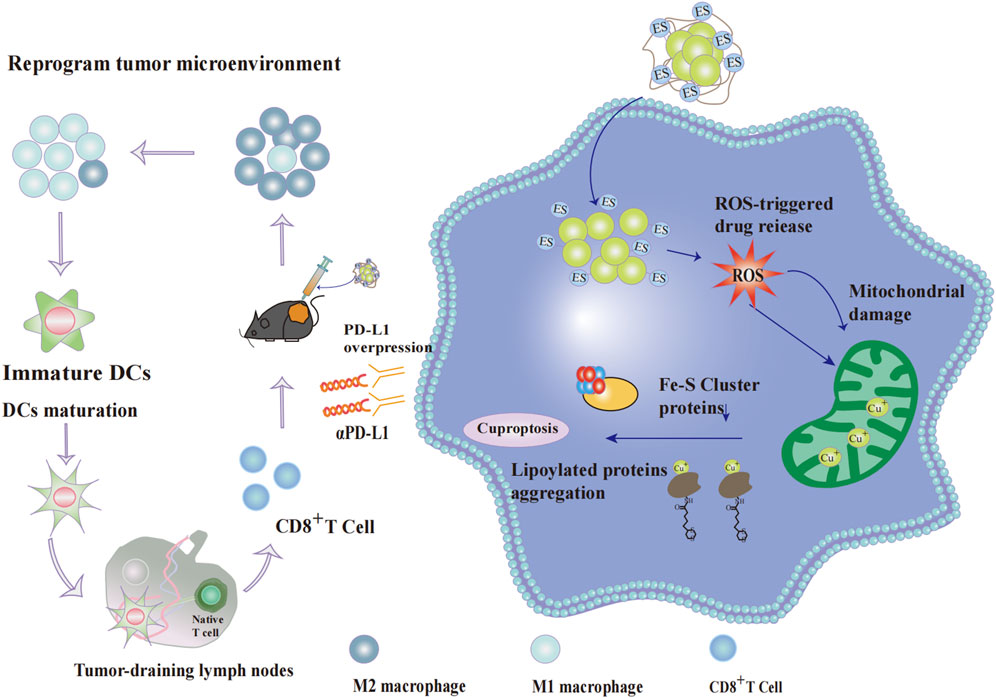
Figure 4. Effect of cupproposis on cancer cells and tumor microenvironment. After cellular uptake, the nanoparticle NP@ESCu dissociates its PHPM polymer chain under ROS stimulation, releasing abundant Cu2+-ES complexes. The complex further elevates intracellular ROS levels, causing severe mitochondrial damage in cancer cells. Simultaneously, mitochondrial Cu+ content rises significantly, reducing Fe-S cluster production and promoting proteotoxic stress through lipoylated protein aggregation, ultimately inducing cancer cell death. NP@ESCu enhances anti-tumor efficacy through multiple immunomodulatory mechanisms: It promotes the polarization of TAMs from the immunosuppressive M2 phenotype to the immunostimulatory M1 phenotype; It induces DC maturation and enhances the infiltration of CD8+ T cells into tumor tissues.; It reprograms the TME, making it more susceptible to immune attack. Additionally, copper overload in cancer cells upregulates PD-L1 expression on the cell surface. The combination of NP@ESCu and αPD-L1 acts synergistically to significantly improve the effectiveness of tumor therapy.Abbreviations: PHPM Poly (N-(2-hydroxypropyl) methacrylamide,ROS reactive oxygen species, Fe-S cluster iron-sulfur cluster, TAMs tumor-associated macrophages, M1 Macrophage M1 phenotype,M2 Macrophage M2 phenotype,DC dendritic cell, CD8+ T cell cluster of differentiation 8-positive T cell,TME tumor microenvironment, PD-L1 programmed death-ligand 1, αPD-L1 anti-Programmed Death-Ligand 1 monoclonal antibod.
6 Discussion
Copper serves as an important cofactor for enzymes in the animal realm and holds a crucial position in numerous physiological processes. Being an essential nutrient, the concentration of copper ions is maintained at extremely low levels. This is to prevent the build-up of free copper ions, which can inflict damage on cells (Ge et al., 2022; Lutsenko, 2010; Rae et al., 1999; Kim et al., 2008). However, even moderately elevated intracellular copper levels can trigger a novel form of cell death known as “cuproptosis” (Tsvetkov et al., 2022). Cancer cells exhibit higher basal oxidative stress and greater reliance on mitochondrial metabolism compared to normal cells (Yang et al., 2023). They also require significantly more copper to support proliferation and metastasis (Chen and Pervaiz, 2010). These characteristics make targeting to induce cuproptosis a highly promising anticancer strategy.
Recent years have witnessed significant breakthroughs in the understanding of cuproptosis mechanisms. Tsvetkov et al. were the first to systematically demonstrate that cuproptosis is a copper-dependent cell death mode induced by mitochondrial respiration and proteotoxic stress, highlighting the crucial role of copper homeostasis in cancer and other diseases (Tsvetkov et al., 2022); Jing Du et al. discovered that copper overload inhibits Fe-S cluster assembly, impairs Fe-S enzyme activity, and disrupts mitochondrial function (Du et al., 2023); Chen Xiong et al. highlighted the significant role of p53 in regulating cuproptosis (Xiong et al., 2023). Studies indicate that cuproptosis strongly depends on mitochondrial metabolic processes. Therefore, copper toxicity can be evaluated by measuring mitochondrial membrane potential, ROS levels, and lipoylated protein content (Harvey and McArdle, 2008).
Additionally, plasma ceruloplasmin activity and the serum Cu/Zn ratio can serve as effective indicators of copper metabolism (Kumar et al., 2007; McCaughern, 2019). When combined with functional markers (e.g., cytochrome c oxidase activity or superoxide dismutase (SOD) activity) (Zhou, 2024), they enable a more comprehensive evaluation of copper metabolic status, thereby supporting the diagnosis and mechanistic investigation of related diseases.
Cuproptosis represents a form of cell death distinct from apoptosis, pyroptosis, and ferroptosis. Its mechanism centers on mitochondrial lipoylated protein aggregation and Fe-S cluster protein inactivation, yet it also exhibits cross-talk with other cell death pathways. For example, copper accumulation induces apoptosis in the mitochondrial pathway (e.g., cytochrome c release) (Tsvetkov et al., 2022); cuproptosis induces proteotoxic stress through direct binding to lipoylated proteins such as DLAT, whereas ferroptosis relies on lipid peroxidation caused by glutathione peroxidase 4 (GPX4) inactivation (Wang et al., 2024). Additionally, copper ions can induce protective autophagy; however, excessive autophagy may synergistically enhance cuproptosis (Li et al., 2024). Studies indicate that targeting key cuproptosis-related genes (such as FDX1) significantly improves the effectiveness of immune checkpoint inhibitors, highlighting the potential for synergistic targeted therapy.
