Abstract
Genome-editing technologies have the potential to correct most genetic defects involved in blood disorders. In contrast to mutation-specific editing, targeted gene insertion can correct most of the mutations affecting the same gene with a single therapeutic strategy (gene replacement) or provide novel functions to edited cells (gene addition). Targeting a selected genomic harbor can reduce insertional mutagenesis risk, while enabling the exploitation of endogenous promoters, or selected chromatin contexts, to achieve specific transgene expression levels/patterns and the modulation of disease-modifier genes. In this review, we will discuss targeted gene insertion and the advantages and limitations of different genomic harbors currently under investigation for various gene therapy applications.
Introduction
Blood genetic disorders are caused by mutations in genes or in their regulatory elements that result in a dysfunctional, dysregulated, or absent protein. Conventional gene therapy approach consists of the addition of a functional copy of a mutated gene to patients' cells using viral vectors, such as adeno-associated virus (AAV) (Mingozzi and High, 2011) and lentivirus (LV)-derived vectors (Naldini, 2011). These modified viruses can deliver the transgene expression cassettes encoded in their genome to the cell nucleus, where the genetic information is used. This gene replacement strategy is mutation-independent and thus can benefit patients with the same condition regardless of their genotype.
Despite its remarkable success for ex vivo and in vivo treatment of several monogenic disorders (Dunbar et al., 2018), there are still major hurdles to overcome to improve therapeutic outcomes and treat challenging monogenic (e.g., hemoglobinopathies, immunodeficiencies, and congenital anemias) as well as multifactorial blood diseases (e.g., cancer, autoimmune, and infectious disorders). Apart from vector-specific issues such as immunogenicity and tropism (Masat et al., 2013; Colella et al., 2018), which are beyond the scope of this review, classic gene replacement has a major limitation: it is hard to faithfully re-create characteristics of endogenous promoters and gene-specific regulation within the context of a viral vector. Tissue-, developmental-, and stimulus-specific gene expression requires the complex interaction of different genomic elements (promoters, enhancers, and silencers) that can be located in distant regions of the genome and span several kilobases (Schoenfelder and Fraser, 2019).
AAV vectors are small viruses (~4.7 kb), limiting the choice of regulatory elements to include in the expression cassette, especially when delivering large transgenes (Li and Samulski, 2020). Moreover, they persist mainly as episomes in non-dividing cells and are progressively lost through cell division (Nakai et al., 2001; Ehrhardt et al., 2003; Bortolussi et al., 2014)—a major obstacle for treating infantile disorders and tissues undergoing rapid proliferation (e.g., hematopoietic and epithelial cells). On the other hand, LV have larger cargo capacity (~8 kb), stably integrate in the genome, and persist through cell replication (Naldini et al., 1996), but they carry the intrinsic risk of insertional mutagenesis and oncogene transactivation (mainly when strong promoters/enhancers are present (Cavazzana et al., 2019; Bushman, 2020)). In addition, their semi-random integration (Schroder et al., 2002) results in transduction mosaicism and heterogeneous transgene expression due to chromatin position effects (Chen et al., 2017; Vansant et al., 2020), making therapeutic levels harder to reach.
When combining AAV and nucleases, both transgene expression cassettes and genomic integration sites contribute to the corrective strategy, dramatically expanding therapeutic possibilities. Primarily, targeting a functional copy of a gene to its endogenous locus, under the control of its own promoter and in the right chromatin context, can result in physiological expression and minimize genotoxic integrations. Alternatively, transgenes can be targeted to safe integration sites or specific genomic elements of interest to engineer cells with novel functions, further improving safety and increasing potential applications of gene replacement/addition therapy (Cox et al., 2015).
Sequence-specific endonucleases (such as ZNF, TALEN, or CRISPR/Cas9) (Gaj et al., 2016) can induce genomic DNA double-strand breaks (DSB) in proximity to pathological mutations and activate cellular DNA repair pathways to correct them. The inclusion of short single-stranded oligodeoxynucleotide (ssODN) donors is a simple and effective approach for precise correction of single-nucleotide mutations (DeWitt et al., 2016; De Ravin et al., 2017; Romero et al., 2019). Although their short size currently limits their application for diseases caused by multiple pathological variants (e.g., β-thalassemia, ~300 different mutations across the β-globin locus), technological advances in long ssODN synthesis would most likely expand their therapeutic potential (Praetorius et al., 2017; Roth et al., 2018).
DSB generated by endonucleases can also facilitate integration of therapeutic transgenes to selected genomic locations (targeted gene replacement). AAV has a tendency to integrate at pre-existing chromosomal breaks that provide free DNA ends for non-homologous end joining (NHEJ) (Miller et al., 2004). To increase efficiency, specificity, and precision of integration, homology arms derived from genomic regions flanking the target site are introduced on each side of the AAV cassette with the aim of leveraging the homologous DNA repair pathway (Hirata et al., 2002). Although effective in proliferating cells, homologous recombination is quite inefficient in quiescent hematopoietic stem cells (HSC) and postmitotic cells or tissues (Nishiyama, 2019; Shin et al., 2020). Therefore, alternative DNA repair mechanisms based on NHEJ or microhomology-mediated end joining (MMEJ) are now being investigated (Suzuki et al., 2016; Banan, 2020). In both cases, AAV are the gold-standard DNA delivery system for gene-targeted integration in vivo (Li et al., 2011) and ex vivo (Wang et al., 2015), though the exact molecular mechanism underpinning this process remains unknown (Deyle and Russell, 2009).
Integration Strategies
Selecting a suitable genomic site for transgene integration depends on many factors, such as the expression level required, the target cells/tissue, and the disease to be treated.
We have subdivided integration sites in four groups according to functional characteristics: (i) endogenous promoters, when promoterless transgenes are inserted under the control of endogenous enhancers/promoters; (ii) safe genomic harbors, when transgenes and their promoters are integrated into genomic regions that allow robust expression without affecting cell physiology; (iii) disease modifier genes, when transgenes integrate into coding sequence of endogenous genes, whose inactivation benefits disease-affected cells; and (iv) specificity exchange, when transgenes are integrated into coding sequence of endogenous genes to change their function.
It is worth noting that this subdivision is only a working framework, as the same integration site can fall into two or more categories, and it is not exhaustive, as new integration strategies are described every day.
Endogenous Promoters
Correction of Dysfunctional Genes
A straightforward approach for targeted gene replacement consists in inserting a functional copy of a gene downstream of its endogenous promoter. This strategy can correct most pathological mutations that are scattered along the gene body (such as substitutions and frameshift mutations), while maintaining physiological gene expression (Table 1A), which can be hard to achieve with artificial promoters used in classical gene therapy vectors (Toscano et al., 2011).
