 Daniela Reyes1
Daniela Reyes1 John Gold
John Gold Rodrigo Vidal
Rodrigo Vidal- 1Laboratory of Molecular Ecology, Genomics and Evolutionary Studies, Department of Biology, Universidad de Santiago, Santiago, Chile
- 2Marine Genomics Laboratory, Department of Life Sciences, Center for Biosystematics and Biodiversity, Harte Research Institute, Texas A and M University, Corpus Christi, TX, USA
Southern hake (Merluccius australis) is an ecological and economically important demersal fish in Chile and Argentina. Notwithstanding, genetic resource for genetic or ecological studies on this species are scarce. Consequently, here we present transcriptome sequencing results (RNA-Seq) for spleen and liver tissues with the 454 FLX titanium platform. The de novo transcriptome assembly generated 10,314 unigenes, with an average length of 510 bp, N50 of 572 bp and 3171 annotated sequences. A specific Gadiform BLAST search, with focus on immune genes, showed 186 (56%) genes homologous to those of Atlantic cod. A total of 2302 microsatellites were detected in 1687 unigenes and 741 presented adequate flanking sequences for primer design. In total, these potential molecular markers and transcriptome characterization represent an important resource for genetic and ecological studies on southern hake.
Introduction
The Merluccius genus is a widely distributed and important group in marine fisheries, which resides in the continental shelf and slope (Quinteiro et al., 2000). Although many taxonomic studies addressing this genus have been published, the most comprehensive ones conclude that this genus contains about 12–13 species and several subspecies (Inada, 1981; Cohen et al., 1990). The southern hake, Merluccius australis, is a circumglobal demersal fish with two described populations or subspecies described (Lloris et al., 2005). One subspecies denominated M. australis polylepis is found primarily from 40°S in the southeast Pacific (Chile) southwards around the southern tip of South America, to 38°S in the southwest Atlantic (Argentina), and another subspecies called M. australis is present in Chatham Rise, Campbell Plateau and South Island northward to the East Cape. This species comprises one the most economically important industrial and artisanal fisheries in the Southern Hemisphere (Alheit and Pitcher, 1995). It also plays a critical ecological role in the marine ecosystem (Ojeda et al., 2011). Moreover, the southern hake is considered a potential farming species and its status corresponds to the phase of demonstration, where its commercial potential is being evaluated (Bricknell et al., 2006). Despite its nutritional, ecological and economic importance, genomics resources and research on this species are scarce. This is illustrated, for example, by the low number of specific molecular markers and genome sequences available for this species (Machado-Schiaffino and Garcia-Vazquez, 2009; Mabragaña et al., 2011; Renshaw et al., 2011). In June 2016, there was no information on southern hake EST available in NCBI dbEST. This shows clear contrast with another relevant hake species, for example, the European hake (Merluccius merluccius), for which a number of transcriptomic and molecular markers resources have been recently developed (Milano et al., 2011, 2014).
In last years, transcriptome analysis was recognized as a key tool for the development and improvement of several aquaculture species (McAndrew and Napier, 2011). Availability of resources in the genome level for southern hake will enhance its genetic and ecologic studies, aquaculture development and contribute to future comparative genomic research on the Merluccius genus. Therefore, in this study we conducted 454 pyrosequencing of one cDNA library isolated from southern hake. Our goals were to (i) characterize the kidney and liver transcriptome of southern hake (M. australis polylepis) and (ii) describe and characterize molecular markers for future genetic analyses. We choose kidney and liver tissues, because both have been described as key organs for immune defense in fishes (Lieschke and Trede, 2009) and its transcriptome characterization will have important utility for both scientific and applied purpose.
