 Victor B. Fenik1,2*
Victor B. Fenik1,2*- 1VA Greater Los Angeles Healthcare System, Los Angeles, CA, United States
- 2Websciences International, Los Angeles, CA, United States
The sleep-related depression of excitability of upper airway motoneurons is a major neurological cause of obstructive sleep apnea whereas a disruption in the inhibition of spinal motoneurons during rapid eye movement (REM) sleep causes the REM sleep behavioral disorder. The large amount of experimental data has been obtained that deal with neurochemical mechanisms that are responsible for sleep-related depression of various motoneuron groups. However, there is a disagreement regarding the outcome of these studies primarily due to the use of different animal models and approaches, as well as due to differences in quantification and interpretation of obtained results. In this study, we sought to apply the same calculation methodology in order to uniformly quantify and compare the relative contribution of excitatory or inhibitory inputs to the decrease of excitability of different motoneuronal pools during REM and/or non-REM sleep. We analyzed only published quantitative data that were obtained by using receptor antagonists or chemogenetic approach to block receptors or silence neuronal populations. The outcomes of this analysis highlight the differences in the neurotransmitter mechanisms of sleep-related motoneuron depression between different motoneuronal pools and demonstrate the consistency of these mechanisms for hypoglossal motoneurons among various animal models.
Introduction
The decrease of upper airway motoneuron excitability during rapid eye movement (REM) sleep and non-REM (NREM) sleep is a major neurological cause of obstructive sleep apnea (OSA), which is recognized as a severe and growing sleep disorder (1, 2). On the other hand, the insufficient inhibition of spinal motoneurons during REM sleep causes REM sleep behavioral disorder (3–5). The OSA is associated with excessive daytime sleepiness, cognitive impairements and decreased quality of life (6–12). The OSA is also linked to hypertension (12–16) and the increased risk of stroke (17). The neurochemical mechanisms that are responsible for REM sleep-related depression of motoneurons have been studied in many laboratories. However, to date, there is no consensus regarding these mechanisms (18–35). The application of receptor antagonists to block neurotransmitter inputs to motoneurons is the main approach in order to assess the involvement of various receptor types and neurotransmitters in the mechanisms of the decrease of motoneuron excitability during both REM sleep and NREM sleep. However, the quantification and interpretation of antagonist effects differed between studies, which contributed to current disagreement of the mechanisms of sleep-related depression of motoneuron activity between various motoneuronal pools.
Recently, a chemogenetic tool was introduced that allows specifically activating or silencing selected groups of neurons by systemic application of clozapine-N-oxide (36–38). The chemogenetic activation of A1C1 catecholaminergic neurons was used to study the involvement of these neurons in sleep-related depression of hypoglossal motoneurons (HM) (39).
In this study, we developed an approach that allows quantifying the contribution of excitatory or inhibitory inputs, which were blocked (or removed) by application of receptor antagonists or chemogenetics, to the decrease of motoneuron excitability during NREM and REM sleep. We applied this approach to uniformly assess the contribution of state-dependent inputs to the three groups of motoneurons—spinal, trigeminal, and hypoglossal—for which published quantitative data are available.
Methodology
An example of a relatively simple case when a receptor antagonist application disfacilitated a motoneuronal activity by blocking only excitatory state-dependent input(s) to motoneurons is shown in Figure 1A. During wakefulness, the antagonist application decreased motoneuron activity from the control level (Wcon) to the level after antagonist application (Want). During sleep, the activity reduced to control level (Scon). The level of motoneuron activity after antagonist during sleep (Sant) is equal to Scon because the antagonist already removed the state-dependent excitatory input that would be removed by sleep under control conditions. The relative effect of sleep (Esleep) on the excitability of the motoneurons is calculated as a difference between levels of activity during wakefulness and sleep normalized by Wcon:
The effect of antagonist (Eant) that removes the excitatory input during wakefulness is
The relative contribution (RC) of the removed excitatory (e) input to the motoneuron excitability compared to the total sleep effect is the following:
When Equations (1) and (2) are combined, the final equation for the RCe is
This example of antagonist action (Figure 1A) is similar to the effect of terazosin, α1-adrenergic receptor antagonist, on the activity of genioglossus muscle during NREM and REM sleep in behaving rats [see Figure 3A in (29)].
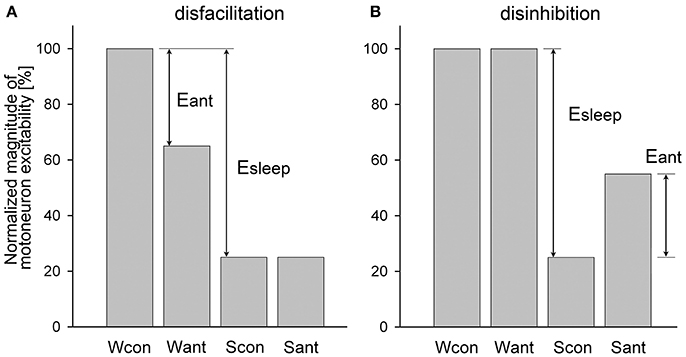
Figure 1. Example of a relatively simple disfacilitatory (A) and disinhibitory (B) effects of receptor antagonists on motoneuronal activity that blocked only state-dependent input(s) to motoneurons. Wcon and Want, levels of motoneuron activity during wakefulness at the control and after antagonist, respectively; Scon and Sant, levels of motoneuron activity during sleep at control and after antagonist, respectively; Eant and Esleep, effects of antagonist and sleep, respectively.
