 Matthew P. Ward
1*
Matthew P. Ward
1* Pedro P. Irazoqui
1,2
Pedro P. Irazoqui
1,2
- 1 School of Biomedical Engineering, Purdue University, West Lafayette, IN, USA
- 2 School of Electrical and Computer Engineering, Purdue University, West Lafayette, IN, USA
Current antidepressant therapies do not effectively control or cure depressive symptoms. Pharmaceutical therapies altogether fail to address an estimated 4 million Americans who suffer from a recurrent and severe treatment-resistant form of depression known as refractory major depressive disorder. Subjective diagnostic schemes, differing manifestations of the disorder, and antidepressant treatments with limited theoretical bases each contribute to the general lack of therapeutic efficacy and differing levels of treatment resistance in the refractory population. Stimulation-based therapies, such as vagus nerve stimulation, transcranial magnetic stimulation, and deep brain stimulation, are promising treatment alternatives for this treatment-resistant subset of patients, but are plagued with inconsistent reports of efficacy and variable side effects. Many of these problems stem from the unknown mechanisms of depressive disorder pathogenesis, which prevents the development of treatments that target the specific underlying causes of the disorder. Other problems likely arise due to the non-specific stimulation of various limbic and paralimbic structures in an open-loop configuration. This review critically assesses current literature on depressive disorder diagnostic methodologies, treatment schemes, and pathogenesis in order to emphasize the need for more stringent depressive disorder classifications, quantifiable biological markers that are suiTable for objective diagnoses, and alternative closed-loop treatment options tailored to well-defined forms of the disorder. A closed-loop neurostimulation device design framework is proposed, utilizing symptom-linked biomarker abnormalities as control points for initiating and terminating a corrective electrical stimulus which is autonomously optimized for correcting the magnitude and direction of observed biomarker abnormality.
Introduction
Depression is among the top predictors of mortality and substandard daily functioning in North America, second only to cardiovascular disorders (Wells et al., 1989 ). Due to conventional symptom-based classification schemes and an incomplete understanding of the disorder, the term “depression” is used to describe a broad set of disparate pathologies sharing a common set of symptoms – pathologies that manifest as abnormal control and expression of mood and emotion (Davidson et al., 2002 ). Depressed individuals may experience a dispirited mood, a lowered sense of enthusiasm or enjoyment with routine tasks, a disrupted sleep schedule, altered behavior, appetite, or weight, a change in the speed of muscle movements, a decreased energy level, an inability to focus, thoughts of worthlessness or guilt, and thoughts of death or suicide over an extended period of time (First and Ross, 2000 ; Kroenke et al., 2001 ). Current treatment measures do not effectively control symptoms in most depressed patients, especially the estimated 4 million Americans with a severe treatment-resistant subtype known as refractory major depressive disorder (refractory MDD) (Kessler et al., 2005 ; Cyberonics, 2007 ).
Refractory MDD is characterized by recurrent, long-lasting cycles of severe, often suicidal depressive episodes that do not remit using multiple types of antidepressant therapies. A depressive episode persists for up to a year (Judd et al., 1998 ), significantly impairing the health and daily activities of the afflicted (Manji et al., 2001 ). In fact, the net loss of productivity stemming from the disorder costs the United States an estimated 83 billion dollars each year(Coyne et al., 1994 ). Even with the best FDA-approved antidepressant treatments, the majority of MDD patients will inevitably suffer from multiple depressive episodes during their lifetime(Mueller et al., 1999 ; Kessler et al., 2003 ). To make matters worse, each recurrent depressive episode puts the patient at a 16% increased risk for developing an additional depressive episode during their lifetime (Solomon et al., 2000 ). More efficacious and durable antidepressant treatments are needed.
Despite considerable efforts to improve pharmaceutical-based antidepressant treatment efficacy, 50–60% of all depressed patients remain partially or fully unresponsive to a first course of properly prescribed therapy (Fava, 2003 ). Up to 20% of these patients require more extreme treatment measures, employing multiple antidepressant medications and/or electroconvulsive therapy (ECT) with variable success rates (Fava, 2003 ; Mayberg et al., 2005 ). The low acute response rate to pharmaceutical-based antidepressant therapy may be explained by selective FDA clinical efficacy report publications, which artificially inflate antidepressant drug efficacy and lead to a false sense of security in physicians prescribing them. In fact, a recent meta-analysis of 74 published and unpublished antidepressant efficacy trials, involving 12 antidepressant drugs and 12,564 participants, showed that only 51% of the trials whose data was submitted to the FDA ended with positive results (While 94% of the published efficacy reports show positive results) (Turner et al., 2008 ). A separate meta-analysis of 47 published and unpublished FDA clinical trial datasets from selective-serotonin reuptake inhibitor (SSRI) efficacy trials showed that six of the seven most-prescribed SSRI antidepressant drugs available within the last 25 years only show clinically significant benefits over placebo for “the upper end of the very severely depressed category” (Kirsch et al., 2008 ). The latter study suggests that SSRIs, which are most often the first class of prescribed drugs for depressive disorders, pose greater risks to patient health than benefits for symptom relief in the majority of patients (Kirsch et al., 2008 ; Turner et al., 2008 ).
Stimulation-based technologies, designed to electrically or chemically modulate abnormal neural activity, are emerging as potential therapeutic options for refractory MDD patients. However, the expected treatment efficacies of these technologies, as with all antidepressant treatments, are burdened by an incomplete understanding of the pathophysiology of depressive disorders and a lack of reproducible and quantifiable biological markers (i.e., biomarkers) of depressed states (antidepressant treatment response is still subjectively evaluated using patient-reported symptom relief, effectively ignoring the prospect of using objectively-quantified, depression-linked biomarker levels to quantify antidepressant responses and to optimize treatment). With modern research tools, additional structural, functional, and genetic abnormalities associated with depression are discovered each year. Concomitantly, several quantifiable genetic, biochemical, and bioelectric diagnostic markers of depression are emerging. Similar discoveries in the epilepsy research field sparked interests in closed-loop neuroprostheses, where biological indicators of an impending seizure are used to determine the time at which an electrical or chemical stimulus must be applied to stop a seizure (Dumitriu et al., 2008 ). This process, known as responsive neurostimulation, is unique to closed-loop devices. It is intended to replace continuous or periodic open-loop stimulation designs so that tailored therapy, based on quantifiable symptom-linked biomarker abnormalities, is provided in a dose-dependent manner only when it is necessary (Sun et al., 2008 ; Goodman and Insel, 2009 ).With time, closed-loop neurostimulation devices for refractory MDD are expected to bridge the knowledge gap that currently exists between subjective antidepressant response evaluations and quantifiable antidepressant responses due to controlled therapeutic stimuli.
Closed-loop treatments are now rapidly emerging for a variety of treatment-resistant disorders with encouraging successes (Sun et al., 2008 ; Wong et al., 2008 ; Behrend et al., 2009 ). Despite the recent advancements in depression research, no closed-loop prosthesis exists for treating refractory MDD. Failing pharmaceutical antidepressants, newly discovered therapeutic targets common to most subtypes of the disorder, emerging biological markers of depression, and advancements in other research fields are paving the road for closed-loop neuroprosthesis development. Using real-time input from simple micro-fabricated biosensors to determine when, where, and what type of stimulus to apply, such near-autonomous devices promise to provide highly efficacious therapy to dysfunctional brain structures in a dose-dependent manner only when it is needed. However, several hurdles must be overcome before any such device can be used in clinical practice.
The purpose of this review is to critically assess the current literature on refractory MDD diagnostic methodologies, theories of depressive disorder pathogenesis, and treatment methodologies in order to define the need for more efficacious closed-loop treatment alternatives and propose means of attaining them. This review first addresses current diagnostic and treatment schemes for depressive disorders, theories of MDD pathogenesis/pathophysiology, and alternative stimulation-based antidepressant therapies. Next, the review presents promising stimulation targets and candidate quantifiable biological markers of depression in context with the current theories of MDD pathophysiology. To conclude, a modular closed-loop neurostimulation device design framework is proposed, utilizing real-time biomarker level measurements as control points for initiating and terminating an autonomously optimized corrective electrical stimulus.
Current MDD Diagnostic and Pharmaceutical Treatment Protocols
Depression is currently diagnosed through an evaluation of a patient’s reported symptoms, clinical history, and full physical examination. A patient is often initially assessed using a depression-specific standardized evaluation such as 9 item Patient Health Questionnaire (PHQ-9), Hamilton Depression Rating Scale (HAM-D or HDRS), or Montgomery–Asberg Depression Rating Scale (MADRS) (Kearns et al., 1982 ; Kroenke et al., 2001 ). Each survey is used to estimate the severity of the symptoms used to diagnose depression in accordance with DSM-IV criteria (refer to the first paragraph of the introduction for a full list of common symptoms). The patient’s clinical history and physical examination are then used to rule out other obvious and treaTable explanations for the symptoms (DepressionGuidelinePanel, 1994 ).
Diagnosing refractory MDD is a lengthy process that often is not in the best interest of the patient’s health due to potentially life-threatening antidepressant side effects (e.g., violent behavior, cardiovascular problems, and/or recurrent thoughts of death/suicide) (Peretti et al., 2000 ; Mann, 2005 ). The most common first line of treatment for an MDD patient is psychotherapy and/or a low-dose SSRI antidepressant therapy. In psychotherapy sessions, a patient is taught to change thinking and behavior patterns in an effort to modulate limbic-cortical pathways in regions of the prefrontal cortex, hippocampus, and cingulate that are associated with normal emotions and behavior (Goldapple et al., 2004 ). After a recommended 6–12 weeks on a particular antidepressant (Quitkin et al., 1986 ; Mann, 2005 ), effectiveness may be assessed using the HAM-D or MADRS questionnaire (Despite the recommended evaluation timeframe, efficacy is typically assessed after 4–6 weeks of treatment). If the patient shows some benefit with zero or non-problematic symptoms, a higher dose of the same medication or a second antidepressant is prescribed. If a patient receives no significant benefit from at least two properly prescribed antidepressants (i.e., correct dose and sufficient evaluation timeframe), he or she is diagnosed with refractory MDD (Dumitriu et al., 2008 ). The level of treatment resistance is then estimated using one of several non-standardized algorithms, most notably the 5 stage model put forth by Thase and Rush (1997) (Dumitriu et al., 2008 ).
The majority of adults visit primary care physicians in lieu of mental health specialists when they believe they are depressed, leading to inaccurate diagnoses in more than 50% of patients (Cepoiu et al., 2008 ). Misdiagnoses likely occur because no quantifiable measures of depression are currently used in clinical medicine, forcing physicians to interpret somewhat unreliable subjective assessments obtained through standardized evaluations such as the PHQ-9. Therefore, the treatment-resistant subset of patients may be smaller than what is reported in literature, because they were inaccurately diagnosed and suffer from another underlying disorder (e.g., heart disease, rheumatoid arthritis, or cancer). Consequently, failed antidepressant treatments may not indicate that a patient is treatment-resistant, but rather that his or her depressive symptoms are secondary to another undetected or developing disease. Objective diagnostic tests based on quantifiable depressive disorder-specific biomarkers are needed to improve diagnostic accuracy and the classifications of differing manifestations of the disorder.
In summary, a major contributor to failing depressive disorder treatments stems from the lack of objective diagnostic criteria, which impedes more accurate distinctions among depressed patients who share the same common symptom profile, but develop depressive disorders through differing circumstances (Lacasse and Leo, 2005 ). With this in mind, antidepressant drug therapy is akin to moderately effective broad-spectrum antibiotic therapy for bacterial infections: the same drug is given to patients who have different underlying disorders in hopes of restoring normal health without determining the specific underlying causes of the illnesses or monitoring the effects on the assumed drug targets. Such a strategy is currently accepTable with broad-spectrum antibiotics due to their general safety, well-defined targets, known mechanisms of action, and proven therapeutic efficacy. Since antidepressant therapies do not have well-defined targets, proven mechanisms of action, and consistent reports of clinical efficacy, it is no surprise that varying levels of treatment resistance are consistently reported (Thase and Rush, 1997 ; Fava, 2003 ; Mann, 2005 ; Belmaker and Agam, 2008 ; Kirsch et al., 2008 ). More individually tailored antidepressant therapies are needed if enhanced therapeutic efficacies are desired in the refractory population. Closed-loop neuroprosthetic devices show great promise for providing such tailored therapies in the foreseeable future. Section “Overview of MDD Pathogenesis” first presents well-known aspects of MDD and concludes with the debated theories of its development and manifestation. The information is intended to highlight the heterogeneous nature of MDD, which drives the need for a heterogeneous set of treatments.
