 Malin Undin
Malin Undin Peter J. Lockhart
Peter J. Lockhart Simon F. K. Hills
Simon F. K. Hills Isabel Castro
Isabel Castro- 1Wildlife and Ecology Group, School of Agriculture and Environment, Massey University, Palmerston North, New Zealand
- 2Wildbase Research, Massey University, Palmerston North, New Zealand
- 3School of Fundamental Sciences, Massey University, Palmerston North, New Zealand
Long-term sustainable and resilient populations is a key goal of conservation. How to best achieve this is controversial. There are, for instance, polarized views concerning the fitness and conservation value of hybrid populations founded through multi-origin translocations. A classic example concerns Apteryx (kiwi) in New Zealand. The A. mantelli of Ponui Island constitute a hybrid population where the birds are highly successful in their island habitat. A key dilemma for managers is understanding the reason for this success. Are the hybrid birds of Ponui Island of “no future conservation value” as recently asserted, or do they represent an outstanding example of genetic rescue and an important resource for future translocations? There has been a paradigm shift in scientific thinking concerning hybrids, but the ecological significance of admixed genomes remains difficult to assess. This limits what we can currently predict in conservation science. New understanding from genome science challenges the sufficiency of population genetic models to inform decision making and suggests instead that the contrasting outcomes of hybridization, “outbreeding depression” and “heterosis,” require understanding additional factors that modulate gene and protein expression and how these factors are influenced by the environment. We discuss these findings and the investigations that might help us to better understand the birds of Ponui, inform conservation management of kiwi and provide insight relevant for the future survival of Apteryx.
Introduction
The arrival of humans to New Zealand and the accompanying deforestation and predation by introduced mammals have decimated many native and endemic species (Holdaway, 1989; Robertson et al., 2016). In responding to this catastrophic development, New Zealand conservation efforts have attracted international recognition for their bold and pioneering methods aimed at rescuing species from the brink of extinction. Most notably, strategies for translocation have influenced practices worldwide (Armstrong and McLean, 1995; Armstrong and Seddon, 2008). Despite this, controversy remains in New Zealand and elsewhere for how to best achieve long-term sustainable and resilient populations (Love Stowell et al., 2017; Ralls et al., 2018, 2020; Von Holdt et al., 2018). Specifically, the outcome of translocations involving the mixing of individuals from genetically distinct populations remains difficult to predict and questions have been raised about the future fate and conservation value of admixed populations originating from such historic translocations.
An iconic example concerns the flightless, nocturnal genus Apteryx, or kiwi birds, which once were common and widespread throughout New Zealand but now are confined mainly to isolated and/or small populations relying on active management to achieve net growth (Innes et al., 2015; Germano et al., 2018). Apteryx face many threats such as habitat degradation, fragmentation, disrupted gene flow, and small population sizes (McLennan and Potter, 1992; McLennan and McCann, 2002; Germano et al., 2018). However, depredation by invasive mammals, in particular stoats (Mustela erminea), ferrets (M. furo), and dogs (Canis lupus familiaris), have been identified as the main cause of their decline (Taborsky, 1988; McLennan et al., 1996; Innes et al., 2010; Germano et al., 2018). For example, in unmanaged populations, about 95% of Apteryx chicks are predated by stoats (McLennan et al., 1996; Innes et al., 2010). Consequently, Apteryx management focuses on trapping and poisoning mammalian predators and on artificial rearing of eggs and chicks collected from the wild under a program referred to as Operation Nest Egg (ONE; Colbourne et al., 2005). In addition, a program called “kohanga kiwi” involves sites where Apteryx pairs are held and allowed to breed in predator-free areas and a proportion of the chicks are “harvested” and used to supplement existing or establish new populations (Innes et al., 2016; Kiwis for Kiwi, 2016). A principle of kohanga kiwi sites is that they are founded by 40 unrelated birds. However, relatedness is inferred based on geographic origin rather than genetic testing (Innes et al., 2016; Kiwis for Kiwi, 2016).
Apteryx genetics have received extensive research attention (Ramstad and Dunning, 2020; Undin et al., in press). Based on findings from these studies, Apteryx is currently split into five extant species (Tennyson et al., 2003) and further into 14 management units based on observed or inferred barriers to gene flow (Powlesland, 1988; Burbidge et al., 2003; Weir et al., 2016; Germano et al., 2018). The exploration of Apteryx diversity has so far focused on describing observations from a taxonomic perspective. Hence, parameters based on population genetic models have not been estimated and there is little understanding of migration between populations, inbreeding within populations, and local adaptation (Shepherd and Lambert, 2008; Germano et al., 2018). Consequently, lack of studies of genetic variation within management units, the nature of the genetic difference between taxa, and the prevalence of in- and outbreeding, means that far-reaching Apteryx conservation decisions and policy are currently being implemented despite a lack of crucial information (McLennan and McCann, 2002; Dussex et al., 2018).
One policy linked to the demarcation of 14 management units is that Apteryx translocations are strictly limited to movement within units (Pierce et al., 2006; Innes et al., 2016; Germano et al., 2018). Another is that several Apteryx populations, regarded as hybrid populations because they originated from translocations involving two or more units, are considered unsuitable sources as well as targets for future translocations (Herbert and Daugherty, 2002; Colbourne, 2005; Shepherd et al., 2012; Germano et al., 2018). One such hybrid population is present on Ponui Island. Concerned for the rapid decline of kiwi on the North Island, in 1964 the landowner Peter Chamberlin on Ponui (Chamberlin's) Island in the Hauraki Gulf had 14 North Island brown kiwi (Apteryx mantelli) translocated to the island (Colbourne, 2005). Six of these birds came from Hauturu-o-Toi (also known as Little Barrier Island; Colbourne, 2005; Figure 1). The remaining eight birds came from the Waipoua Forest in the northernmost part of the North Island mainland (Colbourne, 2005). In turn, the origin of the population on Hauturu-o-Toi was a translocation from Taranaki in the early 1900's, but anecdotal evidence suggests that additional birds of unknown sources may have been present on Hauturu-o-Toi prior to this translocation (Baker et al., 1995; Burbidge et al., 2003; Colbourne, 2005; Figure 1). Because the parental populations belong to different management units, the Ponui birds are classified as hybrids. The initial success of the translocated birds and their Ponui hatched offspring is unknown, but the current high density is evidence for rapid and extensive population growth within a small number of generations. Apteryx typically reach sexual maturity around 4 years of age and live to be over 40 years old (Sales, 2005; Robertson and de Monchy, 2012). Consequently, in the 56 years that have passed since their introduction, the current population on Ponui is likely best described as a hybrid swarm where some individuals birds will be of pure Waipoua or Hauturu-o-Toi origin, others will be F1 hybrids or offspring from crossing and backcrossing between parental types and/or F1 individuals (Hwang et al., 2011; Hamilton and Miller, 2016). While Ponui Island lacks stoats, about one third of the A. mantelli chicks on the island are preyed upon by feral cats (Felis catus) every year and ship rats (Rattus rattus) likely compete with chicks for food (Shapiro, 2005; Dixon, 2015; Strang, 2018). Despite this presence of invasive mammals, the population density on Ponui is suggestive of a population growth rate otherwise only seen in populations in predator-free sanctuaries and/or sites where juvenile mortality is reduced through Operation Nest Egg (Colbourne et al., 2005; Robertson and de Monchy, 2012). The Ponui Apteryx population has been extensively studied over the past 17 years, providing ground-breaking data on habitat utilization, diet, parasite impact, disease, anatomy, social interactions, and causes of chick and egg mortality (e.g., Cunningham and Castro, 2011; Ziesemann, 2011; Hiscox, 2014; Wilson, 2014; Dixon, 2015; Reynolds et al., 2017; Abourachid et al., 2019; Bansal, 2020; Vieco-Galvez et al., 2020). No studies have so far investigated the impact of their mixed heritage on the success of the birds on Ponui.
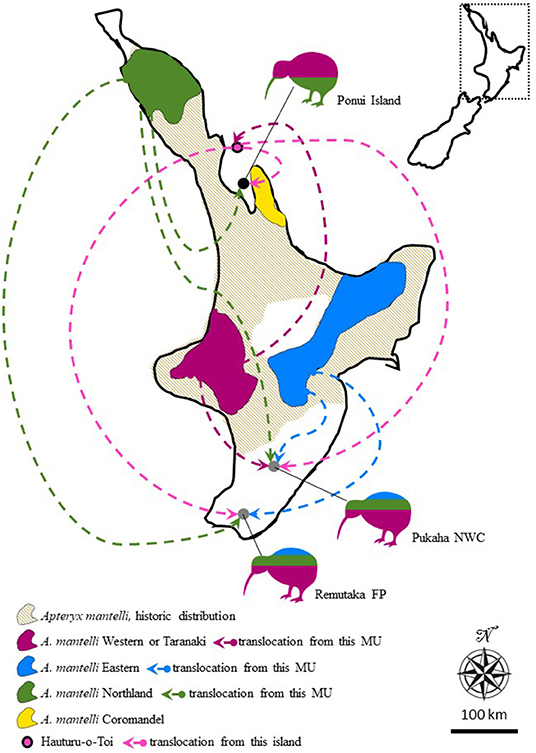
Figure 1. Map of the North Island of New Zealand illustrating the historic, as well as, current distribution of Apteryx mantelli, North Island brown kiwi, and how this species is further split into four geographically disjunct and genetically distinct management units (MU, or taxa Burbidge et al., 2003; Weir et al., 2016; Germano et al., 2018). Arrows and kiwi silhouettes represent three separate cases where multi-origin translocations over the last 100 years have resulted in hybrid populations. Silhouette coloring roughly illustrates the proportional representation of different taxa in the founding population at each site. Translocations from Hauturu-o-Toi (also known as Little Barrier Island) are colored in a different shade of pink for clarity and to highlight the somewhat disputed MU identity of this island. FP, Forest park; NWC, National Wildlife Centre. Inset show the location of the zoomed in map in New Zealand.
Despite the lack of studies, the mixed provenance and the untested assumption that the birds introduced to Ponui Island had limited genetic diversity, it was concluded that the Ponui birds “have no genetic value whatsoever for use in restoration” (Letter to Department of Conservation and the Kiwi Recovery Group shared with Ponui landowners and Ponui Kiwi Research Team, 2016). The suggestion is that the Ponui birds suffer from (1) inbreeding depression resulting from the small number of founding birds and (2) outbreeding depression resulting from anthropogenically-mediated hybridization of birds deemed likely to be adapted to different local conditions (Allendorf et al., 2001). These potential problems have not impacted policymaking around translocations and admixture of geographically adjacent populations. On the contrary, translocations of birds within management units to boost population numbers and attempt to maintain genetic diversity remains a paramount part of ongoing Apteryx conservation (Kiwis for Kiwi, 2016; Germano et al., 2018).
The plight of the Ponui kiwi illustrates the importance of improving our understanding of inbreeding, outbreeding depression, and the sometimes contrasting outcomes of hybridization. This understanding is also needed to realize the full potential of genetic rescue and other forms of translocation involving endangered species. In our contribution, we highlight why study of the Ponui birds will be informative for (1) illuminating the ecological and evolutionary significance of hybrids, (2) establishing evidence-based principles for identifying and matching source and target populations for translocations involving admixture, (3) proposing the most appropriate source(s) of individuals for repopulating areas after locally extinctions, and (4) evaluating the biological relevance of Apteryx management units in New Zealand. We emphasize that addressing these four questions is key for the long-term successful conservation of Apteryx whose populations are highly fragmented and are arguably in need of augmented gene flow given their insular New Zealand habitat. We think that if these questions could be answered for Apteryx in New Zealand, they may be informative for conservation practices elsewhere. We see an important role for integrating genome science in ecological studies of Apteryx that will help us to better understand the evolutionary significance of hybrids and their conservation value.