Materials and Methods
Tissue Collection and RNA Extraction
Spleen and liver tissue were collected by fisherman from 10 commercial caught wild adult individuals (41°28′ S-72°56′ W). All the animal experiments in this study were approved and conformed to the Institutional Ethics Committee, Universidad de Santiago, guidelines. Initially, the tissue were placed in RNAlater buffer (Ambion Inc.) and stored at 4°C by 24 h and then stored at −20°C. Total RNA was extracted according to Cepeda et al. (2011). The concentration and integrity of the RNA were evaluated by spectrophotometry (Nanodrop) and by Agilent Bioanalyser 2100, respectively.
cDNA Library and Sequencing
The MINT-2 kit (Evrogen) was utilized for cDNA synthesis according to the manufacturer's instructions. Double-stranded cDNAs were submitted to McGill University and Genome Québec Innovation Centre for library construction and sequencing. One cDNA library was constructed from pooled equal amounts of RNA from both tissues and sequencing was conducted on a 454 GS FLX titanium instrument.
Sequence Pre-processing and De novo Assembly
The pre-processing of the obtained reads was performed using CLC Genomics Workbench tools version 7.0.3 (CLCBio) and included the trimming of polyA tails, adaptors and low quality fragments (quality score limit = 0.05, ambiguous nucleotides = 2), as well the elimination of duplicated reads and reads with length below 50 base pairs (bp). Processed reads were de novo assembled with CLC de novo assembly tool (minimum contig length = 100, deletion cost = 3, insertion cost = 3, mismatch cost = 2, length fraction = 0.5, similarity fraction = 0.5) and MIRA3 (job = de novo, est, accurate,454, 454_SETTINGS -AL:mo = 40, −SK:pr = 97) (Chevreux et al., 2004) programs. The obtained contigs from both programs were merged and re-assembled with the CAP3 program (default parameters, except to overlap = 40, identity = 97) (Huang and Madan, 1999) to improve the credibility of the assembly (Kumar and Blaxter, 2010).
Sequence Annotation
The functional annotation of the obtained contigs was done through a local alignment against Swiss-Prot database (Bairoch and Apweiler, 2000) and NCBI non-redundant protein database (nr) (June 2016) using the BLASTX program (Camacho et al., 2009), with E-value cut off equal to 1e-3 and 1e-6, respectively. Gene Ontology (GO) terms and enzyme commission (EC) associated to the blast hits were recovered using the BLAST2GO suite tools (Götz et al., 2008), whereas metabolic pathways assignments were performed using tools supplied by Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa and Goto, 2000).
Southern Hake Transcriptome Completeness
Parra et al. (2007) have developed a list of 248 highly conserved core eukaryotic genes (CEGs), which can be considered as an appropriate indicator of completeness of gene cover in eukaryotic organisms. We use BLASTX to map the transcripts obtained of southern hake to the CEGs; hit of e ≤ 1.0e-12 were considered as a match. To further characterize the southern hake transcripts related with immune function, the term corresponding to “immune system” GO level 2 (and its respective children) of Atlantic cod (Gadus morhua; gadMor1) were downloaded from ENSEMBL (BioMart). The ENSEMBL codes were used to download the corresponding protein sequences. Then, we used BLASTX to map the transcripts obtained of southern hake to the Atlantic cod immune system database; hit of e ≤ 1.0e-12 were considered as a match.
Simple Sequence Repeats Identification and Validation
The identification of simple sequence repeats (SSRs) was made using the SSRIT Perl script (Temnykh et al., 2001), which was modified to detect perfect microsatellites from dimers to decamers with at least 5 repeats. Mononucleotide repeats were not considered. These settings are in agreement with the suggested by Doyle et al. (2013).
The program Primer 3.0 (Rozen and Skaletsky, 2000) was utilized for the design of primers. For microsatellite validation, we selected 14 microsatellite markers based on two criteria: (i) availability of sequence with annotation and (ii) prioritization of di or tri-nucleotide loci, which have been described as having a potential for higher mutation rates (Ellegren, 2004). Genomic DNA was extracted in accordance with Vidal et al. (2012) from muscle tissues collected from 18 wild-caught adult individuals (41°28′ S-72°56′ W) associated with commercial bottom fisheries activities. Each microsatellite was amplified using the methodology described in Renshaw et al. (2011). Amplified microsatellites PCR products were genotyped on a ABI 377 Automated Sequencer and output runs were analyzed with Genotyper 2.5 (Perkin Elmer) and Genescan 3.1.2 (Applied Biosystems). Observed (Ho) and expected (He) heterozygosities and Hardy-Weinberg equilibrium (HWE) were calculated for each locus using GenAlex ver 6.5 (Peakall and Smouse, 2012).