Overview of MDD Pathogenesis
Depression as a Multi-System Disorder
The pathophysiological aspects of depressive disorders are not well understood. From prospective heritability studies, it is clear that genetics and environmental factors (i.e., nature and nurture) play synergistic roles in individual susceptibility to developing the disorder (Heils et al., 1996 ; Sullivan et al., 2000 ; Caspi et al., 2003 ; Kendler et al., 2005 ; Bradley et al., 2008 ). Using predominantly qualitative observations of depressive disorders, treatment responses, and assumed functions of various brain regions, researchers found that depressive disorders are often associated with neurotransmitter imbalances (most notably serotonin, norepinephrine, and dopamine, Mann, 2005 ) and reduced blood flow, neurogenesis, and relative amounts of neurotrophic factors in regions of the limbic system (most notably the hippocampus) and prefrontal cortex (Belmaker and Agam, 2008 ). More recent findings implicate stress and a compromised immune system as possible causal factors in depressive disorder pathogenesis (O’Brien et al., 2007 ; Dantzer et al., 2008 ). To date, no clear links between these observations have been established.
Through modern functional neuroimaging studies and post-mortem investigations, researchers discovered numerous structural and functional abnormalities associated with depressive disorders (Mayberg et al., 1999 , 2000 ; Milak et al., 2005 ; Goodman and Insel, 2009 ). An important clue that led to these findings is the commonality of cognitive impairments, or the “diminished ability to think or concentrate,” seen in patients with depressive disorders (Pittenger and Duman, 2008 ). Since the hippocampi are critical for memory and learning, and since they have extensive projections to many brain areas associated with these functions, researchers were able to implicate a dysfunctional prefrontal cortex, hypothalamus, thalamus, amygdala, nucleus accumbens, and subgenual cingulate in depressive disorders (Nestler et al., 2002 ; Pittenger and Duman, 2008 ).
In light of recent advancements, most investigators agree that depression is a multifaceted disorder in which heterogeneous genetic and environmental factors predispose certain individuals to depressed states. These depressed states are brought about by an underlying “system-level disorder affecting discrete, but functionally integrated (biological) pathways” (Mayberg, 1997 ). In the remainder of Section “Overview of MDD Pathogenesis”, several of the major theories of depressive disorder pathogenesis are reviewed in order to establish candidate stimulation targets and biomarkers that can be incorporated into the design of closed-loop neuroprostheses for differing manifestations of refractory MDD.
Monoamine-Deficiency Hypothesis
The dominant hypothesis from 1967 until well into the 1990s was the monoamine-deficiency hypothesis of depression, a hypothesis implicating dysfunctional serotonin, norepinephrine, and dopamine systems in depressive disorder pathogenesis (Coppen, 1967 ). Several observations led to and helped strengthen this hypothesis: (1) experimentally depleting tryptophan in a previously depressed subject treated with a SSRI will cause a relapse into a depressed state, while depleting tryptophan in a non-depressed subject will not (tryptophan is required for normal serotonin production); similarly, experimentally depleting tyrosine hydroxylase in a previously depressed subject treated with a norepinephrine-reuptake inhibitor (NERI) will cause a relapse into a depressed state, while depleting tyrosine hydroxylase in a non-depressed subject will not (tyrosine hydroxylase is required for normal norepinephrine production) (Mann, 2005 ; Ruhe et al., 2007 ; Belmaker and Agam, 2008 ); (2) certain effective antidepressants block monoamine neurotransmitter reuptake in the presynaptic terminals or inhibit monoamine oxidase (an enzyme required for breaking down serotonin and norepinephrine), increasing the availability of serotonin and norepinephrine for postsynaptic neurotransmission (Mann, 2005 ; Belmaker and Agam, 2008 ); and (3) monoamine neurotransmitters are used in most parts of the brain, therefore allowing the possibility of explaining the broad spectrum of underlying pathologies classified as depression (Mann, 2005 ; Belmaker and Agam, 2008 ).
Several recent investigations and literature analyses have discredited this theory by questioning its origin, re-evaluating published and unpublished clinical efficacy data, and elucidating other actions of effective antidepressants (Lacasse and Leo, 2005 ; Kirsch et al., 2008 ; Turner et al., 2008 ). The largest flaw in the monoamine-deficiency hypothesis is in its origin. It originated in 1967 from observations of the mechanisms of action of clinically effective antidepressants, which were known to block serotonin and norepinephrine reuptake at the presynaptic terminals. On average, however, no significant differences in monoamine neurotransmitter levels and dynamics are seen in depressed versus control patients (Belmaker and Agam, 2008 ). Additionally, a meta-analysis conducted by Kirsch and others (on SSRI therapeutic efficacies) demonstrated that 80% of the antidepressant responses seen in the experimental groups are also seen in the control/placebo groups with no significant effect imparted by SSRI dose (Kirsch, 2002 ; Lacasse and Leo, 2005 ; Kirsch et al., 2008 ). As a result, many academics believe that dysfunctional monoamine neurotransmitter systems are likely just downstream effects of another underlying disorder leading to the symptoms of depression (Belmaker and Agam, 2008 ).
Hypothalamic–Pituitary–Adrenal Axis Theory of Depression
A second hypothesis of depression, known as the hypothalamic–pituitary–adrenal (HPA)-axis theory of depression, arose in 1996 after researchers published evidence linking abnormal corticotropin-releasing hormone (CRH), corticotropin (ACTH), and cortisol dynamics/regulation to depressive disorders (Holsboer and Barden, 1996 ). The HPA-axis theory postulates that corticosteroid-receptor signaling is disrupted by some external mechanism, increasing CRH synthesis and release in brain structures implicated in depression and culminating in its pathogenesis (Holsboer, 2000 ). Specifically, prolonged exposure to elevated levels of stress hormones increases atrophy and the susceptibility of neurons and glial cells to damage and death, particularly in the hippocampus (Sapolsky, 2000 ; Duman and Monteggia, 2006 ). Observations leading to and strengthening this theory are: (1) elevated CRH and cortisol levels were observed in the cerebrospinal fluid (Nemeroff et al., 1984 ; Merali et al., 2004 ) and plasma (Burke et al., 2005 ) of depressed patients, respectively; (2) higher levels of cortisol and ACTH secretions were detected from urinalysis in depressed patients (Rubin et al., 1987 ); (3) higher levels of CRH and CRH mRNA were found in depression-associated limbic structures of depressed patients who committed suicide (Merali et al., 2004 ); (4) researchers showed that 50% of severely depressed patients lacked a normal “cortisol-suppression response” through dexamethasone suppression tests (Carroll et al., 2007 ); and (5) HPA-axis function returns to more normal states with successful antidepressant therapies (Carroll et al., 2007 ; Belmaker and Agam, 2008 ).
As with the monoamine-deficiency hypothesis, the HPA-axis theory lacks proof and consistently reproducible results in support of the hypothesis. One major problem with the HPA-axis theory of depression is that it cannot clearly separate many nosologically-distinct neurological disorders from depressive disorders (and healthy patients from depressed patients) due to overlapping mean cortisol and CRH levels in most healthy and depressed patients, the large circadian-rhythm-associated plasma cortisol level fluctuations normally seen within the general population, and the association of the cortisol system with other distinct disorders (Burke et al., 2005 ; Belmaker and Agam, 2008 ). A second major problem with the HPA-axis theory is that the negative effects of long-term exposure to high cortisol levels partially remit when synaptic monoamine neurotransmitter availability is increased (e.g., through antidepressant action, but demonstrated with selective depletion of select monoamine neurotransmitters) (Carroll et al., 2007 ). In light of these observations, Belmaker and Agam (2008) suggest that antidepressants do not directly improve mood; rather, they reduce “secondary stress caused by a painfully dispirited mood,” thereby indirectly elevating mood by reducing an exacerbating cause (i.e., prolonged stress). They propose that such a mechanism of antidepressant action may help explain why antidepressants successfully treat such a variety of non-depressive neurological disorders (e.g., anxiety disorders and post-traumatic stress disorders) (Belmaker and Agam, 2008 ).
Neurotrophic Theory of Depression
A third hypothesis of depressive disorder pathogenesis developed mostly throughout the last decade, building upon the monoamine-deficiency hypothesis and the HPA-axis theory of depression. The neurotrophic theory of depression postulates that (1) reduced brain-derived neurotrophic factor (BDNF) and vascular endothelial growth factor (VEGF) in the hippocampus and associated limbic structures, originating from chronic exposure to stress hormones in susceptible individuals, causes or worsens depressive disorders, and that (2) effective antidepressants contribute to neurotrophic factor upregulation in the dysfunctional limbic structures by restoring normal HPA-axis function (i.e., Antidepressants work to lower stress levels and increase BDNF availability, thereby allowing natural repair mechanisms to correct functional deficits and ultimately reduce symptoms) (Duman and Monteggia, 2006 ; Pittenger and Duman, 2008 ).
Key observations leading to and strengthening this theory are: (1) neurotrophic factors, especially BDNF and VEGF, are under-expressed in limbic structures implicated in depression (especially the hippocampus) and may contribute to their observed atrophy and increased sensitivity to death (Sapolsky, 2000 ; Duman and Monteggia, 2006 ); (2) HPA-axis dysfunction exacerbates this phenomenon, while effective antidepressant treatments counteract it by enhancing hippocampal BDNF mRNA expression through the cAMP-CREB cascade (Duman et al., 1997 , 2000 ); and (3) ECT and MAOI antidepressants, considered to be the most effective at relieving depressive symptoms, were shown to most significantly increase BDNF and trkB (the BDNF receptor) mRNA expression in the hippocampus (Nibuya et al., 1995 ).
Adult Hippocampal Neurogenesis and Cellular Plasticity Theory of Depression
A fourth hypothesis of depressive disorder pathogenesis, known as the adult hippocampal neurogenesis and cellular plasticity theory of depression, closely resembles the neurotrophic theory of depression, but attempts to address the roles of neurotransmitters in depressive disorders. The hypothesis attributes neuronal volume reductions and morphological changes observed in the hippocampi of depressed subjects to a failure of neurogenesis and general cellular plasticity in the granule cell layer of the dentate gyrus (Jacobs et al., 2000 ; Kempermann and Kronenberg, 2003 ). Other than observed structural deficits in key limbic regions, the hypothesis was inspired largely by discovering that elevated serotonin levels, not just elevated neurotrophic factor levels, increases hippocampal neurogenesis (i.e., Successful therapies relieve depressive symptoms through more than one pathway) (Jacobs et al., 2000 ; Kempermann and Kronenberg, 2003 ). Due to the large number of projections from the hippocampus to brain structures implicated in depression, it is proposed that cycles of decreased or increased hippocampal neurogenesis (Jacobs et al., 2000 ) and cellular plasticity (Kempermann and Kronenberg, 2003 ) causes episodes of and remission from depressive episodes and may explain the heterogeneous nature of the disorder.
A major drawback to testing this theory is the difficult nature of accurately quantifying cellular plasticity, especially using non- or minimally-invasive techniques (Kempermann and Kronenberg, 2003 ). The neurotrophic theory involves mostly quantifiable markers of depression, using the effects of the HPA-axis and neurotrophins to explain the pathogenesis of depressive disorders. While the adult hippocampal neurogenesis and cellular plasticity theory of depression considers the roles of stress and BDNF in neurogenesis, they are not strict requirements. The non-specific nature of the hypothesis provides a framework for more specific hypotheses, making it useful even if it does not directly lead to an understanding of depressive disorder pathogenesis.
