 Laura Ylä-Outinen1
Laura Ylä-Outinen1 Juha Heikkilä1
Juha Heikkilä1 Heli Skottman1
Heli Skottman1 Riitta Suuronen1,2,3
Riitta Suuronen1,2,3 Riikka Äänismaa1
Riikka Äänismaa1 Susanna Narkilahti1*
Susanna Narkilahti1*
- 1 Regea – Institute for Regenerative Medicine, University of Tampere and Tampere University Hospital, Tampere, Finland
- 2 Department of Eye, Ear, and Oral Diseases, Tampere University Hospital, Tampere, Finland
- 3 Department of Biomedical Engineering, Tampere University of Technology, Tampere, Finland
At present, most of the neurotoxicological analyses are based on in vitro and in vivo models utilizing animal cells or animal models. In addition, the used in vitro models are mostly based on molecular biological end-point analyses. Thus, for neurotoxicological screening, human cell-based analysis platforms in which the functional neuronal networks responses for various neurotoxicants can be also detected real-time are highly needed. Microelectrode array (MEA) is a method which enables the measurement of functional activity of neuronal cell networks in vitro for long periods of time. Here, we utilize MEA to study the neurotoxicity of methyl mercury chloride (MeHgCl, concentrations 0.5–500 nM) to human embryonic stem cell (hESC)-derived neuronal cell networks exhibiting spontaneous electrical activity. The neuronal cell cultures were matured on MEAs into networks expressing spontaneous spike train-like activity before exposing the cells to MeHgCl for 72 h. MEA measurements were performed acutely and 24, 48, and 72 h after the onset of the exposure. Finally, exposed cells were analyzed with traditional molecular biological methods for cell proliferation, cell survival, and gene and protein expression. Our results show that 500 nM MeHgCl decreases the electrical signaling and alters the pharmacologic response of hESC-derived neuronal networks in delayed manner whereas effects can not be detected with qRT-PCR, immunostainings, or proliferation measurements. Thus, we conclude that human cell-based MEA platform is a sensitive online method for neurotoxicological screening.
Introduction
Neurotoxicity of various chemicals is currently tested solely with in vivo methods due to the lack of proper, validated in vitro cell models. Cells such as primary cultures of rat cerebellar granule cells have been used as a possible in vitro model (Harry et al., 1998; van Vliet et al., 2007; Bal-Price et al., 2009; Hogberg et al., 2009) for both developmental and adult neurotoxicity testing. Recently also human-derived cells, e.g., neural progenitor cells and umbilical cord blood-derived neural stem cells have been tested for neurotoxicity studies (Zeng et al., 2006; Buzanska et al., 2009a,b; Moors et al., 2009; Bal-Price et al., 2010). In addition to the need of developing human cell-based in vitro models that would enable better interpretation of neurotoxicological responses in humans and help to replace and reduce animal tests, more relevant in vitro toxicological models are needed. Currently used in vitro neuronal models are mostly based on molecular biological end-point analyses such as gene expression, cell proliferation and differentiation, and neurite outgrowth. It has, however, become clear that in addition to traditional cell viability testing, the testing pattern should include various general cell function tests and especially concentrate on neuron-specific aspects such as the neuronal network’s electrical functionality and its alterations due to toxic compounds in subcytotoxic levels (van Vliet et al., 2007; Bal-Price et al., 2010). Validation of all these tests is of high importance for development of routinely usable in vitro neurotoxicology testing platforms.
Thus, there is a clear need of human cell-based neurotoxicity models with which the toxicological effects can be monitored online. With non-invasive microelectrode array (MEA), the functional activity of neuronal cell networks can be measured in vitro for long periods of time and can be used as an additional real-time analysis method (Pine, 1980; Ben-Ari, 2001; Wagenaar et al., 2006; Illes et al., 2007; Heikkilä et al., 2009). As MEA technologies offer indeed a proper way to measure the electrical activity of the networks and further allow the detection of network responses to extrinsic factors these technologies are seen as a promising way to rapid and sensitive screening of neurotoxicity testing as discussed in recent review by Johnstone et al. (2010).
Human pluripotent, e.g., embryonic stem cells are an excellent source to produce the main cells types of the human brain: neurons, astrocytes, and oligodendrocytes (Reubinoff et al., 2001; Zhang et al., 2001; Erceg et al., 2008; Lappalainen et al., 2010; Sundberg et al., 2010). These cells follow the in vivo neural development closely and the produced neuronal cells have electrical properties of their brain counter parts (Carpenter et al., 2001; Itsykson et al., 2005; Johnson et al., 2007; Erceg et al., 2008). Importantly, these cells can form spontaneously active functional neuronal networks (Heikkilä et al., 2009) the activity development of which resembles that of rodent primary and embryonic stem cell-derived neuronal cultures (Wagenaar et al., 2006; Illes et al., 2007). Methyl mercury is a well known neurotoxicant for both developing and adult central nervous system (Kaur et al., 2006) affecting microtubule disruption, oxidative stress, and neurotransmitter deregulation (Castoldi et al., 2001) and causing focal damage in particular brain areas leading to manifestation of paresthesia, ataxia, constriction of visual field, and hearing loss in adult humans (Clarkson, 1993). During life span, humans are most sensitive to methyl mercury during the prenatal period which results in several severities range of mental retardation (Clarkson, 1993).
In this study we exposed spontaneously active human embryonic stem cell (hESC)-derived neuronal networks cultured in MEA’s to methyl mercury chloride (MeHgCl) at various concentrations and recorded both acute and delayed networks responses to this toxicant. We show that at subcytotoxic levels, the human neuronal networks’ response to this exposure can be detected with MEA but not with classical molecular biological analyses.
Materials and Methods
Reagents
Methyl mercury II chloride (MeHgCl), polyethyleneimine (PEI), mouse laminin, paraformaldehyde, γ-aminobutyric acid (GABA), and bicuculline methiobromide were purchased from Sigma-Aldrich (St. Louis, MO, USA). 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) and D(-)-2-amino-5-phosphonopentanoic acid (D-AP5) were from Ascent Scientific (Bristol, UK). B27 supplement, N2 supplement, DMEM/F12 and Neurobasal media, Glutamax, and brain-derived neurotrophic factor (BDNF) were from Invitrogen (Carlsbad, CA, USA). Basic fibroblast growth factor (bFGF) was from R&D Systems (Minneapolis, MN, USA). Penicillin/streptomycin and phosphate buffered saline (dPBS) were from Lonza Group Ltd (Switzerland). 48-well and 96-well plates were from Nunc (ThermoFisher Scientific, Rochester, NY, USA).
Rabbit anti-microtubule associated protein 2 polyclonal antibody (MAP-2, 1:600), and monoclonal mouse anti-galactocerebroside (GalC, 1:400) were purchased from Chemicon. Monoclonal mouse anti-beta-tubulin isotype III (β-tubulinIII, 1:1250) was purchased from Sigma and sheep anti-human glial fibrillary acidic protein antibody (GFAP, 1:800) was purchased from R&D systems.
Cell Culture
The hESCs grown as colonies were mechanically cut into aggregates containing approximately 3000 cells and placed into suspension culture in neural differentiation medium as recently reported (Lappalainen et al., 2010). Culture medium consisted of 1:1 DMEM/F12 and Neurobasal media supplemented with 2 mM GlutaMax, 1 × B27, 1 × N2, and 25 U/ml penicillin/streptomycin. During neurosphere culture, constant supply of bFGF (20 ng/ml) was provided. The aggregates were differentiated for 8 weeks during which they formed round, constant neurospheres and expressed typical neural markers as described in the earlier studies (Hicks et al., 2009; Lappalainen et al., 2010). The medium was changed three times per week and growing spheres were manually cut into smaller spheres once a week.
