 Thirukumaran T. Kanagasabapathi1*
Thirukumaran T. Kanagasabapathi1* Davide Ciliberti1
Davide Ciliberti1 Sergio Martinoia2,3
Sergio Martinoia2,3 Wytse J. Wadman4 and Michel M. J. Decré1
Wytse J. Wadman4 and Michel M. J. Decré1
- 1 Minimally Invasive Healthcare Department, Philips Research Laboratories, Eindhoven, Netherlands
- 2 Department of Biophysical and Electronic Engineering, University of Genova, Genova, Italy
- 3 Department of Neuroscience and Brain Technologies, Italian Institute of Technology, Genova, Italy
- 4 Center for NeuroScience, Swammerdam Institute for Life Sciences, University of Amsterdam, Amsterdam, Netherlands
We developed a dual-compartment neurofluidic system with inter-connecting microchannels to connect neurons from their respective compartments, placed on a planar microelectrode arrays. The design and development of the compartmented microfluidic device for neuronal cell culture, protocol for sustaining long-term cultures, and neurite growth through microchannels in such a closed compartment device are presented. Using electrophysiological measurements of spontaneous network activity in the compartments and selective pharmacological manipulation of cells in one compartment, the biological origin of network activity and the fluidic isolation between the compartments are demonstrated. The connectivity between neuronal populations via the microchannels and the crossing-over of neurites are verified using transfection experiments and immunofluorescence staining. In addition to the neurite cross-over to the adjacent compartment, functional connectivity between cells in both the compartments is verified using cross-correlation (CC) based techniques. Bidirectional signal propagation between the compartments is demonstrated using functional connectivity maps. CC analysis and connectivity maps demonstrate that the two neuronal populations are not only functionally connected within each compartment but also with each other and a well connected functional network was formed between the compartments despite the physical barrier introduced by the microchannels.
Introduction
The development of in vitro models of patterned neuronal networks is of significant interest for the neuroscience research community. Considering the multitude of connections that form in un-patterned neuronal cultures, the restraint of neurite outgrowth to specific pathways ensures a considerable control over network complexity. Controlling the topological features of small neuronal networks on planar microelectrode arrays (MEAs) and pre-determining their internal connectivity has been the subject of extensive study in the past decade (Morin et al., 2005, 2006; Chao et al., 2008). Large neuronal ensembles coupled to MEAs represent an interesting means for investigating information processing and dynamics in neuronal systems under controlled condition and by means of applications of external electrical stimuli, chemical stimuli, and/or physical constraints (Jimbo et al., 2003; Martinoia et al., 2005; Berdondini et al., 2006). Compartmentalization of cells and control of the network connectivity may provide an ideal tool for further developing such in vitro neuronal models. Imprisoning neurons in the immediate vicinity of the electrode inside micro-cages on top of electrodes obtained by micro-machining of silicon wafers as in “Neurochip” was demonstrated by Maher et al. (1999) However, the major drawback of Neurochip devices was that neurons tend to migrate from the wells (Morin et al., 2006). The displacement of neurons cultured on planar surfaces is mainly due to the strong pulling forces that the neurites exercise on the soma. “Neurocage” as demonstrated by Tooker et al. (2005) overcame that problem using surface micromachined parylene cages. This approach constrains single neurons in a cage in which signals are recorded by only one electrode. Suzuki et al. (Suzuki et al., 2005; Suzuki and Yasuda, 2007) proposed photothermal etching with an infrared laser beam to create networks of micro-chambers and microchannels inside an agarose gel structure on top of planar MEAs.
Chemical surface patterning, polydimethylsiloxane (PDMS) stencil, microstamping, and microcontact printing of selective substrates have been used to provide successful cell isolation (James et al., 2004; Morin et al., 2006; Nam et al., 2006; Sorkin et al., 2006; Sang Beom et al., 2007). Adhesion and patterning of cortical neurons were investigated on isolated islands of neuron-adhesive polyethylenimine (PEI) surrounded by a neuron-repellent fluorocarbon (FC) layer. Although these techniques make it possible to reconfigure the connectivity of the networks within a culture, a major problem involved in neuronal patterning remains: strict compliance with the underlying geometry, while enabling the study of low-dimensional neural networks under well-controlled conditions, fails to appropriately mimic naturally occurring networks. For these reasons, the integration of microfluidic structures on planar MEAs for the physical isolation of neuronal cells has recently been of significant interest among various research groups (Claverol-Tinture and Pine, 2002; Bani-Yaghoub et al., 2005; Morin et al., 2005, 2006; Berdondini et al., 2006; Ravula et al., 2006; Dworak and Wheeler, 2009). Majumdar et al. (2011) demonstrated a microfluidic platform for co-culturing glia and neurons in a dual-compartment system with capabilities to individually manipulate the microenvironment of different cell types. However, co-culturing subpopulation of dissociated cells for long-term studies and recording electrophysiological activities simultaneously to establish functional connectivity between the sub-populations has not been reported earlier. In this work, based on earlier research from Taylor et al. (2003) we present a technique to integrate closed interconnected micro-chambers on planar MEAs that allow the compartmentalization of neuronal cells and the control of the fluidic environments. This technique when combined with substrate integrated planar MEAs, provides a suitable platform for studying the development and the electrophysiological activity of compartmentalized neuronal networks in culture.
Further, to allow realistic studies of compartmentalized neuronal networks, the device should be capable of sustaining a healthy and stable culture for up to 3 or 4 weeks in vitro for electrophysiological recordings and pharmacological manipulation, while combined with MEAs. Electrophysiological recordings from the compartmented cultures and neuronal network formation through the microchannels connecting the compartments need to be verified. Neurite growth into adjacent compartment through microchannels is a necessary but not sufficient condition for network functional connectivity. Utilizing cross-correlation (CC) based methods, we analyzed the functional connectivity during the spontaneous activity of the network. Statistical analysis on CC data from different devices and the dependency of inter-compartmental correlation with the distance from the microchannels are discussed.