Macrophage Theory of Depression
A fifth, highly-promising hypothesis of depressive disorder pathogenesis considers the role of the immune system in the pathogenesis of depression, specifically drawing upon the parallels between the symptoms and behaviors linked to depression and “sickness behavior” (a term used to describe physically ill patients with various infections and/or diseases that chronically activate the immune system and cause symptoms indicative of depression) (Dantzer et al., 2008 ). Several observations led to the macrophage theory of depression, which states that depressive disorders arise from the effects of abnormally high levels of cytokine production: (1) pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), are soluble inflammatory factors produced by activated immune cells at a site of infection to promote local inflammation and often broader, systemic inflammatory effects. These cytokine-mediated systemic effects can adversely affect brain function, where the cytokine-mediated inflammation is linked to the behaviors typically observed with depressive disorders and severe illnesses (e.g., smaller appetite, impaired memory, irritability, depressed mood, impaired attention, etc.) (Dantzer et al., 2008 ); (2) pro-inflammatory cytokine receptors have been identified in most areas of the brain, although the quantity of receptors per cell is usually small and varies depending on the region of the brain (Konsman et al., 2004 ; Dantzer et al., 2008 ); (3) acutely introducing pro-inflammatory cytokines in a normal subject can chronically disturb hormone dynamics to what is often seen in depressive disorders and can produce sufficient symptoms for a diagnosis of a major depressive episode (Smith, 1991 ; Yirmiya et al., 2000 ); (4) comorbid diseases that are often seen in patients with severe depressive disorders, such as rheumatoid arthritis or heart disease, involve significant macrophage activation (possibly as a result of chronic inflammatory effects, Dantzer et al., 2008 ) (Smith, 1991 ); and (5) Yirmiya et al. (2000) showed that antidepressants can alleviate the symptoms of sickness behavior.
O’Brien et al. (2007) showed that SSRI-resistant MDD patients have significantly higher IL-6 and TNF-α levels in blood plasma than healthy controls and SSRI-resistant MDD patients who successfully responded to another class of antidepressants (O’Brien et al., 2007 ). They conclude that pro-inflammatory cytokine expression (specifically IL-6 and TNF-α) is higher in refractory MDD patients versus normal patients and that a failure of pro-inflammatory cytokine suppression is linked to SSRI resistance in refractory MDD patients. Biomarkers of SSRI-resistant depressive disorders may serve as a means of distinction among refractory MDD patients whose depressive disorders arise through different underlying mechanisms. However, the utility will largely depend on whether similar biomarker-based distinctions are observed in refractory MDD patients with differing drug resistance profiles.
Each theory discussed so far has common features that may be causal in depressive disorders, but none are as far-reaching as the macrophage theory of depression. The macrophage theory of depression is useful for finding quantifiable depressive disorder diagnostic biomarkers, since its general framework can account for many of the assumed underlying causes and/or effects of depressive disorders (e.g., The macrophage theory accounts for the large percentage of chronically ill patients that are diagnosed with comorbid depression) and for certain cases of treatment resistance. However, it does not explain whether a depressive disorder or chronic illness must be present at the same time or whether a depressive disorder leads to chronic illness (or vice versa). Section “Alternative Therapies for Refractory Major Depressive Disorders” discusses non-pharmaceutical, electromagnetic-stimulation-based antidepressant therapies that are most often used when pharmaceutical antidepressant therapy, psychotherapy, and/or ECT fail in a given patient.
Alternative Therapies for Refractory Major Depressive Disorders
If drug-based antidepressant therapies are ineffective in a given patient, few alternative options remain. In severe cases, ECT is most commonly used over several weeks to help control depressive symptoms. This traditional treatment paradigm for treatment-resistant patients involves non-specific, but noninvasive stimulation of broad regions of the cortex. The patients must be lightly anesthetized and/or sedated and often experience significant adverse side effects (e.g., retrograde amnesia that often does not fully improve over time) (Marangell et al., 2007 ; Dumitriu et al., 2008 ). However, despite its inherent limitations, ECT has provided more antidepressant benefits to refractory MDD patients than any other FDA-approved treatment option. ECT will not be further addressed in this review, because it is not a suiTable technology for implantable, closed-loop neuroprosthetic devices. The remainder of Section “Alternative Therapies for Refractory Major Depressive Disorders” discusses current and upcoming treatment alternatives, addressing the limitations of each therapy as they relate to stimulation/therapeutic targets, therapeutic efficacy, and utility in a closed-loop therapeutic device.
Vagus Nerve Stimulation
The prospect of using vagus nerve stimulation (VNS) as an alternative therapy for refractory MDD originated from unexpected patient-reported mood improvements observed in the NeuroCybernetic Prosthesis System trials in the 1990s (Ben-Menachem et al., 1994 ) and the off-label uses of specific seizure medications for stabilizing mood disorders (Ballenger and Post, 1980 ; Post et al., 1986 ; George et al., 2000 ). Several treatment-resistant epileptic patients implanted with the VNS device reported significant mood elevations that researchers could not solely attribute to seizure frequency reduction. Additional positron-emission tomography (PET) studies showed that VNS reduced cingulate activity, the same effect seen from many successful antidepressant therapies (George et al., 2000 ), and altered blood flow and metabolism in limbic structures (Henry et al., 1998 ). Other investigations showed that neurotransmitter levels are altered as a result of VNS (Ben-Menachem et al., 1995 ; Krahl et al., 1998 ). Due to these observations, the first VNS stimulation device for treating unipolar refractory MDD was implanted by Rush and colleagues (1998) (George et al., 2000 ; Rush et al., 2000 ). FDA approval followed in July, 2005 (George et al., 2000 ; Nemeroff et al., 2006 ).
The VNS device implantation procedure is rather straightforward. Under general anesthesia, the VNS device housing is surgically implanted in the left chest wall. A projecting stimulation lead with an attached helical electrode is then wrapped around the left cervical vagus nerve and secured to surrounding tissue. The device is externally activated and programmed using a wand-like device placed over the left chest wall. Stimulation is intermittent and commonly programmed for 30 s of monophasic, constant-current stimulation every 5 min. However, individual parameters are adjusted on a patient-to-patient basis in order to achieve maximal therapeutic efficacy with minimal side effects (Marangell et al., 2007 ).
The efficacy of the current VNS device for treating depression is disputed. In the 2000 paper summarizing the study’s outcome, Rush et al. report antidepressant responses in 40% of the 30 patients who were enrolled. However, statistically significant reductions in depression rating scale scores were not observed for the experimental group as a whole (Rush et al., 2000 ). In 2004, Carpenter et al. showed that monoamine neurotransmitter metabolites, norepinephrine, and GABA levels did not significantly change over a 24-week study, noting that “the results do not suggest a putative antidepressant mechanism of action for VNS” (Carpenter et al., 2004 ). Additional controlled studies with larger sample sizes are needed to make any definitive conclusions.
Transcranial Magnetic Stimulation
Transcranial magnetic stimulation (TMS) was introduced by Barker et al. (1985) (Klein et al., 1999 ). By noninvasively activating target cortical regions, TMS allows investigators to selectively study brain function in a simplified and relatively safe manner (Figiel et al., 1998 ; Klein et al., 1999 ). In the last few decades it has received considerable interest as a therapeutic tool in a variety of neurological disorders, stemming from its favorable spatial selectivity over ECT, noninvasive nature, and generally tolerable side effects (Figiel et al., 1998 ; Klein et al., 1999 ; Janicak et al., 2008 ). As a result, TMS is now used as an FDA-approved treatment option for refractory MDD.
Transcranial magnetic stimulation is typically administered by pulsing a current through a circular or figure-8 coil positioned over the cortical regions of interest. The resulting oriented magnetic field pulses generate an electric field within the superficial layers of cortex (with a maximum depth of 1 cm, Dumitriu et al., 2008 ), depolarizing neurons when a sufficient electric field is generated (Fitzgerald et al., 2002 ).Device size limitations preclude the use of this technology in a fully implanTable closed-loop neuroprosthesis. Current TMS devices are large and typically only accessible through outpatient procedures (such as NeuroStar® TMS Therapy, Neuronetics, 2009 ). TMS device size, which is proportional to the size of the stimulated cortical area, is limited by a tradeoff between coil size and the magnitude of current required to generate the same magnetic field in a smaller device (Cohen and Cuffin, 1991 ). As a result, TMS is not suiTable for use in a fully implanTable neuroprosthesis unless fundamental design changes are made to considerably decrease device size without sacrificing performance.
There are many subtypes of TMS, classified according to stimulation parameters and mode of application. In this review, two traditional subtypes are considered: rapid-rate/repetitive transcranial magnetic stimulation (rTMS – includes any stimulation paradigm using frequencies >1 Hz) and low-frequency/slow transcranial magnetic stimulation (sTMS – includes any stimulation paradigm using frequencies ≤1 Hz). The TMS subtypes produce differing cortical activation properties – depending largely on stimulation parameters, coil shapes and sizes, stimulation sites, and stimulation orientations – and are associated with studies that report conflicting therapeutic efficacies. However, it is believed that rTMS produces more antidepressive effects, as one study of cerebral blood flow showed significant increases in blood supply to prefrontal cortical and limbic regions following rTMS and marked decreases following sTMS (Speer et al., 2000 ).
Deep Brain Stimulation
Deep brain stimulation (DBS) was first used for treating depression in 1954 (Poole, 1954 ; Hardesty and Sackeim, 2007 ). However, DBS gained considerable research interest and momentum in 1987, when Benabid et al. (1987) successfully relieved Parkinsonian tremors in a patient through high-frequency stimulation of one thalamic nucleus ventralis intermedius and removal of the other. Benabid et al.’s paper showed that high-frequency electrical stimulation of a dysfunctional brain structure was as effective as surgically removing the same part of the brain, thereby promoting DBS therapy as a less-invasive and less-extreme alternative to resection surgeries (Benabid et al., 1987 ; Hardesty and Sackeim, 2007 ).
The power of DBS in treating refractory psychiatric disorders has become increasingly apparent throughout the last few decades, largely through unexpected side effects observed in non-depressed DBS patients. For example: in an older woman without any known psychiatric disorders (implanted with a deep brain stimulator for Parkinson’s disease), high-frequency DBS therapy applied to the left substantia nigra caused temporary suicidal depression that reversed once stimulation ceased (The electrical stimulus was inadvertently applied 2 mm below the optimal site of stimulation for Parkinson’s symptom relief) (Bejjani et al., 1999 ; Hardesty and Sackeim, 2007 ). In a separate case study of DBS for treating comorbid obsessive-compulsive disorder with Parkinson’s disease, Fontaine et al. (2004) demonstrated that the choice of stimulation parameters (applied in the sub-thalamic nucleus) largely determined the level of obsessive-compulsive disorder symptom relief. The temporary induction of severe depressive symptoms in the older woman indirectly highlights the power of DBS as a means of rapidly and reversibly modulating neural function. However, it also cautions that the therapeutic efficacy of any treatment heavily depends on the specificity of its delivery, as a small targeting error can induce potentially dangerous nonlinear side effects. The latter case study shows that DBS therapeutic efficacy is largely dose-dependent in addition to site-dependent (Fontaine et al., 2004 ; Hardesty and Sackeim, 2007 ). Hence, despite its invasive nature, DBS is heavily researched due to its promise of rapidly modulating dysfunctional network activity to desired states in a dose-dependent manner only when it is needed. Harnessing the full power of this therapy, however, will require a closed-loop system that autonomously adjusts output stimulus parameters in order to maintain disease- or symptom-linked biomarkers within normal levels.
Stimulation Targets for Refractory MDD
Few neural stimulation targets have been evaluated for treatment efficacy in the refractory MDD population (Table 1 ). In general, proposed stimulation targets are linked to limbic structures and come directly from hypotheses of neural dysfunction in depression, imaging studies, unexpected mood improvements observed in stimulation studies treating other disorders, and areas accessible with a given stimulation technology. The VNS system specifically targets the left cervical vagus nerve (Ben-Menachem et al., 1994 ; George et al., 2000 , 2005 ). TMS studies typically target the left and/or right dorsolateral prefrontal cortex (DLPFC) due to its accessibility with the large stimulation coils and the promising history of its antidepressive effects. Slow TMS (sTMS) has only provided antidepressive effects when used on the right DLPFC (Klein et al., 1999 ; Fitzgerald et al., 2006 ), while repetitive/rapid TMS only has provided antidepressive effects when used on the left DLPFC (Speer et al., 2000 ; Avery et al., 2006 ; Fitzgerald et al., 2006 ). Not surprisingly, DBS studies target deep structures such as the subcallosal cingulate gyrus (SCG) (Mayberg et al., 2000 , 2005 ; Lozano et al., 2008 ), ventral capsule/ventral striatum (VC/VS) (Malone et al., 2009 ), globus pallidus internus (GPi) (Kosel et al., 2007 ), and inferior thalamic peduncle (ITP) (Jimenez et al., 2005 ).
