 Satyakam Bhagavati
Satyakam Bhagavati- Department of Neurology, Downstate Medical Center, State University of New York College of Medicine, New York, NY, United States
Remarkable discoveries over the last two decades have elucidated the autoimmune basis of several, previously poorly understood, neurological disorders. Autoimmune disorders of the nervous system may affect any part of the nervous system, including the brain and spinal cord (central nervous system, CNS) and also the peripheral nerves, neuromuscular junction and skeletal muscle (peripheral nervous system, PNS). This comprehensive overview of this rapidly evolving field presents the factors which may trigger breakdown of self-tolerance and development of autoimmune disease in some individuals. Then the pathophysiological basis and clinical features of autoimmune diseases of the nervous system are outlined, with an emphasis on the features which are important to recognize for accurate clinical diagnosis. Finally the latest therapies for autoimmune CNS and PNS disorders and their mechanisms of action and the most promising research avenues for targeted immunotherapy are discussed.
Introduction
The immune system has evolved to have the extraordinary ability to protect us from a vast multitude of infectious agents. However, in an estimated 4.5% of individuals the immune system attacks the very individual it is designed to protect (1). Abnormal immune responses against self can result in more than 80 autoimmune diseases (2) including about 30 autoimmune disorders of the nervous system.
In this article autoimmune disorders of the central and peripheral nervous system are discussed, including disorders where an auto-antigen has yet to be defined. These include central nervous system demyelinating disorders such as multiple sclerosis and neuromyelitis optica, paraneoplastic, and other autoimmune encephalomyelitis and autoimmune inflammatory myositis and demyelinating neuropathies. Neuroinflammatory disorders due to other causes such as cerebral degeneration or nervous system involvement secondary to systemic autoimmune disorders such as systemic lupus erythematosus are not included in this discussion.
Failure of Self Tolerance and Development of Autoimmunity
What are the factors that lead to the breakdown of tolerance in some individuals?
Genetic Susceptibility
In multiple sclerosis (MS), concordance in monozygotic twins of about 25% compared to ~5% in dizygotic twins (3); and in myasthenia gravis (MG) concordance of ~35% in monozygotic twins compared to ~5% in dizygotic twins (4), suggests the contribution of genetic factors to disease causation. Also higher incidence in females of most autoimmune diseases [~80% of all patients (5); neuro-myelitis optica, NMO, 9:1 (6); MS 3:1, early onset MG 3:1], points to the importance of still poorly understood X chromosome factors in genetic susceptibility (7), although effects of sex hormones on immune responses may also play an important role (8).
The MHC locus makes a greater contribution to risk of autoimmune disease than any other loci. MHCII molecules on antigen presenting cells capture peptides and present them to T cells. The MHC locus is also the most polymorphic in the human genome, with a differential ability to bind and efficiently present peptides. Such variations play an important role in the differences between individuals in their immune response to auto-antigens, and is the basis for the strong association between specific MHC alleles and predisposition to autoimmune disease (as in myasthenia gravis or multiple sclerosis). For example, the HLA-peptide T cell receptor binding geometry may play a crucial role in disease causation (9). An off-center binding topology (as has been demonstrated with a T cell clone isolated from a MS patient), may permit autoreactive T cell clones to escape negative thymic selection but still cause disease (10). In other examples of strong genetic predisposition, anti-LGI1 encephalitis shows a strong HLA association with the DRB1*07:01 allele, which is carried by 90% of patients vs. 13% of healthy controls (11); anti-IgLON5 disease is tightly associated with the DRB1*10.01 allele (12); CASPR2-antibody patients show over representation of HLA-DRB1*11:01 (13); MUSK-Myasthenia gravis is tightly associated with the HLADR14-DQ5 haplotype (14) and early onset anti-AChR MG with HLA-B*8:01 and late onset MG with HLA DRB1* 15:01 (15).
