 Concetta Scimone1,2
Concetta Scimone1,2 Luigi Donato1,2,3
Luigi Donato1,2,3 Zoe Katsarou4
Zoe Katsarou4 Sevasti Bostantjopoulou5
Sevasti Bostantjopoulou5 Rosalia D'Angelo1*
Rosalia D'Angelo1* Antonina Sidoti1,2
Antonina Sidoti1,2- 1Department of Biomedical and Dental Sciences and Morphological and Functional Images, University of Messina, Messina, Italy
- 2Department of Vanguard Medicine and Therapies, Biomolecular Strategies and Neuroscience, I.E.ME.S.T., Palermo, Italy
- 3Department of Chemical, Biological, Pharmaceutical and Environmental Sciences, University of Messina, Messina, Italy
- 4Department of Neurology, Hippokration General Hospital, Thessaloniki, Greece
- 53rd University Department of Neurology, Thessaloniki, Greece
Wide comprehension of genetic features of cerebral cavernous malformations (CCM) represents the starting point to better manage patients and risk rating in relatives. The causative mutations spectrum is constantly growing. KRIT1, CCM2, and PDCD10 are the three loci to date linked to familial CCM development, although germline mutations have also been detected in patients affected by sporadic forms. In this context, the main challenge is to draw up criteria to formulate genotype-phenotype correlations. Clearly, genetic factors determining incomplete penetrance of CCM need to be identified. Here, we report two novel intronic variants probably affecting splicing. Molecular screening of CCM genes was performed on DNA purified by peripheral blood. Coding exons and intron-exon boundaries were sequenced by the Sanger method. The first was detected in a sporadic patient and involves KRIT1. The second affects CCM2 and it is harbored by a woman with familial CCM. Interestingly, molecular analysis extended to both healthy and ill relatives allowed to estimate, for the first time, a penetrance for CCM2 lower than 100%, as to date reported. Moreover, heterogeneity of clinical manifestations among those affected carrying the same genotype further confirms involvement of modifier factors in CCM development.
Introduction
Cerebral cavernous malformations (CCM, OMIM #116860) are vascular lesions affecting brain capillaries. Lesions appear like mulberries, due to the enlarged, tangled vessels which form them. The single layer of endothelial cells is really inhomogeneous as a result of pericyte absence and defective tight and adherens junctions (1) with consequent impairment of the blood-brain barrier. This condition leads to an increased bleeding risk of lesions. Intracerebral hemorrhage (ICH), together with seizures, are the main clinical manifestations appearing in up to 70% of symptomatic patients. Other symptoms include headache, focal neurological deficits, vertigo, and paresis.
However, only about 75% of affected individuals clinically manifest, with no gender predominance (2). CCM arise mainly in the supratentorial region, however, a more severe prognosis is associated to lesions affecting brainstem and pons. Extra-neurological localization was also observed, and others involved organs include kidney, liver, skin (3). Neuro-radiological diagnosis of CCM is performed by magnetic resonance imaging (MRI) with T-2 gradient-echo and susceptibility-weighted imaging. Heterogeneity of clinical manifestations and the age of symptoms onset are linked to incomplete penetrance and variable expressivity of the disease. Due to these conditions, worldwide incidence of CCM is underestimated and ranges between 0.4 and 0.8% (4). Lesion numbers are also quite variable and related to genetic etiology. Usually, single cavernomas are detected in patients affected by sporadic forms while multiple lesions are frequent in familial forms. Sporadic CCM arises mostly from the third decade of life on, due to altered expression of genes involved in regulation of the angiogenetic process, following hypoxia, inflammation, or oxidative stress conditions. Patients affected by familial forms, instead, often show multiple lesions already in childhood, as a consequence of mutations at the three different loci KRIT1/CCM1 (Entrez Gene: 889; 7q11.2-21), MGC4607/CCM2 (Entrez Gene: 83605; 7p13), and PDCD10/CCM3 (Entrez Gene: 11235; 3q26.1). Clinical penetrance is estimated to be about 88% for KRIT1, 100% for CCM2, and 63% for PDCD10 (5). Moreover, mutations at PDCD10 seem to be linked to a more severe phenotype characterized by recurrent ICH (6). Three CCM genes encode for the proteins Krev interaction trapped 1 (krit1), malcavernin and programmed cell death protein 10 (pdcd10), involved in maintaining cell-cell junctions and cell-extra cellular matrix (ECM) adhesion, in the regulation of apoptosis/proliferation switch of endothelial cells and in response to oxidative damage (7). In particular, at cell–cell adherens junctions, the three CCM proteins interact forming a ternary complex in which malcavernin acts as a bridge between krit1 and pdcd10. Therefore, loss of function mutations causes a disassembly of the complex resulting in an increased migratory capacity and in a gain of mesenchymal phenotype of endothelial cells due to perturbation of β-catenin/TGF-β/BMPs signaling (8).
To date, more than 300 mutations at the three CCM loci are reported in the public version of the Human Gene Mutation Database (HGMD®) as disease causing mutations. Of these, 15 and 12 impair splicing of KRIT1 and CCM2 transcripts, respectively. Here, we report two novel intronic variants that affect KRIT1 and CCM2 genes, probably leading to aberrant splicing processes.
Materials and Methods
Case Descriptions
The study involves two patients. The first is a 57-year-old Italian man who accidentally discovered to be affected, being asymptomatic. The single lesion at the right cerebellar hemisphere was diagnosed by an MRI exam performed after an accident (Figure 1a). No evidence of suspected affected relatives was reported by the patient and, therefore, he was classified as sporadic.