Table 1
| Integration strategies | Advantages | Disadvantages | References | ||
|---|---|---|---|---|---|
| A | Endogenous locus | 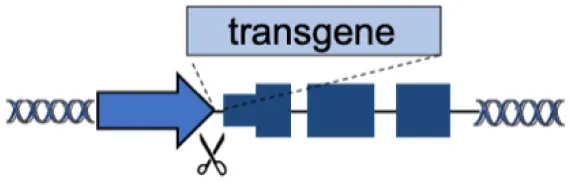 | Physiological transgene expression Corrects multiple mutations | Gene-specific strategy Limited to gene body mutations | Urnov et al., 2005; Lombardo et al., 2007; Li et al., 2011; Genovese et al., 2014; Voit et al., 2014; Dever et al., 2016; Hubbard et al., 2016; Schiroli et al., 2017; Sweeney et al., 2017; Kuo et al., 2018; Wang et al., 2019, 2020a; Rai et al., 2020 |
| B | Superactive promoters (ALB, HBA) | 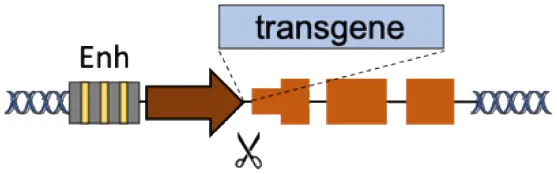 | Accommodates different transgenes Supraphysiological expression Few integrations required | Partial gene disruption Limited to non-cell autonomous disorders Extensive validation required | De Ravin et al., 2016; Diez et al., 2017; Stephens et al., 2018, 2019; Gomez-Ospina et al., 2019; Scharenberg et al., 2020 |
| C | Tolerant to integration (AAVS1, CCR5, Rosa26) | 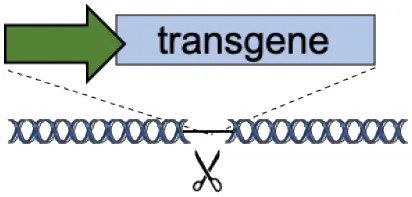 | Accommodates different transgenes | Artificial promoters required Variable expression | De Ravin et al., 2016; Diez et al., 2017; Stephens et al., 2018, 2019; Gomez-Ospina et al., 2019; Scharenberg et al., 2020 |
| D | Chromatin domains (NAD) | 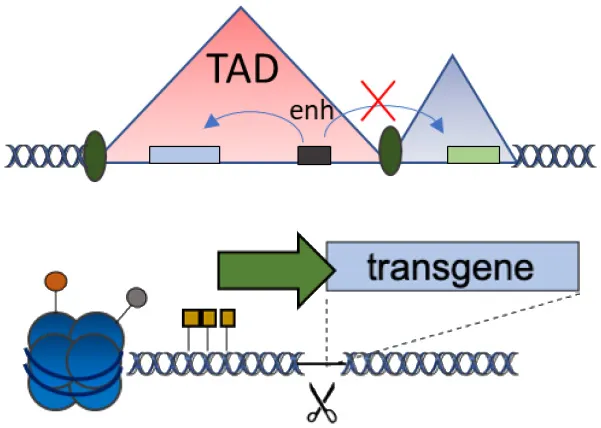 | Fine gene regulation Far from oncogenic genes | No proof-of-principle in clinically relevant models | Schenkwein et al., 2020 |
| E | Disease-modifier genes (CCR5, HBA) | 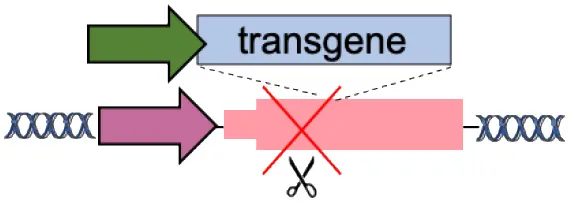 | Improve therapeutic effect Lower therapeutic threshold | Extensive validation required Limited to well-known diseases | Voit et al., 2013; Wiebking et al., 2018 |
| F | Specificity Exchange (TCR, BCR) | 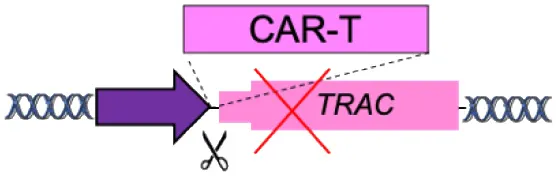 | Improved CAR expression and potency | Off-targets Translocations risk (for multiple edits) | Eyquem et al., 2017; MacLeod et al., 2017; Greiner et al., 2019; Hartweger et al., 2019; Moffett et al., 2019; Voss et al., 2019 |
(A–F) The advantages and disadvantages of different integration strategies.
Scissors: nuclease. Solid arrows indicate promoters, solid ovals indicate histone modifications, and solid squares indicate DNA modifications. Enh, enhancers; TAD, topologically associating, d, domain.
The first proof of concept was obtained using ZFN on primary T cells ex vivo to replace interleukin-2 receptor subunit gamma (IL2RG), whose mutational inactivation causes X-linked severe combined immunodeficiency (X-SCID) (Urnov et al., 2005; Lombardo et al., 2007). X-SCID represents an ideal model for testing this approach, as correction of only a small fraction of treated cells, given their strong growth advantage, should allow expansion and restoration of T cell function in vivo.
However, for effective clinical translation, targeted gene replacement should be performed in hematopoietic stem cells (HSC), the life-long source of all the different blood progenitors. Genovese via ZFN (Genovese et al., 2014) and Schiroli via CRISPR/Cas9 (Schiroli et al., 2017) were the first to report successful integration of a functional copy of IL2RG gene downstream its endogenous promoter in HSC, with the idea of restoring the endogenous lineage specificity and expression level of IL2RG without the risk of insertional mutagenesis (Hacein-Bey-Abina et al., 2003, 2008). Following this example, additional strategies have been developed for many blood diseases, including thalassemia (Voit et al., 2014; Dever et al., 2016), chronic granulomatous disease (De Ravin et al., 2017; Sweeney et al., 2017), hyper-immunoglobulin (Ig) M syndrome (Hubbard et al., 2016; Kuo et al., 2018), and Wiskott–Aldrich Syndrome (Rai et al., 2020).
Beside HSC and terminally differentiated blood cells, like B and T cells (Wang et al., 2016; Hung et al., 2018), AAV and nucleases have been the preferred method to achieve targeted transgene integration in many tissues in vivo (Suzuki et al., 2019; Kohama et al., 2020; Nishiguchi et al., 2020), especially the liver.
Li et al. were the first to demonstrate targeted gene correction in vivo by delivering ZFN and a partial F9 (coagulation factor IX, FIX) cDNA cassette with AAV8 to the liver of a humanized mouse model of hemophilia B (Li et al., 2011). While correction was performed in newborn mice, FIX expression was maintained in adults and even persisted after partial hepatectomy, demonstrating stable genomic integration. This approach was later replicated using CRISPR/Cas9 to integrate a hyperactive FIX variant in the mouse F9 locus (Wang et al., 2019).
Targeted gene replacement can also be combined with classical gene therapy to improve therapeutic outcome. In a neonatal mouse model of ornithine transcarbamylase (OTC) deficiency, an AAV carrying a liver-specific promoter and a human OTC transgene was integrated via CRISPR/Cas9 in the murine OTC locus (Wang et al., 2020a). Prompt, short-term expression from episomal AAV protected newborn mice from fatal hyperammonemia crisis, whereas its genomic integration allowed long-term disease correction.
Although targeting transgenes to their genomic loci is an effective therapeutic approach, it requires the development of countless gene-tailored editing strategies. Moreover, it can be difficult to reach and correct a number of cells that is sufficient to achieve a therapeutic benefit. Finally, its efficacy is limited in the presence of deletions/inversions that affect large portions of the locus or when regulatory elements controlling gene expression are mutated.
Over/Expression by Superactive Promoters
Although gene-editing technologies are evolving at a fast pace, it can be challenging to correct enough cells to reach a clinical benefit even using high doses of nuclease and donor DNA, which increase chances of off-target genomic cleavage, immune responses, and donor random integration. An alternative strategy consists in “hijacking” strong endogenous promoters to overexpress therapeutic cassettes from few modified cells (Table 1B). An elegant example of this approach is the targeted integration of AAV-delivered transgenes under the control of the endogenous albumin promoter in the liver (Barzel et al., 2015; Sharma et al., 2015; Davidoff and Nathwani, 2016). Even with <1% of targeted integration events, the terrific transcriptional activity of this superactive promoter was sufficient to achieve 5–20% of FIX levels and correct bleeding in hemophilia B mice (Barzel et al., 2015). Until today, this strategy has been successfully applied in different preclinical models of hemophilia A and B (Barzel et al., 2015; Sharma et al., 2015; Chen et al., 2019; Conway et al., 2019; Zhang et al., 2019; Wang et al., 2020b) and metabolic disorders (Laoharawee et al., 2018; Conway et al., 2019; De Caneva et al., 2019; Ou et al., 2019). Importantly, this is also the first genome-editing strategy undergoing in vivo testing in humans to treat mucopolysaccharidosis I and II (NTC02702115, NTC03041324).
