 Carolina Sportelli
Carolina Sportelli Daniele Urso
Daniele Urso Peter Jenner3
Peter Jenner3- 1National Parkinson Foundation International Centre of Excellence, King's College Hospital, London, United Kingdom
- 2Institute of Psychiatry, Psychology & Neuroscience, King's College, London, United Kingdom
- 3Institute of Pharmaceutical Sciences, Faculty of Life Sciences and Medicine, King's College, London, United Kingdom
To date, there are no clinically effective neuroprotective or disease-modifying treatments that can halt Parkinson's disease (PD) progression. The current clinical approach focuses on symptomatic management. This failure may relate to the complex neurobiology underpinning the development of PD and the absence of true translational animal models. In addition, clinical diagnosis of PD relies on presentation of motor symptoms which occur when the neuropathology is already established. These multiple factors could contribute to the unsuccessful development of neuroprotective treatments for PD. Prodromal symptoms develop years prior to formal diagnosis and may provide an excellent tool for early diagnosis and better trial design. Patients with idiopathic rapid eye movement behavior disorder (iRBD) have the highest risk of developing PD and could represent an excellent group to include in neuroprotective trials for PD. In addition, repurposing drugs with excellent safety profiles is an appealing strategy to accelerate drug discovery. The anti-diabetic drug metformin has been shown to target diverse cellular pathways implicated in PD progression. Multiple studies have, additionally, observed the benefits of metformin to counteract other age-related diseases. The purpose of this viewpoint is to discuss metformin's neuroprotective potential by outlining relevant mechanisms of action and the selection of iRBD patients for future clinical trials in PD.
Introduction
PD is the fastest growing neurodegenerative age-related disorder with numbers of patients projected to double by 2040 globally (1). The neuropathology is complex and mainly characterized by two features, a selective degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNpc) and the presence of fibrillar aggregates referred to as Lewy bodies (LBs), mainly composed of α-synuclein, which manifest in motor and non-motor features (2–4). According to Braak's hypothesis, the progressive accumulation of α-synuclein-rich LBs begins in the medulla oblongata and anterior olfactory structures and progresses in a stereotypical bottom-up caudo-rostral direction to the neocortex (5). This concept is concordant with the now recognized prodromal state of PD, in which the ongoing pathological process confined to the lower brainstem areas are associated only with some specific non-motor/pre-motor features of PD (6). Many would therefore argue that the emergence of these symptoms along with some genetic markers in susceptible individuals could mark the beginning of PD.
Despite a massive effort involving preclinical investigations and post-mortem studies, the precise pathogenic mechanisms remains unclear and no unifying mechanism has been discovered to account for neurodegeneration in PD (7). It is likely that PD may occur as a result of multiple variable processes involving a range of pathogenic mechanisms. These include, mitochondrial dysfunction, oxidative stress, protein aggregation, abnormal protein degradation due to alterations in proteostasis mechanisms, neuroinflammation, and aging (8–10). While many of these processes might initiate the pathogenic process, by the time motor symptoms become manifest, a cascade of biochemical events leading to cell death will have become engaged. This might explain why clinical trials aimed at early intervention for neuroprotection or neuromodulation, based on preventing discrete components of this cascade, have consistently failed in the face of the already established widespread pathological change and a maelstrom of disruption of cellular functions. At present there are no clinically effective neuroprotective or disease-modifying strategies available for PD and this remains one of the defining unmet needs in the management of this challenging disorder (11, 12). Worryingly, a plethora of potential neuroprotective agents have been produced on the back of in vitro models of dopaminergic cell death and positive effects in in vivo animal models of the presumed pathogenic processes occurring in PD, but none have translated into effective treatments in man (13, 14). Over a billion US dollars have been spent by charity and industry to fund, develop and validate neuroprotective treatment strategies but to no avail.
The Failure of Current Clinical Trial Designs
An alternative explanation for the failure to develop neuroprotective or disease-modifying strategies may rest with the design of clinical studies as these presume that all patients with PD have identical pathogenic mechanisms underlying their disease, which is unlikely to be true. In addition, most neuroprotective trials in PD have enrolled patients either in a “de novo untreated stage” or “early stable treated stage” in attempts to intervene at a point where the rescue of neurons is still feasible—yet the results have been uniformly negative (15). This approach may be flawed as post-mortem studies show that by 4 years after a clinical diagnosis of PD, there is already virtually complete nigrostriatal denervation of the dorsal putamen, profound nigral cell loss and abundant Lewy pathology, which raises the question of the possibility to achieve neuroprotection even in these “early motor stages” (16). It is thus crucial to understand when to intervene, taking into account the natural history pattern of PD, which develops from a pre-prodromal and prodromal stage progressing through stable and unstable phases to a palliative stage (17) (Supplementary Figure 1).
A body of clinical and pathological evidence support the concept of a prodromal stage of PD existing several years, maybe decades, prior to formal PD diagnosis (6, 18). This prodromal stage theoretically represents the ideal time point during which neurodegeneration has just commenced and restoration or protection is still feasible (19). Indeed, recent observations suggest that mitochondrial dysfunction, increased glycolysis and neuroinflammation occur in the prodromal stage of PD (20). In addition, most PD studies continue to mainly focus on motor endpoints despite PD being recognized to also be a non-motor disorder, with a complex range of non-motor symptoms (NMS) that span from prodromal to advance stages of the disease (21). These include dysphagia, autonomic dysfunction, sleep disorders, mood disturbances, cognitive impairment, and dementia (22, 23). This raises the possibility that undertaking clinical trials in the prodromal phase could be an essential step for investigating disease progression and testing agents with putative neuroprotective or disease modifying effects. In addition, drug repurposing could represent an interesting source of candidates to treat or slow diseases as costs are considerably lower than those needed for designing and optimizing a new drug. Furthermore, the safety and tolerability of many repurposed molecules is likely to have been already been established, making clinical trials more cost-effective and in need of smaller samples sizes. To date, drug repurposing have shown many advantages mostly in symptom management of disease progression (24). A drug has yet to be found that can fully revert or prevent the mechanisms of neurodegeneration, however anti-diabetic drugs have been proven to be safe and potentially effective in the treatment of PD.
Association Between Type 2 Diabetes Mellitus and PD
An association between type 2 diabetes mellitus (T2DM) and PD has previously been reported (25), although meta-analyses of prospective cohort studies now suggest that the risk of developing PD among diabetic patients is quite low (26). Patients with both T2DM and PD present aggravated motor symptoms, higher degree of cognitive impairment and earlier onset of motor complications compared to non-diabetic subjects with PD (27–31). In addition, common pathogenic mechanisms also exist between PD and T2DM (32) and are listed in Table 1.

Table 1. List of common pathogenic mechanisms that may exist between PD and T2DM.
Anti-diabetic Drugs and PD Progression
The potential effect of anti-diabetic drugs on the progression of PD has been assessed by several groups. For example, the use of thiazolidinediones was associated with a decreased risk of developing PD in diabetic patients (40) and a reduction of neurodegeneration and neuroinflammation in animal models (41–44). Incretin mimetic agents such as exenatide may also confer some degree of neuroprotection in functional models of PD (45–48). Exenatide has been shown to reduce dopaminergic cell death, improve motor and cognitive functions, decrease neuroinflammation and mitochondrial dysfunction (45, 49–51). A single-center randomized, double-blind, placebo-controlled trial showed that patients with moderate PD treated with exenatide once a week for 48 weeks had a significant 3.5-point advantage compared with those given placebo in the Movement Disorders Society Unified Parkinson's Disease Rating Scale (52). This study also observed some improvement in cognitive decline associated with PD (53, 54). Undoubtedly these initial findings are encouraging and also provide evidence that anti-diabetic agents may have a therapeutic or potentially a neuroprotective role in the treatment of PD. Furthermore, a national multicentre study addressing the potential of exenatide and neuroprotection has now begun in the UK to explore if the findings of the single-center study can be replicated. We posit that another well-established, well-tolerated anti-diabetic drug, metformin, with a long-established safety record should be also investigated in PD. Preclinical studies have shown that metformin may target most pathological mechanisms involved in PD and improve some aging outcomes. In addition to having a pleiotropic action, metformin has the advantage of being orally administered and thus preferential for patients to use over a long period of time, compared to subcutaneous injections needed to administer exenatide.