After differentiation period cells were plated down onto MEA plates (Multi Channel Systems, Germany). Each standard MEA plate contains 59 titanium nitride measurement electrodes in diameter 30 and 200 μm inter-electrode distances. In addition, also 6-well MEA plates were used, which consists of 6 separated 9 electrode areas (diameter 30 mm, inter-electrode distance 200 μm). In both MEA types, electrode areas were separated with in-house designed Poly(dimethyl siloxane) (PDMS) structures (designed and manufactured by J. Kreutzer and P. Kallio, Tampere University of Technology, Department of Automation Science and Engineering, Finland). Cell plating on MEA’s was performed as previously described (Heikkilä et al., 2009). First, MEA plates were coated with two step coating procedure; overnight incubation with PEI (0.05% w/v, incubation +4°C) followed by washing with sterile water, and overnight incubation with mouse laminin (20 μg/ml, incubation +4°C). Thereafter, neurospheres were cut into small aggregates (∅ ∼200 μm) and plated onto coated MEA plates. Neuronal differentiation medium supplemented with 4 ng/ml bFGF and 5 ng/ml BDNF was applied to cells during MEA culture. The cells and networks were grown for 3 weeks before MeHgCl exposure.
MeHgCl Exposure
MeHgCl was dissolved into sterile dPBS in stock concentration 1 mM and thereafter diluted into neuronal differentiation medium (containing bFGF and BDNF) freshly just before use. Next, the medium containing either 0, 0.5, 50, or 500 nM MeHgCl was changed to neuronal networks growing on MEA’s (n = 4/each concentration) or in standard 48- and 96-well plates. The cells were cultured in MeHgCl containing medium for 72 h where after it was washed away.
Electrophysiological Measurements
Measurements were performed with MEA1060-Inv-BC-amplifier with integrated TPC Temperature controller adjusted to +37°C and data was recorded with MC_Rack software (all from Multichannel systems, Reutlingen, Germany). Prior to measurements, MEA plates were sealed with semi-permeable membrane (standard MEAs) or PDMS discs (6-well MEAs) in laminar hood to keep the cultures sterile for repeated measurements as reported earlier (Potter and DeMarse, 2001).
Prior to MeHgCl exposure, the spontaneous baseline electrical activities of each MEA were measured (300 s/MEA) and only MEA cultures demonstrating spike train activity (at least 300 spikes detected at least from one channel per measurement period) were selected to experiment. One MEA from each MeHgCl concentration was recorded for 1 h to detect the acute response. Thereafter, each MEA was recorded at time points of 24, 48, and 72 h for 300 s for delayed response. After 72 h MEA cultures were washed with fresh medium and recovery responses were measured 72 h after washout.
Post-recording data was processed with MC_DataTool (MCS), MATLAB (MathWorks, Natick, MA, USA), Excel (Microsoft), and NeuroExplorer (NexTechnologies, Littleton, MA, USA). Peaks from MEA raw data were detected using threshold limit STD × (-5). From each MEA, total spikes/MEA, total spike number/active channel, and spiking rate/s were calculated. Spike number data was normalized with respect to baseline activity to each MEA separately. For pharmacological response analysis, data was normalized against the mean of baseline activity period. Thereafter, each datapoint was smoothened with Gaussian smoother over ±25 timepoints and plotted into graph.
Pharmacologic Testing
Pharmacological response to specific agonists and antagonists of AMPA/kainite, NMDA, and GABA receptors were tested with CNQX, D-AP5, GABA, and bicuculline in concentrations 30 μM, 20 μM, 100 mM, and 20 μM, respectively. Response test were performed immediately after the 72 h exposure period.
Quantitative RT-PCR Analysis
Samples for qRT-PCR analysis were collected from MEAs immediately after 72 h MeHgCl exposure into lysis buffer and RNA was extracted using NucleoSpin® RNA XS kit according to the manufacturer’s instructions (Machery-Nagel GmbH & Co, Düren, Germany). Next, 200 ng of total RNA was reverse-transcribed into cDNA in reaction volume of 20 μl. The qRT-PCR was performed with standard protocol using ABI Prism 7300 instrument (Applied Biosystems, CA, USA) and TaqMan® Gene Expression assays (all from Applied Biosystems) for musashi (ID Hs01045894), neurofilament-68 (NF-68, ID Hs00196245), and GAPDH (ID 4331182) according to manufacturer’s instructions with TaqMan® Universal PCR master mix. All samples were performed as four technical replicates. Briefly, the expression levels of musashi, NF-68, and GFAP were evaluated using GAPDH as an internal control. Ct values were determined for every reaction and the relative quantification was calculated using 2−ΔΔCt method (Livak and Schmittgen, 2001). Values of the control sample (not exposed to MeHgCl) were used as a calibrator.
Time-Lapse Imaging
For time-lapse imaging of the neuronal cultures during MeHgCl exposure, a subset of neurospheres were replated on mouse laminin (10 μg/ml, Sigma-Aldrich, St. Louis, MO, USA) coated 48-well plates (8 parallel wells/MeHgCl concentration). The cells were allowed to grow as in MEA plates for 3 weeks until MeHgCl exposure. Thereafter, the well plate was transferred into a time-lapse imaging system (Cell-IQ®, Chip-man Technologies Ltd., Tampere, Finland) which allows for continuous monitoring of selected cells as described previously (Narkilahti et al., 2007). Briefly, sealed culture well plate was placed in a chamber (+36.5°C) with 5% CO2 gas supply. The cells of interest were selected for monitoring in 500 μm × 670 μm size areas or in grid areas 2 × 2 (1500 × 2010 μm) using an all-in-focus imaging system (Tarvainen et al., 2002) and gained images were stored for later use in JPEG-format. All areas of interest were imaged at 45-min interval for the total exposure time of 72 h. The cells were then fixed for immunocytochemical analysis.
Immunocytochemistry
The samples for immunocytochemical analyses were fixed with cold 4% paraformaldehyde for 20 min either after 72 h MeHgCl exposure or 72 h after washout. The fixed samples were blocked against non-specific antibody binding for 45 min in room temperature (RT) using 10% normal donkey serum (NDS), 0.1% Triton X-100, and 1% bovine serum albumin (BSA) in phosphate buffered saline (PBS). Next, the cells were washed with 1% NDS, 0.1% Triton X-100, and 1% BSA in PBS and incubated overnight at +4°C with primary antibodies diluted to the same buffer. The used antibodies were MAP-2 and ß-tubulinIII for neuronal cells, GFAP for astrocytes, and GalC for oligodendrocytes. In the following day the cells were washed with 1% BSA in PBS and incubated with secondary antibodies (anti-mouse, anti-rabbit, or anti-sheep conjugated to Alexa Fluor-488 or 568, Molecular Probes, Invitrogen) diluted to the washing buffer for 1 h at RT. Finally, the cells were repeatedly washed with PBS and phosphate buffer, mounted with Vectashield with 4′6-diamidino-2-phenylindole (DAPI, Vector Laboratories, Peterborough, UK), and cover-slipped. For double-labeling, two primary antibodies and two secondary antibodies were applied simultaneously. When primary antibodies were omitted (negative control), no positive labeling was detected. Cells were imaged using an Olympus microscope (IX51, Olympus, Finland) equipped with a fluorescence unit and camera (DP30BW, Olympus, Finland).
