 Rune Enger
Rune Enger Kjell Heuser
Kjell Heuser- 1Letten Centre and GliaLab, Division of Anatomy, Department of Molecular Medicine, Institute of Basic Medical Sciences, University of Oslo, Oslo, Norway
- 2Department of Neurology, Oslo University Hospital, Rikshospitalet, Oslo, Norway
Spreading depolarizations (SD) are slow waves of complete depolarization of brain tissue followed by neuronal silencing that may play a role in seizure termination. Even though SD was first discovered in the context of epilepsy research, the link between SD and epileptic activity remains understudied. Both seizures and SD share fundamental pathophysiological features, and recent evidence highlights the frequent occurrence of SD in experimental seizure models. Human data on co-occurring seizures and SD are limited but suggestive. This mini-review addresses possible roles of SD during epileptiform activity, shedding light on SD as a potential mechanism for terminating epileptiform activity. A common denominator for many forms of epilepsy is reactive astrogliosis, a process characterized by morphological and functional changes to astrocytes. Data suggest that SD mechanisms are potentially perturbed in reactive astrogliosis and we propose that this may affect seizure pathophysiology.
Introduction
Epilepsy is one of the most common neurological disorders—estimated to affect 65 million people worldwide (Hesdorffer et al., 2011; Neligan et al., 2012; Beghi, 2016). It is a chronic disorder, characterized by sudden, violent perturbations of normal brain functions, accounting for much stigma, morbidity and in some cases death for the affected individuals. There is a striking lack of knowledge of the specific cellular mechanisms at play in epilepsy. For instance, the process transforming normal brain matter to a focus for epileptic seizures is elusive. Furthermore, key questions like “What cellular mechanisms set in motion an epileptic seizure?” and “What terminates seizure activity?” remain unanswered. Failure of the mechanisms that curb or prevent hyperexcitation have been increasingly recognized as probable key pathogenic factors of epilepsy.
Spreading depolarizations (SD) are slow waves of complete depolarization of gray matter followed by neuronal silencing, involved in a range of brain disorders. During SD profound changes occur in the brain tissue. For instance, transient hypoxia, cellular swelling, a temporary breakdown of the blood-brain barrier, as well as severely perturbed transmembrane ionic gradients are known to occur (Takano and Nedergaard, 2009; Charles and Baca, 2013). SD was first discovered in 1944 by Aristides Leão while studying seizure activity in rabbits. He discovered a wave-like phenomenon of silencing of brain function that occurred in the seizing brain (Leao, 1943). Since then a large body of literature addresses the role of SD in brain disorders (Dreier, 2011; Lauritzen et al., 2011; Ayata and Lauritzen, 2015). The phenomenon is believed to be the cellular substrate of the migraine aura, and in ischemia, brain trauma and subarachnoid hemorrhage, SD is believed to add insult to injury by increasing the metabolic demand of an already compromised tissue (Takano and Nedergaard, 2009; Dreier, 2011; Lauritzen et al., 2011; Charles and Baca, 2013; Hartings et al., 2020; Dreier et al., 2022). While the role of SD in for instance ischemia and migraine has been quite extensively explored, the role for SD in epilepsy and seizures is much less studied. Seizures and SD are both paroxysmal hyperexcitability phenomena that share elemental pathophysiological features (Rogawski, 2008; Dreier et al., 2012; Wei et al., 2014; Mantegazza and Cestèle, 2018). Importantly recent evidence clearly indicates that SD is a frequent occurrence in a range of acute and chronic experimental seizure models (Aiba and Noebels, 2015; Khoshkhoo et al., 2017; Heuser et al., 2018; Bahari et al., 2020; Tamim et al., 2021). Data from humans are scarce, although a few studies have demonstrated co-occurring seizures and SD following subarachnoid hemorrhage or brain trauma, by intracerebral electrodes (Fabricius et al., 2008; Hartings et al., 2011; Dreier et al., 2012). Even though SD and seizure activity is clearly linked, a surprisingly small number of articles directly address the role of SD in the seizing brain. In this mini review we will address the role of SD as a common denominator of migraine and epilepsy, and discuss SD as a mechanism capable of terminating epileptiform activity. Moreover, we will discuss potential pathophysiological roles of reactive astrocytes in SD in the context of seizure termination. Table 1 summarizes relevant studies and reviews about co-occurrence and shared pathophysiological mechanisms between SD and epileptic activity.
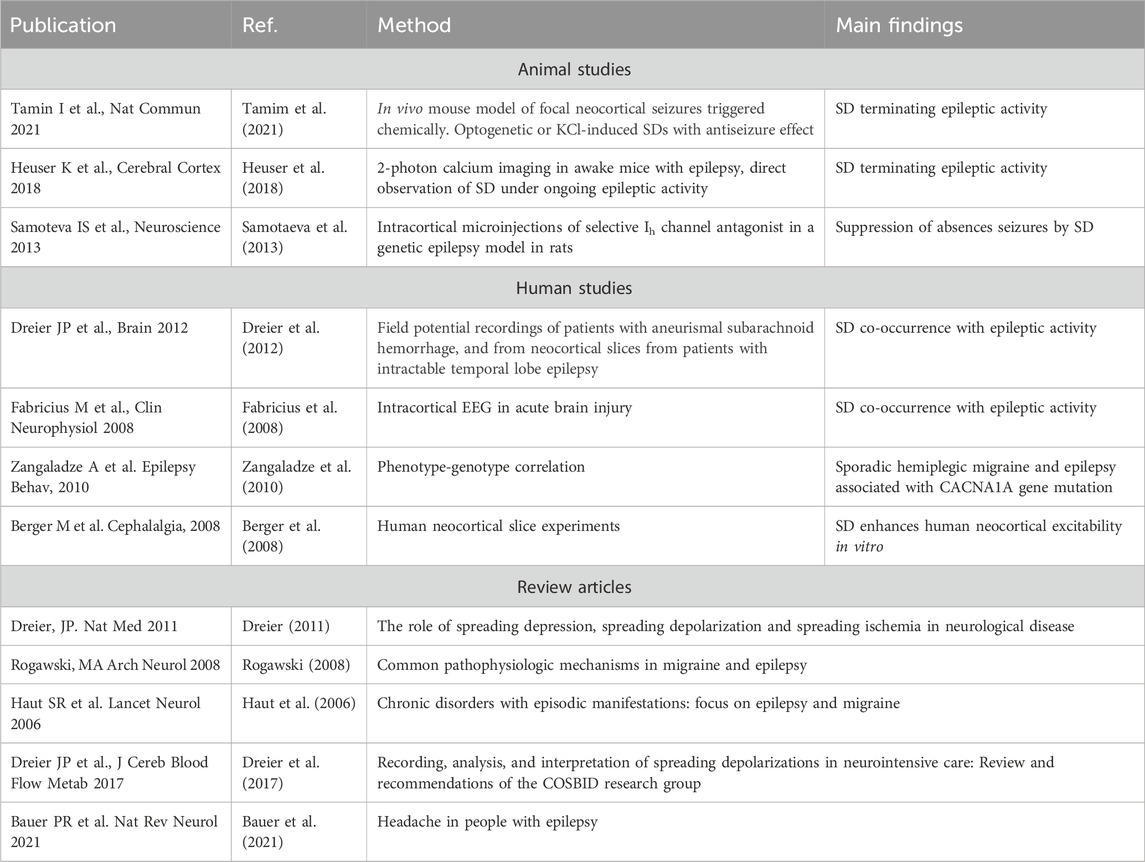
TABLE 1. Key literature on studies and reviews underpinning co-occurrence and shared pathophysiological mechanisms between spreading depolarization and epileptic activity.