In immunotherapy, low PD-L1 expression in tumor cells is often associated with a poor immune response (Iwai et al., 2002; Jiang et al., 2020). Interestingly, copper ions upregulate PD-L1 expression in tumor cells (Cobine and Brady, 2022; Zhang et al., 2023). Delivering copper to tumor cells via copper ionophores not only induces cuproptosis but also enhances the response to αPD-L1 therapy. However, the immunosuppressive TME frequently limits the efficacy of αPD-L1 monotherapy (Jiang et al., 2020; Kudo, 2020). Recent studies have shown that the nanocomplex NP@ESCu, when combined with αPD-L1 therapy, significantly improves the TME and enhances therapeutic efficacy. This combination strategy provides important direction for the development of nanotechnology-based copper-targeted drugs (Guo et al., 2023). In the future, further optimization of nanoparticle structures is needed to enhance targeting capability and biocompatibility, thereby facilitating clinical translation. Additionally, the potential of other types of copper-targeting drugs in combination with immunotherapy should be systematically evaluated.
Traditional medicines have also gained attention due to their multi-target mechanisms and favorable safety profiles. Many natural bioactive compounds (e.g., plant extracts and microbial metabolites) exhibit copper-binding properties and can modulate copper metabolism or induce cuproptosis. For example, fungal-derived pochonin D (PoD) and flavonoid derivatives (HQFs) can induce cancer cell death through copper-binding mechanisms (Feng et al., 2025; Jiang et al., 2025); in contrast, TM depletes tumor copper stores and inhibits the MEK/ERK signaling pathway (Ramchandani et al., 2021). The holistic perspective of traditional medicine has also advanced the development of copper-targeted combination strategies. Examples include NP@ESCu combined with microtubule inhibitors, and copper ionophores paired with PD-1 inhibitors, both aimed at enhancing therapeutic efficacy through ROS generation and remodeling of the immune microenvironment. Based on the screening of traditional medicine components, cuproptosis-related proteins such as peroxiredoxin 1(PRDX1) have been identified, offering new therapeutic targets for difficult-to-treat cancers like triple-negative breast cancer (Feng et al., 2025). Future research should further clarify the specific mechanisms by which traditional medicines regulate copper metabolism and accelerate their clinical translation and application.
Despite promising findings, several key challenges remain in cuproptosis researchi:is there a specific threshold of copper concentration that determines whether a cell will experience toxicity or undergo cuproptosis?The relationship between cytotoxicity and cuproptosis: are they complementary, or does one promote the other? The therapeutic potential of cuproptosis: how can we use the cuproptosis mechanism to develop new drugs for related diseases? Finding the answers to these questions will open up broader prospects for the research on the mechanism of cuproptosis and its clinical applications. Currently, copper ionophores and chelators have shown significant anti-tumor potential. Meanwhile, nanotechnology and copper chaperone-targeting strategies offer promising avenues to improve drug specificity and reduce systemic toxicity. Future efforts should prioritize systematic research into cuproptosis mechanisms, drug development, and clinical translation to open new avenues for cancer therapy.
Author contributions
YJ: Data curation, Writing – original draft, Methodology, Software, Investigation, Writing – review and editing, Conceptualization, Resources. HW: Writing – review and editing, Investigation, Methodology, Supervision, Formal Analysis, Data curation. MZ: Conceptualization, Resources, Visualization, Writing – review and editing, Data curation, Methodology. TL: Validation, Supervision, Visualization, Formal Analysis, Writing – review and editing. YL: Visualization, Conceptualization, Writing – review and editing, Methodology. SY: Visualization, Validation, Writing – review and editing, Supervision. MZ: Writing – review and editing, Data curation, Methodology, Supervision, Investigation, Resources, Funding acquisition. LZ: Supervision, Project administration, Investigation, Data curation, Writing – review and editing, Funding acquisition, Software, Formal Analysis, Resources.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by Science and Technology Program of the Joint Fund of Scientific Research for the Public Hospitals of Inner Mongolia Academy of Medical Sciences (2023GLLH0315) from Minghui Zhang. Additionally, it was supported by the Heilongjiang Provincial Leading Talent Echelon Reserve Leader Funding Fund 2022, from Lei Zhang.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adman, E. T. (1991). “Copper protein structures,”Adv. Protein Chem. Advances in protein chemistry. Editors C. B. Anfinsen, J. T. Edsall, F. M. Richards, and D. S. Eisenberg (Academic Press), 42, 145–197. doi:10.1016/s0065-3233(08)60536-7
Amaravadi, R., Glerum, D. M., and Tzagoloff, A. (1997). Isolation of a cDNA encoding the human homolog of COX17, a yeast gene essential for mitochondrial copper recruitment. Hum. Genet. 99 (3), 329–333. doi:10.1007/s004390050367
Assaily, W., Rubinger, D. A., Wheaton, K., Lin, Y., Ma, W., Xuan, W., et al. (2011). ROS-Mediated p53 induction of Lpin1 regulates fatty acid oxidation in response to nutritional stress. Mol. Cell. 44 (3), 491–501. doi:10.1016/j.molcel.2011.08.038
Aubert, L., Nandagopal, N., Steinhart, Z., Lavoie, G., Nourreddine, S., Berman, J., et al. (2020). Copper bioavailability is a KRAS-Specific vulnerability in colorectal cancer. Nat. Commun. 11 (1), 3701. doi:10.1038/s41467-020-17549-y
Baliakas, P., and Soussi, T. (2025). The TP53 tumor suppressor gene: from molecular biology to clinical investigations. J. Intern Med. 298 (2), 78–96. doi:10.1111/joim.20106
Baltaci, A. K., Dundar, T. K., Aksoy, F., and Mogulkoc, R. (2017). Changes in the serum levels of trace elements before and after the operation in thyroid cancer patients. Biol. Trace Elem. Res. 175 (1), 57–64. doi:10.1007/s12011-016-0768-2