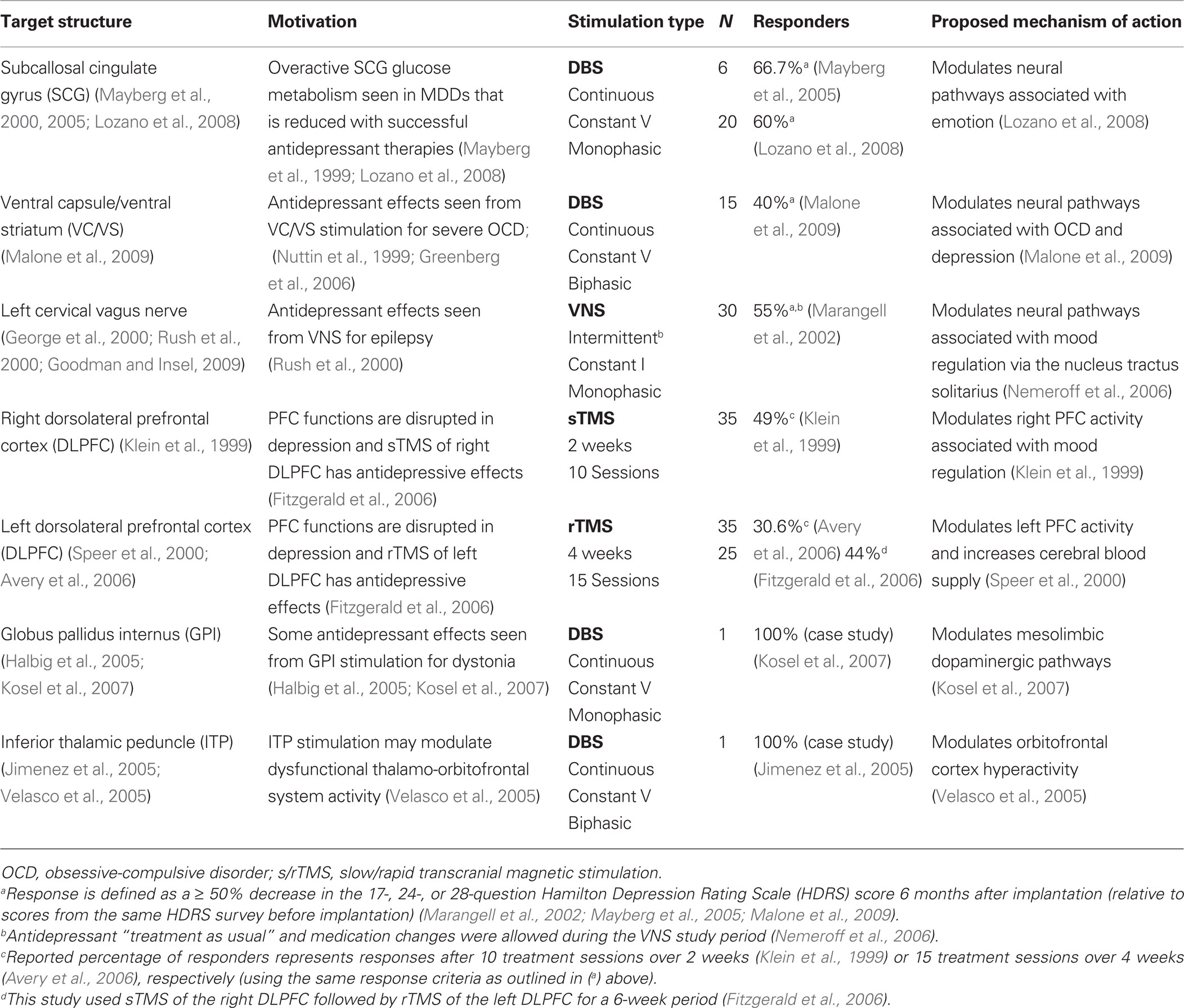
Table 1. Stimulation targets for treating refractory MDD.
Inconsistent study designs, control/sham selections, stimulation parameters, evaluation criteria, and subject selections make it difficult to compare the reported treatment efficacies of the different stimulation technologies and stimulation targets. From Table 1 , it appears that the most promising stimulation-based antidepressant treatment targets the subcallosal cingulate gyrus white matter (SCGwm) using DBS. High-frequency SCGwm stimulation significantly reduced symptoms (i.e., Post-treatment HRSD-17 scores were 50% lower than pre-treatment HRSD-17 scores) in 60% of refractory MDD patients at 6 months and 55% at 12 months (N = 20) (Lozano et al., 2008 ). The SCG has many pathways that project to limbic and paralimbic brain structures, and is likely the most important autonomic regulatory region within the frontal cortex (Ongur et al., 1998 ; Davidson et al., 2002 ). However, due to the aforementioned inconsistencies among studies, it is impossible to definitively determine which technology is superior at this time.
Each stimulation technology uses different sets of stimulation parameters, producing a constant-current- or voltage-based mono- or biphasic waveform with a diverse range of amplitudes, pulse durations, and stimulation frequencies (see Albert et al., 2009 for a comprehensive review of stimulation parameters that have been used for VNS, TMS, and DBS). The respective waveforms stimulate a target structure continuously or intermittently (in open-loop configurations) in hopes of directly or indirectly modulating abnormal activity toward more normal behavior in limbic-associated neural pathways and structures (e.g., VNS technology intermittently stimulates for 30 s every 5 min to indirectly modulate brain activity via the left cervical vagus nerve, Marangell et al., 2007 ). VNS and DBS stimulation parameters are wirelessly programmed approximately 2 weeks after implantation on a patient-specific basis. By using patient-reported symptom relief and side effects, stimulation pulse duration and amplitude are steadily increased over a period of weeks to months (under a constant pulse repetition frequency) to determine a range of parameters that produce the most significant therapeutic benefit with the least side effects (monophasic, constant-current stimulation is typically used in VNS and monophasic, constant-voltage stimulation is typically used in DBS) (Hardesty and Sackeim, 2007 ).TMS devices first measure a patient’s motor threshold (i.e., the magnetic pulse intensity that elicits a motor action potential when applied over the motor cortex) before beginning the procedure (Marangell et al., 2007 ). A percentage of the observed motor threshold is then used as the baseline intensity at which the magnetic pulse is applied for therapy (Albert et al., 2009 ).
Stimulation programming procedures are often uncomforTable for the patient, as severe side effects are often induced due to unintended neural stimulation from poorly placed stimulus transducers, poorly chosen parameters, and/or limited spatial resolution from a given stimulation technology. Increasing the specificity of stimulus delivery to more precisely target the dysfunctional neurons or networks should lead to reduced side effect profiles. For DBS, it will likely require smaller transducers than current FDA-approved DBS electrodes (e.g., microelectrode technology – which may also enable VNS devices to more specifically target afferent vagus nerve fibers whose activation/inhibition are linked to the purported antidepressive benefits). For TMS, increasing the specificity of stimulus delivery will require smaller, more-efficient coil designs. After optimizing the specificity of stimulus delivery, waveform optimization will be needed to determine the safest, most beneficial parameter sets for a given stimulation technology, point of application, and patient. Based on more recent findings, biphasic, constant-current waveforms should be employed due to their proven safety relative to monophasic or constant-voltage waveforms, their enhanced glial scar penetration capabilities, and their reversible, charge-balanced nature (Albert et al., 2009 ; Piallat et al., 2009 ).
Quantifiable Biological Markers of Refractory MDD
Biomarkers are not currently used in depressive disorder diagnostic or treatment paradigms (e.g., Biomarkers are not used to confirm the presence of a depressive disorder, quantify its relative severity, or to assess treatment compliance/resistance). As with stimulation targets, the theories of depressive disorder pathogenesis have led to several candidate biomarkers that can distinguish normal from depressed patients, treatment compliant from treatment-resistant patients, and non-suicidal from suicidal depressed patients (Table 2 ). However, individual biomarkers have not shown sufficient sensitivity and specificity for use in clinical practice. A major hurdle preventing the discovery of depressive disorder-specific biomarkers is the lack of distinctions between MDD patients who share a common set of subjectively defined symptoms, but develop the disorder through different circumstances. As a result, the upcoming DSM-V and ICD-11 criteria for diagnosing MDD may not require biological diagnostic measures of MDDs (Mossner et al., 2007 ).
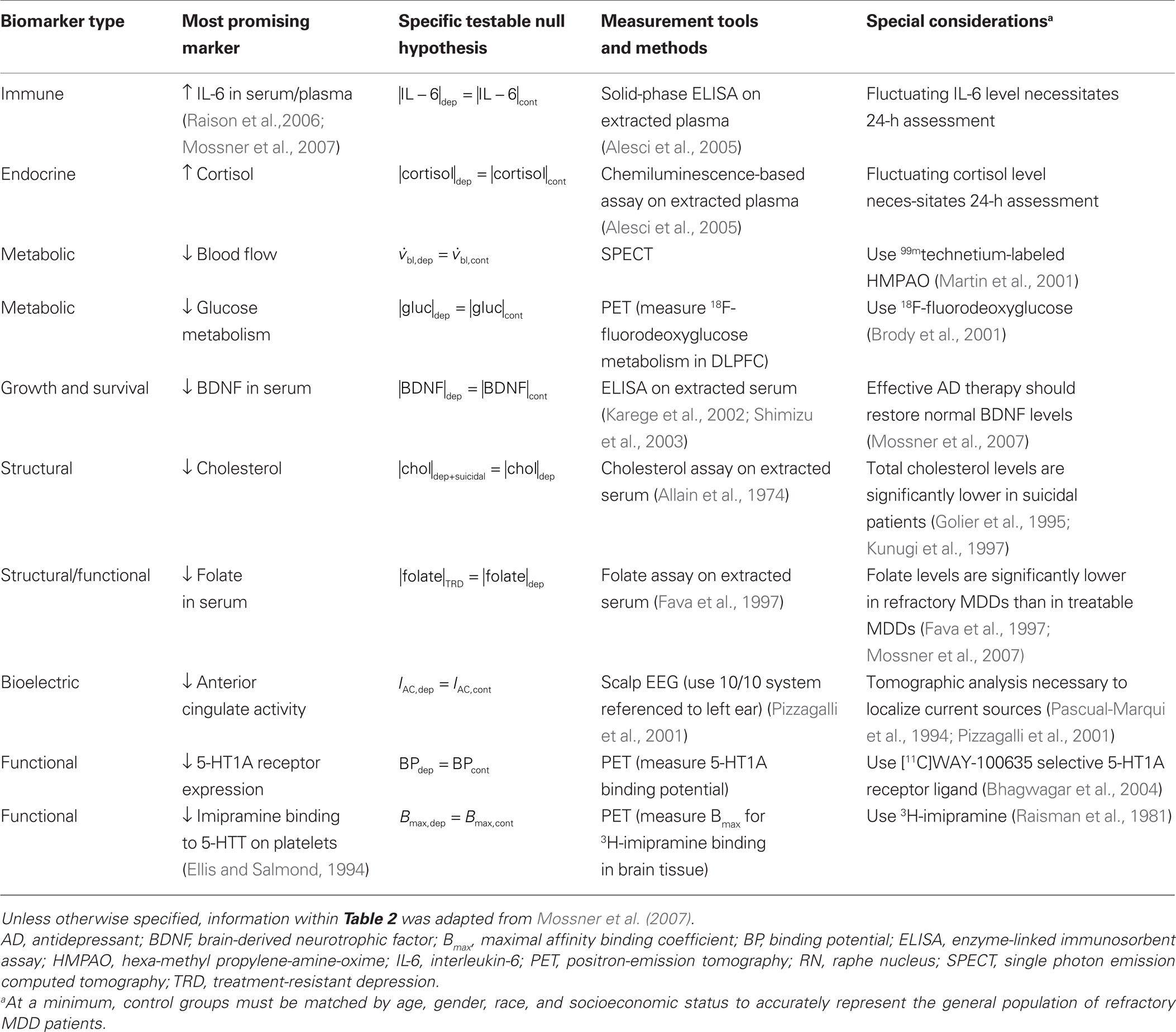
Table 2. Promising biomarkers for objectively distinguishing and quantifying depressive states.