Results and Discussion
Sequence Pre-processing and De novo Assembly
In last years, genomics research in aquaculture and fisheries species have attracted great interest, and a number of genomic resources had been developed. In the present study we performed, for the first time, a comprehensive spleen and liver tissues transcriptome sequencing of southern hake from mixed cDNA libraries of 10 adult samples. The run generated 950,000 raw reads (Short Read Archive SRP081286). Following the processing trimming steps, the sequence sets were reduced to 729,400 reads, with a minimum length of 50 bp and with an average length of 291 bp (Table 1). Currently, there is no reference genome available for this species, thus we used the de novo assembly (Mundry et al., 2012). The pre-processed reads were assembled using CLC de novo assembler and MIRA3 programs, obtaining 10,379 and 7264 unigenes, respectively, which were merged and re-assembled using CAP3 program, obtaining, in turn, a total of 10,314 unigenes with an average length of 510 bp and N50 of 572 bp (Table 2). The higher N50 obtained after CAP3 confirms that merging different assemblies can produce higher N50 values (Kumar and Blaxter, 2010). Although N50 is not an exact parameter to measure the accuracy of contigs (Salzberg et al., 2012), it is one of the most popular reference-free measures (Li et al., 2014). The N50 value was in the range reported for other transcriptome de novo fish studies (Milano et al., 2011; Fan et al., 2014; Schunter et al., 2014).

Table 1. Summary of raw and pre-processed reads.
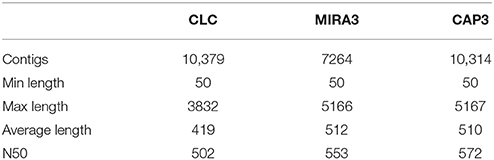
Table 2. Summary of the assemblies done with CLC, MIRA3, and CAP3.
Sequence Annotation and Transcriptome Completeness
To understand the role of the assembled southern hake unigenes, they were annotated using several public databases. All the sequences were aligned against the NCBI non-redundant protein (nr) and Swiss-Prot databases using BLASTX program, 3171 out of the 10,314 unigenes (30.74%) having significant BLAST matches (Supplementary Table S1). Of these, approximately 75% (2401) of the unigenes had significant BLAST matches with fish species, such as Nile tilapia, zebrafish, Atlantic salmon and spotted green pufferfish, among others (Table 3). The remaining 7143 (69.26%) unigenes resulted in non-significant hits, a result similar to other studies of non-model fish species (Shin et al., 2012; Qian et al., 2014). Normally this happens either due to incomplete gene information on non-model species in public database or because sequences of non-coding RNAs among the unigenes are included (Hou et al., 2011). Alternatively, a length effect has been described by Ding et al. (2015), in which short reads, normally obtained in high sequencing approaches, would reduce the BLAST annotation efficiency. However, our results do not support this last suggestion, because unigenes sequences shorter and longer that 572 bp had almost the same rate of annotation (result not shown). Therefore, ours results suggest a huge potential for the disclosure of new genes, species-specific genes or non-coding sequences in this hake species.

Table 3. Top-hit species distribution of BLAST results.