Although promising, this approach still presents some concerns. First, targeted integration can lower serum albumin levels (Zhang et al., 2019; Ou et al., 2020) and albumin mutations have been observed in human hepatocellular carcinoma (Cancer Genome Atlas Research Network, 2017; Rao et al., 2017). Second, long-term AAV-mediated expression of endonucleases can result in off-target editing and unwanted AAV insertions (Li et al., 2019; Breton et al., 2020; Wang et al., 2020c). Finally, pre-existing liver conditions and immune responses against AAV vectors used to deliver transgenes or nucleases severely limit the number of eligible patients (Boutin et al., 2010; Simhadri et al., 2018).
To avoid these issues, we have recently proposed to integrate therapeutic transgenes in the α-globin locus of HSC (Pavani et al., 2020). Similar to albumin targeting, the idea is to combine the strong transcriptional output of the α-globin promoter with the abundance of transgene-expressing erythroblasts to maximize protein production, reducing the number of integration events required to reach therapeutic levels. Moreover, differently from the liver, autologous HSC can be recovered from patients and edited ex vivo before re-administration, thus circumventing immunological issues. Additional experiments in preclinical disease models will elucidate the therapeutic potential of this novel HSC platform for treating genetic diseases.
Following these examples, additional endogenous promoters with specific expression levels/patterns can be exploited for transgene expression. Although promoter hijacking has many advantages over other approaches, it is important to functionally validate the dispensability of the disrupted gene, as nuclease-mediated targeting can result in bi-allelic gene knock out, or to consider safer editing alternatives (e.g., nicking endonucleases Ran et al., 2013).
Safe Genomic Harbors
Tolerant to the Integration of an Expression Cassette
Genomic safe harbors are intragenic or intergenic regions of the human genome that enable stable expression of integrated transgenes without negatively affecting the host cell (Sadelain et al., 2011). Targeting expression cassettes to these loci is an efficient way to develop a “one-fits-all” platform to express different therapeutic transgenes using the same nuclease(s), therefore optimizing efficiency and improving safety.
By far, the most widely targeted genomic loci are AAVS1, CCR5, and Rosa26 (Table 1C).
The AAVS1 locus (chromosome 19 q13.42) was historically identified as the preferential integration site of wild-type AAV in human cell lines (Kotin et al., 1992). It encodes the PPP1R12C gene, a subunit of myosin phosphatase whose functions are not fully elucidated (Surks et al., 2003), but probably redundant (Smith et al., 2008). Stable and corrective editing of patients' HSC at this locus has been obtained by integrating a transgene cassette with (Fanconi anemia (Diez et al., 2017)) or without an exogenous promoter (X-CGD (De Ravin et al., 2016)). It is worth noting that the AAVS1 locus is an extremely gene-rich region and, although the presence of an insulator in the promoter of PPP1R12C could shield the genome from the action of the inserted promoter/enhancer (Ogata et al., 2003; Li et al., 2009), it requires a carefully designed transgene expression cassette to avoid transcriptional perturbation of neighboring genes (Lombardo et al., 2011). Moreover, several studies showed that variable expression and promoter silencing can occur at this site in different cell types (Lamartina et al., 2000; Smith et al., 2008; Ordovas et al., 2018; Bhagwan et al., 2019; Klatt et al., 2020), thus potentially limiting transgene expression.
The CCR5 gene (chromosome 3 p21.31) encodes for the main HIV co-receptor. Since a bi-allelic null mutation of this receptor (CCR5Δ32) confers HIV-1 resistance and is not associated with any major pathology (Hutter et al., 2009), this locus was first targeted/disrupted with nucleases in T cells and HSC to provide protection against AIDS ((Perez et al., 2008; Yu et al., 2020), NCT00842634, NCT02500849, and NCT03164135) and later exploited for targeted gene addition. Therapeutic transgenes involved in lysosomal storage disorders were inserted in the CCR5 gene of human HSC, under the control of exogenous ubiquitous or tissue-specific promoters. Upon transplantation, edited HSC engrafted, differentiated, and corrected the pathological phenotype in mouse models of MPS I (Gomez-Ospina et al., 2019) and Gaucher (Scharenberg et al., 2020). Although promising, the safety of this approach needs to be further validated, as CCR5 deficiency can result in increased susceptibility to West Nile (Lim et al., 2006; Cahill et al., 2018), influenza (Falcon et al., 2015), and Japanese encephalitis viruses (Larena et al., 2012).
The Rosa26 locus (chromosome 3 p25.31) was serendipitously discovered in mice as a reliable site to integrate DNA cassettes for transgenesis (Zambrowicz et al., 1997). This locus was then successfully targeted in vivo with CRISPR/Cas9 to knock-in human alpha-1-antitrypsin or FIX in mouse liver (Stephens et al., 2018, 2019). The human homolog was identified on chromosome 3 (position 3p25.3) (Irion et al., 2007); however, the efficacy and safety of this site for targeted integration is still undetermined.
While genomic safe harbors could represent a universal platform for gene targeting and thus expedite clinical development, so far no site of the human genome has been fully validated. The described loci may be acceptable for research applications, but clinical translation will require extensive validation as they localize in gene-dense areas and in proximity of cancer-related genes.
Chromatin Domains With Specific Expression Patterns
The genomic location of transgene integration can change its transcription up to 1,000-fold, according to some well-studied aspects of large-scale domain organization of chromatin (Akhtar et al., 2013; Brueckner et al., 2016; Corrales et al., 2017). Recent evidence for targeting 3D chromatin domains comes from the work of Schenkwein et al. showing that in primary human T cells genomic regions distant from one another linearly, but near in the three-dimensional genome, became jointly affected when site-specific transgene integration was performed (Schenkwein et al., 2020). In this work, transgenes were targeted to nucleolar-associated domains (NAD), which are distant from protein-encoding genes with oncogenic potential and thus represent safe genomic loci for inserting therapeutic transgenes.
The increasing knowledge of chromatin functions and dynamics (Moore et al., 2020) might soon allow us to select integration sites to obtain a certain transcriptional activity and cell/tissue/developmental specificity, as predicted by the presence/absence of certain histone marks (Talbert et al., 2019), DNA methylation, transcriptional factor binding sites, nuclear lamina interaction (Amendola and van Steensel, 2014), chromatin accessibility, and topology (Zheng and Xie, 2019; Zhang et al., 2020) (Table 1D). We can easily envision that the combination of selected chromatin locations and expression cassettes will allow fine-tuning of therapeutic transgene expression to unprecedented levels.
Disease-Modifier Genes
Inactivation of Pathogen Receptors
A disease-modifier gene alters the expression of another gene involved in a genetic/infectious disorder, therefore changing the penetrance, dominance, and severity of the disease itself (Genin et al., 2008). Novel genome-editing strategies can combine transgene expression with modulation of disease-modifier genes to improve therapeutic outcomes and provide cells with novel functions (Table 1E). Voit et al. were the first to describe the use of ZFN to integrate transgenes encoding for HIV restriction factors into the HIV co-receptor gene CCR5 (Voit et al., 2013). With this strategy, treated T cells were resistant to HIV infection thanks to the concomitant expression of protective transgenes and knockout of CCR5 (disease-modifier).
Restoring Balance in Disease Pathways
A second example of this approach involves β-thalassemias, a group of blood disorders caused by mutations in the β-globin gene. β-globin associates with α-globin to form adult hemoglobin (HbA, α2β2) and, when β-globin chains are absent or limiting, free α-globin precipitates causing hemolysis and ineffective erythropoiesis. Reduction of α-globin has been shown to ameliorate the β-thalassemia phenotype (Mettananda et al., 2015); hence, we and others have proposed to target the integration of a β-globin transgene into the α-globin site (disease-modifier) of HSC to simultaneously express the therapeutic gene while reducing α-globin production in differentiated erythroblasts (Table 1E) (Pavani et al.; Cromer et al.; Molecular Therapy Vol 27 No 4S1, April 2019). The full potential of this combination therapy for these and other genetic diseases will be more clear in the future (Hightower and Alexander, 2018; Rahit and Tarailo-Graovac, 2020).