In light of the current understanding of depressive disorder pathogenesis, Mossner et al. (2007) deduced that decreased BDNF levels, increased IL-6 levels, reduced folate levels, reduced total cholesterol levels, and elevated cortisol levels are among the most promising biomarkers for identifying and distinguishing among MDDs. Relative to healthy control groups, increased IL-6 (Alesci et al., 2005 ) and decreased BDNF levels (Shimizu et al., 2003 ) are among the most promising biomarkers of depression, decreased folate in blood serum is among the most promising biomarkers of treatment resistance (Fava et al., 1997 ), reduced anterior cingulate activity is among the most promising biomarkers for predicting treatment compliance (Pizzagalli et al., 2001 ), and decreased total cholesterol is among the most promising biomarkers of suicidal behavior (Golier et al., 1995 ; Kunugi et al., 1997 ). Several researchers are investigating the predictive power of abnormal electrical activity associated with depression (Tomarken et al., 1992 ; Pizzagalli et al., 2001 ; Garrett et al., 2008 ). However, progress has been limited, likely stemming from the limitations of using low bandwidth electroencephalograms (EEG) (Pizzagalli et al., 2001 ) or evoked response field potentials (Garrett et al., 2008 ) to investigate disorders of specific neural circuits in deeper brain structures.
Toward Closed-Loop Treatment Options for Refractory MDD Patients
As a last-resort invasive treatment option for refractory MDD patients, an implanTable neurostimulation device will ideally (1) remit depressive symptoms in a predictable, reproducible, and quantifiable manner without inducing the variable set of side effects observed with current therapies, (2) apply a focal corrective stimulus (i.e., dose of therapy) only when the device confirms an impending relapse into a depressive episode, (3) autonomously learn and apply the most efficacious corrective stimuli which correct for differing levels of biomarker abnormalities that correlate with patient-specific depressive symptoms, (4) safely function for the duration of a patient’s lifetime without the need for secondary surgeries, and (5) store and transmit peri-stimulus biomarker responses for off-line analyses. An open-loop system can meet criteria 4–5, but a closed-loop system is needed to meet all five. The remainder of Section “Toward Closed-Loop Treatment Options for Refractory MDD Patients” first discusses the algorithm requirements for autonomously translating abnormal biomarker signals into optimized corrective stimuli. It concludes with a closed-loop neurostimulation device design framework.
There are two general requirements for an algorithm to autonomously optimize closed-loop treatment efficacy. First, an autonomous closed-loop optimization algorithm requires a reproducible relationship between the applied stimulus parameters and there sulting disorder-specific biomarker responses (i.e., The applied therapeutic dose of charge and its rate of delivery, using electrical stimulation, or the applied drug dose and its rate of delivery, using chemical stimulation, should lead to predicTable and reproducible restorative changes in the observed abnormal biomarker levels). Secondly, an autonomous closed-loop optimization algorithm requires a reproducible relationship between the magnitude and direction of biomarker abnormality correction (due the preceding applied stimuli) and the level of symptom remission (Behrend et al., 2009 ). While the feasibility of the former requirement was recently demonstrated (refer to the following paragraph), significant challenges remain with the latter due to a knowledge gap that remains between subjective reports of depressive symptoms and quantifiable levels of symptom-linked biomarker abnormality. However, groups seeking such knowledge for optimizing treatments in other neurological disorders have demonstrated promising progress toward this end (Wong et al., 2008 ; Behrend et al., 2009 ; Smith et al., 2009 ).
Behrend et al. (2009) successfully demonstrated the feasibility of the first of the autonomous closed-loop optimization algorithm requirements by using closed-loop STN stimulation to maintain defined glutamate levels in the same structure. To do this, a transfer function was first derived to model the relationship between randomly patterned high-frequency stimulation pulse trains and extracellular glutamate dynamics in rat STN (The investigators applied pseudorandom binary sequences of stimulation pulses to the STN and used an autoregressive exogenous model to derive the transfer function). They found that (1) the derived transfer function accurately predicts the dynamic glutamate responses to unique pulse trains and that (2) on-demand STN stimulation sufficiently maintained the desired glutamate levels (i.e., Continuous or pre-programmed periodic stimulation is not necessary for effective control of biomarker levels; the desired biomarker levels in the STN can be maintained with on-demand stimulation using pulse train patterns with predicTable effects on the biomarker level(s)) (Behrend et al., 2009 ). If a predicTable and reproducible relationship can be established between biomarker abnormality correction and depressive symptom relief, then Behrend et al.’s second finding also implies that withdrawal of the corrective stimulus will not necessarily lead to a relapse.
To address the feasibility of the second autonomous closed-loop optimization algorithm requirement, reproducible and reliable relationships between the magnitude and direction of stimulus-associated biomarker abnormality correction and stimulus-associated depressive symptom relief must be established on a patient-to-patient basis. One proposed approach to surmounting this barrier is to subjectively assist (i.e., train) the treatment-optimization algorithm after the implantation recovery period. For example: after recovering from device implantation and before turning on the stimulation circuitry, a physician can program the device to measure the biomarker(s) of interest whenever the patient externally triggers it (e.g., an external piece of hardware, such as the wand-like device that a VNS implantee uses to externally trigger stimulation (Marangell et al., 2007 ), can be used to wirelessly signal the implanted device to measure the biomarker of interest).To subjectively assist the stimulus-optimization algorithm and provide insight to the physician, the physician can instruct the patient to routinely assess their depressive symptom severity (e.g., using a standardized depression rating scale) while simultaneously signaling the device to measure the biomarker(s) of interest. Over time, a rough relationship between the biomarker levels and symptom severity in a particular patient can be established. Then, starting with stimulus parameter combinations that have shown therapeutic efficacy in other patients, the process can be repeated to derive a rough relationship between the magnitude/direction of stimulus-associated biomarker abnormality correction and the level of stimulus-associated depressive symptom relief. If such relationships are established, investigators can begin to bridge the knowledge gap that currently exists between subjective reports of depressive symptoms and quantifiable abnormalities in biomarker levels. With time, the device can autonomously fine-tune the most efficacious parameter sets that most efficiently lead to the desired outcomes (i.e., minimal dose, maximal antidepressant response, minimal side effects, maximal antidepressant durability, etc.).
Figure 1 presents a closed-loop device design framework based on topics addressed within the review. Figure 1 A shows a generic closed-loop control system, which runs in parallel to the dysfunctional structure of interest. In each cycle, the four basic processes involve (I) measuring the biomarker(s) of interest from the stimulation target or downstream from the stimulation target, (II) comparing the measured biomarker levels to non-depressed biomarker levels to determine whether a corrective stimulus is needed, (III) determining the appropriate set of corrective stimulus parameters based on the magnitude of biomarker abnormality and previous stimulus/biomarker response data (represented as a multi-dimensional stimulus response surface in Figure 1 B), and (IV) applying the appropriate corrective stimulus to the stimulation target. This generic closed-loop control system design supports any of the alternative stimulation-based therapies, accessible stimulation targets, and depression-linked biomarkers (refer to Tables 1 and 2 ).
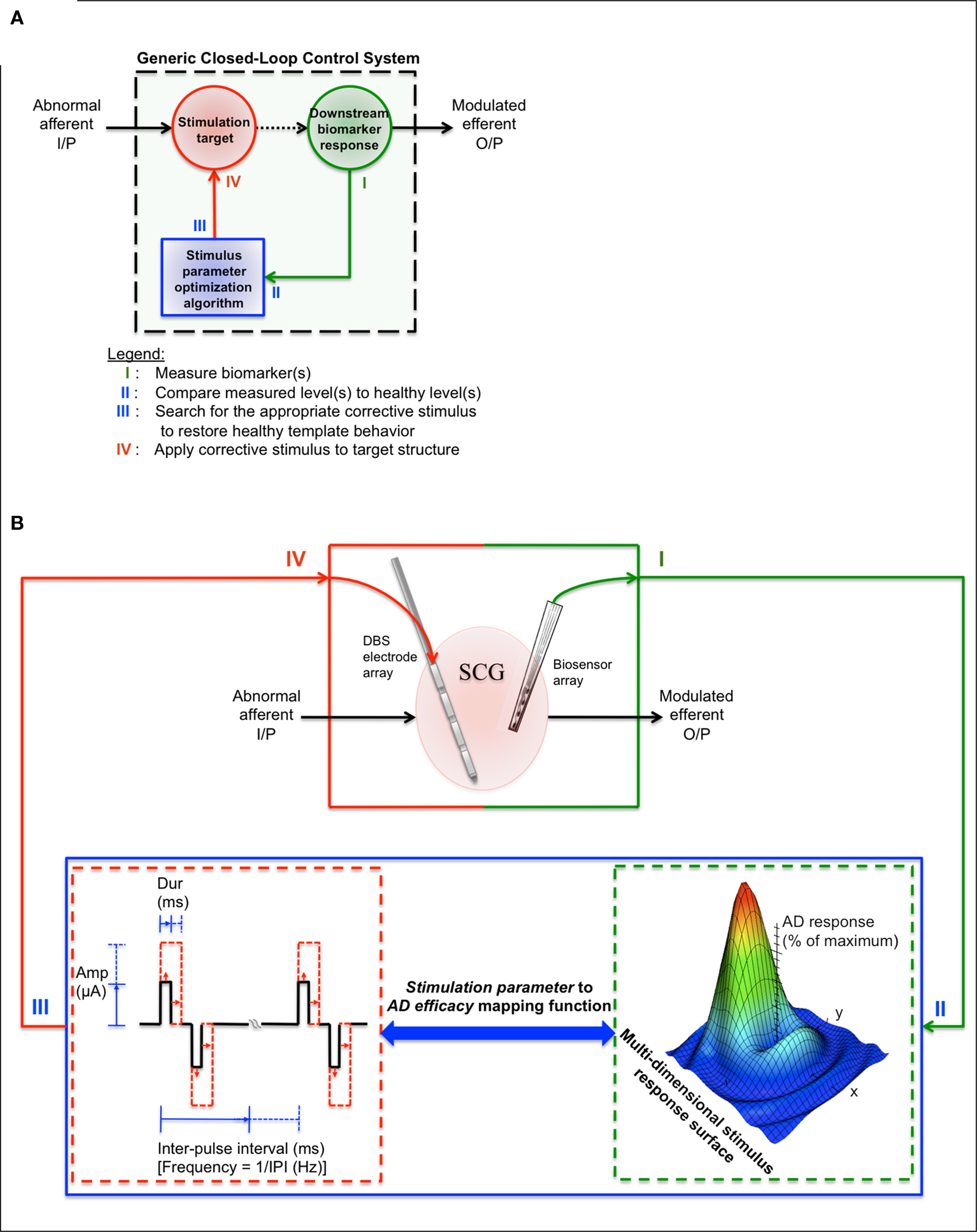
Figure 1. Proposed closed-loop treatment design framework for refractory MDD therapeutics. (A) Generic closed-loop control system. In each cycle, the four basic processes involve (I) measuring the biomarker(s) of interest from the stimulation target or downstream from the stimulation target, (II) comparing the measured biomarker levels to non-depressed biomarker levels in order to determine whether a corrective stimulus is needed, (III) determining the appropriate set of corrective stimulus parameters based on the magnitude of biomarker abnormality and previous stimulus/response data (represented as a multi-dimensional stimulus response surface in (B)), and (IV) applying the appropriate corrective stimulus. This generic design supports any of the alternative stimulation-based therapies, accessible stimulation targets, and depression-linked biomarkers (refer to Tables 1 and 2 ). (B) A proposed closed-loop device design framework. Table 1 can be used to find stimulation targets that have shown promising treatment efficacy in the refractory MDD population (In this example, the SCGwm serves as the stimulation target). Table 2 can be used to guide the initial biomarker selection process (in this example, downstream single neuron activity is measured using a microelectrode array). A charge-balanced, constant-current stimulation waveform is used as the corrective stimulus. Pulse width, amplitude, pulse train duration, and/or pulse repetition frequency is varied for the next SCGwm stimulation based on the type of biomarker abnormality detected and previous stimulus/biomarker response data (this data is represented as a multi-dimensional stimulus response surface). The cycle continues until the maximum antidepressant response is achieved.