In addition, genome wide association studies (GWAS) have identified hundreds of single nucleotide polymorphisms (SNP; variations in the genome at the single nucleotide level) which increase the risk of developing autoimmune disease by small to moderate amounts. Many of these SNPs are in non-coding regions of the genome and the molecular and cellular consequences of these subtle variations are complex and largely undefined. But it is known that autoimmune disease risk SNPs cluster in genes in key immunological pathways. A few examples of how inherited genetic variations can predispose individuals to autoimmune disease are: (1) A recent whole-genome sequence study showed that deletion of complement component C4A gene in the MHC class III region heightens risk for NMO and also SLE (16); (2) Interleukin-2 receptor (IL2R) signaling is essential for optimal stability and function of immunosuppressive Treg cells. Polymorphisms in genes in this pathway such as the IL2R locus leads to decreased responsiveness to IL2 and increased susceptibility to MS (17); (3) Nucleotide variations in the promoter of the muscle acetyl choline receptor (AChR) gene, associated with early onset of myasthenia gravis, prevent binding of interferon 8 which leads to decreased expression of AChR gene in thymic medullary cells (18). This may raise the threshold and impair clonal deletion of AChR auto-reactive T cells in the thymus; (4) Rare polymorphisms in coding sequences of genes important in immunity (for example in perforin, important in granzyme mediated lymphocyte cytotoxicity) increase MS risk (19).
Factors Which May Trigger Autoimmune Disease
Infections
Although not fully understood, epidemiological and clinical evidence suggests that autoimmune disease may be triggered by infections (20). In MS, it has been proposed that lifelong infection with EB virus or CMV infection creates a pool of auto-reactive memory T cells which may be reactivated when exposed to antigens released after CNS injury (21). Similarly high rates of preceding infection have been noted before MOG- antibody associated disease. Molecular mimicry between host self-antigens and microbial antigens is a leading hypothesis to explain this association (22). Low avidity, circulating auto-reactive T cells which may have unusual binding conformations with self-MHC and do not cause pathology, may be activated by cross-reaction with high affinity or high abundance pathogenic peptides derived from infectious agents. Cross reactivity is plausible as a single T cell receptor can recognize a broad range of different epitopes (TCR degeneracy) (23, 24) and importantly, because the peripheral T cell activation threshold is lower than that for negative selection in the thymus (25). Classically, molecular mimicry was postulated on the basis of shared epitopes between Group A streptococci and cardiac glycoproteins leading to rheumatic fever. In the nervous system a strong case has been made for the development of Guillain Barre syndrome (GBS) after Campylobacter jejuni infection. Antibodies against C. jejuni cell wall lipo-oligosaccharide antigens cross-react against gangliosides (nearly identical self-antigens), which are major constituents of the nerve cell membrane. An animal model in rabbits in which immunization with C. jejuni lipo-oligosaccharide results in a neuropathy similar to GBS provides support for this hypothesis (26). It is important to note, however, that <0.1% of C. jejuni infected individuals develop cross-reactive antibodies and Guillain Barre syndrome (27), raising questions about the broader relevance of the molecular mimicry model for autoimmune disease causation.
Alternatively, primary CNS tissue damage (after Herpes simplex encephalitis, for example, which has been known to trigger NMDA encephalitis) (28), may lead to the release and export of self- antigens that drain into the CNS draining deep cervical nodes. The discovery of lymphatics in the dura mater draining to cervical lymph nodes has pointed to a path by which CNS antigens may reach peripheral immune tissue (29). In support of this, in experimental autoimmune encephalomyelitis (EAE), myelin antigens associated with dendritic cells have been found in the cervical lymph nodes of non-human primates (30). In this model, on presentation of CNS antigens to quiescent, auto-reactive lymphocytes in CNS draining lymph nodes, T or B cells could be activated. Activated CD4+T cells could then provide help to activate naïve B cells and also produce high amounts of cytokines like interferon λ and granulocyte macrophage colony stimulating factor (GM-CSF), which promote local inflammation and loss of blood-brain barrier integrity. Activated memory B cells migrating back to the CNS could undergo antigen driven affinity maturation (possibly in ectopic tertiary lymphoid structures in the meninges as has been described in MS and NMOSD) and differentiate into antibody producing plasma cells, explaining the observed intrathecal synthesis of autoantibodies.
In addition accumulation of somatic point mutations in immunoglobulin variable (V) gene of B cells proliferating in response to pathogens, provides the basis for extraordinary antibody diversity and affinity maturation in response to foreign antigens. A genealogical analysis of antibodies secreted by antigen specific B cell clones from autoimmune disease patients has shown that unmutated common ancestors of auto-antibodies (for example to the autoantigen desmoglein3 in pemphigus) (31) or to nuclear autoantigens in SLE (32) do not bind to the respective self-antigen. Thus, somatic mutations in immunoglobulin (V) genes in B cell clones, proliferating in response to infection, while generating a massive diversity of antibodies, may randomly and inadvertently generate antibodies that occasionally bind and cross-react with self-antigens, even in the absence of structural molecular mimicry with pathogenic antigens (33). This may be another mechanism for breach of self-tolerance.
