 Mohamed Aborageh
Mohamed Aborageh Peter Krawitz
Peter Krawitz Holger Fröhlich
Holger Fröhlich- 1Bonn-Aachen International Center for Information Technology (B-IT), Rheinische Friedrich-Wilhelms-Universität Bonn, Bonn, Germany
- 2Institute for Genomic Statistics and Bioinformatics, University Hospital Bonn, Bonn, Germany
- 3Department of Bioinformatics, Fraunhofer Institute for Algorithms and Scientific Computing (SCAI), Sankt Augustin, Germany
Parkinson’s Disease (PD) is a neurodegenerative disorder with highly heterogeneous phenotypes. Accordingly, it has been challenging to robustly identify genetic factors associated with disease risk, prognosis and therapy response via genome-wide association studies (GWAS). In this review we first provide an overview of existing statistical methods to detect associations between genetic variants and the disease phenotypes in existing PD GWAS. Secondly, we discuss the potential of machine learning approaches to better quantify disease phenotypes and to move beyond disease understanding towards a better-personalized treatment of the disease.
1 Introduction
Parkinson’s Disease (PD) is a neurodegenerative disorder (NDD) affecting 7–10 Million patients worldwide. PD patients suffer from motor symptoms like bradykinesia, rigidity, tremor, and postural instability. Speech impairments, characterized by hypokinetic dysarthria, are among the first symptoms (including disruptions in prosody, articulation and, phonation). In addition, non-motor symptoms include cognitive impairment, sleep disorders as well as autonomic and mood dysfunction. The cause of idiopathic PD is unknown, and all currently available treatments (e.g. l-DOPA) are symptomatic. PD has a high subject-to-subject variability of symptoms reflecting disease progression (Poewe et al., 2017).
In recent years, genome-wide association studies (GWAS) have shed light on the polygenic nature of Parkinson’s Disease (PD) (Simón-Sánchez et al., 2009; Satake et al., 2009; Kara et al., 2014; Siitonen et al., 2017; Bandres-Ciga et al., 2019; Nalls et al., 2019). First GWAS aimed to identify mutations in coding regions that could be linked to each neurodegenerative trait. Accordingly, variants associated with α-synuclein were detected (Mata et al., 2010), one of the hallmark proteins of the disease. However, a meta-analysis of several studies found more variants with smaller effects to be more common in patients than fully penetrant variants (Tran et al., 2020). In addition, larger cohorts now open the possibility to identify less frequent variants and study the interaction with environmental factors. An example is the 23andMe PD cohort, which was able to identify 17 new risk loci for idiopathic PD (Chang et al., 2017; Nalls et al., 2014). Another example is United Kingdom Biobank (UKB), where other authors were able to demonstrate novel gene-environment interactions (Jacobs et al., 2020).
Despite these successes, unraveling the genetic basis of PD, specifically in its sporadic form, remains challenging:
• PD demonstrates a highly heterogeneous phenotype with different long-term outcomes (Aasly, 2020). Accordingly, it is difficult to find genetic associations. So far most research has focused on risk factors for PD diagnosis, but less attention has been paid to identifying genetic variants associated with different long-term outcomes. Notably, a few papers report on genetic risk factors for cognitive impairment in idiopathic PD (Collins and Williams-Gray, 2016; Amer et al., 2018; Planas-Ballvé and Vilas, 2021).
• Sizes of existing cohorts still impose a statistical challenge to identify rare variants.
• Many genetic variants jointly contribute to the phenotype, possibly in a non-linear manner via gene-gene interactions. Finding the true causal subset of variants is still difficult due to the high dimensionality of the GWAS data, the existence of linkage disequilibrium, and statistically low contributions of rare genetic variants on the population level.
• While in a recent meta-study more than 70 single-nucleotide polymorphisms (SNPs) have been associated with the risk to develop PD, most of them are located in non-coding regions and thus difficult to interpret (Ho et al., 2022).
In this context the goal of this review is two-fold: First, we provide an overview of existing statistical methods that have been employed to detect associations between genetic variants and the disease phenotype as shown in Figure 1 and Table 1. The second goal of this review is to discuss the potential of machine learning approaches, which could allow to better quantify complex phenotypes and to move beyond disease understanding towards a better personalized treatment of PD in the future. While previous reviews focused on the genetic architecture of PD and discuss associated risk factors (Billingsley et al., 2018), gene-specific polymorphisms (Jiménez-Jiménez et al., 2016), gene-gene and gene-environment interactions (Singh et al., 2014; Dunn et al., 2019), our review has thus a distinguishable methodological focus.
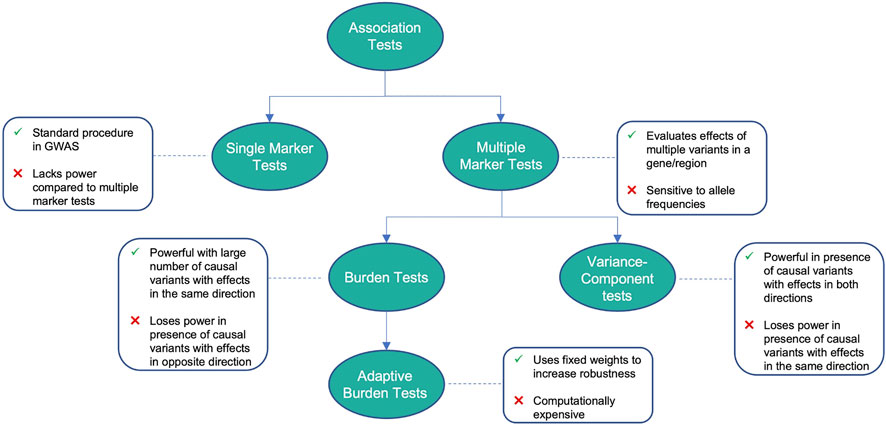
FIGURE 1. Advantages and disadvantages of different association tests.

TABLE 1. Advantages and disadvantages of methods for variant association testing.
2 Variant association tests
In 2011, Sun et al. (2011) considered rare variants as single-nucleotide polymorphisms with minor allele frequencies (MAF) less than 0.01, and have larger effects than common variants. However, when combined, the number of low-frequency variants makes them common. According to the multiple rare variant (MRV) hypothesis, cases of common inherited diseases are due to the combined effects of highly-penetrant variants (Bodmer and Bonilla, 2008). The genetic composition of PD is often described by two non-mutually exclusive hypotheses: the common disease common variant (CDCV) hypothesis which describes the genetic basis of PD as a result of a large number of common variants with relatively small effects but combined confer significant disease risk (Pritchard and Cox, 2002), and the common disease rare variant (CDRV) hypothesis which speculates that risk components for complex diseases will be rare genetic variants of small or large effects where highly functional or deleterious alleles may exist. This may be noticeable in late-onset diseases like PD where selective pressures are not profound (Billingsley et al., 2018).
Typically, GWA studies focus on variants with MAF greater than 1–5%, and while they were able to identify several variants with evidence of association to disease risk, these common variants only explain 5%–10% of the disease heritability. This led to the conclusion that disease risk is comprised of both common and rare variants (Schork et al., 2009). Variants located near SNCA, MAPT genes and low frequency coding variants in GBA are validated by GWAS to be statistically significant signals associated with PD (Spencer et al., 2011; Lill et al., 2012; Nalls et al., 2014; Chang et al., 2017).
2.1 Single-marker tests
Single-marker testing involves the application of a univariate test for each variant and assessing their significance while using a scaled p-value threshold to account for multiple testing (Asimit and Zeggini, 2010). These tests include X2, Fisher’s test, Cochran-Armitage (CA) test for trend and regression analysis, be it logistic regression for testing binary traits or linear regression for quantitative traits. Since each variant is tested independently, corrections for multiple testing should be accounted for to control the family-wise error (FWE) which may result in a loss of power. Instead, controlling the false-discovery rate (FDR) by allowing a small proportion of incorrect null hypotheses may result in a gain of power, especially at a larger number of tests.
If we assume m number of variants within an n number of samples, a regression model can be fit at each of the m variants to test their association with a trait. Assuming that yi is the phenotype for sample i and xij is the minor allele count of variant j for sample i, the relationship of variant i can be explained by a linear regression model with the following formula:
Where zj is a matrix of covariates that may be present, and ɛi is an error term representing independent random variables with a mean of 0. For that model, a value of βj = 0 represents the null hypothesis of no association at variant j. For a logistic regression model, yi is replaced by
The X2, Fisher’s test, and CA tests construct a 2 × 3 contingency matrix to compare the genotype frequencies between cases and controls, where rows represent disease status and columns represent the three possible genotypes. For X2 and Fisher’s tests, a null hypothesis of equal genotype frequencies for both the cases and controls is considered. Usually, Fisher’s test is preferred since it provides exact results of significance, while X2 test approximates the results with an accuracy that depends on the sample size, which is not ideal in the case of small samples.
If we represent the genotypes as ordered categories AA, Aa, and aa, the CA test is considered a modification of the X2 test to introduce a suspected ordering of the genotype effects and aims to test a linear effect of the minor allele’s copy counts (Slager and Schaid, 2001), which is defined as follows:
As mentioned earlier, multiple testing needs to be corrected to control the family-wise error (FWE). The Bonferroni correction is used to test an m number of variants while assuming the significance level for the m independent hypothesis tests is α, using α/m to calculate the test-specific significance level (Ranstam, 2016). To control the FDR for independent tests, Benjamini & Hochberg (Benjamini and Hochberg, 1995) developed a sequential Bonferroni procedure, where the m p-values from the individual tests are first ranked: p(1) ≤ p(2) ≤ ⋯ ≤ p(m). At FDR level q, assume k to be the largest i such that
A study by Mata et al. (2017) used linear regression to identify genetic variants that may lead to a cognitive decline in PD patients. Eighteen common variants in thirteen genomic regions exceeded the significance threshold for one cognitive test each. However, rare variant analysis did not yield any significance. Another study by Simón-Sánchez et al. (2009) used the Cochran-Armitage test for trend to test associations with PD in European patients. Four SNPs at the SNCA locus and three at the MAPT locus exceeded Bonferroni-corrected GWAS significance thresholds. An overview about further PD studies and employed statistical tests is provided in Table 2.