The presented framework for designing depression-specific closed-loop devices narrows the design space to a smaller subset of promising treatment strategies. In one possible implementation (Figure 1 B), a DBS electrode array is placed in the SCGwm in order to modulate dysfunctional downstream limbic and paralimbic network activity to more “healthy” states when an onboard algorithm detects a depressed state (IV). A biosensing microelectrode array (or a second DBS electrode array, since these are already FDA-approved, Han and McCreery, 2009 ) is placed downstream from the stimulation target in order to monitor symptom-linked biomarker changes in response to the applied stimulus (I, II). Using stored stimulus/biomarker response data, a stochastic search algorithm is used to predict an optimal set of output stimulus parameters that have the highest likelihood of correcting for the magnitude of abnormality that was detected (III). Many algorithms can accomplish this process, such as stochastic search algorithm, genetic algorithm or a clustering algorithm designs, but each will require some form of training to maximize sensitivity and specificity for unique applications.
A stochastic search algorithm design called a “Gur Game” can in theory optimize stimulation parameters based on a measured set of stimulus/biomarker response data from previous stimulations (II–III) (Tung and Kleinrock, 1996 ; Wong et al., 2008 ). Such an algorithm treats parameters independently, and autonomously varies parameters in order to maximize a global response variable (the downstream biomarker response to stimulation, which is mapped as a percent of the maximum antidepressant response). The stochastic nature of the search algorithm enables it to rapidly find parameter sets that maximize (i.e., optimize) the global response variable within a large multi-dimensional parameter search space. It is well-suited for closed-loop applications, because (1) the underlying mechanisms of the antidepressant therapy are largely irrelevant to the algorithm, as it works solely to maximize a global response variable by manipulating parameter values within predefined safety limits, (2) the performance does not significantly decrease with additional parameters, enabling non-biological properties such as power consumption to be included in the optimization scheme, and (3) the algorithm does not get trapped in local maxima (Tung and Kleinrock, 1996 ; Feng et al., 2007 ; Wong et al., 2008 ).
Conclusions
An inadequate understanding of depressive disorder pathophysiology impedes investigators from developing reliable diagnostic measures and more effective, durable antidepressant therapeutics. Consequently, the current questionnaire- and physical-exam-based evaluations fail to address the specific biological causes of depressive disorders, leading to incorrect diagnoses and ineffective open-loop therapies (Kearns et al., 1982 ; Mossner et al., 2007 ). Furthermore, most current antidepressant treatments aim to correct unproven neurotransmitter imbalances that are believed to cause a subset of general patient-reported symptoms, whereas the biological cause of the depressed state likely stems from one or more seemingly unrelated physiological or genetic causes (Lacasse and Leo, 2005 ).This review presents a framework for designing closed-loop refractory MDD treatments, which are expected to overcome many of the challenges that stem from open-loop treatments. These include autonomous antidepressant response optimization, therapeutic dose minimization, side effect minimization, and treatment durability maximization, among others.
In current clinical practice, alternative open-loop treatments such as VNS, TMS, and DBS provide more focal therapy for refractory MDD patients than pharmaceuticals or ECT, but have unsubstantiated therapeutic values. Furthermore, implanted devices require months of stimulus waveform adjustments that can induce variable adverse side effects and significant discomfort for the patients (Hardesty and Sackeim, 2007 ; Dumitriu et al., 2008 ). Case studies of DBS implantees indicate that variable therapeutic efficacies and many side effects stem from non-specific stimulation of neural circuits using stimulation parameters with no established theoretical bases and large surface area DBS macroelectrodes (Bejjani et al., 1999 ; Fontaine et al., 2004 ; Hardesty and Sackeim, 2007 ). It is also likely that the continuous or intermittent high-frequency stimulation schemes employed by DBS and VNS contribute to the reported side effects and the need for larger stimulus amplitudes over time (e.g., long-term, high-frequency stimulation in this manner may enhance phenomena such as stimulation-induced depression of neuronal excitability, a phenomenon in which a neuron develops a tolerance to a constant stimulus by increasing its threshold for activation, McCreery et al., 1997 ).It is clear that more efficacious treatment alternatives with lower side effect profiles are needed for refractory MDD patients.
A closed-loop neuroprosthesis that focally stimulates specific dysfunctional neuron populations in response to abnormal biomarker level(s) detected by onboard algorithms is hypothesized to lead to enhanced therapeutic efficacy and reduced side-effect profiles. Prior to use in humans, closed-loop neuroprostheses should be evaluated in animals and used to systematically test theories of depressive disorder pathogenesis, objectively define and delineate specific subtypes of the disorder, and locate biomarkers or combinations of biomarkers that reliably predict or detect depressive episodes, suicidal tendencies, and/or levels of treatment response/resistance. Since animal models of depression are highly debated and often evaluated subjectively, it would be useful to also study depressive disorders in human subjects. If appropriate data use guidelines and permissions are in place, patient-specific electrical and/or chemical data can be downloaded (from the closed-loop device) and stored in a database for data mining purposes. Investigators can use such a database to help establish more stringent, objective classifications of depressive disorders based on quantifiable biological markers of the disorder, not just traditional symptom-based classification schemes. Objective measures of depression will lead to more accurate diagnoses, with the additional benefit of allowing physicians to tailor all treatments (pharmaceutical, psychotherapeutic, and alternative) to specific, well-defined forms of depression. Furthermore, companies and researchers will have greater insights into the disorder, allowing them to develop more focal therapies with higher therapeutic efficacies and reduced symptom profiles.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
The authors would like to thank members of the Center for ImplanTable Devices (CID) for their help with refining this review document.
References
Albert, G. C., Cook, C. M., Prato, F. S., and Thomas, A. W. (2009). Deep brain stimulation, vagal nerve stimulation and transcranial magnetic stimulation: an overview of stimulation parameters and neurotransmitter release. Neurosci. Biobehav. Rev. 33, 1042–1060.
Alesci, S., Martinez, P. E., Kelkar, S., Ilias, I., Ronsaville, D. S., Listwak, S. J., Ayala, A. R., Licinio, J., Gold, H. K., Kling, M. A., Chrousos, G. P., and Gold, P. W. (2005). Major depression is associated with significant diurnal elevations in plasma interleukin-6 levels, a shift of its circadian rhythm, and loss of physiological complexity in its secretion: clinical implications. J. Clin. Endocrinol. Metab. 90, 2522–2530.
Allain, C. C., Poon, L. S., Chan, C. S. C., Richmond, W., and Fu, P. C. (1974). Enzymatic determination of total serum cholesterol. Clin. Chem. 20, 470–475.
Avery, D. H., Holtzheimer III, P. E., Fawaz, W., Russo, J., Neumaier, J., Dunner, D. L., Haynor, D. R., Claypoole, K. H., Wajdik, C., and Roy-Byrne, P. (2006). A controlled study of repetitive transcranial magnetic stimulation in medication-resistant major depression. Biol. Psychiatry 59, 187–194.
Ballenger, J. C., and Post, R. M. (1980). Carbamazepine in manic-depressive illness: a new treatment. Am. J. Psychiatry 137, 782–790.
Barker, A. T., Jalinous, R., and Freeston, I. L. (1985). Non-invasive magnetic stimulation of human motor cortex. Lancet 325, 1106–1107.
Behrend, C. E., Cassim, S. M., Pallone, M. J., Daubenspeck, J. A., Hartov, A., Roberts, D. W., and Leiter, J. C. (2009). Toward feedback controlled deep brain stimulation: dynamics of glutamate release in the subthalamic nucleus in rats. J. Neurosci. Methods 180, 278–289.
Bejjani, B. P., Damier, P., Arnulf, I., Thivard, L., Bonnet, A. M., Dormont, D., Cornu, P., Pidoux, B., Samson, Y., and Agid, Y. (1999). Transient acute depression induced by high-frequency deep-brain stimulation. N. Engl. J. Med. 340, 1476–1480.
Benabid, A. L., Pollak, P., Louveau, A., Henry, S., and de Rougemont, J. (1987). Combined (thalamotomy and stimulation) stereotactic surgery of the VIM thalamic nucleus for bilateral Parkinson disease. Appl. Neurophysiol. 50, 344–346.
Ben-Menachem, E., Hamberger, A., Hedner, T., Hammond, E. J., Uthman, J. S., Treig, T., Stefan, H., Ramsay, R. E., Wernicke, J. F., and Wilder, B. J. (1995). Effects of vagus nerve stimulation on amino acids and other metabolites in the CSF of patients with partial seizures. Epilepsy Res. 20, 221–227.
Ben-Menachem, E., Manon-Espaillat, R., Ristanovic, R., Wilder, B. J., Stefan, H., Mirza, W., Tarver, W. B., and Wernicke, J. F. (1994). Vagus nerve stimulation for treatment of partial seizures: 1. A controlled study of effects on seizures. First International Vagus Nerve Stimulation Study Group. Epilepsia 35, 616–626.
Bhagwagar, Z., Rabiner, E. A., Sargent, P. A., Grasby, P. M., and Cowen, P. J. (2004). Persistent reduction in brain serotonin1A receptor binding in recovered depressed men measured by positron emission tomography with [11C]WAY-100635. Mol. Psychiatry 9, 386–392.
Bradley, R. G., Binder, E. B., Epstein, M. P., Tang, Y., Nair, H. P., Liu, W., Gillespie, C. F., Berg, T., Evces, M., Newport, D. J., Stowe, Z. N., Heim, C. M., Nemeroff, C. B., Schwartz, A., Cubells, J. F., and Ressler, K. J. (2008). Influence of child abuse on adult depression: moderation by the corticotropin-releasing hormone receptor gene. Arch. Gen. Psychiatry 65, 190–200.
Brody, A. L., Saxena, S., Stoessel, P., Gillies, L. A., Fairbanks, L. A., Alborzian, S., Phelps, M. E., Huang, S. C., Wu, H. M., Ho, M. L., Ho, M. K., Au, S. C., Maidment, K., and Baxter, L. R. (2001). Regional brain metabolic changes in patients with major depression treated with either paroxetine or interpersonal therapy. Arch. Gen. Psychiatry 58, 631–640.
Burke, H. M., Davis, M. C., Otte, C., and Mohr, D. C. (2005). Depression and cortisol responses to psychological stress: a meta-analysis. Psychoneuroendocrinology 30, 846–856.
Carpenter, L. L., Moreno, F. A., Kling, M. A., Anderson, G. M., Regenold, W. T., Labiner, D. M., and Price, L. H. (2004). Effect of vagus nerve stimulation on cerebrospinal fluid monoamine metabolites, norepinephrine, and gamma-aminobutyric acid concentrations in depressed patients. Biol. Psychiatry 56, 418–426.
Carroll, B. J., Cassidy, F., Naftolowitz, D., Tatham, N. E., Wilson, W. H., Iranmanesh, A., Liu, P. Y., and Veldhuis, J. D. (2007). Pathophysiology of hypercortisolism in depression. Acta Psychiatr. Scand. 115, 90–103.
Caspi, A., Sugden, K., Moffitt, T. E., Taylor, A., Craig, I. W., Harrington, H., McClay, J., Mill, J., Martin, J., Braithwaite, A., and Poulton, R. (2003). Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 301, 386–389.
Cepoiu, M., McCusker, J., Cole, M. G., Sewitch, M., Belzile, E., and Ciampi, A. (2008). Recognition of depression by non-psychiatric physicians – a systematic literature review and meta-analysis. J. Gen. Intern. Med. 23, 25–36.
Cohen, D., and Cuffin, B. N. (1991). Developing a more focal magnetic stimulator. Part I: some basic principles. Clin. Neurophysiol. 8, 102–111.
Coyne, J. C., Fechner-Bates, S., and Schwenk, T. L. (1994). Prevalence, nature, and comorbidity of depressive disorders in primary care. Gen. Hosp. Psychiatry 16, 267.
Cyberonics (2007). About Treatment Resistant Depression. VNS Therapy. Cyberonics. <http://www.vnstherapy.com/depression/vnsrightforme/abouttrd.asp >
Dantzer, R., O’Connor, J. C., Freund, G. G., Johnson, R. W., and Kelley, K. W. (2008). From inflammation to sickness and depression: when the immune system subjugates the brain. Nat. Rev. Neurosci. 9, 46–56.
Davidson, R. J., Pizzagalli, D., Nitschke, J. B., and Putnam, K. (2002). Depression: perspectives from affective neuroscience. Annu. Rev. Psychol. 53, 545–574.
DepressionGuidelinePanel (1994). Depression in primary care: detection, diagnosis, and treatment. J. Am. Acad. Nurse Pract. 6, 224–238.