An important challenge in de novo transcriptome assemblies' research consists in selecting an adequate sequencing depth to cover an appropriate range of transcripts expressed in a particular sample and organism. There is a limited number of approaches available for evaluating the amount of reads to produce an adequate depth coverage of transcript expressed in a determined species and sample. An appropriate metric was developed by Parra et al. (2007) based on 248 CEG, of which 72% were covered by the southern hake transcripts obtained in this study. Moreover, we extracted 184 GO terms associated with immune system and 358 corresponding immune proteins for Atlantic cod. BLAST search against this immune database showed that southern hake unigenes matched 52.2% (186) of the entire Atlantic cod database. In comparison, the percentage of all the Atlantic cod genes available in public database with hits to the southern hake unigenes was noticeably less (30%). We consider that these results are due to the prevailing immune function of liver and spleen tissue in fish (Lieschke and Trede, 2009). To further characterize the assembled unigenes, the BLAST results were submitted to BLAST2GO suite for annotation of Gene Ontology (GO) terms and enzyme commission (EC) numbers. A total of 16,728 GO terms were associated with the 3171 unigenes, with the higher number of GO terms linked to Biological process (9406, 45%) followed by Cellular component (7430, 36%) and Molecular function (4090, 20%). The distribution and composition of the GO terms obtained were similar to those reported in another fish, including another hake species, European hake (Milano et al., 2011; Liao et al., 2013; Li et al., 2015). In the Biological process category, proteins involved in metabolic and cellular processes were dominant, while in the categories of Cellular component and Molecular function, cell and cell part, and catalytic activity and binding proteins, respectively, were dominant (Figure 1). Of the 3171 sequences with GO terms related, 776 had a total of 845 EC numbers. KEGG analysis linked the annotated sequences to 10,428 metabolic pathways, being a large number of sequences related to carbohydrate metabolism (205), translation (252), signal transduction (270), cancers (360), neurodegenerative diseases (262) and infectious diseases (680) (Table 4). Moreover, several transcripts (119) associated with key immune response (GO:0002376) were identified. For example, we have identified various major histocompatibility antigens transcripts, which is consistent with the great variability reported for this gene group (Cohen et al., 2006). Likewise, we have detected several transcript with crucial roles in the immune and stress response such as cytokines (tumor necrosis alpha, interleukin), complement component and heat shock proteins, among others.
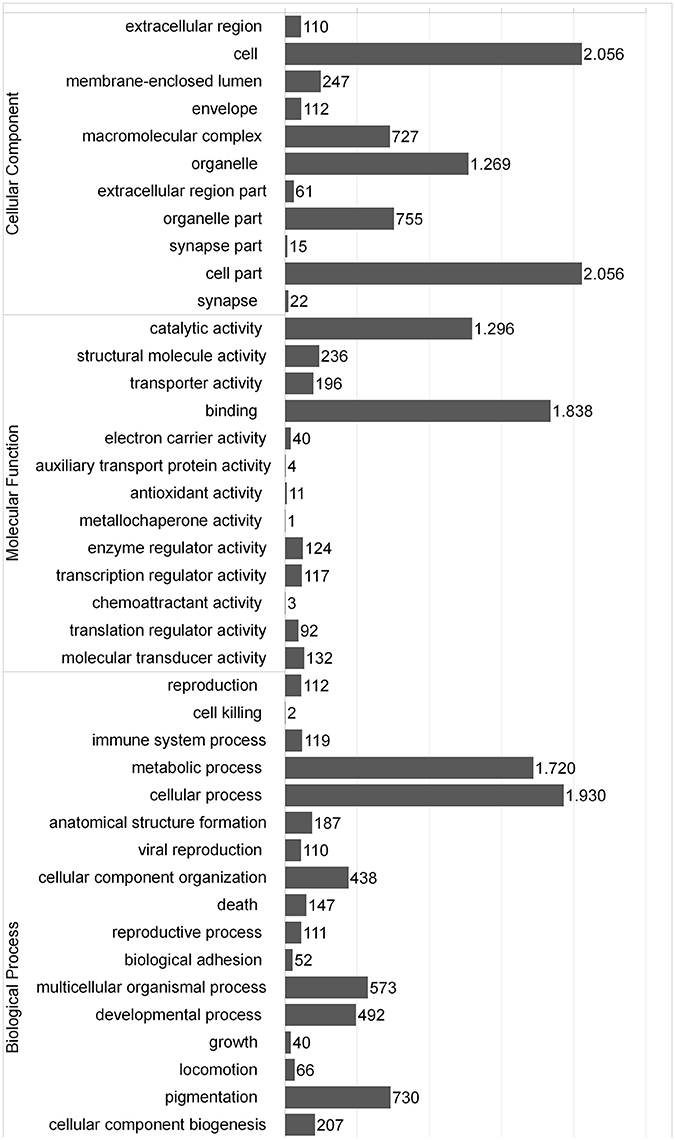
Figure 1. Gene ontology classification (level 2).