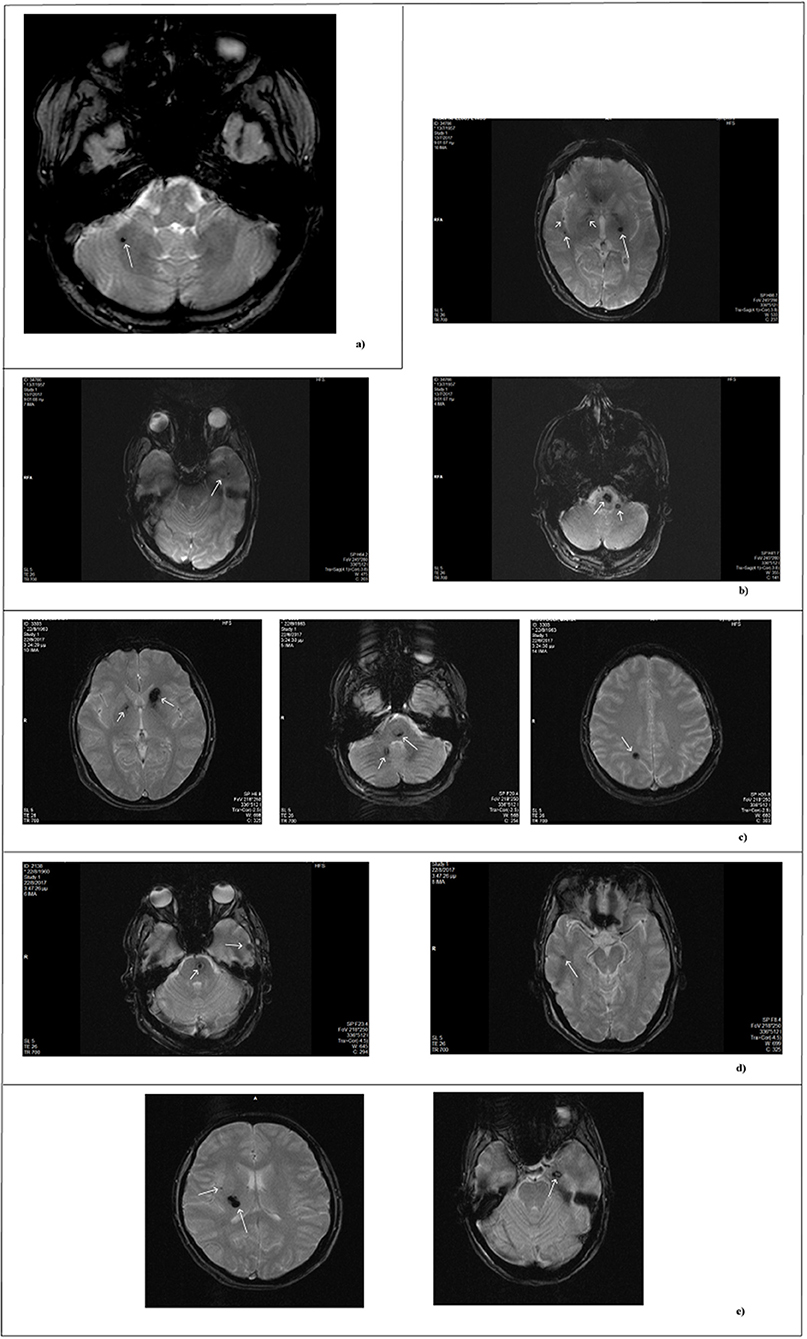
Figure 1. MRI scan of patients. MRI images obtained by T2- weighted gradient echo sequences. Each panel refers to a single patient. (a) Single lesion detected in the sporadic patient. (b–e) Lesions of affected family members. In reference to Figure 2: (b) II-1; (c) II-3; (d) II-4; (e) III-13.
The second patient is an 84 year-old woman affected by hereditary CCM (Figure 2; I-2) diagnosed about 20 years ago following recurrent epileptic seizures. No MRI scan images are available; however, from medical reports read, the presence of four cavernous lesions emerged and, particularly, two at the pontine region and two at the right parietal lobe. The woman could not provide anamnesis information on her parents. The study was also extended to other members of the family and, particularly, to three of her sons and five grandchildren. Three of her sons, as reported in Figure 2, are affected. The first born is a 60-year-old man (Figure 2; II-1) who suffers from global transient amnesia without neurological deficits. MRI highlighted at least seven lesions, distributed among brainstem and both cerebellar hemispheres (Figure 1b). Diagnosis in one of the two second-born (a twin) Figure 2; II-3) was performed subsequently to familial history. Despite MRI showing the presence of four CCM lesions at brainstem (Figure 1c) the 58-year-old patient is, to date, asymptomatic. No information is available about his twin. The younger daughter (Figure 2; II-4) is 48 years old and manifests recurrent headaches, right hemiparesis, and hemi-hyperesthesia. These symptoms are due to five CCM lesions affecting brainstem and both cerebral hemispheres (Figure 1d). Regarding the five grandchildren, MRI was positive only for one of them (Figure 2; III-13) who, however, is asymptomatic. In detail, she carries three lesions, two in the area of the posterior limb of the right internal capsule and one in the left temporal lobe (Figure 1e). The patients involved in the study were fully informed and informed consent was obtained in accordance with the Declaration of Helsinki. Written informed consent for the publication of this study was obtained from the participants. The study was approved by our local Ethical Committee A.O.U. G. Martino Messina.
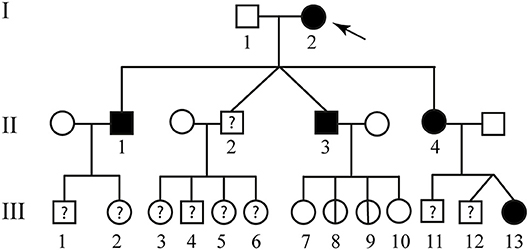
Figure 2. Familial pedigree of case report 2. The arrow indicates the proband. Healthy members are indicated by empty symbols. Empty circles vertically crossed refer to healthy carrier mutation. Affected ones are represented by black filled. By question mark are indicated consanguineous of which no genotype or phenotype data are available.