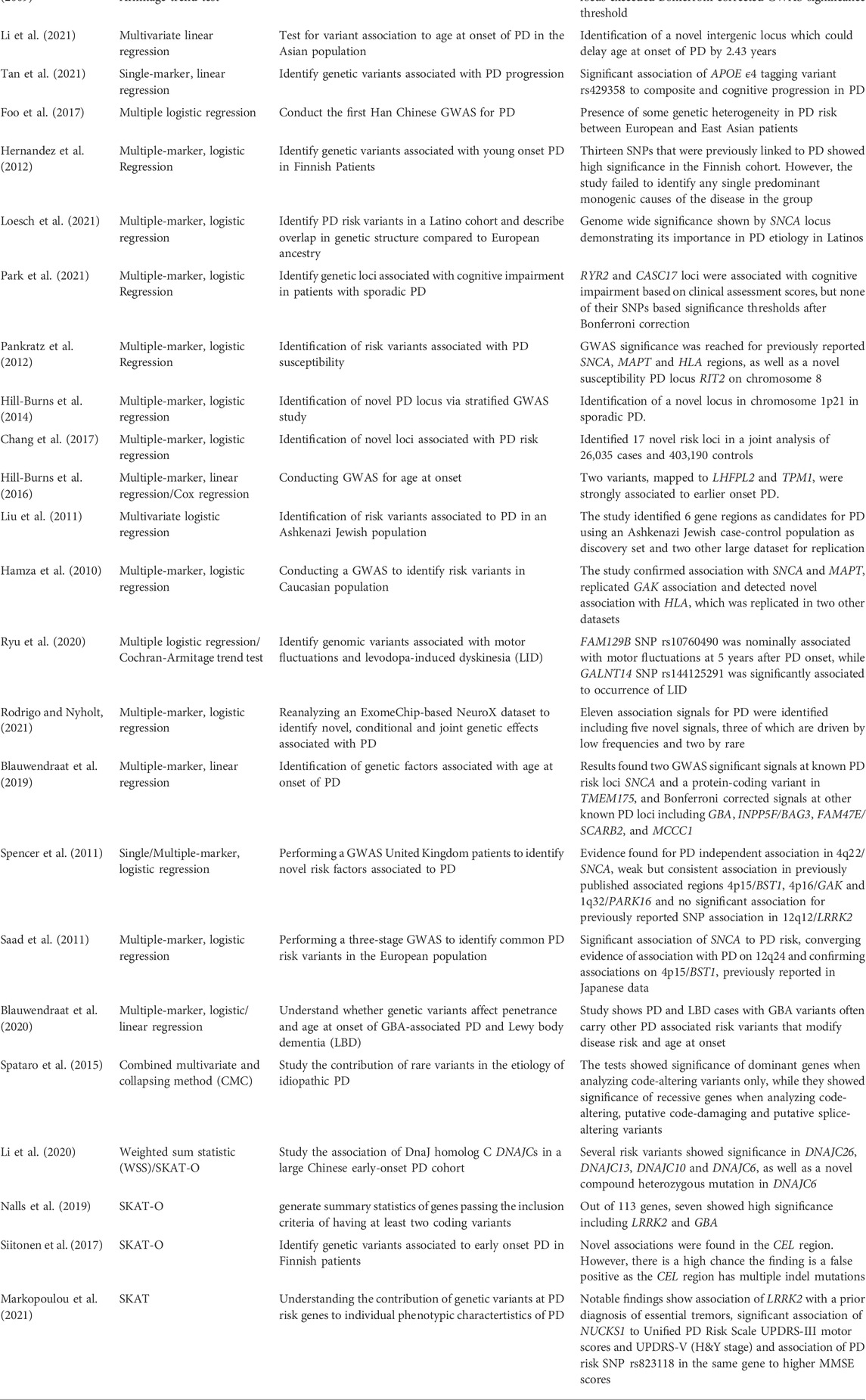
TABLE 2. Selected studies on risk variant association utilizing multiple techniques.
2.2 Multiple-marker tests
Multivariate methods can be used as an alternative to testing variants individually by combining information across the variants and testing the multiple variant sites simultaneously. In that case, a multiple-marker test’s power will be higher than that of single-marker tests for multiple moderate SNP effects. Such approaches include Fisher’s method, Hotelling’s T2 test, and multiple logistic or linear regression. These tests may be less powerful as they require multiple degrees of freedom.
Fisher’s test combines the results of all m single-marker tests, and the test statistic can be represented by
Multiple regression can be used to test for the association between the variants and the phenotype in tandem instead of fitting m regression models at each of the rare variants separately. A simple regression model with no covariates for a binary trait can be represented as follows:
where X is an n × m matrix of the minor allele counts for n subjects at m variants, and β is the m vector of regression coefficients. By estimating the associations at each variant collectively, the fit requires m degrees of freedom for the test statistics of each null hypothesis with βj = 0 to have n − m degrees of freedom rather than n − 1 as in single-marker tests.
Hotelling’s two-sample T2 is a multivariate generalization of the Student’s t-test (Xiong et al., 2002) which can be used for case-control studies. Assume we have NA affected and
and Yij is defined similarly for
and under the null hypothesis,
A drawback to multiple-marker tests is their sensitivity to allele frequencies. A simulation study on rare variants by Li & Leal (Li and Leal, 2008) shows that Hotelling’s T2 test is greatly affected by MAF, and shows a reduction in power in cases of increased numbers of rare causal variants.
Li et al. (2021) used multivariate linear regression to test for variant association to age at onset of PD in the Asian population. Results showed a significant effect of a novel intergenic locus rs9783733 that could delay the age at onset in patients by 2.43 years. Another study by Pankratz et al. (2012) used logistic regression to identify genetic variants associated with pD. Genome-wide significance was reached for variants in SNCA, MAPT, GAK/DGKQ, HLA region and RIT2. Additional tests can be found in Table 2.
2.3 Burden tests
Aggregation tests can be used to evaluate the combined effects of multiple variants in a gene or region, rather than testing each of them individually. One class of such tests is called burden tests, which collapse information of multiple variants into a single genetic score and test for its association to a trait (Morgenthaler and Thilly, 2007; Li and Leal, 2008; Zawistowski et al., 2010; Morris and Zeggini, 2010; Asimit et al., 2012). By counting minor alleles across all variants in a set, we can summarize the genotype information, and the statistic is represented by:
where Gij represents the allele counts of subject i at variant j, and wj is the weight for variant j.
The summary genetic score Ci can adapt to different assumptions about disease mechanisms. The MZ test (Morris and Zeggini, 2010) utilizes a dominant genetic model instead of an additive one to calculate Ci, which is the number of rare variants for which individual i carries at least a single copy of the minor allele. As for the cohort allelic sums test (CAST) (Morgenthaler and Thilly, 2007), it assumes an increase in disease risk with the presence of any rare variant, and sets the genetic score Ci = 0 if there are no minor alleles in the region and Ci = 1 otherwise.
We can focus on rare variants by assuming wj = 1 when the MAF of the variant j MAFj is smaller than a preset threshold or wj = 0 if otherwise. We can upweight rare variants by using a continuous weight function. Madsen and Browning (Madsen and Browning, 2009) proposed
Outside of the regression framework, several burden approaches have been presented. The combined multivariate and collapsing method (CMC) (Li and Leal, 2008) collapses rare variants as in CAST, but in different MAF categories and calculates the combined effects of the variants using Hoteling’s t test. The Madsen and Browning weighted-sum test (WST) (Madsen and Browning, 2009) uses Wilcoxon’s rank-sum test and obtains the p-values by permutation.
All rare variants in a set are assumed to be causal and related to a trait with the same direction and magnitude by burden techniques. Breaking such assumptions can result in a significant loss of power (Neale et al., 2011; Lee et al., 2012a).
Spataro et al. (2015) used different collapsing methods, including the CMC and weighted sum tests, to study the contribution of rare variants in the etiology of idiopathic pD. The tests showed high significance in a Mendelian group of genes that comprise genes of dominant and recessive inheritance. In dominant genes, the tests showed high significance only when analyzing code-altering variants. As for recessive genes, the tests showed significance for code-altering, putative code-damaging, and putative splice-altering variants. Another study by Li et al. (2020) used the weighted sum statistic (WSS) to study the associations of the DNAJC proteins family by genetic analysis to early onset PD in a large Chinese cohort. The study identified 61 rare variants, two of which showed significance after Bonferroni correction in DNAJC26, two in DNAJC13, one in DNAJC10 and one more in DNAJC6, as well as a novel compound heterozygous mutation in DNAJC6. An overview of further studies using burden tests can be found in Table 2.
2.4 Adaptive burden tests
Adaptive methods were developed to address the limitations posed by the traditional burden tests. These methods are robust in presence of null variants and allow for train-increasing or trait-decreasing variants. Han et al. (Han and Pan, 2010) developed a data-adaptive sum test (aSum) that performs a burden test with estimated directions after first estimating the direction of effect for each variant in a marginal model. It assigns wj = −1 when βj is likely to be negative and wj = 1 if not. This approach requires permutation for the p-values to be calculated. This procedure is improved in the step-up test (Hoffmann et al., 2010), which uses a model-selection framework that assigns wj = 0 when a variant is unlikely to be associated, removing it from consideration.
A more direct approach is utilized by the estimated regression coefficient test (EREC) (Lin and Tang, 2011), which uses estimated regression coefficients for each variant as weights. This is based on the assumption that the true regression coefficient βj is an optimal weight to maximize power. When minor allele counts (MAC) are small, βj estimates are unstable, and hence the EREC test stabilizes the estimates by adding a small constant to the estimated βj, which might reduce the test’s optimality. The test uses parametric bootstrap to estimate p-values because asymptotic approximation of the test statistic is only accurate for very large samples.