An example of the disinhibitory effect of a receptor antagonist is illustrated in Figure 1B. In this example, the antagonist removes only one inhibitory state-dependent input that appears only during sleep. Using the same logic as for the disfacilitation case, the contribution of the removed inhibitory (i) input relative to the total effect of sleep is calculated as following:
However, in most experiments, antagonists often block both state-dependent and non-state-dependent inputs to motoneurons, which make it challenging to calculate the magnitude of removed state-dependent input. An example of such dual disfacilitatory effect of an antagonist is shown in Figure 2A. In this example the additional antagonist effect during sleep (Eant2) indicates that the antagonist also removed some non-state-dependent inputs. One approach to qualitatively estimate if antagonist removes any state-dependent input is to compare the relative effects of sleep in control and after antagonist application. For example, if Scon/Wcon < Sant/Want then it would suggest that some state-dependent excitatory input has been removed by antagonist, whereas a condition of Scon/Wcon ≥ Sant/Want would hint that only non-state-dependent input was removed (Figure 2A). Similarly, when the application of an antagonist produces the dual disinhibition, which is apparent by disinhibition of motoneuron activity during both wakefulness and sleep (Figure 2B), the condition of Scon/Wcon ≥ Sant/Want would also indicate that no state-dependent input were removed by antagonist (Figure 2B).
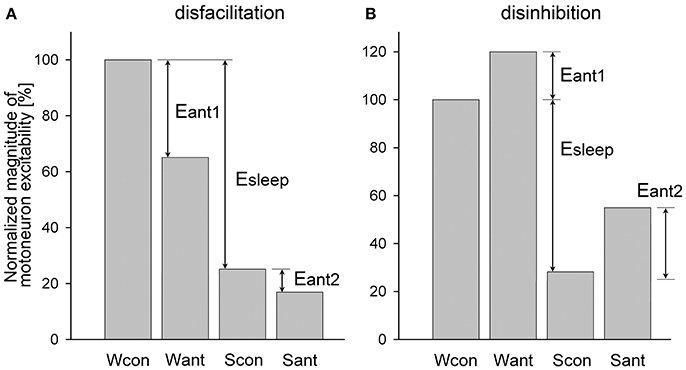
Figure 2. Example of a dual disfacilitatory (A) and disinhibitory (B) effects of receptor antagonist on motoneuronal activity that blocked both state-dependent and non-state-dependent inputs to motoneurons. Abbreviations are the same as in Figure 1 except for Eant1 and Eant2, which are the effects of antagonists during wakefulness and sleep, respectively.
We developed an approach that allows quantifying the relative contribution of a state-dependent input to the total decrease of motoneuron excitability during sleep when receptor antagonist(s) produce the dual effects as discussed above. The approach consists of upgrading the Formulas (3, 4) to include the elimination of the non-state-dependent effects, as following. For the disfacilitatory mixed effect of an antagonist (Figure 2A), i.e., the removal of excitatory state-dependent and excitatory non-state-dependent inputs, the relative contribution of the state-dependent input to depression of motoneuron activity during sleep is
The relative contribution of an inhibitory state-dependent input to the sleep-related depression of motoneuron activity for the disinhibitory antagonist mixed effects (Figure 2B), i.e., removal of inhibitory state-dependent and inhibitory non-state-dependent inputs, is calculated as following:
Both Formulas (5, 6) work well for either the simple (Figure 1) or the dual (Figure 2) antagonist effects. We applied these formulas to uniformly assess the relative contribution of the removed excitatory or inhibitory state-dependent inputs to the sleep-related decrease of excitability of different motoneuron groups using published data. The contribution to the decrease of motoneuron excitability during NREM sleep was calculated relative to wakefulness, i.e., levels of motoneuron excitability during wakefulness and NREM sleep were used for calculations; whereas the contribution during REM sleep was always assessed relative to NREM sleep, i.e., numbers obtained during NREM sleep and REM sleep were used for the formulas. If the numerical data were not explicitly reported within the text of published manuscripts, we estimated the required numbers from corresponding figures. In cases when Formulas (5, 6) produced numbers that were negative or exceeded 100 (see below), then respectively 0 or 100 were taken as final results.
Results
Spinal Motoneurons
In the pioneering work conducted by Michael Chase group (40), intracellular recording of lumbar motoneurons were performed during sleep and wakefulness in unanesthetized head-restrained cats. It has been found that the REM sleep-induced decrease of the motoneuron excitability (membrane hyperpolarization and the increase in rheobase) were abolished by strychnine, a glycinergic receptor antagonist, applied iontophoretically to the vicinity of recorded motoneurons. In that study, intracellular parameters of motoneurons were measured during NREM sleep in control (Ncon) and after the antagonist (Nant) and during REM sleep before (Rcon) and after the antagonist (Rant). Corresponding averaged membrane potentials were Ncon = −57.4 mV and Nant = −55.6 mV during NREM sleep and Rcon = −66.5 mV and Rant = −56.9 mV during REM sleep. By the Formula (6), the relative contribution of glycine to the membrane hyperpolarization was RCi = 85.3% (Figure 3). Similarly, for rheobase: Ncon = 9.4 nA; Nant = 9.7 nA; Rcon = 14.9 nA; and Rant = 9.7 nA. The contribution of glycine to the REM sleep-induced increase in rheobase was RCi = 100% (Figure 3).