Rationale for Selecting Metformin
Metformin is a cheap yet highly effective drug which has been used for over 50 years for the management of T2DM (55, 56). It is a synthetic dimethyl biguanide, orally administrated, which has been shown to reduce total mortality compared to other diabetes agents (57, 58). Metformin has a global safety record, is well-tolerated by the majority of patients and is used by roughly 125 million people worldwide (59). Metformin does not undergo significant metabolism and is excreted unchanged via the organic cation transporter-2 in the kidney (59, 60). Metformin does not have significant adverse effects and it has low risk for hypoglycaemia, however, it may cause vitamin B-12 deficiency (61) and lactic acidosis, mainly in patients with significant renal function impairment (62, 63). It is of important note that reported incidence of lactic acidosis in patients receiving metformin is very low and in over 20,000 patients exposed to metformin in clinical trials, there were no reports of lactic acidosis (64); diarrhea, nausea and stomach upset represent more common side effects (65).
Beyond its anti-diabetic properties, metformin has a pleiotropic action and potentially slows aging by targeting mitochondrial metabolism and insulin signaling (66). Recent studies have demonstrated that metformin can rapidly penetrate the blood–brain barrier (67) and confer neuroprotection against stroke, cognitive impairment, Huntington's disease and potentially prevent dementia (68–73). Metformin can reduce α-synuclein phosphorylation and aggregation, influence cellular processes associated with age-related conditions including inflammation and autophagy, all of which are associated with PD pathogenesis. These actions are described in detail below as they may represent the potential of metformin to be neuroprotective or disease modifying in PD (74) (Figure 1).
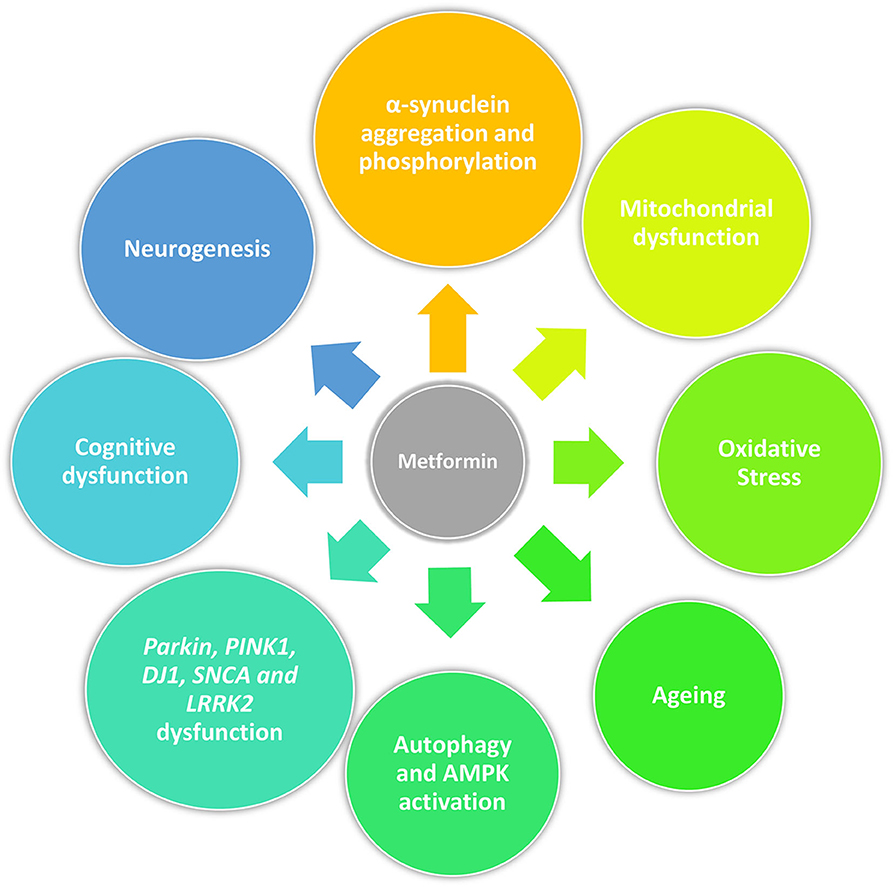
Figure 1. Pleiotropic action of metformin in PD. Beyond its anti-diabetic properties, metformin may act as a neuroprotective drug by reducing α-synuclein phosphorylation and aggregation, mitigating mitochondrial dysfunction and oxidative stress, influencing cellular processes associated with age-related conditions including cellular senescence and autophagy, and promoting neurogenesis. Furthermore, it could restore physiological molecular functions disrupted by genetic mutations related with PD (Parkin, PINK1, DJ1, SNCA, and LRRK2) and have an effect on cognition.
Mechanisms of Action of Metformin
Mitochondrial Dysfunction
Mitochondrial dysfunction is commonly accepted as a key component of the pathogenesis of PD—through the inhibition of complex I and oxidative stress in sporadic disease and the linkage to SNCA, parkin, PINK1, DJ-1, and LRRK2 mediated genetic forms of the illness (9, 75–77). Metformin also acts on mitochondria to alter the activity of the respiratory chain and to decrease reactive oxygen species (ROS) (78–81). This may have functional significance as metformin can protect dopaminergic cells against MPP+ toxicity in vitro by attenuating mitochondrial dysfunction and oxidative stress (82). Whether this translates in vivo is unclear as metformin has not consistently protected dopaminergic cells against toxin induced damage although it may reduce markers of oxidative stress—such as superoxide dismutase (83) and alter the expression of key mitochondrial proteins in basal ganglia (84). Metformin also restores the mitochondrial integrity of dopaminergic neurons disrupted by parkin or LRRK2 mutations in fruit flies (85). Importantly, metformin promotes the expression of peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α) (84), a key regulator of mitochondrial biogenesis (86). PGC-1α is downregulated in the brain in PD and it protects dopaminergic neurones in animal models of PD (87). Although not directly focused on PD, metformin can alter mitochondrial fission and fusion protein expression and mitochondrial fragmentation in experimental systems linked to diabetes-induced oxidative stress, endothelial dysfunction, atherosclerosis development and Down's syndrome (88, 89). All of this being supportive of a potential neuroprotective role.
It is of important note that most of metformin's effects are an indirect result of complex I inhibition, although its exact mechanism warrants further investigation. Complex I alterations are widely associated with mitochondrial dysfunction and PD risk, as previously described in MPTP and rotenone models (90, 91). Despite this contradiction, however, sub-lethal concentrations of complex I inhibitors, such as metformin, which do not generate ROS (or produce a reduced amount), could still be beneficial and have a neuroprotective action (74).