The variable threshold (VT) (Price et al., 2010) is an adaptive modification that chooses the best frequency thresholds for rare variant burden testing and calculates p-values analytically or by permutation. Using kernel-based adaptive weighting, the kernel-based adaptive cluster (KBAC) (Liu and Leal, 2010) method combines variant classification of non-risk and risk variants with association tests.
As referenced in the previous section, Li et al. (2020) included the aSUM and KBAC tests with the WSS test to study the associations of the DNAJC proteins family to early onset pD. Further information and results of the study can be found in Table2.
2.5 Variance-component tests
This type of association tests uses a variance-component test within a random-effects model and tests for the association of a group of variants by evaluating the distribution of their genetic effects. These tests include the C-alpha test (Neale et al., 2011), the sequence kernel association test (SKAT) (Wu et al., 2010; Wu et al., 2011), and the sum of squared score test (Pan, 2009). These tests evaluate the distribution of aggregated score test statistics of the individual variants.
SKAT is a non-burden test that uses mixed models and includes the C-alpha test in special cases when covariates are absent, and can also accommodate SNP-SNP interactions. SKAT assumes the regression coefficients βj are independent and follow a distribution with mean 0 and variance
which is a weighted sum of squares of the single-variant score statistic Sj. Similar to burden tests, SKAT is robust to groups that include variants with both positive and negative effects, as it collapses
While burden tests are not powerful when the target region has several noncausal variants or causal variants of different associations, they can outperform SKAT in cases where a high proportion of causal variants with effects in a similar direction are present. Lee et al, (2012b) proposed a unified test that is optimal in both scenarios and combines both burden tests and SKAT in a single framework. The test statistic of the unified test is
which is a weighted average of SKAT and burden tests, which reduces to SKAT when ρ = 0 or the burden test when ρ = 1.
In their meta genome-wide association study, Nalls et al. (2019) used SKAT-O to generate summary statistics of genes with rare coding variants which had an imputation quality larger than 0.8%. 113 genes passed the inclusion criteria of having at least two coding variants. After Bonferroni correction for the 113 genes, seven significant genes were identified including LRRK2 and GBA. Siitonen et al. (2017) also used SKAT-O in their study to analyze variants associated with early onset PD in Finnish patients. The results showed significant associations to PD in the CEL locus which were not previously identified. However, the validity of the result is questioned by the fact that the CEL region has several indel mutations (Taylor et al., 1991; Siitonen et al., 2017).
2.6 Linkage disequilibrium score regression
Linkage Disequilibrium score regression (LDSC) is a method developed by Bulik-Sullivan et al. (2015) that determines if the distribution of a test statistic in GWAS is inflated due to confounding biases or polygenicity. The idea behind LDSC is that variants in linkage disequilibrium (LD) with a causal variant in an association analysis will show elevated test statistics that are proportional to the LD with the causal variant, while elevations due to confounders like cryptic relatedness or population stratification will not correlate with the LD score. LDSC involves using regression techniques to study the relationship between LD scores and test statistics of SNPs obtained from GWAS studies.
Nalls et al. (2019) used LDSC in their GWAS to examine correlations of PD genetics with that of other traits and diseases using data obtained from GWAS available via LD Hub (Zheng et al., 2017) and biomarker GWAS summary statistics on c-reactive protein and cytokine measures. p-values obtained from the LDSC were adjusted for FDR to account for multiple testing. The authors found four significant correlations, two of which were positive correlations with intracranial volume and putamen volume, and two negative correlations with tobacco use and educational attainment.
Tirozzi et al. (2020) wanted to investigate the genetic overlap between PD and platelet parameters since associations between both have been established but not thoroughly investigated on a genetic level. The authors applied LDSC to summary statistics of a large independent GWAS conducted on Alzheimer’s disease (AD), PD, and platelet parameters including mean platelet volume (MPV), platelet count (PLT), and platelet distribution width (PDW) (Jansen et al., 2019). The results showed a significant correlation between PDW and PD risk suggesting the existence of genetic overlap and presenting PDW as a new potential biomarker for PD.
Another study by Andersen et al. (2021) investigates how the immune system contributes to pathogenesis in PD, by studying the enrichment of common variant heritability for PD stratified by immune and brain cell types. The authors performed a stratified LDSC (s-LDSC) analysis using full summary statistics from the meta-analysis of PD GWAS by Nalls et al. (2019) and an earlier meta-analysis by Chang et al. (2017). The results found significant enrichment in open chromatin regions of microglia, with further investigation of expression quantitative locus (eQTL) databases showing the P2RY12 locus to be the most interesting, suggesting it as a microglial gene with PD association signal.
2.7 Mendelian randomization
Mendelian Randomization (MR) is a method that uses measured variation in genes of known function to study the causal effects of a modifiable exposure on disease or health-related outcomes (Lawlor et al., 2008). MR studies use genetic variants as instrumental variables (IV) which can be defined as variables that are associated with an outcome only through their robust association with an intermediary variable.
In this context, the aim of MR studies is not to identify genetic variants that are directly associated with the disease but to use the variants as IVs for the modifiable exposure of interest. The genetic variants need to satisfy three assumptions to be considered as IVs in MR studies:
• The variant is associated with the modifiable exposure
• The variant is independent of confounding factors that confound the association of the modifiable exposure to the outcome
• The variant is independent of the outcome given the modifiable exposure and the confounding factors
Therefore, genetic variants that explain variations in an exposure can be used as a proxy to explain how changes in that exposure can influence the outcome of a disease of interest. An illustration of the MR framework is shown in Figure 2.
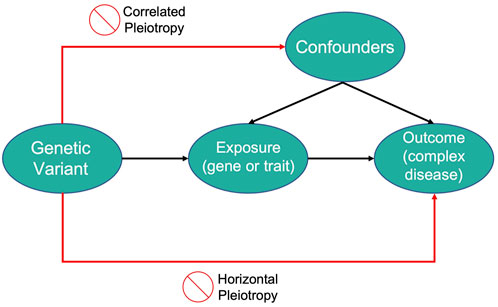
FIGURE 2. An illustration of Mendelian Randomization framework.
MR was used by Simon et al. (2014) to investigate whether genetic variants that can predict serum urate levels can predict the rate of progression in patients with early PD, on the basis that higher serum urate levels lower the risk of developing pD. In this study, the authors used SLC2A9 gene as an IV, which explains most of the genetically specified variability in serum urate levels but does not have any known direct associations with the central nervous system. The authors then estimated the association between genetically determined urate levels and PD progression using two-stage regression, where they first fitted a generalized linear regression model with urate levels as the dependent variable, and a SLC2A9 score based on the number of minor alleles at three selected loci, along with potential confounders, as independent variables. Then, a Cox proportional hazards model used the predicted urate levels from the first stage regression as a continuous independent variable to determine its association with PD progression. The results showed that an increase in the number of SLC2A9 minor alleles is associated with a decreased serum urate level. Also, the rate of PD progression increased with the number of minor SLC2A9 alleles associated with lower serum urate levels. Genetic variants other than SLC2A9 did not show any significant association to lower serum urate levels or rapid PD progression. The results suggest that high serum urate levels are protective of rapid progression in early PD.
Similarly, a study by Domenighetti et al. (2022) uses MR to investigate the association between genetically predicted dairy intake and higher PD risk by using the LCT lactase gene’s minor allele rs4988235 as an IV, where TT/TC genotypes are associated with lactase persistence and the ability to digest lactose and CC genotype with non-persistence. The authors then used logistic regression to compare the frequency of rs4988235-TC+TT genotypes in patients and controls of European ancestry. Results showed that rs4988235-TC+TT genotypes were more frequent in PD patients than controls, suggesting that higher dairy intake increases PD risk.
Another study by Storm et al. (2021) uses MR to investigate several druggable genes and predict their efficacy as PD drug targets. In this study, the authors considered the expression levels of the druggable genes as the modifiable exposure, while variants associated with expression levels of the genes, called eQTLs, were used as the IVs. The authors sought to use openly available eQTL data for genes under investigation to mimic exposure to corresponding medications (Finan et al., 2017). First, the authors used the cohort collected for the meta-analysis by Nalls et al. (2014). The causal estimates, known as the Wald ratio, were calculated for each SNP, and the ratios were weighted by inverse-variance (IVW) for genes with more than one eQTL available. This identified 31 genes with genetically-determined expression that is highly associated with PD risk. The authors then attempted to replicate the genes with significant association with PD risk in an independent cohort that does not overlap with the original cohort. The authors then used several meta-analysis methods to look for pleiotropy due to confounders including IVW, the MR-Egger intercept test, Cochran’s Q test, and the I2 test. Based on the results, the authors propose the genes CTSB, GPNMB, CD38, RHD, IRAK3 and LMAN1 as drug targets with the strongest MR evidence.
As previously discussed, Nalls et al. (2019) identified correlations of PD genetics with tobacco consumption, educational attainment, and brain volumes using LDSC. The authors used MR to assess the existence of a causal relationship between PD and the traits. The results showed that cognitive performance and educational attainment had a large causal effect on PD risk, while smoking and brain volumes did not have any significant causal relationship.
2.8 Multiple testing corrections
Multiple testing is one of the major concerns regarding high-dimensional data which results from simultaneous testing of multiple hypotheses, which if not taken into consideration, may lead to rejecting a true null hypothesis by chance, known as a false discovery. This can be accounted for by controlling an appropriate error rate such as the family-wise error rate (FWE) which is the probability of one or more false discoveries. The classical method of controlling FWE is the Bonferroni method (Bland and Altman, 1995), which is an adjustment made to p-values when several tests are performed. To perform a Bonferroni correction, assume the critical p-value to be α, then divide it by the number of tests made n. The new critical p-value would then be α/n, and the statistical power of the study is then calculated based on the newly modified p-value.