Banci, L., Bertini, I., Ciofi-Baffoni, S., Kozyreva, T., Zovo, K., and Palumaa, P. (2010). Affinity gradients drive copper to cellular destinations. Nature 465 (7298), 645–648. doi:10.1038/nature09018
Basu, S., Singh, M. K., Singh, T. B., Bhartiya, S. K., Singh, S. P., and Shukla, V. K. (2013). Heavy and trace metals in carcinoma of the gallbladder. World J. Surg. 37 (11), 2641–2646. doi:10.1007/s00268-013-2164-9
Bergsbaken, T., Fink, S. L., and Cookson, B. T. (2009). Pyroptosis: host cell death and inflammation. Nat. Rev. Microbiol. 7 (2), 99–109. doi:10.1038/nrmicro2070
Berkenblit, A., Eder, J. P., Ryan, D. P., Seiden, M. V., Tatsuta, N., Sherman, M. L., et al. (2007). Phase I clinical trial of STA-4783 in combination with paclitaxel in patients with refractory solid tumors. Clin. Cancer Res. 13 (2 Pt 1), 584–590. doi:10.1158/1078-0432.CCR-06-0964
Blackman, R. K., Cheung-Ong, K., Gebbia, M., Proia, D. A., He, S., Kepros, J., et al. (2012). Mitochondrial electron transport is the cellular target of the oncology drug elesclomol. PLoS One 7 (1), e29798. doi:10.1371/journal.pone.0029798
Brancaccio, D., Gallo, A., Piccioli, M., Novellino, E., Ciofi-Baffoni, S., and Banci, L. (2017). [4Fe-4S] cluster assembly in Mitochondria and its impairment by copper. J. Am. Chem. Soc. 139 (2), 719–730. doi:10.1021/jacs.6b09567
Carneiro, B. A., and El-Deiry, W. S. (2020). Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 17 (7), 395–417. doi:10.1038/s41571-020-0341-y
Chaintreuil, P., Kerreneur, E., Bourgoin, M., Savy, C., Favreau, C., Robert, G., et al. (2023). The generation, activation, and polarization of monocyte-derived macrophages in human malignancies. Front. Immunol. 14, 1178337. doi:10.3389/fimmu.2023.1178337
Chang, W., Li, H., Zhong, L., Zhu, T., Chang, Z., Ou, W., et al. (2022). Development of a copper metabolism-related gene signature in lung adenocarcinoma. Front. Immunol. 13, 1040668. doi:10.3389/fimmu.2022.1040668
Chen, Z. X., and Pervaiz, S. (2010). Involvement of cytochrome c oxidase subunits Va and Vb in the regulation of cancer cell metabolism by Bcl-2. Cell. Death Differ. 17 (3), 408–420. doi:10.1038/cdd.2009.132
Chen, W. H., Chen, Q. W., Chen, Q., Cui, C., Duan, S., Kang, Y., et al. (2022). Biomedical polymers: synthesis, properties, and applications. Sci. China Chem. 65 (6), 1010–1075. doi:10.1007/s11426-022-1243-5
Chillappagari, S., Seubert, A., Trip, H., Kuipers, O. P., Marahiel, M. A., and Miethke, M. (2010). Copper stress affects iron homeostasis by destabilizing iron-sulfur cluster formation in Bacillus subtilis. J. Bacteriol. 192 (10), 2512–2524. doi:10.1128/JB.00058-10
Choi, S. G., Olivet, J., Cassonnet, P., Vidalain, P. O., Luck, K., Lambourne, L., et al. (2019). Maximizing binary interactome mapping with a minimal number of assays. Nat. Commun. 10 (1), 3907. doi:10.1038/s41467-019-11809-2
Cobine, P. A., and Brady, D. C. (2022). Cuproptosis: cellular and molecular mechanisms underlying copper-induced cell death. Mol. Cell. 82 (10), 1786–1787. doi:10.1016/j.molcel.2022.05.001
Contractor, T., and Harris, C. R. (2012). p53 negatively regulates transcription of the pyruvate dehydrogenase kinase Pdk2. Cancer Res. 72 (2), 560–567. doi:10.1158/0008-5472.CAN-11-1215
Culotta, V. C., Klomp, L. W., Strain, J., Casareno, R. L., Krems, B., and Gitlin, J. D. (1997). The copper chaperone for superoxide dismutase. J. Biol. Chem. 272 (38), 23469–23472. doi:10.1074/jbc.272.38.23469
Denoyer, D., Masaldan, S., La Fontaine, S., and Cater, M. A. (2015). Targeting copper in cancer therapy: 'copper that Cancer. Metallomics. 7 (11), 1459–1476. doi:10.1039/c5mt00149h
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 149 (5), 1060–1072. doi:10.1016/j.cell.2012.03.042
Du, J., Huang, Z., Li, Y., Ren, X., Zhou, C., Liu, R., et al. (2023). Copper exerts cytotoxicity through inhibition of iron-sulfur cluster biogenesis on ISCA1/ISCA2/ISCU assembly proteins. Free Radic. Biol. Med. 204, 359–373. doi:10.1016/j.freeradbiomed.2023.05.017
Elmore, S. (2007). Apoptosis: a review of programmed cell death. Toxicol. Pathol. 35 (4), 495–516. doi:10.1080/01926230701320337
Eriksson, M., Ambroise, G., Ouchida, A. T., Lima Queiroz, A., Smith, D., Gimenez-Cassina, A., et al. (2017). Effect of mutant p53 proteins on glycolysis and mitochondrial metabolism. Mol. Cell. Biol. 37 (24), e00328-17. doi:10.1128/MCB.00328-17
Fagherazzi, P., Stixová, L., and Bartova, E. (2025). Specific TP53 mutations impair the recruitment of 53BP1 to DNA double-strand breaks underlying the mechanism of radioresistance. Eur. Biophys. J. doi:10.1007/s00249-025-01774-8
Feng, J. F., Lu, L., Zeng, P., Yang, Y. H., Luo, J., Yang, Y. W., et al. (2012). Serum total oxidant/antioxidant status and trace element levels in breast cancer patients. Int. J. Clin. Oncol. 17 (6), 575–583. doi:10.1007/s10147-011-0327-y
Feng, L., Wu, T. Z., Guo, X. R., Wang, Y. J., Wang, X. J., Liu, S. X., et al. (2025). Discovery of natural resorcylic acid lactones as novel potent copper ionophores covalently targeting PRDX1 to induce cuproptosis for triple-negative breast cancer therapy. ACS Cent. Sci. 11 (2), 357–370. doi:10.1021/acscentsci.4c02188
Festa, R. A., and Thiele, D. J. (2011). Copper: an essential metal in biology. Curr. Biol. 21 (21), R877–R883. doi:10.1016/j.cub.2011.09.040
Gaetke, L. M., and Chow, C. K. (2003). Copper toxicity, oxidative stress, and antioxidant nutrients. Toxicology 189 (1-2), 147–163. doi:10.1016/s0300-483x(03)00159-8
Gao, W., Huang, Z., Duan, J., Nice, E. C., Lin, J., and Huang, C. (2021). Elesclomol induces copper-dependent ferroptosis in colorectal cancer cells via degradation of ATP7A. Mol. Oncol. 15 (12), 3527–3544. doi:10.1002/1878-0261.13079
Garber, K. (2015). Cancer's copper connections. Science. 349 (6244), 129. doi:10.1126/science.349.6244.129
Ge, E. J., Bush, A. I., Casini, A., Cobine, P. A., Cross, J. R., DeNicola, G. M., et al. (2022). Connecting copper and cancer: from transition metal signalling to metalloplasia. Nat. Rev. Cancer 22 (2), 102–113. doi:10.1038/s41568-021-00417-2
Gehrmann, M. (2006). Drug evaluation: STA-4783--enhancing taxane efficacy by induction of Hsp70. Curr. Opin. Investig. Drugs 7 (6), 574–580.