Cell Proliferation Measurements
BrdU
The proliferation rate of the neuronal cultures was measured 72 h after MeHgCl washout using a colorimetric enzyme-linked immunosorbent assay (ELISA) for 5-bromo-2′-deoxyuridine (BrdU) (Roche, Basel, Switzerland), which is based on the BrdU incorporation into the DNA during strand synthesis. The manufacturer’s protocol was used with small modifications. Briefly, the networks were incubated with BrdU labeling reagent (100 μM) for 20 h at +37°C in 5% CO2. The cells were then collected and replated into 96-well plate wells (Nunc) at a density of 5000 cells/well. Ten parallel samples/group were prepared. The well plates were centrifuged at 300 × g for 10 min at RT after which the medium was tapped off the wells. The cells were dried for 15 min and fixed with FixDenat solution for 30 min at RT. The FixDenat solution was tapped off and anti-BrdU monoclonal antibody conjugated with peroxidase was added and incubated for 90 min at RT. The plates were then washed three times with dPBS and the substrate solution was added for 5 min. The reaction was stopped with H2SO4 and absorbance was measured at a 450-nm wavelength using a Viktor2 1420 Multilabel Counter (PerkinElmer-Wallac). The background absorbance (measured from negative controls) was subtracted from the measured sample absorbances.
Wst-1 analysis
Cell proliferation capacity was also measured with WST-1 reagent by evaluating succinate-tetrazolium reductase activity. Wst-1 Cell proliferation Assay (Takara Bio Inc., Shiga, Japan) was performed with ELISA from 12 parallel 96-wells/sample. Briefly, cells were once washed with PBS and ready-to-use solution of WST-1 was diluted into PBS and added to the cells. Cells were incubated for 4 h in +37°C, 5% CO2 after which the measurement was performed with Viktor2 1420 Multilabel Counter at a 450-nm wavelength. The background absorbance (measured from negative controls) was subtracted from the measured sample absorbances.
Results
Human Embryonic Stem Cell-Derived Neural Cells Spontaneously Form Electrically Active Networks
HESC-derived neuronal cell networks stay viable and active in MEA plates for several months and their activity can be measured repeatedly (Heikkilä et al., 2009). The spontaneous activity develops in 3 weeks to a stage where the networks exhibit train-like activity repeatedly from several channels. Figure 1A shows MEA plate with spontaneously active neuronal network. A raster plot showing prominent training activity of the network is represented in Figure 1B. For MeHgCl exposure only MEA plates with spontaneously active networks were selected.
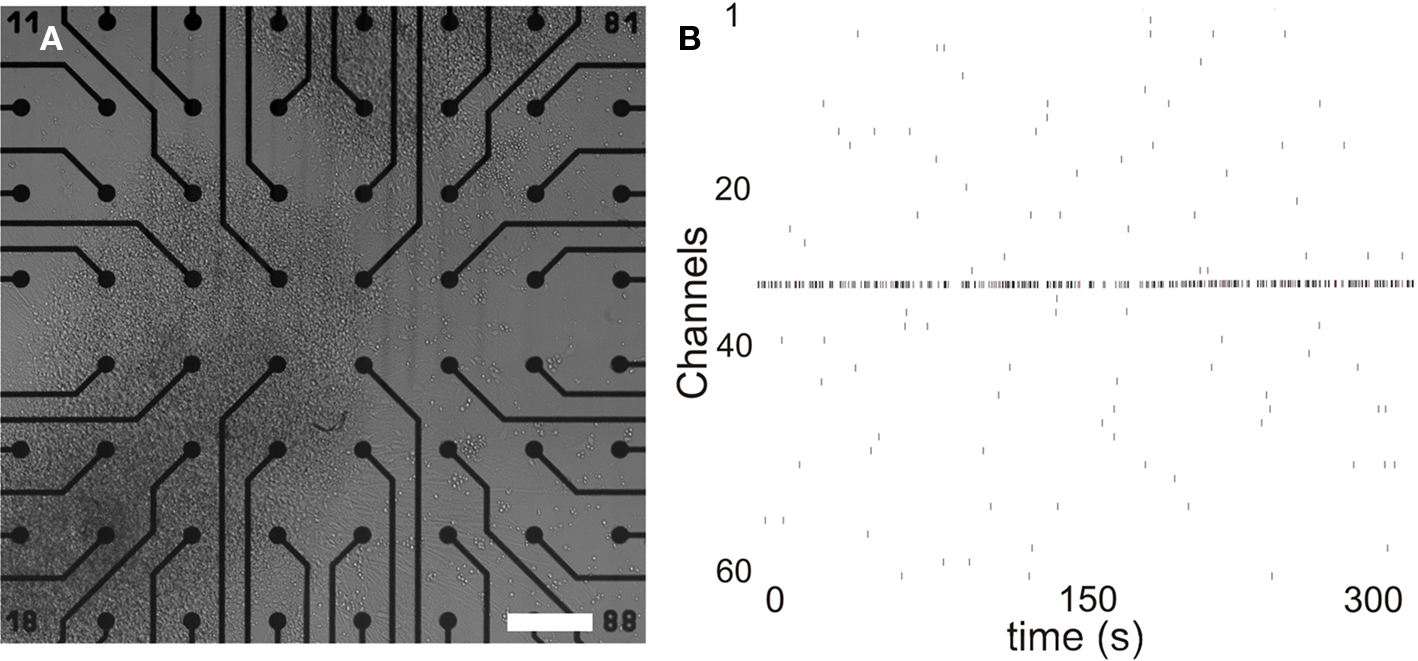
Figure 1. Spontaneously active neuronal networks in MEA. (A) HESC-derived neuronal cells form networks in vitro in MEA. (B) Formed networks show training activity within 3 weeks of culturing. Scale 200 μm. x-Axis time: 300 s, y-axis represents each channel.
MeHgCl does not have acute effect on network activity
Immediately after adding the cell culture medium containing 0, 0.5, 50, or 500 nM MeHgCl one MEA/each group was measured for the first hour continuously to detect possible acute effects. No acute toxic effects were observed. The same natural fluctuation on spiking activity was detected in all MEAs and MeHgCl concentration-dependent alteration could not be observed during the measurement period (Figure 2).