Materials and Methods
Neurofluidic Device Preparation
The 3-mm-thick polydimethylsiloxane (PDMS) devices used for this study have two microfluidic compartments of 100 μm height, 1.5 mm width, and 8 mm length interconnected with microchannels of 10 μm width, 3 μm height, and 150 μm length spaced at regular intervals of 60 μm (Figure 1A; Taylor et al., 2003). The small size of microchannels prevents migration of cells between compartments while allowing only neurites to pass through (Taylor et al., 2003). Conventional soft lithographic techniques as pioneered by Whitesides and his collaborators (McDonald et al., 2000; McDonald and Whitesides, 2002) were utilized in the fabrication of the device. PDMS stamps were replicated using Si master molds and the fabrication techniques were described in an earlier work (Kanagasabapathi et al., 2009). Four 6-mm-diameter reservoir holes were laser cut into the fabricated device. The fabricated PDMS stamps were rinsed thoroughly in an ultrasonic bath, stored in Milli-Q water for 24 h and decontaminated in 70% ethanol. Each PDMS device was then sterilized in a dry oven at 120°C for 20 min. To selectively hydrophilize the microcompartments and the microchannels, the PDMS devices were placed on cover-slips with the microchannels facing downward for oxygen–plasma treatment (Owen and Smith, 1994; Katzenberg, 2005; Bodas and Khan-Malek, 2007). This treatment renders the microfluidic compartments and the microchannels hydrophilic, while preserving hydrophobicity of the contact surface, thereby preventing leakage. The PDMS devices were then reversibly bonded onto a commercially available planar microelectrode array (Multichannel Systems, Germany) with 60 electrodes of 30 μm diameter and 200 μm spacing placed in an 8 × 8 array (no corner electrodes) as shown in Figure 1B. While bonding, PDMS device was aligned on the MEA to include 30 electrodes each per compartment. Prior to the placement of the PDMS devices, MEAs were sterilized in a vacuum oven, coated overnight with a solution of Polyethylenimine (PEI; Sigma-Aldrich, The Netherlands) at a concentration of 40 μg/ml and rinsed thoroughly in sterile water (GIBCO, Invitrogen, CA, USA) on the day of culture.
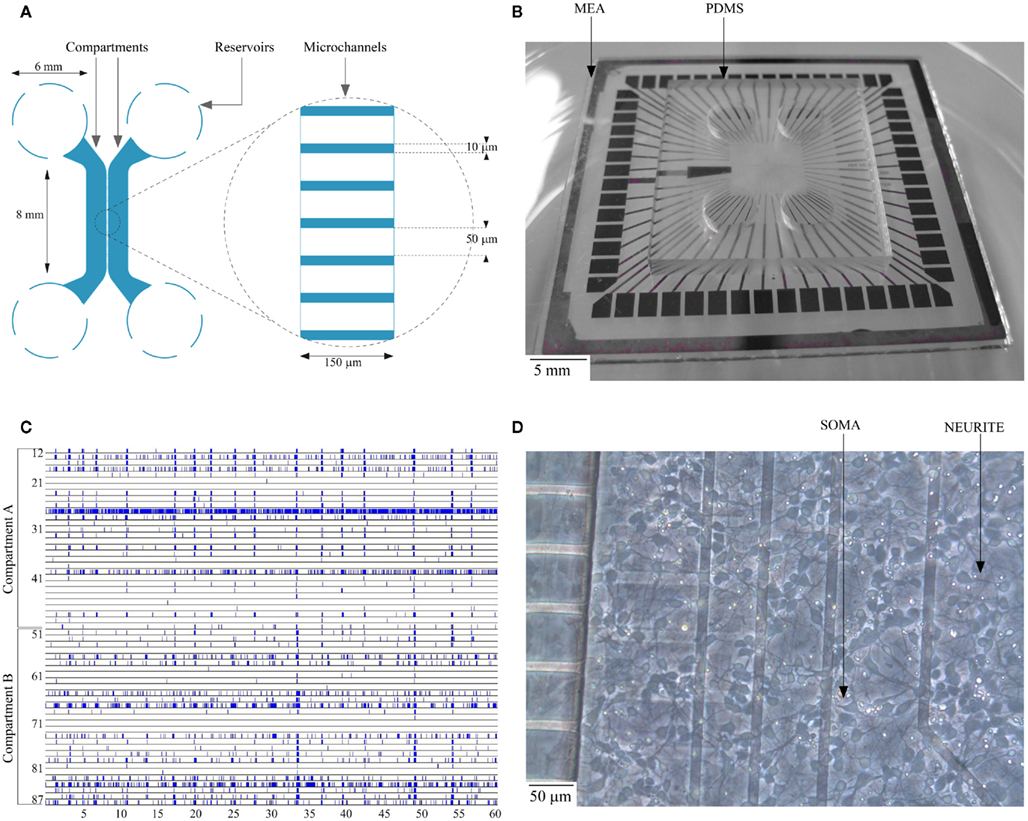
Figure 1. (A) Schematic layout of the dual-compartment device; (B) Planar MEA substrate with dual-compartment PDMS device; (C) Spontaneous activity of a culture on DIV 14 (the electrode number along the y-axis run from 11 through 88 with the first digit representing column and the second digit its row respectively. Each dot represents an action potential recorded by one of the MEA channels); (D) Typical cortical cell culture in a compartment on DIV 4.
Device Cleaning and Reuse
After a complete set of experiments with a culture, planar MEA substrates were reused for subsequent experiments. The reversible sealing of PDMS on MEAs facilitated easy disposal of PDMS devices while retaining MEA substrates without damage. MEAs were rinsed in deionized (DI) water and placed in 1% Tergazyme (Alconox Inc., USA) solution at 37°C overnight to enhance tissue break-down and removal of cell debris. The MEAs were then rinsed in DI water overnight and prepared as described in the device preparation protocol earlier. Freshly prepared sets of PDMS devices were used for each experiment.
Cortical Cell Culture
Primary neuron dissociated cells were prepared by trypsin digestion of day-18 embryonic rat whole cortices (Wistar rat, Harlan Laboratories, NL, USA), as per the approved protocols for the care and use of lab animals in The Netherlands. The dissociated cortical cells were cultured in neurobasal medium supplemented with 1% fresh, stable L-Glutamine (GIBCO, Invitrogen, CA, USA), 1% Penicillin–Streptomycin (GIBCO, Invitrogen, CA, USA), 2% B27 Supplement™ (GIBCO, Invitrogen, CA, USA), and 10 nM Triiodothyronine (Ramakers et al., 1990, 1991). Cultured cells were plated on PEI coated MEA substrate at a concentration of ∼20 × 106 cells/ml which resulted in a plating density of 2 × 105 cells/cm2. PEI coated substrate ensure proper adhesion of cell bodies and also avoids migration of cells during the developmental period. While bonding PDMS to MEA substrate, care was taken to ensure proper device sealing by examining the contact surfaces and to avoid any access path underneath the PDMS other than the microchannels. The device was then incubated in a humidified incubator at 37°C supplied with 5% CO2 and 95% O2. The cell culture was checked at regular intervals and complete medium change was facilitated three times a week [on days-in vitro (DIV) 4, 7, 9, 11, 14 etc.,] to compensate for the evaporation losses and to provide sufficient supplements for the culture. Medium was refreshed by completely removing the medium from all four reservoirs and by adding ∼80 μl of freshly prepared medium to one reservoir per compartment. The osmotic pressure difference between the reservoirs connected to the compartment resulted in medium refreshing within the compartment due to capillary force. Once the level of medium in the two reservoirs connected to a compartment saturates, an additional ∼40 μl of medium was added to compensate for the initial volume removed. The chosen medium refreshing frequency offered optimal cell survival thus far resulting in active cultures up to DIV 35.