Patient Genotyping
Molecular screening of CCM genes was performed on DNA purified by peripheral blood. Coding exons and intron-exon boundaries were screened using primer pairs designed according to the KRIT1, CCM2, and PDCD10 published nucleotide sequences of GenBank (accession numbers NG_012964.1, NG_016295.1, and NG_008158.1, respectively) and are available upon request. The sequence variations detected were described according to the recommendations by the Human Genome Variation Society, having the A of the ATG translation starting codon as +1 at the cDNA level. Amplified products were sequenced by Sanger method using the BigDye Terminator v3.1 chemistry (Applied Biosystems®, Thermofisher Scientific) and run on a 3500 Genetic Analyser (Applied Biosystems®, Thermofisher Scientific). Raw data analysis was performed by Applied Biosystems Sequencing Analysis Software® v6.0.
Multiplex Ligation-Dependent Probe Amplification (MLPA) Assay
In order to exclude the presence of large genomic rearrangements, MLPA was performed according to the manufacturer's instructions using two MLPA kits (SALSA MLPA Kits, P130 and P131 CCM, MRC-Holland). For visual inspection, peak heights were compared between the sample and controls to find any alteration in relative peak heights within the test sample. For the normalized peak-area calculations, each peak area was normalized by dividing the individual peak area by the total peak area of all peaks for that sample.
Characterization of Novel Variants and of Mutated Proteins Features by Coding Sequences Study
Two novel intronic variants were detected by DNA direct sequencing in KRIT1 and CCM2 genes. To evaluate any splicing impairment, the transcripts of both genes were retro-transcribed, amplified, and sequenced. Total RNA was purified by whole peripheral blood using TRIzol® reagent (Thermo Fisher Scientific), following manufacturer protocol. Selective retro-transcription of mRNA was performed using GeneAmp RNA PCR Core Kit® (Thermo Fisher Scientific), applying the two-step protocol. Retro-transcription was followed by amplification of the partial KRIT1 and CCM2 coding sequences. In particular, the region between exons 13–15 was sequenced for KRIT1, using the primer pairs: 5′-GAA ATT CCT ACT TAT GGA GCA GC-3′ and 3′-GCA TTA ACT GTC CAT TTA GCT TCA-5′. Obtained amplicon has a length of 301 bp. Regarding CCM2, primer pairs 5′-ACT TTC TGC TTC CCT GAA TCT GT-3′ and 3′-AGT CCT GGT CCA TGC TGC AGC-5′ were used and a fragment of 529 bp between exons 8–10 was amplified. Detailed protocols are available upon request.
Allele Frequency Estimation
Frequency in the population of these two novel variants was estimated by DNA direct sequencing and by restriction fragment length polymorphism (PCR-RFLP) analysis, performed on two different 150 members of healthy control groups, recruited from Italian and Greek populations. The groups were heterogeneous for age and sex.
In particular, the novel KRIT1 variant does not affect any restriction site therefore it was screened by direct sequencing. Regarding CCM2, the novel variant leads to loss of a restriction site for PstI. The wild-type 499 base pairs (bp) genomic fragment has two restriction sites for PstI and digestion produces three fragments of 153, 259, and 87 bp. In the presence of the variant, the first restriction site is lost resulting in formation of only two fragments of 412 and 87 bp (not shown). Five PstI units were used for overnight digestion of 0.5 μg of DNA, incubating the reaction at 37°C. Controls consisted of two Italian and Greek groups, each made up of 150 healthy, unrelated, randomly selected subjects. The Italian group was made up of 72 males and 78 females, average age 41.8. The Greek group was formed by 77 males and 73 females, average age 46.3. All controls were recruited after having undergone MRI for non-specific reasons, such as headache, dizziness, or mental confusion and resulted negative.
In-silico Prediction of Structural Alterations and Loss of Interaction
As subsequently reported, only the CCM2 variant affects splicing. Comparison between wild-type and mutated secondary structures was performed by “Structure Prediction” module of RaptorX tool (9), giving as input native and mutated malcavernin amino acid sequences in FASTA format.
Results
Patient Genotyping
Direct sequencing of coding regions and of intron-exon boundaries of the three CCM genes allowed to detect two novel intronic variants that may affect the splicing process. Specifically, the first patient affected by the sporadic form of CCM carries the nucleotide substitution IVS15-66A>T(c.2026-66A>T), located in the intron between coding exon 14 and 15 (10), affecting the adenine located 66 nucleotide upstream of the exon junction (Figure 3); the nomenclature refers to coding exons only.
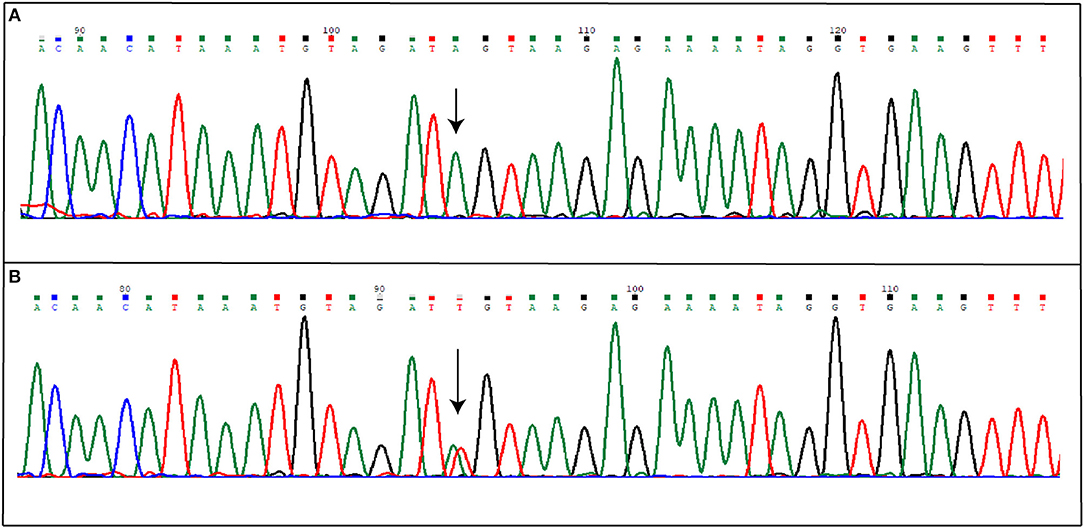
Figure 3. Novel IVS15-66A>T mutation detected in KRIT1 gene. The figure shows both wild-type (A) and mutated (B) electropherograms. The heterozygous substitution is indicated by the arrow.