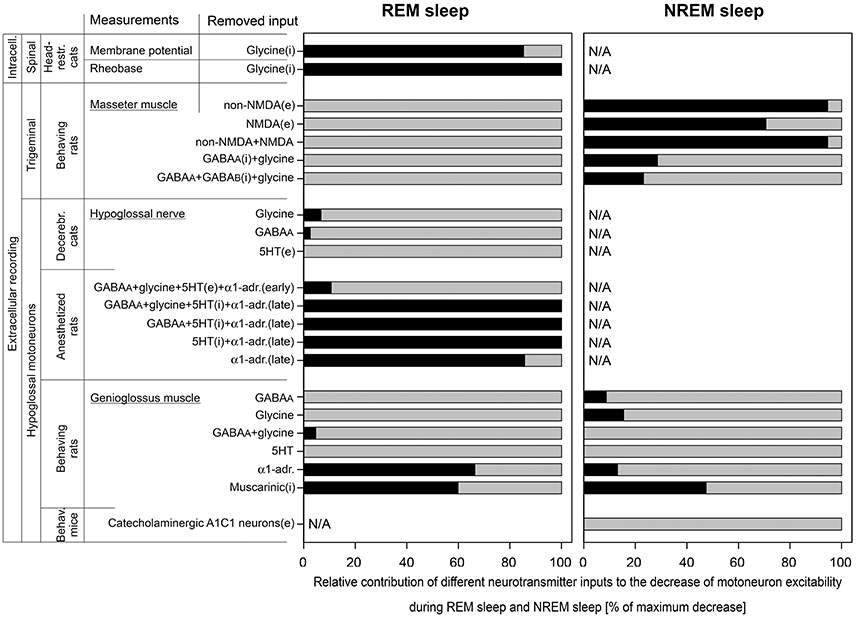
Figure 3. The relative contributions of excitatory (e) and inhibitory (i) inputs to the decrease of motoneuron excitability during REM sleep and NREM sleep that were calculated in this study. Data were used from experiments that were conducted with two approaches: intracellular (intracell.) and extracellular recording; on three motoneuron pools: spinal, trigeminal, and hypoglossal; using different animal models: head-restrained cats, behaving rats, decerebrated cats, anesthetized rats, and behaving mice. The length of black bars shows the relative contribution of tested receptors to the total depression of motoneuron excitability (gray bars) during REM sleep (left panel) or NREM sleep (right panel). Note that 5HT behaved as excitatory input in “early” and as inhibitory input in “late” carbachol responses in anesthetized rats.
Trigeminal Motoneurons
The role of glutamatergic, GABAergic, and glycinergic receptors in the decrease of excitability of trigeminal motoneurons during both NREM sleep and REM sleep has been studied in behaving rats (41, 42). Receptor antagonists were applied to the motor trigeminal nucleus using the reverse microdialysis technique and the electromyogram (EMG) of ipsilateral masseter muscle was quantified (41, 42). The glutamatergic antagonists were mostly effective in disfacilitating trigeminal motoneurons during NREM sleep compared to wakefulness but not during REM sleep as compared NREM sleep. For the CNQX, a non-NMDA glutamatergic receptor antagonist, we estimated the following average numbers from Figure 7A in (41), in arbitrary units (AU): Wcon = 47.0; Want = 7.49; Ncon = 5.69; Nant = 5.39; Rcon = 1.35; and Rant = 1.20. Using the Formula (5), we calculated the contribution of non-NMDA receptors RCe = −8.61 = 0% for REM sleep and, for NREM sleep, RCe = 94.6% (Figure 3). Similarly for D-AP5, a NMDA glutamatergic receptor antagonist, we estimated average magnitudes from Figure 7D in (41), in AU: Wcon = 28.3; Want = 13.9; Ncon = 5.93; Nant = 6.60; Rcon = 1.67; and Rant = 1.67. For REM sleep, the calculated contribution was RCe = −15.7 = 0% and, for NREM sleep, RCe = 70.7% (Figure 3). The combined effect of CNQX and D-AP5 was similar to the CNQX effect [Figure 5B in (41)], in AU: Wcon = 37.0; Want = 6.66; Ncon = 5.26; Nant = 5.02; Rcon = 1.33; and Rant = 1.15. For REM sleep, the calculated RCe was −13.9 = 0% and, for NREM sleep, RCe was 94.6% (Figure 3).
The GABAergic and glycinergic antagonists were also more effective in disinhibition of trigeminal motoneurons during NREM sleep than REM sleep. For the effect of combined antagonism of GABAA and glycine receptors on trigeminal motoneurons, we estimated average numbers of masseter EMG from Figure 6B in (42) as following, in AU: Wcon = 2.37; Want = 6.04; Ncon = 0.963; Nant = 3.48; Rcon = 0.664; and Rant = 0.664. Using the Formula (6), for REM sleep, the calculated combined contribution of GABAA and glycinergic receptors RCi = −160 = 0% and, for NREM sleep, RCi was 28.6% (Figure 3). The simultaneous antagonism of GABAA, glycine, and GABAB receptors on trigeminal motoneurons produced similar effects on masseter muscle activity. The average numbers of masseter EMG were estimated from Figure 8B in (42), in AU: Wcon = 2.37; Want = 6.96; Ncon = 0.977; Nant = 3.82; Rcon = 0.672; and Rant = 1.00. For REM sleep, the calculated RCi = −136 = 0% and, for NREM sleep, RCi = 23.2% (Figure 3).