Transfection and Immunofluorescence Labeling
Cultures were transfected with a thy1-green fluorescent protein (eGFP) construct in one compartment, using lipofectamine 2000 (Invitrogen, CA, USA). One milligram DNA was mixed with 100 ml neurobasal medium and mixed with 3 mg lipofectamine 2000 diluted in 100 ml neurobasal medium (without supplement), and left at room temperature for 20 min. After diluting the transfection mixture with four volumes of neurobasal medium with supplement, it was gently flushed into one of the compartments. Images of transfected neurons (typically only a few neurons were stained per compartment) were recorded after fixation of the cultures with 4% paraformaldehyde in phosphate buffered saline at DIV 18–21. Similarly fixated cultures during the second week of development (DIV 14) were histologically stained with primary monoclonal anti-NeuN (Sigma-Aldrich, USA) and secondary Goat anti-mouse AlexaFluor488 (Chemicon International Inc., Germany) to post-mitotic neuronal soma and polyclonal anti-Neurofilament 200 kDa (Sigma-Aldrich, USA) and secondary goat anti-rabbit Alexa-Fluor 546 (Sigma-Aldrich, USA) to axonal or generally neurite processes.
Pharmacological Experiment
A pharmacological neuromodulator was used to confirm the biological origin of network activity and to show fluidic separation between the compartments. Tetrodotoxin (TTX), a sodium channel blocker prevents the generation of action potentials in the neurons at a concentration of 100 nM (Ramakers and Boer, 1991). Freshly prepared TTX solution in neurobasal medium was applied to one compartment to suppress the spiking activity of the cells in that compartment. Before applying TTX, spiking activity in the device was recorded for 2 min as a control condition. Neurobasal medium from one of the compartment (i.e., compartment B) was completely removed and 100 μl of TTX at a concentration of 100 nM was added to one of the reservoirs in Compartment “B,” the fluid level between the two reservoirs are allowed to equalize for 15 min and electrophysiological activity from both the compartments was recorded for 2 min. The slightly higher volume of neurobasal medium in compartment A (120 μl) acts against leakage or diffusion of TTX from compartment B (Taylor et al., 2003). After recording the electrophysiological activities, TTX in the compartment was rinsed three times with supplement enriched neurobasal medium and the activities were recorded after each wash cycle to observe the recovery of spiking.
Electrical Recordings
The spontaneous neuronal activity was recorded twice per week using MEA1060 system (Multichannel Systems, Germany) equipped with 60 recording channels at 10 or 25 kHz sampling frequency with a hardware filter of 10–3000 Hz. Each recording session usually lasted 10 min and started at least 5 min after the device was moved from the incubator in order to soften the effects of mechanical perturbation on the activity (Wagenaar et al., 2006). Digitalized signals were filtered at 200 Hz (high-pass Butterworth). MCRack software was used for data recording, while offline data analysis was performed in MATLAB (The Mathworks, Natick, MA, USA) on data imported in MCD format from MCRack. Devices were maintained at 37°C and recordings were performed in a custom-built enclosure provided with 5% CO2 supply and 100% relative humidity to replicate incubator conditions within a laminar flow hood. The setup avoided contamination issues and ensures sterile condition during the entire recording procedure. A custom-built glass-lid with reference electrodes for electrophysiological recordings was used to prevent excessive evaporation loss during the recording sessions.
Data Analysis
An amplitude based threshold detection was used to detect the neuronal spikes (Maccione et al., 2009). The peak-to-peak threshold was set to eight times the RMS noise level (Eytan et al., 2003). No attempt was made to discriminate and sort spikes collected from a single recording site (Chiappalone et al., 2006) and hence the individual spike train may include spikes from one or more neurons. A raster plot of the electrophysiological recording of a typical spontaneous activity within the culture is shown in Figure 1C. The raster plot shows the spontaneous activity of a culture in a closed compartmented device on DIV 14. The typical activity is characterized by both local random spiking and bursting behavior as shown in Figure 1C.
Functional connectivity can be defined as the temporal correlation between spatially remote neurophysiological events, expressed as a deviation from the statistical independence across these events in distributed neuronal groups and areas (Friston et al., 1993). Functional connectivity was estimated by applying CC based methods (Knox, 1981; Melssen and Epping, 1987). In order to have a valid reference for the analysis of the genuineness of the network connectivity across the microchannels, surrogate spike trains were generated. Given two spike trains (one per compartment), the spike surrogation aimed at destroying any temporal relationship between them. It consisted of a shuffling procedure in which the spikes were shifted by randomly sliding the original zero instant (t0, org) to a new temporal instant (t0,surr): t0,surr was set to a different value for each of the recording channels (no phase lockings were possible). Noisy or silent channels during the recording phase were disregarded from the analysis. The procedure preserved the statistical features of the original spike train, namely average spiking rate and inter-spike intervals distribution, and any overestimation of the significance of the correlation was thus avoided. Correlation in network activities between two compartments was analyzed using the tools developed by Bologna et al. (2010). The bin size for the computation of the CC was set to the optimal value of 0.1 ms (Garofalo et al., 2009), while the half-window size was set to 5 ms. The strength of a functional connection between two recording sites was measured with the Cpeak value. The Cpeak value corresponds to the area centered around the CC peak subtended by the cross-correlogram in a time span of 300 μs (Berdondini et al., 2009). CC between an original and a surrogate spike train can be used to evaluate the contribution of randomness to the estimated functional connectivity.
Since the placement of PDMS device on MEA ensured a symmetrical distribution of the electrode array with 30 electrodes per compartment, the electrode columns were segregated into four regions (i.e., Region 1 with electrodes 41 through 58, Region 2 with electrodes 31 through 38 and 61 through 68, Region 3 with electrodes 21 through 28 and 71 through 78, and Region 4 with electrodes from 12 through 18 and 82 through 88). A vector of Cpeak values for each electrode under each of the four circumstances such as (i) with all the electrodes in the same compartment (“Intra”), (ii) with all the electrodes in the other compartment (“Inter”), (iii) with all the electrodes in both compartment (“Global”), and (iv) with all the electrodes of the other compartment with surrogate spike trains (“Inter_Shuffled”) was created. A two-step statistical analysis was performed utilizing the Cpeak vectors. In the first step, for each of the four circumstances the median of the Cpeak values was calculated, thus extracting a number from each vector. In the second step, the median of these numbers within each region was calculated. The 16 measures (“Intra,” “Inter,” “Global,” and “Inter_Shuffled” per each of the four regions) are used for the evaluation of network connectivity for each experiment and they were used for comparisons across different devices. Non-parametrical tests such as the paired sign test (Whitley and Bai, 2002) and the Friedman’s test (Sprent and Smeeton, 2001) were adopted on these overall estimates to confirm the statistical consistency of our results related to functional connectivity across different experiments.