Another mechanism to consider is secondary to the presence of natural autoantibodies, found in individuals even without prior antigenic experience. The sera of healthy individuals contain an enormous number of such antibodies that are poly-reactive and bind with low affinity with various self and non-self-antigens. By chronically reacting with self-antigens autoreactive B cells achieve non-responsiveness to them (anergy) but, it has been speculated that they may be susceptible to activation by infection or tissue damage leading to the production of high affinity antibodies and autoimmune disease (34, 35).
Microbiota
The microbiota is an ecosystem of trillions of microrganisms that reside on mucosal surfaces and skin in beneficial co-existence with the host. Recently the surprising connection between disordered intestinal microbiota and brain autoimmunity has been made (36). This was based on the finding that germ free mice were resistant to developing EAE (the classic animal model for multiple sclerosis), but susceptibility to EAE was restored in mice exposed to commensal bacteria (37). Also, fecal transplants from MS patients into germ free mice have been shown to result in a higher incidence or severity of EAE (38, 39). Another recent study of the antigen reactivity of a CD4+ T cell clone isolated from brain tissue of a multiple sclerosis patient showed reactivity to an ubiquitous auto-antigen guanosine diphosphate (GDP)–L- fucose synthetase, an evolutionarily conserved enzyme also used by gut microbiota (40). Forty percent of T cell clones isolated from CSF of MS patients reacted to this antigen and, notably, also to myelin. Thus, in this hypothesis, CD4+ T cells could be activated in the gut against microbial gut GDP-L-fucose synthetase, travel to the CNS and cross-react with myelin or other CNS antigens resulting in disease (41). Similarly, in another study of a mouse model of autoimmune uveitis, it was shown that non-cognate antigen from commensal intestinal microbes can activate eye specific auto-reactive T cells resulting in autoimmune uveitis (42).
Epitope Spreading
An alternative explanation for the finding described above could be that after the initial CNS inflammation induced by a specific CNS autoantigen, CNS injury results in the release of cryptic antigens which provoke a secondary CD4+ T cell response. In a similar manner, CSF immunoglobulin oligoclonal bands, produced by clonally expanded CSF B cells suggests intra-thecal antibody synthesis and is a hallmark of multiple sclerosis. The CNS antigenic target of these antibodies has been long sought to elucidate MS pathogenesis. However, a recent study based on aligning patient specific CSF oligoclonal peptides to patient specific immunoglobulin transcriptome derived from CSF B cells, showed that the B cell response in MS is heterogenous and non-specific and directed against ubiquitous intracellular auto-antigens (43) released during tissue destruction, implying unmasking of cryptic epitopes and cascading inflammatory damage.
Tumors
Ectopic expression of neural proteins (onconeural antigens) in cancer can trigger an aberrant autoimmune response misdirected against the nervous system (para-neoplastic disorders, see Table 1). For example in ovarian teratoma (associated with NMDA encephalitis), the tumor itself contains mature or immature neural tissue (52). Antigens released by dying tumor cells may be taken up, processed and presented by antigen presenting cells in regional lymph nodes to activate T cells. Furthermore, ectopic germinal centers have been described within ovarian teratomas in some patients who present with NMDAR encephalitis (53). These could be a source of continued auto-antibody production.