Clinical overlap between migraine and epilepsy
The association between migraine and epilepsy was already recognized more than a century ago (Gowers, 1906; Noebels, 2012; Rogawski, 2012). Migraineurs exhibit a significantly elevated risk of epilepsy compared to non-migraineurs, mirroring the increased prevalence of migraine in populations with epilepsy (Ottman and Lipton, 1994; Haut et al., 2006; Bagheri et al., 2020). Both migraine and epilepsy manifest as chronic disorders with recurrent episodic attacks and interictal wellbeing (Ryan and Ptácek, 2010). In some instances, the attacks persist, leading to status epilepticus or status migrainosus, respectively. Both migraine attacks and epileptic seizures go through distinct phases, including prodromal and/or aura phases, the attack itself, and a postdromal or postictal phase.
While inducing a seizure through migraine is uncommon, headaches often accompany seizures. Although preictal and ictal headaches are infrequent, postictal headaches are prevalent and may exhibit migraine-like features (Silberstein et al., 2008). Both migraine and epilepsy may present with a range of symptoms involving visual, auditory, somatosensory, or motor features. In some cases, migraine aura may precede seizures, a phenomenon termed migralepsy (Lennox, 1960; Cianchetti et al., 2013; Headache Classification Committee of the International Headache Society IHS, 2018). While migraine boasts a broader array of triggering factors compared to epilepsy, numerous shared triggers exist, such as sleep deprivation, alcohol, bright light, stress or stress relief, and hormonal changes.
Strong support for a shared genetic basis emerges from the identification of genes implicated in both epilepsy and migraine. Phenotypic-genotypic correlations, particularly mutations in the genes CACNA1A (P/Q-type voltage-gated Ca2+ channel), ATP1A2 (Na+-K+ ATPase), SCN1A (voltage-gated Na+ channel), are observed in familial hemiplegic migraine, as well as in generalized and focal epilepsies (Chioza et al., 2001; Zangaladze et al., 2010).
Also in terms of treatment, a clear overlap exists between migraine and epilepsy. Robust evidence, including findings from randomized controlled clinical trials, supports the effectiveness of anti-seizure medications such as valproate, topiramate, gabapentin, pregabalin, levetiracetam, zonisamide, and lamotrigine as preventive drugs for migraine (Olesen and Ramadan, 2008; D’Amico, 2010; Calandre et al., 2010; Bermejo and Dorado, 2009; Villani et al., 2011; Lampl et al., 2005). Furthermore, case reports suggest that sumatriptan may be effective in treating postictal headaches (Jacob et al., 1996). The extensive overlap across clinical presentation, genetic predisposition and therapeutic response is suggestive for a common pathophysiological framework.
An elegant way to study the spatiotemporal co-occurrence of SD and epileptic activity is by intracranial electrocorticography (ECoG). In neurocritical care, this method may be an important future tool for prognostication and personalized treatment (Dreier et al., 2017). In human ECoG recordings, SD are easily distinguishable from ictal activity because the negative DC shift of SD is several times larger than the negative DC shift of an ictal event, and the propagation rate is usually much faster in the latter (Dreier et al., 2012). Both events are often co-occurring in acute brain conditions, including in patients with acute status epilepticus (Fabricius et al., 2008). However, SD in human epilepsy is understudied. Whether SD has a role in epileptogenesis, defined as the transformation of normal brain matter to one that is prone to generate epileptic activity, remains to be investigated. SD facilitates neuronal death, which is one key feature of epileptogenesis. Notably, early SD showed a significant association with the development of late epilepsy in patients with acute subarachnoid hemorrhage (Dreier et al., 2012). A recent small prospective study performed in patients with serious traumatic brain injury and malignant ischemic stroke found no association between SD events and epileptogenesis (Sueiras et al., 2021).
Spreading depolarization waves as local emergency brakes
The brain operates on a fine balance between excitation and inhibition, and potentially, only small relative reductions in inhibition or increases in excitation may cause synchronization of neural networks and resultant seizure activity (Isaacson and Scanziani, 2011). Generalized seizures, particularly in an evolutionary perspective, are detrimental as they render the organism incapable of reacting to external stimuli and evading predation, in addition to potential injuries from falling, or death. Hence, potent mechanisms should be in place to prevent seizure activity. Since SDs are known to co-occur with seizures, can be elicited by some of the same mechanisms as seizures, and strongly suppress neuronal activity for minutes, it is tempting to speculate that SD could be one such mechanism.
The exact mechanistic underpinnings underlying SD initiation and propagation are not known. However, it is believed that increases in extracellular K+ concentration, and potentially an interplay with extracellular glutamate, are key events, setting in motion a self-regenerating wave-like depolarization. Triggering of SD experimentally can be achieved in many ways, but likely a common denominator of all these methods is an increase of K+ above a threshold level of ∼15 mM in a sufficiently large extracellular volume (Tang et al., 2014; Wei et al., 2014), an increase of local extracellular glutamate (Parker et al., 2021), or an interplay between these two factors. For the propagation of SD, we have demonstrated that elevations in extracellular K+ seems to precede any other significant local cellular event by several seconds (Enger et al., 2015). This is in line with one of the earliest hypotheses about SD, namely that spread of extracellular K+ released by depolarized neurons serves to depolarize neighboring cells, leading to more release of K+ and a self-regenerating wave of depolarization (Grafstein, 1956). The exact mechanisms are, however, still unclear.