More variants at the three CCM genes were, instead, detected in the second patient presenting a familial form. Results are summarized in Table 1. As shown, three single nucleotide polymorphisms (SNPs) were detected in KRIT1 gene. Of these, rs17164451 and rs2027950 are intronic, while rs11542682is a synonymous substitution known to lead splicing impairment and, thus, associated to CCM lesion development (11). Another five SNPs were identified in the CCM2 gene and, among these, c.358G>A (rs11552377) is reported in HGMD as associated to CCM development (12). Furthermore, a novel heterozygous intronic variant was detected and is located at the G nucleotide at the splice-acceptor site adjacent to the 10th exon. This is a single nucleotide substitution G>A that, according to the Human Genome Variation Society guidelines, was named IVS10-1G>A(c.1055-1G>A) (Figures 4A,B). Mutational analysis was also performed on proband's relatives and genotyping results are reported in Table 2.

Table 1. Variants detected in the 84-years old woman affected by familial CCM.
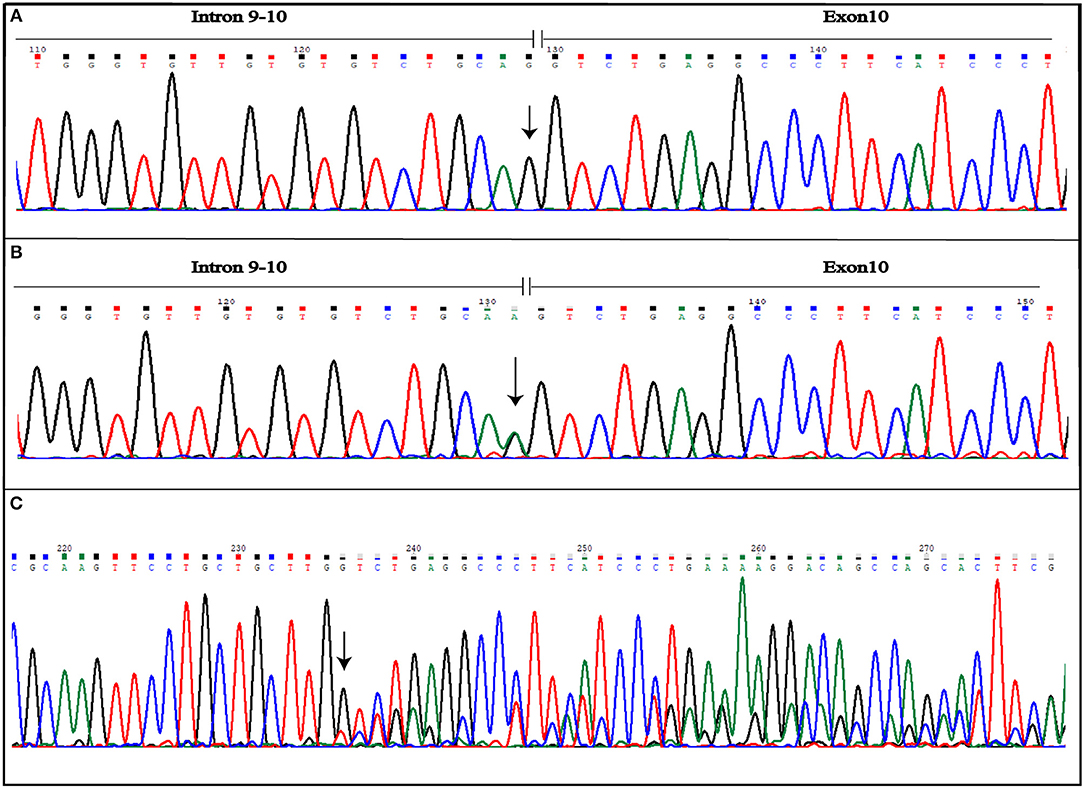
Figure 4. Novel IVS10-1G>A mutation detected in CCM2 gene. The figure shows both wild-type (A) and mutated (B) electropherograms. The heterozygous substitution is indicated by the arrow. The novel heterozygous variant causes splicing alteration that occurs including also the first nucleotide of exon 10, as shown by frameshift observed in electropherogram of the coding sequence (C). The arrow indicates the point of the mutation.