Another method is the Benjamini–Hochberg method which controls the false discovery rate (FDR) (Benjamini and Hochberg, 1995), known as the expected proportion of false rejections out of all rejections. The Benjamini–Hochberg procedure involves ordering all p-values from smallest to largest then assigning a ranking to each one, then calculating the critical p-value as (i/m)Q, where i is the rank of the p-value, m is the total number of tests and Q is the chosen FDR. The method then checks the largest p-value below the critical rate, and considers any smaller values as significant.
3 Polygenic risk score
The risk of polygenic disorders such as PD cannot be assessed by information conferred from a single variant, but the total set of risk variants that comprise its genetic architecture is required to provide enough information that can help identify individuals at high-risk (Lewis and Vassos, 2020). An individual’s risk can be assessed using polygenic risk scores (PRS), calculated as the sum of risk alleles an individual carries, each weighted by their relative effect sizes obtained from the GWAS summary statistics (Ibanez et al., 2019), where the result is a score that represents the individual’s genetic load for the disease or trait in question.
In this context linkage disequilibrium (LD) and p-values thresholds for individual SNPs have to be considered. Simpler approaches, such as PRSice (Choi and O’Reilly, 2019) and PLINK (Purcell et al., 2007; Gaunt et al., 2007; Chang et al., 2015), only use p-value thresholds (clumping + thresholding), whereas more advanced methods, including LDPred (Vilhjálmsson et al., 2015), PRS-CS (Ge et al., 2019), JAMPred (Newcombe et al., 2019), and Lassosum (Mak et al., 2017) additionally take into account based on reference data.
While PRS can provide a simple estimate of the genetic architecture of complex disorders, its additive model generally does not take into account gene-gene interactions (Aschard, 2016). Moreover, the typically required pre-filtering of SNPs implies a focus on more common genetic variants.
The largest meta-analysis was performed by Nalls et al. (2014) and was considered the reference for PD-related PRS before including more data from the 23andME (Chang et al., 2017) meta-analysis. The included PRS were associated with PD status, faster motor and cognitive decline (Paul et al., 2018) and age at onset of disease. Another study by Escott-Price et al. (2015) mentions that only PRS built from SNPs with p-values below the significant thresholds were associated with PD, suggesting that the genetic architecture of PD includes several common variants with small effects. Another study by Ibanez et al. (2017) shows that PRS from more significant SNPs are also associated with PD risk. Furthermore, PRS were used to show a higher genetic burden in early-onset PD than in late-onset PD (Escott-Price et al., 2015). More studies with established PRS in the PD field can be found in Table 3. A more detailed review of PRS in the PD field can be found in (Dehestani et al., 2021).
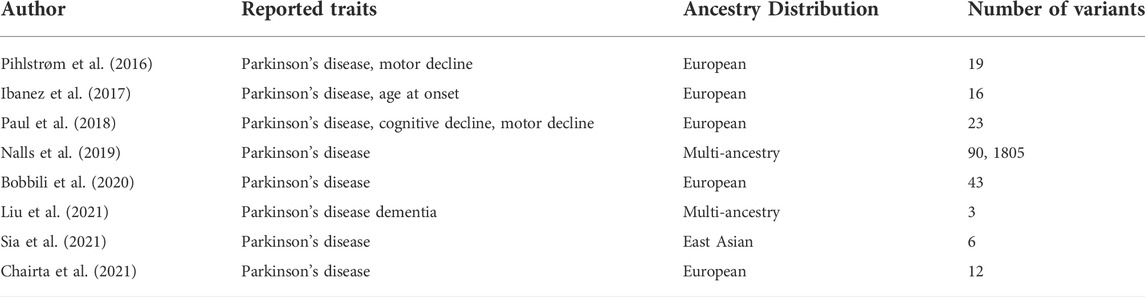
TABLE 3. Polygenic Risk Scores listed in the Polygenic Score Catalog (Lambert et al., 2021).
4 The perspective of machine learning
4.1 Multi-modal data integration
There is an increasing awareness that PD has to be understood as a complex disease, in which aging, (epi-)genetic variants, environmental pollutants/toxins, lifestyle, and comorbidities jointly contribute to the observed phenotype (Espay et al., 2017; Titova and Chaudhuri, 2017). Whereas variants association tests and PRS have helped to gain a better understanding of the genetic basis of PD, developing algorithms for accurate disease risk assessment, diagnosis, prognosis, and treatment response in the context of precision medicine require combining PRS as well as relevant genetic variants with further data modalities. Hence, predictive machine learning models are needed, which can potentially also overcome one of the typical limitations of PRS, namely lacking variant interactions and thus non-linearities. A recent study shows the combined role of PRS, rare high-impact variants, and family history in PD risk (Hassanin et al., 2021). Cope et al. demonstrated that a non-linear machine learning algorithm purely trained on genetic variants can result in dramatically improved prediction performances compared to a classical PRS (Cope et al., 2021). Notably, analysis of the model allowed us to identify an interaction between variants in TMEM175 (coding for a potassium channel in late endosomes) and GAPDHP25 (glyceraldehyde-3 phosphate dehydrogenase pseudogene 25), which have been linked to PD (Nalls et al., 2014). Another study by Prashanth et al. (2016) used multimodal features to classify early PD subjects from controls using machine learning models. The authors used non-motor features of Rapid Eye Movement (REM) sleep Behaviour Disorder (RBD) and olfactory loss as well as cerebrospinal fluid (CSF) measurements and dopaminergic imaging markers to classify the patients using Naive Bayes, Support Vector Machine (SVM), Boosted Trees and Random Forest classifiers, where SVM gave the highest performance. Based on the results, the authors suggest that the combination of non-motor, CSF, and imaging features can help in the preclinical diagnosis of PD.
A further example is the use of non-linear unsupervised machine learning algorithms by Emon et al. (2020) to identify patient subgroups by exploring the genetic burden by SNPs in genes that have been previously associated with AD and PD, which allowed for a molecular mechanism based stratification of AD and PD patient sub-types. The authors further investigated clinical outcome measures of the patients to confirm whether the patient clusters were disease-associated or reflected general genetic variations in the population and found the clusters to be associated with different clinical symptoms, pathophysiological brain differences, and biological processes that were enriched only in each of the clusters.
Experiences from neurological conditions other than PD suggest that combinations of PRS, (non-linear) combinations of genetic variants, pathway-level burden scores and a detailed description of the clinical phenotype could allow for a rather accurate prediction of disease risk (Khanna et al., 2018; Birkenbihl et al., 2020) and even clinical drug response (de Jong et al., 2021). Interestingly, in both cases, genetic factors played a comparably small role in the prediction of the clinical outcome. In another study, Makarious et al. (2022) demonstrate the benefits of using multiple data modalities by integrating clinical, genetic, and transcriptomic data in a predictive machine learning framework. Their results showed that integrating multiple data modalities improved PD prediction in mixed populations of cases and controls. They also demonstrated the benefits of using machine learning approaches and the ability to tune the models’ parameters and accommodate nonlinearities, as well as identifying important features that contributed the most to the models’ predictive performance using model explanation methods such as SHAPley Additive exPlanations (SHAP).
4.2 Deep phenotyping
A few studies have started to focus on genetic risk factors associated to symptoms of idiopathic PD, including cognitive impairment (Collins and Williams-Gray, 2016; Amer et al., 2018; Planas-Ballvé and Vilas, 2021). In this context, it has to be re-emphasized that PD patients suffer from a whole spectrum of motor and non-motor symptoms. Traditionally, these symptoms are assessed via questionnaires, such as the Unified Parkinson’s Disease Rating Scale (UPDRS), during a patient’s visit to a medical specialist center. The assessment is dependent on the experience of the individual examiner and can thus be subjectively biased. Therefore, during the last years, there has been a strongly growing interest in remote monitoring techniques (RMTs), including wearable sensors and devices (measuring e.g. gait) and smartphone apps (measuring e.g. cognitive abilities). Compared to established questionnaire-based assessments, RMTs offer several potential benefits:
1) They are patient-centric and not biased by a rater’s experience.
2) They allow for monitoring disease symptoms within a patient’s natural at-home environment, potentially 24/7, hence considering the fact that PD symptoms are variable over the daytime. RMT signals can thus be viewed as real-world data.
3) Digital sensing techniques provide an objective measure of a clinical symptom.
Notably, processing of RMT signals requires advanced data analytical techniques, including machine learning (Fröhlich et al., 2022). The outcome is an abstract set of features representing a patient’s phenotype. Following sufficient validation, within clinical studies, these features can result in digital biomarkers, which provide an accurate and quantitative description of PD symptoms. The combination with genetic data thus opens completely new opportunities to identify risk factors for specific PD symptoms, such as cognitive impairment or sleep disturbances. Moreover, machine learning algorithms could potentially be used to combine digital biomarkers with genetic features and other data modalities, including electronic health records, to predict disease risk, prognosis, and response to treatment.
4.3 Parkinson’s disease prediction
Multiple studies have used machine learning models to predict PD using different data modalities, analyzing hidden information in data that cannot be interpreted in clinical diagnosis. Wang et al. (2020) investigated the diagnosis of PD based on vowel phonation. Features were obtained from the mPower dataset and improved with additional novel features using a Bayesian correlated t-test. The features were then used as input for an SVM model which performed with moderate accuracy. Bhurane et al. (2022) used SVM with a cubic kernel to classify PD patients and healthy controls. Using features extracted from Electroencephalography (ECG) signals, the proposed approach performed with high accuracy.
Chakraborty et al. (2020) used features extracted from 3T T1-MRI scans to detect neurodegeneration in pD. Using atlas-based segmentation, eight subcortical structures were segmented from the MRI scans, on which feature extraction was performed to extract textural, morphological, and statistical features. The features were then used to train four different machine learning algorithms: an artificial neural network (ANN), XGBoost model, random forest classifier, and an SVM, where the ANN model performed with the highest accuracy. In another study, Ali et al. (2019) used neural networks to detect PD using features obtained from acoustic analysis of voice signals. Linear discriminant analysis (LDA) was used for dimensionality reduction, and a genetic algorithm (GA) to optimize the hyperparameters of the neural network. Initially, the model performed with accuracy which falls after excluding gender-dependent features to eliminate bias.