While the possibility of combining gene replacement and endogenous gene regulation could attain unparalleled additive or synergic therapeutic effects, it is limited to the treatment of diseases for which a deep knowledge of the underlying molecular mechanism is available, and it requires careful examination.
Providing Novel Functions
Targeted integration can also provide cells with novel functions, such as a “safety-switch” for cell therapy applications. Transgene integration can be directed to inactivate an essential metabolic enzyme, the uridine monophosphate synthetase, which makes T cells dependent on supplemented uridine for their growth and survival (Wiebking et al., 2018). This approach could help therapies based on chimeric antigen receptor T cells by introducing a metabolic control of their proliferation and persistence. Further experiments are required to evaluate the clinical readiness of the approach.
Specificity Exchange
A special case of gene targeting is represented by the “specificity exchange” (Table 1F). Chimeric antigen receptors (CARs) are synthetic receptors that redirect and reprogram T cells to recognize specific antigens for tumor rejection (June and Sadelain, 2018). Initially, CARs were introduced in T cells using retroviral and lentiviral vectors (gene addition), with the risk of insertional mutagenesis. In addition, these CAR-T cells had two antigen specificities, the engineered one and the physiological one encoded by the endogenous αβ T cell receptor (TCR) chains, which may induce graft-vs-host disease when allogenic T cells are used (Torikai et al., 2012).
New CAR-T cells are generated by targeting the integration of the CAR transgene under the transcriptional control of TCR α-chain gene promoter to simultaneously achieve physiological expression of CAR and disruption of the endogenous TCR, thus maintaining only CAR antigen specificity (specificity exchange) (Eyquem et al., 2017; MacLeod et al., 2017). Overall, this strategy allows uniform CAR expression in human T cells and enhances T cell potency, outperforming conventional CAR-T cells.
A similar strategy has also been described to integrate and express a sequence encoding for a defined monoclonal antibody (Ab) of interest under the control of the heavy or light immunoglobulin chain promoter to reprogram B cells to secrete broadly neutralizing Ab against pathogens, for which no protective Ab has been isolated (Greiner et al., 2019; Hartweger et al., 2019; Moffett et al., 2019; Voss et al., 2019).
Conclusions
Over the past decades, gene therapy for blood disorders has mainly focused on the optimization of transgenes and synthetic promoters to improve expression and achieve therapeutic effects using gene replacement. However, this strategy is associated with the risk of insertional mutagenesis (LV) and episomal vector loss (AAV). The advent of the first generation of DNA endonucleases allowed the integration of transgenes in few selected genomic loci, mainly to achieve stable expression while minimizing insertional mutagenesis risk. Now, thanks to easily programmable nucleases such as CRISPR/Cas9, we have dramatically expanded our integration options and can creatively exploit different genomic locations to finely tune transgene expression or modulate disease-modifier genes to improve gene therapy outcomes.
A common strategy to target transgene integration combines nucleases with a donor DNA template (generally AAV) and leverages the homologous recombination pathway. However, before clinical translation, strict functional validation will be necessary to reduce potential adverse events associated with each individual component of this system. In particular, nucleases can induce potential off-targets (Kleinstiver et al., 2016; Carroll, 2019) and chromosomal alterations induced by on-target cleavage (Adikusuma et al., 2018; Kosicki et al., 2018; Cullot et al., 2019; Ledford, 2020); nucleases and AAV activate p53 response and trigger cell cycle arrest (Schwartz et al., 2007; Haapaniemi et al., 2018; Ihry et al., 2018); donor DNA integration can occur by different DNA repair mechanisms with outcomes sometimes difficult to predict (Canaj et al., 2019; Hanlon et al., 2019; Nelson et al., 2019); the target site needs to be functionally validated for safety and disposability (Papapetrou and Schambach, 2016).
Additional studies and further optimization of existing editing technologies will remove these hurdles and allow a broad clinical application of the described strategies to treat both monogenic and multifactorial blood diseases.
Statements
Author contributions
GP and MA wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Bayer (Hemophilia Awards Program), AFM-Telethon, INSERM, Genopole (Chaire Fondagen), the European Union's Horizon 2020 (SCIDNET No 666908), and the Agence nationale de la recherche (ANR-16-CE18 STaHR).
Acknowledgments
MA thanks the members of his laboratory at the Genethon Institute.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
AdikusumaF.PiltzS.CorbettM. A.TurveyM.McCollS. R.HelbigK. J.et al. (2018). Large deletions induced by Cas9 cleavage. Nature560, E8–E9. 10.1038/s41586-018-0380-z
2
AkhtarW.de JongJ.PindyurinA. V.PagieL.MeulemanW.de RidderJ.et al. (2013). Chromatin position effects assayed by thousands of reporters integrated in parallel. Cell154, 914–927. 10.1016/j.cell.2013.07.018
3
AmendolaM.van SteenselB. (2014). Mechanisms and dynamics of nuclear lamina-genome interactions. Curr. Opin. Cell Biol. 28, 61–68. 10.1016/j.ceb.2014.03.003
4
BananM. (2020). Recent advances in CRISPR/Cas9-mediated knock-ins in mammalian cells. J. Biotechnol. 308, 1–9. 10.1016/j.jbiotec.2019.11.010
5
BarzelA.PaulkN. K.ShiY.HuangY.ChuK.ZhangF.et al. (2015). Promoterless gene targeting without nucleases ameliorates haemophilia B in mice. Nature517, 360–364. 10.1038/nature13864
6
BhagwanJ. R.CollinsE.MosqueiraD.BakarM.JohnsonB. B.ThompsonA.et al. (2019). Variable expression and silencing of CRISPR-Cas9 targeted transgenes identifies the AAVS1 locus as not an entirely safe harbour. F1000Res8:1911. 10.12688/f1000research.19894.1
7
BortolussiG.ZentillinL.VanikovaJ.BockorL.BellarosaC.MancarellaA.et al. (2014). Life-long correction of hyperbilirubinemia with a neonatal liver-specific AAV-mediated gene transfer in a lethal mouse model of Crigler-Najjar Syndrome. Hum. Gene Ther. 25, 844–855. 10.1089/hum.2013.233