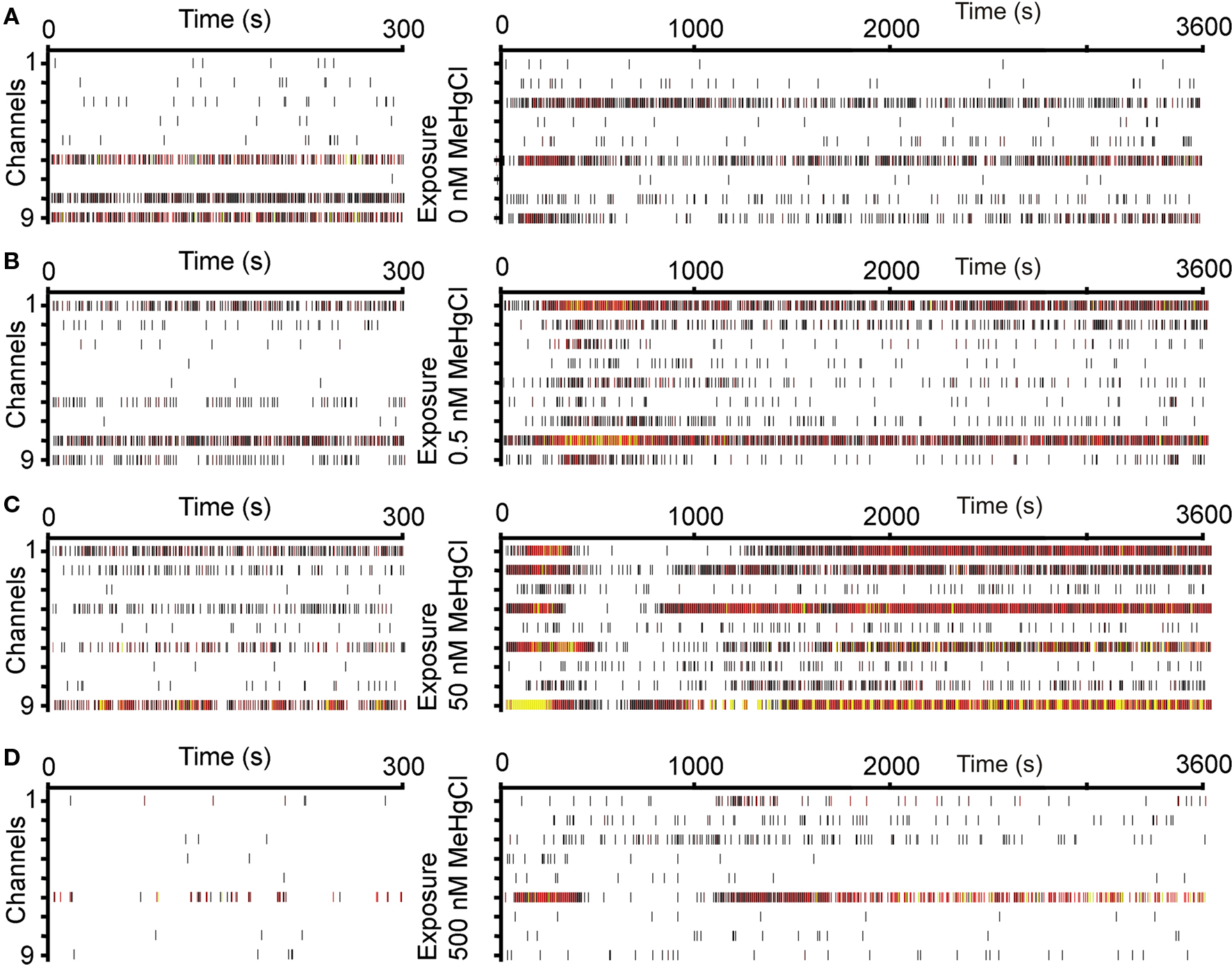
Figure 2. Acute effect of MeHgCl exposure to neuronal network activity. The baseline activity (on the left) of one MEA in each group: (A) 0 nM MeHgCl, (B) 0.5 nM MeHgCl, (C) 50 nM MeHgCl, (D) 500 nM MeHgCl exposure, and activity immediately after MeHgCl exposure (1 h, on the right). x-Axis time: baseline 300 s, acute response 600 s, y-axis represents each channel. Spikes are marked as rasters and heated colors represent increased activity rate.
MeHgCl Exposure Decreases Network Activity in a Delayed Manner
The used subtoxic doses of MeHgCl did not have acute effects but during longer exposure clear effects on spontaneous activity was detected. The activity of neuronal networks prominently decreased with each MeHgCl concentration when compared to 0 nM MeHgCl controls after 72 h exposure (Figures 3A,C).
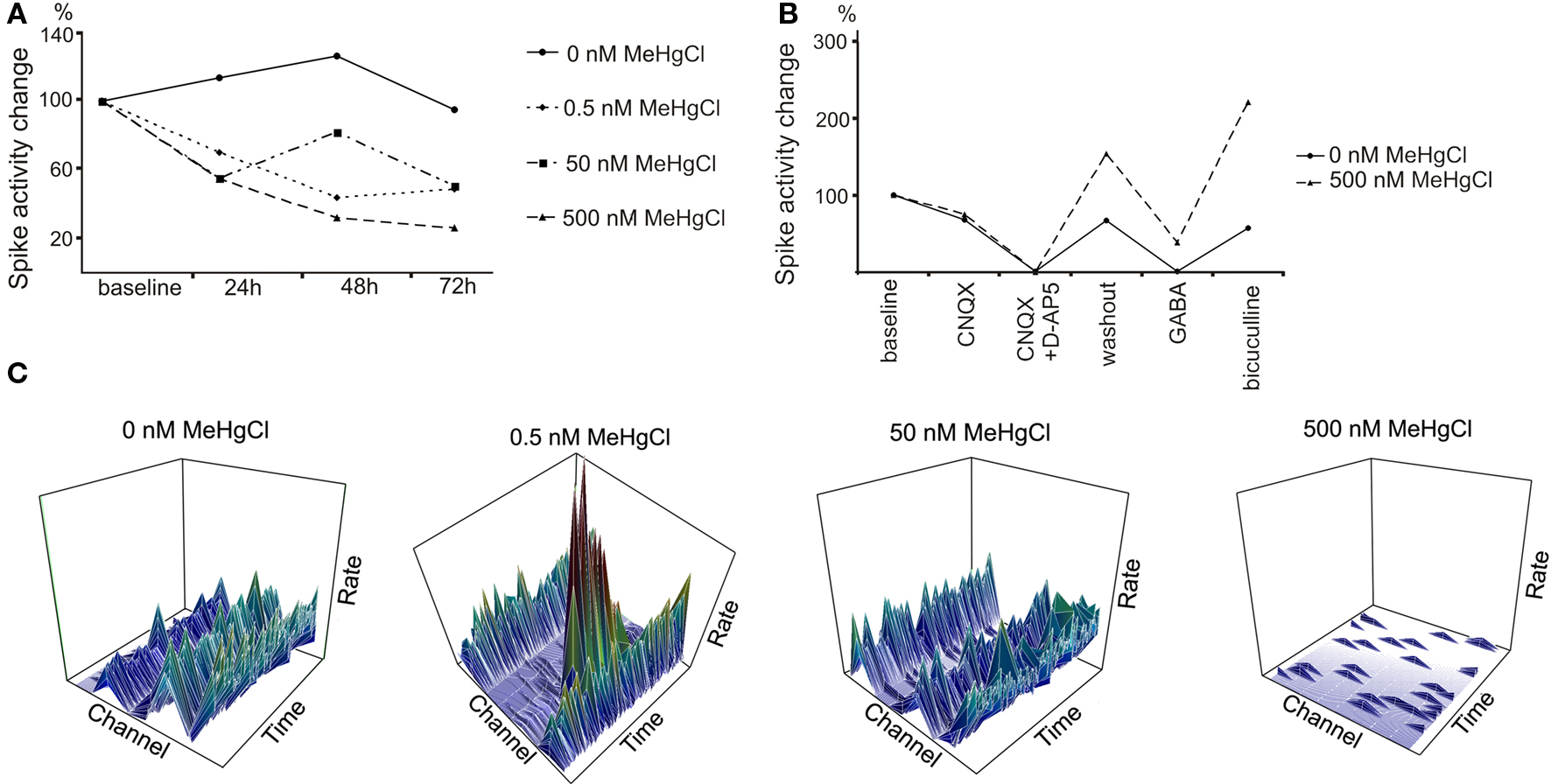
Figure 3. Delayed effect of MeHgCl exposure to neuronal network activity. (A) The activity of neuronal networks decreased toward 72 h of MeHgCl exposure in a concentration-dependent manner. The activity was measured from four parallel MEAs for 300 s at each timepoint. (B) 500 nM MeHgCl exposure altered the networks’ response to pharmacological substances. (C) Representative 3D plots of the activity rate/s (z-axis)/time (x-axis)/channel (y-axis) from one MEA/MeHgCl concentration at 72 h timepoint. Scales: activity rate/s 0–7 (bin = 1 s), time 300 s, channels 1–9. All data was gained as number of spikes/MEA which was normalized to the number of spikes at baseline.