The changes associated with SD in the brain tissue are dramatic, and it has been clearly shown that SD waves are detrimental to already compromised brain tissue (Lauritzen et al., 2011). Hence, most studies on SD currently focus on the damaging effects of SD. However, the role of SD in migraine may suggest that given an uncompromised metabolic supply, SD may not be such a detrimental event (Hadjikhani et al., 2001; Lauritzen et al., 2011). After all, migraineurs may experience hundreds of auras throughout a lifetime with strikingly few realized long-term consequences. Our data, directly visualizing SD in cortex in awake behaving mice with two-photon microscopy (Enger et al., 2017) (Figure 1), corroborate this idea as we find that in the awake state with unperturbed physiological conditions, brain tissue regenerates from SD events considerably faster than what has been reported in anesthetized mice (Enger et al., 2017). SDs are relatively frequent occurrences in the population. For instance, the lifetime prevalence of migraine has been estimated to be ∼30%, of which about a quarter experience aura symptoms (Russell and Olesen, 1996; Kim et al., 2022). Recordings in humans suggest that SDs outside primary sensory areas are difficult to detect for the patient, or give subtle unusual symptoms pertaining to association cortices (Bowyer et al., 2001; Vincent and Hadjikhani, 2007; Hadjikhani and Vincent, 2021). Hence, it could be that SD are more frequent than what patients report in the form of symptomatic migraine auras (Hadjikhani and Vincent, 2021).
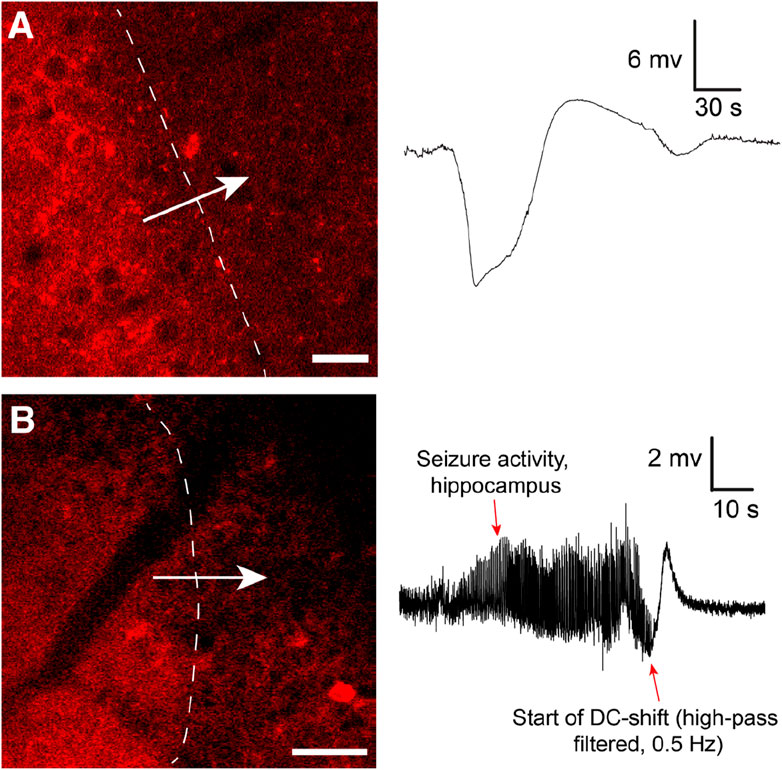
FIGURE 1. (A) Cortical spreading depression wave induced by KCl imaged by in vivo two-photon microscopy using the genetically encoded fluorescent sensor jRCaMP1a in unanesthetized mice. The sensor was expressed under the human synapsin1 promoter. In the awake unanesthetized state these waves move with a speed of 4.5–6 mm/min. Note typical shape of recorded DC shift. Adapted from Enger et al., 2017 Cerebral Cortex (Enger et al., 2017). Scale bar 25 μm. (B) Similar waves were imaged during Kainate-induced seizures in the hippocampus. In this experiment neuronal Ca2+ dynamics were imaged with jRGECO1a under the human synapsin1 promoter. To record seizure activity the hippocampal local field potential electrode signal had to be high-pass filtered to compensate for signal drift and saturation of signal. This distorts the classical DC shift, as also reported in (Dreier et al., 2017). Adapted from Heuser et al., 2018 Cerebral Cortex (Heuser et al., 2018). Scale bar 50 μm.
Little focus has been given to the potential beneficial roles of SD, and why this seemingly pathological event is so very well conserved through phylogeny (Ayata and Lauritzen, 2015). SD can be induced in a broad range of species, from grasshoppers and mudpuppies to man (Pietrobon and Moskowitz, 2014; Ayata and Lauritzen, 2015; Kramer et al., 2016). Because of this ability of the nervous tissue to sustain such activity, and its apparent prevalence at least in humans, it is tempting to speculate that SD may serve beneficial roles. In the aftermath of spreading depolarizations the neurons are silenced for several minutes, and potentially curbing brain hyperexcitation is a beneficial physiological role for SD. SDs may serve as an emergency brake that prevent localized hyperexcitation to transform into a full-blown seizure, and potentially SD plays a role in ending seizure activity in the brain.
Spreading depolarization silence neuronal activity during seizures
The spontaneous termination of seizures remains a fundamental question in the field of epileptology without a definitive answer (Lado and Moshé, 2008; Löscher and Köhling, 2010). As proposed by Kramer et al. brain seizure activity undergoes a critical transition when approaching termination, that can be observed in EEG (Kramer et al., 2012). Especially prolonged seizures, such as those seen in status epilepticus, repeatedly approach but do not surpass this critical transition (Kramer et al., 2012). Several mechanisms have been proposed to play a role in seizure termination, including increased GABAergic signaling, neurotransmitter or ATP depletion, ionic imbalance, or release of adenosine, which has an inhibitory effect on neurons (Lado and Moshé, 2008; Kramer et al., 2012). Could it be that SD in the seizure generating circuitry plays a role in seizure termination?
A phenomenon sharing both clinical and electrophysiological features with SD is postictal depression (PD). PD refers to the period of altered brain activity that follows a seizure (So and Blume, 2010). Electrophysiologically it is characterized by suppression and neuronal activity similar to what is observed in SD. PD is defined as abnormal slow-wave activity or EGG amplitudes of <10 µV within 30 s of seizure cessation, lasting more than 2 s (So and Blume, 2010; Bateman et al., 2019). It has been found in 84% of seizures and in 94% of epilepsy patients (Bateman et al., 2019). Key clinical features of PD are mood changes, fatigue, cognitive impairment, and also physical symptoms like headache and muscle aches, all phenomena also observed in migraine patients (Pottkämper et al., 2020). While the exact mechanisms of postictal depression are not understood, researchers have proposed several factors that may contribute (Bruno and Richardson, 2020; Pottkämper et al., 2020). Although PD often manifests as a generalized phenomenon, which is difficult to reconcile with the relatively localized nature of SD, SDs in key parts of the seizure circuitry, like the hippocampus, may play a role.