Peng et al. (2019) used a three-step method for PD gene prediction. The method, called N2A-SVM, uses the Node2vec algorithm to extract vector representations of each gene in the protein-protein interaction (PPI) network. Then it uses an autoencoder to reduce the dimensions of the obtained vector, and an SVM for classification. The performance of N2A-SVM was tested in comparison to the other methods: random walk with restart (RWR) (Li and Patra, 2010), shortest path length (SPL) (Krauthammer et al., 2004) and Euclidean distance (ED) (Díaz-Uriarte and Alvarez de Andrés, 2006), where N2A-SVM showed the highest performance.
Another study by Rastegar et al. (Ahmadi Rastegar et al., 2019) used machine learning models to assess if serum cytokine levels can be used to predict PD progression. The authors used data from the Michael J Fox Foundation LRRK2 clinical cohort consortium to assess the variability of inflammatory cytokine levels in patients over a one-year period. Then, the authors used the cytokine measurements with elastic net and random forest models to predict longitudinal clinical outcomes. Using baseline cytokine measurements, random forest models of motor severity showed the best predictive performance, with cytokines MIP1α and MCP1 contributing the most to the predictive model.
5 Discussion
The heterogeneous nature of PD imposes specific challenges for finding the underlying genetic causes. We briefly discussed several association tests that were used to identify genetic variants associated with disease risk. Single-marker tests are the simplest approach to studying associations by applying a univariate test to each variant and assessing their significance. However, their statistical power is low for small datasets and requires corrections for multiple testing. These issues were addressed by developing statistical methods that evaluate the associations of multiple variants in specific regions or genes. They are used as a standard method to test for variant association in GWAS, and helped identify several variants associated with PD including SNCA, MAPT, GBA and HLA loci as well as others associated with cognitive decline in PD including APOEϵ4, RYR2 and CASC17 loci.
Burden tests collapse multiple genetic variants into a single genetic score, which is used to test the association to a trait. Since these tests assume all collapsed rare variants to be causal and associated with the trait under study in a similar direction and magnitude of effect, any changes in said assumptions lead to a loss in their statistical power. Adaptive burden tests address these limitations as they require fewer assumptions about the genetic architecture at each locus, and hence they are suitable in the presence of null variants and trait-increasing or decreasing variants. However, adaptive tests are two-step procedures that may require regression coefficient estimation of individual variants as a first step and can be unstable for rare variants. They also estimate p-values by computationally intensive permutation. The use of burden tests helped us understand the role of different variant types in the etiology of idiopathic PD, and the identified four mendelian mutations of LRRK2 and PARK2 loci in idiopathic PD cases (Spataro et al., 2015). Adaptive tests were also used to study the associations of DNAJC proteins family with early onset PD (Li et al., 2020).
Variance-component tests evaluate the distribution of genetic effects for groups of variants to test for their association. Instead of aggregating the variants, they assess the distribution of each of the variants’ aggregated score test statistics. Variance-component tests are more powerful than burden tests if the genetic region under study has many non-causal variants or variants with different directions of association, while burden tests are more powerful when there are more causal variants with the same direction of association. SKAT-O combines both burden tests and SKAT in a single framework but can be less powerful than any of its components if their underlying assumptions are largely true. Nalls et al. (2019) used SKAT-O in their meta GWAS to identify genes with two or more rare coding variants, and 7 significant genes: LRRK2, GBA, CATSPER3, LAMB2, LOC442028, NFKB2 and SCARB2. SKAT has also been used to study the association of genetic variants to individual phenotypic characteristics of PD, including motor and cognitive functions (Markopoulou et al., 2021).
LD score regression helped researchers distinguish whether inflated GWAS test statistic distributions are due to variants in LD with a causal variant or due to confounding bias or polygenicity. LDSC has been used to examine correlations of PD genetics with different traits, including brain measurements, blood measurements, habitual behaviors, and immune system activity in different cell types (Nalls et al., 2019; Tirozzi et al., 2020; Andersen et al., 2021).
Mendelian Randomization helped understand the causal effects of modifiable exposures on pD. The method uses the genetic variants as instrumental variables in statistical analysis to describe the relationship between the disease and the modifiable exposure of interest. MR was used to investigate the relationship between PD and serum urate levels, suggesting that elevated urate levels are protective of rapid progression in early PD (Simon et al., 2014). MR was used as well to investigate the relationship with lactose tolerance in different PD patient populations, suggesting that high tolerance and increased dairy intake elevate PD risk (Domenighetti et al., 2022). MR also helped propose druggable targets by investigating the expression levels of druggable genes and using them as the modifiable exposure of interest (Storm et al., 2021).
PRS have opened the possibility to assess disease risk on an individual basis rather than purely on the average population level. Limitations of PRS include their additive nature, which neglects gene-gene interactions, and the focus on more common genetic variants. Machine learning models can mitigate this limitation and additionally include further data modalities, such as other molecular and phenotypic data. In this context, electronic health records, as well as digital biomarkers, could help to longitudinally and more objectively characterize disease symptoms. The main challenge with the use of machine learning models is, however, their difficult interpretation, specifically in the case of neural networks. Novel approaches coming from the field of Explainable AI (XAI) could here provide a solution (Linardatos et al., 2020; Arrieta et al., 2020).
In summary, novel methodological developments are necessary to deepen the understanding of the genetic basis of PD and to transfer these insights into better individualized treatment of PD in the future.
Author contributions
MA, HF and PK contributed to arrangement of the literature review. MA wrote the first draft of the manuscript, HF and PK wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmmed.2022.933383/full#supplementary-material
References
Aasly, J. O. (2020). Long-term outcomes of genetic Parkinson's disease. J. Mov. Disord. 13 (2), 81–96. doi:10.14802/jmd.19080
Ahmadi Rastegar, D., Ho, N., Halliday, G. M., and Dzamko, N. (2019). Parkinson's progression prediction using machine learning and serum cytokines. NPJ Park. Dis. 5, 14. doi:10.1038/s41531-019-0086-4
Ali, L., Zhu, C., Zhang, Z., and Liu, Y. (2019). Automated detection of Parkinson's disease based on multiple types of sustained phonations using linear discriminant analysis and genetically optimized neural network. IEEE J. Transl. Eng. Health Med. 7, 2000410. doi:10.1109/JTEHM.2019.2940900
Amer, H., Shehata, H., Rashed, L. A., Helmy, H., El-Jaafary, S., Sabbah, A., et al. (2018). Genetic influences on cognition in idiopathic Parkinson's disease. Neurol. Res. Int. 2018, 5603571. doi:10.1155/2018/5603571
Andersen, M. S., Bandres-Ciga, S., Reynolds, R. H., Hardy, J., Ryten, M., Krohn, L., et al. (2021). Heritability enrichment implicates microglia in Parkinson's disease pathogenesis. Ann. Neurol. 89 (5), 942–951. doi:10.1002/ana.26032
Arrieta, A. B., Díaz-Rodríguez, N., Ser, J. D., Bennetot, A., Tabik, S., Barbado, A., et al. (2020). Explainable Artificial Intelligence (XAI): Concepts, taxonomies, opportunities and challenges toward responsible AI. Inf. Fusion 58, 82–115. doi:10.1016/j.inffus.2019.12.012
Aschard, H. (2016). A perspective on interaction effects in genetic association studies. Genet. Epidemiol. 40 (8), 678–688. doi:10.1002/gepi.21989
Asimit, J., and Zeggini, E. (2010). Rare variant association analysis methods for complex traits. Annu. Rev. Genet. 44, 293–308. doi:10.1146/annurev-genet-102209-163421
Asimit, J. L., Day-Williams, A. G., Morris, A. P., and Zeggini, E. (2012). ARIEL and AMELIA: Testing for an accumulation of rare variants using next-generation sequencing data. Hum. Hered. 73 (2), 84–94. doi:10.1159/000336982