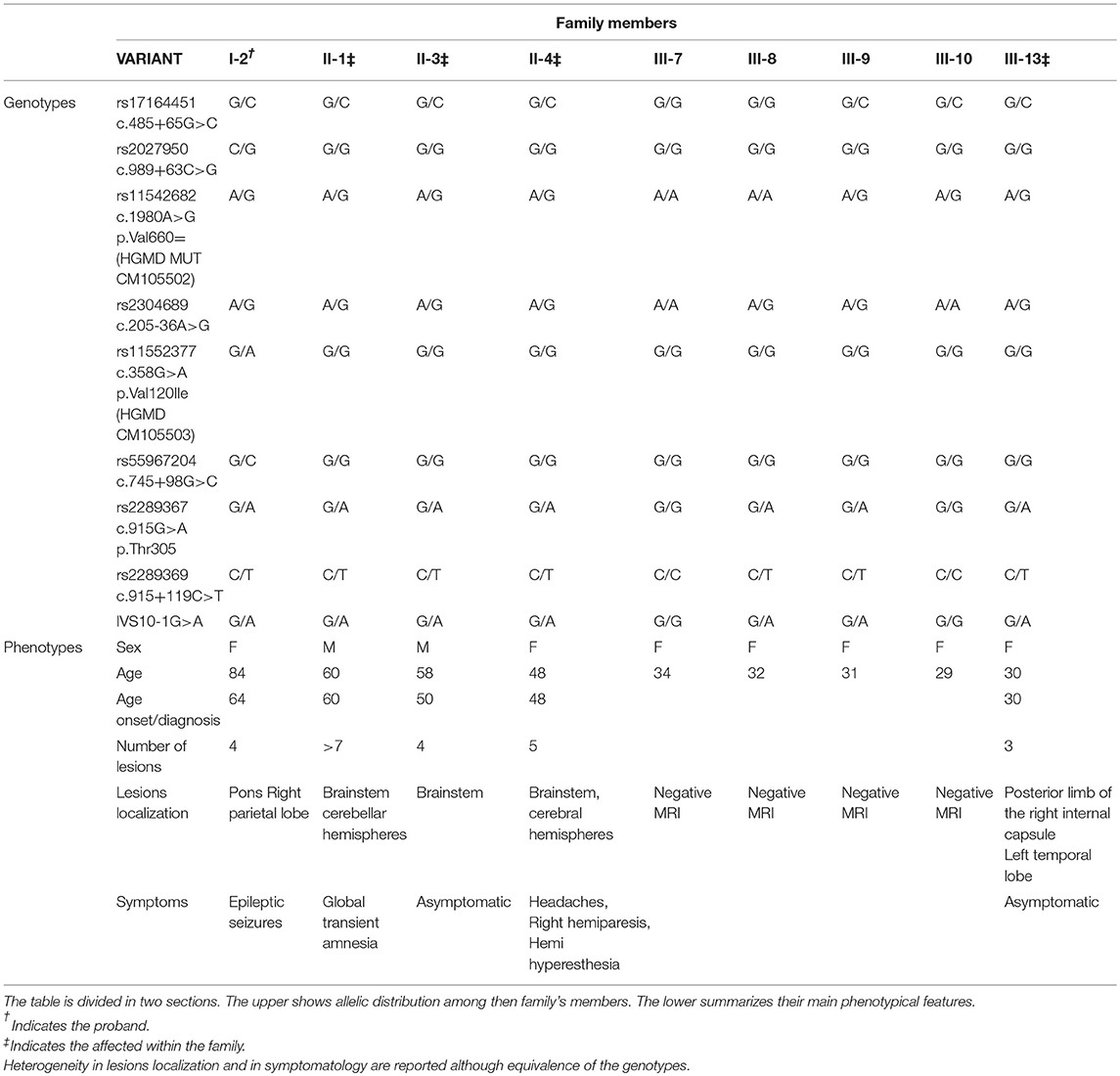
Table 2. Intra-familial genotype-phenotype comparison.
No large genomic rearrangements were detected by MLPA analysis.
Characterization of Novel Variants and of Mutated Proteins Features
Variants were found neither in HGMD, ExAC nor 1000G databases. As predicted, the novel variants detected in KRIT1 and CCM2 genes do not affect coding sequencing.
Hypothesis of normal splicing process alteration due to IVS15-66A>T variant was tested by sequencing of partial KRIT1 cDNA. Analysis did not reveal any change in the nucleotide sequence, compared with the wild-type one (not shown). To date, therefore, its involvement in CCM development cannot be confirmed.
Aberrant splicing, instead, results as a consequence of IVS10-1G>A variant affecting the CCM2 gene. The substitution causes loss of splicing acceptor site and the activation of an intronic cryptic acceptor site one nucleotide downstream of the wild-type, resulting in the deletion of the first nucleotide of exon 10 (c.1055delG) (Figure 4C). This leads to the change of amino acid at position 352 followed by a stop codon in position 353 (p.G352Vfs*2). Mutated protein is truncated by 92 amino acids, if compared with 444 amino acid wild-type ones.
Population Study
To estimate the frequency of KRIT1 IVS15-66A>T and CCM2 IVS10-1G>A variants in the population we selected two Italian and Greek control groups, each consisting of 150 healthy subjects. None of the controls carried the variants that, therefore, were defined as mutations.
As described, CCM2 IVS10-1G>A substitution impairs splicing of 10th exon leading to a 352 amino acid truncated protein. In-silico prediction of secondary structure performed at RaptorX tool shows how Harmonin Homology Domain (HHD) in C-terminus is partially disrupted (Figure 5).
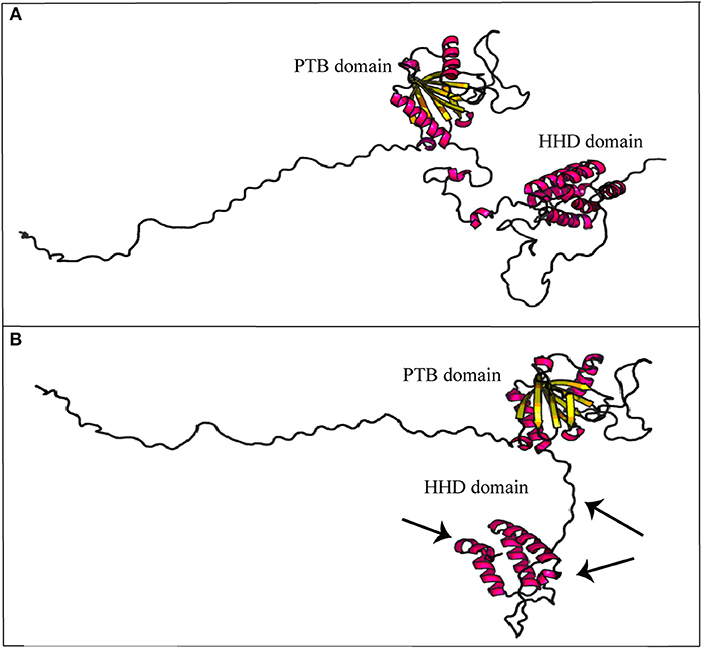
Figure 5. Secondary structure prediction of mutated malcavernin. The image compares native (A) and mutated (B) malcavernin secondary structures. As indicated by the arrows, the main structural modifications affect harmonin-homology domain (HHD) at C-terminus with consequent loss of two helices. Images were obtained by “Structure Prediction” module of RaptorX tool.