MeHgCl Exposed Networks Show Alteration in their Response to Pharmacological Substances
To evaluate the neuronal receptor actions in neuronal networks the pharmaceutical testing was performed. Control networks (0 nM MeHgCl) responded as expected as D-AP5, CNQX, and GABA inhibited but bicuculline excited the network activity. MeHgCl at concentrations 0.5 and 50 nM did not have effect on pharmaceutical response (data not shown). Alteration was detected on GABA and bicuculline response, however, with networks exposed to 500 nM MeHgCl as the signaling of the exposed networks was not completely silenced by GABA and the response to bicuculline was stronger when compared to the control networks. (Figure 3B).
Traditional Molecular Biological Analyses did not Show Mehgcl-Dependent Effects on the Neuronal Networks
QRT-PCR analysis of the cells collected at the end of the MeHgCl exposure (72 h) did not show MeHgCl-related variation. Musashi was equally expressed by the cells treated with 0.5, 50, or 500 nM MeHgCl (Figure 4A). NF-68 was prominently expressed by the cells exposed to 0.5 or 50 nM MeHgCl (Figure 4A) but overall the expression of NF-68 in cells exposed to 500 nM MeHgCl was similar as in unexposed cells.
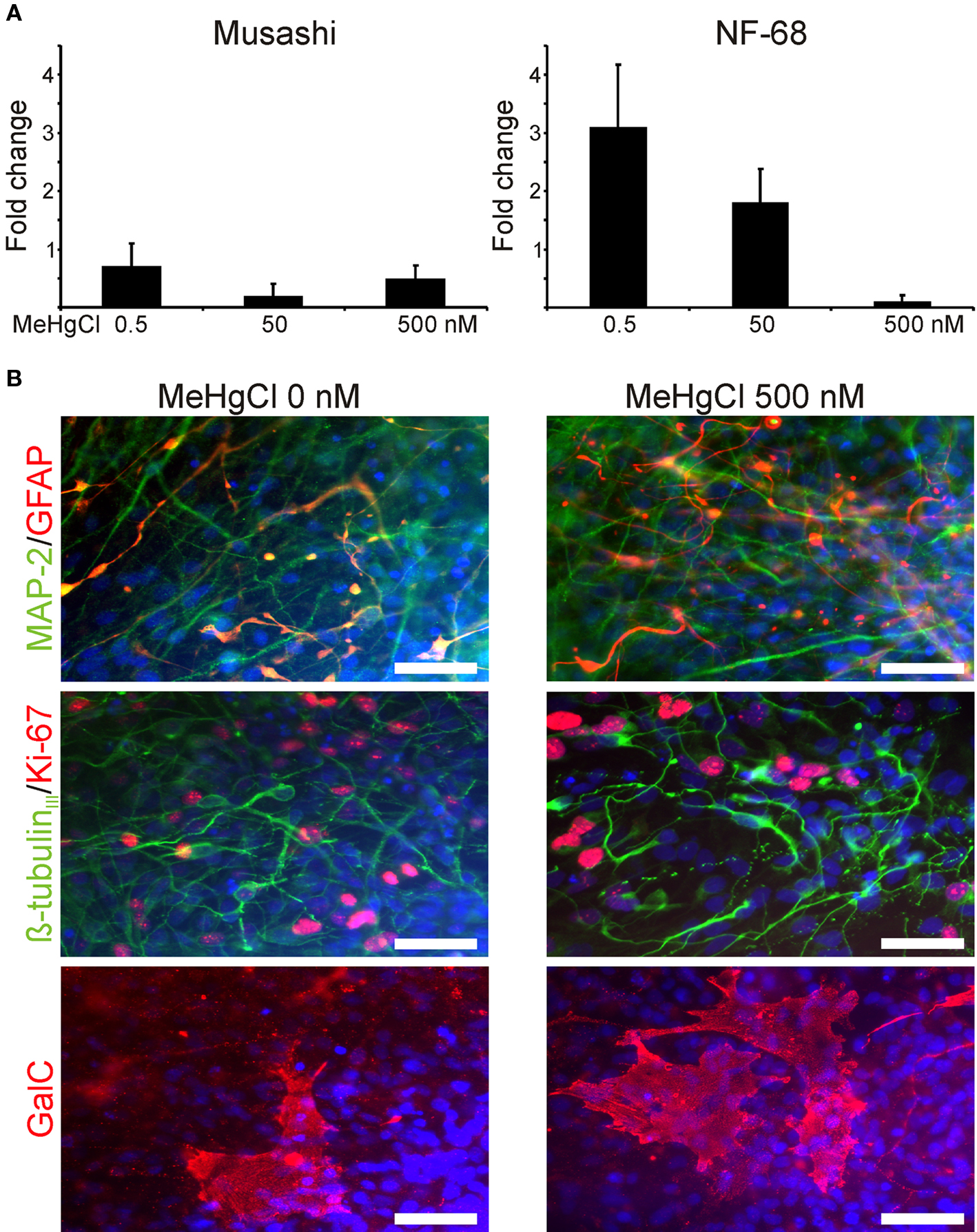
Figure 4. Molecular biological analyses of the cells after MeHgCl exposure. (A) The relative musashi and NF-68 mRNA expression of neuronal cells exposed to 0, 0.5, 50, or 500 nM MeHgCl did not show prominent variation. The data was normalized to the expression of house-keeping gene GAPDH, and values of unexposed cells were used as a calibrator set to 0 at y-axis. (B) The neuronal cell phenotypes appeared similar regardless of the MeHgCl concentration and stained positive for MAP-2, ß-tubulinIII, GFAP, and GalC (B). Scale 50 μm.
Immunocytochemical staining and analysis verified the similarity of the neuronal networks regardless of the used MeHgCl concentration. MAP-2 and ß-tubulinIII positive neuronal cells were detected throughout the network (Figure 4B). Also, some GFAP positive astrocytes were detected in each group. In addition to neuronal cells and astrocytes few cells among the networks stained positive for oligodendrocytic marker GalC (Figure 4B).
Time-lapse imaging was performed during MeHgCl exposure and it further verified that the unexposed cells or cells exposed to MeHgCl grew and proliferated in a similar fashion (Figure 5A).