α-Synuclein Aggregation and Phosphorylation
The relationship between α-synuclein accumulation in LBs and neuronal toxicity is strong (92, 93). The evidence from familial SNCA mutations and PD in man coupled to the clear toxicity of fibrils and protofibrils of α-synuclein presents an opportunity for interfering in the final pathogenic process. Based on a range of experimental models of PD, metformin acts to counter the toxicity of the protein. In MPTP-treated mice, metformin reduced α-synuclein expression and the number of α-synuclein positive cells (82). In C. elegans, metformin reduced the loss of dopaminergic neurons and decreased α-synuclein aggregation induced by 6-hydroxydopamine (94). Recently, metformin treatment was shown to attenuate dopaminergic cell loss and α-synuclein accumulation in the SN of rotenone-treated mice (95). How metformin alters α-synuclein toxicity is not clear but the drug is able reduce the phosphorylation of the protein that is key to the mediation of its toxicity (96). This may relate to the ability of metformin to increase the activity of phosphatases involved in α-synuclein dephosphorylation as shown in the SN of MPTP-treated mice (97).
Autophagy
Autophagy is a key cellular mechanism in protein homeostasis on which many of the pathogenic pathways involved in PD eventually converge (98). Autophagy plays a role in α-synuclein handling, mitochondrial function and oxidative stress, emphasizing its potential as a target for neuroprotection and disease modification (99, 100). Metformin has actions in experimental models of PD related to alterations in autophagy. For example, in MPTP-treated mice, metformin treatment prevented dopaminergic cell death and reduced motor impairment while decreasing α-synuclein aggregation, autophagic impairment, and ROS (82). The possible mechanism of this effect is not clear but may involve activation of AMP-activated protein kinase (AMPK) through the mitochondrial effects of metformin which in turn leads to an induction of autophagy involving in part, autophagosome formation and lysosomal biogenesis (101–104). Support for a role of AMPK comes from a study in MPTP-treated mice where downstream effectors of AMPK prevented dopaminergic cell death and motor impairment (105). This is supported by data from other areas, for example ischemia, where metformin exerts neuroprotective actions through manipulation of autophagy (106–108).
Neurogenesis
Dopamine modulates ontogenetic neurogenesis (109). Post-mortem studies suggest that dopamine depletion may impair neuronal precursor cell proliferation in PD (109, 110) and thus negatively impact neurogenesis. While the relationships are not fully understood, impaired neurogenesis in the subgranular zone of the hippocampus and olfactory bulb of PD patients (109) likely contributes to memory deficits (111), depression (112), and olfactory dysfunction (113), commonly present in PD. Wang et al. (114) showed that metformin treatment could stimulate neurogenesis via an atypical PKC-CBP (Protein kinase C-CREB-binding protein) pathway. In particular, the transcription factor CREB (c-AMP response element-binding protein), was found to be a key component for neurodevelopment, cell survival, plasticity, memory and learning (115). Additionally, CREB was shown to regulate TH gene expression (116). At the molecular level, metformin may also upregulate the expression of the brain-derived neurotrophic factor (BDNF) by activating AMPK/CREB-mediated histone acetylation improving the ability of mice to resist stress (117). Therefore, activation of CREB by metformin could boost compensatory and regenerative mechanisms in the brain.
Studies on Metformin Targeting Age-Related Diseases
Beyond the positive effects on multiple PD underlying mechanisms, metformin has also been shown to delay aging and extend lifespan in nematodes and rodents models (118–121). Metformin has, additionally, been considered to be effective in multiple human studies targeting age-related diseases. It was shown to delay cardiovascular disease, providing the rationale for metformin's designation as first-line therapy for most patients with T2DM UK Prospective Diabetes Study (UKPDS) Group (122). A recent study has shown lower mortality in patients with T2DM on metformin compared with non-diabetics despite the fact that the diabetic patients were more obese and had greater co-morbidities at baseline (123). Preliminary data support the concept that metformin may reduce the risk of cognitive impairment and dementia in both T2DM and non-T2DM (69, 72, 124). Long-term metformin therapy was also associated with lower incidence of neurodegenerative disorders among elderly veterans with T2DM (125). An ongoing trial called “Targeting Aging with Metformin” is validating metformin's ability to delay the onset of comorbidities related to aging (126). In relation to PD, clinical studies have mainly investigated the effects of metformin in comparison to, or in combination with other anti-hyperglycaemic agents and taken together all the studies look at different medications and are hardly comparable (40, 127). There is thus a lack of studies specifically evaluating the neuroprotective effects of metformin on PD development and/or progression.
The Selection of an “Enriched” Cohort of Prodromal PD for a Metformin Trial
To confer neuroprotection against PD, in addition to understanding the cellular mechanisms involved in PD pathogenesis, it is crucial to perform studies in a group of patients that reside in the prodromal stage of the disease and who will most certainly phenoconvert to clinical PD within a reasonable time. The identification of such group would allow the testing of a putative neuroprotective or disease-modifying agent, such as metformin, in the ideal time frame to maximize the possible beneficial impacts (18).
The definition of this stage requires working with the probability of conversion to overt PD in large populations of “at-risk” subjects. These include patients with idiopathic rapid eye movement behavior disorder (iRBD), olfactory dysfunction, autonomic dysfunction, depression, excessive daytime sleepiness, constipation, or carriers of a known PD mutation such as LRRK2 or GBA. iRBD is defined as apparent acting out of dreams during REM sleep, associated with a loss of normal REM sleep atonia (128). iRBD has a strong evidence on being a predictor of synucleinopathies as multiple single-center prospective cohort studies have documented that iRBD phenoconverts to PD, dementia with Lewy bodies or multiple system atrophy in 80% of cases over a 6–10 years and possibly to a higher rate over 12 years (129–133). This risk is higher if a person with iRBD also displays other prodromal features of PD (olfactory dysfunction or constipation), shows a reduced uptake on presynaptic dopamine transporters (DaTSCAN) (134–137) or has mild bradykinesia. It is impossible to give a definitive conversion rate in iRBD patients as data are only available from a few highly selected cohort studies (136, 138). However, a likelihood ratio (LR) of several motor and non-motor features of phenoconversion to clinical PD is available (132, 139, 140). The LR is the highest for RBD, followed by a positive DaTSCAN and hyposmia (140). Thus, “enriched” iRBD cases (with hyposmia, or positive DaTSCAN) may provide an earlier window of phenoconversion and an optimal group to study neuroprotection (141). In addition, if such patients have mild bradykinesia, the time of phenoconversion is likely to be within 4 years as shown by a controlled study by Schrag et al. (142). We therefore would propose a randomized double blind placebo-controlled trial with metformin in an enriched iRBD cohort where other risk factors are also comorbid (hyposmia, abnormal DaTSCAN and/or bradykinesia) with a follow up period of 4–6 years to allow for the maximal possibility of phenoconversion to PD.
Conclusion
To date, all clinical disease-modifying trials in PD have been performed in individuals in the manifested “in-life” motor stage, targeting either early “de novo” PD or treated PD and have all failed to show any convincing effects on neuroprotection. Investigating the prodromal phase of the disease could offer greater promise of success, assuming the less-advanced pathology and the greater potential to intervene at key points of the molecular pathogenesis. There are currently no studies using metformin to evaluate a possible protective effect on motor and non-motor functions on PD, although the idea seems compelling. Given its properties and the fact that mitochondrial dysfunction, autophagy, α-synuclein, aging, have all been proposed to be involved in PD pathophysiological processes, and the potential benefits of metformin to counteract age-related disorders (cancer, cardiovascular and neurodegenerative diseases), metformin seems a reasonable pluripotent agent to try given its safety, ease of use and wide availability. The above evidence encourages us to pilot a study investigating the role of metformin as a potentially neuroprotective agent in prodromal PD. iRBD subjects have a high likelihood to convert to PD or a related synucleinopathy and may therefore represent an ideal group for neuroprotective trials, enabling the field to push into investigating the prodromal stage of the disease and hopefully prevent or slow the development of PD.
Data Availability Statement
All datasets generated for this study are included in the article/ Supplementary Material.