One reason discussion of hybridization in conservation can be problematic, and often fruitless, stems from a lack of agreement concerning the many criteria for delimiting species (Mallet, 2013). The prevalence of hybridization in plant evolution and the challenges of delimiting plant species has recently led to emphasis being placed on what occurs when taxa from distinct evolutionary lines are brought together, rather than on whether or not these taxa are named as distinct species (Winkworth et al., 2005; Abbott et al., 2013; Hoffmann et al., 2015; Canestrelli et al., 2016; Chan et al., 2019). We adopt this approach, and this review accepts the definition of hybridization used by Canestrelli et al. (2016) as “mating between individuals from genetically distinct populations that produces offspring.” We also adopt the perspective that it may not be helpful to distinguish hybridization that results from human intervention or otherwise since, for philosophical as well as practical reasons, the causes of some hybridization events are almost impossible to disentangle (Allendorf et al., 2001).
Population Genetic Models and Predicting Hybrid Success
Hybridization will impact genetic diversity and plasticity, however, predicting the success of managed gene flow requires understanding not only the genetic diversity and plasticity of species, but also of the extent and directionality of the gene flow and how admixture between genetically diverged populations and lineages affect reproductive success (Abbott et al., 2013). Models for the accumulative effects of particular alleles (gene variants) and overall allelic diversity in the genomes of organisms have been used since the early 1900's to help explain the relative fitness (reproductive success) of offspring when the parents are genetically similar (inbreeding) as well as genetically dissimilar (outbreeding or hybridization; Roff, 2002; Wright et al., 2008; Hochholdinger and Baldauf, 2018). The Partial Dominance hypothesis (Davenport, 1908) predicts that mating between close relatives increases the number of genes in offspring where the same sub-optimal (deleterious) variant of a gene is inherited from both parents. This is said to decrease the fitness of the offspring. The Overdominance hypothesis (East, 1908) proposes that it is not deleterious genes per se, but that an increased number of genes with the same variant inherited from both parents reduces the fitness of the offspring. These alternative explanations for inbreeding depression – reduced fitness of offspring relative to parents – are still largely unresolved (e.g., Roff, 2002; Wright et al., 2008). While there is good reason that population genetic models based on inheritance of gene variants remain a leading explanation for heterosis and genetic rescue, as well as inbreeding and outbreeding depression, after a 100 years of empirical testing, there is now a chorus of voices questioning the sufficiency of these models (Groszmann et al., 2013; O'Dea et al., 2016; Hochholdinger and Baldauf, 2018; Bell et al., 2019; Rey et al., 2020).
Heterosis or hybrid vigor is expressed as a release from inbreeding depression inferred from relatively higher fitness of offspring compared to its genetically distinct parents (Whitlock et al., 2000; Hochholdinger and Baldauf, 2018; Bell et al., 2019). The superior performance of first-generation (F1) hybrids is a well-documented phenomenon of ecological and agricultural advantage. It has evolutionary significance for adaptation of populations and generation of novel species (Johansen-Morris and Latta, 2006; Janes and Hamilton, 2017; Hochholdinger and Baldauf, 2018; Junaid et al., 2018; Koide et al., 2019; Miryeganeh and Saze, 2019; Taylor and Larson, 2019). When differences in phenotype and underlying gene expression are much greater or much less between progeny and parents than the additive difference between the parents, it is sometimes referred to as transgressive segregation (Stelkens et al., 2014; Koide et al., 2019). However, whether the mechanisms of heterosis and transgressive segregation are different, the same, or whether one is a subset of the other remains unclear. According to some definitions, the difference lies in that while transgressive segregation results in individuals that “express trait values that fall outside the range of both parent species” in either direction (Stelkens et al., 2014), heterosis only refers to an “increased fitness relative to more ‘pure-bred’ individuals” (Whitlock et al., 2000). Others suggest that the difference lies in having a population vs. an individual perspective, for instance, heterosis is “usually ascribed to the average fitness of the hybrid offspring” while transgressive segregation refers to “the presence of extreme phenotypes (in either a negative or a positive direction) relative to the parental phenotypes” that make particular individuals “more fit […] than either of the parents” (Johansen-Morris and Latta, 2006). Another perspective is that the difference concerns at what point in time the positive fitness following hybridization is observed. Heterosis is said to be evident in the “observation that cross-pollinated hybrids are more vigorous than their parents [….] calculated as the difference in the phenotypic performance of a trait between a hybrid and the average of its two distinct parents” (Hochholdinger and Baldauf, 2018) or the “elevated fitness of F1 hybrids relative to their parents” (Bell et al., 2019). In contrast transgressive segregation produces “hybrid progeny phenotypes that exceed the parental phenotypes […which are] heritably stable” (Koide et al., 2019).
Outbreeding depression, on the other hand, is the reduced fitness of admixed offspring compared to their genetically distinct parents (Marr et al., 2002; Goldberg et al., 2005; Frankham et al., 2011; Barmentlo et al., 2018). Population genetic models have also been used to explain this phenomenon (Lynch, 1991; Marr et al., 2002; Goldberg et al., 2005; Frankham et al., 2011; Whitlock et al., 2013; Kardos et al., 2016). Hybrid offspring will be heterozygous for many genes, and it is suggested this could cause an averaging or intermediate effect that makes the progeny adapted to neither parental habitat (Edmands and Timmerman, 2003). In subsequent generations of interbreeding it has been suggested that the depression could worsen, because particular combinations of gene variants which have been of adaptive value to parental lineages in their environments may no longer co-occur in the genomes of the hybrid offspring (Lynch, 1991; Allendorf et al., 2001; Edmands, 2007). In the worst cases, where parental species have evolved differences in their chromosome karyotypes (the packaged form of their DNA sequences) it is possible that the chromosomes will not pair properly during meiosis and this will affect the fertility of the hybrid offspring (e.g., the famous case of donkeys and horses producing sterile mules; Rieseberg, 2001).
A growing number of studies support the view that outbreeding depression is more likely to occur when genetic differences are linked with local adaptation to specific environments. In contrast, isolation per se even for many generations, under similar selection pressure rarely leads to symptoms of outbreeding depression upon admixture (Barton, 1996; Orr and Smith, 1998; Hendry et al., 2000; Rundle et al., 2000; Nosil et al., 2002; Rundle, 2003; Frankham et al., 2011). However, disruption of adapted phenotypes cannot explain some reported instances of outbreeding depression. For example, adaptive differences between parental linages fail to explain why several crosses between the same lineages can generate very different outcomes for offspring fitness and phenotypes (Johansen-Morris and Latta, 2006; Escobar et al., 2008; Barmentlo et al., 2018). The importance of local adaptation in Apteryx evolution remains untested. However, there are examples of both within and between species hybrids in kiwi that are vital, reproducing, and even very successful (Herbert and Daugherty, 2002; Cunningham and Castro, 2011; Shepherd et al., 2012). There are also cases of long-distance translocations of Apteryx that are reported to be successful (Colbourne, 2005; Robertson et al., 2019). While more rigorous investigations need to be conducted, these observations may suggest a limited role of local adaptation in the disjunct management units.
Despite considerable effort, finding the so-called “sweet spot” of genetic and/or phenotypic distance between taxa that will produce heterosis and not outbreeding depression has not been possible, raising the question of whether the concept of a sweet spot is useful at all (Tallmon et al., 2004; Edmands, 2007; Escobar et al., 2008; Stelkens et al., 2014; Kardos et al., 2016; Barmentlo et al., 2018; Bell et al., 2019; Koide et al., 2019). Further complicating the picture, several studies have found that crossing of different lineages within the same species has sometimes resulted in outbreeding depression and sometimes in heterosis (Edmands, 1999; Rundle et al., 2000; Marr et al., 2002; Escobar et al., 2008; Whitlock et al., 2013). Occasionally, these outcomes have differed depending on which lineage was maternal and which was paternal (Escobar et al., 2008; Barmentlo et al., 2018). While at other times, hybridization has produced some traits with both negative and positive consequences for fitness (Johansen-Morris and Latta, 2006; Escobar et al., 2008). Similarly, Whitlock et al. (2013), have also reported that the frequency and magnitude of outbreeding depression have differed depending on what trait was the subject of study. A further anomaly not explained by population genetic models is the resilience toward inbreeding in some species (Jamieson, 2015). This is perhaps most striking in recovery success following population bottlenecks (Heber et al., 2013; Ramstad et al., 2013; Frankham, 2015; Jamieson, 2015). This success has been attributed to the purging of deleterious gene variants, but empirical evidence for this speculation of losing the worst alleles due to homozygous expression has not yet been forthcoming, and an alternative explanation might be needed (Crnokrak and Barrett, 2002; Kennedy et al., 2014; López-Cortegano et al., 2018). Also anomalous is the observation that inbreeding depression can manifest differently between environments, suggesting that understanding genetic variation alone is insufficient to predict fitness outcomes (Keller et al., 2002; Cheptou and Donohue, 2013). These examples suggest that critical information is missing, and an improved conceptual framework is needed to understand the fitness of populations that we wish to manage (Escobar et al., 2008; Hochholdinger and Baldauf, 2018; Rey et al., 2020). With this increasing realization, attention has recently turned to the phenomenon of epigenetics and the extent to which chemical modification of the DNA in response to environmental signals also contributes to an organism's fitness.
Gene variants (alleles), specifically for transcription factors and in regulatory regions, are thought to play a major role in altering the dynamics of an organism's transcriptome, with consequences for the dynamics of its proteome, morphology, physiology, and behavior (Johnston et al., 2019; Lai et al., 2019). However, the expression of genes is also modulated through chemical modification of DNA, RNA, and proteins in processes linked to environmental signals - a phenomenon known as epigenetics (Donohue, 2014; Junaid et al., 2018). Mechanisms of epigenetic regulation are thought to have a role in the plastic (varied) expression of genes and phenotypes (Bonduriansky et al., 2012; Groszmann et al., 2013; Li et al., 2018; Thiebaut et al., 2019). Such regulation has been linked to the phenotypic divergence of populations and can affect the width of a niche and the capacity to fulfill roles in an ecosystem (Miryeganeh and Saze, 2019; Thiebaut et al., 2019; Rey et al., 2020). Epigenetic change has also been linked to altered patterns of gene expression associated with transgressive segregation and heterosis in hybrid offspring (Groszmann et al., 2013; Junaid et al., 2018; Botet and Keurentjes, 2020). Recent work in plants suggests that epigenetic regulation plays a crucial role in hybrid vigor and that non-additive and yet not random differences in the patterns of chemical DNA modification (methylation of Cytosine bases) between parents and hybrid offspring contributes to phenotypic differences (Kawanabe et al., 2016; Junaid et al., 2018; Lauss et al., 2018; Miryeganeh and Saze, 2019; Sinha et al., 2020). Other studies have also found that inbred and outbred lines exhibit different epigenetic profiles and that manipulating these profiles can revert symptoms of inbreeding depression (Vergeer et al., 2012). Taken together, these findings strongly suggest that accounting for both allelic and epigenetic variation is likely to be necessary to predict fitness outcomes for hybrid populations (Rey et al., 2020). While most studies to date involve plants, common mechanisms of eukaryotic gene expression suggest similar studies in animals will reveal similar results.