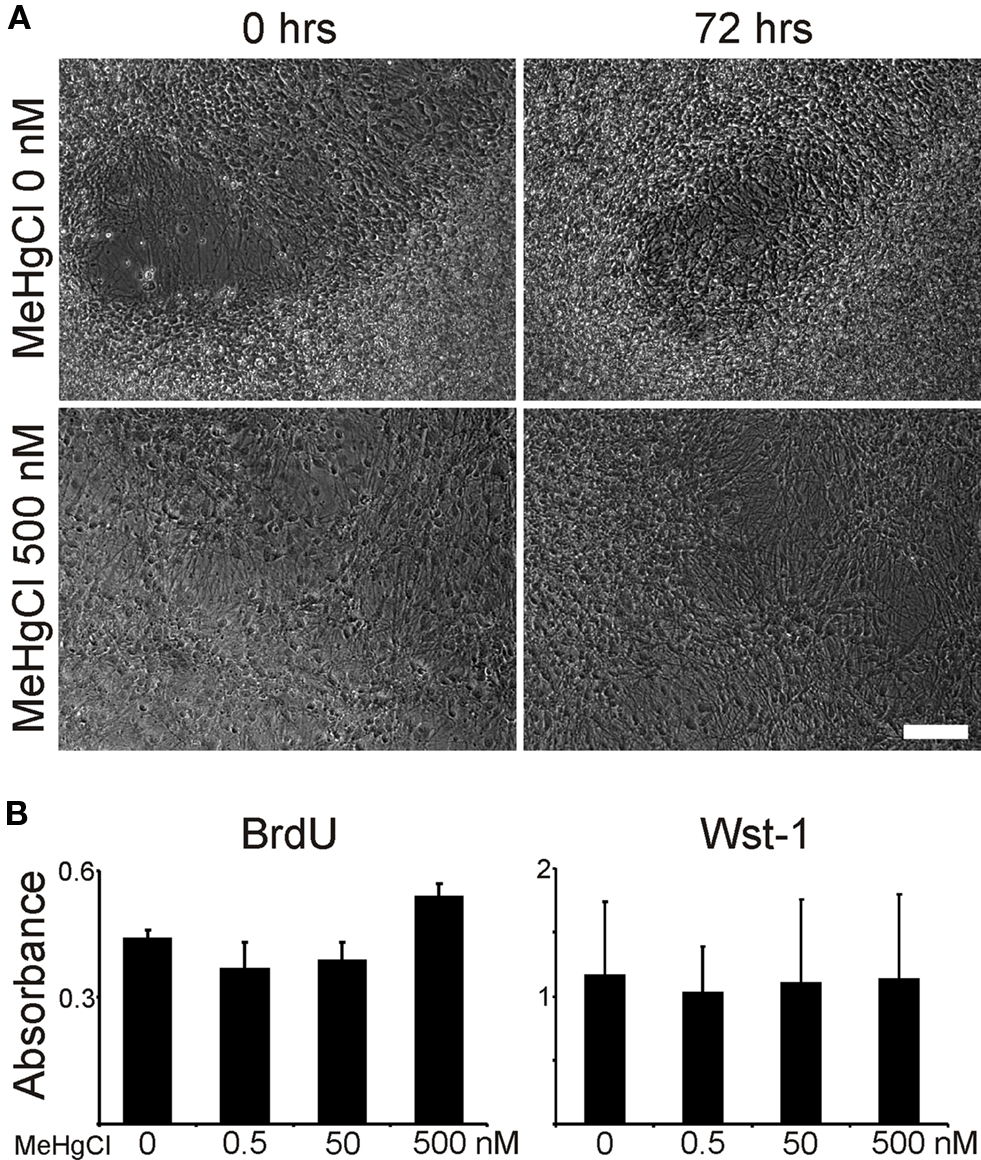
Figure 5. Time-lapse and proliferation analyses. (A) Time-lapse imaging verified the viability of the neuronal networks as no differences could be observed on morphologies during the 72 h of MeHgCl exposure. (B) Both BrdU and Wst-1 analyses asserted that all neuronal networks proliferated similarly 72 h after MeHgCl washout. Scale 100 μm.
Due to not detecting molecular biological differences between the unexposed or exposed cells we maintained a subset of cells for additional 72 h after removing the MeHgCl. These cells were analyzed for their proliferation capacity and protein expression using immunostaining. Proliferation of unexposed or MeHgCl exposed cells 72 h after MeHgCl washout was similar according to BrdU and WST-1 analyses (Figure 5B). Moreover, the network phenotype was similar as in the previous staining (data not shown).
Discussion
Here we have provided the first proof-of-principle that human ESC-derived neuronal cells are suitable for neurotoxicological screening in MEA platform. The known neurotoxic substance, MeHgCl, caused alterations in functionality in spontaneously active human neuronal networks under subcytotoxic levels. This study underlines the need to develop more sensitive methods for neurotoxicology testing platforms and shows that MEA-based technologies are suitable for that purpose.
The development of human-based cell models is highly needed for toxicological testing and drug screening studies. For neurotoxicological studies, fetal human brain-derived cells have been used (Fritsche et al., 2005; Moors et al., 2009). These cells hold, however, limiting features such as origin and culturing time which makes their large scale use challenging. Other possible human-based cell sources considered for neurotoxicological studies are, e.g., SH-SY5Y human neuroblastoma cells (Biedler et al., 1973) or NT2 human teratocarcinoma cells (Bliss et al., 2006) which can be differentiated toward CNS cell types. These cells are, however, different from “normal” healthy cells by genetics and physiologics which raise questions on their behavior when exposed to toxic substances. The development of hESC culture methods (Thomson et al., 1998) and their neural differentiation (Carpenter et al., 2001; Reubinoff et al., 2001; Zhang et al., 2001) opened up a new era for neurotoxicological field. Neuronal cells derived from hESCs most likely resemble the counterpart cells in the developing and adult human brain (Zeng et al., 2006). Thus, these cells could be used both for developmental and adult neurotoxicity testing as shown for 1-methyl-4-phenylpyridinium (MPP+) toxicity to hESC-derived dopaminergic neurons (Zeng et al., 2006). Here, we used hESC-derived neurosphere cultures which have been characterized for expression of neural markers previously (Heikkilä et al., 2009; Hicks et al., 2009; Sundberg et al., 2009; Lappalainen et al., 2010). In addition, these cultures were shown to contain MAP-2 or β-tubulinIII positive neurons and GFAP positive astrocytes after 4 week culturing on MEA dishes.
Here, we focused to study the MeHgCl effects on reported subcytotoxic levels (>1 μM in vitro) (van Vliet et al., 2008) to spontaneously active hESC-derived neuronal networks. The results showed that MeHgCl at 0.5–500 nM range caused concentration depended decrease in total spike activity during 72 h exposure compared to the control cultures. Previous, it has been reported that MeHgCl exposure caused decrease the evoked field potential amplitudes in rat primary telencephalon cultures in a concentration depended manner at 1–100 μM range whereas the effects on the spontaneous network activities were not reported (van Vliet et al., 2007). In addition to changes observed in functional level, we did not detected major changes caused by MeHgCl on cell morphology, proliferation, gene or protein expression on the studied concentrations. It has been shown that MeHgCl at a slightly higher concentration (750 nM) caused decrease in amount of differentiating β-tubulinIII positive cells, and cell migration at 500 nM to 10 μM in human fetal brain cells which showed cytotoxicity to MeHgCl at 10 μM concentration (Moors et al., 2009). Although we did not study cell migration in more detail, the time-lapse imaging of treated cultures did not implicate that hESC-derived neuronal cell migration would be affected at studied MgHgCl concentrations (0.5–500 nM).