Author Contributions
CS conceptualized the paper, drafted, and revised the manuscript. DU drafted and revised the manuscript. PJ revised the manuscript. KC conceptualized the paper and revised the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2020.00556/full#supplementary-material
References
1. Dorsey ER, Bloem BR. The Parkinson Pandemic—A Call to ActionThe Parkinson PandemicThe Parkinson Pandemic. JAMA Neurol. (2018) 75:9–10. doi: 10.1001/jamaneurol.2017.3299
2. Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with lewy bodies. Proc Natl Acad Sci USA. (1998) 95:6469–73.
3. Chaudhuri KR, Healy DG, Schapira AH. Non-motor symptoms of Parkinson's disease: diagnosis and management. Lancet Neurol. (2006) 5:235–45. doi: 10.1016/s1474-4422(06)70373-8
4. Schapira AH. Etiology and pathogenesis of Parkinson disease. Neurol Clin. (2009) 27:583–603, v. doi: 10.1016/j.ncl.2009.04.004
5. Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. (2003) 24, 197–211. doi: 10.1016/S0197-4580(02)00065-9
6. Mahlknecht P, Seppi K, Poewe W. The concept of prodromal parkinson's disease. J Parkinsons Dis. (2015) 5:681–97. doi: 10.3233/jpd-150685
7. Obeso JA, Rodriguez-Oroz MC, Goetz CG, Marin C, Kordower JH, Rodriguez M, et al. Missing pieces in the Parkinson's disease puzzle. Nat Med. (2010) 16:653–61. doi: 10.1038/nm.2165
8. Maiti P, Manna J, Dunbar GL. Current understanding of the molecular mechanisms in Parkinson's disease: Targets for potential treatments. Transl Neurodeg. (2017) 6:28. doi: 10.1186/s40035-017-0099-z
9. Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, et al. Parkinson disease. Nat Rev Dis Primers. (2017) 3:17013. doi: 10.1038/nrdp.2017.13
10. Zeng X-S, Geng W-S, Jia J-J, Chen L, Zhang P-P. Cellular and molecular basis of neurodegeneration in parkinson disease. Front Aging Neurosci. (2018) 10:109. doi: 10.3389/fnagi.2018.00109
11. Athauda D, Foltynie T. The ongoing pursuit of neuroprotective therapies in Parkinson disease. Nat Rev Neurol. (2015) 11:25–40. doi: 10.1038/nrneurol.2014.226
12. Kalia LV, Kalia SK, Lang AE. Disease-modifying strategies for Parkinson's disease. Mov Disord. (2015) 30:1442–50. doi: 10.1002/mds.26354
13. Olanow CW, Kieburtz K, Schapira AH. Why have we failed to achieve neuroprotection in Parkinson's disease? Ann Neurol. (2008) 64 Suppl 2:S101–110. doi: 10.1002/ana.21461
14. Sarkar S, Raymick J, Imam S. Neuroprotective and therapeutic strategies against parkinson's disease: recent perspectives. Int J Mol Sci. (2016) 17:904. doi: 10.3390/ijms17060904
15. Salamon A, Zádori D, Szpisjak L, Klivényi P, Vécsei L. Neuroprotection in Parkinson's disease: facts and hopes. J Neural Transm. (2019) 16:9772–93. doi: 10.1007/s00702-019-02115-8
16. Kordower JH, Olanow CW, Dodiya HB, Chu Y, Beach TG, Adler CH, et al. Disease duration and the integrity of the nigrostriatal system in Parkinson's disease. Brain. (2013) 136(Pt 8):2419–31. doi: 10.1093/brain/awt192
17. Kalia LV, Lang AE. Parkinson's disease. Lancet. (2015) 386:896–912. doi: 10.1016/S0140-6736(14)61393-3
18. Olanow CW, Obeso JA. The significance of defining preclinical or prodromal Parkinson's disease. Mov Disord. (2012) 27:666–9. doi: 10.1002/mds.25019
19. Foffani G, Trigo-Damas I, Pineda-Pardo JA, Blesa J, Rodriguez-Rojas R, Martinez-Fernandez R, et al. Focused ultrasound in Parkinson's disease: a twofold path toward disease modification. Mov Disord. (2019) 34:1262–73. doi: 10.1002/mds.27805
20. Smith AM, Depp C, Ryan BJ, Johnston GI, Alegre-Abarrategui J, Evetts S, et al. Mitochondrial dysfunction and increased glycolysis in prodromal and early Parkinson's blood cells. Mov Disord. (2018) 33:1580–90. doi: 10.1002/mds.104
21. Chaudhuri KR, Sauerbier A. Unraveling the nonmotor mysteries of Parkinson disease. Nat Rev Neurol. (2016) 12:10–1. doi: 10.1038/nrneurol.2015.236
22. Todorova A, Jenner P, Ray Chaudhuri K. Non-motor Parkinson's: integral to motor Parkinson's, yet often neglected. Pract Neurol. (2014) 14:310–22. doi: 10.1136/practneurol-2013-000741
23. Titova N, Padmakumar C, Lewis SJG, Chaudhuri KR. Parkinson's: a syndrome rather than a disease? J Neural Transm (Vienna). (2017) 124:907–14. doi: 10.1007/s00702-016-1667-6
24. Shim JS, Liu JO. Recent advances in drug repositioning for the discovery of new anticancer drugs. Int J Biol Sci. (2014) 10:654–63. doi: 10.7150/ijbs.9224
25. Hu G, Jousilahti P, Bidel S, Antikainen R, Tuomilehto J. Type 2 diabetes and the risk of parkinson's disease. Diabetes Care. (2007) 30:842–7. doi: 10.2337/dc06-2011
26. Cereda E, Barichella M, Pedrolli C, Klersy C, Cassani E, Caccialanza R, et al. Diabetes and risk of Parkinson's disease: a systematic review and meta-analysis. Diabetes Care. (2011) 34:2614–23. doi: 10.2337/dc11-1584
27. Bohnen NI, Kotagal V, Müller MLTM, Koeppe RA, Scott PJH, Albin RL, et al. Diabetes mellitus is independently associated with more severe cognitive impairment in Parkinson disease. Parkinsonism Related Disorders. (2014) 20:1394–8. doi: 10.1016/j.parkreldis.2014.10.008
28. Petrou M, Davatzikos C, Hsieh M, Foerster BR, Albin RL, Kotagal V, et al. Diabetes, gray matter loss, and cognition in the setting of parkinson disease. Acad Radiol. (2016) 23:577–81. doi: 10.1016/j.acra.2015.07.014
29. Ong M, Foo H, Chander RJ, Wen M-C, Au WL, Sitoh YY, et al. Influence of diabetes mellitus on longitudinal atrophy and cognition in Parkinson's disease. J Neurol Sci. (2017) 377:122–6. doi: 10.1016/j.jns.2017.04.010
30. Mohamed Ibrahim N, Ramli R, Koya Kutty S, Shah SA. Earlier onset of motor complications in Parkinson's patients with comorbid diabetes mellitus. Movement Disorders. (2018) 33:1967–8. doi: 10.1002/mds.27526
31. Pagano G, Polychronis S, Wilson H, Giordano B, Ferrara N, Niccolini F, et al. Diabetes mellitus and Parkinson disease. Neurology. (2018) 90:e1654–e1662. doi: 10.1212/wnl.0000000000005475
32. Camargo Maluf F, Feder D, Alves de Siqueira Carvalho A. Analysis of the relationship between type II diabetes mellitus and parkinson's disease: a systematic review. Parkinsons Dis. (2019) 2019:4951379. doi: 10.1155/2019/4951379
33. Morris JK, Bomhoff GL, Gorres BK, Davis VA, Kim J, Lee PP, et al. Insulin resistance impairs nigrostriatal dopamine function. Exp Neurol. (2011) 231:171–80. doi: 10.1016/j.expneurol.2011.06.005
34. Aviles-Olmos I, Limousin P, Lees A, Foltynie T. Parkinson's disease, insulin resistance and novel agents of neuroprotection. Brain. (2012) 136:374–84. doi: 10.1093/brain/aws009
35. Wang L, Zhai Y-Q, Xu L-L, Qiao C, Sun X-L, Ding J-H, et al. Metabolic inflammation exacerbates dopaminergic neuronal degeneration in response to acute MPTP challenge in type 2 diabetes mice. Exp Neurol. (2014) 251:22–9. doi: 10.1016/j.expneurol.2013.11.001