Table 4. Biochemical pathway mapping (KEGG) for southern hake unigenes.
Simple Sequence Repeats Identification and Validation
Generally, genetic markers, such as SSRs, have been considered useful tool for evaluating and studying genetic structure, diversity and quantitative traits loci discovery (Chauhan and Rajiv, 2010) in fishes. Moreover, SSRs developed from transcriptome sequences are meaningful for the evaluation of functional variation and represent a useful level of transferability among close species.
A total of 2302 SSRs were identified on 1687 unigenes, dimers and trimers being the most abundant class, representing together near 95% of the total. The most abundant dimers motifs (36%) were AC, CA, and GT, present on 304, 265, and 276 unigenes respectively, while the motifs most abundant in trimers (4%) were CCT, GAT and GGA identified on 30, 35, and 29 sequences. Among all detected SSRs, it was possible to design appropriate PCR primers for 741 (32.19%) (Supplementary Table S1). Out of the 14 SSRs selected for validation, 4 (28.56%) had very low polymorphism (a single allele across samples) and 1 (7.14%) did not amplify (Table 5).

Table 5. Summary of the subgroup of SSRs (14) evaluated.
In conclusion, we have conducted a transcriptome sequencing study of spleen and liver tissues in southern hake and identified 3171 genes, of which 186 were predicted to be related to immune pathways. Moreover, we also identified 741 microsatellites and evaluated a subgroup of them. Altogether, these results provide an excellent ground for the development of platforms to investigate important genetics and ecological aspects of southern hake populations.
Author Contributions
RV: Participated in the conception and design of the study and drafted the manuscript; JG: Participated in the conception, design, and coordination of the study and helped write the manuscript; DR and RG: Performed the bench work, data analysis and drafted the manuscript.
Funding
This work was supported by CORFO-INNOVA Chile 12IDL2-16192.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmars.2016.00216/full#supplementary-material
Supplementary Table S1. This is a xlsx archive that contains additional results from microsatellite PCR primers research and gene annotation.
References
Alheit, J., and Pitcher, T. J. (1995). Hake. Fisheries, Ecology and Markets, Fish and Fisheries Series 15. London: Chapman and Hall.
Bairoch, A., and Apweiler, R. (2000). The SWISS-PROT protein sequence database its supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48. doi: 10.1093/nar/28.1.45.
Bricknell, I. R., Bron, J. E., and Bowden, T. J. (2006). Diseases of gadoid fish in cultivation: a review. ICES J. Mar. Sci. 63, 253–266. doi: 10.1016/j.icesjms.2005.10.009
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Cepeda, V., Cofre, C., González, R., MacKenzie, S., and Vidal, R. (2011). Identification of genes involved in immune response of Atlantic salmon (Salmo salar) to IPN virus infection, using expressed sequence tag (EST) analysis. Aquaculture 318, 54–60. doi: 10.1016/j.aquaculture.2011.04.045
Chauhan, T., and Rajiv, K. (2010). Molecular markers and their applications in fisheries and aquaculture. Adv. Biosci. Biotechnol. 1, 281–291. doi: 10.4236/abb.2010.14037
Chevreux, B., Pfisterer, T., Drescher, B., Driesel, A. J., Müller, W. E. G., Wetter, T., et al. (2004). Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 14, 1147–1159. doi: 10.1101/gr.1917404
Cohen, D. M., Inada, T., Iwamoto, T., and Scialabba, N. (1990). FAO Species Catalogue, Vol. 10. Gadiform Fishes of the World (Order Gadiformes). An Annotated and Illustrated Catalogue of Cods, Hakes, Grenadiers and Other Gadiform Fishes Known to Date. Rome: FAO Fisheries Synopsis.