Results
Viability of Neurons and Electrophysiological Activity
Cell viability and neurite formation were observed at regular intervals over the developmental period (up to DIV 35). The development of a typical culture in a compartment on DIV 4 is shown in Figure 1D. Cells were observed to be restricted to the compartments except on rare occasions when cells smaller than 10 μm were observed to migrate inside the microchannels during initial cell plating stage. In our initial experiments, we observed that the cells in the center of the compartment started to deteriorate after approximately DIV 7, probably from oxygen and nutrients depletion and/or waste accumulation. This resulted in cell death in the center of the compartment propagating toward the periphery. To circumvent the issue of low medium availability to the cells and to provide cells with sufficient oxygen, we developed an optimal medium change protocol for the dual-compartment device as explained earlier. With the change in protocol, viability of cells in dual-compartment device was increased substantially, cells were observed to deteriorate only from around DIV 30 and the cultures were sustained for up to DIV 35 successfully.
A raster plot of the electrophysiological recording of a typical spontaneous activity within the culture is shown in Figure 1C. The raster plot shows the spontaneous activity of a culture in a closed compartmented device on DIV 14. The observed typical activity is very similar to that of non-compartmentalized networks and it is indeed characterized by both local random spiking and network bursting behavior. The evolution of network activity over the developmental period from DIV 11 to DIV 35 was recorded at regular intervals (on DIV 11, 14, 17, 21, 24 etc.). An increase in network activity was observed from DIV 14, with a maturation phase in the spontaneous activity during the third week in culture and the activity subsided after DIV 25 resulting in very few spiking neurons by DIV 30, similar to those reported in literature (Chiappalone et al., 2006). Disappearance of network bursts signaling the deterioration of the culture was observed after DIV 30 (Mukai et al., 2003). During third week in culture (∼DIV 21), cortical culture in devices showed an extensive network bursting activity. Pharmacological experiments were performed during the most active phase of the culture starting from DIV 14. To ensure the biological origin of the spiking activity, experiments with TTX were performed as explained earlier. When TTX was added to one compartment (i.e., Compartment B), neuronal activity in that compartment was immediately suppressed without any significant impact on the spiking behavior of neuronal population in the other compartment (Figure 2). Neuronal spiking activities were recovered when TTX in the compartment was washed out with supplement enriched neurobasal medium. Recovery of activity from few electrodes after 1× wash cycle and from majority of the electrodes with 3× wash cycles from a sample experiment is as shown in Figure 2.
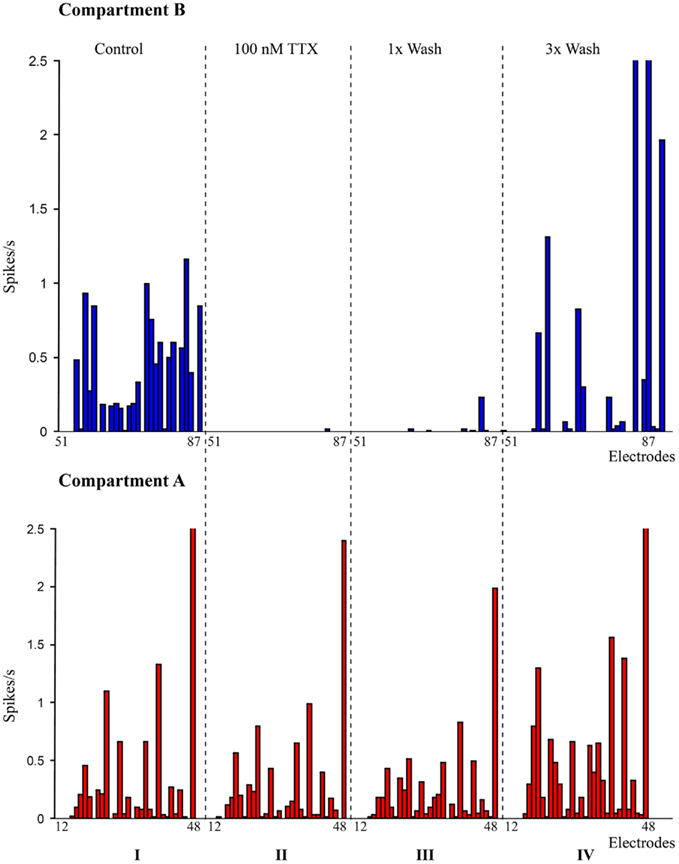
Figure 2. Pharmacological manipulation of spontaneous activity in the dual-compartment device. I: spike rate recorded at individual electrodes in compartment A (bottom – red) and compartment B (Top – blue). II: spike rate recorded with 100 nM TTX in compartment B (suppression of network activity in compartment B); III: spike rate after first wash cycle; IV: spike rates after three wash cycles.
Neurite Growth through Microchannels
Visual observation of the culture over the developmental period indicates neurite growth across the compartment from DIV 3 onward. Neurites were observed to cross-over to the adjacent compartment through the microchannels along the full length of the compartment. Phase contrast imaging of cell bodies isolated within a compartment and neurites crossing-over to the adjacent compartment confirmed the physical confinement obtained by means of the microchannel structure (Figure 3).
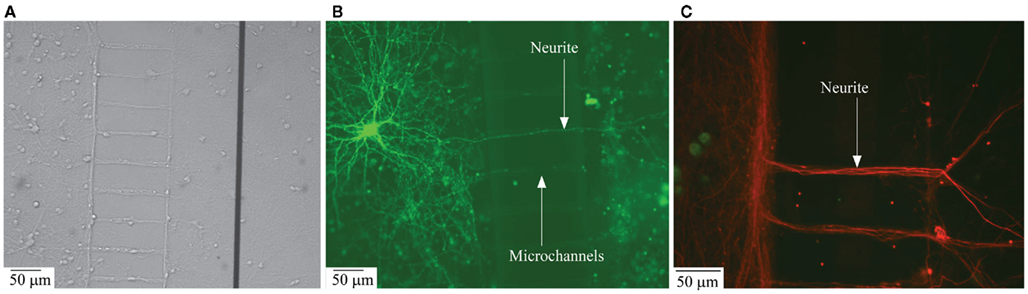
Figure 3. Structural connectivity between the two compartmentalized neuronal sub-populations. (A) Phase contrast image of neurite ladder structure intact after the removal of PDMS structures from the MEA surface; (B) Transfection image of a neurite grown across the microchannels connecting the compartments; (C) Immunofluorescence image of neurite structure following the microchannel placement.
Structural connectivity
Transfection and immunofluorescence imaging. To visualize neurite growth across the compartment, transfection of individual neurons with eGFP was performed on DIV 11. Cultures in one compartment were transfected as explained earlier resulting in staining about 1:1000 neurons. Figure 3B shows extensive neuritic arborization of a single neuron within the compartment of origin, as well as neurites crossing-over to the other compartment through the microchannels.