8
BoutinS.MonteilhetV.VeronP.LeborgneC.BenvenisteO.MontusM. F.et al. (2010). Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum. Gene Ther. 21, 704–712. 10.1089/hum.2009.182
9
BretonC.ClarkP. M.WangL.GreigJ. A.WilsonJ. M. (2020). ITR-Seq, a next-generation sequencing assay, identifies genome-wide DNA editing sites in vivo following adeno-associated viral vector-mediated genome editing. BMC Genom. 21:239. 10.1186/s12864-020-6655-4
10
BruecknerL.van ArensbergenJ.AkhtarW.PagieL.van SteenselB. (2016). High-throughput assessment of context-dependent effects of chromatin proteins. Epigenet. Chromatin9:43. 10.1186/s13072-016-0096-y
11
BushmanF. D. (2020). Retroviral insertional mutagenesis in humans: evidence for four genetic mechanisms promoting expansion of cell clones. Mol. Ther. 28, 352–356. 10.1016/j.ymthe.2019.12.009
12
CahillM. E.ConleyS.DeWanA. T.MontgomeryR. R. (2018). Identification of genetic variants associated with dengue or West Nile virus disease: a systematic review and meta-analysis. BMC Infect. Dis. 18:282. 10.1186/s12879-018-3186-6
13
CanajH.HussmannJ. A.LiH.BeckmanK. A.GoodrichL.ChoN. H.et al. (2019). Deep profiling reveals substantial heterogeneity of integration outcomes in CRISPR knock-in. bioRxiv [Preprint]. bioRxiv: 841098. 10.1101/841098
14
Cancer Genome Atlas Research Network (2017). Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell169, 1327–1341 e23. 10.1016/j.cell.2017.05.046
15
CarrollD. (2019). Collateral damage: benchmarking off-target effects in genome editing. Genome Biol. 20:114. 10.1186/s13059-019-1725-0
16
CavazzanaM.BushmanF. D.MiccioA.Andre-SchmutzI.SixE. (2019). Gene therapy targeting haematopoietic stem cells for inherited diseases: progress and challenges. Nat. Rev. Drug Discov. 18, 447–462. 10.1038/s41573-019-0020-9
17
ChenH.ShiM.GilamA.ZhengQ.ZhangY.AfrikanovaI.et al. (2019). Hemophilia A ameliorated in mice by CRISPR-based in vivo genome editing of human factor. Sci. Rep. 9:16838. 10.1038/s41598-019-53198-y
18
ChenH. C.MartinezJ. P.ZoritaE.MeyerhansA.FilionG. J. (2017). Position effects influence HIV latency reversal. Nat. Struct. Mol. Biol. 24, 47–54. 10.1038/nsmb.3328
19
ColellaP.RonzittiG.MingozziF. (2018). Emerging issues in AAV-mediated in vivo gene therapy. Mol. Ther. Methods Clin. Dev. 8, 87–104. 10.1016/j.omtm.2017.11.007
20
ConwayA.MendelM.KimK.McGovernK.BoykoA.ZhangL.et al. (2019). Non-viral delivery of zinc finger nuclease mRNA enables highly efficient in vivo genome editing of multiple therapeutic gene targets. Mol. Ther. 27, 866–877. 10.1016/j.ymthe.2019.03.003
21
CorralesM.RosadoA.CortiniR.van ArensbergenJ.van SteenselB.FilionG. J. (2017). Clustering of Drosophila housekeeping promoters facilitates their expression. Genome Res. 27, 1153–1161. 10.1101/gr.211433.116
22
CoxD. B.PlattR. J.ZhangF. (2015). Therapeutic genome editing: prospects and challenges. Nat. Med. 21, 121–131. 10.1038/nm.3793
23
CullotG.BoutinJ.ToutainJ.PratF.PennamenP.RooryckC.et al. (2019). CRISPR-Cas9 genome editing induces megabase-scale chromosomal truncations. Nat. Commun. 10:1136. 10.1038/s41467-019-09006-2
24
DavidoffA. M.NathwaniA. C. (2016). Genetic targeting of the albumin locus to treat Hemophilia. N. Engl. J. Med. 374, 1288–1290. 10.1056/NEJMcibr1600347
25
De CanevaA.PorroF.BortolussiG.SolaR.LisjakM.BarzelA.et al. (2019). Coupling AAV-mediated promoterless gene targeting to SaCas9 nuclease to efficiently correct liver metabolic diseases. JCI Insight. 5:128863. 10.1172/jci.insight.128863
26
De RavinS. S.LiL.WuX.ChoiU.AllenC.KoontzS.et al. (2017). CRISPR-Cas9 gene repair of hematopoietic stem cells from patients with X-linked chronic granulomatous disease. Sci. Transl. Med. 9:aah3480. 10.1126/scitranslmed.aah3480
27
De RavinS. S.ReikA.LiuP. Q.LiL.WuX.SuL.et al. (2016). Targeted gene addition in human CD34(+) hematopoietic cells for correction of X-linked chronic granulomatous disease. Nat. Biotechnol. 34, 424–429. 10.1038/nbt.3513
28
DeverD. P.BakR. O.ReinischA.CamarenaJ.WashingtonG.NicolasC. E.et al. (2016). CRISPR/Cas9 beta-globin gene targeting in human haematopoietic stem cells. Nature539, 384–389. 10.1038/nature20134
29
DeWittM. A.MagisW.BrayN. L.WangT.BermanJ. R.UrbinatiF.et al. (2016). Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci. Transl. Med. 8:360ra134. 10.1126/scitranslmed.aaf9336
30
DeyleD. R.RussellD. W. (2009). Adeno-associated virus vector integration. Curr Opin Mol Ther. 11, 442–447.
31
DiezB.GenoveseP.Roman-RodriguezF. J.AlvarezL.SchiroliG.UgaldeL.et al. (2017). Therapeutic gene editing in CD34(+) hematopoietic progenitors from Fanconi anemia patients. EMBO Mol. Med. 9, 1574–1588. 10.15252/emmm.201707540
32
DunbarC. E.HighK. A.JoungJ. K.KohnD. B.OzawaK.SadelainM. (2018). Gene therapy comes of age. Science359:aan4672. 10.1126/science.aan4672
33
EhrhardtA.XuH.KayM. A. (2003). Episomal persistence of recombinant adenoviral vector genomes during the cell cycle in vivo. J. Virol. 77, 7689–7695. 10.1128/JVI.77.13.7689-7695.2003
34
EyquemJ.Mansilla-SotoJ.GiavridisT.van der StegenS. J.HamiehM.CunananK. M.et al. (2017). Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature543, 113–117. 10.1038/nature21405
35
FalconA.CuevasM. T.Rodriguez-FrandsenA.ReyesN.PozoF.MorenoS.et al. (2015). CCR5 deficiency predisposes to fatal outcome in influenza virus infection. J. Gen. Virol. 96, 2074–2078. 10.1099/vir.0.000165
36
GajT.SirkS. J.ShuiS. L.LiuJ. (2016). Genome-editing technologies: principles and applications. Cold Spring Harb. Perspect. Biol. 8:a023754. 10.1101/cshperspect.a023754
37
GeninE.FeingoldJ.Clerget-DarpouxF. (2008). Identifying modifier genes of monogenic disease: strategies and difficulties. Hum. Genet. 124, 357–368. 10.1007/s00439-008-0560-2
38
GenoveseP.SchiroliG.EscobarG.TomasoT. D.FirritoC.CalabriaA.et al. (2014). Targeted genome editing in human repopulating haematopoietic stem cells. Nature510, 235–240. 10.1038/nature13420
39
Gomez-OspinaN.ScharenbergS. G.MostrelN.BakR. O.MantriS.QuadrosR. M.et al. (2019). Human genome-edited hematopoietic stem cells phenotypically correct Mucopolysaccharidosis type. Nat. Commun. 10:4045. 10.1038/s41467-019-11962-8
40