Cohen, S., Tirindelli, J., Gomez-Chiarri, M., and Nacci, D. (2006). Functional implications of major histocompatibility (MH) variation using estuarine fish populations. Integr. Comp. Biol. 46, 1016–1029. doi: 10.1093/icb/icl044
Ding, J., Zhao, L., Chang, Y., Zhao, W., Du, Z., and Hao, Z. (2015). Transcriptome sequencing and characterization of Japanese scallop Patinopecten yessoensis from different shell color lines. PLoS One 10:e0116406. doi: 10.1371/journal.pone.0116406
Doyle, J. M., Siegmund, G., Ruhl, J. D., Eo, S. H., Hale, M. C., Marra, N. J., et al. (2013). Microsatellite analyses across three diverse vertebrate transcriptomes (Acipenser fulvescens, Ambystoma tigrinum, and Dipodomys spectabilis). Genome 56, 407–414. doi: 10.1139/gen-2013-0056
Ellegren, H. (2004). Microsatellites: simple sequences with complex evolution. Nat. Rev. Genet. 5, 435–445. doi: 10.1038/nrg1348
Fan, Z., You, F., Wang, L., Weng, S., Wu, Z., Hu, J., et al. (2014). Gonadal transcriptome analysis of male and female olive flounder (Paralichthys olivaceus). Biomed Res. Int. 2014:291067. doi: 10.1155/2014/291067
Götz, S., García-Gómez, J. M., Terol, J., Williams, T. D., Nagaraj, S. H., Nueda, M. J., et al. (2008). High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36, 3420–3435. doi: 10.1093/nar/gkn176
Hou, R., Bao, Z., Wang, S., Su, H., Li, Y., Du, H., et al. (2011). Transcriptome sequencing and de novo analysis for Yesso scallop (Patinopecten yessoensis) using 454 GS FLX. PLoS ONE 6:e21560. doi: 10.1371/journal.pone.021560
Huang, X., and Madan, A. (1999). CAP3: A DNA sequence assembly program. Genome Res. 9, 868–877. doi: 10.1101/gr.9.9.868
Kanehisa, M., and Goto, S. (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Kumar, S., and Blaxter, M. L. (2010). Comparing de novo assemblers for 454 transcriptome data. BMC Genomics 16:571. doi: 10.1186/1471-2164-11-571
Li, B., Fillmore, N., Bai, Y., Collins, M., Thomson, J. A., Stewart, R., et al. (2014). Evaluation of de novo transcriptome assemblies from RNA-Seq data. Genome Biol. 15:553. doi: 10.1186/s13059-014-0553-5
Li, F. G., Chen, J., Jiang, X. Y., and Zou, S. M. (2015). Transcriptome analysis of blunt snout bream (Megalobrama amblycephala) reveals putative differential expression genes related to growth and hypoxia. PLoS ONE 10:e0142801. doi: 10.1371/journal.pone.0142801
Liao, X., Cheng, L., Xu, P., Lu, G., Wachholtz, M., Sun, X., et al. (2013). Transcriptome analysis of crucian carp (Carassius auratus), an important aquaculture and hypoxia-tolerant species. PLoS ONE 8:e62308. doi: 10.1371/journal.pone.0062308
Lieschke, G. J., and Trede, N. S. (2009). Fish immunology. Curr. Biol. 19, R678–R682. doi: 10.1016/j.cub.2009.06.068
Lloris, D., Matallanas, J., and Oliver, P. (2005). Hakes of the world (Family Merlucciidae). An Annotated and Illustrated Catalogue of Hake Species Known to Date. FAO Species Catalogue for Fishery. Rome: Food and Agriculture Organization of the United Nations.