Bandres-Ciga, S., Ahmed, S., Sabir, M. S., Blauwendraat, C., Adarmes-Gómez, A. D., Bernal-Bernal, I., et al. (2019). The genetic architecture of Parkinson disease in Spain: Characterizing population-specific risk, differential haplotype structures, and providing etiologic insight. Mov. Disord. 34 (12), 1851–1863. doi:10.1002/mds.27864
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 57, 289–300. doi:10.1111/j.2517-6161.1995.tb02031.x
Bhurane, A. A., Dhok, S., Sharma, M. D., Rajamanickam, Y., Murugappan, M., and Acharya, U. R. (2022). Diagnosis of Parkinson's disease from electroencephalography signals using linear and self‐similarity features. Expert Syst. 39. doi:10.1111/exsy.12472
Billingsley, K. J., Bandres-Ciga, S., Saez-Atienzar, S., and Singleton, A. B. (2018). Genetic risk factors in Parkinson's disease. Cell Tissue Res. 373 (1), 9–20. doi:10.1007/s00441-018-2817-y
Birkenbihl, C., Emon, M. A., Vrooman, H., Westwood, S., Lovestone, S., Hofmann-Apitius, M., et al. (2020). Differences in cohort study data affect external validation of artificial intelligence models for predictive diagnostics of dementia - lessons for translation into clinical practice. EPMA J. 11 (3), 367–376. doi:10.1007/s13167-020-00216-z
Bland, J. M., and Altman, D. G. (1995). Multiple significance tests: The Bonferroni method. BMJ 310 (6973), 170. doi:10.1136/bmj.310.6973.170
Blauwendraat, C., Heilbron, K., Vallerga, C. L., Bandres-Ciga, S., von Coelln, R., Pihlstrøm, L., et al. (2019). Parkinson's disease age at onset genome-wide association study: Defining heritability, genetic loci, and α-synuclein mechanisms. Mov. Disord. 34 (6), 866–875. doi:10.1002/mds.27659
Blauwendraat, C., Reed, X., Krohn, L., Heilbron, K., Bandres-Ciga, S., Tan, M., et al. (2020). Genetic modifiers of risk and age at onset in GBA associated Parkinson's disease and Lewy body dementia. Brain 143 (1), 234–248. doi:10.1093/brain/awz350
Bobbili, D. R., Banda, P., Krüger, R., and May, P. (2020). Excess of singleton loss-of-function variants in Parkinson's disease contributes to genetic risk. J. Med. Genet. 57 (9), 617–623. doi:10.1136/jmedgenet-2019-106316
Bodmer, W., and Bonilla, C. (2008). Common and rare variants in multifactorial susceptibility to common diseases. Nat. Genet. 40 (6), 695–701. doi:10.1038/ng.f.136
Bulik-Sullivan, B. K., Loh, P. R., Finucane, H. K., Ripke, S., Yang, J., Patterson, N., et al. (2015). LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47 (3), 291–295. doi:10.1038/ng.3211
Chairta, P. P., Hadjisavvas, A., Georgiou, A. N., Loizidou, M. A., Yiangou, K., Demetriou, C. A., et al. (2021). Prediction of Parkinson's disease risk based on genetic profile and established risk factors. Genes 12 (8), 1278. doi:10.3390/genes12081278
Chakraborty, S., Aich, S., and Kim, H. C. (2020). 3D textural, morphological and statistical analysis of voxel of interests in 3T MRI scans for the detection of Parkinson's disease using artificial neural networks. Healthc. (Basel) 8 (1), 34. doi:10.3390/healthcare8010034
Chang, C. C., Chow, C. C., Tellier, L. C., Vattikuti, S., Purcell, S. M., and Lee, J. J. (2015). Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 4, 7. doi:10.1186/s13742-015-0047-8
Chang, D., Nalls, M. A., Hallgrímsdóttir, I. B., Hunkapiller, J., van der Brug, M., Cai, F., et al. (2017). A meta-analysis of genome-wide association studies identifies 17 new Parkinson's disease risk loci. Nat. Genet. 49 (10), 1511–1516. doi:10.1038/ng.3955
Choi, S. W., and O’Reilly, P. F. (2019). PRSice-2: Polygenic Risk Score software for biobank-scale data. GigaScience 8, giz082. giz082. doi:10.1093/gigascience/giz082
Collins, L. M., and Williams-Gray, C. H. (2016). The genetic basis of cognitive impairment and dementia in Parkinson's disease. Front. Psychiatry 7, 89. doi:10.3389/fpsyt.2016.00089
Cope, J. L., Baukmann, H. A., Klinger, J. E., Ravarani, C. N. J., Böttinger, E. P., Konigorski, S., et al. (2021). Interaction-based feature selection algorithm outperforms polygenic risk score in predicting Parkinson's disease status. Front. Genet. 12, 744557. doi:10.3389/fgene.2021.744557
de Jong, J., Cutcutache, I., Page, M., Elmoufti, S., Dilley, C., Fröhlich, H., et al. (2021). Towards realizing the vision of precision medicine: AI based prediction of clinical drug response. Brain. 144 (6), 1738–1750. doi:10.1093/brain/awab108
Dehestani, M., Liu, H., and Gasser, T. (2021). Polygenic risk scores contribute to personalized medicine of Parkinson's disease. J. Pers. Med. 11 (10), 1030. doi:10.3390/jpm11101030
Díaz-Uriarte, R., and Alvarez de Andrés, S. (2006). Gene selection and classification of microarray data using random forest. BMC Bioinforma. 7, 3. doi:10.1186/1471-2105-7-3
Domenighetti, C., Sugier, P. E., Ashok Kumar Sreelatha, A., Schulte, C., Grover, S., Mohamed, O., et al. (2022). Dairy intake and Parkinson's disease: A mendelian randomization study. Mov. Disord. 37 (4), 857–864. doi:10.1002/mds.28902
Dunn, A. R., O'Connell, K. M. S., and Kaczorowski, C. C. (2019). Gene-by-environment interactions in Alzheimer's disease and Parkinson's disease. Neurosci. Biobehav. Rev. 103, 73–80. doi:10.1016/j.neubiorev.2019.06.018
Emon, M. A., Heinson, A., Wu, P., Domingo-Fernández, D., Sood, M., Vrooman, H., et al. (2020). Clustering of Alzheimer's and Parkinson's disease based on genetic burden of shared molecular mechanisms. Sci. Rep. 10 (1), 19097. doi:10.1038/s41598-020-76200-4
Escott-Price, V., Nalls, M. A., Morris, H. R., Lubbe, S., Brice, A., Gasser, T., et al. (2015). Polygenic risk of Parkinson disease is correlated with disease age at onset. Ann. Neurol. 77 (4), 582–591. doi:10.1002/ana.24335
Espay, A. J., Brundin, P., and Lang, A. E. (2017). Precision medicine for disease modification in Parkinson disease. Nat. Rev. Neurol. 13 (2), 119–126. doi:10.1038/nrneurol.2016.196
Finan, C., Gaulton, A., Kruger, F. A., Lumbers, R. T., Shah, T., Engmann, J., et al. (2017). The druggable genome and support for target identification and validation in drug development. Sci. Transl. Med. 9 (383), eaag1166. doi:10.1126/scitranslmed.aag1166
Foo, J. N., Tan, L. C., Irwan, I. D., Au, W. L., Low, H. Q., Prakash, K. M., et al. (2017). Genome-wide association study of Parkinson's disease in East Asians. Hum. Mol. Genet. 26 (1), 226–232. doi:10.1093/hmg/ddw379
Fröhlich, H., Bontridder, N., Petrovska-Delacréta, D., Glaab, E., Kluge, F., Yacoubi, M. E., et al. (2022). Leveraging the potential of digital technology for better individualized treatment of Parkinson's disease. Front. Neurol. 13, 788427. doi:10.3389/fneur.2022.788427
Gaunt, T. R., Rodríguez, S., and Day, I. N. (2007). Cubic exact solutions for the estimation of pairwise haplotype frequencies: Implications for linkage disequilibrium analyses and a web tool 'CubeX. BMC Bioinforma. 8, 428. doi:10.1186/1471-2105-8-428
Ge, T., Chen, C. Y., Ni, Y., Feng, Y. A., and Smoller, J. W. (2019). Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat. Commun. 10 (1), 1776. doi:10.1038/s41467-019-09718-5
Hamza, T. H., Zabetian, C. P., Tenesa, A., Laederach, A., Montimurro, J., Yearout, D., et al. (2010). Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson's disease. Nat. Genet. 42 (9), 781–785. doi:10.1038/ng.642
Han, F., and Pan, W. (2010). A data-adaptive sum test for disease association with multiple common or rare variants. Hum. Hered. 70 (1), 42–54. doi:10.1159/000288704
Hassanin, E., May, P., Aldisi, R., Krawitz, P., Maj, C., and Bobbili, D. R. (2021)“Assessing the role of polygenic background on the penetrance of monogenic forms in Parkinson’s disease. MedRxiv [Preprint]. Available at: https://doi.org/10.1101/2021.06.06.21253270 (Accessed September 10, 2022).
Hernandez, D. G., Nalls, M. A., Ylikotila, P., Keller, M., Hardy, J. A., Majamaa, K., et al. (2012). Genome wide assessment of young onset Parkinson's disease from Finland. PLoS One 7 (7), e41859. doi:10.1371/journal.pone.0041859
Hill-Burns, E. M., Ross, O. A., Wissemann, W. T., Soto-Ortolaza, A. I., Zareparsi, S., Siuda, J., et al. (2016). Identification of genetic modifiers of age-at-onset for familial Parkinson's disease. Hum. Mol. Genet. 25 (17), 3849–3862. doi:10.1093/hmg/ddw206
Hill-Burns, E. M., Wissemann, W. T., Hamza, T. H., Factor, S. A., Zabetian, C. P., and Payami, H. (2014). Identification of a novel Parkinson's disease locus via stratified genome-wide association study. BMC Genomics 15, 118. doi:10.1186/1471-2164-15-118
Ho, D., Schierding, W., Farrow, S. L., Cooper, A. A., Kempa-Liehr, A. W., and O'Sullivan, J. M. (2022). Machine learning identifies six genetic variants and alterations in the heart atrial appendage as key contributors to PD risk predictivity. Front. Genet. 12, 785436. doi:10.3389/fgene.2021.785436
Hoffmann, T. J., Marini, N. J., and Witte, J. S. (2010). Comprehensive approach to analyzing rare genetic variants. PLoS One 5 (11), e13584. doi:10.1371/journal.pone.0013584