36. Horvath I, Wittung-Stafshede P. Cross-talk between amyloidogenic proteins in type-2 diabetes and Parkinson's disease. Proc Nat Acad Sci USA. (2016) 113:12473–7. doi: 10.1073/pnas.1610371113
37. Rodriguez-Araujo G, Nakagami H, Takami Y, Katsuya T, Akasaka H, Saitoh S, et al. Low alpha-synuclein levels in the blood are associated with insulin resistance. Sci Rep. (2015) 5:12081. doi: 10.1038/srep12081
38. Jain D, Jain R, Eberhard D, Eglinger J, Bugliani M, Piemonti L, et al. Age- and diet-dependent requirement of DJ-1 for glucose homeostasis in mice with implications for human type 2 diabetes. J Mol Cell Biol. (2012) 4:221–30. doi: 10.1093/jmcb/mjs025
39. Deas E, Piipari K, Machhada A, Li A, Gutierrez-del-Arroyo A, Withers DJ, et al. PINK1 deficiency in β-cells increases basal insulin secretion and improves glucose tolerance in mice. Open Biol. (2014) 4:140051. doi: 10.1098/rsob.140051
40. Brakedal B, Flones I, Reiter SF, Torkildsen O, Dolle C, Assmus J, et al. Glitazone use associated with reduced risk of Parkinson's disease. Mov Disord. (2017) 32:1594–9. doi: 10.1002/mds.27128
41. Barbiero JK, Santiago RM, Persike DS, da Silva Fernandes MJ, Tonin FS, da Cunha C, et al. Neuroprotective effects of peroxisome proliferator-activated receptor alpha and gamma agonists in model of parkinsonism induced by intranigral 1-methyl-4-phenyl-1,2,3,6-tetrahyropyridine. Behav Brain Res. (2014) 274:390–9. doi: 10.1016/j.bbr.2014.08.014
42. Martinez AA, Morgese MG, Pisanu A, Macheda T, Paquette MA, Seillier A, et al. Activation of PPAR gamma receptors reduces levodopa-induced dyskinesias in 6-OHDA-lesioned rats. Neurobiol Dis. (2015) 74:295–304. doi: 10.1016/j.nbd.2014.11.024
43. Ren Z, Yang N, Ji C, Zheng J, Wang T, Liu Y, et al. Neuroprotective effects of 5-(4-hydroxy-3-dimethoxybenzylidene)-thiazolidinone in MPTP induced Parkinsonism model in mice. Neuropharmacology. (2015) 93:209–18. doi: 10.1016/j.neuropharm.2015.01.030
44. Pinto M, Nissanka N, Peralta S, Brambilla R, Diaz F, Moraes CT. Pioglitazone ameliorates the phenotype of a novel Parkinson's disease mouse model by reducing neuroinflammation. Mol Neurodegener. (2016) 11:25. doi: 10.1186/s13024-016-0090-7
45. Cao L, Li D, Feng P, Li L, Xue G-F, Li G, et al. A novel dual GLP-1 and GIP incretin receptor agonist is neuroprotective in a mouse model of Parkinson's disease by reducing chronic inflammation in the brain. NeuroReport. (2016) 27:384–91. doi: 10.1097/wnr.0000000000000548
46. Hansen HH, Fabricius K, Barkholt P, Mikkelsen JD, Jelsing J, Pyke C, et al. Characterization of liraglutide, a glucagon-like peptide-1 (GLP-1) receptor agonist, in rat partial and full nigral 6-hydroxydopamine lesion models of Parkinson's disease. Brain Res. (2016) 1646:354–65. doi: 10.1016/j.brainres.2016.05.038
47. Jalewa J, Sharma MK, Hölscher C. Novel incretin analogues improve autophagy and protect from mitochondrial stress induced by rotenone in SH-SY5Y cells. J Neurochem. (2016) 139:55–67. doi: 10.1111/jnc.13736
48. Zhang L, Zhang L, Li L, Hölscher C. Semaglutide is neuroprotective and reduces α-synuclein levels in the chronic MPTP mouse model of parkinson's disease. J Parkinsons Dis. (2019) 9:157–71. doi: 10.3233/jpd-181503
49. Bertilsson G, Patrone C, Zachrisson O, Andersson A, Dannaeus K, Heidrich J, et al. Peptide hormone exendin-4 stimulates subventricular zone neurogenesis in the adult rodent brain and induces recovery in an animal model of parkinson's disease. J Neurosci Res. (2008) 86:326–38. doi: 10.1002/jnr.21483
50. Harkavyi A, Abuirmeileh A, Lever R, Kingsbury AE, Biggs CS, Whitton PS. Glucagon-like peptide 1 receptor stimulation reverses key deficits in distinct rodent models of Parkinson's disease. J Neuroinfl. (2008) 5:19. doi: 10.1186/1742-2094-5-19
51. Aviles-Olmos I, Dickson J, Kefalopoulou Z, Djamshidian A, Ell P, Soderlund T, et al. Exenatide and the treatment of patients with Parkinson's disease. J Clin Invest. (2013) 123:2730–6. doi: 10.1172/JCI68295
52. Athauda D, Maclagan K, Skene SS, Bajwa-Joseph M, Letchford D, Chowdhury K, et al. Exenatide once weekly versus placebo in Parkinson's disease: a randomised, double-blind, placebo-controlled trial. Lancet. (2017) 390:1664–75. doi: 10.1016/S0140-6736(17)31585-4
53. Athauda D, Wyse R, Brundin P, Foltynie T. Is exenatide a treatment for parkinson's disease? J Parkinson's Dis. (2017) 7:451–8. doi: 10.3233/JPD-171192
54. Jankovic J. Exenatide – a drug for diabetes and Parkinson disease? Nat Rev Neurol. (2017) 13:643–4. doi: 10.1038/nrneurol.2017.140
55. Goodarzi MO, Bryer-Ash M. Metformin revisited: re-evaluation of its properties and role in the pharmacopeia of modern antidiabetic agents. Diab Obesity Metab. (2005) 7:654–65. doi: 10.1111/j.1463-1326.2004.00448.x
56. Rojas LBA, Gomes MB. Metformin: an old but still the best treatment for type 2 diabetes. Diabetol Metab Syndrome. (2013) 5:6. doi: 10.1186/1758-5996-5-6
57. Johnson JA, Simpson SH, Toth EL, Majumdar SR. Reduced cardiovascular morbidity and mortality associated with metformin use in subjects with Type 2 diabetes. Diabetic Med. (2005) 22:497–502. doi: 10.1111/j.1464-5491.2005.01448.x
58. Campbell JM, Bellman SM, Stephenson MD, Lisy K. Metformin reduces all-cause mortality and diseases of ageing independent of its effect on diabetes control: a systematic review and meta-analysis. Ageing Res Rev. (2017) 40:31–44. doi: 10.1016/j.arr.2017.08.003
59. Triggle CR, Ding H. Metformin is not just an antihyperglycaemic drug but also has protective effects on the vascular endothelium. Acta Physiol (Oxf). (2017) 219:138–51. doi: 10.1111/apha.12644
60. Kimura N, Masuda S, Tanihara Y, Ueo H, Okuda M, Katsura T, et al. Metformin is a superior substrate for renal organic cation transporter OCT2 rather than hepatic OCT1. Drug Metab Pharmacokinet. (2005) 20:379–86. doi: 10.2133/dmpk.20.379
61. de Jager J, Kooy A, Lehert P, Wulffelé MG, van der Kolk J, Bets D, et al. (2010). Long term treatment with metformin in patients with type 2 diabetes and risk of vitamin B-12 deficiency: randomised placebo controlled trial. BMJ. 340:c2181. doi: 10.1136/bmj.c2181
62. Lalau JD. Lactic acidosis induced by metformin: incidence, management and prevention. Drug Saf. (2010) 33:727–40. doi: 10.2165/11536790-000000000-00000
63. Scheen AJ, Paquot N. Metformin revisited: a critical review of the benefit-risk balance in at-risk patients with type 2 diabetes. Diabetes Metab. (2013) 39:179–90. doi: 10.1016/j.diabet.2013.02.006
64. Crowley MJ, Diamantidis CJ, McDuffie JR, Cameron B, Stanifer J, Mock CK, et al. VA evidence-based synthesis program reports. In: Metformin Use in Patients With Historical Contraindications or Precautions. Washington, DC: Department of Veterans Affairs (2016).