Mabragaña, E., Díaz de Astarloa, J. M., Hanner, R., Zhang, J., and González Castro, M. (2011). DNA barcoding identifies argentine fishes from marine and brackish waters. PLoS ONE 6:e28655. doi: 10.1371/journal.pone.0028655
Machado-Schiaffino, G., and Garcia-Vazquez, E. (2009). Isolation and characterization of microsatellite loci in Merluccius australis and cross-species amplification. Mol. Ecol. Resour. 9, 585–587. doi: 10.1111/j.1755-0998.2008.02442.x
McAndrew, B., and Napier, J. (2011). Application of genetics and genomics to aquaculture development: current and future directions. J. Agric. Sci. 149, 143–151. doi: 10.1017/S0021859610001152
Milano, I., Babbucci, M., Cariani, A., Atanassova, M., Bekkevold, D., Carvalho, G. R., et al. (2014). Outlier SNP markers reveal fine-scale genetic structuring across European hake populations (Merluccius merluccius). Mol. Ecol. 23, 118–135. doi: 10.1111/mec.12568
Milano, I., Babbucci, M., Panitz, F., Ogden, R., Nielsen, R. O., Taylor, M. I., et al. (2011). Novel tools for conservation genomics: comparing two high-throughput approaches for SNP discovery in the transcriptome of the European hake. PLoS ONE 6:e28008. doi: 10.1371/journal.pone.0028008
Mundry, M., Bornberg-Bauer, E., Sammeth, M., and Feulner, P. G. D. (2012). Evaluating characteristics of de novo assembly software on 454 transcriptome data: a simulation approach. PLoS ONE 7:e31410. doi: 10.1371/journal.pone.0031410
Ojeda, J., Suazo, C. G., and Rau, J. R. (2011). Seasonal seabird assemblages in the artisanal long-line fishery of austral hake. RBMO 46, 443–451. doi: 10.4067/S0718-19572011000300013
Parra, G., Bradnam, K., and Korf, I. (2007). CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067. doi: 10.1093/bioinformatics/btm071
Peakall, R., and Smouse, P. E. (2012). GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28, 2537–2539. doi: 10.1093/bioinformatics/bts460
Qian, X., Ba, Y., Zhuang, Q., and Zhong, G. (2014). RNA-Seq technology and its application in fish transcriptomics. OMICS 18, 98–110. doi: 10.1089/omi.2013.0110
Quinteiro, J., Vidal, R., and Rey Méndez, M. (2000). Phylogeny and biogeographic history of hake (genus Merluccius), inferred from mitochondrial DNA control-region sequences. Mar. Biol. 136, 163–174. doi: 10.1007/s002270050019
Renshaw, M. A., Portnoy, D. S., Vidal, R., and Gold, J. (2011). Isolation and characterization of microsatellite markers in the southern hake, Merluccius australis. Conserv. Genet. Resour. 3, 91–94. doi: 10.1007/s12686-010-9298-y
Rozen, S., and Skaletsky, H. (2000). Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 132, 365–386. doi: 10.1385/1-59259-192-2:365
Salzberg, S. L., Phillippy, A. M., Zimin, A., Puiu, D., Magoc, T., Koren, S., et al. (2012). GAGE: a critical evaluation of genome assemblies and assembly algorithms. Genome Res. 22, 557–567. doi: 10.1101/gr.131383.111
Schunter, C., Vollmer, S. V., Macpherson, E., and Pascual, M. (2014). Transcriptome analyses and differential gene expression in a non-model fish species with alternative mating tactics. BMC Genomics 15:167. doi: 10.1186/1471-2164-15-167
Shin, S. C., Kim, S. J., Lee, J. K., Ahn, D. H., Kim, M. G., Lee, H., et al. (2012). Transcriptomics and comparative analysis of three antarctic notothenioid fishes. PLoS ONE 7:e43762. doi: 10.1371/journal.pone.0043762
Temnykh, S., DeClerck, G., Lukashova, A., Lipovich, L., Cartinhour, S., and McCouch, S. (2001). Computational and experimental analysis of microsatellites in rice (Oryza sativa L.): frequency, length variation, transposon associations, and genetic marker potential. Genome Res. 11, 1441–1452. doi: 10.1101/gr.184001
Keywords: microsatellite, southern hake, RNA-Seq, transcriptome, spleen, kidney
Citation: Reyes D, Gold J, González R and Vidal R (2016) De novo Assembly, Characterization and Functional Annotation of Southern Hake (Merluccius australis) Transcriptome. Front. Mar. Sci. 3:216. doi: 10.3389/fmars.2016.00216
Received: 13 August 2016; Accepted: 20 October 2016;
Published: 10 November 2016.
Edited by:
Çetin Keskin, Istanbul University, TurkeyReviewed by:
Celia Schunter, King Abdullah University of Science and Technology, Saudi ArabiaAndrey Tatarenkov, University of California, Irvine, USA
Copyright © 2016 Reyes, Gold, González and Vidal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rodrigo Vidal, ruben.vidal@usach.cl