Discussion
Genetic bases of CCM are to date not well-defined, particularly for sporadic forms. Common features for these and hereditary cavernomatosis include neuro-radiological and clinically incomplete penetrance and variable expressivity (13). A low percentage of patients affected by the sporadic form of the disease, instead, carry germ-line mutations at one of the three CCM loci. Usually these patients show multiple lesions that, however, can remain asymptomatic during the course of life (14).
Conversely, familial forms tend to be symptomatic already in childhood (15).
The reason for this heterogeneity has not yet been elucidated. Different hypotheses have been proposed and among them, the existence of biallelic somatic mutations was proven by mutational analyses conducted on specimens derived from both inherited and sporadic lesions. Therefore, the Knudsonian two-hit mutation mechanism is one of the most accredited hypotheses to explain CCM pathogenesis (16). The cases here described report two affected patients with a not-completely overlapping clinical history. The first one is a man presenting a single CCM asymptomatic lesion that was discovered by chance. The patient carries the novel IVS15-66A>T germ-line mutation in the KRIT1 gene that does not seem to compromise normal splicing, as shown by sequencing of the transcript. Despite its association with CCM development cannot be confirmed, also its total exclusion in pathogenesis cannot be considered correct. It may be due to a premature degradation of mutated transcript that did not allow its detection or an impairment of an alternative splicing form of the same transcript. None of these hypotheses have been demonstrated here. Moreover, the existence of somatic mutations cannot now be evaluated. Regarding the second case, this is an 84 year-old woman who developed many lesions which remained silent until the age of 60. The patient is affected by a familial form and more relatives are also affected. From genetic analysis, more variants associated to the onset of lesion were detected and, among these, the novel CCM2 IVS10-1G>A. As reported, IVS10-1G>A causes the loss of a conventional splice site and gain of a cryptic one that includes the first nucleotide of exon 10. This results in a truncated protein, lacking 92 amino acids at C-terminus. In its structure, malcavernin contains a phosphotyrosine binding (PTB) domain by which interaction between krit1 and pdcd10 (17) is mediated, and a HHD (18) which interacts with TrkA inducing cell death (19), and with N-terminus of MEKK3 (20). CCM2-MEKK3 interaction is essential for maintaining endothelial barrier properties (21). Loss of this interaction causes an increased MEKK3 autophosphorylation resulting in an enhanced endothelial permeability due to Rho-/ROCK-dependent signaling perturbation (22). Therefore, CCM2–MEKK3 interaction needs negative regulation of MEKK3 activity (23).
In detail, Fisher et al. (18) proposed a crystallographic model of wild-type malcavernin in which HHD is formed by the S283-K375 amino acidic region folded to organize five α-helix motifs. In this region, amino acidic residuesP355-F356 are conserved and essential to stabilize the structure. As highlighted in Figure 5, the novel mutation here reported leads to the synthesis of a truncated and misfolded protein lacking in two helices of HHD. The consequence is an impairment in MEKK3 binding that triggers pathogenic signaling resulting in CCM phenotype.
Besides the complex molecular mechanisms that lead to lesion development, another important feature of CCM is the heterogeneity of the phenotypes. To date, only few data are available on genotype-phenotype correlation due to high clinical variability among patients. Variable expressivity is also observed within affected individuals belonging to the same family and linked to the same mutation. Together with allelic constitution, Table 2 reports phenotypical manifestations of the family here described. Despite lesion numbers tending to be homogeneous, they were distributed both at supratentorial and infratentorial regions as well as clinical manifestations which substantially differed among patients. Regarding genotypes, all affected individuals showed the same allelic assortment, with the exception of the proband (Table 2; I-2) who had a heterozygous condition for the two SNPs rs11552377 (c.358G>A p.Val120Ile) and rs55967204 (c.745+98G>C) in the CCM2 gene. Only the carrier condition for rs11552377 could support the epileptic seizures that have arisen as the single clinical manifestation. As we previously described in a case-control study, indeed, this SNP is associated with both increased risk to develop CCM and a more severe symptomatology (12). An important data regards the segregation of the novel IVS10-1G>A mutation within the family. In Table 2, the proband is marked with a single asterisk while the affected relatives are twice starred. All examined 2nd generation members (Figure 2; II-1; II-3; II-4) were affected and manifested different symptoms although they carried the same variants. This is in accordance with the typical variable expressivity observed in CCM patients. Regarding the five members of the 3rd generation, only one (Figure 2; III-13) was positive at MRI scan. However, by comparison of their genotypes, the presence of the novel mutation IVS10-1G>A can be observed in three of them and, particularly, in the young affected woman (Figure 2; III-13) and in two of her healthy cousins (Figure 2; III-8 and III-9). This observation could be explained by two different hypotheses. The first is the lack of linkage between the pathological phenotype onset and the presence of the novel mutation. However, it produces deleterious effects on the malcavernin structure, therefore its involvement in CCM pathogenesis cannot be excluded. The second hypothesis, instead, regards the penetrance of the CCM2 locus. Among CCM loci, CCM2 is the only one reported with 100% of clinical and neuroradiological penetrance. However, in this case, seven members of the family carried the mutation but only five of them showed lesions at MRI scan. Therefore, for the first time, a penetrance for CCM2 locus >100% is here reported and, to be precise, equivalent to 70%. Moreover, members II-1, II-3, II-4, III-9, and III-13 carried the same variants and, among these, III-9 is healthy. Based on the dynamic nature of familial CCM, if compared with 2nd generation members, absence of lesions in III-9 may be imputed to her younger age, and the probability that she will become illcannot be excluded. However, her coetaneous cousin already carries three lesions. As clearly evident by genotypes comparison, c.205-36A>G(rs2304689), c.915G>A (rs2289367), c.915+119C>T (rs2289369), and IVS10-1G>A in CCM2 were in cis configuration, therefore other genetic or environmental factors influenced the phenotype. Moreover, the role of c.1980A>G p.Val660 = (rs11542682, HGMD MUT CM105502) SNP in KRIT1remains controversial. Despite being a synonymous substitution, it seems to impair splicing (11) and, therefore, it was associated to an increased risk of developing CCM. However, as in this case, it is frequently represented in families with hereditary CCM. The absence of the pathological phenotype in III-9 and III-10 despite them also harboring this variant could be attributed to not yet identified modifier factors that contribute to incomplete penetrance of CCM.