In this study we could show that MeHgCl at subcytotoxic levels clearly decreased the electrical functionality of hESC-derived networks. Harmful effects were not detected with traditional molecular biological methods. This clearly clarifies the advantage of MEA setup which is more sensitive to detect differences between groups treated with subcytotoxic concentrations. These results underline the need to study the subcytotoxic effects of different substances more carefully since they may reveal effects on functional properties which may or may not be acute, long-lasting, or even permanent. The MeHgCl exposed hESC-derived neuronal networks also showed distinct responses to pharmacologic substances. The network signaling decreased when treated with AMPA/kainate (CNQX) and NMDA (D-AP5) receptor antagonist and GABA agonist treatments as in control networks whereas the response for GABAA receptor antagonist bicuculline was substantially stronger when compared to untreated networks. Thus it is possible, that 72 h MeHgCl exposure had an effect on GABA receptor subunit expression on the hESC-derived neuronal cells. Interestingly, Bal-Price and colleagues reported also changed responses to bicuculline in their study where primary rat cortical cultures were exposed to domoic acid (Hogberg et al., 2010). Unfortunately, due to small amounts of RNA gained from MEAs more detailed GABA receptor subunit analysis with qRT-PCR could not be performed here. Thus, this will require further studies in the future.
Here we have showed that hESC-derived neuronal networks are indeed suitable for toxicological screening studies. The MEA platform is especially eligible for screening functional responses in spontaneously active neuronal networks that are missed with traditional molecular biology end-point analyses. We have shown that hESC-derived neural cells are capable of forming spontaneously active networks exhibiting signaling from sporadic single spiked to more mature synchronous bursting activity (Heikkilä et al., 2009). This setup is in routine use in our laboratory, thus it is highly applicable also for neurotoxicological studies. Regardless of the usability and enormous research possibilities MEA setup also holds its cons. The controllability of the networks is plating-dependable thus batch to batch variation is expected. Also, standard glass MEA plates are not favored by hESC-derived neuronal cells and coatings are crucial for network forming capability. Also, the gained data is a large package to handle and analysis of it is challenging (Wagenaar et al., 2006; Illes et al., 2007; Heikkilä et al., 2009) and more standardized analysis methods should be developed. Also more structured networks gained by different surface patterning and guiding applications would enable more standardized functional networks instead of the used spontaneously self organizing cultures that would increase the repeatability of the tests. In the future, the MEA setup offers an intriguing possibility to study neuronal networks derived from human patients’ induced pluripotent stem cells (iPSCs) as we recently reported (äänismaa et al., 2010). In deed, there are almost limitless possibilities to model genetic neurological deficiencies in vitro as well as perform, e.g., drug screening with these iPSC-derived neural cells. We conclude that MEA platform is highly suitable for neurotoxicological testing and would be a valuable addition to current analysis patterns.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was funded by Academy of Finland (projects 122959 and 123233), BioneXt Tampere, and Competitive Research Funding of Pirkanmaa Hospital District. We want to acknowledge the personnel of Regea for the support in stem cell research and J. Kreutzer and P. Kallio, Tampere University of Technology, for PDMS designs.
References
äänismaa, R., Ylä-Outinen, L., Heikkilä, J., Suuronen, R., Skottman, H., and Narkilahti, S. (2010). Neural Differentiation of Human Induced Pluripotent Stem Cells: Formation of Functional Neuronal Networks. Conference Proceedings of the 7th International Meeting on Substrate-Integrated Micro Electrode Arrays, Reutlingen, 90–91.
Bal-Price, A. K., Hogberg, H. T., Buzanska, L., and Coecke, S. (2010). Relevance of in vitro neurotoxicity testing for regulatory requirements: challenges to be considered. Neurotoxicol. Teratol. 32, 36–41.
Bal-Price, A. K., Hogberg, H. T., Buzanska, L., Lenas, P., van Vliet, E., and Hartung, T. (2009). In vitro developmental neurotoxicity (DNT) testing: relevant models and endpoints. Neurotoxicology. doi: 10.1016/j.neuro.2009.11.006.
Biedler, J. L., Helson, L., and Spengler, B. A. (1973). Morphology and growth, tumorigenicity, and cytogenetics of human neuroblastoma cells in continuous culture. Cancer Res. 33, 2643–2652.
Bliss, T. M., Kelly, S., Shah, A. K., Foo, W. C., Kohli, P., Stokes, C., Sun, G. H., Ma, M., Masel, J., Kleppner, S. R., Schallert, T., Palmer, T., and Steinberg, G. K. (2006). Transplantation of hNT neurons into the ischemic cortex: cell survival and effect on sensorimotor behavior. J. Neurosci. Res. 83, 1004–1014.
Buzanska, L., Ruiz, A., Zychowicz, M., Rauscher, H., Ceriotti, L., Rossi, F., Colpo, P., Domanska-Janik, K., and Coecke, S. (2009a). Patterned growth and differentiation of human cord blood-derived neural stem cells on bio-functionalized surfaces. Acta Neurobiol. Exp. (Wars) 69, 24–36.
Buzanska, L., Sypecka, J., Nerini-Molteni, S., Compagnoni, A., Hogberg, H. T., del Torchio, R., Domanska-Janik, K., Zimmer, J., and Coecke, S. (2009b). A human stem cell-based model for identifying adverse effects of organic and inorganic chemicals on the developing nervous system. Stem Cells 27, 2591–2601.
Carpenter, M. K., Inokuma, M. S., Denham, J., Mujtaba, T., Chiu, C. P., and Rao, M. S. (2001). Enrichment of neurons and neural precursors from human embryonic stem cells. Exp. Neurol. 172, 383–397.
Castoldi, A. F., Coccini, T., Ceccatelli, S., and Manzo, L. (2001). Neurotoxicity and molecular effects of methylmercury. Brain Res. Bull. 55, 197–203.
Clarkson, T. W. (1993). Mercury: major issues in environmental health. Environ. Health Perspect. 100, 31–38.
Erceg, S., Lainez, S., Ronaghi, M., Stojkovic, P., Perez-Arago, M. A., Moreno-Manzano, V., Moreno-Palanques, R., Planells-Cases, R., and Stojkovic, M. (2008). Differentiation of human embryonic stem cells to regional specific neural precursors in chemically defined medium conditions. PLoS ONE 3, e2122. doi: 10.1371/journal.pone.0002122.
Fritsche, E., Cline, J. E., Nguyen, N. H., Scanlan, T. S., and Abel, J. (2005). Polychlorinated biphenyls disturb differentiation of normal human neural progenitor cells: clue for involvement of thyroid hormone receptors. Environ. Health Perspect. 113, 871–876.
Harry, G. J., Billingsley, M., Bruinink, A., Campbell, I. L., Classen, W., Dorman, D. C., Galli, C., Ray, D., Smith, R. A., and Tilson, H. A. (1998). In vitro techniques for the assessment of neurotoxicity. Environ. Health Perspect. 106(Suppl. 1), 131–158.
Heikkilä, T. J., Ylä-Outinen, L., Tanskanen, J. M., Lappalainen, R. S., Skottman, H., Suuronen, R., Mikkonen, J. E., Hyttinen, J. A., and Narkilahti, S. (2009). Human embryonic stem cell-derived neuronal cells form spontaneously active neuronal networks in vitro. Exp. Neurol. 218, 109–116.