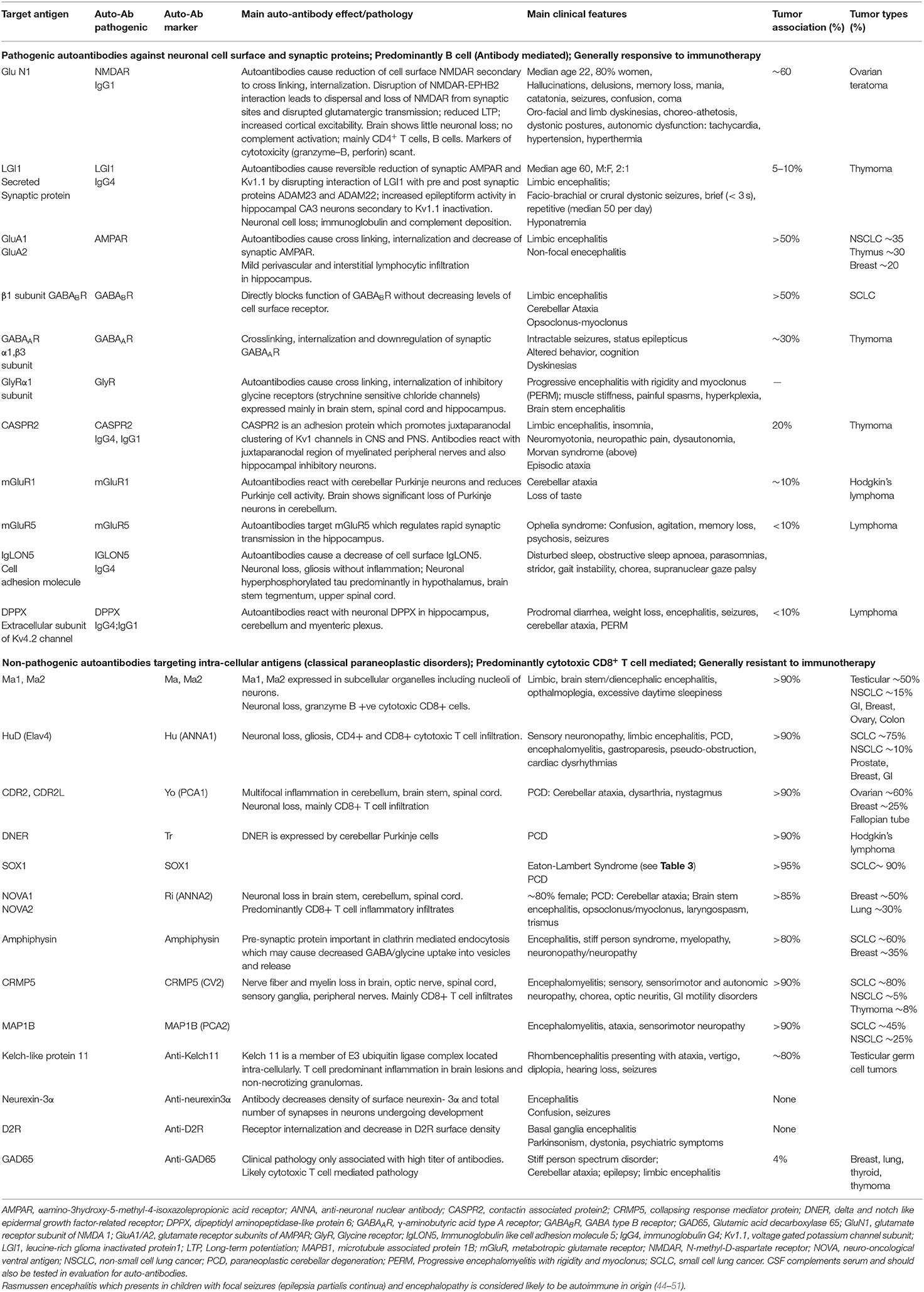
Table 1. Main features of autoimmune diseases of the central nervous system.
It is important to note that even though onconeural antigens occur frequently, only a small minority of cancer patients (<1%) develop paraneoplastic syndromes (54). This may be because, for auto-reactive T cells to be activated, mutations may have to be present in tumor antigens (55) (to be seen by the immune system as “foreign” antigens) and secondly, effective antigen presentation of the tumor peptide to T cells also requires compatibility with the patient's MHC molecules.
Vitamin D
Apart from its role in calcium homeostasis Vitamin D plays an important immunomodulatory, anti-inflammatory and anti-oxidant role (56). Several studies suggest that Vitamin D has an effect on the immune system by inhibiting B cell proliferation and antibody production and also by reducing T cell proliferation, Th1 and Th17 differentiation, increasing Treg cells and decreasing the secretion of pro-inflammatory cytokines (57).
Vitamin D is mainly produced in the skin after exposure to ultraviolet light and hypovitaminosis D has been linked to reduced exposure to sunlight in northern latitudes, where MS is more common, sparking interest in studying Vitamin D levels in MS. However, these studies have produce conflicting results. Vitamin D levels have been reported to correlate with decreased MS incidence (58) and lower levels have also been reported to increase susceptibility to the development of MS in pediatric patients (59). Other studies have, however, shown no difference in the level of Vitamin D between patients and controls (60) and a Cochrane review concluded no benefit from Vitamin D administration in reducing relapses, disability or MRI lesions in MS patients (61). The role of Vitamin D in nervous system autoimmune diseases is a subject of ongoing investigation.