Gong, L., Pan, X., Yuan, Z. M., Peng, J., and Chen, J. (2016). p53 coordinates with Δ133p53 isoform to promote cell survival under low-level oxidative stress. J. Mol. Cell. Biol. 8 (1), 88–90. doi:10.1093/jmcb/mjv069
Gourdoupis, S., Nasta, V., Calderone, V., Ciofi-Baffoni, S., and Banci, L. (2018). IBA57 recruits ISCA2 to form a [2Fe-2S] cluster-mediated complex. J. Am. Chem. Soc. 140 (43), 14401–14412. doi:10.1021/jacs.8b09061
Gu, M., Cooper, J. M., Butler, P., Walker, A. P., Mistry, P. K., Dooley, J. S., et al. (2000). Oxidative-phosphorylation defects in liver of patients with Wilson's disease. Lancet 356 (9228), 469–474. doi:10.1016/s0140-6736(00)02556-3
Gu, Y., Wang, H., Xue, W., Zhu, L., Fu, C., Zhang, W., et al. (2025). Endoplasmic reticulum stress related super-enhancers suppress cuproptosis via glycolysis reprogramming in lung adenocarcinoma. Cell. Death Dis. 16 (1), 316. doi:10.1038/s41419-025-07613-0
Guan, D., Zhao, L., Shi, X., Ma, X., and Chen, Z. (2023). Copper in cancer: from pathogenesis to therapy. Biomed. Pharmacother. 163, 114791. doi:10.1016/j.biopha.2023.114791
Gunshin, H., Mackenzie, B., Berger, U. V., Gunshin, Y., Romero, M. F., Boron, W. F., et al. (1997). Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 388 (6641), 482–488. doi:10.1038/41343
Guo, B., Yang, F., Zhang, L., Zhao, Q., Wang, W., Yin, L., et al. (2023). Cuproptosis induced by ROS responsive nanoparticles with Elesclomol and copper combined with αPD-L1 for enhanced cancer immunotherapy. Adv. Mater 35 (22), e2212267. doi:10.1002/adma.202212267
Guo, Z., Chen, D., Yao, L., Sun, Y., Li, D., Le, J., et al. (2025). The molecular mechanism and therapeutic landscape of copper and cuproptosis in cancer. Signal Transduct. Target Ther. 10 (1), 149. doi:10.1038/s41392-025-02192-0
Guowei, L., Xiufang, L., Qianqian, X., and Yanping, J. (2023). The FDX1 methylation regulatory mechanism in the malignant phenotype of glioma. Genomics 115 (2), 110601. doi:10.1016/j.ygeno.2023.110601
Gupta, S. K., Shukla, V. K., Vaidya, M. P., Roy, S. K., and Gupta, S. (1993). Serum and tissue trace elements in colorectal cancer. J. Surg. Oncol. 52 (3), 172–175. doi:10.1002/jso.2930520311
Gupte, A., and Mumper, R. J. (2009). Elevated copper and oxidative stress in cancer cells as a target for cancer treatment. Cancer Treat. Rev. 35 (1), 32–46. doi:10.1016/j.ctrv.2008.07.004
Guthrie, L. M., Soma, S., Yuan, S., Silva, A., Zulkifli, M., Snavely, T. C., et al. (2020). Elesclomol alleviates Menkes pathology and mortality by escorting Cu to cuproenzymes in mice. Science 368 (6491), 620–625. doi:10.1126/science.aaz8899
Harvey, L. J., and McArdle, H. J. (2008). Biomarkers of copper status: a brief update. Br. J. Nutr. 99 (Suppl. 3), S10–S13. doi:10.1017/S0007114508006806
Hasinoff, B. B., Yadav, A. A., Patel, D., and Wu, X. (2014). The cytotoxicity of the anticancer drug elesclomol is due to oxidative stress indirectly mediated through its complex with Cu(II). J. Inorg. Biochem. 137, 22–30. doi:10.1016/j.jinorgbio.2014.04.004
Helsel, M. E., and Franz, K. J. (2015). Pharmacological activity of metal binding agents that alter copper bioavailability. Dalton Trans. 44 (19), 8760–8770. doi:10.1039/c5dt00634a
Herzig, S., and Shaw, R. J. (2018). AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell. Biol. 19 (2), 121–135. doi:10.1038/nrm.2017.95
Huang, Z., Li, N., Gao, Z., Chen, J., Xue, Z., Wu, C., et al. (2025). Cuproptosis improves CAR-T cell therapy in osteosarcoma. Cell. Biomater., 100148. doi:10.1016/j.celbio.2025.100148
Hunsaker, E. W., and Franz, K. J. (2019). Emerging opportunities to manipulate metal trafficking for therapeutic benefit. Inorg. Chem. 58 (20), 13528–13545. doi:10.1021/acs.inorgchem.9b01029
Ishida, S., Andreux, P., Poitry-Yamate, C., Auwerx, J., and Hanahan, D. (2013). Bioavailable copper modulates oxidative phosphorylation and growth of tumors. Proc. Natl. Acad. Sci. U. S. A. 110 (48), 19507–19512. doi:10.1073/pnas.1318431110
Iwai, Y., Ishida, M., Tanaka, Y., Okazaki, T., Honjo, T., and Minato, N. (2002). Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. U. S. A. 99 (19), 12293–12297. doi:10.1073/pnas.192461099
Jiang, C., Liu, R., and Li, Z. (2025). A phosphatidylinositol 3-Kinase gamma inhibitor enhances anti-programmed Death-1/Programmed death Ligand-1 antitumor effects by remodeling the tumor immune microenvironment of ovarian cancer. MedComm 6 (8), e70223. doi:10.1002/mco2.70223
Jiang, J., Chen, G., Zhang, W., Qin, S., Li, M., Zhong, S., et al. (2025). Pseudonatural flavonols as novel copper ionophores for NAFLD intervention via synergistic copper delivery and flavonoid activity. J. Med. Chem. 68 (6), 6450–6461. doi:10.1021/acs.jmedchem.4c02927
Jin, Y., Zhang, C., Xu, H., Xue, S., Wang, Y., Hou, Y., et al. (2011). Combined effects of serum trace metals and polymorphisms of CYP1A1 or GSTM1 on non-small cell lung cancer: a hospital based case-control study in China. Cancer Epidemiol. 35 (2), 182–187. doi:10.1016/j.canep.2010.06.004
Jones, R. G., Plas, D. R., Kubek, S., Buzzai, M., Mu, J., Xu, Y., et al. (2005). AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell. 18 (3), 283–293. doi:10.1016/j.molcel.2005.03.027
Kahlson, M. A., and Dixon, S. J. (2022). Copper-induced cell death. Science 375 (6586), 1231–1232. doi:10.1126/science.abo3959
Kawauchi, K., Araki, K., Tobiume, K., and Tanaka, N. (2008). p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat. Cell. Biol. 10 (5), 611–618. doi:10.1038/ncb1724
Kciuk, M., Kołat, D., Kałuzińska-Kołat, Ż., Gawrysiak, M., Drozda, R., Celik, I., et al. (2023). Doxorubicin-An agent with multiple mechanisms of anticancer activity. Cells 12 (4), 659. doi:10.3390/cells12040659
Kim, B. E., Nevitt, T., and Thiele, D. J. (2008). Mechanisms for copper acquisition, distribution and regulation. Nat. Chem. Biol. 4 (3), 176–185. doi:10.1038/nchembio.72
Kirshner, J. R., He, S., Balasubramanyam, V., Kepros, J., Yang, C. Y., Zhang, M., et al. (2008). Elesclomol induces cancer cell apoptosis through oxidative stress. Mol. Cancer Ther. 7 (8), 2319–2327. doi:10.1158/1535-7163.MCT-08-0298
Kodama, H., Fujisawa, C., and Bhadhprasit, W. (2012). Inherited copper transport disorders: biochemical mechanisms, diagnosis, and treatment. Curr. Drug Metab. 13 (3), 237–250. doi:10.2174/138920012799320455
Kudo, M. (2020). Limited impact of Anti-PD-1/PD-L1 monotherapy for hepatocellular carcinoma. Liver Cancer 9 (6), 629–639. doi:10.1159/000512170
Kumar, N., Butz, J. A., and Burritt, M. F. (2007). Clinical significance of the laboratory determination of low serum copper in adults. Clin. Chem. Lab. Med. 45 (10), 1402–1410. doi:10.1515/CCLM.2007.292
Kuo, H. W., Chen, S. F., Wu, C. C., Chen, D. R., and Lee, J. H. (2002). Serum and tissue trace elements in patients with breast cancer in Taiwan. Biol. Trace Elem. Res. 89 (1), 1–11. doi:10.1385/BTER:89:1:1
Lai, S., Chen, Y., Yang, F., Xiao, W., Liu, Y., and Wang, C. (2022). Quantitative site-specific chemoproteomic profiling of protein lipoylation. J. Am. Chem. Soc. 144 (23), 10320–10329. doi:10.1021/jacs.2c01528
Lee, J., Peña, M. M., Nose, Y., and Thiele, D. J. (2002). Biochemical characterization of the human copper transporter Ctr1. J. Biol. Chem. 277 (6), 4380–4387. doi:10.1074/jbc.M104728200
Lener, M. R., Scott, R. J., Wiechowska-Kozłowska, A., Serrano-Fernández, P., Baszuk, P., Jaworska-Bieniek, K., et al. (2016). Serum concentrations of selenium and copper in patients diagnosed with pancreatic cancer. Cancer Res. Treat. 48 (3), 1056–1064. doi:10.4143/crt.2015.282
Li, Z. H., Lu, X., Li, S. W., Chen, J. T., and Jia, J. (2019). Expression of ATP7A in esophageal squamous cell carcinoma (ESCC) and its clinical significance. Int. J. Clin. Exp. Pathol. 12 (9), 3521–3525.