GreinerV.Bou PuertoR.LiuS.HerbelC.CarmonaE. M.GoldbergM. S. (2019). CRISPR-mediated editing of the B cell receptor in primary human B cells. iScience12, 369–378. 10.1016/j.isci.2019.01.032
41
HaapaniemiE.BotlaS.PerssonJ.SchmiererB.TaipaleJ. (2018). CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 24, 927–930. 10.1038/s41591-018-0049-z
42
Hacein-Bey-AbinaS.GarrigueA.WangG. P.SoulierJ.LimA.MorillonE.et al. (2008). Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Invest. 118, 3132–3142. 10.1172/JCI35700
43
Hacein-Bey-AbinaS.Von KalleC.SchmidtM.McCormackM. P.WulffraatN.LeboulchP.et al. (2003). LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science302, 415–419. 10.1126/science.1088547
44
HanlonK. S.KleinstiverB. P.GarciaS. P.ZaborowskiM. P.VolakA.SpirigS. E.et al. (2019). High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat. Commun. 10:4439. 10.1038/s41467-019-12449-2
45
HartwegerH.McGuireA. T.HorningM.TaylorJ. J.DosenovicP.YostD.et al. (2019). HIV-specific humoral immune responses by CRISPR/Cas9-edited B cells. J. Exp. Med. 216, 1301–1310. 10.1084/jem.20190287
46
HightowerR. M.AlexanderM. S. (2018). Genetic modifiers of Duchenne and facioscapulohumeral muscular dystrophies. Muscle Nerve. 57, 6–15. 10.1002/mus.25953
47
HirataR.ChamberlainJ.DongR.RussellD. W. (2002). Targeted transgene insertion into human chromosomes by adeno-associated virus vectors. Nat. Biotechnol. 20, 735–738. 10.1038/nbt0702-735
48
HubbardN.HaginD.SommerK.SongY.KhanI.CloughC.et al. (2016). Targeted gene editing restores regulated CD40L function in X-linked hyper-IgM syndrome. Blood127, 2513–2522. 10.1182/blood-2015-11-683235
49
HungK. L.MeitlisI.HaleM.ChenC. Y.SinghS.JacksonS. W.et al. (2018). Engineering protein-secreting plasma cells by homology-directed repair in primary human B cells. Mol. Ther. 26, 456–467. 10.1016/j.ymthe.2017.11.012
50
HutterG.NowakD.MossnerM.GanepolaS.MussigA.AllersK.et al. (2009). Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N. Engl. J. Med. 360, 692–698. 10.1056/NEJMoa0802905
51
IhryR. J.WorringerK. A.SalickM. R.FriasE.HoD.TheriaultK.et al. (2018). p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat. Med. 24, 939–946. 10.1038/s41591-018-0050-6
52
IrionS.LucheH.GadueP.FehlingH. J.KennedyM.KellerG. (2007). Identification and targeting of the ROSA26 locus in human embryonic stem cells. Nat. Biotechnol. 25, 1477–1482. 10.1038/nbt1362
53
JuneC. H.SadelainM. (2018). Chimeric antigen receptor therapy. N. Engl. J. Med. 379, 64–73. 10.1056/NEJMra1706169
54
KlattD.ChengE.HoffmannD.SantilliG.ThrasherA. J.BrendelC.et al. (2020). Differential transgene silencing of myeloid-specific promoters in the AAVS1 safe harbor locus of induced pluripotent stem cell-derived myeloid cells. Hum. Gene Ther. 31, 199–210. 10.1089/hum.2019.194
55
KleinstiverB. P.PattanayakV.PrewM. S.TsaiS. Q.NguyenN. T.ZhengZ.et al. (2016). High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature529, 490–495. 10.1038/nature16526
56
KohamaY.HigoS.MasumuraY.ShibaM.KondoT.IshizuT.et al. (2020). Adeno-associated virus-mediated gene delivery promotes S-phase entry-independent precise targeted integration in cardiomyocytes. Sci. Rep. 10:15348. 10.1038/s41598-020-72216-y
57
KosickiM.TombergK.BradleyA. (2018). Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 36, 765–771. 10.1038/nbt.4192
58
KotinR. M.LindenR. M.BernsK. I. (1992). Characterization of a preferred site on human chromosome 19q for integration of adeno-associated virus DNA by non-homologous recombination. EMBO J. 11, 5071–5078. 10.1002/j.1460-2075.1992.tb05614.x
59
KuoC. Y.LongJ. D.Campo-FernandezB.de OliveiraS.CooperA. R.RomeroZ.et al. (2018). Site-specific gene editing of human hematopoietic stem cells for X-linked hyper-IgM syndrome. Cell Rep. 23, 2606–2616. 10.1016/j.celrep.2018.04.103
60
LamartinaS.SporenoE.FattoriE.ToniattiC. (2000). Characteristics of the adeno-associated virus preintegration site in human chromosome 19: open chromatin conformation and transcription-competent environment. J. Virol. 74, 7671–7677. 10.1128/JVI.74.16.7671-7677.2000
61
LaoharaweeK.DeKelverR. C.Podetz-PedersenK. M.RohdeM.SproulS.NguyenH. O.et al. (2018). Dose-dependent prevention of metabolic and neurologic disease in murine MPS II by ZFN-mediated in vivo genome editing. Mol. Ther. 26, 1127–1136. 10.1016/j.ymthe.2018.03.002
62
LarenaM.RegnerM.LobigsM. (2012). The chemokine receptor CCR5, a therapeutic target for HIV/AIDS antagonists, is critical for recovery in a mouse model of Japanese encephalitis. PLoS ONE7:e44834. 10.1371/journal.pone.0044834
63
LedfordH. (2020). CRISPR gene editing in human embryos wreaks chromosomal mayhem. Nature583, 17–18. 10.1038/d41586-020-01906-4
64
LiA.LeeC. M.HurleyA. E.JarrettK. E.De GiorgiM.LuW.et al. (2019). A self-deleting AAV-CRISPR system for in vivo genome editing. Mol. Ther. Methods Clin. Dev. 12, 111–122. 10.1016/j.omtm.2018.11.009
65
LiC.HirschM.CarterP.AsokanA.ZhouX.WuZ.et al. (2009). A small regulatory element from chromosome 19 enhances liver-specific gene expression. Gene Ther. 16, 43–51. 10.1038/gt.2008.134
66
LiC.SamulskiR. J. (2020). Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 21, 255–272. 10.1038/s41576-019-0205-4
67
LiH.HaurigotV.DoyonY.LiT.WongS. Y.BhagwatA. S.et al. (2011). In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature475, 217–221. 10.1038/nature10177
68
LimJ. K.GlassW. G.McDermottD. H.MurphyP. M. (2006). CCR5: no longer a “good for nothing” gene–chemokine control of West Nile virus infection. Trends Immunol. 27, 308–312. 10.1016/j.it.2006.05.007
69
LombardoA.CesanaD.GenoveseP.Di StefanoB.ProvasiE.ColomboD. F.et al. (2011). Site-specific integration and tailoring of cassette design for sustainable gene transfer. Nat. Methods8, 861–869. 10.1038/nmeth.1674
70
LombardoA.GenoveseP.BeausejourC. M.ColleoniS.LeeY. L.KimK. A.et al. (2007). Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat. Biotechnol. 25, 1298–1306. 10.1038/nbt1353
71
MacLeodD. T.AntonyJ.MartinA. J.MoserR. J.HekeleA.WetzelK. J.et al. (2017). Integration of a CD19 CAR into the TCR alpha chain locus streamlines production of allogeneic gene-edited CAR T cells. Mol. Ther. 25, 949–961. 10.1016/j.ymthe.2017.02.005
72
MasatE.PavaniG.MingozziF. (2013). Humoral immunity to AAV vectors in gene therapy: challenges and potential solutions. Discov. Med. 15, 379–389.