66. Valencia WM, Palacio A, Tamariz L, Florez H. Metformin and ageing: improving ageing outcomes beyond glycaemic control. Diabetologia. (2017) 60:1630–8. doi: 10.1007/s00125-017-4349-5
67. Labuzek K, Suchy D, Gabryel B, Bielecka A, Liber S, Okopien B. Quantification of metformin by the HPLC method in brain regions, cerebrospinal fluid and plasma of rats treated with lipopolysaccharide. Pharmacol Rep. (2010) 62:956–65. doi: 10.1016/s1734-1140(10)70357-1
68. Jiang T, Yu J-T, Zhu X-C, Wang H-F, Tan M-S, Cao L, et al. Acute metformin preconditioning confers neuroprotection against focal cerebral ischaemia by pre-activation of AMPK-dependent autophagy. Br J Pharmacol. (2014) 171:3146–57. doi: 10.1111/bph.12655
69. Ng TP, Feng L, Yap KB, Lee TS, Tan CH, Winblad B. Long-term metformin usage and cognitive function among older adults with diabetes. J Alzheimers Dis. (2014) 41:61–8. doi: 10.3233/jad-131901
70. Palleria C, Leporini C, Maida F, Succurro E, De Sarro G, Arturi F, et al. Potential effects of current drug therapies on cognitive impairment in patients with type 2 diabetes. Front Neuroendocrinol. (2016) 42:76–92. doi: 10.1016/j.yfrne.2016.07.002
71. Vazquez-Manrique RP, Farina F, Cambon K, Dolores Sequedo M, Parker AJ, Millan JM, et al. AMPK activation protects from neuronal dysfunction and vulnerability across nematode, cellular and mouse models of Huntington's disease. Hum Mol Genet. (2016) 25:1043–58. doi: 10.1093/hmg/ddv513
72. Campbell JM, Stephenson MD, de Courten B, Chapman I, Bellman SM, Aromataris E. Metformin use associated with reduced risk of dementia in patients with diabetes: a systematic review and meta-analysis. J Alzheimer's Dis. (2018) 65:1225–36. doi: 10.3233/JAD-180263
73. Lin Y, Wang K, Ma C, Wang X, Gong Z, Zhang R, et al. Evaluation of metformin on cognitive improvement in patients with non-dementia vascular cognitive impairment and abnormal glucose metabolism. Front Aging Neurosci. (2018) 10:227. doi: 10.3389/fnagi.2018.00227
74. Rotermund C, Machetanz G, Fitzgerald JC. The therapeutic potential of metformin in neurodegenerative diseases. Front Endocrinol. (2018) 9:400. doi: 10.3389/fendo.2018.00400
75. Schapira AH. Mitochondrial dysfunction in Parkinson's disease. Cell Death Differ. (2007) 14:1261–6. doi: 10.1038/sj.cdd.4402160
76. Bose A, Beal MF. Mitochondrial dysfunction in Parkinson's disease. J Neurochem. (2016) 139 (Suppl. 1):216–31. doi: 10.1111/jnc.13731
77. Lill CM. Genetics of Parkinson's disease. Mol Cell Probes. (2016) 30:386–96. doi: 10.1016/j.mcp.2016.11.001
78. El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. (2000) 275:223–8. doi: 10.1074/jbc.275.1.223
79. Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. (2000) 348:607–14. doi: 10.1042/0264-6021:3480607
80. Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. (2001) 108:1167–74. doi: 10.1172/jci13505
81. Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, et al. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. (2005) 2:21–33. doi: 10.1016/j.cmet.2005.06.005
82. Lu M, Su C, Qiao C, Bian Y, Ding J, Hu G. Metformin prevents dopaminergic neuron death in mptp/p-induced mouse model of parkinson's disease via autophagy and mitochondrial ROS clearance. Int J Neuropsychopharmacol. (2016) 19:pyw047. doi: 10.1093/ijnp/pyw047
83. Patil SP, Jain PD, Ghumatkar PJ, Tambe R, Sathaye S. Neuroprotective effect of metformin in MPTP-induced Parkinson's disease in mice. Neuroscience. (2014) 277:747–54. doi: 10.1016/j.neuroscience.2014.07.046
84. Kang H, Khang R, Ham S, Jeong GR, Kim H, Jo M, et al. Activation of the ATF2/CREB-PGC-1α pathway by metformin leads to dopaminergic neuroprotection. Oncotarget. (2017) 8:48603–18. doi: 10.18632/oncotarget.18122
85. Ng C-H, Guan MSH, Koh C, Ouyang X, Yu F, Tan E-K, et al. AMP kinase activation mitigates dopaminergic dysfunction and mitochondrial abnormalities in Drosophila models of Parkinson's disease. J Neurosci. (2012) 32:14311–7. doi: 10.1523/JNEUROSCI.0499-12.2012
86. Mud ò G, Mäkel ä J, Di Liberto V, Tselykh TV, Olivieri M, Piepponen P, et al. Transgenic expression and activation of PGC-1α protect dopaminergic neurons in the MPTP mouse model of Parkinson's disease. Cell Mol Life Sci. (2012) 69:1153–65. doi: 10.1007/s00018-011-0850-z
87. Zheng B, Liao Z, Locascio JJ, Lesniak KA, Roderick SS, Watt ML, et al. PGC-1α, a potential therapeutic target for early intervention in Parkinson's disease. Sci Transl Med. (2010) 2:52ra73. doi: 10.1126/scitranslmed.3001059
88. Izzo A, Nitti M, Mollo N, Paladino S, Procaccini C, Faicchia D, et al. Metformin restores the mitochondrial network and reverses mitochondrial dysfunction in Down syndrome cells. Hum Mol Genet. (2017) 26:1056–69. doi: 10.1093/hmg/ddx016
89. Wang Q, Zhang M, Torres G, Wu S, Ouyang C, Xie Z, et al. Metformin suppresses diabetes-accelerated atherosclerosis via the inhibition of Drp1-mediated mitochondrial fission. Diabetes. (2017) 66:193–205. doi: 10.2337/db16-0915
90. Nicklas WJ, Vyas I, Heikkila RE. Inhibition of NADH-linked oxidation in brain mitochondria by 1-methyl-4-phenyl-pyridine, a metabolite of the neurotoxin, 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. Life Sci. (1985) 36:2503–8.