Genetic Rescue
Prolonged periods of low (effective) population size and/or restricted gene flow are a growing concern among conservationists and there is increasing interest to utilize genetically motivated management to address the loss of genetic diversity and its negative effects (IUCN/SSC, 2013; Frankham, 2015; Hoffmann et al., 2015; Whiteley et al., 2015; Frankham et al., 2017; Ralls et al., 2018; Bell et al., 2019; Chan et al., 2019; Taylor and Larson, 2019). In practice, management interventions to increase genetic diversity usually entail translocation of individuals from a source to a genetically distinct target population with the aim of intentionally generating intermixed (hybrid) populations of the same species (Armstrong and McLean, 1995; Weeks et al., 2011; IUCN/SSC, 2013; Pierson et al., 2016; Wennerström et al., 2017; Flanagan et al., 2018). Efforts to increase genetic diversity have also entailed management of connectivity between distinct populations or different forms of guided mate choice (Soulé, 1985; Pierson et al., 2016; Wennerström et al., 2017; Flanagan et al., 2018). These translocations involving genetically distinct populations differ from more classic supplementary translocations (also referred to as reinforcement translocations) which have the goal to increase population size directly by adding more individuals, and differ from reintroduction translocations where the purpose and focus are to re-establish populations with genetic stock obtained from within its historical range (IUCN/SSC, 2013; Armstrong et al., 2019). All the above-mentioned types of interventions have been suggested for Apteryx. However, these proposals have been made without studies having been conducted to quantify inbreeding and/or inbreeding depression in neither source nor target populations (Innes et al., 2015; Kiwis for Kiwi, 2016; Germano et al., 2018).
The interbreeding of individuals from populations with the aim of increasing fitness is now commonly referred to as genetic rescue (Hedrick, 1995; Johnson et al., 2010; Frankham, 2015; Bell et al., 2019; Ochoa et al., 2019). “Rescue,” in this case, refers to decreasing the extinction risk in the target population and is commonly evaluated by an observed population growth rate increase after genetic admixture (Ingvarsson, 2001; Hedrick et al., 2011; Frankham, 2015; Whiteley et al., 2015; Bell et al., 2019). A growing number of authors argue that this prospect of alleviating extinction risk by augmented gene flow is underutilized (Frankham et al., 2011; Frankham, 2015; Whiteley et al., 2015; Love Stowell et al., 2017; Ralls et al., 2018, 2020; Bell et al., 2019). Since 1964, the 14 birds released on Ponui Island have produced an estimated population of over 1,700 birds, or one bird per hectare, which makes the Ponui Island brown kiwi population one of the densest in the world having experienced an equivalent of on average 9% annual population growth (Potter, 1990; McLennan and Potter, 1992). Thus, even though this was not an intention of the original translocations, the Ponui population might be one of New Zealand's best examples of genetic rescue.
Conceptually, genetic rescue and hybrid vigor likely describe the same biological phenomenon resulting from hybridization. However, in conservation, genetic rescue is rarely or never promoted as hybridization due to the negative connotations of this word (Allendorf et al., 2001; Wayne and Shaffer, 2016; Love Stowell et al., 2017; Bell et al., 2019; Chan et al., 2019; Taylor and Larson, 2019). Hitherto, most attempts of genetic rescue have focused on utilizing source populations with smallest possible genetic difference to the target population (Edmands, 2007; Frankham et al., 2017; Ralls et al., 2018, 2020). However, discussion about the trade-off between increasing genetic diversity and maintaining genetic integrity is becoming increasingly relevant. Recently, several authors have discussed the ambiguity and sometimes mismatch in genetically motivated conservation interventions. For instance, Von Holdt et al. (2018) highlighted that there is a need for debate around our understanding of the evolutionary significance of hybridization and its implications for conservation management, and Ralls et al. (2018) called for a “paradigm shift in the genetic management of fragmented populations.”
As for hybrid vigor, the allele centered modeling of genetic rescue (Davenport, 1908) is still the commonly suggested explanation for successful population growth, leading to the prediction that the magnitude of genetic rescue is a function of the severity of the genetic load in the target population (Bell et al., 2019). A variation on this model, while still having an allele focus, acknowledges the role of the size of the source population (the bigger the better) and the amount of adaptive genetic difference between target and source population (the less the better, Whiteley et al., 2015; Bell et al., 2019). In reviewing 156 published cases, Frankham (2015) identified that in practice the two most important conditions for successful genetic rescue were (1) that the source population was not inbred, and (2) that the target population experienced some level of environmental stress. Interestingly, the review of Frankham (2015) found that severe inbreeding in both target and source population did not have to exclude observed genetic rescue, such as in the case of Mexican wolves (Canis lupus baileyi, Fredrickson et al., 2007), however, the magnitude of rescue was larger when outbred sources were used. The noticeable discrepancy between theory and empirical finding highlights the need to identify what is missing from our understanding of inbreeding depression, outbreeding depression and heterosis. Quantification of these phenomena requires a baseline for comparison, and in cases where species are confined to small isolated populations and have been so for a long time, what can be learnt from comparing these populations may be limited (Hedrick and Fredrickson, 2010; Heber et al., 2013; Ramstad et al., 2013; Taylor, 2014). An exemplary case concerns A. owenii. This is the only Apteryx species in which inbreeding has been thoroughly studied. Results show evidence of several bottlenecks and extremely low variation in the MHC genes (Ramstad et al., 2010; Miller et al., 2011; Shepherd et al., 2012; Taylor, 2014). However, the fact that extant members of this species originate from as few as five founders, means that findings might not be applicable to other Apteryx species (Ramstad et al., 2013).
While debate remains surrounding the degree to which genetic load contributes to extinction risk, there will remain debate as to the magnitude of threat aversion or loss of extinction risk that can be expected from genetic rescue (Bell et al., 2019). One challenge for evaluating the success of translocations involving birds from multiple sources is that long term outcomes might not be immediately apparent. While some studies have suggested that the effect of genetic rescue is greatest in the first- and/or second-generation (Johansen-Morris and Latta, 2006; Bell et al., 2019), other studies have shown that initial generations can suffer reduced fitness as a consequence of outbreeding depression. In such cases, crossing, backcrossing, and genomic recombination can later generate a hybrid swarm with higher average fitness than the parental populations (Hwang et al., 2011). While these findings are somewhat contradictory, they caution against drawing conclusions too soon after translocations involving interpopulation crosses. The Ponui island birds, resulting from a mixed translocation that occurred more than 50 years ago, offer the opportunity to investigate the longer-term fitness consequences of admixture.
The successful introduction of diversity resulting in phenotypic diversity on which selection can act and adaptations evolve in response to environmental conditions is said to diffusely depend on the genomic makeup of the introduced individuals (Bell et al., 2019). It will also depend on how the genome is packaged and expressed in individuals, and how this affects the fitness of individuals (Hochholdinger and Baldauf, 2018; Li et al., 2018; Botet and Keurentjes, 2020). This is an area of research where there are many outstanding questions, but where knowledge is increasing rapidly (Hochholdinger and Baldauf, 2018; Rey et al., 2020). Genome Science that links genetic variation, genome expression and local adaptation will be key, and this may require rethinking our measures of biodiversity.
Assessing Biodiversity
Since the ratification of the Convention on Biological Diversity (CBD) in 1992, the world has agreed to acknowledge and preserve three levels of biodiversity: ecosystem diversity, species diversity and genetic diversity (UN, 1992). The latter is mainly justified because standing genetic diversity – allelic variation – is thought to contribute most to evolutionary potential of species and thus to their capacity to adapt to a rapidly changing environment (e.g., Haenel et al., 2019; Lai et al., 2019). In practice, this has led to genetic differences being used as an objective criterion to measure diversity, delimit and identify species, subspecies, taxa, lineages, evolutionary significant units (ESUs), or management units, often driven by improving opportunity for legal protection, funding and/or threat acknowledgment (Rojas, 1992; Godfray et al., 2004; Palsbøll et al., 2007; Lohman et al., 2010; Wayne and Shaffer, 2016; Cobley, 2017; Groves et al., 2017; Taylor et al., 2017a,b). The prevailing standpoint in such cases has been that preserving genetic diversity means maintaining the observed differences (see for example Palsbøll et al., 2007; Weir et al., 2016; Richmond et al., 2017; Taylor et al., 2017c; Germano et al., 2018). As mentioned above, the Apteryx genus serves as a typical example of both these tendencies (Tennyson et al., 2003; Weir et al., 2016; Germano et al., 2018).
Numerous genetic studies that have identified populations or other groups within species and genera, such as Apteryx, as genetically distinct have been based on markers such as microsatellites, allozymes, mitochondrial DNA sequence, or sequence fragments of a small number of selected genes (Baker et al., 1995; Cooper et al., 2001; Haddrath and Baker, 2001, 2012; Burbidge et al., 2003; Shepherd et al., 2013). These traditional markers have successfully been used to provide measures of genetic diversity and infer evolutionary relationships (Mitchell et al., 2014; Weir et al., 2016). However, this approach can be limiting when only a small number of gene loci are investigated and/or when loci evolve at different rates of evolutionary change within and between taxa (Allendorf et al., 2010; Funk et al., 2012; Steiner et al., 2013; Putman and Carbone, 2014). Low resolution can lead to underestimation and/or miss interpretation of the complex history of species and populations (Goldstein et al., 1996; Hudson and Turelli, 2003; Zink and Barrowclough, 2008; Funk et al., 2012; Steiner et al., 2013; Jeffries et al., 2016; Kardos et al., 2016; Hodel et al., 2017; Richmond et al., 2017; Galla et al., 2020). Another limitation of the most commonly used genetic markers is that they are not linked to traits of functional adaptive significance and have restricted ability for determining the cause and nature of ecological distinctiveness of taxa (Allendorf et al., 2010; Funk et al., 2012; Wennerström et al., 2017; Leroy et al., 2018). Hence, there are limitations to the ability of such markers to inform and evaluate the consequences of hybridization and translocation success based on possible outbreeding and inbreeding effects (Funk et al., 2012; Hess et al., 2013; Frankham, 2015; Whiteley et al., 2015; Flanagan et al., 2018; Bell et al., 2019; Taylor and Larson, 2019).
Newer molecular tools allow researchers to reduce these limitations. For example, genomic methods, such as reduced-representation-sequencing (for instance through protocols like genotyping-by-sequencing) (1) provide information from thousands of loci spread throughout the entire genome, and (2) cover both neutral, non-coding sequences and genes under selection (Elshire et al., 2011; Funk et al., 2012; Hess et al., 2013; Narum et al., 2013; Reitzel et al., 2013; Hunter et al., 2018; Leroy et al., 2018; Picq et al., 2018). This resolution allows for much more detailed genetic characterization of closely related taxa, and thus also of historic as well as recent hybridization and introgression between them and the population growth effects over time such admixture has had (Elshire et al., 2011; Weeks et al., 2011; Narum et al., 2013; Escudero et al., 2014; Leaché et al., 2014; Gaughran et al., 2017; Schmickl et al., 2017; Zhen et al., 2017; Picq et al., 2018; Taylor and Larson, 2019). In addition, genome-wide association studies (GWAS), provide insight into the nature of the physiological and behavioral differences of populations (Hess et al., 2013; Flanagan et al., 2018; Hunter et al., 2018). Methodological advances mean that genomics is likely to take on a larger role in conservation biology, in delimiting management units, in identifying suitable source populations and in evaluating translocation outcomes (Funk et al., 2012; Flanagan et al., 2018; Von Holdt et al., 2018; Galla et al., 2019; Ramstad and Dunning, 2020; Russello et al., 2020). Even so, the potential for genomics to contribute to conservation science may not be fully realized while focus remains on genetic variation alone which in many cases will be insufficient to make reliable predictions for managing biodiversity.