Li, Y., Ma, J., Wang, R., Luo, Y., Zheng, S., and Wang, X. (2024). Zinc transporter 1 functions in copper uptake and cuproptosis. Cell. Metab. 36 (9), 2118–29.e6. doi:10.1016/j.cmet.2024.07.009
Lill, R., and Freibert, S. A. (2020). Mechanisms of mitochondrial iron-sulfur protein biogenesis. Annu. Rev. Biochem. 89, 471–499. doi:10.1146/annurev-biochem-013118-111540
Lill, R. (2009). Function and biogenesis of iron-sulphur proteins. Nature 460 (7257), 831–838. doi:10.1038/nature08301
Lin, C. H., Chin, Y., Zhou, M., Sobol, R. W., Hung, M. C., and Tan, M. (2024). Protein lipoylation: Mitochondria, cuproptosis, and beyond. Trends Biochem. Sci. 49 (8), 729–744. doi:10.1016/j.tibs.2024.04.002
Lippard, S. J., and Berg, J. M. (1994). Principles of bioinorganic chemistry. University Science Books.
Lippard, S. J. (1999). Free copper ions in the cell? Science 284 (5415), 748–749. doi:10.1126/science.284.5415.748
Liu, Y., He, Y., Jin, A., Tikunov, A. P., Zhou, L., Tollini, L. A., et al. (2014). Ribosomal protein-Mdm2-p53 pathway coordinates nutrient stress with lipid metabolism by regulating MCD and promoting fatty acid oxidation. Proc. Natl. Acad. Sci. U. S. A. 111 (23), E2414–E2422. doi:10.1073/pnas.1315605111
Liu, Y., Su, Z., Tavana, O., and Gu, W. (2024). Understanding the complexity of p53 in a new era of tumor suppression. Cancer Cell. 42 (6), 946–967. doi:10.1016/j.ccell.2024.04.009
Liu, G., Ren, H., Li, J., Dong, W., Li, S., Zhang, N., et al. (2025). Covalent organic framework eliciting a click-based intracellular copper accumulation cascade for cuproptosis cancer therapy. Cell. Biomater. 1 (7), 100126. doi:10.1016/j.celbio.2025.100126
Lopez, J., Ramchandani, D., and Vahdat, L. (2019). Copper depletion as a therapeutic strategy in cancer. Met. Ions Life Sci. 19. doi:10.1515/9783110527872-018
Lossow, K., Schwarz, M., and Kipp, A. P. (2021). Are trace element concentrations suitable biomarkers for the diagnosis of cancer? Redox Biol. 42, 101900. doi:10.1016/j.redox.2021.101900
Lucena-Valera, A., Ruz-Zafra, P., and Ampuero, J. (2023). Wilson's disease: overview. Med. Clin. Barc. 160 (6), 261–267. doi:10.1016/j.medcli.2022.12.016
Lutsenko, S., Barnes, N. L., Bartee, M. Y., and Dmitriev, O. Y. (2007). Function and regulation of human copper-transporting ATPases. Physiol. Rev. 87 (3), 1011–1046. doi:10.1152/physrev.00004.2006
Lutsenko, S. (2010). Human copper homeostasis: a network of interconnected pathways. Curr. Opin. Chem. Biol. 14 (2), 211–217. doi:10.1016/j.cbpa.2010.01.003
Macomber, L., and Imlay, J. A. (2009). The iron-sulfur clusters of dehydratases are primary intracellular targets of copper toxicity. Proc. Natl. Acad. Sci. U. S. A. 106 (20), 8344–8349. doi:10.1073/pnas.0812808106
Maio, N., and Rouault, T. A. (2020). Outlining the complex pathway of Mammalian Fe-S cluster biogenesis. Trends Biochem. Sci. 45 (5), 411–426. doi:10.1016/j.tibs.2020.02.001
Mantovani, F., Collavin, L., and Del Sal, G. (2019). Mutant p53 as a guardian of the cancer cell. Cell. Death Differ. 26 (2), 199–212. doi:10.1038/s41418-018-0246-9
Matoba, S., Kang, J. G., Patino, W. D., Wragg, A., Boehm, M., Gavrilova, O., et al. (2006). p53 regulates mitochondrial respiration. Science 312 (5780), 1650–1653. doi:10.1126/science.1126863
Matsui, M. S., Petris, M. J., Niki, Y., Karaman-Jurukovska, N., Muizzuddin, N., Ichihashi, M., et al. (2015). Omeprazole, a gastric proton pump inhibitor, inhibits melanogenesis by blocking ATP7A trafficking. J. Investig. Dermatol. 135 (3), 834–841. doi:10.1038/jid.2014.461
McCaughern, J. H. (2019). Optimising the health and performance of dairy cows through improved copper nutrition (Doctoral dissertation). Newport, Shropshire: Harper Adams University Repository.