Hypoglossal Motoneurons
The decrease of excitability of HM during NREM and REM sleep was investigated in several studies using various animal models. The increased interest to HM was mainly due to their innervation of upper airway muscles including the genioglossus, which play a critical role in maintaining the upper airway patency in OSA patients (43–46).
In early studies, the REM sleep-related decrease of excitability of HM was studied in a quantitative manner using decerebrated cats and anesthetized rats (19, 25). In both animal models, the REM sleep-like state was triggered by microinjections of carbachol, a cholinergic agonist, into dorsolateral pontine tegmentum and receptor antagonists were microinjected into the hypoglossal motor nucleus while spontaneous inspiratory activity of HM was recorded and quantified in ipsilateral hypoglossal nerve. In decerebrated cats, antagonizing GABAA and glycinergic receptors within the hypoglossal nucleus disinhibited HM during the baseline NREM sleep-like condition. The averaged normalized values of hypoglossal nerve activity during antagonism of glycinergic receptors (19) were the following, in %: Ncon = 107; Nant = 132; Rcon = 14; and Rant = 25. By the Formula (6), the contribution of glycine to the decrease of excitability of HM during REM sleep-like state was RCi = 6.74% (Figure 3). Similarly, for the antagonism of GABAA receptors (19), the hypoglossal nerve activity was, in %: Ncon = 69; Nant = 138; Rcon = 10.5; and Rant = 24. The calculated RCi was 2.56% (Figure 3). The contribution of serotonergic (5HT) effects to carbachol-induced depression of HM activity has been studied with methysergide, a broad-spectrum 5HT antagonist, that decreased HM activity (47). For the calculations, we used the following averaged normalized magnitudes of hypoglossal nerve activity, in %: Ncon = 100; Nant = 54; Rcon = 10; and Rant = 5. By the Formula (5), the contribution of 5HT receptors to the reduction of HM excitability during carbachol-induced REM sleep-like state was calculated as RCe = −8.89 = 0% (Figure 3).
In the REM sleep model using anesthetized rats, pontine carbachol could repeatedly elicit REM sleep-like state in the same animals, which helped to study neurochemical mechanisms of the depressant effect of REM sleep on HM excitability. In one study, a mix containing four antagonists—bicuculline, strychnine, methysergide, and prazosin—to antagonize GABAA, glycine, 5HT and α1-adrenergic receptors, respectively, was microinjected into hypoglossal nucleus (25). Early after the injections, the spontaneous inspiratory activity in ipsilateral hypoglossal nerve was disinhibited. Averaged normalized amplitudes of the nerve activity were estimated from the Figure 3B in (25) and were the following, in %: Ncon = 100; Nant = 140; Rcon = 18.6; and Rant = 38.2. The calculated RCi for the “early” effect by the Formula (6) was 10.7% (Figure 3). In 30–60 min after the injection of antagonists, hypoglossal nerve activity was disfacilitated and decreased below the control level most likely due to diffusion of the antagonists (26), which led to the abolition of the carbachol-induced depression of the nerve activity. The average nerve activity for these “late” carbachol trials was estimated from the Figure 3B in (25) as following, in %: Ncon = 100; Nant = 40; Rcon = 18.6; and Rant = 46.8. By the Formula (5), the calculated RCe for the “late” effect was 103 = 100% (Figure 3).
The abolition of the carbachol-induced depression of HM that occurred ~30 min after microinjections of the four antagonists into hypoglossal nucleus prompted us for additional studies to determine the role of each antagonist in this effect (26, 27). Microinjections of three antagonists to simultaneously antagonize GABAA, 5HT,and α1-adrenergic receptors resulted in the following numbers of the average normalized nerve activity for the “late” antagonist mix effect (26), in %: Ncon = 100; Nant = 32.8; Rcon = 28.6; and Rant = 37.7 (32.8*1.15). By the Formula (5), the calculated RCe was 105 = 100% (Figure 3). When only two monoaminergic 5HT and α1-adrenergic receptors were simultaneously antagonized, the depression of HM during REM sleep-like state was also abolished (27), in %: Ncon = 100; Nant = 27.0; Rcon = 24.9; and Rant = 25.2. The calculated RCe for the “late” antagonist mix effect was 100% (Figure 3). Further single injections of antagonists proved that the simultaneous blocking of 5HT and α1-adrenergic receptors were required for the abolition effect, while the separate antagonism of either α1-adrenergic or 5HT receptors was insufficient. The corresponding numbers for the “late” effect of the antagonism of α1-adrenergic receptors were the following (27), in %: Ncon = 100; Nant = 20.8; Rcon = 26.1; and Rant = 14.8. The RCe calculated by the Formula (5) was 85.7% (Figure 3). Antagonizing only 5HT receptors had moderate effect on carbachol-induced depression of HM activity. In addition, the methysergide effect was complicated by changes of the activity of HM that had opposite directions. Early after methysergide injection, the HM activity was disfacilitated for a long time. However, approximately after 30 min, the HM activity was disinhibited on the top of the long-lasting disfacilitation that started earlier [Figure 3C in (27)]. This mixed effect of methysergide was masked by a stronger effect of blocking α1-adrenoceptors by prazosin when it was present in the antagonist mix. Due to the opposite effects of methysergide, either Formula (5, 6) could not be used for the calculation of 5HT contribution. However, the disinhibitory contribution of 5HT receptors during the “late” effect could be estimated as difference between the full REM sleep-like depression of HM activity and the effect of α1-adrenergic receptors (see above) as following: 100 – 85.7 = 14.3%.