While our understanding of diversity, resilience and the contrasting outcomes of hybridization is informed by genetic variation, the importance of epigenetic variation is increasingly being recognized (Cheptou and Donohue, 2013; Groszmann et al., 2013; Biémont and Vieira, 2014). Epigenetic changes accompanying environmental change play a key role in plastic responses that occur at a faster rate than mutational change and the sorting of allelic variation (Hochholdinger and Baldauf, 2018; Miryeganeh and Saze, 2019; Rey et al., 2020). Epigenetic factors could explain why some natural populations are more fit than others, and also why some species that have crashed to very low levels of genetic diversity and adaptive potential can still rebound successfully (Heber et al., 2013; Ramstad et al., 2013; Frankham, 2015; Jamieson, 2015). The contribution of epigenetic processes to evolutionary and ecological success requires more study and understanding, but the potential of “conservation epigenetics” is both exciting and promising (Rey et al., 2020). Differences in epigenetic markers between populations could help us to identify and categorize evolutionary significant units (ESUs) and whether species with low genetic variation might nevertheless be resilient to environmental change. This in turn will help us to better predict the viability and differences between populations – information that could be crucial for identifying suitable source- and target populations for translocations. Epigenetic monitoring, as a complement to genetic investigations, could provide more ecologically significant information than studies of genetic variation alone and improve prediction of what interventions are likely to be most successful (Hochholdinger and Baldauf, 2018; Miryeganeh and Saze, 2019; Rey et al., 2020).
Investigation of epigenetic processes will involve studies of DNA, RNA, and proteins. However, a place to start at the population level is with DNA methylation profiles (Sepers et al., 2019; Rey et al., 2020). Of interest for conservation planning would be understanding variation among individuals and between populations, and whether ESUs are categorized appropriately, determining whether source populations show differences from translocated and admixed populations and whether there is evidence of population fragmentation sooner than is appreciated from studies of genetic variation (Rey et al., 2020). Variant protocols of reduced-representation-sequencing, such as DREAM, EpiGBS and bsRADseq offer the potential to characterize partial methylation profiles (Jelinek and Madzo, 2016; Trucchi et al., 2016; van Gurp et al., 2016). However, these protocols have limitations since they capture only a small subset of the entire epigenome. A more complete methylome profile is possible using whole-genome bisulfite sequencing (Lister et al., 2009; Hansen et al., 2012). Locus specific bisulfite sequencing is also possible (Hernández et al., 2013; Lam et al., 2020). Another alternative is the so-called assay for transposase-accessible chromatin using sequencing (ATAC-seq) which maps genome-wide chromatin accessibility, which is tightly linked to gene expression (Miskimen et al., 2017). However, both the latter methods introduce the cost of whole-genome sequencing. Whatever might be the methodological developments to come in the fast-developing field of high throughput sequencing technologies, a focus on both genomics and epigenetics is likely to provide the insight that is needed to understand hybridization, and its contribution to biological diversity and successful conservation strategies (Goulet et al., 2017; Von Holdt et al., 2018; Chan et al., 2019; Taylor and Larson, 2019; Rey et al., 2020).
The Hybrid Birds of Ponui
In 2016, the hybrid birds of Ponui were said to be of no value for kiwi conservation other than as specimens for public display. However, this conclusion needs to be reconsidered in light of recent findings and modern tools of genome science. There is no doubt that allelic diversity and the genetic background of individuals is important for individual fitness, population sustainability and genetic rescue. However, knowledge of genetic variation alone appears insufficient to fully understand the link between genotype, transcriptome, phenotype, and fitness. Focusing solely on allelic variation makes for attractive and accessible models. However, the poor performance of these models to predict outcomes has the potential to damage the relationship between researchers and practitioners. This motivates the need for research investigating the interactions and relative contributions of genotype, epigenetics, and the environment for understanding phenotypic diversity, reproductive success, and adaptive potential.
The Ponui birds, which are highly successful in their island habitat, provide an exciting model system to investigate ecological success and potentially the evolutionary and ecological significance of hybridization. In addition, this system could help to inform how conservation translocations can effectively utilize genome-level data to achieve their goals. Ecological factors might help explain the success of this population relative to the mainland and other island populations. For instance, compared to unmanaged populations the juvenile kiwi on Ponui experience much lower mortality from predation (Shapiro, 2005; Dixon, 2015; Strang, 2018). However, this mortality is still higher than observed in the most extensively managed populations (Colbourne et al., 2005; Robertson and de Monchy, 2012). Thus, even if juvenile mortality contributes to population success it may still be important to determine other significant factors. Is the secret to their success the loss of recessive deleterious effects? Is it phenotypic diversity – and if so, how did that come about? Mainly through allelic admixture? Or via epigenetics and thus transcriptome variation? To gain understanding, the next step will be to compare the genomic diversity of Ponui birds with their parent populations and with other mainland populations whose history has not involved extreme genetic bottlenecks associated with the founding of island populations by a small number of translocated birds. Epigenetic studies might initially focus on methylation profiles and their density (Rey et al., 2020). A study could seek to answer questions such as: Is there epigenetic variation within and between populations of A. mantelli? How do the epigenetic profiles of Ponui birds compare with other populations? How do the methylation profiles change over generations on Ponui? How do they compare between parents and offspring? Does the genetic distance between parents influence the non-additivity of the epigenetic profile of offspring? What are the methylation patterns for genes of potential adaptive value? Following this investigation, important studies would compare genomic and epigenetic diversity in relation to transcriptome variation, and to how the resulting phenotypic variation relates to fitness, and measures of inbreeding-, outbreeding depression and/or hybrid vigor. A key for meaningful interpretation of transcriptome analyses will be detailed knowledge of individual birds (including information on generation, age, health, and sex) and here again, Ponui could prove suitable after 17 years of extensive studies on this population.
The questions that could be addressed by investigations of the Ponui birds are not only relevant to Apteryx, improved understanding of genomics and epigenetics and thus of the nature of population differences is key for conservation of all fragmented populations in need of augmented genetic influx (Tallmon et al., 2004; Edmands, 2007; Escobar et al., 2008; Stelkens et al., 2014; Barmentlo et al., 2018; Von Holdt et al., 2018; Bell et al., 2019; Koide et al., 2019; Rey et al., 2020). Worldwide, habitat and population fragmentation has rendered a situation where focusing solely on species-level conservation may lead to either inbreeding or homogenization, both of which results in loss of genetic diversity. Retaining evolutionary potential is arguably one of the main challenges for conservationists across the globe, not only because of the intrinsic value of diversity but also for providing populations with the ability to adapt to our changing environment. Because of this challenge, we support the call for a paradigm shift in conservation that includes redefining admixture and hybridization (Canestrelli et al., 2016; Ralls et al., 2018; Von Holdt et al., 2018; Taylor and Larson, 2019). We believe that it is wrong to dismiss a prolific population of a threatened species as unimportant in an unsubstantiated way when there remains uncertainty as to the best way of managing the species. This is even more so when these populations have the potential to serve as sources for ongoing, translocation focused interventions – a literal source for individuals and a source of increased knowledge. Lastly, we recognize the importance of multidisciplinary studies that are needed to help better understand and predict hybridization outcomes.
The questions that could be addressed with genetic and epigenetic investigations of the Ponui birds are not only relevant for Apteryx. Making meaningful measurements of population differences is important for determining what interventions are appropriate to ensure the sustainability of fragmented populations (Tallmon et al., 2004; Edmands, 2007; Escobar et al., 2008; Stelkens et al., 2014; Barmentlo et al., 2018; Von Holdt et al., 2018; Bell et al., 2019; Koide et al., 2019; Rey et al., 2020). These measurements require embracing the complexity of epigenetic phenomena and understanding how it interacts with genetic variation in affecting the fitness of individuals in different environments. Improved understanding will in turn help us to better understand the adaptive potential and resilience of species to environmental stresses and change. We support the call being made by others for a paradigm shift in conservation to rethink the negative connotations of admixture and hybridization (Canestrelli et al., 2016; Ralls et al., 2018; Von Holdt et al., 2018; Taylor and Larson, 2019). Measures of fitness of individuals and populations need to consider both temporal and environmental factors. Furthermore, until interactions between the environment of Ponui island and the epigenomes of its kiwi are better understood, we believe that it would be wrong to dismiss a prolific population of a threatened species. This is even more important when this population has the potential to serve as a source for ongoing, translocation focused interventions – a literal source of individuals and a source of increased knowledge to be drawn upon in decision making. Lastly, we acknowledge the importance of integrating contributions from other disciplines when using novel tools from genome science to improve understanding and better predict intervention outcomes. An important goal is to inform and develop practices that meet conservation aspirations.
Author Contributions
MU and PL performed the literature search. MU wrote the first draft of the manuscript. All authors commented on previous versions of the manuscript, read, approved the final manuscript, and jointly developed the concept for the article.
Funding
Funding was also provided by Kiwi Rescue's MBIE Programme C09X1609 and the New Zealand Marsden Fund MAU1707.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
First, we want to acknowledge the people of the land, in particular Te Patukeha, Ngati Kuta Hapū, and Richard Witehira – without you, this project would never have started. We thank the Chamberlin family for allowing us to have a program of research to study the Ponui kiwi for the last 21 years. We are also very grateful to Bernard Sabrair and Elka Gouzer-Waechter who funded part of this research review through the Massey Foundation. Lastly, we would like to thank Doug Armstrong and reviewers for extremely valuable comments on previous versions of this manuscript.