Mo, Z., Liu, D., Rong, D., and Zhang, S. (2021). Hypoxic characteristic in the immunosuppressive microenvironment of hepatocellular carcinoma. Front. Immunol. 12, 611058. doi:10.3389/fimmu.2021.611058
Moon, S. H., Huang, C. H., Houlihan, S. L., Regunath, K., Freed-Pastor, W. A., Morris, J. P., et al. (2019). p53 represses the mevalonate pathway to mediate tumor suppression. Cell. 176 (3), 564–80.e19. doi:10.1016/j.cell.2018.11.011
Mukha, D., Fokra, M., Feldman, A., Sarvin, B., Sarvin, N., Nevo-Dinur, K., et al. (2022). Glycine decarboxylase maintains mitochondrial protein lipoylation to support tumor growth. Cell. Metab. 34 (5), 775–82.e9. doi:10.1016/j.cmet.2022.04.006
Nagai, M., Vo, N. H., Shin Ogawa, L., Chimmanamada, D., Inoue, T., Chu, J., et al. (2012). The oncology drug elesclomol selectively transports copper to the mitochondria to induce oxidative stress in cancer cells. Free Radic. Biol. Med. 52 (10), 2142–2150. doi:10.1016/j.freeradbiomed.2012.03.017
Nevitt, T., Ohrvik, H., and Thiele, D. J. (2012). Charting the travels of copper in eukaryotes from yeast to mammals. Biochim. Biophys. Acta 1823 (9), 1580–1593. doi:10.1016/j.bbamcr.2012.02.011
Ni, M., Solmonson, A., Pan, C., Yang, C., Li, D., Notzon, A., et al. (2019). Functional assessment of Lipoyltransferase-1 deficiency in cells, mice, and humans. Cell. Rep. 27 (5), 1376–86.e6. doi:10.1016/j.celrep.2019.04.005
Nose, Y., Rees, E. M., and Thiele, D. J. (2006). Structure of the Ctr1 copper trans'PORE'ter reveals novel architecture. Trends Biochem. Sci. 31 (11), 604–607. doi:10.1016/j.tibs.2006.09.003
Oliveri, V. (2020). Biomedical applications of copper ionophores. Coord. Chem. Rev. 422, 213474. doi:10.1016/j.ccr.2020.213474
Pan, Q., Kleer, C. G., van Golen, K. L., Irani, J., Bottema, K. M., Bias, C., et al. (2002). Copper deficiency induced by tetrathiomolybdate suppresses tumor growth and angiogenesis. Cancer Res. 62 (17), 4854–4859.
Pan, J., Meyers, R. M., Michel, B. C., Mashtalir, N., Sizemore, A. E., Wells, J. N., et al. (2018). Interrogation of Mammalian protein complex structure, function, and membership using genome-scale fitness screens. Cell. Syst. 6 (5), 555–68.e7. doi:10.1016/j.cels.2018.04.011
Patteson, J. B., Putz, A. T., Tao, L., Simke, W. C., Bryant, L. H., Britt, R. D., et al. (2021). Biosynthesis of fluopsin C, a copper-containing antibiotic from Pseudomonas aeruginosa. Science 374 (6570), 1005–1009. doi:10.1126/science.abj6749
Polishchuk, E. V., Concilli, M., Iacobacci, S., Chesi, G., Pastore, N., Piccolo, P., et al. (2014). Wilson disease protein ATP7B utilizes lysosomal exocytosis to maintain copper homeostasis. Dev. Cell. 29 (6), 686–700. doi:10.1016/j.devcel.2014.04.033
Qu, Y., Wang, J., Sim, M. S., Liu, B., Giuliano, A., Barsoum, J., et al. (2010). Elesclomol, counteracted by Akt survival signaling, enhances the apoptotic effect of chemotherapy drugs in breast cancer cells. Breast Cancer Res. Treat. 121 (2), 311–321. doi:10.1007/s10549-009-0470-6
Que, E. L., Domaille, D. W., and Chang, C. J. (2008). Metals in neurobiology: probing their chemistry and biology with molecular imaging. Chem. Rev. 108 (5), 1517–1549. doi:10.1021/cr078203u
Rae, T. D., Schmidt, P. J., Pufahl, R. A., Culotta, V. C., and O'Halloran, T. V. (1999). Undetectable intracellular free copper: the requirement of a copper chaperone for superoxide dismutase. Science 284 (5415), 805–808. doi:10.1126/science.284.5415.805
Raffa, N., Won, T. H., Sukowaty, A., Candor, K., Cui, C., Halder, S., et al. (2021). Dual-purpose isocyanides produced by Aspergillus fumigatus contribute to cellular copper sufficiency and exhibit antimicrobial activity. Proc. Natl. Acad. Sci. U. S. A. 118 (8), e2015224118. doi:10.1073/pnas.2015224118
Ramchandani, D., Berisa, M., Tavarez, D. A., Li, Z., Miele, M., Bai, Y., et al. (2021). Copper depletion modulates mitochondrial oxidative phosphorylation to impair triple negative breast cancer metastasis. Nat. Commun. 12 (1), 7311. doi:10.1038/s41467-021-27559-z
Resh, M. D. (2016). Fatty acylation of proteins: the long and the short of it. Prog. Lipid Res. 63, 120–131. doi:10.1016/j.plipres.2016.05.002
Roberts, E. A., Robinson, B. H., and Yang, S. (2008). Mitochondrial structure and function in the untreated Jackson toxic milk (tx-j) mouse, a model for Wilson disease. Mol. Genet. Metab. 93 (1), 54–65. doi:10.1016/j.ymgme.2007.08.127
Roberts, R. A., Laskin, D. L., Smith, C. V., Robertson, F. M., Allen, E. M., Doorn, J. A., et al. (2009). Nitrative and oxidative stress in toxicology and disease. Toxicol. Sci. 112 (1), 4–16. doi:10.1093/toxsci/kfp179
Roelofsen, H., Wolters, H., Van Luyn, M. J., Miura, N., Kuipers, F., and Vonk, R. J. (2000). Copper-induced apical trafficking of ATP7B in polarized hepatoma cells provides a mechanism for biliary copper excretion. Gastroenterology 119 (3), 782–793. doi:10.1053/gast.2000.17834
Rowland, E. A., Snowden, C. K., and Cristea, I. M. (2018). Protein lipoylation: an evolutionarily conserved metabolic regulator of health and disease. Curr. Opin. Chem. Biol. 42, 76–85. doi:10.1016/j.cbpa.2017.11.003
Ruiz, L. M., Libedinsky, A., and Elorza, A. A. (2021). Role of copper on mitochondrial function and metabolism. Front. Mol. Biosci. 8, 711227. doi:10.3389/fmolb.2021.711227
Saporito-Magriñá, C. M., Musacco-Sebio, R. N., Andrieux, G., Kook, L., Orrego, M. T., Tuttolomondo, M. V., et al. (2018). Copper-induced cell death and the protective role of glutathione: the implication of impaired protein folding rather than oxidative stress. Metallomics. 10 (12), 1743–1754. doi:10.1039/c8mt00182k
Saleem, A., Adhihetty, P. J., and Hood, D. A. (2009). Role of p53 in mitochondrial biogenesis and apoptosis in skeletal muscle. Physiol. Genomics 37 (1), 58–66. doi:10.1152/physiolgenomics.90346.2008
Sanford, J. D., Jin, A., Grois, G. A., and Zhang, Y. (2023). A role of cytoplasmic p53 in the regulation of metabolism shown by bat-mimicking p53 NLS mutant mice. Cell. Rep. 42 (1), 111920. doi:10.1016/j.celrep.2022.111920