To further substantiate structural inter-compartment connectivity by neuritic crossover to the adjacent compartment, immunolabeling of cells in compartment was performed. Dissociated cells at concentrations similar to those in the experimental conditions were cultured in control cover-slips with PDMS. After DIV 14, the PDMS structure was removed carefully without damaging the neuritic crossover across the microchannels. As shown in the phase-contrast image in Figure 3A, a ladder like structure was retained even after the removal of PDMS, this confirms neuritic growth through the individual microchannels. Neurites accumulate along the walls of the compartment near the microchannels and were observed to propagate only through the channel openings into the adjacent compartment. Further, the confocal microscopic image in Figure 3C confirms the extensive neuritic cross-over to the adjacent compartment and structural connectivity between the compartments with neurite projections across the microchannels. From Figure 3, it may also be observed that the specific aspect ratio of the microchannels (i.e., width = 10 μm and height = 3 μm) prevents cell bodies from entering the microchannels and thereby confines them to the individual compartments.
Functional connectivity
Cross-correlation analysis. As in Figure 1C, synchronized bursting activity is present between the compartments suggesting the existence of coordination in the activity between the two cultured populations. However, in order to bolster the suitability of the device to study the interactions between different neuronal populations, not only structural connectivity but also functional connectivity between the two cultures was investigated. To assess this, we analyzed the CC between individual electrode spike trains. The correlation in the spontaneous activity of all electrodes with respect to a particular electrode was analyzed utilizing CC based methods (Garofalo et al., 2009). Figure 4A shows a sample cross-correlogram obtained during an experimental session, between a sample electrode in compartment A and all the electrodes in both compartments. Autocorrelogram is also included in this case and it corresponds to the maximum peak value. For each couple X → Y (where X = Electrode index 17), the correlograms depict the electrode index (y-axis) and the spike count (z-axis) divided by the time bin of 5 ms, in which electrode X and Y fire a spike with a precise time delay (x-axis). The correlograms when compared with surrogate peak trains in the other compartment showed a significant loss in correlation across the microchannels (Figure 4B).
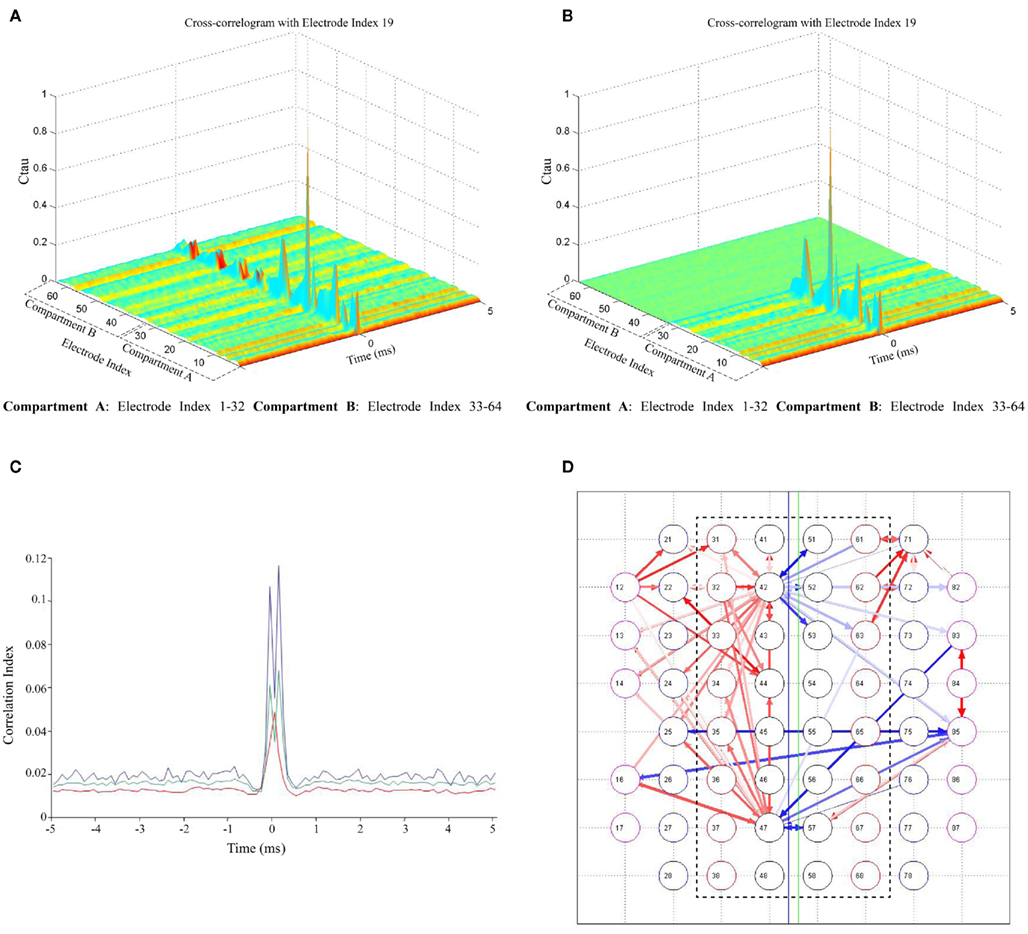
Figure 4. Cross-correlation of spontaneous activity in two compartments. (A) Cross-correlograms between a sample electrode in compartment A (Electrode index # 19) and all electrodes in both the compartments; (B) Cross-correlograms with surrogate peak trains in compartment B (between a sample electrode in compartment A (Electrode index # 19) and all electrodes in both the compartments); (C) Comparison of CC between global mean CC averaged over all 60 electrodes (red), mean CC of a sample electrode (Electrode index # 19) with compartment B (green) and CC of most correlated electrodes in both the compartments (Electrode index # 19in compartment A with a sample electrode [(Electrode index # 43) in compartment B (blue)]. Half-window size = 5 ms and Bin size (temporal resolution) = 0.1 ms; (D) Functional connectivity map showing the strongest 100 connections in the network. Red color arrows (intensity coded) represent the functional connections within a compartment; the blue color arrows (intensity coded) represent the functional connectivity between two compartments (inter-compartment connections). Inter-compartment connections are compared with surrogate spike trains and proven to be genuine (Connectivity map with surrogate spike train data not shown). The bidirectionality in network connectivity can be inferred from the direction of the arrows.