Among candidate loci that could act as modifier genes, surely CCM2L (C20orf160) can be considered. This is a CCM2 homologous identified in zebrafish some years ago. Although knowledge about its functions is still poor and controversial, its involvement in cardiovascular development has been confirmed by several studies, as well as its participation in CCM protein signaling. Expression studies showed that CCM2L silencing in zebrafish embryos develop CCM-like phenotypes, comparable with knock-down KRIT1 or HEG1 ones. Like malcavernin, also ccm2l interacts with the N-terminus region of krit1 and, furthermore if one of these is deficient, it can be replaced by the other (24).
CCM2L expression was also observed in endothelial cells where, similar to CCM2, its C-terminus region binds N-terminus of MEKK3 (23) and, in particular, CCM2L can rescue CCM2 deficiency. Therefore, in the case we presented, healthy CCM2 IVS10-1G>A mutation carriers might not have developed CCM phenotype due to a greater compensatory activity of CCM2L. This hypothesis is being evaluated.
Conclusions
In conclusion, here we describe two novel intronic mutations detected in KRIT1 and in CCM2 genes. Reported data are part of a wider project that aims to increase the spectrum of mutations associated to CCM development. In this context, we previously published data collected on more numerous cohorts (12, 25, 26). Regarding the novel mutations detected, we can affirm no evidence of association between KRIT1 IVS15-66A>T, and therefore CCM development in a sporadic patient could not be a consequence of synthesis of an aberrant KRIT1 transcript. Regarding IVS10-1G>A in CCM2, instead, it causes the formation of a truncated protein due to loss of the first nucleotide of the 10th exon that is deleted together with intron 9–10. This mutation was detected in several members belonging to a family affected by hereditary CCM. However, not all subjects harboring the mutation show lesions at MRI; this allows us to report, for the first time, a penetrance for CCM2 locus lower than 100% and to enrich the spectrum of mutations responsible for CCM etiopathogenesis.
Author Contributions
CS designed the study and wrote the manuscript. LD carried out the genetic analysis and validation. ZK and SB performed the clinical evaluation and MRI examination of patients. RD participated in the validation of novel mutation and the sequence alignment, as well as drafting of manuscript. AS oversaw design and conceptualization of the study, and revising the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank Apostolia Papalexandri for her contribution in the experimental phase procedures and the patients who participated to the study.
Abbreviations
CCM, Cerebral cavernous malformations; ICH, Intracerebral hemorrhage; MRI, Magnetic Resonance Imaging,; Krit1, Krev interaction trapped 1; Pdcd10, Programmed cell death protein 10; ECM, cell-extra cellular matrix; HGMD, Human Gene Mutation Database; MLPA, Multiplex Ligation-Dependent Probe Amplification; PCR-RFLP, Polymerase Chain Reaction- Restriction Fragment Length Polymorphism; SNPs, single nucleotide polymorphisms; PTB, phosphotyrosine binding; HHD, Harmonin Homology Domain; MEKK3, mitogen-activated protein 3 kinase.
References
1. Spiegler S, Rath M, Paperlein C, Felbor U. Cerebral cavernous malformations: an update on prevalence, molecular genetic analyses, and genetic counselling. Mol Syndromol. (2018) 9:60–9. doi: 10.1159/000486292
2. Haasdijk RA, Cheng C, Maat-Kievit AJ, Duckers HJ. Cerebral cavernous malformations: from molecular pathogenesis to genetic counselling and clinical management. Eur J Hum Genet. (2012) 20:134–40. doi: 10.1038/ejhg.2011.155
3. Alvarez H, Castillo M. Genetic markers and their influence on cerebrovascular malformations. Neuroimag Clin N Am (2015) 25:69–82. doi: 10.1016/j.nic.2014.09.009
4. Mouchtouris N, Chalouhi N, Chitale A, Starke RM, Tjoumakaris SI, Rosenwasser RH, et al. Management of cerebral cavernous malformations: from diagnosis to treatment. Sci World J. (2015) 2015:808314. doi: 10.1155/2015/808314
5. de Vos IJ, Vreeburg M, Koek GH, van Steensel MA. Review of familial cerebral cavernous malformations and report of seven additional families. J Med Genet A (2017) 173:338–51. doi: 10.1002/ajmg.a.38028
6. Shenkar R, Shi C, Rebeiz T, Stockton RA, McDonald DA, Mikati AG, et al. Exceptional aggressiveness of cerebral cavernous malformation disease associated with PDCD10 mutations. Genet Med. (2015) 17:188–96. doi: 10.1038/gim.2014.97
7. Baranoski JF, Kalani MY, Przybylowski C, Zabramski JM. Cerebral cavernous malformations: review of the genetic and protein-protein interactions resulting in disease pathogenesis. Front Surg. (2017) 3:60. doi: 10.3389/fsurg.2016.00060
8. Cuttano R, Rudini N, Bravi L, Corada M, Giampietro C, Papa E, et al. KLF4 is a key determinant in the development and progression of cerebral cavernous malformations. EMBO Mol Med. (2016) 8:6–24. doi: 10.15252/emmm.201505433