Scheiber, I., Dringen, R., and Mercer, J. F. (2013). Copper: effects of deficiency and overload. Met. Ions Life Sci. 13, 359–387. doi:10.1007/978-94-007-7500-8_11
Schwartzenberg-Bar-Yoseph, F., Armoni, M., and Karnieli, E. (2004). The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 64 (7), 2627–2633. doi:10.1158/0008-5472.can-03-0846
Seidel, S. A., Dijkman, P. M., Lea, W. A., van den Bogaart, G., Jerabek-Willemsen, M., Lazic, A., et al. (2013). Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions. Methods. 59 (3), 301–315. doi:10.1016/j.ymeth.2012.12.005
Semenza, G. L. (2010). HIF-1: upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 20 (1), 51–56. doi:10.1016/j.gde.2009.10.009
Shanbhag, V., Jasmer-McDonald, K., Zhu, S., Martin, A. L., Gudekar, N., Khan, A., et al. (2019). ATP7A delivers copper to the lysyl oxidase family of enzymes and promotes tumorigenesis and metastasis. Proc. Natl. Acad. Sci. U. S. A. 116 (14), 6836–6841. doi:10.1073/pnas.1817473116
Sheftel, A. D., Stehling, O., Pierik, A. J., Elsässer, H. P., Mühlenhoff, U., Webert, H., et al. (2010). Humans possess two mitochondrial ferredoxins, Fdx1 and Fdx2, with distinct roles in steroidogenesis, heme, and Fe/S cluster biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 107 (26), 11775–11780. doi:10.1073/pnas.1004250107
Simanshu, D. K., Nissley, D. V., and McCormick, F. (2017). RAS proteins and their regulators in human disease. Cell. 170 (1), 17–33. doi:10.1016/j.cell.2017.06.009
Simon-Molas, H., Vallvé-Martínez, X., Caldera-Quevedo, I., Fontova, P., Arnedo-Pac, C., Vidal-Alabró, A., et al. (2021). TP53-Induced Glycolysis and Apoptosis Regulator (TIGAR) is upregulated in lymphocytes stimulated with Concanavalin A. Int. J. Mol. Sci. 22 (14), 7436. doi:10.3390/ijms22147436
Sokol, R. J., Twedt, D., McKim, J. M., Devereaux, M. W., Karrer, F. M., Kam, I., et al. (1994). Oxidant injury to hepatic mitochondria in patients with Wilson's disease and Bedlington terriers with copper toxicosis. Gastroenterology 107 (6), 1788–1798. doi:10.1016/0016-5085(94)90822-2
Solmonson, A., and DeBerardinis, R. J. (2018). Lipoic acid metabolism and mitochondrial redox regulation. J. Biol. Chem. 293 (20), 7522–7530. doi:10.1074/jbc.TM117.000259
Solomon, E. I., Sundaram, U. M., and Machonkin, T. E. (1996). Multicopper oxidases and oxygenases. Chem. Rev. 96 (7), 2563–2606. doi:10.1021/cr950046o
Stambolsky, P., Weisz, L., Shats, I., Klein, Y., Goldfinger, N., Oren, M., et al. (2006). Regulation of AIF expression by p53. Cell. Death Differ. 13 (12), 2140–2149. doi:10.1038/sj.cdd.4401965
Su, Y., Zhang, X., Li, S., Xie, W., and Guo, J. (2022). Emerging roles of the Copper-CTR1 axis in tumorigenesis. Mol. Cancer Res. 20 (9), 1339–1353. doi:10.1158/1541-7786.MCR-22-0056
Suwara, J., and Hartman, M. L. (2025). Balancing between cuproplasia and copper-dependent cell death: molecular basis and clinical implications of ATOX1 in cancer. J. Exp. Clin. Cancer Res. 44 (1), 222. doi:10.1186/s13046-025-03486-5
Tan, G., Cheng, Z., Pang, Y., Landry, A. P., Li, J., Lu, J., et al. (2014). Copper binding in IscA inhibits iron-sulphur cluster assembly in Escherichia coli. Mol. Microbiol. 93 (4), 629–644. doi:10.1111/mmi.12676
Tang, D., Kang, R., Berghe, T. V., Vandenabeele, P., and Kroemer, G. (2019). The molecular machinery of regulated cell death. Cell. Res. 29 (5), 347–364. doi:10.1038/s41422-019-0164-5
Tang, X., Yan, Z., Miao, Y., Ha, W., Li, Z., Yang, L., et al. (2023). Copper in cancer: from limiting nutrient to therapeutic target. Front. Oncol. 13, 1209156. doi:10.3389/fonc.2023.1209156
Tian, Z., Jiang, S., Zhou, J., and Zhang, W. (2023). Copper homeostasis and cuproptosis in mitochondria. Life Sci. 334, 122223. doi:10.1016/j.lfs.2023.122223
Trachootham, D., Alexandre, J., and Huang, P. (2009). Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat. Rev. Drug Discov. 8 (7), 579–591. doi:10.1038/nrd2803
Tsvetkov, P., Coy, S., Petrova, B., Dreishpoon, M., Verma, A., Abdusamad, M., et al. (2022). Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 375 (6586), 1254–1261. doi:10.1126/science.abf0529
Tsymbal, S., Refeld, A., Zatsepin, V., and Kuchur, O. (2023). The p53 protein is a suppressor of Atox1 copper chaperon in tumor cells under genotoxic effects. PLoS One 18 (12), e0295944. doi:10.1371/journal.pone.0295944
Wang, Y., Zhu, S., Hodgkinson, V., Prohaska, J. R., Weisman, G. A., Gitlin, J. D., et al. (2012). Maternofetal and neonatal copper requirements revealed by enterocyte-specific deletion of the Menkes disease protein. Am. J. Physiol. Gastrointest. Liver Physiol. 303 (11), G1236–G1244. doi:10.1152/ajpgi.00339.2012
Wang, L., Xiong, H., Wu, F., Zhang, Y., Wang, J., Zhao, L., et al. (2014). Hexokinase 2-mediated Warburg effect is required for PTEN- and p53-deficiency-driven prostate cancer growth. Cell. Rep. 8 (5), 1461–1474. doi:10.1016/j.celrep.2014.07.053
Wang, T., Yu, H., Hughes, N. W., Liu, B., Kendirli, A., Klein, K., et al. (2017). Gene essentiality profiling reveals gene networks and synthetic lethal interactions with oncogenic ras. Cell. 168 (5), 890–903.e15. doi:10.1016/j.cell.2017.01.013
Wang, L., Yu, Y., Wei, D., Zhang, L., Zhang, X., Zhang, G., et al. (2021). A systematic strategy of combinational blow for overcoming Cascade drug resistance via NIR-Light-Triggered hyperthermia. Adv. Mater 33 (20), e2100599. doi:10.1002/adma.202100599
Wang, X., Zhou, M., Liu, Y., and Si, Z. (2023). Cope with copper: from copper linked mechanisms to copper-based clinical cancer therapies. Cancer Lett. 561, 216157. doi:10.1016/j.canlet.2023.216157
Wang, J., Li, J., Liu, J., Chan, K. Y., Lee, H. S., Lin, K. N., et al. (2024). Interplay of ferroptosis and cuproptosis in cancer: dissecting metal-driven mechanisms for therapeutic potentials. Cancers (Basel) 16 (3), 512. doi:10.3390/cancers16030512
Wang, Y., Yao, X., Lu, Y., Ruan, J., Yang, Z., Wang, C., et al. (2025). A PROTAC-Based cuproptosis sensitizer in lung cancer therapy. Adv. Mat. 37 (34), e2501435. doi:10.1002/adma.202501435