Important data has been obtained during natural sleep and wakefulness in behaving rats that has greatly advanced our understanding of the mechanisms of sleep-related depression of HM (28, 29, 33, 48, 35). The reverse microdialysis was used to deliver antagonists into the hypoglossal nucleus and the EMG of the genioglossus (GG) muscle was quantified to assess the antagonist effects during sleep and wakefulness. Bicuculline and strychnine were used to antagonize GABAA and glycine receptors, respectively (48, 35). For the effect of bicuculline, we estimated the average magnitude of respiratory EMG of GG at room air using Figure 4 in (48), in AU: Wcon = 18.4; Want = 54.4; Ncon = 3.61; Nant = 14.5; Rcon = 1.35; and Rant = 2.03. The calculated relative contribution of GABAA receptors the sleep-related depression of GG EMG by the Formula (6) was RCi = −37.4 = 0% for REM sleep and RCi = 8.75% for NREM sleep (Figure 3). For the antagonizing glycinergic receptors by strychnine, the average magnitude of respiratory activity of GG at room air was estimated from Figure 4 in (35), in AU: Wcon = 35.2; Want = 54.2; Ncon = 7.29; Nant = 17.9; Rcon = 1.10; and Rant = 2.34. The calculated relative contribution of glycinergic receptors was RCi = −2.37 = 0% for REM sleep and RCi = 15.5% for NREM sleep (Figure 3). For the combined effect of bicuculline and strychnine, numbers of the average magnitude of respiratory activity of the GG muscle were estimated from Figure 8 in (35), in AU: Wcon = 25.8; Want = 101; Ncon = 6.52; Nant = 17.1; Rcon = 1.12; and Rant = 3.60. The relative contribution of combined GABAA and glycine receptors was RCi = 4.68% for REM sleep and RCi = −11.2 = 0% for NREM sleep (Figure 3).
The effect of 5HT receptor antagonism on the GG muscle EMG have been studied using mianserin, a broad-spectrum 5HT antagonist (28). There was no visible effect of mianserin in the room air trials during NREM sleep or REM sleep and both Formulas (5, 6) produced negative results; therefore, zeros were entered in Figure 3.
Terazosin was used to study the role of α1-adrenoceptors in sleep-related decrease of GG activity in behaving rats (29). The disfacilitatory effect of terazosin was assessed using estimated numbers of the average magnitude of respiratory EMG of the GG muscle from Figure 3A in (29), in AU: Wcon = 44.2; Want = 24.5; Ncon = 29.7; Nant = 17.2; Rcon = 7.74; and Rant = 8.80. By the Formula (5), the relative contribution of α1-adrenergic receptors was RCe = 66.4% for REM sleep and RCe = 13.1% for NREM sleep (Figure 3).
The involvement of muscarinic receptor in the decrease of the GG muscle activity during NREM and REM sleep has been studied with scopolamine, a broad-spectrum muscarinic receptor antagonist (33). The antagonist had a disinhibitory effect on the GG muscle and we estimated numbers for average respiratory GG muscle activity during wakefulness, NREM sleep and REM (–) sleep from Figure 1D in (33), in AU: Wcon = 22.7; Want = 26.6; Ncon = 9.06; Nant = 18.2; Rcon = 0; and Rant = 10.9. By the Formula (6), the relative contribution of muscarinic receptors was RCi = 59.9% during REM (–) sleep and RCi = 47.5% for NREM sleep (Figure 3).
The role of A1C1 catecholaminergic neurons in control of HM has been studied using chemogenetics in behaving transgenic mice. Inhibitory DREADD (designer receptors activated by designer drug) has been used to study the effect of inhibition of A1C1 neurons on EMG of the GG muscle during NREM sleep and wakefulness (39). The silencing of A1C1 catecholaminergic neurons disfacilitated the GG muscle activity with the following numbers for averaged magnitude of GG EMG, in AU: Wcon = 142; Want = 120; Ncon = 37.2; Nant = 28.1. The relative contribution of A1C1 neurons to the GG muscle depression during NREM sleep calculated by Formula (5) was RCe = −16.1 = 0% (Figure 3).
Discussion
The major advantage of our analysis is that the single approach was used to uniformly quantify the contribution of excitatory and inhibitory inputs to the sleep-induced decrease of motoneuron excitability in different motoneuron pools. The relative contribution of state-dependent excitatory inputs to the total depressing effect of sleep was calculated using the Formula (5) whereas the contribution of state-dependent inhibitory inputs was calculated using Formula (6). These formulas can produce reliable results only when both state-dependent and non-state-dependent inputs that are removed by antagonists are of the same type, either excitatory or inhibitory; otherwise, the formulas produce false results. For example, methysergide had both disfacilitatory and disinhibitory effects on HM activity at the same time [see Figure 7A in (27)] and therefore we could not use either formula. In addition, the variability of data greatly affected the results of both formulas, which often exceeded the expected range of results 0–100%.