References
Abbott, R., Albach, D., Ansell, S., Arntzen, J. W., Baird, S. J., Bierne, N., et al. (2013). Hybridization and speciation. J. Evol. Biol. 26, 229–246. doi: 10.1111/j.1420-9101.2012.02599.x
Abourachid, A., Castro, I., and Provini, P. (2019). How to walk carrying a huge egg? Trade-offs between locomotion and reproduction explain the special pelvis and leg anatomy in kiwi (Aves; Apteryx spp.). J. Anat. 235, 1045–1056. doi: 10.1111/joa.13072
Allendorf, F. W., Hohenlohe, P. A., and Luikart, G. (2010). Genomics and the future of conservation genetics. Nat. Rev. Genet. 11, 697–709. doi: 10.1038/nrg2844
Allendorf, F. W., Leary, R. F., Spruell, P., and Wenburg, J. K. (2001). The problems with hybrids: setting conservation guidelines. Trends Ecol. Evol. 16, 613–622. doi: 10.1016/S0169-5347(01)02290-X
Armstrong, D. P., and McLean, I. G. (1995). New Zealand translocations: theory and practice. Pac. Conserv. Biol. 2, 39–54. doi: 10.1071/PC950039
Armstrong, D. P., and Seddon, P. J. (2008). Directions in reintroduction biology. Trends Ecol. Evol. 23, 20–25. doi: 10.1016/j.tree.2007.10.003
Armstrong, D. P., Seddon, P. J., and Moehrenschlager, A. (2019). “Reintroduction,” in Encyclopedia of Ecology, 2nd Edn, Vol. 1. ed. B. D. Fath (Oxford: Elsevier), 458–466. doi: 10.1016/B978-0-12-409548-9.10589-5
Baker, A. J., Daugherty, C. H., Colbourne, R., and McLennan, J. (1995). Flightless brown kiwis of New Zealand possess extremely subdivided population structure and cryptic species like small mammals. Proc. Natl. Acad. Sci. U.S.A. 92, 8254–8258. doi: 10.1073/pnas.92.18.8254
Bansal, N. (2020). Exploring the Host-Parasite Relationship Between Brown Kiwi (Apteryx mantelli), Kiwi Ticks (Ixodes anatis) and Kiwi Tick-Borne Haemoparasites. Doctor of philosophy in ecology (Doctoral thesis). Manawatu: Massey University.
Barmentlo, S. H., Meirmans, P. G., Luijten, S. H., Triest, L., and Oostermeijer, J. G. B. (2018). Outbreeding depression and breeding system evolution in small, remnant populations of Primula vulgaris: consequences for genetic rescue. Conserv. Genet. 19, 545–554. doi: 10.1007/s10592-017-1031-x
Barton, N. H. (1996). Natural selection and random genetic drift as causes of evolution on islands. Philos. Trans. R. Soc. Lond. B Biol. Sci. 351, 785–795. doi: 10.1098/rstb.1996.0073
Bell, D. A., Robinson, Z. L., Funk, W. C., Fitzpatrick, S. W., Allendorf, F. W., Tallmon, D. A., et al. (2019). The exciting potential and remaining uncertainties of genetic rescue. Trends Ecol. Evol. 34, 1070–1079. doi: 10.1016/j.tree.2019.06.006
Biémont, C., and Vieira, C. (2014). Could interallelic interactions be a key to the epigenetic aspects of fitness-trait inbreeding depression? Heredity 112, 219–220. doi: 10.1038/hdy.2013.80
Bonduriansky, R., Crean, A. J., and Day, T. (2012). The implications of nongenetic inheritance for evolution in changing environments. Evol. Appl. 5, 192–201. doi: 10.1111/j.1752-4571.2011.00213.x
Botet, R., and Keurentjes, J. J. (2020). The role of transcriptional regulation in hybrid vigor. Front. Plant Sci. 11:410. doi: 10.3389/fpls.2020.00410
Burbidge, M. L., Colbourne, R. M., Robertson, H. A., and Baker, A. J. (2003). Molecular and other biological evidence supports the recognition of at least three species of brown kiwi. Conserv. Genet. 4, 167–177. doi: 10.1023/A:1023386506067
Canestrelli, D., Porretta, D., Lowe, W. H., Bisconti, R., Carere, C., and Nascetti, G. (2016). The tangled evolutionary legacies of range expansion and hybridization. Trends Ecol. Evol. 31, 677–688. doi: 10.1016/j.tree.2016.06.010
Chan, W. Y., Hoffmann, A. A., and van Oppen, M. J. (2019). Hybridization as a conservation management tool. Conserv. Lett. 12:e12652. doi: 10.1111/conl.12652
Cheptou, P., and Donohue, K. (2013). Epigenetics as a new avenue for the role of inbreeding depression in evolutionary ecology. Heredity 110, 205–206. doi: 10.1038/hdy.2012.66
Cobley, J. (2017). Cultivating the cultural memory of Ranunculus paucifolius T. Kirk, a South Island subalpine buttercup. Int. Rev. Environ. Hist. 3, 43–62. doi: 10.22459/IREH.03.01.2017.06
Colbourne, R. (2005). Kiwi (Apteryx spp.) on offshore New Zealand islands. Depart. Conserv. Res. Dev. Ser. 208, 1176–8886.
Colbourne, R., Bassett, S., Billing, T., McCormick, H., McLennan, J., Nelson, A., et al. (2005). The development of operation nest egg as a tool in the conservation management of kiwi. Sci. Conserv. 259:24.
Cooper, A., Lalueza-Fox, C., Anderson, S., Rambaut, A., Austin, J., and Ward, R. (2001). Complete mitochondrial genome sequences of two extinct moas clarify ratite evolution. Nature 409, 704–707. doi: 10.1038/35055536
Crnokrak, P., and Barrett, S. C. (2002). Perspective: purging the genetic load: a review of the experimental evidence. Evolution 56, 2347–2358. doi: 10.1111/j.0014-3820.2002.tb00160.x
Cunningham, S. J., and Castro, I. (2011). The secret life of wild brown kiwi: studying behaviour of a cryptic species by direct observation. N. Z. J. Ecol. 35, 209–219.
Davenport, C. B. (1908). Degeneration, albinism and inbreeding. Science 28, 454–455. doi: 10.1126/science.28.718.454-b
Dixon, T. (2015). What They do in the Shadows: Habitat Utilisation and Diet of Brown Kiwi (Apteryx mantelli) Adults Within a High-Density Island Population. Master of Science in Ecology (Master thesis). Palmerston North: Massey University.
Donohue, K. (2014). The epigenetics of adaptation: focusing on epigenetic stability as an evolving trait. Evolution 68, 617–619. doi: 10.1111/evo.12347
Dussex, N., Taylor, H. R., Irestedt, M., and Robertson, B. C. (2018). When genetic and phenotypic data do not agree: the conservation implications of ignoring inconvenient taxonomic evidence. N. Z. J. Ecol. 42, 284–290. doi: 10.20417/nzjecol.42.13
East, E. (1908). “Inbreeding in corn,” in Reports of the Connecticut Agricultural Experiment Station for Years 1907–1908. Annual Report 31, ed E. H. Jenkins (New Haven, CT: The Tuttle, Morehouse & Taylor Press), 419–428.
Edmands, S. (1999). Heterosis and outbreeding depression in interpopulation crosses spanning a wide range of divergence. Evolution 53, 1757–1768. doi: 10.1111/j.1558-5646.1999.tb04560.x
Edmands, S. (2007). Between a rock and a hard place: evaluating the relative risks of inbreeding and outbreeding for conservation and management. Mol. Ecol. 16, 463–475. doi: 10.1111/j.1365-294X.2006.03148.x
Edmands, S., and Timmerman, C. C. (2003). Modeling factors affecting the severity of outbreeding depression. Conserv. Biol. 17, 883–892. doi: 10.1046/j.1523-1739.2003.02026.x
Elshire, R. J., Glaubitz, J. C., Sun, Q., Poland, J. A., Kawamoto, K., Buckler, E. S., et al. (2011). A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6:e19379. doi: 10.1371/journal.pone.0019379
Escobar, J. S., Nicot, A., and David, P. (2008). The different sources of variation in inbreeding depression, heterosis and outbreeding depression in a metapopulation of Physa acuta. Genetics 180, 1593–1608. doi: 10.1534/genetics.108.092718
Escudero, M., Eaton, D. A., Hahn, M., and Hipp, A. L. (2014). Genotyping-by-sequencing as a tool to infer phylogeny and ancestral hybridization: a case study in Carex (Cyperaceae). Mol. Phylogen. Evol. 79, 359–367. doi: 10.1016/j.ympev.2014.06.026
Flanagan, S. P., Forester, B. R., Latch, E. K., Aitken, S. N., and Hoban, S. (2018). Guidelines for planning genomic assessment and monitoring of locally adaptive variation to inform species conservation. Evol. Appl. 11, 1035–1052. doi: 10.1111/eva.12569
Frankham, R. (2015). Genetic rescue of small inbred populations: meta-analysis reveals large and consistent benefits of gene flow. Mol. Ecol. 24, 2610–2618. doi: 10.1111/mec.13139
Frankham, R., Ballou, J. D., Eldridge, M. D., Lacy, R. C., Ralls, K., Dudash, M. R., et al. (2011). Predicting the probability of outbreeding depression. Conserv. Biol. 25, 465–475. doi: 10.1111/j.1523-1739.2011.01662.x
Frankham, R., Ballou, J. D., Ralls, K., Eldridge, M., Dudash, M. R., Fenster, C. B., et al. (2017). Genetic Management of Fragmented Animal and Plant Populations. Oxford: Oxford University Press. doi: 10.1093/oso/9780198783398.001.0001
Fredrickson, R. J., Siminski, P., Woolf, M., and Hedrick, P. W. (2007). Genetic rescue and inbreeding depression in Mexican wolves. Proc. R. Soc. B 274, 2365–2371. doi: 10.1098/rspb.2007.0785
Funk, W. C., McKay, J. K., Hohenlohe, P. A., and Allendorf, F. W. (2012). Harnessing genomics for delineating conservation units. Trends Ecol. Evol. 27, 489–496. doi: 10.1016/j.tree.2012.05.012
Galla, S. J., Forsdick, N. J., Brown, L., Hoeppner, M. P., Knapp, M., Maloney, R. F., et al. (2019). Reference genomes from distantly related species can be used for discovery of single nucleotide polymorphisms to inform conservation management. Genes 10:9. doi: 10.3390/genes10010009
Galla, S. J., Moraga, R., Brown, L., Cleland, S., Hoeppner, M. P., Maloney, R. F., et al. (2020). A comparison of pedigree, genetic and genomic estimates of relatedness for informing pairing decisions in two critically endangered birds: implications for conservation breeding programmes worldwide. Evol. Appl. 13, 991–1008. doi: 10.1111/eva.12916
Gaughran, S. J., Quinzin, M. C., Miller, J. M., Garrick, R. C., Edwards, D. L., Russello, M. A., et al. (2017). Theory, practice, and conservation in the age of genomics: the galápagos giant tortoise as a case study. Evol. Appl. 11, 1084–1093. doi: 10.1111/eva.12551
Germano, J., Barlow, S., Castro, I., Colbourne, R., Cox, M., Gillies, C., et al. (2018). Kiwi Recovery Plan 2018–2028 Mahere Whakaora Kiwi 2018–2028. Threatened Species Recovery Plan 64. Wellington: Department of Conservation.