Clinical Features and Scientific Basis of Different CNS and PNS Autoimmune Diseases
Important discoveries over the last two decades have identified many new autoimmune diseases of the nervous system and illuminated their pathogenesis. The clinical and pathophysiological features of CNS autoimmune diseases are outlined in Tables 1, 2 and Figure 1 and of the PNS in Table 3 and Figure 2 (44–50, 52, 62, 63, 67, 72).
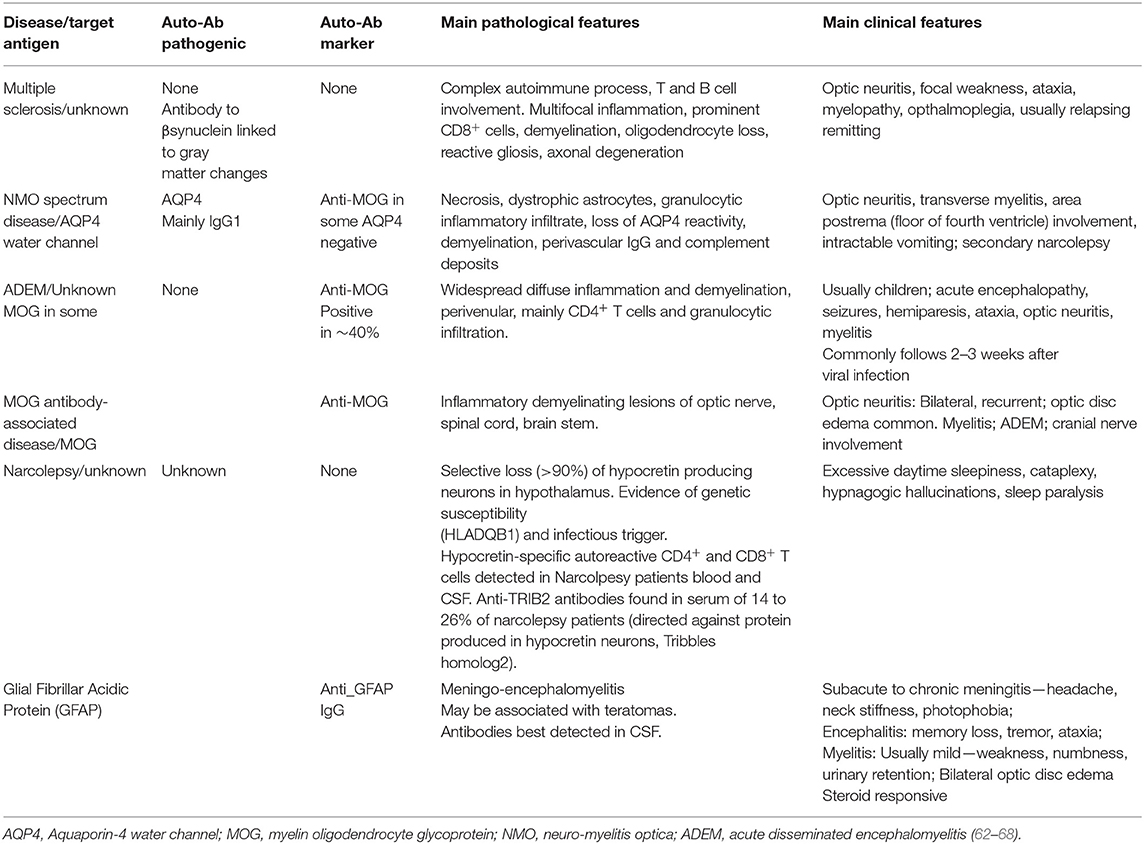
Table 2. Classic and new CNS autoimmune diseases.