Wangpaichitr, M., Wu, C., You, M., Maher, J. C., Dinh, V., Feun, L. G., et al. (2009). N',N'-Dimethyl-N',N'-bis(phenylcarbonothioyl) propanedihydrazide (Elesclomol) selectively kills Cisplatin resistant lung cancer cells through Reactive Oxygen Species (ROS). Cancers (Basel) 1 (1), 23–38. doi:10.3390/cancers1010023
Weiler, B. D., Brück, M. C., Kothe, I., Bill, E., Lill, R., and Mühlenhoff, U. (2020). Mitochondrial [4Fe-4S] protein assembly involves reductive [2Fe-2S] cluster fusion on ISCA1-ISCA2 by electron flow from ferredoxin FDX2. Proc. Natl. Acad. Sci. U. S. A. 117 (34), 20555–20565. doi:10.1073/pnas.2003982117
Weinlich, R., Oberst, A., Beere, H. M., and Green, D. R. (2017). Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell. Biol. 18 (2), 127–136. doi:10.1038/nrm.2016.149
Wu, M., Huang, Q., Xie, Y., Wu, X., Ma, H., Zhang, Y., et al. (2022). Improvement of the anticancer efficacy of PD-1/PD-L1 blockade via combination therapy and PD-L1 regulation. J. Hematol. Oncol. 15 (1), 24. doi:10.1186/s13045-022-01242-2
Xing, J., Qiao, G., Luo, X., Liu, S., Chen, S., Ye, G., et al. (2023). Ferredoxin 1 regulates granulosa cell apoptosis and autophagy in polycystic ovary syndrome. Clin. Sci. (Lond). 137 (6), 453–468. doi:10.1042/CS20220408
Xiong, C., Ling, H., Hao, Q., and Zhou, X. (2023). Cuproptosis: p53-regulated metabolic cell death? Cell. Death Differ. 30 (4), 876–884. doi:10.1038/s41418-023-01125-0
Yadav, A. A., Patel, D., Wu, X., and Hasinoff, B. B. (2013). Molecular mechanisms of the biological activity of the anticancer drug elesclomol and its complexes with Cu(II), Ni(II) and Pt(II). J. Inorg. Biochem. 126, 1–6. doi:10.1016/j.jinorgbio.2013.04.013
Yang, M., Li, J., Gu, P., and Fan, X. (2021). The application of nanoparticles in cancer immunotherapy: targeting tumor microenvironment. Bioact. Mater 6 (7), 1973–1987. doi:10.1016/j.bioactmat.2020.12.010
Yang, Y., Li, M., Chen, G., Liu, S., Guo, H., Dong, X., et al. (2023). Dissecting copper biology and cancer treatment: ‘activating cuproptosis or suppressing Cuproplasia. Coord. Chem. Rev. 495, 215395. doi:10.1016/j.ccr.2023.215395
Yang, Z., Su, W., Wei, X., Pan, Y., Xing, M., Niu, L., et al. (2025). Hypoxia inducible factor-1α drives cancer resistance to cuproptosis. Cancer Cell. 43 (5), 937–54.e9. doi:10.1016/j.ccell.2025.02.015
Zhang, C., Lin, M., Wu, R., Wang, X., Yang, B., Levine, A. J., et al. (2011). Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc. Natl. Acad. Sci. U. S. A. 108 (39), 16259–16264. doi:10.1073/pnas.1113884108
Zhang, C., Liu, J., Xu, D., Zhang, T., Hu, W., and Feng, Z. (2020). Gain-of-function mutant p53 in cancer progression and therapy. J. Mol. Cell. Biol. 12 (9), 674–687. doi:10.1093/jmcb/mjaa040
Zhang, Z., Ma, Y., Guo, X., Du, Y., Zhu, Q., Wang, X., et al. (2021). FDX1 can impact the prognosis and mediate the metabolism of lung adenocarcinoma. Front. Pharmacol. 12, 749134. doi:10.3389/fphar.2021.749134
Zhang, L., Wang, Y., Karges, J., Tang, D., Zhang, H., Zou, K., et al. (2023). Tetrahedral DNA nanostructure with Interferon stimulatory DNA delivers highly potent toxins and activates the cGAS-STING pathway for robust chemotherapy and immunotherapy. Adv. Mater 35 (8), e2210267. doi:10.1002/adma.202210267
Zhang, R., Tan, Y., Xu, K., Huang, N., Wang, J., Liu, M., et al. (2025). Cuproplasia and cuproptosis in hepatocellular carcinoma: mechanisms, relationship and potential role in tumor microenvironment and treatment. Cancer Cell. Int. 25 (1), 137. doi:10.1186/s12935-025-03683-4
Zhao, H. J., and Zhao, X. H. (2019). Modulatory effect of the supplemented copper ion on in vitro activity of bovine Lactoferrin to Murine Splenocytes and RAW264.7 macrophages. Biol. Trace Elem. Res. 189 (2), 519–528. doi:10.1007/s12011-018-1472-1
Zheng, P., Zhou, C., Lu, L., Liu, B., and Ding, Y. (2022). Elesclomol: a copper ionophore targeting mitochondrial metabolism for cancer therapy. J. Exp. Clin. Cancer Res. 41 (1), 271. doi:10.1186/s13046-022-02485-0
Zhou, J., Yu, Q., Song, J., Li, S., Li, X. L., Kang, B. K., et al. (2023). Photothermally triggered copper payload release for Cuproptosis-Promoted cancer synergistic therapy. Angew. Chem. Int. Ed. Engl. 62 (12), e202213922. doi:10.1002/anie.202213922
Zhou, S., Kachhap, S., and Singh, K. K. (2003). Mitochondrial impairment in p53-deficient human cancer cells. Mutagenesis 18 (3), 287–292. doi:10.1093/mutage/18.3.287
Zhou, Y., Niu, W., Luo, Y., Li, H., Xie, Y., Wang, H., et al. (2019). p53/Lactate dehydrogenase A axis negatively regulates aerobic glycolysis and tumor progression in breast cancer expressing wild-type p53. Cancer Sci. 110 (3), 939–949. doi:10.1111/cas.13928
Zhou, W. Q., Niu, Y. Y., Zhu, S. Y., and Yu, C. (2024). Research progress on association of copper homeostasis dysregulation with kidney fibrosis. J. Tongji Univ. (Med. Sci.) 45 (1), 1–7. doi:10.12289/j.issn.1008-0392.2024.0001
Zimnicka, A. M., Tang, H., Guo, Q., Kuhr, F. K., Oh, M. J., Wan, J., et al. (2014). Upregulated copper transporters in hypoxia-induced pulmonary hypertension. PLoS One 9 (3), e90544. doi:10.1371/journal.pone.0090544
Keywords: cuproptosis, copper-overload, tumor microenvironment, FDX1, protein lipoylation
Citation: Jiao Y, Wang H, Zhu M, Liu T, Li Y, Yang S, Zhang M and Zhang L (2025) Copper-induced cell death mechanisms and their role in the tumor microenvironment. Front. Cell Death 4:1628470. doi: 10.3389/fceld.2025.1628470
Received: 14 May 2025; Accepted: 09 October 2025;
Published: 20 October 2025.
Edited by:
Paola Maycotte, Instituto Mexicano del Seguro Social, MexicoReviewed by:
Walaa Keshk, Tanta University, EgyptVictor E. López-Guerrero, National Polytechnic Institute, Mexico
Copyright © 2025 Jiao, Wang, Zhu, Liu, Li, Yang, Zhang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Minghui Zhang, Y2Z6aGFuZ21pbmdodWlAMTYzLmNvbQ==; Lei Zhang, emhhbmdsZWlAaHJibXUuZWR1LmNu
†These authors have contributed equally to this work