Few investigators have looked into the actual role of SD in seizure termination. In Heuser et al. 2018, we showed that kainate-induced seizure activity in the hippocampus appeared to be terminated by SD waves (Heuser et al., 2018) (Figure 1): After SD, no epileptic activity was recorded locally for several minutes, before seizure activity typically re-emerged. Similar observations were made in a study presenting a model for inducing seizures by optogenetically stimulating cortical interneurons, where they observed fluorescence waves characteristic of SD waves (Khoshkhoo et al., 2017). In Samotaeva et al. 2013 they observed that SD induced by cortical sham microinjections suppressed spike-wave discharges in WAG/Rij rats, a genetic model for absence epilepsy, for up to 90 min (Samotaeva et al., 2013). In another study in the same rat model that also displays audiogenic seizures, they found that audiogenic seizures associated with SD were associated with a suppression of spike-wave discharges for over 1 h (Vinogradova et al., 2005). Recently, in Tamim et al. 2021 they elegantly demonstrated that SD in addition to often occuring during seizure activity also played a prominent role in terminating seizure activity, and even prevented generalization of seizures (Tamim et al., 2021). They showed that focal seizures elicited by 4-aminopyridine, penicillin and bicuculline all triggered SD events, in particular if the seizure activity spread widely. Moreover, by inducing SD experimentally during seizure activity they were able to both prevent seizure generalization and terminate seizure activity (Tamim et al., 2021). In the same study they also showed that inhibiting SDs pharmacologically led to more severe seizure activity and a higher chance of generalization of seizure activity. In another study using Kv1.1 potassium channel knockout mice or mice with a knock-in mutation in the Scn1a gene known to cause Dravet Syndrome in humans, brainstem SD were found during induced seizure activity (Aiba and Noebels, 2015).
These studies were performed in acute seizure models, or in induced seizures in seizure susceptible animal models. Since seizures and SD can be triggered by many of the same agents, another question is to what extent SDs also occur in relation to spontaneous seizures in chronic seizure models. A recent preprint provides evidence suggesting that indeed SD is a hallmark of chronic epilepsy models as well (Bahari et al., 2020). They demonstrate that in a chronic tetanus toxin model of temporal lobe epilepsy in rats, and in a post cerebral malaria model of epilepsy around one-third of spontaneous seizures were associated with SD (Bahari et al., 2020). Data from humans demonstrating SDs co-occurring with seizures are scarce, likely because standard scalp EEG does not pick up SDs (Dreier et al., 2017). Moreover, intracerebral recordings are often slightly high pass-filtered obscuring the classical direct current shifts associated with SD (Dreier et al., 2017). A handful of studies have demonstrated co-occurring seizures and SD following subarachnoid hemorrhage or brain trauma by intracerebral electrodes (Fabricius et al., 2008; Hartings et al., 2011; Dreier et al., 2012). In these studies typically SDs are recorded in a subset of seizures. However, since SDs are relatively localized events, it could be that the reported rates are underestimating the true occurrence.
Complicating the interpretation of SD in curbing hyperexcitation is that experimental data suggest that neurons both locally and in other parts of the brain are hyperexcitable in the aftermath of a SD wave, even 45 min after the event (Wernsmann et al., 2006; Berger et al., 2008; Ghadiri et al., 2012). Potentially these findings could explain hyperexcitation symptoms in migraineurs like photophobia. In Bahari et al. 2020 they also reported that the interval between seizures for seizure bouts associated with SD was shorter than for seizures without SD (Bahari et al., 2020). However, that does not necessarily mean that SD promotes seizures: In Tamim et al. 2021, Samotaeva et al. 2013 and Vinogradova et al. 2005, a clear suppression in epileptiform activity for 30–90 min following SD was observed (Samotaeva et al., 2013).
Another intriguing aspect is that many pharmacological agents, including anti-seizure medication, have been shown to impede SD (Klass et al., 2018). However, these drugs also attenuate excitability in general and by this also target SD. The differential effects on pharmacological agents on epileptiform activity versus SD is to the best of our knowledge not investigated.
Possible role of spreading depolarization in preventing generalization of seizure activity
We have here hypothesized and presented data from reports suggesting that SD may be a mechanism for the brain to curb epileptiform activity. Could SD also play a role in preventing spread of localized spontaneously occurring interictal epileptiform activity before it spreads to larger neuronal networks and becomes a clinical seizure?
A range of brain lesions and brain disorders can trigger epilepsy (Balestrini et al., 2021). Examples include structural changes associated with stroke and traumatic brain injury. A common denominator of many of these insults is that they are associated with reactive astrogliosis (as reviewed in (Pekny and Pekna, 2016; Patel et al., 2019; Escartin et al., 2021)). Reactive astrocytes are characterized by morphological, expressional and functional changes, and can exist in mild reversible forms, or in the extreme case, as glial scar tissue (Escartin et al., 2021). There are multiple lines of evidence suggesting that reactive astrogliosis could be pro-epileptic, although the precise mechanisms are unknown (Robel et al., 2015; Zhu et al., 2016; Patel et al., 2019; Sano et al., 2019; Heuser et al., 2021; Çarçak et al., 2023). An illustrative example of reactive gliosis in epilepsy is mesial temporal lobe epilepsy with progressive gliotic transformation of the hippocampi (Bedner et al., 2015; Walker, 2015; Balestrini et al., 2021). This disease entity is relatively well characterized in terms of the histopathological changes in the tissue. Interestingly, investigations of resectates from patients undergoing surgery for MTLE have shown a range of molecular changes in reactive astrocytes, including loss of the potassium channel Kir4.1 (Heuser et al., 2012), changes in gap junction subtypes (Fonseca et al., 2002) and loss of gap junctional coupling (Bedner et al., 2015), loss of glutamine synthetase (Eid et al., 2004) and aquaporin-4 (Lee et al., 2004). In preclinical studies a range of other mechanisms, including astrocyte signaling has been implicated (Shigetomi et al., 2019; Heuser and Enger, 2021). The exact mechanisms by which the molecular and morphological changes in reactive astrocytes affect propensity to develop epileptiform activity is unknown. Likely some of the changes observed could also be protective, hindering hyperexcitation.
If one takes the perspective that SD may have beneficial effects in hyperexcitation disorders by acting as emergency brakes for excitation, one could also conjecture that a higher threshold for SD would be pro-epileptogenic. Astrocytes and astrocytic molecules are known to affect SD propagation and initiation (Seidel et al., 2016; Enger et al., 2017). Importantly, astrocytes are crucial for clearance of extracellular K+ and glutamate, as discussed above in the context of reactive astrogliosis, which are key mechanisms involved in SD initiation and propagation. One illustrative example is familial hemiplegic migraine caused by mutations in the ATP1A2 gene that encodes the predominant Na+-K+ ATPase in astrocytes (Gritz and Radcliffe, 2013).