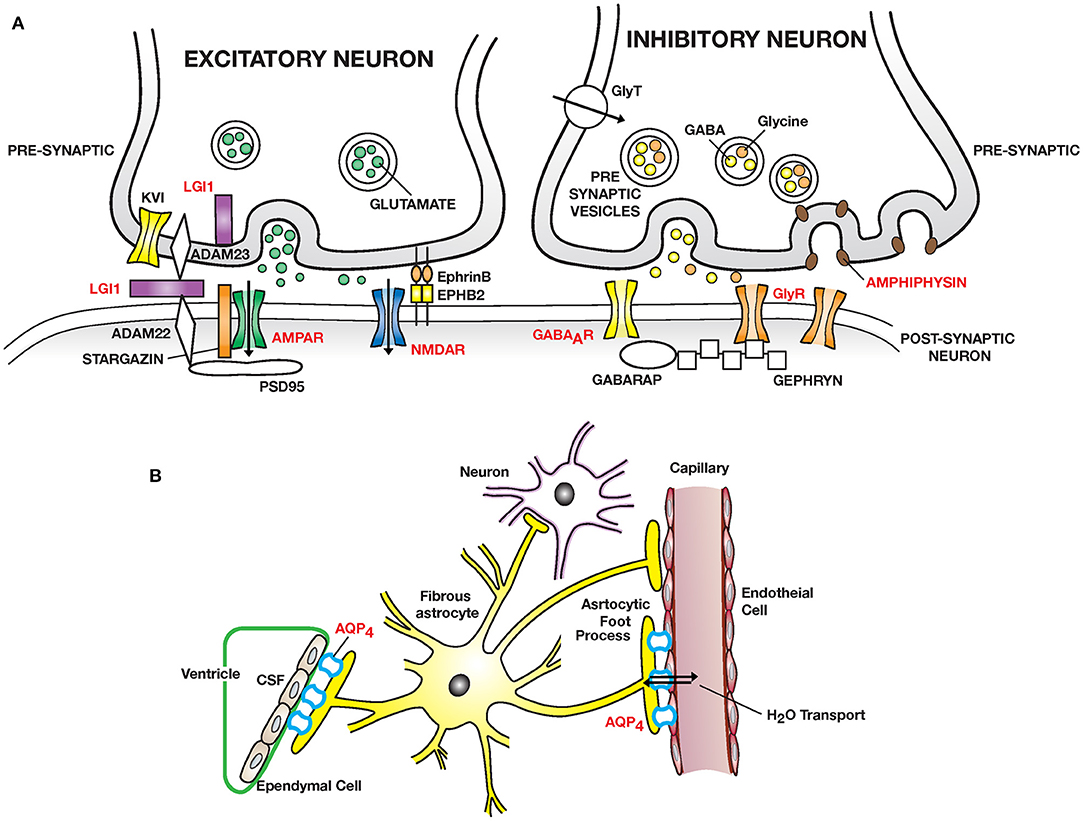
Figure 1. Autoimmmune diseases of the CNS: (A) Excitatory synapses: NMDAR encephalitis is caused by anti–NMDA receptor (NMDAR) antibodies binding and cross-linking GluN1 subunits, disrupting interaction of NMDAR with ephrin type B receptor 2 (EPHB2) causing internalization of NMDAR and impaired glutamergic transmission. LGI1 encephalitis is caused by auto-antibodies to leucine rich glioma inactivated protein 1 (LGI1), a neuronal glycoprotein secreted into the synapse. LGI1 interacts with presynaptic disintegrin and metalloproteinase domain-containing protein 2 (ADAM23) and postsynaptic ADAM22, modulates AMPA receptor trafficking and presynaptic Kv1, voltage gated potassium channel function. PSD95, post-synaptic density protein 95. Other receptors targeted: AMPAR; mGluR1/5. Inhibitory synapses: Autoantibodies to GABA type A receptor (GABAAR) on inhibitory synapses may lead to encephalitis with intractable seizures. Autoantibodies to the glycine receptor (GlyR) cause painful spasms; progressive encephalitis with rigidity and myoclonus (PERM). Stiff person syndrome can also be caused by auto-antibodies to amphiphysin, a presynaptic protein expressed in all synapses, important in clathrin mediated endocytosis leading to a decreased number of presynaptic vesicles filled with neurotransmitter available for exocytosis. Higher tonic activity may make inhibitory synapses (release of GABA and glycine) especially vulnerable. GABARAP, GABA associated receptor protein. GAD, decarboxylates glutamate to GABA (not shown). (B) Neuromyelitis optica (NMO) is caused by auto-antibodies to Aquaporin 4 (AQP4) water channel expressed widely in the body including on astrocytic foot processes abutting capillary endothelial cells and ependymal cells. Predominant pathology is in spinal cord, optic nerve and brain peri-ependymal regions.