Duman, R. S., Heninger, G. R., and Nestler, E. J. (1997). A molecular and cellular theory of depression. Arch. Gen. Psychiatry 54, 597.
Duman, R. S., Malberg, J., Nakagawa, S., and D’Sa, C. (2000). Neuronal plasticity and survival in mood disorders. Biol. Psychiatry 48, 732–739.
Duman, R. S., and Monteggia, L. M. (2006). A neurotrophic model for stress-related mood disorders. Biol. Psychiatry 59, 1116–1127.
Dumitriu, D., Collins, K., Alterman, R., and Mathew, S. J. (2008). Neurostimulatory therapeutics in management of treatment-resistant depression with focus on deep brain stimulation. Mt. Sinai J. Med. 75, 263–275.
Ellis, P. M., and Salmond, C. (1994). Is platelet imipramine binding reduced in depression? A meta-analysis. Biol. Psychiatry 36, 292–299.
Fava, M. (2003). Diagnosis and definition of treatment-resistant depression. Biol. Psychiatry 53, 649–659.
Fava, M., Borus, J. S., Alpert, J. E., Nierenberg, A. A., Rosenbaum, J. F., and Bottiglieri, T. (1997). Folate, vitamin B12, and homocysteine in major depressive disorder. Am. J. Psychiatry 154, 426–428.
Feng, X. J., Greenwald, B., Rabitz, H., Shea-Brown, E., and Kosut, R. (2007). Toward closed-loop optimization of deep brain stimulation for Parkinson’s disease: concepts and lessons from a computational model. J. Neuroeng. Rehabil. 4, L14–L21.
Figiel, G. S., Epstein, C. M., McDonald, W. M., Amazon-Leece, J., Figiel, L., Saldivia, A., and Glover, S. (1998). The use of rapid-rate transcranial magnetic stimulation (rTMS) in refractory depressed patients. J. Neuropsychiatry Clin. Neurosci. 10, 20–25.
First, M. B., and Ross, R. (eds) (2000). American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders, Text Revision. Washington, DC: R.R. Donnelly and Sons Company.
Fitzgerald, P. B., Benitez, J., de Castella, A., Daskalakis, Z. J., Brown, T. L., and Kulkarni, J. (2006). A randomized, controlled trial of sequential bilateral repetive transcranial magnetic stimulation for treatment-resistant depression. Am. J. Psychiatry 163, 88–94.
Fitzgerald, P. B., Brown, T. L., and Daskalakis, Z. J. (2002). The application of transcranial magnetic stimulation in psychiatry and neurosciences research. Acta Psychiatr. Scand. 105, 324–340.
Fontaine, D., Mattei, V., Borg, M., von Langsdorff, D., Magnie, M. N., Chanalet, S., Robert, P., and Paquis, P. (2004). Effect of subthalamic nucleus stimulation on obsessive-compulsive disorder in a patient with Parkinson disease. J. Neurosurg. 100, 1084–1086.
Garrett, A., Lithgow, B. J., Gurvich, C., and Fitzgerald, P. (2008). EVestG(TM): Responses in Depressed Patients. 30th Annual International IEEE EMBS Conference, Vancouver, BC.
George, M. S., Rush, A. J., Marangell, L. B., Sackeim, H. A., Brannan, S. K., Davis, S. M., Howland, R., Kling, M. A., Moreno, F., Rittberg, B., Dunner, D., Schwartz, T., Carpenter, L., Burke, M., Ninan, P., and Goodnick, P. (2005). A one-year comparison of vagus nerve stimulation with treatment as usual for treatment-resistant depression. Biol. Psychiatry 58, 364–373.
George, M. S., Sackeim, H. A., Rush, A. J., Marangell, L. B., Nahas, Z., Husain, M. M., Lisanby, S., Burt, T., Goldman, J., and Ballenger, J. C. (2000). Vagus nerve stimulation: a new tool for brain research and therapy. Biol. Psychiatry 47, 287–295.
Goldapple, K., Segal, Z., Garson, C., Lau, M., Bieling, P., Kennedy, S., and Mayberg, H. (2004). Modulation of cortical-limbic pathways in major depression: treatment specific effects of cognitive behavior therapy. Arch. Gen. Psychiatry 61, 34–41.
Golier, J. A., Marzuk, P. M., Leon, A. C., Weiner, C., and Tardiff, K. (1995). Low serum cholesterol level and attempted suicide. Am. J. Psychiatry 152, 419–423.
Goodman, W. K., and Insel, T. R. (2009). Deep brain stimulation in psychiatry: concentrating on the road ahead. Biol. Psychiatry 65, 263–266.
Greenberg, B. D., Malone, D. A., Friehs, G. M., Rezai, A. R., Kubu, C. S., Malloy, P. F., Salloway, S. P., Okun, M. S., Goodman, W. K., and Rassmussen, S. A. (2006). Three-year outcomes in deep brain stimulation for highly resistant obsessive-compulsive disorder. Neuropsychopharmacology 31, 2384–2393.
Halbig, T. D., Gruber, D., Kopp, U. A., Schneider, G. H., Trottenberg, T., and Kupsch, A. (2005). Pallidal stimulation in dystonia: effects on cognition, mood, and quality of life. J. Neurol. Neurosurg. Psychiatr. 76, 1713–1716.
Han, M., and McCreery, D. B. (2009). Microelectrode Technologies for Deep Brain Stimulation. ImplanTable Neural Prostheses 1. New York: Springer, 195–219.
Hardesty, D. E., and Sackeim, H. A. (2007). Deep brain stimulation in movement and psychiatric disorders. Biol. Psychiatry 61, 831–835.
Heils, A., Teufel, A., Petri, S., Stoeber, G., Riederer, P., Bengel, D., and Lesch, K. P. (1996). Allelic variation of human serotonin transporter gene expression. J. Neurochem. 66, 2621–2624.
Henry, T. R., Bakay, R. A. E., Votaw, J. R., Pennell, P. B., Epstein, C. M., Faber, T. L., Grafton, S. T., and Hoffman, J. M. (1998). Brain blood flow alterations induced by therapeutic vagus nerve stimulation in partial epilepsy: I. Acute effects at high and low levels of stimulation. Epilepsia 39, 983–990.
Holsboer, F. (2000). The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology 23, 477–500.
Holsboer, F., and Barden, N. (1996). Antidepressants and hypothalamic–pituitary–adrenocortical regulation. Endocr. Rev. 17, 187–205.
Jacobs, B. L., van Praag, H., and Gage, F. H. (2000). Adult brain neurogenesis and psychiatry: a novel theory of depression. Mol. Psychiatry 5, 262–269.
Janicak, P. G., O’Reardon, J. P., Sampson, S. M., Husain, M. M., Lisanby, S. H., Rado, J. T., Heart, K. L., and Demitrack, M. A. (2008). Transcranial magnetic stimulation in the treatment of major depressive disorder: a comprehensive summary of safety experience from acute exposure, extended exposure, and during reintroduction treatment. J. Clin. Psychiatry 69, 222–232.
Jimenez, F., Velasco, F., Salin-Pascual, R., Hernandez, J., Velasco, M., Criales, J. L., and Nicolini, H. (2005). A patient with a resistant major depression disorder treated with deep brain stimulation in the inferior thalamic peduncle. Neurosurgery 57, 585–593.
Judd, L. L., Akiskal, H. S., Maser, J. D., Zeller, P. J., Endicott, J., Coryell, W., Paulus, M. P., Kunovac, J. L., Leon, A. C., Mueller, T. I., Rice, J. A., and Keller, M. B. (1998). A prospective 12-year study of subsyndromal and syndromal depressive symptoms in unipolar major depressive disorders. Arch. Gen. Psychiatry 55, 694–700.
Karege, F., Perret, G., Bondolfi, G., Schwald, M., Bertschy, G., and Aubry, J. M. (2002). Decreased serum brain-derived neurotrophic factor levels in major depressed patients. Psychiatry Res. 109, 143–148.
Kearns, N. P., Cruickshank, C. A., McGuigan, K. J., Riley, S. A., Shaw, S. P., and Snaith, R. P. (1982). A comparison of depression rating scales. Br. J. Psychiatry 141, 45–49.
Kempermann, G., and Kronenberg, G. (2003). Depressed new neurons? – Adult hippocampal neurogenesis and a cellular plasticity hypothesis of major depression. Biol. Psychiatry 54, 499–503.
Kendler, K. S., Kuhn, J. W., Vittum, J., Prescott, C. A., and Riley, B. (2005). The interaction of stressful life events and a serotonin transporter polymorphism in the prediction of episodes of major depression: a replication. Arch. Gen. Psychiatry 62, 353–529.
Kessler, R. C., Berglund, P., Demler, O., Jin, R., Koretz, D., Merikangas, K. R., Rush, A. J., Walters, E. E., and Wang, P. S. (2003). The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA 289, 3095–3105.
Kessler, R. C., Berglund, P., Demler, O., Jin, R., Merikangas, K. R., and Walters, E. E. (2005). Lifetime prevalance and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch. Gen. Psychiatry 62, 593–602.
Kirsch, I. (2002). The emporer’s new drugs: an analysis of antidepressant medication data submitted to the U.S. Food and Drug Administration. Prev. Treat. 5, 1–11.
Kirsch, I., Deacon, B. J., Huedo-Medina, T. B., Scoboria, A., Moore, T. J., and Johnson, B. T. (2008). Initial severity and antidepressant benefits: a meta-analysis of data submitted to the Food and Drug Administration. PLoS Med. 5, 0260–0268. doi: 10.1371/journal.pmed.0050045.
Klein, E., Kreinin, I., Chistyakov, A., Koren, D., Mecz, L., Marmur, S., Ben-Shachar, D., and Feinsod, M. (1999). Therapeutic efficacy of right prefrontal slow repetitive transcranial magnetic stimulation in major depression. Arch. Gen. Psychiatry 56, 315–320.
Konsman, J. P., Vigues, S., Mackerlova, L., Bristow, A., and Blomqvist, A. (2004). Rat brain vascular distribution of interleukin-1 type-1 receptor immunoreactivity: relationship to patterns of inducible cyclooxygenase expression by peripheral inflammatory stimuli. J. Comp. Neurol. 472, 113–129.
Kosel, M., Sturm, V., Frick, C., Lenartz, D., Zeidler, G., Brodesser, D., and Schlaepfer, T. E. (2007). Mood improvement after deep brain stimulation of the internal globus pallidus for tardive dyskinesia in a patient suffering from major depression. J. Psychiatr. Res. 41, 801–803.
Krahl, S. E., Clark, K. B., Smith, D. C., and Browning, R. A. (1998). Locus coeruleus lesions suppress the seizure-attenuating effects of vagus nerve stimulation. Epilepsia 39, 709–714.
Kroenke, K., Spitzer, R. L., and Williams, J. B. W. (2001). The PHQ-9: validity of a brief depression severity measure. J. Gen. Intern. Med. 16, 606–613.
Kunugi, H., Takei, N., Aoki, H., and Nanko, S. (1997). Low serum cholesterol in suicide attempters. Biol. Psychiatry 41, 196–200.
Lacasse, J. R., and Leo, J. (2005). Serotonin and depression: a disconnect between the advertisements and the scientific literature. PLoS Med. 2, 1211–1216. doi: 10.1371/journal.pmed.0020392.
Lozano, A. M., Mayberg, H. S., Giacobbe, P., Hamani, C., Craddock, R. C., and Kennedy, S. H. (2008). Subcallosal cingulate gyrus deep brain stimulation for treatment-resistant depression. Biol. Psychiatry 64, 461–467.
Malone, D. A., Dougherty, D. D., Rezai, A. R., Carpenter, L. L., Friehs, G. M., Eskandar, E. N., Rauch, S. L., Rassmussen, S. A., Machado, A. G., Kubu, C. S., Tyrka, A. R., Price, L. H., Stypulkowski, P. H., Giftakis, J. E., Rise, M. T., Malloy, P. F., Salloway, S. P., and Greenberg, B. D. (2009). Deep brain stimulation of the ventral capsule/ventral striatum for treatment-resistant depression. Biol. Psychiatry 65, 267–275.
Manji, H. K., Drevets, W. C., and Charney, D. S. (2001). The cellular neurobiology of depression. Nat. Med. 7, 541–547.
Marangell, L. B., Martinez, M., Jurdi, R. A., and Zboyan, H. (2007). Neurostimulation therapies in depression: a review of new modalities. Acta Psychiatr. Scand. 116, 174–181.