Interestingly, recent work demonstrates that reactive astrogliosis increases the threshold for SD (Seidel et al., 2015; Seidel et al., 2016). Here they used lentiviral transfection of neurons so that they constitutively express ciliary neurotrophic factor, a cytokine that induces a reactive astrocyte phenotype (Escartin et al., 2006; Escartin et al., 2007). Using an in vitro model for SD by application of K+ on acute brain slices they observed a much higher threshold for eliciting SD in slices with reactive astrocytes, despite otherwise normal neuronal excitability. Potentially this effect was mediated through augmented astrocytic K+ uptake mechanisms in the reactive astrocytes. Another in vivo study clearly demonstrates that SD susceptibility is lower after repeated SD induction which leads to a reactive astrogliosis. However, they did not establish a causal relationship (Sukhotinsky et al., 2011). These are interesting observations, but more research is needed to establish whether attenuated SD susceptibility in reactive astrogliosis also occurs in epilepsy, and whether such mechanisms contribute to seizure propensity.
Conclusion
The brain operates on a fine balance between excitation and inhibition, and potent mechanisms of curbing hyperexcitation are essential for survival. Accruing evidence indicates that SDs frequently occur in seizures, and potentially have a role in curbing hyperexcitation. If SD has such a role, perturbed SD mechanisms in reactive astrogliosis could potentially contribute to seizure propensity in epilepsy. Further studies are needed to elucidate the role of SD in epilepsy, and to leverage this potential anti-seizure mechanism to treat epilepsy.
Author contributions
RE: Conceptualization, Investigation, Methodology, Visualization, Writing–review and editing. KH: Conceptualization, Investigation, Methodology, Visualization, Writing–review and editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aiba, I., and Noebels, J. L. (2015). Spreading depolarization in the brainstem mediates sudden cardiorespiratory arrest in mouse SUDEP models. Sci. Transl. Med. 7, 282ra46. doi:10.1126/scitranslmed.aaa4050
Ayata, C., and Lauritzen, M. (2015). Spreading depression, spreading depolarizations, and the cerebral vasculature. Physiol. Rev. 95, 953–993. doi:10.1152/physrev.00027.2014
Bagheri, M. H., Jalli, R., and Hoseyni Moghadam, A. (2020). New MRI finding in migraineurs: mesial temporal sclerosis. J. Biomed. Phys. Eng. 10, 459–466. doi:10.31661/jbpe.v0i0.887
Bahari, F., Ssentongo, P., Liu, J., Kimbugwe, J., Curay, C., Schiff, S. J., et al. (2020). Seizure-associated spreading depression is a major feature of ictal events in two animal models of chronic epilepsy. bioRxiv, 455519. doi:10.1101/455519
Balestrini, S., Arzimanoglou, A., Blümcke, I., Scheffer, I. E., Wiebe, S., Zelano, J., et al. (2021). The aetiologies of epilepsy. Epileptic Disord. 23, 1–16. doi:10.1684/epd.2021.1255
Bauer, P. R., Tolner, E. A., Keezer, M. R., Ferrari, M. D., and Sander, J. W. (2021). Headache in people with epilepsy. Nat. Rev. Neurol. 17, 529–544. doi:10.1038/s41582-021-00516-6
Bateman, L. M., Mendiratta, A., Liou, J. Y., Smith, E. J., Bazil, C. W., Choi, H., et al. (2019). Postictal clinical and electroencephalographic activity following intracranially recorded bilateral tonic-clonic seizures. Epilepsia 60, 74–84. doi:10.1111/epi.14621
Bedner, P., Dupper, A., Hüttmann, K., Müller, J., Herde, M. K., Dublin, P., et al. (2015). Astrocyte uncoupling as a cause of human temporal lobe epilepsy. Brain 138, 1208–1222. doi:10.1093/brain/awv067
Beghi, E. (2016). Addressing the burden of epilepsy: many unmet needs. Pharmacol. Res. 107, 107 79–84. Preprint at. doi:10.1016/j.phrs.2016.03.003
Berger, M., Speckmann, E.-J., Pape, H. C., and Gorji, A. (2008). Spreading depression enhances human neocortical excitability in vitro. Cephalalgia 28, 558–562. doi:10.1111/j.1468-2982.2008.01556.x
Bermejo, P. E., and Dorado, R. (2009). Zonisamide for migraine prophylaxis in patients refractory to topiramate. Clin. Neuropharmacol. 32, 103–106. doi:10.1097/WNF.0B013E318170577F
Bowyer, S. M., Aurora, K. S., Moran, J. E., Tepley, N., and Welch, K. M. (2001). Magnetoencephalographic fields from patients with spontaneous and induced migraine aura. Ann. Neurol. 50, 582–587. doi:10.1002/ana.1293
Bruno, E., and Richardson, M. P. (2020). Postictal generalized EEG suppression and postictal immobility: what do we know? Epileptic Disord. 22, 245–251. doi:10.1684/epd.2020.1158
Calandre, E. P., Garcia-Leiva, J. M., Rico-Villademoros, F., Vilchez, J. S., and Rodriguez-Lopez, C. M. (2010). Pregabalin in the treatment of chronic migraine: an open-label study. Clin. Neuropharmacol. 33, 35–39. doi:10.1097/WNF.0b013e3181bf1dbe
Çarçak, N., Onat, F., and Sitnikova, E. (2023). Astrocytes as a target for therapeutic strategies in epilepsy: current insights. Front. Mol. Neurosci. 16, 1183775. doi:10.3389/fnmol.2023.1183775
Charles, A. C., and Baca, S. M. (2013). Cortical spreading depression and migraine. Nat. Rev. Neurol. 9, 637–644. doi:10.1038/nrneurol.2013.192
Chioza, B., Wilkie, H., Nashef, L., Blower, J., McCormick, D., Sham, P., et al. (2001). Association between the alpha(1a) calcium channel gene CACNA1A and idiopathic generalized epilepsy. Neurology 56, 1245–1246. doi:10.1212/wnl.56.9.1245
Cianchetti, C., Pruna, D., and Ledda, M. (2013). Epileptic seizures and headache/migraine: a review of types of association and terminology. Seizure 22, 679–685. doi:10.1016/j.seizure.2013.05.017