91. Betarbet R, Sherer T, MacKenzie G, Garcia-Osuna M, Panov A, Greenamyre J. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. (2000) 3:1301–6. doi: 10.1038/81834
92. Jakes R, Spillantini MG, Goedert M. Identification of two distinct synucleins from human brain. FEBS Lett. (1994) 345:27–32.
93. Wakabayashi K, Tanji K, Mori F, Takahashi H. The Lewy body in Parkinson's disease: molecules implicated in the formation and degradation of alpha-synuclein aggregates. Neuropathology. (2007) 27:494−506. doi: 10.1007/s12035-012-8280-y
94. Saewanee N, Praputpittaya T, Malaiwong N, Chalorak P, Meemon K. Neuroprotective effect of metformin on dopaminergic neurodegeneration α-synuclein aggregation in C. elegans model of Parkinson's disease. Neurosci Res. (2019). doi: 10.1016/j.neures.2019.12.017. [Epub ahead of print].
95. Ozbey G, Nemutlu-Samur D, Parlak H, Yildirim S, Aslan M, Tanriover G, et al. Metformin protects rotenone-induced dopaminergic neurodegeneration by reducing lipid peroxidation. Pharmacol Rep. (2020) doi: 10.1007/s43440-020-00095-1. [Epub ahead of print].
96. Pérez-Revuelta BI, Hettich MM, Ciociaro A, Rotermund C, Kahle PJ, Krauss S, et al. Metformin lowers Ser-129 phosphorylated α-synuclein levels via mTOR-dependent protein phosphatase 2A activation. Cell Death Dis. (2014) 5:e1209. doi: 10.1038/cddis.2014.175
97. Katila N, Bhurtel S, Shadfar S, Srivastav S, Neupane S, Ojha U, et al. Metformin lowers alpha-synuclein phosphorylation and upregulates neurotrophic factor in the MPTP mouse model of Parkinson's disease. Neuropharmacology. (2017) 125:396–407. doi: 10.1016/j.neuropharm.2017.08.015
98. Lehtonen Š, Sonninen T-M, Wojciechowski S, Goldsteins G, Koistinaho J. Dysfunction of Cellular Proteostasis in Parkinson's Disease. Front Neurosci. (2019) 13:457. doi: 10.3389/fnins.2019.00457
99. Lynch-Day MA, Mao K, Wang K, Zhao M, Klionsky DJ. The role of autophagy in Parkinson's disease. Cold Spring Harb Perspect Med. (2012) 2:a009357. doi: 10.1101/cshperspect.a009357
100. Moors TE, Hoozemans JJM, Ingrassia A, Beccari T, Parnetti L, Chartier-Harlin M-C, et al. Therapeutic potential of autophagy-enhancing agents in Parkinson's disease. Mol Neurodegener. (2017) 12:11. doi: 10.1186/s13024-017-0154-3
101. Russell RC, Tian Y, Yuan H, Park HW, Chang Y-Y, Kim J, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. (2013) 15:741–50. doi: 10.1038/ncb2757
102. Young NP, Kamireddy A, Van Nostrand JL, Eichner LJ, Shokhirev MN, Dayn Y, et al. AMPK governs lineage specification through Tfeb-dependent regulation of lysosomes. Genes Dev. (2016) 30:535–52. doi: 10.1101/gad.274142.115
103. Zhang CS, Li M, Ma T, Zong Y, Cui J, Feng JW, et al. Metformin activates AMPK through the lysosomal pathway. Cell Metab. (2016) 24:521–2. doi: 10.1016/j.cmet.2016.09.003
104. Collodet C, Foretz M, Deak M, Bultot L, Metairon S, Viollet B, et al. AMPK promotes induction of the tumor suppressor FLCN through activation of TFEB independently of mTOR. FASEB J. (2019) 33:12374–91. doi: 10.1096/fj.201900841R
105. Choi J-S, Park C, Jeong J-W. AMP-activated protein kinase is activated in Parkinson's disease models mediated by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Biochem Biophys Res Commun. (2010) 391:147–51. doi: 10.1016/j.bbrc.2009.11.022
106. Ashabi G, Khodagholi F, Khalaj L, Goudarzvand M, Nasiri M. Activation of AMP-activated protein kinase by metformin protects against global cerebral ischemia in male rats: interference of AMPK/PGC-1α pathway. Metab Brain Dis. (2014) 29:47–58. doi: 10.1007/s11011-013-9475-2
107. Ashabi G, Khalaj L, Khodagholi F, Goudarzvand M, Sarkaki A. Pre-treatment with metformin activates Nrf2 antioxidant pathways and inhibits inflammatory responses through induction of AMPK after transient global cerebral ischemia. Metab Brain Dis. (2015) 30:747–54. doi: 10.1007/s11011-014-9632-2
108. Sarkaki A, Farbood Y, Badavi M, Khalaj L, Khodagholi F, Ashabi G. Metformin improves anxiety-like behaviors through AMPK-dependent regulation of autophagy following transient forebrain ischemia. Metab Brain Dis. (2015) 30:1139–50. doi: 10.1007/s11011-015-9677-x
109. Höglinger GU, Rizk P, Muriel MP, Duyckaerts C, Oertel WH, Caille I, et al. Dopamine depletion impairs precursor cell proliferation in Parkinson disease. Nat Neurosci. (2004) 7:726–35. doi: 10.1038/nn1265
110. O'Keeffe GC, Tyers P, Aarsland D, Dalley JW, Barker RA, Caldwell MA. Dopamine-induced proliferation of adult neural precursor cells in the mammalian subventricular zone is mediated through EGF. Proc Natl Acad Sci USA. (2009) 106:8754–9. doi: 10.1073/pnas.0803955106
111. Carlesimo G, Piras F, Assogna F, Pontieri F, Caltagirone C, Spalletta G. Hippocampal abnormalities and memory deficits in Parkinson disease A multimodal imaging study. Neurology. (2012) 78:1939–45. doi: 10.1212/WNL.0b013e318259e1c5
112. Ballanger B, Klinger H, Eche J, Lerond J, Vallet AE, Le Bars D, et al. Role of serotonergic 1A receptor dysfunction in depression associated with Parkinson's disease. Mov Disord. (2012) 27:84–9. doi: 10.1002/mds.23895
113. Bohnen NI, Gedela S, Herath P, Constantine GM, Moore RY. Selective hyposmia in Parkinson disease: association with hippocampal dopamine activity. Neurosci Lett. (2008) 447:12–6. doi: 10.1016/j.neulet.2008.09.070
114. Wang J, Gallagher D, DeVito LM, Cancino GI, Tsui D, He L, et al. Metformin activates an atypical PKC-CBP pathway to promote neurogenesis and enhance spatial memory formation. Cell Stem Cell. (2012) 11:23–35. doi: 10.1016/j.stem.2012.03.016
115. Sakamoto K, Karelina K, Obrietan K. CREB: a multifaceted regulator of neuronal plasticity and protection. J Neurochem. (2011) 116:1–9. doi: 10.1111/j.1471-4159.2010.07080.x
116. Piech-Dumas KM, Tank AW. CREB mediates the cAMP-responsiveness of the tyrosine hydroxylase gene: use of an antisense RNA strategy to produce CREB-deficient PC12 cell lines. Mol Brain Res. (1999) 70:219–30. doi: 10.1016/S0169-328X(99)00149-7
117. Fang W, Zhang J, Hong L, Huang W, Dai X, Ye Q, et al. Metformin ameliorates stress-induced depression-like behaviors via enhancing the expression of BDNF by activating AMPK/CREB-mediated histone acetylation. J Affect Disord. (2020) 260:302–13. doi: 10.1016/j.jad.2019.09.013
118. Anisimov VN, Berstein LM, Egormin PA, Piskunova TS, Popovich IG, Zabezhinski MA, et al. Metformin slows down aging and extends life span of female SHR mice. Cell Cycle. (2008) 7:2769–73. doi: 10.4161/cc.7.17.6625
119. Anisimov VN, Egormin PA, Piskunova TS, Popovich IG, Tyndyk ML, Yurova MN, et al. Metformin extends life span of HER-2/neu transgenic mice and in combination with melatonin inhibits growth of transplantable tumors in vivo. Cell Cycle. (2010) 9:188–97. doi: 10.4161/cc.9.1.10407
120. Cabreiro F, Au C, Leung KY, Vergara-Irigaray N, Cochemé HM, Noori T, et al. Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell. (2013) 153:228–39. doi: 10.1016/j.cell.2013.02.035
121. De Haes W, Frooninckx L, Van Assche R, Smolders A, Depuydt G, Billen J, et al. Metformin promotes lifespan through mitohormesis via the peroxiredoxin PRDX-2. Proc Natl Acad Sci USA. (2014) 111:E2501–9. doi: 10.1073/pnas.1321776111
122. UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet. (1998) 352:854–65.
123. Bannister CA, Holden SE, Jenkins-Jones S, Morgan CL, Halcox JP, Schernthaner G, et al. Can people with type 2 diabetes live longer than those without? A comparison of mortality in people initiated with metformin or sulphonylurea monotherapy and matched, non-diabetic controls. Diabetes Obes Metab. (2014) 16:1165–73. doi: 10.1111/dom.12354
124. Cheng C, Lin CH, Tsai YW, Tsai CJ, Chou PH, Lan TH. Type 2 diabetes and antidiabetic medications in relation to dementia diagnosis. J Gerontol A Biol Sci Med Sci. (2014) 69:1299–305. doi: 10.1093/gerona/glu073
125. Shi Q, Liu S, Fonseca VA, Thethi TK, Shi L. Effect of metformin on neurodegenerative disease among elderly adult US veterans with type 2 diabetes mellitus. BMJ Open. (2019) 9:e024954. doi: 10.1136/bmjopen-2018-024954
126. Barzilai N, Crandall JP, Kritchevsky SB, Espeland MA. Metformin as a Tool to Target Aging. Cell metabolism. (2016) 23:1060–5. doi: 10.1016/j.cmet.2016.05.011
127. Wahlqvist ML, Lee MS, Hsu CC, Chuang SY, Lee JT, Tsai HN. Metformin-inclusive sulfonylurea therapy reduces the risk of Parkinson's disease occurring with Type 2 diabetes in a Taiwanese population cohort. Parkinsonism Relat Disord. (2012) 18:753–8. doi: 10.1016/j.parkreldis.2012.03.010
128. Schenck CH, Montplaisir JY, Frauscher B, Hogl B, Gagnon JF, Postuma R, et al. Rapid eye movement sleep behavior disorder: devising controlled active treatment studies for symptomatic and neuroprotective therapy–a consensus statement from the International Rapid Eye Movement Sleep Behavior Disorder Study Group. Sleep Med. (2013) 14:795–806. doi: 10.1016/j.sleep.2013.02.016
129. Postuma RB, Gagnon JF, Vendette M, Fantini ML, Massicotte-Marquez J, Montplaisir J. Quantifying the risk of neurodegenerative disease in idiopathic REM sleep behavior disorder. Neurology. (2009) 72:1296–300. doi: 10.1212/01.wnl.0000340980.19702.6e
130. Iranzo A, Tolosa E, Gelpi E, Molinuevo JL, Valldeoriola F, Serradell M, et al. Neurodegenerative disease status and post-mortem pathology in idiopathic rapid-eye-movement sleep behaviour disorder: an observational cohort study. Lancet Neurol. (2013) 12:443–53. doi: 10.1016/s1474-4422(13)70056-5
131. Schenck CH, Boeve BF, Mahowald MW. Delayed emergence of a parkinsonian disorder or dementia in 81% of older men initially diagnosed with idiopathic rapid eye movement sleep behavior disorder: a 16-year update on a previously reported series. Sleep Med. (2013) 14:744–8. doi: 10.1016/j.sleep.2012.10.009
132. Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W, et al. MDS clinical diagnostic criteria for Parkinson's disease. Mov Disord. (2015) 30:1591–601. doi: 10.1002/mds.26424
133. Postuma RB, Gagnon J-F, Bertrand J-A, Génier Marchand D, Montplaisir JY. Parkinson risk in idiopathic REM sleep behavior disorder: preparing for neuroprotective trials. Neurology. (2015) 84:1104–13. doi: 10.1212/WNL.0000000000001364
134. Mahlknecht P, Iranzo A, Högl B, Frauscher B, Müller C, Santamaría J, et al. Olfactory dysfunction predicts early transition to a Lewy body disease in idiopathic RBD. Neurology. (2015) 84:654–8. doi: 10.1212/wnl.0000000000001265
135. Li Y, Kang W, Zhang L, Zhou L, Niu M, Liu J. Hyposmia Is Associated with RBD for PD Patients with Variants of SNCA. Front Aging Neurosci. (2017) 9:303–303. doi: 10.3389/fnagi.2017.00303
136. Fereshtehnejad S-M, Yao C, Pelletier A, Montplaisir JY, Gagnon J-F, Postuma RB. Evolution of prodromal Parkinson's disease and dementia with Lewy bodies: a prospective study. Brain. (2019) 142:2051–67. doi: 10.1093/brain/awz111
137. Postuma RB, Iranzo A, Hu M, Högl B, Boeve BF, Manni R, et al. Risk and predictors of dementia and parkinsonism in idiopathic REM sleep behaviour disorder: a multicentre study. Brain. (2019) 142:744–59. doi: 10.1093/brain/awz030%JBrain
138. Postuma RB, Iranzo A, Hogl B, Arnulf I, Ferini-Strambi L, Manni R, et al. Risk factors for neurodegeneration in idiopathic rapid eye movement sleep behavior disorder: a multicenter study. Ann Neurol. (2015) 77:830–9. doi: 10.1002/ana.24385
139. Postuma R, Berg D. MDS research criteria for prodromal pakrinson's disease (I1.011). Neurology. (2016) 86:I1.011. doi: 10.1002/mds.26431
140. Heinzel S, Berg D, Gasser T, Chen H, Yao C, Postuma RB. Update of the MDS research criteria for prodromal Parkinson's disease. Mov Disord. (2019) 34:1464–70. doi: 10.1002/mds.27802
141. Torres N, Molet J, Moro C, Mitrofanis J, Benabid AL. Neuroprotective surgical strategies in parkinson's disease: role of preclinical data. Int J Mol Sci. (2017) 18:2190. doi: 10.3390/ijms18102190
Keywords: Parkinson's disease, prodromal, metformin, neuroprotection, idiopathic REM behavior disorder
Citation: Sportelli C, Urso D, Jenner P and Chaudhuri KR (2020) Metformin as a Potential Neuroprotective Agent in Prodromal Parkinson's Disease—Viewpoint. Front. Neurol. 11:556. doi: 10.3389/fneur.2020.00556
Received: 03 April 2020; Accepted: 15 May 2020;
Published: 12 June 2020.
Edited by:
Pedro J. Garcia-Ruiz, University Hospital Fundación Jiménez Díaz, SpainReviewed by:
Juan Carlos Martinez Castrillo, Hospital Universitario Ramón y Cajal, SpainMicaela Morelli, University of Cagliari, Italy
Copyright © 2020 Sportelli, Urso, Jenner and Chaudhuri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carolina Sportelli, carolina.sportelli@nhs.net; Daniele Urso, daniele.urso@kcl.ac.uk