Godfray, H. C. J., Knapp, S., and Mace, G. M. (2004). The role of taxonomy in species conservation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 359, 711–719. doi: 10.1098/rstb.2003.1454
Goldberg, T. L., Grant, E. C., Inendino, K. R., Kassler, T. W., Claussen, J. E., and Philipp, D. P. (2005). Increased infectious disease susceptibility resulting from outbreeding depression. Conserv. Biol. 19, 455–462. doi: 10.1111/j.1523-1739.2005.00091.x
Goldstein, D. B., Zhivotovsky, L. A., Nayar, K., Linares, A. R., Cavalli-Sforza, L. L., and Feldman, M. W. (1996). Statistical properties of the variation at linked microsatellite loci: implications for the history of human Y chromosomes. Mol. Biol. Evol. 13, 1213–1218. doi: 10.1093/oxfordjournals.molbev.a025686
Goulet, B. E., Roda, F., and Hopkins, R. (2017). Hybridization in plants: old ideas, new techniques. Plant Physiol. 173, 65–78. doi: 10.1104/pp.16.01340
Groszmann, M., Greaves, I. K., Fujimoto, R., Peacock, W. J., and Dennis, E. S. (2013). The role of epigenetics in hybrid vigour. Trends Genet. 29, 684–690. doi: 10.1016/j.tig.2013.07.004
Groves, C. P., Cotterill, F. P. D., Gippoliti, S., Robovský, J., Roos, C., Taylor, P. J., et al. (2017). Species definitions and conservation: a review and case studies from African mammals. Conserv. Genet. 18, 1247–1256. doi: 10.1007/s10592-017-0976-0
Haddrath, O., and Baker, A. J. (2001). Complete mitochondrial DNA genome sequences of extinct birds: ratite phylogenetics and the vicariance biogeography hypothesis. Proc. R. Soc. B 268, 939–945. doi: 10.1098/rspb.2001.1587
Haddrath, O., and Baker, A. J. (2012). Multiple nuclear genes and retroposons support vicariance and dispersal of the palaeognaths, and an early cretaceous origin of modern birds. Proc. Biol. Sci 279, 4617–25. doi: 10.1098/rspb.2012.1630
Haenel, Q., Roesti, M., Moser, D., MacColl, A. D., and Berner, D. (2019). Predictable genome-wide sorting of standing genetic variation during parallel adaptation to basic versus acidic environments in stickleback fish. Evol. Lett. 3, 28–42. doi: 10.1002/evl3.99
Hamilton, J. A., and Miller, J. M. (2016). Adaptive introgression as a resource for management and genetic conservation in a changing climate. Conserv. Biol. 30, 33–41. doi: 10.1111/cobi.12574
Hansen, K. D., Langmead, B., and Irizarry, R. A. (2012). BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol. 13:R83. doi: 10.1186/gb-2012-13-10-r83
Heber, S., Varsani, A., Kuhn, S., Girg, A., Kempenaers, B., and Briskie, J. (2013). The genetic rescue of two bottlenecked South Island robin populations using translocations of inbred donors. Proc. R. Soc. B 280:20122228. doi: 10.1098/rspb.2012.2228
Hedrick, P. W. (1995). Gene flow and genetic restoration: the florida panther as a case study. Conserv. Biol. 9, 996–1007. doi: 10.1046/j.1523-1739.1995.9050988.x-i1
Hedrick, P. W., Adams, J. R., and Vucetich, J. A. (2011). Reevaluating and broadening the definition of genetic rescue. Conserv. Biol. 25, 1069–1070. doi: 10.1111/j.1523-1739.2011.01751.x
Hedrick, P. W., and Fredrickson, R. (2010). Genetic rescue guidelines with examples from Mexican wolves and florida panthers. Conserv. Genet. 11, 615–626. doi: 10.1007/s10592-009-9999-5
Hendry, A. P., Wenburg, J. K., Bentzen, P., Volk, E. C., and Quinn, T. P. (2000). Rapid evolution of reproductive isolation in the wild: evidence from introduced salmon. Science 290, 516–518. doi: 10.1126/science.290.5491.516
Herbert, J., and Daugherty, C. H. (2002). “Genetic variation, systematics and management of kiwi (Apteryx spp.), some early 1990s studies in kiwi,” in Science and Research Internal Report 19, ed F. Overmars (Wellington: Department of Conservation Science Publishing).
Hernández, H. G., Tse, M. Y., Pang, S. C., Arboleda, H., and Forero, D. A. (2013). Optimizing methodologies for PCR-based DNA methylation analysis. BioTechniques 55, 181–197. doi: 10.2144/000114087
Hess, J. E., Campbell, N. R., Close, D. A., Docker, M. F., and Narum, S. R. (2013). Population genomics of Pacific lamprey: adaptive variation in a highly dispersive species. Mol. Ecol. 22, 2898–2916. doi: 10.1111/mec.12150
Hiscox, J. D. (2014). Can Microbes be Contributing to the Decline of the North Island Brown Kiwi (Apteryx mantelli)? Palmerston North: Master of Science in Zoology at Massey University.
Hochholdinger, F., and Baldauf, J. A. (2018). Heterosis in plants. Curr. Biol. 28, R1089–R1092. doi: 10.1016/j.cub.2018.06.041
Hodel, R. G., Chen, S., Payton, A. C., McDaniel, S. F., Soltis, P., and Soltis, D. E. (2017). Adding loci improves phylogeographic resolution in red mangroves despite increased missing data: comparing microsatellites and RAD-Seq and investigating loci filtering. Sci. Rep. 7:17598. doi: 10.1038/s41598-017-16810-7
Hoffmann, A., Griffin, P., Dillon, S., Catullo, R., Rane, R., Byrne, M., et al. (2015). A framework for incorporating evolutionary genomics into biodiversity conservation and management. Clim. Change Responses 2:1. doi: 10.1186/s40665-014-0009-x
Holdaway, R. (1989). New Zealand's pre-human avifauna and its vulnerability. N. Z. J. Ecol. 12, 11–25.
Hudson, R. R., and Turelli, M. (2003). Stochasity overrules the “three-times rule”: genetic drift, genetic draft, and coalescence time for nuclear versus mitochondrial DNA. Evolution 57, 182–190. doi: 10.1111/j.0014-3820.2003.tb00229.x
Hunter, M. E., Hoban, S. M., Bruford, M. W., Segelbacher, G., and Bernatchez, L. (2018). Next-generation conservation genetics and biodiversity monitoring. Evol. Appl. 11, 1029–1034. doi: 10.1111/eva.12661
Hwang, A., Northrup, S., Alexander, J., Vo, K., and Edmands, S. (2011). Long-term experimental hybrid swarms between moderately incompatible Tigriopus californicus populations: hybrid inferiority in early generations yields to hybrid superiority in later generations. Conserv. Genet. 12, 895–909. doi: 10.1007/s10592-011-0193-1
Ingvarsson, P. K. (2001). Restoration of genetic variation lost–the genetic rescue hypothesis. Trends Ecol. Evol. 16, 62–63. doi: 10.1016/S0169-5347(00)02065-6
Innes, J., Eppink, F., Andreson, D., and Robertson, H. A. (2016). Roles for Kõhanga in Kiwi Conservation a Review. Landcare Research/Manaaki Whenua Contract Report LC2504 for the Department of Conservation, Lincoln, New Zealand.
Innes, J., Kelly, D., Overton, J. M., and Gillies, C. (2010). Predation and other factors currently limiting New Zealand forest birds. N. Z. J. Ecol. 34, 86–114.
Innes, J., Eppink, F. V., and Robertson, H. A. (2015). Saving a National Icon: Preliminary Estimation of the Additional Cost of Achieving Kiwi Population Stability or 2% Growth. Auckland: Landcare Research Manaaki Whenua.
IUCN/SSC. (2013). Guidelines for Reintroductions and Other Conservation Translocations. Gland: IUCN Species Survival Commission.
Jamieson, I. G. (2015). Significance of population genetics for managing small natural and reintroduced populations in New Zealand. N. Z. J. Ecol. 39, 1–18.
Janes, J., and Hamilton, J. (2017). Mixing it up: the role of hybridization in forest management and conservation under climate change. Forests 8:237. doi: 10.3390/f8070237
Jeffries, D. L., Copp, G. H., Handley, L. L., Olsén, K. H., Sayer, C. D., and Hänfling, B. (2016). Comparing RADseq and microsatellites to infer complex phylogeographic patterns, an empirical perspective in the Crucian carp, Carassius carassius, L. Mol. Ecol. 25, 2997–3018. doi: 10.1111/mec.13613
Jelinek, J., and Madzo, J. (2016). “DREAM: a simple method for DNA methylation profiling by high-throughput sequencing,” in Chronic Myeloid Leukemia, eds S. Li and H. Zhang (New York, NY: Springer), 111–127. doi: 10.1007/978-1-4939-4011-0_10
Johansen-Morris, A., and Latta, R. G. (2006). Fitness consequences of hybridization between ecotypes of Avena barbata: hybrid breakdown, hybrid vigor, and transgressive segregation. Evolution 60, 1585–1595. doi: 10.1111/j.0014-3820.2006.tb00503.x
Johnson, W. E., Onorato, D. P., Roelke, M. E., Land, E. D., Cunningham, M., Belden, R. C., et al. (2010). Genetic restoration of the florida panther. Science 329, 1641–1645. doi: 10.1126/science.1192891
Johnston, A. D., Simões-Pires, C. A., Thompson, T. V., Suzuki, M., and Greally, J. M. (2019). Functional genetic variants can mediate their regulatory effects through alteration of transcription factor binding. Nat. Commun. 10:3472. doi: 10.1038/s41467-019-11412-5
Junaid, A., Kumar, H., Rao, A., Patil, A., Singh, N., and Gaikwad, K. (2018). Unravelling the epigenomic interactions between parental inbreds resulting in an altered hybrid methylome in pigeonpea. DNA Res. 25, 361–373. doi: 10.1093/dnares/dsy008
Kardos, M., Taylor, H. R., Ellegren, H., Luikart, G., and Allendorf, F. W. (2016). Genomics advances the study of inbreeding depression in the wild. Evol. Appl. 9, 1205–1218. doi: 10.1111/eva.12414
Kawanabe, T., Ishikura, S., Miyaji, N., Sasaki, T., Wu, L. M., Itabashi, E., et al. (2016). Role of DNA methylation in hybrid vigor in Arabidopsis thaliana. Proc. Natl. Acad. Sci. U.S.A. 113, E6704–E6711. doi: 10.1073/pnas.1613372113
Keller, L. F., Grant, P. R., Grant, B. R., and Petren, K. (2002). Environmental conditions affect the magnitude of inbreeding depression in survival of darwin's finches. Evolution 56, 1229–1239. doi: 10.1111/j.0014-3820.2002.tb01434.x
Kennedy, E. S., Grueber, C. E., Duncan, R. P., and Jamieson, I. G. (2014). Severe inbreeding depression and no evidence of purging in an extremely inbred wild species—the Chatham Island black robin. Evolution 68, 987–995. doi: 10.1111/evo.12315
Kiwis for Kiwi (2016). Target 2% Working With New Zealanders to Grow Kiwi Populations. Auckland: Kiwis for Kiwi Investment Strategy.