To better appreciate the differences in the cross-correlogram shape and strength, correlation functions obtained between a sample electrode (electrode index # 19)in compartment A and an electrode (electrode index # 43) in compartment B, in comparison to the correlation with all the electrodes in compartment B and correlation in the entire network averaged over all electrodes (global mean CC, introduced by Chiappalone et al., 2007) is shown in Figure 4C. The shape of the obtained function clearly shows the correlation between electrodes in the two compartments. Further, in addition to the correlation analysis, the functional connectivity in the network was estimated based on the techniques described by Garofalo et al. (2009). Figure 4D shows the connectivity map with strongest 200 connections in a network, red colored (intensity coded) for connections within a compartment with few connections between the two compartments (blue colored, intensity coded). The bidirectionality in network connectivity can be inferred from the direction of the arrows (Figure 4D). When the functional connectivity maps obtained using original data were compared with maps obtained using surrogate spike trains in the opposite compartment (with surrogate spike trains as explained earlier), it was observed that there were no connections between the compartments even with an increased threshold, thereby confirming the functional connectivity between the compartments (data not shown).
Statistical Analysis
The electrode columns were segregated into four regions as in Figure 5A. The 16 values obtained through the two-step statistical analysis previously described can be plotted as in Figure 5B. It may be noted that: (i) the intra-compartmental connectivity is higher than inter-compartmental connectivity (ii) intra compartment and inter-compartment connectivity are higher than the non-genuine (namely due to randomness) connectivity contribution obtained with surrogate spike trains and (iii) no clear differences can be observed among the different spatial regions (R1–R4). To assess the robustness of our observations, we analyzed 17 devices that contained cortical cells in both compartment plated with similar cell densities but from different DIV (from DIV 17 to DIV 32). Each device had at least 52 active electrodes (an electrode was considered to be active if the average spiking frequency was at least 0.2 Hz; Shahaf and Marom, 2001).
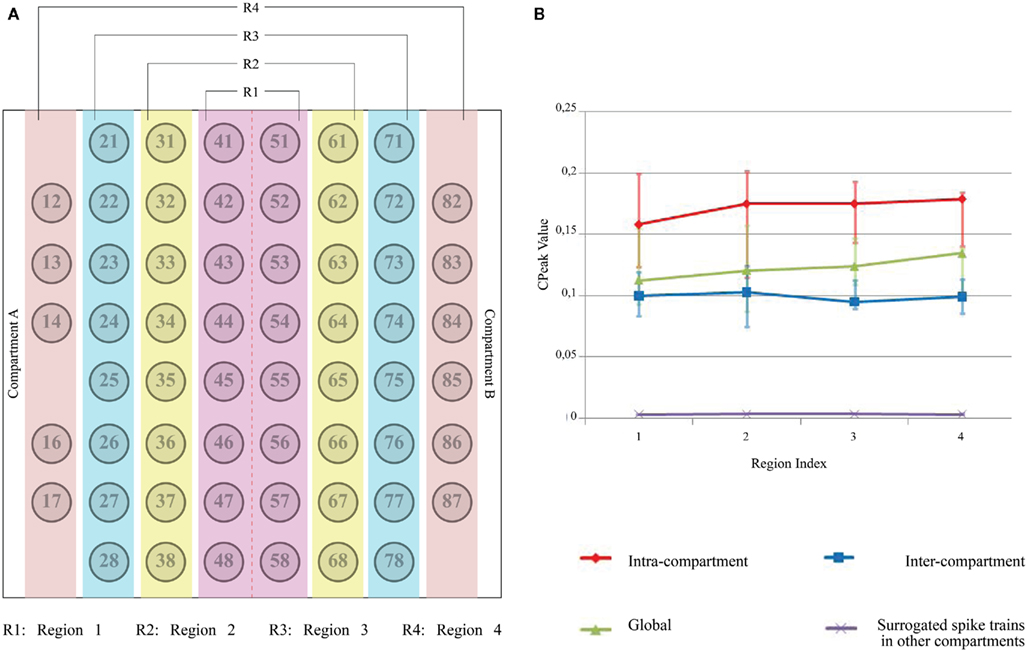
Figure 5. Two-step statistical analysis of an individual device. (A) MEA electrode layout and region separation across both the compartments (red color line between electrode column 4 and 5 represents the microchannel separation); (B) In the first step, “median” of Cpeak (t = 1 bin) for all the electrodes under four circumstances (i.e., Intra-compartment, Inter-compartment, Both compartments (Global) and Surrogated spike train in the other compartment) is computed and the electrodes are then segregated into separate regions as described earlier. In the second step, another statistical parameter (“median,” in this case) within each region is computed. Positive error bars are equal to the differences between the 75th percentile and the median, while negative error bars are equal to the differences between the median and the 25th percentile. The two percentiles were computed along with the “median” in the second step of the statistical analysis described in the text. Error bars for Surrogated spike trains in the other compartment are not visible because of their very low amplitude.
Box plots of correlation levels obtained from original spike trains in the same compartment (“Intra”) and the other compartment (“Inter”) as shown in Figure 6 in comparison to the surrogate spike train in the other compartment (“Inter_Shuffled”) reveal very clearly that a genuine inter-compartmental connectivity is present in the considered devices. A paired sign test on “Inter” and “Inter_Shuffled” of each region confirmed the existence of a significant difference between such values for each of the four identified regions of electrodes (p < 0.01). The above mentioned box plots do not show a clear fall off of the inter-compartmental connectivity with the distance from the barrier of microchannels. The Friedman’s test on the four groups of “Inter” values revealed that they are not statistically different from each other (p = 0.1843). This high degree of connectivity may be explained by the fact that cortical neurons usually develop long neurites.
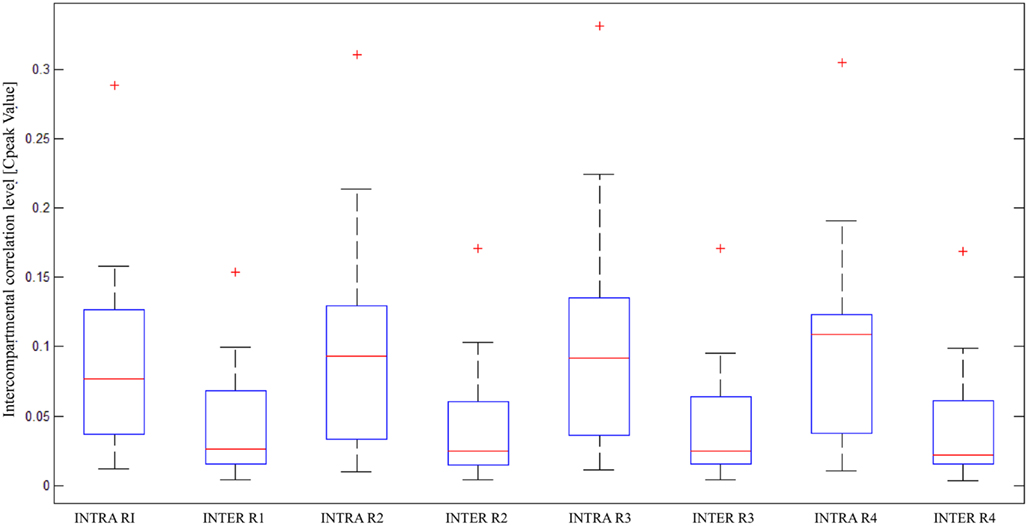
Figure 6. Statistical analysis of correlation on multiple devices; box plots of intra and inter-compartmental correlation levels, divided per region, in 17 devices (statistical parameters used in the two-step analysis: “median” for both the steps).