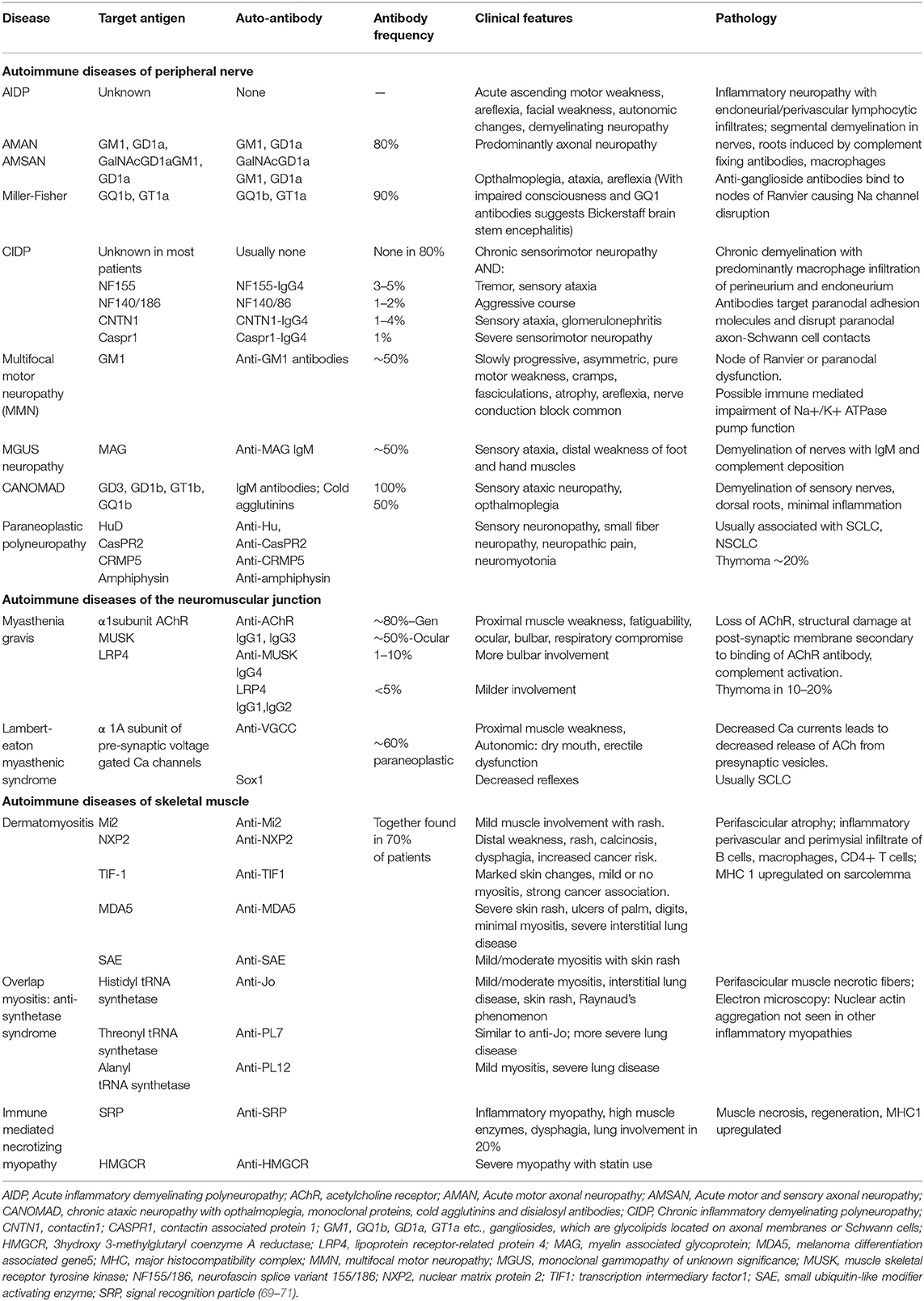
Table 3. Autoimmune diseases of the peripheral nervous system.