73
MettanandaS.GibbonsR. J.HiggsD. R. (2015). alpha-Globin as a molecular target in the treatment of beta-thalassemia. Blood125, 3694–3701. 10.1182/blood-2015-03-633594
74
MillerD. G.PetekL. M.RussellD. W. (2004). Adeno-associated virus vectors integrate at chromosome breakage sites. Nat. Genet. 36, 767–773. 10.1038/ng1380
75
MingozziF.HighK. A. (2011). Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat. Rev. Genet. 12, 341–355. 10.1038/nrg2988
76
MoffettH. F.HarmsC. K.FitzpatrickK. S.TooleyM. R.BoonyaratanakornkitJ.TaylorJ. J. (2019). B cells engineered to express pathogen-specific antibodies protect against infection. Sci. Immunol. 4:aax0644. 10.1126/sciimmunol.aax0644
77
MooreJ. E.PurcaroM. J.PrattH. E.EpsteinC. B.ShoreshN.AdrianJ.et al. (2020). Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature583, 699–710. 10.1038/s41586-020-2493-4
78
NakaiH.YantS. R.StormT. A.FuessS.MeuseL.KayM. A. (2001). Extrachromosomal recombinant adeno-associated virus vector genomes are primarily responsible for stable liver transduction in vivo. J. Virol. 75, 6969–6976. 10.1128/JVI.75.15.6969-6976.2001
79
NaldiniL. (2011). Ex vivo gene transfer and correction for cell-based therapies. Nat. Rev. Genet. 12, 301–315. 10.1038/nrg2985
80
NaldiniL.BlomerU.GallayP.OryD.MulliganR.GageF. H.et al. (1996). In vivo gene delivery and stable transduction of non-dividing cells by a lentiviral vector. Science272, 263–267. 10.1126/science.272.5259.263
81
NelsonC. E.WuY.GemberlingM. P.OliverM. L.WallerM. A.BohningJ. D.et al. (2019). Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 25, 427–432. 10.1038/s41591-019-0344-3
82
NishiguchiK. M.FujitaK.MiyaF.KatayamaS.NakazawaT. (2020). Single AAV-mediated mutation replacement genome editing in limited number of photoreceptors restores vision in mice. Nat. Commun. 11:482. 10.1038/s41467-019-14181-3
83
NishiyamaJ. (2019). Genome editing in the mammalian brain using the CRISPR-Cas system. Neurosci. Res. 141, 4–12. 10.1016/j.neures.2018.07.003
84
OgataT.KozukaT.KandaT. (2003). Identification of an insulator in AAVS1, a preferred region for integration of adeno-associated virus DNA. J. Virol. 77, 9000–9007. 10.1128/JVI.77.16.9000-9007.2003
85
OrdovasL.BoonR.PistoniM.ChenY.WolfsE.GuoW.et al. (2018). Efficient recombinase-mediated cassette exchange in hPSCs to study the hepatocyte lineage reveals AAVS1 locus-mediated transgene inhibition. Stem Cell Rep. 10:673. 10.1016/j.stemcr.2018.01.034
86
OuL.DeKelverR. C.RohdeM.TomS.RadekeR.St MartinS. J.et al. (2019). ZFN-mediated in vivo genome editing corrects murine hurler syndrome. Mol. Ther. 27, 178–187. 10.1016/j.ymthe.2018.10.018
87
OuL.PrzybillaM. J.AhlatO.KimS.OvernP.JarnesJ.et al. (2020). A highly efficacious PS gene editing system corrects metabolic and neurological complications of Mucopolysaccharidosis type I. Mol. Ther. 28, 1442–1454. 10.1016/j.ymthe.2020.03.018
88
PapapetrouE. P.SchambachA. (2016). Gene insertion into genomic safe harbors for human gene therapy. Mol. Ther. 24, 678–684. 10.1038/mt.2016.38
89
PavaniG.LaurentM.FabianoA.CantelliE.SakkalA.CorreG.et al. (2020). Ex vivo editing of human hematopoietic stem cells for erythroid expression of therapeutic proteins. Nat. Commun. 11:3778. 10.1038/s41467-020-17552-3
90
PerezE. E.WangJ.MillerJ. C.JouvenotY.KimK. A.LiuO.et al. (2008). Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat. Biotechnol. 26, 808–816. 10.1038/nbt1410
91
PraetoriusF.KickB.BehlerK. L.HonemannM. N.Weuster-BotzD.DietzH. (2017). Biotechnological mass production of DNA origami. Nature552, 84–87. 10.1038/nature24650
92
RahitK.Tarailo-GraovacM. (2020). Genetic modifiers and rare mendelian disease. Genes. 11:30239. 10.3390/genes11030239
93
RaiR.RomitoM.RiversE.TurchianoG.BlattnerG.VetharoyW.et al. (2020). Targeted gene correction of human hematopoietic stem cells for the treatment of Wiskott - Aldrich Syndrome. Nat. Commun. 11:4034. 10.1038/s41467-020-17626-2
94
RanF. A.HsuP. D.LinC. Y.GootenbergJ. S.KonermannS.TrevinoA. E.et al. (2013). Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell154, 1380–1389. 10.1016/j.cell.2013.08.021
95
RaoC. V.AschA. S.YamadaH. Y. (2017). Frequently mutated genes/pathways and genomic instability as prevention targets in liver cancer. Carcinogenesis38, 2–11. 10.1093/carcin/bgw118
96
RomeroZ.LomovaA.SaidS.MiggelbrinkA.KuoC. Y.Campo-FernandezB.et al. (2019). Editing the sickle cell disease mutation in human hematopoietic stem cells: comparison of endonucleases and homologous donor templates. Mol. Ther. 27, 1389–1406. 10.1016/j.ymthe.2019.05.014
97
RothT. L.Puig-SausC.YuR.ShifrutE.CarnevaleJ.LiP. J.et al. (2018). Reprogramming human T cell function and specificity with non-viral genome targeting. Nature559, 405–409. 10.1038/s41586-018-0326-5
98
SadelainM.PapapetrouE. P.BushmanF. D. (2011). Safe harbours for the integration of new DNA in the human genome. Nat. Rev. Cancer. 12, 51–58. 10.1038/nrc3179
99
ScharenbergS. G.PolettoE.LucotK. L.ColellaP.SheikaliA.MontineT. J.et al. (2020). Engineering monocyte/macrophage-specific glucocerebrosidase expression in human hematopoietic stem cells using genome editing. Nat. Commun. 11:3327. 10.1038/s41467-020-17148-x
100
SchenkweinD.AfzalS.NousiainenA.SchmidtM.Yla-HerttualaS. (2020). Efficient nuclease-directed integration of lentivirus vectors into the human ribosomal DNA locus. Mol. Ther. 28, 1858–1875. 10.1016/j.ymthe.2020.05.019
101
SchiroliG.FerrariS.ConwayA.JacobA.CapoV.AlbanoL.et al. (2017). Preclinical modeling highlights the therapeutic potential of hematopoietic stem cell gene editing for correction of SCID-X1. Sci. Transl. Med. 9:aan0820. 10.1126/scitranslmed.aan0820
102
SchoenfelderS.FraserP. (2019). Long-range enhancer-promoter contacts in gene expression control. Nat. Rev. Genet. 20, 437–455. 10.1038/s41576-019-0128-0
103
SchroderA. R.ShinnP.ChenH.BerryC.EckerJ. R.BushmanF. (2002). HIV-1 integration in the human genome favors active genes and local hotspots. Cell110, 521–529. 10.1016/S0092-8674(02)00864-4
104
SchwartzR. A.PalaciosJ. A.CassellG. D.AdamS.GiaccaM.WeitzmanM. D. (2007). The Mre11/Rad50/Nbs1 complex limits adeno-associated virus transduction and replication. J. Virol. 81, 12936–12945. 10.1128/JVI.01523-07
105
SharmaR.AnguelaX. M.DoyonY.WechslerT.DeKelverR. C.SproulS.et al. (2015). In vivo genome editing of the albumin locus as a platform for protein replacement therapy. Blood126, 1777–1784. 10.1182/blood-2014-12-615492
106
ShinJ. J.SchroderM. S.CaiadoF.WymanS. K.BrayN. L.BordiM.et al. (2020). Controlled cycling and quiescence enables efficient HDR in engraftment-enriched adult hematopoietic stem and progenitor cells. Cell Rep. 32:108093. 10.1016/j.celrep.2020.108093
107
SimhadriV. L.McGillJ.McMahonS.WangJ.JiangH.SaunaZ. E. (2018). Prevalence of pre-existing antibodies to CRISPR-associated nuclease Cas9 in the USA population. Mol. Ther. Methods Clin. Dev. 10, 105–112. 10.1016/j.omtm.2018.06.006
108