The performed analysis of the state-dependent inputs, which contribute to the decrease of excitability of motoneurons during NREM and REM sleep, reveals that the neurochemical mechanisms of sleep-induced motoneuron depression is considerably different between the three motoneuronal pools (Figure 3). The major points of this finding are the following. (1) In agreement with the author conclusion (40), our calculations indicate that the spinal motoneurons are inhibited by ~100% by glycine during REM sleep (depression of the motoneurons was not studied during NREM sleep in this study). (2) Our analysis suggests that the trigeminal motoneurons are not inhibited by either GABA or glycine, nor disfacilitated by glutamatergic mechanisms, during REM sleep. Although, the glutamatergic disfacilitation plays a major role in depression of trigeminal motoneurons during NREM sleep while either GABAA or glycine receptors, or both, have a moderate contribution to NREM sleep-induced depression of trigeminal motoneurons (41, 42). (3) Our analysis also suggests the following: (a) However, both GABA and glycine may play a role during NREM sleep-induced depression of HM (19, 25, 27, 28, 47, 48, 35); (b) Noradrenergic excitatory input plays a major role in HM depression during REM sleep-like state in anesthetized rats (27); (c) However, during natural REM sleep, noradrenergic and muscarinic mechanisms contribute ~50/50% to the depression of HM activity (29, 33). In addition, our analysis revealed some contribution of noradrenergic disfacilitation and approximately 50% of muscarinic inhibition during natural NREM sleep-induced depression of HM (29, 33); (d) Our analysis also confirmed no contribution of A1C1 neurons to HM depression during NREM sleep as was concluded by authors (39).
Conclusion
Our calculations confirmed that during natural REM sleep the decrease of excitability of spinal motoneurons is mediated only by glycinergic receptors (100%). The depression of trigeminal motoneurons during NREM sleep could be fully explained by excitatory glutamatergic input (~70%) and inhibitory GABAergic and/or glycinergic inputs (30%) whereas the contribution of these inputs during REM sleep was not detected by using either Formulas (5, 6). The depression of HM during natural REM sleep is approximately equally mediated by α1-adrenoceptors and muscarinic receptors (50/50%). In anesthetized animal model of REM sleep, α1-adrenoceptors mainly contributed to depression of HM during the “late” carbachol-induced REM sleep-like episodes (with some 5HT receptor contribution), which together fully (100%) accounted for the depressant effect of REM sleep-like state on HM. During the “early” carbachol-induced REM sleep-like episodes, the combined GABAA, glycinergic, 5HT, and α1-adrenergic inputs minimally contributed to the depression of HM. Other inputs must be responsible for HM depression during “early” REM sleep-like episodes, e.g., muscarinic, but this possibility was not tested. The neurotransmitters GABA, glycine and 5HT had minimal or no contribution to REM sleep-induced depression of HM activity regardless of used animal model. The contribution of A1C1 catecholaminergic neurons to NREM sleep-related depression of HM was not detected using the Formula (5).
Author Contributions
VF developed the calculation approach, quantitatively analyzed available published data and wrote the manuscript.
Funding
This study was supported by National Heart, Lung, and Blood Institute grants HL116845 and HL133847.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Arias M, Sánchez A. Obstructive sleep apnea and its relationship to cardiac arrhythmias. J Cardiovasc Electrophysiol. (2007) 18:1006–14. doi: 10.1111/j.1540-8167.2007.00891.x
2. Motamedi KK, McClary AC, Amedee RG. Obstructive sleep apnea: a growing problem. Ochsner J. (2009) 9:149–53.
3. Schenck C, Mahowald M. REM sleep behavior disorder: clinical, developmental, and neuroscience perspectives 16 years after its formal identification in sleep. Sleep (2002) 25:120–38. doi: 10.1093/sleep/25.2.120
4. Boeve BF, Silber MH, Saper CB, Ferman TJ, Dickson DW, Parisi JE, et al. Pathophysiology of REM sleep behavior disorder and relevance to neurodegenerative disease. Brain (2007) 130:2770–88. doi: 10.1093/brain/awm056
5. Mahowald M, Schenck C, Bornemann M. Pathophysiologic mechanisms in REM sleep behavior disorder. Curr Neurol Neurosci Rep. (2007) 7:167–72. doi: 10.1007/s11910-007-0013-7
6. Cheshire K, Engleman H, Deary I, Shapiro C, Douglas N. Factors impairing daytime performance in patients with sleep apnea/hypopnea syndrome. Arch Intern Med. (1992) 152:538–41. doi: 10.1001/archinte.1992.00400150068012