Koide, Y., Sakaguchi, S., Uchiyama, T., Ota, Y., Tezuka, A., Nagano, A. J., et al. (2019). Genetic properties responsible for the transgressive segregation of days to heading in rice. G3 9, 1655–1662. doi: 10.1534/g3.119.201011
Lai, Y.-T., Yeung, C. K., Omland, K. E., Pang, E.-L., Hao, Y., Liao, B.-Y., et al. (2019). Standing genetic variation as the predominant source for adaptation of a songbird. Proc. Natl. Acad. Sci. U.S.A. 116, 2152–2157. doi: 10.1073/pnas.1813597116
Lam, D., Luu, P.-L., Song, J. Z., Qu, W., Risbridger, G. P., Lawrence, M. G., et al. (2020). Comprehensive evaluation of targeted multiplex bisulphite PCR sequencing for validation of DNA methylation biomarker panels. Clin. Epigenet. 12, 1–16. doi: 10.1186/s13148-020-00880-y
Lauss, K., Wardenaar, R., Oka, R., van Hulten, M. H., Guryev, V., Keurentjes, J. J., et al. (2018). Parental DNA methylation states are associated with heterosis in epigenetic hybrids. Plant Physiol. 176, 1627–1645. doi: 10.1104/pp.17.01054
Leaché, A. D., Fujita, M. K., Minin, V. N., and Bouckaert, R. R. (2014). Species delimitation using genome-wide SNP data. Syst. Biol. 63, 534–542. doi: 10.1093/sysbio/syu018
Leroy, G., Carroll, E. L., Bruford, M. W., DeWoody, J. A., Strand, A., Waits, L., et al. (2018). Next-generation metrics for monitoring genetic erosion within populations of conservation concern. Evol. Appl. 11, 1066–1083. doi: 10.1111/eva.12564
Li, H., Yuan, J., Wu, M., Han, Z., Li, L., Jiang, H., et al. (2018). Transcriptome and DNA methylome reveal insights into yield heterosis in the curds of broccoli (Brassica oleracea L var. italica). BMC Plant Biol. 18:168. doi: 10.1186/s12870-018-1384-4
Lister, R., Pelizzola, M., Dowen, R. H., Hawkins, R. D., Hon, G., Tonti-Filippini, J., et al. (2009). Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462, 315–322. doi: 10.1038/nature08514
Lohman, D. J., Ingram, K. K., Prawiradilaga, D. M., Winker, K., Sheldon, F. H., Moyle, R. G., et al. (2010). Cryptic genetic diversity in “widespread” Southeast Asian bird species suggests that Philippine avian endemism is gravely underestimated. Biol. Conserv. 143, 1885–1890. doi: 10.1016/j.biocon.2010.04.042
López-Cortegano, E., Bersabé, D., Wang, J., and García-Dorado, A. (2018). Detection of genetic purging and predictive value of purging parameters estimated in pedigreed populations. Heredity 121, 38–51. doi: 10.1038/s41437-017-0045-y
Love Stowell, S. M., Pinzone, C. A., and Martin, A. P. (2017). Overcoming barriers to active interventions for genetic diversity. Biodivers. Conserv. 26, 1753–1765. doi: 10.1007/s10531-017-1330-z
Lynch, M. (1991). The genetic interpretation of inbreeding depression and outbreeding depression. Evolution 45, 622–629. doi: 10.1111/j.1558-5646.1991.tb04333.x
Mallet, J. (2013). Species, concepts of. Encyclopedia Biodiversity 6, 679–691. doi: 10.1016/B978-0-12-384719-5.00131-3
Marr, A. B., Keller, L. F., and Arcese, P. (2002). Heterosis and outbreeding depression in descendants of natural immigrants to an inbred population of song sparrows (Melospiza melodia). Evolution 56, 131–142. doi: 10.1111/j.0014-3820.2002.tb00855.x
McLennan, J., and Potter, M. (1992). Distribution, population changes and management of brown kiwi in hawke's bay. N. Z. J. Ecol. 16, 91–102.
McLennan, J., Potter, M., Robertson, H., Wake, G., Colbourne, R., Dew, L., et al. (1996). Role of predation in the decline of kiwi, Apteryx spp., in New Zealand. N. Z. J. Ecol. 20, 27–35.
McLennan, J., and McCann, T. (2002). Genetic variability, distribution and abundance of great spotted kiwi (Apteryx haastii). Sci. Res. Intern. Rep. 191, 35–56.
Miller, H. C., Bowker-Wright, G., Kharkrang, M., and Ramstad, K. (2011). Characterisation of class II B MHC genes from a ratite bird, the little spotted kiwi (Apteryx owenii). Immunogenetics 63, 223–233. doi: 10.1007/s00251-010-0503-7
Miryeganeh, M., and Saze, H. (2019). Epigenetic inheritance and plant evolution. Popul. Ecol. 62, 17–27. doi: 10.1002/1438-390X.12018
Miskimen, K. L. S., Chan, E. R., and Haines, J. L. (2017). Assay for transposase-accessible chromatin using sequencing (ATAC-seq) data analysis. Curr. Protoc. Hum. Genet. 92, 20.4.1–20.4.13. doi: 10.1002/cphg.32
Mitchell, K. J., Llamas, B., Soubrier, J., Rawlence, N. J., Worthy, T. H., Wood, J., et al. (2014). Ancient DNA reveals elephant birds and kiwi are sister taxa and clarifies ratite bird evolution. Science 344, 898–900. doi: 10.1126/science.1251981
Narum, S. R., Buerkle, C. A., Davey, J. W., Miller, M. R., and Hohenlohe, P. A. (2013). Genotyping-by-sequencing in ecological and conservation genomics. Mol. Ecol. 22, 2841–2847. doi: 10.1111/mec.12350
Nosil, P., Crespi, B. J., and Sandoval, C. P. (2002). Host-plant adaptation drives the parallel evolution of reproductive isolation. Nature 417:440. doi: 10.1038/417440a
Ochoa, A., Onorato, D. P., Fitak, R. R., Roelke-Parker, M. E., and Culver, M. (2019). Evolutionary and functional mitogenomics associated with the genetic restoration of the Florida panther. J. Hered. 108, 449–455. doi: 10.1093/jhered/esx015
O'Dea, R. E., Noble, D. W., Johnson, S. L., Hesselson, D., and Nakagawa, S. (2016). The role of non-genetic inheritance in evolutionary rescue: epigenetic buffering, heritable bet hedging and epigenetic traps. Environ. Epigenet. 2:dvv014. doi: 10.1093/eep/dvv014
Orr, M. R., and Smith, T. B. (1998). Ecology and speciation. Trends Ecol. Evol. 13, 502–506. doi: 10.1016/S0169-5347(98)01511-0
Palsbøll, P. J., Berube, M., and Allendorf, F. W. (2007). Identification of management units using population genetic data. Trends Ecol. Evol. 22, 11–16. doi: 10.1016/j.tree.2006.09.003
Picq, S., Keena, M., Havill, N., Stewart, D., Pouliot, E., Boyle, B., et al. (2018). Assessing the potential of genotyping-by-sequencing-derived single nucleotide polymorphisms to identify the geographic origins of intercepted gypsy moth (Lymantria dispar) specimens: a proof-of-concept study. Evol. Appl. 11, 325–339. doi: 10.1111/eva.12559
Pierce, R., Gardiner, C., Moodie, H., Robertson, H., and Sporle, W. (2006). Sustainable Management of Brown Kiwi and Other Threatened Birds in Northland. Whangarei: Wildland Consultants Report No. 1193
Pierson, J. C., Coates, D. J., Oostermeijer, J. G. B., Beissinger, S. R., Bragg, J. G., Sunnucks, P., et al. (2016). Genetic factors in threatened species recovery plans on three continents. Front. Ecol. Environ. 14, 433–440. doi: 10.1002/fee.1323
Potter, M. (1990). Movement of North Island brown kiwi (Apteryx australis mantelli) between forest remnants. N. Z. J. Ecol. 14, 17–24.
Powlesland, R. (1988). Kiwi Research and Conservation: An Account of a NZ Wildlife Service Workshop, 20–21 May 1986. Science and Research Internal Report No. 2. Science and Research Directorate, Department of Conservation, Wellington, New Zealand.
Putman, A. I., and Carbone, I. (2014). Challenges in analysis and interpretation of microsatellite data for population genetic studies. Ecol. Evol. 4, 4399–4428. doi: 10.1002/ece3.1305
Ralls, K., Ballou, J. D., Dudash, M. R., Eldridge, M. D. B., Fenster, C. B., Lacy, R. C., et al. (2018). Call for a paradigm shift in the genetic management of fragmented populations. Conserv. Lett. 11:e12412. doi: 10.1111/conl.12412
Ralls, K., Sunnucks, P., Lacy, R. C., and Frankham, R. (2020). Genetic rescue: a critique of the evidence supports maximizing genetic diversity rather than minimizing the introduction of putatively harmful genetic variation. Biol. Conserv. 251:108784. doi: 10.1016/j.biocon.2020.108784
Ramstad, K. M., Colbourne, R. M., Robertson, H. A., Allendorf, F. W., and Daugherty, C. H. (2013). Genetic consequences of a century of protection: serial founder events and survival of the little spotted kiwi (Apteryx owenii). Proc. R. Soc. B 280:20130576. doi: 10.1098/rspb.2013.0576
Ramstad, K. M., Pfunder, M., Robertson, H. A., Colbourne, R. M., Allendorf, F. W., and Daugherty, C. H. (2010). Fourteen microsatellite loci cross-amplify in all five kiwi species (Apteryx spp.) and reveal extremely low genetic variation in little spotted kiwi (A. owenii). Conserv. Genet. Resour. 2, 333–336. doi: 10.1007/s12686-010-9233-2
Ramstad, K. M., and Dunning, L. T. (2020). “Population genomics advances and opportunities in conservation of kiwi (Apteryx spp.),” in Population Genomics: Wildlife, eds P. A. Hohenlohe and O. P. Rajora (New York, NY: Springer International Publishing), 493–521. doi: 10.1007/13836_2019_71
Reitzel, A., Herrera, S., Layden, M., Martindale, M., and Shank, T. (2013). Going where traditional markers have not gone before: utility of and promise for RAD sequencing in marine invertebrate phylogeography and population genomics. Mol. Ecol. 22, 2953–2970. doi: 10.1111/mec.12228
Rey, O., Eizaguirre, C., Angers, B., Baltazar-Soares, M., Sagonas, K., Prunier, J. G., et al. (2020). Linking epigenetics and biological conservation: towards a conservation epigenetics perspective. Funct. Ecol. 34, 414–427. doi: 10.1111/1365-2435.13429
Reynolds, S. M., Castro, I., Alley, M. R., and Cunningham, S. J. (2017). Apteryx spp. (Kiwi) possess an uropygial gland: anatomy and pathology. Eur. J. Anat. 21, 125–139.
Richmond, J. Q., Wood, D. A., Westphal, M. F., Vandergast, A. G., Leaché, A. D., Saslaw, L. R., et al. (2017). Persistence of historical population structure in an endangered species despite near-complete biome conversion in California's San Joaquin Desert. Mol. Ecol. 26, 3618–3635. doi: 10.1111/mec.14125
Rieseberg, L. H. (2001). Chromosomal rearrangements and speciation. Trends Ecol. Evol. 16, 351–358. doi: 10.1016/S0169-5347(01)02187-5
Robertson, H. A., Baird, K., Dowding, J. E., Elliott, G. P., Hitchmough, R. A., Miskelly, C. M., et al. (2016). Conservation Status of New Zealand birds, 2016 (Vol. 19). Wellington: Department of Conservation.
Robertson, H. A., Coad, N. B., Colbourne, R. M., and Fraser, J. R. (2019). Translocations of Little Spotted Kiwi (Apteryx owenii) for Genetic Management, 2016–17. Department of Conservation Research and Development Series 358. Clelland L, Publishing Team, Department of Conservation, Wellington, New Zealand.