SmithJ. R.MaguireS.DavisL. A.AlexanderM.YangF.ChandranS.et al. (2008). Robust, persistent transgene expression in human embryonic stem cells is achieved with AAVS1-targeted integration. Stem Cells26, 496–504. 10.1634/stemcells.2007-0039
109
StephensC. J.KashentsevaE.EverettW.KaliberovaL.CurielD. T. (2018). Targeted in vivo knock-in of human alpha-1-antitrypsin cDNA using adenoviral delivery of CRISPR/Cas9. Gene Ther. 25, 139–156. 10.1038/s41434-018-0003-1
110
StephensC. J.LauronE. J.KashentsevaE.LuZ. H.YokoyamaW. M.CurielD. T. (2019). Long-term correction of hemophilia B using adenoviral delivery of CRISPR/Cas9. J. Contr. Release. 298, 128–141. 10.1016/j.jconrel.2019.02.009
111
SurksH. K.RichardsC. T.MendelsohnM. E. (2003). Myosin phosphatase-Rho interacting protein. A new member of the myosin phosphatase complex that directly binds RhoA. J. Biol. Chem. 278, 51484–51493. 10.1074/jbc.M305622200
112
SuzukiK.TsunekawaY.Hernandez-BenitezR.WuJ.ZhuJ.KimE. J.et al. (2016). In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature540, 144–149. 10.1038/nature20565
113
SuzukiK.YamamotoM.Hernandez-BenitezR.LiZ.WeiC.SoligallaR. D.et al. (2019). Precise in vivo genome editing via single homology arm donor mediated intron-targeting gene integration for genetic disease correction. Cell Res. 29, 804–819. 10.1038/s41422-019-0213-0
114
SweeneyC. L.ZouJ.ChoiU.MerlingR. K.LiuA.BodanskyA.et al. (2017). Targeted repair of CYBB in X-CGD iPSCs requires retention of intronic sequences for expression and functional correction. Mol. Ther. 25, 321–330. 10.1016/j.ymthe.2016.11.012
115
TalbertP. B.MeersM. P.HenikoffS. (2019). Old cogs, new tricks: the evolution of gene expression in a chromatin context. Nat. Rev. Genet. 20, 283–297. 10.1038/s41576-019-0105-7
116
TorikaiH.ReikA.LiuP. Q.ZhouY.ZhangL.MaitiS.et al. (2012). A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood119, 5697–5705. 10.1182/blood-2012-01-405365
117
ToscanoM. G.RomeroZ.MunozP.CoboM.BenabdellahK.MartinF. (2011). Physiological and tissue-specific vectors for treatment of inherited diseases. Gene Ther. 18, 117–127. 10.1038/gt.2010.138
118
UrnovF. D.MillerJ. C.LeeY. L.BeausejourC. M.RockJ. M.AugustusS.et al. (2005). Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature435, 646–651. 10.1038/nature03556
119
VansantG.ChenH. C.ZoritaE.TrejbalovaK.MiklikD.FilionG.et al. (2020). The chromatin landscape at the HIV-1 provirus integration site determines viral expression. Nucl. Acids Res. 48, 7801–7817. 10.1093/nar/gkaa536
120
VoitR. A.HendelA.Pruett-MillerS. M.PorteusM. H. (2014). Nuclease-mediated gene editing by homologous recombination of the human globin locus. Nucl. Acids Res. 42, 1365–1378. 10.1093/nar/gkt947
121
VoitR. A.McMahonM. A.SawyerS. L.PorteusM. H. (2013). Generation of an HIV resistant T-cell line by targeted “stacking” of restriction factors. Mol. Ther. 21, 786–795. 10.1038/mt.2012.284
122
VossJ. E.Gonzalez-MartinA.AndrabiR.FullerR. P.MurrellB.McCoyL. E.et al. (2019). Reprogramming the antigen specificity of B cells using genome-editing technologies. Elife8:42995. 10.7554/eLife.42995
123
WangH.LuH.LeiY. S.GongC. Y.ChenZ.LuanY. Q.et al. (2020c). Development of a self-restricting CRISPR-Cas9 system to reduce off-target effects. Mol. Ther. Methods Clin. Dev. 18, 390–401. 10.1016/j.omtm.2020.06.012
124
WangJ.DeClercqJ. J.HaywardS. B.LiP. W.ShivakD. A.GregoryP. D.et al. (2016). Highly efficient homology-driven genome editing in human T cells by combining zinc-finger nuclease mRNA and AAV6 donor delivery. Nucl. Acids Res. 44:e30. 10.1093/nar/gkv1121
125
WangJ.ExlineC. M.DeClercqJ. J.LlewellynG. N.HaywardS. B.LiP. W.et al. (2015). Homology-driven genome editing in hematopoietic stem and progenitor cells using ZFN mRNA and AAV6 donors. Nat. Biotechnol. 33, 1256–1263. 10.1038/nbt.3408
126
WangL.YangY.BretonC.BellP.LiM.ZhangJ.et al. (2020a). A mutation-independent CRISPR-Cas9-mediated gene targeting approach to treat a murine model of ornithine transcarbamylase deficiency. Sci. Adv. 6:eaax5701. 10.1126/sciadv.aax5701
127
WangL.YangY.BretonC. A.WhiteJ.ZhangJ.CheY.et al. (2019). CRISPR/Cas9-mediated in vivo gene targeting corrects hemostasis in newborn and adult factor IX-knockout mice. Blood133, 2745–2752. 10.1182/blood.2019000790
128
WangQ.ZhongX.LiQ.SuJ.LiuY.MoL.et al. (2020b). CRISPR-Cas9-mediated in vivo gene integration at the albumin locus recovers hemostasis in neonatal and adult hemophilia B mice. Mol. Ther. Methods Clin. Dev. 18, 520–531. 10.1016/j.omtm.2020.06.025
129
WiebkingV.PattersonJ. O.MartinR.ChandaM. K.LeeC. M.SrifaW.et al. (2018). Metabolic engineering generates a transgene-free safety switch for cell therapy. Nat. Biotechnol2020:6. 10.1038/s41587-020-0580-6
130
YuS.OuY.XiaoH.LiJ.AdahD.LiuS.et al. (2020). Experimental treatment of SIV-infected macaques via autograft of CCR5-disrupted hematopoietic stem and progenitor cells. Mol. Ther. Methods Clin. Dev. 17, 520–531. 10.1016/j.omtm.2020.03.004
131
ZambrowiczB. P.ImamotoA.FieringS.HerzenbergL. A.KerrW. G.SorianoP. (1997). Disruption of overlapping transcripts in the ROSA beta geo 26 gene trap strain leads to widespread expression of beta-galactosidase in mouse embryos and hematopoietic cells. Proc. Natl. Acad. Sci. U. S. A. 94, 3789–3794. 10.1073/pnas.94.8.3789
132
ZhangD.HuangP.SharmaM.KellerC. A.GiardineB.ZhangH. (2020). Alteration of genome folding via contact domain boundary insertion. Nat. Genet. 52, 1076–1087. 10.1038/s41588-020-0680-8
133
ZhangJ. P.ChengX. X.ZhaoM.LiG. H.XuJ.ZhangF.et al. (2019). Curing hemophilia A by NHEJ-mediated ectopic F8 insertion in the mouse. Genome Biol. 20:276. 10.1186/s13059-019-1907-9
134
ZhengH.XieW. (2019). The role of 3D genome organization in development and cell differentiation. Nat. Rev. Mol. Cell Biol. 20, 535–550. 10.1038/s41580-019-0132-4
Summary
Keywords
genome editing, gene therapy, nuclease, CRISPR, targeted integration (TI), knock-in, safe harbor, homologous recombination (HR)
Citation
Pavani G and Amendola M (2021) Targeted Gene Delivery: Where to Land. Front. Genome Ed. 2:609650. doi: 10.3389/fgeed.2020.609650
Received
23 September 2020
Accepted
16 December 2020
Published
20 January 2021
Volume
2 - 2020
Edited by
Pietro Genovese, Boston Children's Hospital and Harvard Medical School, United States
Reviewed by
Christian Brendel, Boston Children's Hospital, United States; Giulia Schiroli, Massachusetts General Hospital and Harvard Medical School, United States
Updates
Copyright
© 2021 Pavani and Amendola.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mario Amendola mamendola@genethon.fr
†Present address: Giulia Pavani, The Children's Hospital of Philadelphia, Raymond G. Perelman Center for Cellular and Molecular Therapeutics, Philadelphia, PA, United States
This article was submitted to Genome Editing in Blood Disorders, a section of the journal Frontiers in Genome Editing
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.