D’Amico, D. (2010). Pharmacological prophylaxis of chronic migraine: a review of double-blind placebo-controlled trials. Neurol. Sci. 31 (1), S23–S28. doi:10.1007/s10072-010-0268-7
Dreier, J. P. (2011). The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat. Med. 17, 439–447. doi:10.1038/nm.2333
Dreier, J. P., Fabricius, M., Ayata, C., Sakowitz, O. W., Shuttleworth, C. W., Dohmen, C., et al. (2017). Recording, analysis, and interpretation of spreading depolarizations in neurointensive care: review and recommendations of the COSBID research group. J. Cereb. Blood Flow. Metab. 37, 1595–1625. doi:10.1177/0271678X16654496
Dreier, J. P., Major, S., Pannek, H. W., Woitzik, J., Scheel, M., Wiesenthal, D., et al. (2012). Spreading convulsions, spreading depolarization and epileptogenesis in human cerebral cortex. Brain 135, 259–275. doi:10.1093/brain/awr303
Dreier, J. P., Winkler, M. K. L., Major, S., Horst, V., Lublinsky, S., Kola, V., et al. (2022). Spreading depolarizations in ischaemia after subarachnoid haemorrhage, a diagnostic phase III study. Brain 145, 1264–1284. doi:10.1093/brain/awab457
Eid, T., Thomas, M. J., Spencer, D. D., Rundén-Pran, E., Lai, J. C. K., Malthankar, G. V., et al. (2004). Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet 363, 28–37. doi:10.1016/s0140-6736(03)15166-5
Enger, R., Dukefoss, D. B., Tang, W., Pettersen, K. H., Bjørnstad, D. M., Helm, P. J., et al. (2017). Deletion of aquaporin-4 curtails extracellular glutamate elevation in cortical spreading depression in awake mice. Cereb. Cortex 27, 24–33. doi:10.1093/cercor/bhw359
Enger, R., Tang, W., Vindedal, G. F., Jensen, V., Johannes Helm, P., Sprengel, R., et al. (2015). Dynamics of ionic shifts in cortical spreading depression. Cereb. Cortex 25, 4469–4476. doi:10.1093/cercor/bhv054
Escartin, C., Brouillet, E., Gubellini, P., Trioulier, Y., Jacquard, C., Smadja, C., et al. (2006). Ciliary neurotrophic factor activates astrocytes, redistributes their glutamate transporters GLAST and GLT-1 to raft microdomains, and improves glutamate handling in vivo. J. Neurosci. 26, 5978–5989. doi:10.1523/JNEUROSCI.0302-06.2006
Escartin, C., Galea, E., Lakatos, A., O'Callaghan, J. P., Petzold, G. C., Serrano-Pozo, A., et al. (2021). Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 24, 312–325. doi:10.1038/s41593-020-00783-4
Escartin, C., Pierre, K., Colin, A., Brouillet, E., Delzescaux, T., Guillermier, M., et al. (2007). Activation of astrocytes by CNTF induces metabolic plasticity and increases resistance to metabolic insults. J. Neurosci. 27, 7094–7104. doi:10.1523/JNEUROSCI.0174-07.2007
Fabricius, M., Fuhr, S., Willumsen, L., Dreier, J. P., Bhatia, R., Boutelle, M. G., et al. (2008). Association of seizures with cortical spreading depression and peri-infarct depolarisations in the acutely injured human brain. Clin. Neurophysiol. 119, 1973–1984. doi:10.1016/j.clinph.2008.05.025
Fonseca, C. G., Green, C. R., and Nicholson, L. F. B. (2002). Upregulation in astrocytic connexin 43 gap junction levels may exacerbate generalized seizures in mesial temporal lobe epilepsy. Brain Res. 929, 105–116. doi:10.1016/s0006-8993(01)03289-9
Ghadiri, M. K., Kozian, M., Ghaffarian, N., Stummer, W., Kazemi, H., Speckmann, E. J., et al. (2012). Sequential changes in neuronal activity in single neocortical neurons after spreading depression. Cephalalgia 32, 116–124. doi:10.1177/0333102411431308
Gowers, W. R. (1906). Clinical lectures on the borderland of epilepsy: vertigo: delivered at the national hospital for the paralysed and epileptic. Br. Med. J. 2 (2377), 128–31. doi:10.1136/bmj.2.2377.128
Grafstein, B. (1956). Mechanism of spreading cortical depression. J. Neurophysiol. 19, 154–171. doi:10.1152/jn.1956.19.2.154
Gritz, S. M., and Radcliffe, R. A. (2013). Genetic effects of ATP1A2 in familial hemiplegic migraine type II and animal models. Hum. Genomics 7, 8. doi:10.1186/1479-7364-7-8
Hadjikhani, N., Sanchez Del Rio, M., Wu, O., Schwartz, D., Bakker, D., Fischl, B., et al. (2001). Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc. Natl. Acad. Sci. U. S. A. 98, 4687–4692. doi:10.1073/pnas.071582498
Hadjikhani, N., and Vincent, M. (2021). Can you have a migraine aura without knowing it? Curr. Opin. Neurol. 34, 350–355. doi:10.1097/WCO.0000000000000924
Hartings, J. A., Andaluz, N., Bullock, M. R., Hinzman, J. M., Mathern, B., Pahl, C., et al. (2020). Prognostic value of spreading depolarizations in patients with severe traumatic brain injury. JAMA Neurol. 77, 489–499. doi:10.1001/jamaneurol.2019.4476
Hartings, J. A., Bullock, M. R., Okonkwo, D. O., Murray, L. S., Murray, G. D., Fabricius, M., et al. (2011). Spreading depolarisations and outcome after traumatic brain injury: a prospective observational study. Lancet Neurol. 10, 1058–1064. doi:10.1016/S1474-4422(11)70243-5
Haut, S. R., Bigal, M. E., and Lipton, R. B. (2006). Chronic disorders with episodic manifestations: focus on epilepsy and migraine. Lancet Neurol. 5, 148–157. doi:10.1016/S1474-4422(06)70348-9
Headache Classification Committee of the International Headache Society (IHS) (2018). Headache classification committee of the international headache society (IHS) the international classification of headache disorders, 3rd edition. Cephalalgia 38, 1–211. doi:10.1177/0333102417738202
Hesdorffer, D. C., Logroscino, G., Benn, E. K. T., Katri, N., Cascino, G., and Hauser, W. A. (2011). Estimating risk for developing epilepsy: a population-based study in Rochester, Minnesota. Neurology 76, 76 23–27. Preprint at. doi:10.1212/wnl.0b013e318204a36a
Heuser, K., de Curtis, M., and Steinhäuser, C. (2021). Editorial: glial dysfunction in epileptogenesis. Front. Neurol. 12, 716308. doi:10.3389/fneur.2021.716308