Marangell, L. B., Rush, A. J., George, M. S., Sackeim, H. A., Johnson, C. R., Husain, M. M., Nahas, Z., and Lisanby, S. (2002). Vagus nerve stimulation (VNS) for major depressive episodes: one year outcomes. Biol. Psychiatry 51, 280–287.
Martin, S. D., Martin, E., Rai, S. S., Richardson, M. A., and Royall, R. (2001). Brain blood flow changes in depressed patients treated with interpersonal psychotherapy or venlafaxine hydrochloride. Arch. Gen. Psychiatry 58, 641–648.
Mayberg, H. S. (1997). Limbic-cortical dysregulation: a proposed model of depression. J. Neuropsychiatry Clin. Neurosci. 9, 471–481.
Mayberg, H. S., Brannan, S. K., Tekell, J. L., Silva, J. A., Mahurin, R. K., McGinnis, S., and Jerabek, P. A. (2000). Regional metabolic effects of fluoxetine in major depression: serial changes and relationship to clinical response. Biol. Psychiatry 48, 830–843.
Mayberg, H. S., Liotti, M., Brannan, S. K., McGinnis, S., Mahurin, R. K., Jerabek, P. A., Silva, J. A., Tekell, J. L., Martin, C. C., Lancaster, J. L., and Fox, P. T. (1999). Reciprocal limbic-cortical function and negative mood: converging PET findings in depression and normal sadness. Am. J. Psychiatry 156, 675–682.
Mayberg, H. S., Lozano, A. M., Voon, V., McNeely, H. E., Seminowicz, D., Hamani, C., Schwalb, J. M., and Kennedy, S. H. (2005). Deep brain stimulation for treatment-resistant depression. Neuron 45, 651–660.
McCreery, D. B., Yuen, T. G. H., Agnew, W. F., and Bullara, L. A. (1997). A characterization of the effects of neuronal excitability due to prolonged microstimulation with chronically implanted microelectrodes. IEEE Trans. Biomed. Eng. 44, 931–939.
Merali, Z., Du, L., Hrdina, P., Palkovitz, M., Faludi, G., Poulter, M. O., and Anisman, H. (2004). Dysregulation in the suicide brain: mRNA expression of corticotropin-releasing hormone receptors and GABA(A) receptor subunits in frontal cortical brain region. J. Neurosci. 24, 1478–1485.
Milak, M. S., Parsey, R. V., Keilp, J., Oquendo, M. A., Malone, K. M., and Mann, J. J. (2005). Neuroanatomic correlates of psychopathologic components of major depressive disorder. Arch. Gen. Psychiatry 62, 397–408.
Mossner, R., Mikova, O., Koutsilieri, E., Saoud, M., Ehlis, A. C., Muller, N., Fallgatter, A. J., and Riederer, P. (2007). Consensus of the WFSBP Task Force on biological markers: biological markers in depression. World J. Biol. Psychiatry 8, 141–174.
Mueller, T. I., Leon, A. C., Keller, M. B., Solomon, D. A., Endicott, J., Coryell, W., Warshaw, M., and Maser, J. D. (1999). Recurrence after recovery from major depressive disorder during 15 years of observational follow-up. Am. J. Psychiatry 156, 1000–1006.
Nemeroff, C. B., Mayberg, H. S., Krahl, S. E., McNamara, J., Frazer, A., Henry, T. R., George, M. S., Charney, D. S., and Brannan, S. K. (2006). VNS therapy in treatment-resistant depression: clinical evidence and putative neurobiological mechanisms. Neuropsychopharmacology 31, 1345–1355.
Nemeroff, C. B., Widerlov, E., Bissette, G., Walleus, H., Karlsson, I., Eklund, K., Kilts, C. D., Loosen, P. T., and Vale, W. (1984). Elevated concentrations of CSF corticotropin-releasing factor-like immunoreactivity in depressed patients. Science 226, 1342–1344.
Nestler, E. J., Barrot, M., DiLeone, R. J., Eisch, A. J., Gold, S. J., and Monteggia, L. M. (2002). Neurobiology of depression. Neuron 34, 13–25.
Neuronetics (2009). About TMS Therapy. NeuroStar TMS Therapy, Retrieved August 6, 2009, from http://www.neurostartms.com .
Nibuya, M., Morinobu, S., and Duman, R. S. (1995). Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J. Neurosci. 15, 7539–7547.
Nuttin, B., Cosyns, P., Demeulemeester, H., Gybels, J., and Meyerson, B. (1999). Electrical stimulation in anterior limbs of internal capsules in patients with obsessive-compulsive disorder. Lancet 354, 1526.
O’Brien, S. M., Scully, P., Fitzgerald, P., Scott, L. V., and Dinan, T. G. (2007). Plasma cytokine profiles in depressed patients who fail to respond to selective serotonin reuptake inhibitor therapy. J. Psychiatr. Res. 41, 326–331.
Ongur, D., An, X., and Price, J. L. (1998). Prefrontal cortical projections to the hypothalamus in macaque monkeys. J. Comp. Neurol. 401, 480–505.
Pascual-Marqui, R. D., Michel, C. M., and Lehmann, D. (1994). Low resolution electromagnetic tomography: a new method for localizing electrical activity in the brain. Int. J. Psychophysiol. 18, 49–65.
Peretti, S., Judge, R., and Hindmarch, I. (2000). Safety and tolerability considerations: tricyclic antidepressants vs. selective serotonin reuptake inhibitors. Acta Psychiatr. Scand. 101, 17–25.
Piallat, B., Chabardes, S., Devergnas, A., Torres, N., Allain, M., Barrat, E., and Benabid, A. L. (2009). Monophasic but not biphasic pulses induce brain tissue damage during monopolar high-frequency deep brain stimulation. Neurosurgery 64, 156–163.
Pittenger, C., and Duman, R. S. (2008). Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology 33, 88–109.
Pizzagalli, D., Pascual-Marqui, R. D., Nitschke, J. B., Oakes, T. R., Larson, C. L., Abercrombie, H. C., Schaefer, S. M., Koger, J. V., Benca, R. M., and Davidson, R. J. (2001). Anterior cingulate activity as a predictor of degree of treatment response in major depression: evidence from brain electrical tomography analysis. Am. J. Psychiatry 158, 405–415.
Post, R. M., Uhde, T. W., Roy-Byrne, P. P., and Joffe, R. T. (1986). Antidepressant effects of carbamazepine. Am. J. Psychiatry 143, 29–34.
Quitkin, F. M., Rabkin, J. G., Stewart, J. W., McGrath, P. J., and Harrison, W. (1986). Study duration in antidepressant research: advantages of a 12-week trial. J. Psychiatr. Res. 20, 211–216.
Raisman, R., Sechter, D., Briley, M. S., Zarifan, E., and Langer, S. Z. (1981). High-affinity 3H-imipramine binding in platelets from untreated and treated depressed patients compared to healthy volunteers. Psychopharmacology (Berl.) 75, 368–371.
Raison, C. L., Capuron, L., and Miller, A. H. (2006). Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol. 27, 24–31.
Rubin, R. T., Poland, R. E., Lesser, I. M., Winston, R. A., and Blodgett, A. L. (1987). Neuroendocrine aspects of primary endogenous depression. I. Cortisol secretory dynamics in patients and matched controls. Arch. Gen. Psychiatry 44, 328–336.
Ruhe, H. G., Mason, N. S., and Schene, A. H. (2007). Mood is indirectly related to serotonin, norepinephrine, and dopamine levels in humans: a meta-analysis of monoamine depletion studies. Mol. Psychiatry 12, 331–359.
Rush, A. J., George, M. S., Sackeim, H. A., Marangell, L. B., Mustafa, M. H., Giller, C., Nahas, Z., Haines, S., Simpson, R. K., Jr., and Goodman, R. (2000). Vagus Nerve Stimulation (VNS) for treatment-resistant depressions: a multicenter study. Biol. Psychiatry 47, 276–286.
Sapolsky, R. M. (2000). Glucocorticoids and hippocampal atrophy in neuropsychiatric disorders. Arch. Gen. Psychiatry 57, 925–935.
Shimizu, E., Hashimoto, K., Okamura, N., Koike, K., Komatsu, N., Kumakiri, C., Nakazato, M., Watanabe, H., Shinoda, N., Okada, S., and Iyo, M. (2003). Alterations of serum levels of brain-derived neurotrophic factor (BDNF) in depressed patients with or without antidepressants. Biol. Psychiatry 54, 70–75.
Smith, A. C., Shah, S. A., Hudson, A. E., Purpura, K. P., Victor, J. D., Brown, E. N., and Schiff, N. D. (2009). A bayesian statistical analysis of behavioral facilitation associated with deep brain stimulation. J. Neurosci. Methods 183, 267–276.
Solomon, D. A., Keller, M. B., Leon, A. C., Mueller, T. I., Lavori, P. W., Shea, T., Coryell, W., Warshaw, M., Turvey, C., Maser, J. D., and Endicott, J. (2000). Multiple recurrences of major depressive disorder. Am. J. Psychiatry 157, 229–233.
Speer, A. M., Kimbrell, T. A., Wassermann, E. M., Repella, J. D., Willis, M. W., Herscovitch, P., and Post, R. M. (2000). Opposite effects of high and low frequency rTMS on regional brain activity in depressed patients. Biol. Psychiatry 48, 1133–1141.
Sullivan, P. F., Neale, M. C., and Kendler, K. S. (2000). Genetic epidemiology of major depression: review and meta-analysis. Am. J. Psychiatry 157, 1552–1562.
Sun, F. T., Morrell, M. J., and Wharen, R. E. (2008). Responsive cortical stimulation for the treatment of epilepsy. Neurotherapeutics 5, 68–74.
Thase, M. E., and Rush, A. J. (1997). When at first you don’t succeed: sequential strategies for antidepressant nonresponders. J. Clin. Psychiatry 53, 649–659.
Tomarken, A. J., Davidson, R. J., Wheeler, R. E., and Doss, R. C. (1992). Individual differences in anterior brain asymmetry and fundamental dimensions of emotion. J. Pers. Soc. Psychol. 62, 676–687.
Tung, B., and Kleinrock, L. (1996). Using finite state automata to produce self-optimization and self-control. IEEE Trans. Parallel Distrib. Syst. 7, 439–448.
Turner, E. H., Matthews, A. M., Linardatos, E., Tell, R. A., and Rosenthal, R. (2008). Selective publication of antidepressant trials and its influence on apparent efficacy. N. Engl. J. Med. 358, 252–260.
Velasco, F., Velasco, M., Jimenez, F., Velasco, A. L., and Salin-Pascual, R. (2005). Neurobiological background for performing surgical intervention in the inferor thalamic peduncle for treatment of major depression disorders. Neurosurgery 57, 439–448.
Wells, K. B., Stewart, A., Hays, R. D., Burnam, M. A., Rogers, W., Daniels, M., Berry, S., Greenfield, S., and Ware, J. (1989). The functioning and well-being of depressed patients: results from the medical outcomes study. JAMA 262, 914–919.
Wong, P. K., Yu, F., Shahangian, A., Cheng, G., Sun, R., and Ho, C. M. (2008). Closed-loop control of cellular functions using combinatory drugs guided by a stochastic search algorithm. Proc. Natl. Acad. Sci. U.S.A. 105, 5105–5110.
Keywords: major depressive disorder, closed-loop therapies, neurostimulation
Citation: Ward MP and Irazoqui PP (2010) Evolving refractory major depressive disorder diagnostic and treatment paradigms: toward closed-loop therapeutics. Front. Neuroeng. 3:7. doi: 10.3389/fneng.2010.00007
Received: 30 December 2009;
Paper pending published: 01 March 2010;
Accepted: 05 May 2010;
Published online: 31 May 2010
Edited by:
Martin Stelzle, University of Tübingen, GermanyReviewed by:
Hansjürgen Volkmer, Universität Tübingen, GermanyJohn P. O'Reardon, University of Pennsylvania, USA
Copyright: © 2010 Ward and Irazoqui. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Matthew P. Ward, Center for ImplanTable Devices, School of Biomedical Engineering, Purdue University, South Martin Jischke Drive 206, Rm. 2083, West Lafayette, IN 47907, USA. e-mail:bXB3YXJkQG1hYy5jb20=