Discussion and Conclusion
A dual-compartment device was designed, fabricated and validated with microfluidic channels capable of physically segregating neuronal population and providing fluidic isolation between the compartments. The microfluidic channels also provided the necessary access path for neurites to cross-over to the adjacent compartment in either direction and connect the two neuronal populations across the barrier.
With the microchannels offering the necessary physical and fluidic isolation between the compartments due to their small aspect ratio (width = 10 μm and height = 3 μm), cells in the compartment were observed to be plated uniformly. We addressed the issue of low volume medium availability to the cell culture, by implementing a medium change protocol. Medium change of three times per week offered good culture stability and viable neurons for up to DIV 35. Inter-compartment neurite growth and connectivity were observed using transfection and fluorescence imaging.
From the functional point of view, experiments with TTX injected in one compartment showed the suppression of spontaneous activity in that particular compartment and the neuronal activity was recovered after three wash cycles, confirming the biological origin of network activity and fluidic isolation between the compartments. The results presented in this work show that the device supports healthy neuronal cultures with good fluidic and somatic separation. The microchannels provided an access path for the neurites to propagate to the adjacent compartment and to form a functional neuronal network. This configuration facilitates independent pharmacological manipulation of the two compartments as demonstrated with TTX compound. Simultaneous recording of spontaneous activity in both compartments showed strong network CC revealing functional connectivity between cells in the compartments. Use of surrogate data and statistical analysis of correlation reveal that the two neuronal populations are not only functionally connected within each compartment but also with each other. Moreover, the dependency of the inter-compartmental correlation was not statistically significant, suggesting that well connected networks are formed despite the presence of the physical barrier of microchannels. In a dual-compartment device, with functionally connected network between compartments, the neurons and the synaptic connections within a compartment can be individually manipulated with pharmacological compounds without influencing the other compartment. The presented dual-compartment devices could also be efficiently used for co-culturing different neuronal populations (e.g., thalamus–cortex; hippocampus–cortex) and studying interaction between the two segregated sub-networks in both spontaneous and pharmacologically manipulated conditions.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work is supported by a Point-One grant (Brain Mimic project) from the Dutch government, and by Philips Research Europe. The authors thank Dr. Mariateresa Tedesco (DIBE, University of Genoa, Italy) for her valuable assistance in immunocytochemistry and for providing images for Figures 3A,C and Frenk van Kan and Carlijn van Helvert (Life Sciences Facilities, MiPlaza Technologies) for their assistance in cell culture work.
References
Bani-Yaghoub, M., Tremblay, R., Voicu, R., Mealing, G., Monette, R., Py, C., Faid, K., and Sikorska, M. (2005). Neurogenesis and neuronal communication on micropatterned neurochips. Biotechnol. Bioeng. 92, 336–345.
Berdondini, L., Chippalone, M., Van Der Wal, P. D., Imfeld, K., De Rooij, N. F., Koudelka-Hep, M., Tedesco, M., Martinoia, S., Van Pelt, J., and Le Masson, G. (2006). A microelectrode array (MEA) integrated with clustering structures for investigating in vitro neurodynamics in confined interconnected sub-populations of neurons. Sen. Actuators B Chem. 114, 530–541.
Berdondini, L., Massobrio, P., Chiappalone, M., Tedesco, M., Imfeld, K., Maccione, A., Gandolfo, M., Koudelka-Hep, M., and Martinoia, S. (2009). Extracellular recordings from locally dense microelectrode arrays coupled to dissociated cortical cultures. J. Neurosci. Methods 177, 386–396.
Bodas, D., and Khan-Malek, C. (2007). Hydrophilization and hydrophobic recovery of PDMS by oxygen plasma and chemical treatment – An SEM investigation. Sens. Actuators B Chem. 123, 368–373.
Bologna, L., Pasquale, L., Bove, V., Vato, M., Tedesco, A. M., and Martinoia, S. (2010). Investigating neuronal activity by SPYCODE multi-channel data analyzer. Neural Netw. 23, 685–697.
Chao, Z. C., Bakkum, D. J., and Potter, S. M. (2008). Shaping embodied neural networks for adaptive goal-directed behavior. PLoS Comput. Biol. 4(3), e1000042. doi: 10.1371/journal.pcbi.1000042
Chiappalone, M., Bove, M., Vato, A., Tedesco, M., and Martinoia, S. (2006). Dissociated cortical networks show spontaneously correlated activity patterns during in vitro development. Brain Res. 1093, 41–53.
Chiappalone, M., Vato, A., Berdondini, L., Koudelka-Hep, M., and Martinoia, S. (2007). Network dynamics and synchronous activity in cultured cortical neurons. Int. J. Neural Syst. 17, 87–103.
Claverol-Tinture, E., and Pine, J. (2002). Extracellular potentials in low-density dissociated neuronal cultures. J. Neurosci. Methods 117, 13–21.
Dworak, B. J., and Wheeler, B. C. (2009). Novel MEA platform with PDMS microtunnels enables the detection of action potential propagation from isolated axons in culture. Lab Chip 9, 404–410.
Eytan, D., Brenner, N., and Marom, S. (2003). Selective adaptation in networks of cortical neurons. J. Neurosci. 23, 9349–9356.
Friston, K. J., Frith, C. D., Liddle, P. F., and Frackowiak, R. S. (1993). Functional connectivity – the principal-component analysis of large (pet) data sets. J. Cereb. Blood Flow Metab. 13, 5–14.
Garofalo, M., Nieus, T., Massobrio, P., and Martinoia, S. (2009). Evaluation of the performance of information theory-based methods and cross-correlation to estimate the functional connectivity in cortical networks. PLoS ONE 4, e6482. doi: 10.1371/journal.pone.0006482
James, C. D., Spence, A. J. H., Dowell-Mesfin, N. M., Hussain, R. J., Smith, K. L., Craighead, H. G., Isaacson, M. S., Shain, W., and Turner, J. N. (2004). Extracellular recordings from patterned neuronal networks using planar microelectrode arrays. IEEE Trans. Biomed. Eng. 51, 1640–1648.
Jimbo, Y., Kasai, N., Torimitsu, K., Tateno, T., and Robinson, H. P. (2003). A system for MEA-based multisite stimulation. IEEE Trans. Biomed. Eng. 50, 241–248.
Kanagasabapathi, T. T., Wang, K., Mellace, M., Ramakers, G. J., and Decre, M. M. (2009). Dual compartment neurofluidic system for electrophysiological measurements in physically isolated neuronal cell cultures. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2009, 1655–1658.