Heuser, K., Eid, T., Lauritzen, F., Thoren, A. E., Vindedal, G. F., Taubøll, E., et al. (2012). Loss of perivascular Kir4.1 potassium channels in the sclerotic hippocampus of patients with mesial temporal lobe epilepsy. J. Neuropathol. Exp. Neurol. 71, 814–825. doi:10.1097/NEN.0b013e318267b5af
Heuser, K., and Enger, R. (2021). Astrocytic Ca2+ signaling in epilepsy. Front. Cell. Neurosci. 15, 695380. doi:10.3389/fncel.2021.695380
Heuser, K., Nome, C. G., Pettersen, K. H., Åbjørsbråten, K. S., Jensen, V., Tang, W., et al. (2018). Ca2+ signals in astrocytes facilitate spread of epileptiform activity. Cereb. Cortex 28, 4036–4048. doi:10.1093/cercor/bhy196
Isaacson, J. S., and Scanziani, M. (2011). How inhibition shapes cortical activity. Neuron 72, 231–243. doi:10.1016/j.neuron.2011.09.027
Jacob, J., Goadsby, P. J., and Duncan, J. S. (1996). Use of sumatriptan in post-ictal migraine headache. Neurology 47, 1104. doi:10.1212/wnl.47.4.1104
Khoshkhoo, S., Vogt, D., and Sohal, V. S. (2017). Dynamic, cell-type-specific roles for GABAergic interneurons in a mouse model of optogenetically inducible seizures. Neuron 93, 291–298. doi:10.1016/j.neuron.2016.11.043
Kim, K. M., Kim, B. K., Lee, W., Hwang, H., Heo, K., and Chu, M. K. (2022). Prevalence and impact of visual aura in migraine and probable migraine: a population study. Sci. Rep. 12, 426. doi:10.1038/s41598-021-04250-3
Klass, A., Sánchez-Porras, R., and Santos, E. (2018). Systematic review of the pharmacological agents that have been tested against spreading depolarizations. J. Cereb. Blood Flow. Metab. 38, 1149–1179. doi:10.1177/0271678X18771440
Kramer, D. R., Fujii, T., Ohiorhenuan, I., and Liu, C. Y. (2016). Cortical spreading depolarization: pathophysiology, implications, and future directions. J. Clin. Neurosci. 24, 22–27. doi:10.1016/j.jocn.2015.08.004
Kramer, M. A., Truccolo, W., Eden, U. T., Lepage, K. Q., Hochberg, L. R., Eskandar, E. N., et al. (2012). Human seizures self-terminate across spatial scales via a critical transition. Proc. Natl. Acad. Sci. U. S. A. 109, 21116–21121. doi:10.1073/pnas.1210047110
Lado, F. A., and Moshé, S. L. (2008). How do seizures stop? Epilepsia 49, 1651–1664. doi:10.1111/j.1528-1167.2008.01669.x
Lampl, C., Katsarava, Z., Diener, H.-C., and Limmroth, V. (2005). Lamotrigine reduces migraine aura and migraine attacks in patients with migraine with aura. J. Neurol. Neurosurg. Psychiatry 76, 1730–1732. doi:10.1136/jnnp.2005.063750
Lauritzen, M., Dreier, J. P., Fabricius, M., Hartings, J. A., Graf, R., and Strong, A. J. (2011). Clinical relevance of cortical spreading depression in neurological disorders: migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J. Cereb. Blood Flow. Metab. 31, 17–35. doi:10.1038/jcbfm.2010.191
Lee, T. S., Eid, T., Mane, S., Kim, J. H., Spencer, D. D., Ottersen, O. P., et al. (2004). Aquaporin-4 is increased in the sclerotic hippocampus in human temporal lobe epilepsy. Acta Neuropathol. 108, 493–502. doi:10.1007/s00401-004-0910-7
Löscher, W., and Köhling, R. (2010). Functional, metabolic, and synaptic changes after seizures as potential targets for antiepileptic therapy. Epilepsy Behav. 19, 105–113. doi:10.1016/j.yebeh.2010.06.035
Mantegazza, M., and Cestèle, S. (2018). Pathophysiological mechanisms of migraine and epilepsy: similarities and differences. Neurosci. Lett. 667, 92–102. doi:10.1016/j.neulet.2017.11.025
Neligan, A., Hauser, W. A., and Sander, J. W. (2012). The epidemiology of the epilepsies. Handb. Clin. Neurology 107, 113–133. Preprint at. doi:10.1016/b978-0-444-52898-8.00006-9
Olesen, J., and Ramadan, N. (2008). Innovative drug development for headache disorders. Frontiers in Headache Research Series (Oxford, 2008). doi:10.1093/med/9780199552764.001.0001
Ottman, R., and Lipton, R. B. (1994). Comorbidity of migraine and epilepsy. Neurology 44, 2105–2110. doi:10.1212/wnl.44.11.2105
Parker, P. D., Suryavanshi, P., Melone, M., Sawant-Pokam, P. A., Reinhart, K. M., Kaufmann, D., et al. (2021). Non-canonical glutamate signaling in a genetic model of migraine with aura. Neuron 109, 611–628.e8. doi:10.1016/j.neuron.2020.11.018
Patel, D. C., Tewari, B. P., Chaunsali, L., and Sontheimer, H. (2019). Neuron-glia interactions in the pathophysiology of epilepsy. Nat. Rev. Neurosci. 20, 282–297. doi:10.1038/s41583-019-0126-4
Pekny, M., and Pekna, M. (2016). Reactive gliosis in the pathogenesis of CNS diseases. Biochim. Biophys. Acta 1862, 483–491. doi:10.1016/j.bbadis.2015.11.014
Pietrobon, D., and Moskowitz, M. A. (2014). Chaos and commotion in the wake of cortical spreading depression and spreading depolarizations. Nat. Rev. Neurosci. 15, 379–393. doi:10.1038/nrn3770
Pottkämper, J. C. M., Hofmeijer, J., van Waarde, J. A., and van Putten, M. J. A. M. (2020). The postictal state - what do we know? Epilepsia 61, 1045–1061. doi:10.1111/epi.16519
Robel, S., Buckingham, S. C., Boni, J. L., Campbell, S. L., Danbolt, N. C., Riedemann, T., et al. (2015). Reactive astrogliosis causes the development of spontaneous seizures. J. Neurosci. 35, 3330–3345. doi:10.1523/JNEUROSCI.1574-14.2015
Rogawski, M. A. (2008). Common pathophysiologic mechanisms in migraine and epilepsy. Arch. Neurol. 65, 709–714. doi:10.1001/archneur.65.6.709
Rogawski, M. A. (2012). “Migraine and epilepsy—shared mechanisms within the family of episodic disorders,” in Jasper’s basic mechanisms of the epilepsies [Internet]. 4th ed. Bethesda (MD). Editors J. L. Noebels, M. Avoli, M. A. Rogawski, R. W. Olsen, and A. V. Delgado-Escueta (United States: National Center for Biotechnology Information).