7. Ferini-Strambi L, Baietto C, Di Gioia M, Castaldi P, Castronovo C, Zucconi M, et al. Cognitive dysfunction in patients with obstructive sleep apnea (OSA): partial reversibility after continuous positive airway pressure (CPAP). Brain Res Bull. (2003) 61:87–92. doi: 10.1016/S0361-9230(03)00068-6
8. Parish J, Lyng P. Quality of life in bed partners of patients with obstructive sleep apnea or hypopnea after treatment with continuous positive airway pressure. Chest (2003) 124:942–7. doi: 10.1378/chest.124.3.942
9. Naismith S, Winter V, Gotsopoulos H, Hickie I, Cistulli P. Neurobehavioral functioning in obstructive sleep apnea: differential effects of sleep quality, hypoxemia and subjective sleepiness. J Clin Exp Neuropsychol. (2004) 26:43–54. doi: 10.1076/jcen.26.1.43.23929
10. Sforza E, Haba-Rubio J, De Bilbao F, Rochat T, Ibanez V. Performance vigilance task and sleepiness in patients with sleep-disordered breathing. Eur Respir J. (2004) 24:279–85. doi: 10.1183/09031936.04.00091903
11. Fleisher K, Krieger A. Current trends in the treatment of obstructive sleep apnea. J Oral Maxillofac Surg. (2007) 65:2056–68. doi: 10.1016/j.joms.2006.11.058
12. Norman D, Loredo J. Obstructive sleep apnea in older adults. Clin Geriatr Med. (2008) 24:151–65. doi: 10.1016/j.cger.2007.08.006
13. Young T, Peppard P, Palta M, Hla K, Finn L, Morgan B, et al. Population-based study of sleep-disordered breathing as a risk factor for hypertension. Arch Intern Med. (1997) 157:1746–52. doi: 10.1001/archinte.1997.00440360178019
14. Shamsuzzaman A, Gersh B, Somers V. Obstructive sleep apnea: implications for cardiac and vascular disease. JAMA (2003) 290:1906–14. doi: 10.1001/jama.290.14.1906
15. Wolk R, Somers V. Cardiovascular consequences of obstructive sleep apnea. Clin Chest Med. (2003) 24:195–205. doi: 10.1016/S0272-5231(03)00020-0
16. Nieto F, Young T, Lind B, Shahar E, Samet J, Redline S, et al. Association of sleep-disordered breathing, sleep apnea, and hypertension in a large community-based study. Sleep Heart Health study. JAMA (2000) 283:1829–36. doi: 10.1001/jama.283.14.1829
17. Yaggi H, Concato J, Kernan W, Lichtman J, Brass L, Mohsenin V. Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med. (2005) 353:2034–41. doi: 10.1056/NEJMoa043104
18. Kubin L, Tojima H, Davies RO, Pack AI. Serotonergic excitatory drive to hypoglossal motoneurons in the decerebrate cat. Neurosci Lett. (1992) 139:243–8. doi: 10.1016/0304-3940(92)90563-M
19. Kubin L, Kimura H, Tojima H, Davies R, Pack A. Suppression of hypoglossal motoneurons during the carbachol-induced atonia of REM sleep is not caused by fast synaptic inhibition. Brain Res. (1993) 611:300–12. doi: 10.1016/0006-8993(93)90517-Q
20. Parkis MA, Bayliss DA, Berger AJ. Actions of norepinephrine on rat hypoglossal motoneurons. J Neurophysiol. (1995) 74:1911–9. doi: 10.1152/jn.1995.74.5.1911
21. Bellingham MC, Berger AJ. Presynaptic depression of excitatory synaptic inputs to rat hypoglossal motoneurons by muscarinic M2 receptors. J Neurophysiol. (1996) 76:3758–70. doi: 10.1152/jn.1996.76.6.3758
22. Yamuy J, Fung S, Xi M, Morales FR, Chase MH. Hypoglossal motoneurons are postsynaptically inhibited during carbachol-induced rapid eye movement sleep. Neuroscience (1999) 94:11–5. doi: 10.1016/S0306-4522(99)00355-3
23. Lai YY, Kodama T, Siegel JM. Changes in monoamine release in the ventral horn and hypoglossal nucleus linked to pontine inhibition of muscle tone: an in vivo microdialysis study. J Neurosci. (2001) 21:7384–91. doi: 10.1523/JNEUROSCI.21-18-07384.2001
24. Kodama T, Lai YY, Siegel JM. Changes in inhibitory amino acid release linked to pontine-induced atonia: an in vivo microdialysis study. J Neurosci. (2003) 23:1548–54. doi: 10.1523/JNEUROSCI.23-04-01548.2003
25. Fenik V, Davies RO, Kubin L. Combined antagonism of aminergic excitatory and amino acid inhibitory receptors in the XII nucleus abolishes REM sleep-like depression of hypoglossal motoneuronal activity. Arch Ital Biol. (2004) 142:237–49. doi: 10.4449/aib.v142i3.373
26. Fenik V, Davies RO, Kubin L. Noradrenergic, serotonergic and GABAergic antagonists injected together into the XII nucleus abolish the REM sleep-like depression of hypoglossal motoneuronal activity. J Sleep Res. (2005) 14:419–29. doi: 10.1111/j.1365-2869.2005.00461.x
27. Fenik VB, Davies RO, Kubin L. REM sleep-like atonia of hypoglossal (XII) motoneurons is caused by loss of noradrenergic and serotonergic inputs. Am J Respir Crit Care Med. (2005) 172:1322–30. doi: 10.1164/rccm.200412-1750OC
28. Sood S, Morrison JL, Liu H, Horner RL. Role of endogenous serotonin in modulating genioglossus muscle activity in awake and sleeping rats. Am J Respir Crit Care Med. (2005) 172:1338–47. doi: 10.1164/rccm.200502-258OC