Knox, C. K. (1981). Detection of neuronal interactions using correlation-analysis. Trends Neurosci. 4, 222–225.
Maccione, A., Gandolfo, M., Massobrio, P., Novellino, A., Martinoia, S., and Chiappalone, M. (2009). A novel algorithm for precise identification of spikes in extracellularly recorded neuronal signals. J. Neurosci. Methods 177, 241–249.
Maher, M. P., Pine, J., Wright, J., and Tai, Y. C. (1999). The neurochip: a new multielectrode device for stimulating and recording from cultured neurons. J. Neurosci. Methods 87, 45–56.
Majumdar, D., Gao, Y. D., Li, D., and Webb, D. J. (2011). Co-culture of neurons and glia in a novel microfluidic platform. J. Neurosci. Methods 196, 38–44.
Martinoia, S., Bonzano, L., Chiappalone, M., and Tedesco, M. (2005). Electrophysiological activity modulation by chemical stimulation in networks of cortical neurons coupled to microelectrode arrays: a biosensor for neuropharmacological applications. Sens. Actuators B Chem. 108, 589–596.
McDonald, J. C., Duffy, D. C., Anderson, J. R., Chiu, D. T., Wu, H., Schueller, O. J., and Whitesides, G. M. (2000). Fabrication of microfluidic systems in poly(dimethylsiloxane). Electrophoresis 21, 27–40.
McDonald, J. C., and Whitesides, G. M. (2002). Poly(dimethylsiloxane) as a material for fabricating microfluidic devices. Acc. Chem. Res. 35, 491–499.
Melssen, W. J., and Epping, W. J. M. (1987). Detection and estimation of neural connectivity based on cross-correlation analysis. Biol. Cybern. 57, 403–414.
Morin, F., Nishimura, N., Griscom, L., Lepioufle, B., Fujita, H., Takamura, Y., and Tamiya, E. (2006). Constraining the connectivity of neuronal networks cultured on microelectrode arrays with microfluidic techniques: a step towards neuron-based functional chips. Biosens Bioelectron 21, 1093–1100.
Morin, F. O., Takamura, Y., and Tamiya, E. (2005). Investigating neuronal activity with planar microelectrode arrays: achievements and new perspectives. J. Biosci. Bioeng. 100, 131–143.
Mukai, Y., Shiina, T., Moriguchi, H., and Jimbo, Y. (2003). Continuous monitoring of developmental activity changes in cultured cortical networks. Electr. Eng. Jpn. 145, 28–37.
Nam, Y., Musick, K., and Wheeler, B. C. (2006). Application of a PDMS microstencil as a replaceable insulator toward a single-use planar microelectrode array. Biomed. Microdevices 8, 375–381.
Owen, M. J., and Smith, P. J. (1994). Plasma treatment of Polydimethylsiloxane. J. Adhes. Sci. Technol. 8, 1063–1075.
Ramakers, G. J. A., and Boer, G. J. (1991). Chronic suppression of bioelectric activity and cell-survival in primary cultures of rat cerebral-cortex – biochemical observations. Eur. J. Neurosci. 3, 154–161.
Ramakers, G. J. A., Corner, M. A., and Habets, A. M. (1990). Development in the absence of spontaneous bioelectric activity results in increased stereotyped burst firing in cultures of dissociated cerebral-cortex. Exp. Brain Res. 79, 157–166.
Ramakers, G. J. A., Raadsheer, F. C., Corner, M. A., Ramaekers, F. C., and Van Leeuwen, F. W. (1991). Development of neurons and glial-cells in cerebral-cortex, cultured in the presence or absence of bioelectric activity – morphological observations. Eur. J. Neurosci. 3, 140–153.
Ravula, S. K., McClain, M. A., Wang, M. S., Glass, J. D., and Frazier, A. B. (2006). A multielectrode microcompartment culture platform for studying signal transduction in the nervous system. Lab Chip 6, 1530–1536.
Sang Beom, J., Hynd, M. R., Dowell-Mesfin, N., Smith, K. L., Turner, J. N., Shain, W., and Kim, S. J. (2007). Low-density neuronal networks cultured using patterned poly-L-lysine on microelectrode arrays. J. Neurosci. Methods 160, 317–326.
Shahaf, G., and Marom, S. (2001). Learning in networks of cortical neurons. J. Neurosci. 21, 8782–8788.
Sorkin, R., Gabay, T., Blinder, P., Baranes, D., Ben-Jacob, E., and Hanein, Y. (2006). Compact self-wiring in cultured neural networks. J. Neural Eng. 3, 95–101.
Sprent, P., and Smeeton, N. (2001). Applied Nonparametric Statistical Methods: Three or More Samples, London: Chapman and Hall/CRC.
Suzuki, I., Sugio, Y., Jimbo, Y., and Yasuda, K. (2005). Stepwise pattern modification of neuronal network in photo-thermally-etched agarose architecture on multi-electrode array chip for individual-cell-based electrophysiological measurement. Lab. Chip 5, 241–247.
Suzuki, I., and Yasuda, K. (2007). Constructive formation and connection of aligned micropatterned neural networks by stepwise photothermal etching during cultivation. Jpn. J. Appl. Phys. 46, 6398–6403.
Taylor, A. M., Rhee, S. W., Tu, C. H., Cribbs, D. H., Cotman, C. W., and Jeon, N. L. (2003). Microfluidic multicompartment device for neuroscience research. Langmuir 19, 1551–1556.
Tooker, A., Meng, E., Erickson, J., Tai, Y. C., and Pine, J. (2005). Biocompatible parylene neurocages. IEEE Eng. Med. Biol. Mag. 24, 30–33.
Wagenaar, D. A., Pine, J., and Potter, S. M. (2006). An extremely rich repertoire of bursting patterns during the development of cortical cultures. BMC Neurosci. 7, 11. doi: 10.1186/1471-2202-7-11
Keywords: compartmented device, invitro model, neuron cell culture, functional connectivity, pharmacological manipulation
Citation: Kanagasabapathi TT, Ciliberti D, Martinoia S, Wadman WJ and Decré MMJ (2011) Dual-compartment neurofluidic system for electrophysiological measurements in physically segregated and functionally connected neuronal cell culture. Front. Neuroeng. 4:13. doi: 10.3389/fneng.2011.00013
Received: 30 July 2011; Paper pending published: 09 September 2011;
Accepted: 03 October 2011; Published online: 19 October 2011.
Edited by:
Jay Nadeau, McGill University, CanadaCopyright: © 2011 Kanagasabapathi, Ciliberti, Martinoia, Wadman and Decré. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Thirukumaran T. Kanagasabapathi, Minimally Invasive Healthcare Department, Philips Research Laboratories, High Tech Campus 34.2.023, 5656 AE Eindhoven, Netherlands. e-mail:dGhpcnUua2FuYWdhc2FiYXBhdGhpQHBoaWxpcHMuY29t