Robertson, H. A., and de Monchy, P. J. (2012). Varied success from the landscape-scale management of kiwi Apteryx spp. in five sanctuaries in New Zealand. Bird Conserv. Int. 22, 429–444. doi: 10.1017/S0959270912000044
Roff, D. A. (2002). Inbreeding depression: tests of the overdominance and partial dominance hypotheses. Evolution 56, 768–775. doi: 10.1111/j.0014-3820.2002.tb01387.x
Rojas, M. (1992). The species problem and conservation: what are we protecting? Conserv. Biol. 6, 170–178. doi: 10.1046/j.1523-1739.1992.620170.x
Rundle, H. D. (2003). Divergent environments and population bottlenecks fail to generate premating isolation in Dorosophila pseudoobscura. Evolution 57, 2557–2565. doi: 10.1111/j.0014-3820.2003.tb01499.x
Rundle, H. D., Nagel, L., Boughman, J. W., and Schluter, D. (2000). Natural selection and parallel speciation in sympatric sticklebacks. Science 287, 306–308. doi: 10.1126/science.287.5451.306
Russello, M., Amato, G., DeSalle, R., and Knapp, M. (2020). “Conservation genetics and genomics,” in Multidisciplinary, ed P. W. Young (Basel: Digital Publishing Institute). doi: 10.3390/genes11030318
Schmickl, R., Marburger, S., Bray, S., and Yant, L. (2017). Hybrids and horizontal transfer: introgression allows adaptive allele discovery. J. Exp. Bot. 68, 5453–5470. doi: 10.1093/jxb/erx297
Sepers, B., van den Heuvel, K., Lindner, M., Viitaniemi, H., Husby, A., and van Oerset, K. (2019). Avian ecological epigenetics: pitfalls and promises. J. Ornithol. 160, 1183–1203. doi: 10.1007/s10336-019-01684-5
Shapiro, L. M. (2005). Diet overlap and potential competition between North Island brown kiwi chicks (Apteryx mantelli) and ship rats (Rattus rattus) for limited resources on Ponui Island, New Zealand. Master of science in ecology (Master thesis). Massey University: Palmerston North.
Shepherd, L. D., and Lambert, D. M. (2008). Ancient DNA and conservation: lessons from the endangered kiwi of New Zealand. Mol. Ecol. 17, 2174–2184. doi: 10.1111/j.1365-294X.2008.03749.x
Shepherd, L. D., Tennyson, A. J. D., and Lambert, D. M. (2013). Using ancient DNA to enhance museum collections: a case study of rare kiwi (Apteryx spp.) specimens. J. R. Soc. N. Z. 43, 119–127. doi: 10.1080/03036758.2012.732585
Shepherd, L. D., Worthy, T. H., Tennyson, A. J., Scofield, R. P., Ramstad, K. M., and Lambert, D. M. (2012). Ancient DNA analyses reveal contrasting phylogeographic patterns amongst kiwi (Apteryx spp.) and a recently extinct lineage of spotted kiwi. PLoS ONE 7:e42384. doi: 10.1371/journal.pone.0042384
Sinha, P., Singh, V. K., Saxena, R. K., Kale, S. M., Li, Y., Garg, V., et al. (2020). Genome-wide analysis of epigenetic and transcriptional changes associated with heterosis in pigeonpea. Plant Biotechnol. J. 18, 1697–1710. doi: 10.1111/pbi.13333
Soulé, M. E. (1985). What is conservation biology? Bioscience 35, 727–734. stable/1310054 doi: 10.2307/1310054
Steiner, C. C., Putnam, A. S., Hoeck, P. E., and Ryder, O. A. (2013). Conservation genomics of threatened animal species. Annu. Rev. Anim. Biosci. 1, 261–281. doi: 10.1146/annurev-animal-031412-103636
Stelkens, R. B., Brockhurst, M., Hurst, G., Miller, E. L., and Greig, D. (2014). The effect of hybrid transgression on environmental tolerance in experimental yeast crosses. J. Evol. Biol. 27, 2507–2519. doi: 10.1111/jeb.12494
Strang, K. E. (2018). The Ecology of Feral Cats (Felis catus) on a New Zealand Offshore Island: Considerations for Management. Doctor of Philosophy in Ecology (Doctoral thesis). Massey University: Manawatu.
Taborsky, M. (1988). Kiwis and dog predation: observations in Waitangi State Forest. Notornis 35, 197–202.
Tallmon, D. A., Luikart, G., and Waples, R. S. (2004). The alluring simplicity and complex reality of genetic rescue. Trends Ecol. Evol. 19, 489–496. doi: 10.1016/j.tree.2004.07.003
Taylor, B. L., Archer, F. I., Martien, K. K., Rosel, P. E., Hancock-Hanser, B. L., Lang, A. R., et al. (2017a). Guidelines and quantitative standards to improve consistency in cetacean subspecies and species delimitation relying on molecular genetic data. Mar. Mamm. Sci. 33, 132–155. doi: 10.1111/mms.12411
Taylor, B. L., Perrin, W. F., Reeves, R. R., Rosel, P. E., Wang, J. Y., Cipriano, F., et al. (2017b). Why we should develop guidelines and quantitative standards for using genetic data to delimit subspecies for data-poor organisms like cetaceans. Mar. Mamm. Sci. 33, 12–26. doi: 10.1111/mms.12413
Taylor, H. R. (2014). Detecting Inbreeding Depression in a Severely Bottlenecked, Recovering Species: the Little Spotted Kiwi (Apteryx owenii) (Doctoral theses). Victoria Univeristy of Wellington: New Zealand.
Taylor, H. R., Dussex, N., and van Heezik, Y. (2017c). Bridging the conservation genetics gap by identifying barriers to implementation for conservation practitioners. Glob. Ecol. Conserv. 10, 231–242. doi: 10.1016/j.gecco.2017.04.001
Taylor, S. A., and Larson, E. L. (2019). Insights from genomes into the evolutionary importance and prevalence of hybridization in nature. Nat. Ecol. Evol. 3, 170–177. doi: 10.1038/s41559-018-0777-y
Tennyson, A. J., Palma, R. L., Robertson, H. A., Worthy, T. H., and Gill, B. (2003). A new species of kiwi (Aves, Apterygiformes) from Okarito, New Zealand. Rec. Auckl. Museum 40, 55–64.
Thiebaut, F., Hemerly, A. S., and Ferreira, P. C. G. (2019). A role for epigenetic regulation in the adaptation and stress responses of non-model plants. Front. Plant Sci. 10:246. doi: 10.3389/fpls.2019.00246
Trucchi, E., Mazzarella, A. B., Gilfillan, G. D., Lorenzo, M. T., Schönswetter, P., and Paun, O. (2016). Bs RAD seq: screening DNA methylation in natural populations of non-model species. Mol. Ecol. 25, 1697–1713. doi: 10.1111/mec.13550
Undin, M., Hills, S. F. K., Lockhart, P. J., and Castro, I. (in press). Gaps in genetic knowledge affect conservation management of Kiwi, Apteryx spp. IBIS.
van Gurp, T. P., Wagemaker, N. C., Wouters, B., Vergeer, P., Ouborg, J. N., and Verhoeven, K. J. (2016). epiGBS: reference-free reduced representation bisulfite sequencing. Nat. Methods 13, 322–324. doi: 10.1038/nmeth.3763
Vergeer, P., Wagemaker, N., and Ouborg, N. J. (2012). Evidence for an epigenetic role in inbreeding depression. Biol. Lett. 8, 798–801. doi: 10.1098/rsbl.2012.0494
Vieco-Galvez, D., Castro, I., Morel, P., Chua, W., and Loh, M. (2020). Eggshell structure in Apteryx: Form, function, and adaptation. Authorea doi: 10.22541/au.160027528.87752141. [Epub ahead of print].
Von Holdt, B. M., Brzeski, K. E., Wilcove, D. S., and Rutledge, L. Y. (2018). Redefining the role of admixture and genomics in species conservation. Conserv. Lett. 11:e12371. doi: 10.1111/conl.12371
Wayne, R. K., and Shaffer, H. B. (2016). Hybridization and endangered species protection in the molecular era. Mol. Ecol. 25, 2680–2689. doi: 10.1111/mec.13642
Weeks, A. R., Sgro, C. M., Young, A. G., Frankham, R., Mitchell, N. J., Miller, K. A., et al. (2011). Assessing the benefits and risks of translocations in changing environments: a genetic perspective. Evol. Appl. 4, 709–725. doi: 10.1111/j.1752-4571.2011.00192.x
Weir, J. T., Haddrath, O., Robertson, H. A., Colbourne, R. M., and Baker, A. J. (2016). Explosive ice age diversification of kiwi. Proc. Natl. Acad. Sci. U.S.A. 113, E5580–E5587. doi: 10.1073/pnas.1603795113
Wennerström, L., Jansson, E., and Laikre, L. (2017). Baltic sea genetic biodiversity: current knowledge relating to conservation management. Aquat. Conserv. Mar. Freshwat. Ecosyst. 27, 1069–1090. doi: 10.1002/aqc.2771
Whiteley, A. R., Fitzpatrick, S. W., Funk, W. C., and Tallmon, D. A. (2015). Genetic rescue to the rescue. Trends Ecol. Evol. 30, 42–49. doi: 10.1016/j.tree.2014.10.009
Whitlock, M. C., Ingvarsson, P. K., and Hatfield, T. (2000). Local drift load and the heterosis of interconnected populations. Heredity 84, 452–457. doi: 10.1046/j.1365-2540.2000.00693.x
Whitlock, R., Stewart, G. B., Goodman, S. J., Piertney, S. B., Butlin, R. K., Pullin, A. S., et al. (2013). A systematic review of phenotypic responses to between-population outbreeding. Environ. Evid. 2:13. doi: 10.1186/2047-2382-2-13
Wilson, A. L. (2014). The Triumphs, Challenges and Failures of Young North Island Brown Kiwi (Apteryx mantelli). Palmerston North: Master of Science in Zoology at Massey University.
Winkworth, R. C., Wagstaff, S. J., Glenny, D., and Lockhart, P. J. (2005). Evolution of the New Zealand mountain flora: origins, diversification and dispersal. Org. Divers. Evol. 5, 237–247. doi: 10.1016/j.ode.2004.12.001
Wright, L. I., Tregenza, T., and Hosken, D. J. (2008). Inbreeding, inbreeding depression and extinction. Conserv. Genet. 9:833. doi: 10.1007/s10592-007-9405-0
Zhen, Y., Harrigan, R. J., Ruegg, K. C., Anderson, E. C., Ng, T. C., Lao, S., et al. (2017). Genomic divergence across ecological gradients in the Central African rainforest songbird (Andropadus virens). Mol. Ecol. 26, 4966–4977. doi: 10.1111/mec.14270
Ziesemann, B. (2011). The Social Organisation and Mating System of the Brown Kiwi (Apteryx mantelli). Doctor of philosophy in ecology (Doctoral thesis). Albany: Massey University.
Keywords: hybridization, outbreeding depression, heterosis, translocation, conservation management, evolutionary potential, epigenetics, Apteryx
Citation: Undin M, Lockhart PJ, Hills SFK and Castro I (2021) Genetic Rescue and the Plight of Ponui Hybrids. Front. Conserv. Sci. 1:622191. doi: 10.3389/fcosc.2020.622191
Received: 27 October 2020; Accepted: 18 December 2020;
Published: 15 January 2021.
Edited by:
Cristiano Vernesi, Fondazione Edmund Mach, ItalyReviewed by:
Jocelyn P. Colella, University of Kansas, United StatesYelin Huang, Sun Yat-sen University, China
Copyright © 2021 Undin, Lockhart, Hills and Castro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Malin Undin, bS51bmRpbkBtYXNzZXkuYWMubno=; Peter J. Lockhart, cC5qLmxvY2toYXJ0QG1hc3NleS5hYy5ueg==