Hicks, A. U., Lappalainen, R. S., Narkilahti, S., Suuronen, R., Corbett, D., Sivenius, J., Hovatta, O., and Jolkkonen, J. (2009). Transplantation of human embryonic stem cell-derived neural precursor cells and enriched environment after cortical stroke in rats: cell survival and functional recovery. Eur. J. Neurosci. 29, 562–574.
Hogberg, H. T., Kinsner-Ovaskainen, A., Hartung, T., Coecke, S., and Bal-Price, A. K. (2009). Gene expression as a sensitive endpoint to evaluate cell differentiation and maturation of the developing central nervous system in primary cultures of rat cerebellar granule cells (CGCs) exposed to pesticides. Toxicol. Appl. Pharmacol. 235, 268–286.
Hogberg, H. T., Novellino, A., Sobanski, T., and Bal-Price, A. K. (2010). Application of Micro Electrode Arrays (MEAs) as an Emerging Technology for Developmental Neurotoxicity. Conference Proceeding of the 7th International Meeting on Substrate-Integrated Micro Electrode Arrays, Reutlingen, 142–144.
Illes, S., Fleischer, W., Siebler, M., Hartung, H. P., and Dihne, M. (2007). Development and pharmacological modulation of embryonic stem cell-derived neuronal network activity. Exp. Neurol. 207, 171–176.
Itsykson, P., Ilouz, N., Turetsky, T., Goldstein, R. S., Pera, M. F., Fishbein, I., Segal, M., and Reubinoff, B. E. (2005). Derivation of neural precursors from human embryonic stem cells in the presence of noggin. Mol. Cell. Neurosci. 30, 24–36.
Johnson, M. A., Weick, J. P., Pearce, R. A., and Zhang, S. C. (2007). Functional neural development from human embryonic stem cells: accelerated synaptic activity via astrocyte coculture. J. Neurosci. 27, 3069–3077.
Johnstone, A. F., Gross, G. W., Weiss, D. G., Schroeder, O. H., Gramowski, A., and Shafer, T. J. (2010). Microelectrode arrays: a physiologically based neurotoxicity testing platform for the 21st century. Neurotoxicology 31, 331–350.
Kaur, P., Aschner, M., and Syversen, T. (2006). Glutathione modulation influences methyl mercury induced neurotoxicity in primary cell cultures of neurons and astrocytes. Neurotoxicology 27, 492–500.
Lappalainen, R. S., Salomäki, M., Ylä-Outinen, L., Heikkilä, T. J., Hyttinen, J. A., Pihlajamäki, H., Suuronen, R., Skottman, H., and Narkilahti, S. (2010). Similarly derived and cultured hESC lines show variation in their developmental potential towards neuronal cells in long-time culture. Regen. Med. (in press).
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 4, 402–408.
Moors, M., Rockel, T. D., Abel, J., Cline, J. E., Gassmann, K., Schreiber, T., Schuwald, J., Weinmann, N., and Fritsche, E. (2009). Human neurospheres as three-dimensional cellular systems for developmental neurotoxicity testing. Environ. Health Perspect. 117, 1131–1138.
Narkilahti, S., Rajala, K., Pihlajamaki, H., Suuronen, R., Hovatta, O., and Skottman, H. (2007). Monitoring and analysis of dynamic growth of human embryonic stem cells: comparison of automated instrumentation and conventional culturing methods. Biomed. Eng. Online 6, 11.
Pine, J. (1980). Recording action potentials from cultured neurons with extracellular microcircuit electrodes. J. Neurosci. Methods 2, 19–31.
Potter, S. M., and DeMarse, T. B. (2001). A new approach to neural cell culture for long-term studies. J. Neurosci. Methods 110, 17–24.
Reubinoff, B. E., Itsykson, P., Turetsky, T., Pera, M. F., Reinhartz, E., Itzik, A., and Ben-Hur, T. (2001). Neural progenitors from human embryonic stem cells. Nat. Biotechnol. 19, 1134–1140.
Sundberg, M., Jansson, L., Ketolainen, J., Pihlajamaki, H., Suuronen, R., Skottman, H., Inzunza, J., Hovatta, O., and Narkilahti, S. (2009). CD marker expression profiles of human embryonic stem cells and their neural derivatives, determined using flow-cytometric analysis, reveal a novel CD marker for exclusion of pluripotent stem cells. Stem Cell Res. 2, 113–124.
Sundberg, M., Skottman, H., Suuronen, R., and Narkilahti, S. (2010). Production and isolation of NG2(+) oligodendrocyte precursors from human embryonic stem cells in defined serum-free medium. Stem Cell Res. 5, 91–103.
Tarvainen, J., Saarinen, M., Laitinen, J., Korpinen, J., and Viitanen, J. (2002). Creating images with high data contents for microworld applications. Ind. Syst. Rev. 1, 17–23.
Thomson, J. A., Itskovitz-Eldor, J., Shapiro, S. S., Waknitz, M. A., Swiergiel, J. J., Marshall, V. S., and Jones, J. M. (1998). Embryonic stem cell lines derived from human blastocysts. Science 282, 1145–1147.
van Vliet, E., Morath, S., Eskes, C., Linge, J., Rappsilber, J., Honegger, P., Hartung, T., and Coecke, S. (2008). A novel in vitro metabolomics approach for neurotoxicity testing, proof of principle for methyl mercury chloride and caffeine. Neurotoxicology 29, 1–12.
van Vliet, E., Stoppini, L., Balestrino, M., Eskes, C., Griesinger, C., Sobanski, T., Whelan, M., Hartung, T., and Coecke, S. (2007). Electrophysiological recording of re-aggregating brain cell cultures on multi-electrode arrays to detect acute neurotoxic effects. Neurotoxicology 28, 1136–1146.
Wagenaar, D. A., Pine, J., and Potter, S. M. (2006). An extremely rich repertoire of bursting patterns during the development of cortical cultures. BMC Neurosci. 7, 11. doi: 10.1186/1471-2202-7-11.
Zeng, X., Chen, J., Deng, X., Liu, Y., Rao, M. S., Cadet, J. L., and Freed, W. J. (2006). An in vitro model of human dopaminergic neurons derived from embryonic stem cells: MPP+ toxicity and GDNF neuroprotection. Neuropsychopharmacology 31, 2708–2715.
Keywords: human embryonic stem cell, neuronal network, microelectrode array, neurotoxicology, methyl mercury chloride
Citation: Ylä-Outinen L, Heikkilä J, Skottman H, Suuronen R, äänismaa R and Narkilahti S (2010) Human cell-based micro electrode array platform for studying neurotoxicity. Front. Neuroeng. 3:111. doi: 10.3389/fneng.2010.00111
Received: 28 July 2010;
Paper pending published: 29 July 2010;
Accepted: 30 August 2010;
Published online: 30 September 2010.
Edited by:
Antonio Novellino, ett s.r.l., ItalyReviewed by:
Leandro Lorenzelli, Fondazione Bruno Kessler, ItalyHari S. Sharma, Uppsala University, Sweden
Copyright: © 2010 Ylä-Outinen, Heikkilä, Skottman, Suuronen, äänismaa and Narkilahti. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Susanna Narkilahti, Regea – Institute for Regenerative Medicine, University of Tampere, Biokatu 12, 33520 Tampere, Finland. e-mail:c3VzYW5uYS5uYXJraWxhaHRpQHJlZ2VhLmZp