Ibanez, L., Dube, U., Saef, B., Budde, J., Black, K., Medvedeva, A., et al. (2017). Parkinson disease polygenic risk score is associated with Parkinson disease status and age at onset but not with alpha-synuclein cerebrospinal fluid levels. BMC Neurol. 17 (1), 198. doi:10.1186/s12883-017-0978-z
Ibanez, L., Farias, F. H. G., Dube, U., Mihindukulasuriya, K. A., and Harari, O. (2019). Polygenic risk scores in neurodegenerative diseases: A review. Curr. Genet. Med. Rep. 7, 22–29. doi:10.1007/s40142-019-0158-0
Jacobs, B. M., Belete, D., Bestwick, J., Blauwendraat, C., Bandres-Ciga, S., Heilbron, K., et al. (2020). Parkinson's disease determinants, prediction and gene-environment interactions in the UK Biobank. J. Neurol. Neurosurg. Psychiatry 91 (10), 1046–1054. doi:10.1136/jnnp-2020-323646
Jansen, I. E., Savage, J. E., Watanabe, K., Bryois, J., Williams, D. M., Steinberg, S., et al. (2019). Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat. Genet. 51 (3), 404–413. doi:10.1038/s41588-018-0311-9
Jiménez-Jiménez, F. J., Alonso-Navarro, H., García-Martín, E., and Agúndez, J. A. (2016). NAT2 polymorphisms and risk for Parkinson's disease: A systematic review and meta-analysis. Expert Opin. Drug Metab. Toxicol. 12 (8), 937–946. doi:10.1080/17425255.2016.1192127
Kara, E., Xiromerisiou, G., Spanaki, C., Bozi, M., Koutsis, G., Panas, M., et al. (2014). Assessment of Parkinson's disease risk loci in Greece. Neurobiol. Aging 35 (2), e9–442. e16. doi:10.1016/j.neurobiolaging.2013.07.011
Khanna, S., Domingo-Fernández, D., Iyappan, A., Emon, M. A., Hofmann-Apitius, M., and Fröhlich, H. (2018). Using multi-scale genetic, neuroimaging and clinical data for predicting alzheimer's disease and reconstruction of relevant biological mechanisms. Sci. Rep. 8 (1), 11173. doi:10.1038/s41598-018-29433-3
Krauthammer, M., Kaufmann, C. A., Gilliam, T. C., and Rzhetsky, A. (2004). Molecular triangulation: Bridging linkage and molecular-network information for identifying candidate genes in alzheimer's disease. Proc. Natl. Acad. Sci. U. S. A. 101 (42), 15148–15153. doi:10.1073/pnas.0404315101
Lambert, S. A., Gil, L., Jupp, S., Ritchie, S. C., Xu, Y., Buniello, A., et al. (2021). The Polygenic Score Catalog as an open database for reproducibility and systematic evaluation. Nat. Genet. 53 (4), 420–425. doi:10.1038/s41588-021-00783-5
Lawlor, D. A., Harbord, R. M., Sterne, J. A., Timpson, N., and Davey Smith, G. (2008). Mendelian randomization: Using genes as instruments for making causal inferences in epidemiology. Stat. Med. 27 (8), 1133–1163. doi:10.1002/sim.3034
Lee, S., Emond, M. J., Bamshad, M. J., Barnes, K. C., Rieder, M. J., Nickerson, D. A., et al. (2012b). Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am. J. Hum. Genet. 91 (2), 224–237. doi:10.1016/j.ajhg.2012.06.007
Lee, S., Wu, M. C., and Lin, X. (2012a). Optimal tests for rare variant effects in sequencing association studies. Biostatistics 13 (4), 762–775. doi:10.1093/biostatistics/kxs014
Lewis, C. M., and Vassos, E. (2020). Polygenic risk scores: From research tools to clinical instruments. Genome Med. 12 (1), 44. doi:10.1186/s13073-020-00742-5
Li, B., and Leal, S. M. (2008). Methods for detecting associations with rare variants for common diseases: Application to analysis of sequence data. Am. J. Hum. Genet. 83 (3), 311–321. doi:10.1016/j.ajhg.2008.06.024
Li, C., Ou, R., Chen, Y., Gu, X., Wei, Q., Cao, B., et al. (2020). Mutation analysis of DNAJC family for early-onset Parkinson's disease in a Chinese cohort. Mov. Disord. 35 (11), 2068–2076. doi:10.1002/mds.28203
Li, C., Ou, R., Chen, Y., Gu, X., Wei, Q., Cao, B., et al. (2021). Genetic modifiers of age at onset for Parkinson's disease in asians: A genome-wide association study. Mov. Disord. 36 (9), 2077–2084. doi:10.1002/mds.28621
Li, Y., and Patra, J. C. (2010). Genome-wide inferring gene-phenotype relationship by walking on the heterogeneous network. Bioinformatics 26 (9), 1219–1224. doi:10.1093/bioinformatics/btq108
Lill, C. M., Roehr, J. T., McQueen, M. B., Kavvoura, F. K., Bagade, S., Schjeide, B. M., et al. (2012). Comprehensive research synopsis and systematic meta-analyses in Parkinson's disease genetics: The PDGene database. PLoS Genet. 8 (3), e1002548. doi:10.1371/journal.pgen.1002548
Lin, D. Y., and Tang, Z. Z. (2011). A general framework for detecting disease associations with rare variants in sequencing studies. Am. J. Hum. Genet. 89 (3), 354–367. doi:10.1016/j.ajhg.2011.07.015
Linardatos, P., Papastefanopoulos, V., and Kotsiantis, S. (2020). Explainable AI: A review of machine learning interpretability methods. Entropy (Basel) 23 (1), 18. doi:10.3390/e23010018
Liu, D. J., and Leal, S. M. (2010). A novel adaptive method for the analysis of next-generation sequencing data to detect complex trait associations with rare variants due to gene main effects and interactions. PLoS Genet. 6 (10), e1001156. doi:10.1371/journal.pgen.1001156
Liu, G., Peng, J., Liao, Z., Locascio, J. J., Corvol, J. C., Zhu, F., et al. (2021). Genome-wide survival study identifies a novel synaptic locus and polygenic score for cognitive progression in Parkinson's disease. Nat. Genet. 53 (6), 787–793. doi:10.1038/s41588-021-00847-6
Liu, X., Cheng, R., Verbitsky, M., Kisselev, S., Browne, A., Mejia-Sanatana, H., et al. (2011). Genome-wide association study identifies candidate genes for Parkinson's disease in an Ashkenazi Jewish population. BMC Med. Genet. 12, 104. doi:10.1186/1471-2350-12-104
Loesch, D. P., Horimoto, A. R. V. R., Heilbron, K., Sarihan, E. I., Inca-Martinez, M., Mason, E., et al. (2021). Characterizing the genetic architecture of Parkinson's disease in latinos. Ann. Neurol. 90 (3), 353–365. doi:10.1002/ana.26153
Madsen, B. E., and Browning, S. R. (2009). A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 5 (2), e1000384. doi:10.1371/journal.pgen.1000384
Mak, T. S. H., Porsch, R. M., Choi, S. W., Zhou, X., and Sham, P. C. (2017). Polygenic scores via penalized regression on summary statistics. Genet. Epidemiol. 41 (6), 469–480. doi:10.1002/gepi.22050
Makarious, M. B., Leonard, H. L., Vitale, D., Iwaki, H., Sargent, L., Dadu, A., et al. (2022). Multi-modality machine learning predicting Parkinson's disease. NPJ Park. Dis. 8 (1), 35. doi:10.1038/s41531-022-00288-w
Markopoulou, K., Chase, B. A., Premkumar, A. P., Schoneburg, B., Kartha, N., Wei, J., et al. (2021). Variable effects of PD-risk associated SNPs and variants in parkinsonism-associated genes on disease phenotype in a community-based cohort. Front. Neurol. 12, 662278. doi:10.3389/fneur.2021.662278
Mata, I. F., Johnson, C. O., Leverenz, J. B., Weintraub, D., Trojanowski, J. Q., Van Deerlin, V. M., et al. (2017). Large-scale exploratory genetic analysis of cognitive impairment in Parkinson's disease. Neurobiol. Aging 56, 211e1–211. e7. doi:10.1016/j.neurobiolaging.2017.04.009
Mata, I. F., Shi, M., Agarwal, P., Chung, K. A., Edwards, K. L., Factor, S. A., et al. (2010). SNCA variant associated with Parkinson disease and plasma alpha-synuclein level. Arch. Neurol. 67 (11), 1350–1356. doi:10.1001/archneurol.2010.279
Morgenthaler, S., and Thilly, W. G. (2007). A strategy to discover genes that carry multi-allelic or mono-allelic risk for common diseases: A cohort allelic sums test (CAST). Mutat. Res. 615 (1-2), 28–56. doi:10.1016/j.mrfmmm.2006.09.003
Morris, A. P., and Zeggini, E. (2010). An evaluation of statistical approaches to rare variant analysis in genetic association studies. Genet. Epidemiol. 34 (2), 188–193. doi:10.1002/gepi.20450
Nalls, M. A., Blauwendraat, C., Vallerga, C. L., Heilbron, K., Bandres-Ciga, S., Chang, D., et al. (2019). Identification of novel risk loci, causal insights, and heritable risk for Parkinson's disease: A meta-analysis of genome-wide association studies. Lancet. Neurol. 18 (12), 1091–1102. doi:10.1016/S1474-4422(19)30320-5
Nalls, M. A., Pankratz, N., Lill, C. M., Do, C. B., Hernandez, D. G., Saad, M., et al. (2014). Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson's disease. Nat. Genet. 46 (9), 989–993. doi:10.1038/ng.3043
Neale, B. M., Rivas, M. A., Voight, B. F., Altshuler, D., Devlin, B., Orho-Melander, M., et al. (2011). Testing for an unusual distribution of rare variants. PLoS Genet. 7 (3), e1001322. doi:10.1371/journal.pgen.1001322
Newcombe, P. J., Nelson, C. P., Samani, N. J., and Dudbridge, F. (2019). A flexible and parallelizable approach to genome-wide polygenic risk scores. Genet. Epidemiol. 43 (7), 730–741. doi:10.1002/gepi.22245
Pan, W. (2009). Asymptotic tests of association with multiple SNPs in linkage disequilibrium. Genet. Epidemiol. 33 (6), 497–507. doi:10.1002/gepi.20402
Pankratz, N., Beecham, G. W., DeStefano, A. L., Dawson, T. M., Doheny, K. F., Factor, S. A., et al. (2012). Meta-analysis of Parkinson's disease: Identification of a novel locus, RIT2. Ann. Neurol. 71 (3), 370–384. doi:10.1002/ana.22687