29. Chan E, Steenland HW, Liu H, Horner RL. Endogenous excitatory drive modulating respiratory muscle activity across sleep–wake states. Am J Respir Crit Care Med. (2006) 174:1264–73. doi: 10.1164/rccm.200605-597OC
30. Brooks PL, Peever JH. Unraveling the mechanisms of REM sleep atonia. Sleep (2008) 31:1492–1497. doi: 10.1093/sleep/31.11.1492
31. Steenland HW, Liu H, Horner RL. Endogenous glutamatergic control of rhythmically active mammalian respiratory motoneurons in vivo. J Neurosci. (2008) 28:6826–35. doi: 10.1523/JNEUROSCI.1019-08.2008
32. Lydic R. The motor atonia of REM sleep: a critical topics forum. Sleep (2008) 31:1471–2. doi: 10.1093/sleep/31.11.1471
33. Grace KP, Hughes SW, Horner RL. Identification of the mechanism mediating genioglossus reactivation muscle suppression in REM sleep. Am J Respir Crit Care Med. (2013) 187:311–9. doi: 10.1164/rccm.201209-1654OC
34. Fenik VB. Revisiting antagonist effects in hypoglossal nucleus: brainstem circuit for the state-dependent control of hypoglossal motoneurons: a hypothesis. Front Neurol. (2015) 6:254. doi: 10.3389/fneur.2015.00254
35. Morrison JL, Sood S, Liu H, Park E, Liu X, Nolan P, Horner RL. Role of inhibitory amino acids in control of hypoglossal motor outflow to genioglossus muscle in naturally sleeping rats J. Physiol. (2003) 552:975–91. doi: 10.1113/jphysiol.2003.052357
36. Anaclet C, Ferrari L, Arrigoni E, Bass CE, Saper CB, Lu J, et al. GABAergic parafacial zone is a medullary slow-wave-sleep promoting center. Nat Neurosci. (2014) 9:1217–24. doi: 10.1038/nn.3789
37. Anaclet C, Pedersen NP, Ferrari LL, Venner A, Bass CE, Arrigoni E, et al. Basal forebrain control of wakefulness and cortical rhythms. Nat Commun. (2015) 6:8744. doi: 10.1038/ncomms9744
38. Venner A, Anaclet C, Broadhurst RY, Saper CB, Fuller PM. A novel population of wake-promoting GABAergic neurons in the ventral lateral hypothalamus. Curr Biol. (2016) 26:2137–43. doi: 10.1016/j.cub.2016.05.078
39. Rukhadze I, Carballo NJ, Bandaru SS, Malhotra A, Fuller PM, Fenik VB. Catecholaminergic A1/C1 neurons contribute to the maintenance of upper airway muscle tone but may not participate in NREM sleep-related depression of these muscles. Resp Physiol Neurobiol. (2017) 244:41–50. doi: 10.1016/j.resp.2017.07.001
40. Soja P, López-Rodríguez F, Morales F, Chase M. The postsynaptic inhibitory control of lumbar motoneurons during the atonia of active sleep: effect of strychnine on motoneuron properties. J Neurosci. (1991) 11:2804–11. doi: 10.1523/JNEUROSCI.11-09-02804.1991
41. Burgess C, Lai D, Siegel J, Peever J. An endogenous glutamatergic drive onto somatic motoneurons contributes to the stereotypical pattern of muscle tone across the sleep-wake cycle. J Neurosci. (2008) 28:4649–60. doi: 10.1523/JNEUROSCI.0334-08.2008
42. Brooks PL, Peever JH. Identification of the transmitter and receptor mechanisms responsible for REM sleep paralysis. J Neurosci. (2012) 32:9785–95. doi: 10.1523/JNEUROSCI.0482-12.2012
43. Remmers JE, deGroot WJ, Sauerland EK, Anch AM. Pathogenesis of upper airway occlusion during sleep. J Appl Physiol. (1978) 44:931–8. doi: 10.1152/jappl.1978.44.6.931
44. Mezzanotte WS, Tangel DJ, White DP. Waking genioglossal electromyogram in sleep apnea patients versus normal controls (a neuromuscular compensatory mechanism). J Clin Invest. (1992) 89:1571–9. doi: 10.1172/JCI115751
45. Saboisky JP, Butler JE, Fogel RB, Taylor JL, Trinder JA, White DP, et al. Tonic and phasic respiratory drives to human genioglossus motoneurons during breathing. J Neurophysiol. (2006) 95:2213–21. doi: 10.1152/jn.00940.2005
46. Saboisky JP, Jordan AS, Eckert DJ, White DP, Trinder JA, Nicholas CL, et al. Recruitment and rate-coding strategies of the human genioglossus muscle. J Appl Physiol. (2010) 109:1939–49. doi: 10.1152/japplphysiol.00812.2010
47. Kubin L, Tojima H, Reignier C, Pack AI, Davies RO. Interaction of serotonergic excitatory drive to hypoglossal motoneurons with carbachol-induced, REM sleep-like atonia. Sleep (1996) 19:187–95. doi: 10.1093/sleep/19.3.187
Keywords: spinal motoneurons, trigeminal motoneurons, hypoglossal motoneurons, neurotransmitters, genioglossus
Citation: Fenik VB (2018) Contribution of Neurochemical Inputs to the Decrease of Motoneuron Excitability During Non-REM and REM Sleep: A Systematic Review. Front. Neurol. 9:629. doi: 10.3389/fneur.2018.00629
Received: 20 May 2018; Accepted: 12 July 2018;
Published: 31 July 2018.
Edited by:
Birendra N. Mallick, Jawaharlal Nehru University, IndiaReviewed by:
Giancarlo Vanini, University of Michigan, United StatesMathias Dutschmann, Florey Institute of Neuroscience and Mental Health, Australia
Copyright © 2018 Fenik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Victor B. Fenik, dmljdG9yLmZlbmlrQHZhLmdvdg==