Russell, M. B., and Olesen, J. (1996). A nosographic analysis of the migraine aura in a general population. Brain 119 (2), 355–361. doi:10.1093/brain/119.2.355
Ryan, D. P., and Ptácek, L. J. (2010). Episodic neurological channelopathies. Neuron 68, 282–292. doi:10.1016/j.neuron.2010.10.008
Samotaeva, I. S., Tillmanns, N., van Luijtelaar, G., and Vinogradova, L. V. (2013). Intracortical microinjections may cause spreading depression and suppress absence seizures. Neuroscience 230, 50–55. doi:10.1016/j.neuroscience.2012.11.013
Sano, F., Shigetomi, E., Shinozaki, Y., Tsuzukiyama, H., Saito, K., Mikoshiba, K., et al. (2019). Reactive astrocyte-driven epileptogenesis is induced by microglia initially activated following status epilepticus. bioRxiv. Preprint at. doi:10.1101/806398
Seidel, J. L., Escartin, C., Ayata, C., Bonvento, G., and Shuttleworth, C. W. (2016). Multifaceted roles for astrocytes in spreading depolarization: a target for limiting spreading depolarization in acute brain injury? Glia 64, 5–20. doi:10.1002/glia.22824
Seidel, J. L., Faideau, M., Aiba, I., Pannasch, U., Escartin, C., Rouach, N., et al. (2015). Ciliary neurotrophic factor (CNTF) activation of astrocytes decreases spreading depolarization susceptibility and increases potassium clearance. Glia 63, 91–103. doi:10.1002/glia.22735
Shigetomi, E., Saito, K., Sano, F., and Koizumi, S. (2019). Aberrant calcium signals in reactive astrocytes: a key process in neurological disorders. Int. J. Mol. Sci. 20, 996. doi:10.3390/ijms20040996
Silberstein, S. D., Lipton, R. B., and Haut, S. (2008). “Migraine,” in Epilepsy: a comprehensive textbook. Editor P. T. A. Engel 2nd ed. (Wolters Kluwer Health/Lippincott Williams & Wilkins.), 2733–2743.
So, N. K., and Blume, W. T. (2010). The postictal EEG. Epilepsy Behav. 19, 121–126. doi:10.1016/j.yebeh.2010.06.033
Sueiras, M., Thonon, V., Santamarina, E., Sánchez-Guerrero, Á., Riveiro, M., Poca, M. A., et al. (2021). Is spreading depolarization a risk factor for late epilepsy? A prospective study in patients with traumatic brain injury and malignant ischemic stroke undergoing decompressive craniectomy. Neurocrit. Care 34, 876–888. doi:10.1007/s12028-020-01107-x
Sukhotinsky, I., Dilekoz, E., Wang, Y., Qin, T., Eikermann-Haerter, K., Waeber, C., et al. (2011). Chronic daily cortical spreading depressions suppress spreading depression susceptibility. Cephalalgia 31, 1601–1608. doi:10.1177/0333102411425865
Takano, T., and Nedergaard, M. (2009). Deciphering migraine. J. Clin. investigation 119, 119 16–19. doi:10.1172/JCI38051
Tamim, I., Chung, D. Y., de Morais, A. L., Loonen, I. C. M., Qin, T., Misra, A., et al. (2021). Spreading depression as an innate antiseizure mechanism. Nat. Commun. 12, 2206. doi:10.1038/s41467-021-22464-x
Tang, Y. T., Mendez, J. M., Theriot, J. J., Sawant, P. M., López-Valdés, H. E., Ju, Y. S., et al. (2014). Minimum conditions for the induction of cortical spreading depression in brain slices. J. Neurophysiol. 112, 2572–2579. doi:10.1152/jn.00205.2014
Villani, V., Ciuffoli, A., Prosperini, L., and Sette, G. (2011). Zonisamide for migraine prophylaxis in topiramate-intolerant patients: an observational study. Headache 51, 287–291. doi:10.1111/j.1526-4610.2010.01842.x
Vincent, M. B., and Hadjikhani, N. (2007). Migraine aura and related phenomena: beyond scotomata and scintillations. Cephalalgia 27, 1368–1377. doi:10.1111/j.1468-2982.2007.01388.x
Vinogradova, L. V., Kuznetsova, G. D., and Coenen, A. M. L. (2005). Audiogenic seizures associated with a cortical spreading depression wave suppress spike-wave discharges in rats. Physiol. Behav. 86, 554–558. doi:10.1016/j.physbeh.2005.08.017
Walker, M. C. (2015). Hippocampal sclerosis: causes and prevention. Semin. Neurol. 35, 193–200. doi:10.1055/s-0035-1552618
Wei, Y., Ullah, G., and Schiff, S. J. (2014). Unification of neuronal spikes, seizures, and spreading depression. J. Neurosci. 34, 11733–11743. doi:10.1523/JNEUROSCI.0516-14.2014
Wernsmann, B., Pape, H.-C., Speckmann, E.-J., and Gorji, A. (2006). Effect of cortical spreading depression on synaptic transmission of rat hippocampal tissues. Eur. J. Neurosci. 23, 1103–1110. doi:10.1111/j.1460-9568.2006.04643.x
Zangaladze, A., Asadi-Pooya, A. A., Ashkenazi, A., and Sperling, M. R. (2010). Sporadic hemiplegic migraine and epilepsy associated with CACNA1A gene mutation. Epilepsy Behav. 17, 293–295. doi:10.1016/j.yebeh.2009.12.017
Keywords: astrocyte, spreading depression, spreading depolarization, epilepsy, migraine, seizure termination
Citation: Enger R and Heuser K (2024) Astrocytes as critical players of the fine balance between inhibition and excitation in the brain: spreading depolarization as a mechanism to curb epileptic activity. Front. Netw. Physiol. 4:1360297. doi: 10.3389/fnetp.2024.1360297
Received: 22 December 2023; Accepted: 25 January 2024;
Published: 09 February 2024.
Edited by:
Kerstin Lenk, Graz University of Technology, AustriaReviewed by:
Mani Ratnesh Singh Sandhu, The University of Iowa, United StatesCopyright © 2024 Enger and Heuser. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kjell Heuser, a2hldXNlckBvdXMtaGYubm8=; Rune Enger, cnVuZS5lbmdlckBtZWRpc2luLnVpby5ubw==