9. Källberg M, Wang H, Wang S, Peng J, Wang Z, Lu H, et al. Template-based protein structure modeling using the RaptorX web server. Nat Protoc. (2012) 7:1511–22. doi: 10.1038/nprot.2012.085
10. Zhang J, Clatterbuck RE, Rigamonti D, Dietz HC. Cloning of the murine Krit1 cDNA reveals novel mammalian 5' coding exons. Genomics (2000) 70:392–5. doi: 10.1006/geno.2000.6410
11. Pileggi S, Buscone S, Ricci C, Patrosso MC, Marocchi A, Brunori P, et al. Genetic variations within KRIT1/CCM1, MGC4607/CCM2 and PDCD10/CCM3 in a large Italian family harbouring a Krit1/CCM1 mutation. J Mol Neurosci. (2010) 42:235–42. doi: 10.1007/s12031-010-9360-y
12. Scimone C, Bramanti P, Alafaci C, Granata F, Piva F, Rinaldi C, et al. Update on novel CCM gene mutations in patients with cerebral cavernous malformations. J Mol Neurosci. (2017) 61:189–98. doi: 10.1007/s12031-016-0863-z
13. Battistini S, Rocchi R, Cerase A, Citterio A, Tassi L, Lando G, et al. Clinical, magnetic resonance imaging, and genetic study of 5 Italian families with cerebral cavernous malformation. Arch Neurol. (2007) 64:843–8. doi: 10.1001/archneur.64.6.843
14. Denier C, Labauge P, Bergametti F, Marchelli F, Riant F, Arnoult M, et al. Genotype-phenotype correlations in cerebral cavernous malformations patients. Ann Neurol. (2006) 60:550–6. doi: 10.1002/ana.20947
15. Dammann P, Wrede K, Zhu Y, Matsushige T, Maderwald S, Umutlu L, et al. Correlation of the venous angioarchitecture of multiple cerebral cavernous malformations with familial or sporadic disease: a susceptibility-;weighted imaging study with 7-Tesla MRI. J Neurosurg. (2017) 126:570–7. doi: 10.3171/2016.2.JNS152322
16. McDonald DA, Shi C, Shenkar R, Gallione CJ, Akers AL, Li S, et al. Lesions from patients with sporadic cerebral cavernous malformations harbor somatic mutations in the CCM genes: evidence for a common biochemical pathway for CCM pathogenesis. Hum Mol Genet. (2014) 23:4357–70. doi: 10.1093/hmg/ddu153
17. Zawistowski JS, Stalheim L, Uhlik MT, Abell AN, Ancrile BB, Johnson GL, et al. CCM1 and CCM2 protein interactions in cell signaling: implications for cerebral cavernous malformations pathogenesis. Hum Mol Genet. (2005) 14:2521–31. doi: 10.1093/hmg/ddi256
18. Fisher OS, Zhang R, Li X, Murphy JW, Demeler B, Boggon TJ. Structural studies of cerebral cavernous malformations 2 (CCM2) reveal a folded helical domain at its C-terminus. FEBS Lett. (2013) 587:272–7. doi: 10.1016/j.febslet.2012.12.011
19. Harel L, Costa B, Tcherpakov M, Zapatka M, Oberthuer A, Hansford LM, et al. CCM2 mediates death signaling by the TrkA receptor tyrosine kinase. Neuron (2009) 63:585–91. doi: 10.1016/j.neuron.2009.08.020
20. Richardson BT, Dibble CF, Borikova AL, Johnson GL. Cerebral cavernous malformation is a vascular disease associated with activated RhoA signaling. Biol Chem. (2013) 394:35–42. doi: 10.1515/hsz-2012-0243
21. Wang X, Hou Y, Deng K, Zhang Y, Wang DC, Ding J. Structural insights into the molecular recognition between cerebral cavernous malformation 2 and mitogen;activated protein kinase kinase kinase 3. Structure (2015) 23:1087–96. doi: 10.1016/j.str.2015.04.003
22. Fisher OS, Deng H, Liu D, Zhang Y, Wei R, Deng Y, et al. Structure and vascular function of MEKK3; cerebral cavernous malformations 2 complex. Nat Commun. (2015) 6:7937. doi: 10.1038/ncomms8937
23. Cullere X, Plovie E, Bennett PM, MacRae CA, Mayadas TN. The cerebral cavernous malformation proteins CCM2L and CCM2 prevent the activation of the MAP kinase MEKK3. Proc Natl Acad Sci USA. (2015) 112:14284–91. doi: 10.1073/pnas.1510495112
24. Rosen JN, Sogah VM, Ye LY, Mably JD. ccm2-like is required for cardiovascular development as a novel component of the Heg.-CCM pathway. Dev Biol. (2013) 376:74–85. doi: 10.1016/j.ydbio.2013.01.006
25. D'Angelo R, Marini V, Rinaldi C, Origone P, Dorcaratto A, Avolio M, et al. Mutation analysis of CCM1, CCM2 and CCM3 genes in a cohort of Italian patients with cerebral cavernous malformation. Brain Pathol. (2011) 21:215–24. doi: 10.1111/j.1750-3639.2010.00441.x
Keywords: CCM, splicing variants, CCM2 penetrance, pathogenesis, CCM genotype-phenotype correlation
Citation: Scimone C, Donato L, Katsarou Z, Bostantjopoulou S, D'Angelo R and Sidoti A (2018) Two Novel KRIT1 and CCM2 Mutations in Patients Affected by Cerebral Cavernous Malformations: New Information on CCM2 Penetrance. Front. Neurol. 9:953. doi: 10.3389/fneur.2018.00953
Received: 26 July 2018; Accepted: 24 October 2018;
Published: 14 November 2018.
Edited by:
Ioannis Dragatsis, University of Tennessee Health Science Center, United StatesReviewed by:
Jun Zhang, Texas Tech University Health Sciences Center, United StatesSouvik Kar, Internationales Neurowissenschaftliches Institut Hannover, Germany
Copyright © 2018 Scimone, Donato, Katsarou, Bostantjopoulou, D'Angelo and Sidoti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rosalia D'Angelo, rdangelo@unime.it