Park, K. W., Jo, S., Kim, M. S., Jeon, S. R., Ryu, H. S., Kim, J., et al. (2021). Genomic association study for cognitive impairment in Parkinson's disease. Front. Neurol. 11, 579268. doi:10.3389/fneur.2020.579268
Paul, K. C., Schulz, J., Bronstein, J. M., Lill, C. M., and Ritz, B. R. (2018). Association of polygenic risk score with cognitive decline and motor progression in Parkinson disease. JAMA Neurol. 75 (3), 360–366. doi:10.1001/jamaneurol.2017.4206
Peng, J., Guan, J., and Shang, X. (2019). Predicting Parkinson's disease genes based on Node2vec and autoencoder. Front. Genet. 10, 226. doi:10.3389/fgene.2019.00226
Pihlstrøm, L., Morset, K. R., Grimstad, E., Vitelli, V., and Toft, M. (2016). A cumulative genetic risk score predicts progression in Parkinson's disease. Mov. Disord. 31 (4), 487–490. doi:10.1002/mds.26505
Planas-Ballvé, A., and Vilas, D. (2021). Cognitive impairment in genetic Parkinson's disease. Park. Dis. 2021, 8610285. doi:10.1155/2021/8610285
Poewe, W., Seppi, K., Tanner, C. M., Halliday, G. M., Brundin, P., Volkmann, J., et al. (2017). Parkinson disease. Nat. Rev. Dis. Prim. 3, 17013. doi:10.1038/nrdp.2017.13
Prashanth, R., Dutta Roy, S., Mandal, P. K., and Ghosh, S. (2016). High-accuracy detection of early Parkinson's disease through multimodal features and machine learning. Int. J. Med. Inf. 90, 13–21. doi:10.1016/j.ijmedinf.2016.03.001
Price, A. L., Kryukov, G. V., de Bakker, P. I., Purcell, S. M., Staples, J., Wei, L. J., et al. (2010). Pooled association tests for rare variants in exon-resequencing studies. Am. J. Hum. Genet. 86 (6), 832–838. doi:10.1016/j.ajhg.2010.04.005
Pritchard, J. K., and Cox, N. J. (2002). The allelic architecture of human disease genes: Common disease-common variant.or not? Hum. Mol. Genet. 11 (20), 2417–2423. doi:10.1093/hmg/11.20.2417
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A., Bender, D., et al. (2007). Plink: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81 (3), 559–575. doi:10.1086/519795
Ranstam, J. (2016). Multiple P-values and Bonferroni correction. Osteoarthr. Cartil. 24 (5), 763–764. doi:10.1016/j.joca.2016.01.008
Rodrigo, L. M., and Nyholt, D. R. (2021). Imputation and reanalysis of ExomeChip data identifies novel, conditional and joint genetic effects on Parkinson's disease risk. Genes (Basel) 12 (5), 689. doi:10.3390/genes12050689
Ryu, H. S., Park, K. W., Choi, N., Kim, J., Park, Y. M., Jo, S., et al. (2020). Genomic analysis identifies new loci associated with motor complications in Parkinson's disease. Front. Neurol. 11, 570. doi:10.3389/fneur.2020.00570
Saad, M., Lesage, S., Saint-Pierre, A., Corvol, J. C., Zelenika, D., Lambert, J. C., et al. (2011). Genome-wide association study confirms BST1 and suggests a locus on 12q24 as the risk loci for Parkinson's disease in the European population. Hum. Mol. Genet. 20 (3), 615–627. doi:10.1093/hmg/ddq497
Satake, W., Nakabayashi, Y., Mizuta, I., Hirota, Y., Ito, C., Kubo, M., et al. (2009). Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat. Genet. 41 (12), 1303–1307. doi:10.1038/ng.485
Schork, N. J., Murray, S. S., Frazer, K. A., and Topol, E. J. (2009). Common vs. rare allele hypotheses for complex diseases. Curr. Opin. Genet. Dev. 19 (3), 212–219. doi:10.1016/j.gde.2009.04.010
Sia, M. W., Foo, J. N., Saffari, S. E., Wong, A. S., Khor, C. C., Yuan, J. M., et al. (2021). Polygenic risk scores in a prospective Parkinson's disease cohort. Mov. Disord. 36 (12), 2936–2940. doi:10.1002/mds.28761
Siitonen, A., Nalls, M. A., Hernandez, D. G., Gibbs, J. R., Ding, J., Ylikotila, P., et al. (2017). Genetics of early-onset Parkinson's disease in Finland: Exome sequencing and genome-wide association study. Neurobiol. Aging 53, e7. e7-195e10. doi:10.1016/j.neurobiolaging.2017.01.019
Simon, K. C., Eberly, S., Gao, X., Oakes, D., Tanner, C. M., Shoulson, I., et al. Parkinson Study Group (2014). Mendelian randomization of serum urate and Parkinson disease progression. Ann. Neurol. 76 (6), 862–868. doi:10.1002/ana.24281
Simón-Sánchez, J., Schulte, C., Bras, J. M., Sharma, M., Gibbs, J. R., Berg, D., et al. (2009). Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat. Genet. 41 (12), 1308–1312. doi:10.1038/ng.487
Singh, N. K., Banerjee, B. D., Bala, K., Chhillar, M., and Chhillar, N. (2014). Gene-gene and gene-environment interaction on the risk of Parkinson's disease. Curr. Aging Sci. 7 (2), 101–109. doi:10.2174/1874609807666140805123621
Slager, S. L., and Schaid, D. J. (2001). Case-control studies of genetic markers: Power and sample size approximations for armitage's test for trend. Hum. Hered. 52 (3), 149–153. doi:10.1159/000053370
Spataro, N., Calafell, F., Cervera-Carles, L., Casals, F., Pagonabarraga, J., Pascual-Sedano, B., et al. (2015). Mendelian genes for Parkinson's disease contribute to the sporadic forms of the disease. Hum. Mol. Genet. 24 (7), 2023–2034. doi:10.1093/hmg/ddu616
Spencer, C. C., Plagnol, V., Strange, A., Gardner, M., Paisan-Ruiz, C., Band, G., et al. (2011). Dissection of the genetics of Parkinson's disease identifies an additional association 5' of SNCA and multiple associated haplotypes at 17q21. Hum. Mol. Genet. 20 (2), 345–353. doi:10.1093/hmg/ddq469
Storm, C. S., Kia, D. A., Almramhi, M. M., Bandres-Ciga, S., Finan, C., Hingorani, A. D., et al. (2021). Finding genetically-supported drug targets for Parkinson's disease using Mendelian randomization of the druggable genome. Nat. Commun. 12 (1), 7342. doi:10.1038/s41467-021-26280-1
Sun, X., Namkung, J., Zhu, X., and Elston, R. C. (2011). Capability of common SNPs to tag rare variants. BMC Proc. 5 (9), S88. doi:10.1186/1753-6561-5-S9-S88
Tan, M. M. X., Lawton, M. A., Jabbari, E., Reynolds, R. H., Iwaki, H., Blauwendraat, C., et al. (2021). Genome-wide association studies of cognitive and motor progression in Parkinson's disease. Mov. Disord. 36 (2), 424–433. doi:10.1002/mds.28342
Taylor, A. K., Zambaux, J. L., Klisak, I., Mohandas, T., Sparkes, R. S., Schotz, M. C., et al. (1991). Carboxyl ester lipase: A highly polymorphic locus on human chromosome 9qter. Genomics 10 (2), 425–431. doi:10.1016/0888-7543(91)90328-c
Tirozzi, A., Izzi, B., Noro, F., Marotta, A., Gianfagna, F., Hoylaerts, M. F., et al. (2020). Assessing genetic overlap between platelet parameters and neurodegenerative disorders. Front. Immunol. 11, 02127. doi:10.3389/fimmu.2020.02127
Titova, N., and Chaudhuri, K. R. (2017). Personalized medicine in Parkinson's disease: Time to be precise. Mov. Disord. 32 (8), 1147–1154. doi:10.1002/mds.27027
Tran, J., Anastacio, H., and Bardy, C. (2020). Genetic predispositions of Parkinson's disease revealed in patient-derived brain cells. NPJ Park. Dis. 6, 8. doi:10.1038/s41531-020-0110-8
Vilhjálmsson, B. J., Yang, J., Finucane, H. K., Gusev, A., Lindström, S., Ripke, S., et al. (2015). Modeling linkage disequilibrium increases accuracy of polygenic risk scores. Am. J. Hum. Genet. 97 (4), 576–592. doi:10.1016/j.ajhg.2015.09.001
Wang, M., Ge, W., Apthorp, D., and Suominen, H. (2020). Robust feature engineering for Parkinson disease diagnosis: New machine learning techniques. JMIR Biomed. Eng. 5 (1), e13611. doi:10.2196/13611
Wu, M. C., Kraft, P., Epstein, M. P., Taylor, D. M., Chanock, S. J., Hunter, D. J., et al. (2010). Powerful SNP-set analysis for case-control genome-wide association studies. Am. J. Hum. Genet. 86 (6), 929–942. doi:10.1016/j.ajhg.2010.05.002
Wu, M. C., Lee, S., Cai, T., Li, Y., Boehnke, M., and Lin, X. (2011). Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 89 (1), 82–93. doi:10.1016/j.ajhg.2011.05.029
Xiong, M., Zhao, J., and Boerwinkle, E. (2002). Generalized T2 test for genome association studies. Am. J. Hum. Genet. 70 (5), 1257–1268. doi:10.1086/340392
Zawistowski, M., Gopalakrishnan, S., Ding, J., Li, Y., Grimm, S., and Zöllner, S. (2010). Extending rare-variant testing strategies: Analysis of noncoding sequence and imputed genotypes. Am. J. Hum. Genet. 87 (5), 604–617. doi:10.1016/j.ajhg.2010.10.012
Zheng, J., Erzurumluoglu, A. M., Elsworth, B. L., Kemp, J. P., Howe, L., Haycock, P. C., et al. (2017). LD Hub: A centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinformatics 33 (2), 272–279. doi:10.1093/bioinformatics/btw613
Keywords: Parkinson disease, risk, genome-wide association study, machine learning, polygenic risk score
Citation: Aborageh M, Krawitz P and Fröhlich H (2022) Genetics in parkinson’s disease: From better disease understanding to machine learning based precision medicine. Front. Mol. Med. 2:933383. doi: 10.3389/fmmed.2022.933383
Received: 30 April 2022; Accepted: 30 August 2022;
Published: 03 October 2022.
Edited by:
Alessandra Luchini, George Mason University, United StatesReviewed by:
Nathan Harmston, Yale-NUS College, SingaporeRuey-Meei Wu, National Taiwan University, Taiwan
Copyright © 2022 Aborageh, Krawitz and Fröhlich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Holger Fröhlich, aG9sZ2VyLmZyb2VobGljaEBzY2FpLmZyYXVuaG9mZXIuZGU=