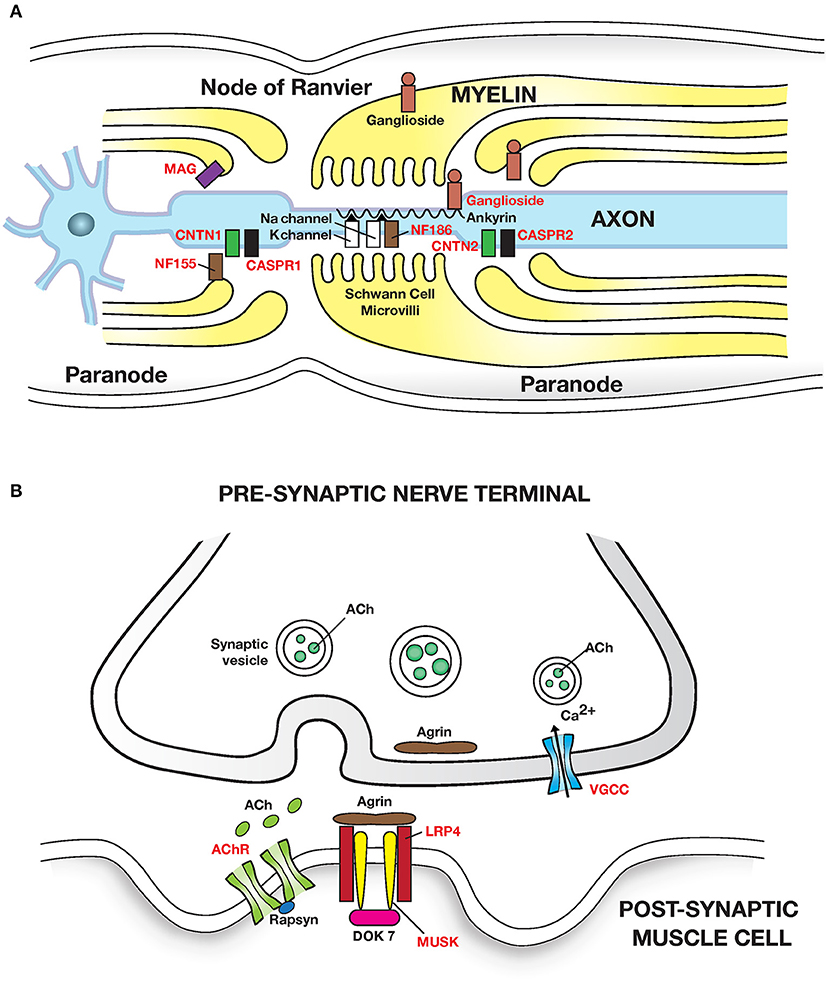
Figure 2. Autoimmune diseases of peripheral nerve and neuromuscular junction: (A) Peripheral nerve: Guillain Barre syndrome and its variants have been linked to autoantibodies to gangliosides (glycosphingolipids) which are abundant in peripheral nerves. Autoantibodies cause segmental demyelination and disruption of Na channel clustering. GM1 and GalNAc-GD1a ganglioside are most abundant in axolemma of motor nerves and nodes of Ranvier and are associated with motor phenotypes. GQ1b is enriched in oculomotor cranial nerves and is linked with Miller Fisher syndrome. Multifocal motor neuropathy (MMN) is linked to anti-GM1 ganglioside antibodies. MGUS neuropathy is linked to autoantibodies to myelin associated glycoprotein (MAG). Chronic inflammatory demyelinating polyneuropathy (CIDP) is linked to autoantibodies (in <10% patients) to the nodal and paranodal adhesion proteins neurofascin 186 and 155 (NF186, NF155); contactin 1 (CNTN1); contactin associated protein (CASPR1 and 2) which disrupt axon-Schwann cell contacts. Adhesion molecules are attached to the cytoskeleton by proteins such as Ankyrin/Spectrin. (B) Neuromuscular junction: Myasthenia gravis caused by autoantibodies to the (i) post synaptic Acetylcholine receptor (AChR); IgG1 and IgG3 autoantibodies activate the complement cascade leading to structural damage to postsynaptic membrane and loss of AChR. (ii) Muscle skeletal receptor tyrosine protein kinase (MUSK). MUSK autoantibodies (Fab- arm exchanged monovalent IgG4) block agrin (released from motor nerve terminals) induced MUSK—LRP4 (low density lipoprotein receptor related protein 4) interaction, and disrupt clustering of AChR. Dok7, Downstream of tyrosine kinase 7: cytoplasmic protein that activates MUSK in concert with agrin and LRP4. Lambert-Eaton myasthenic syndrome is caused by autoantibodies to presynaptic P/Q type voltage gated calcium channels (VGCC) disrupting Ca2+ influx; Ca2+ is essential for release of ACh from presynaptic vesicles. Antigenic targets of autoantibodies are shown in red text in all figures. Autoantibodies associated with inflammatory myopathies are shown in Table 3.
Nervous System Autoimmune Diseases With Intracellular or Neuronal Cell Surface Antigenic Targets
Autoimmune disorders of the nervous system may have: (1) Autoantibodies directed against intra-cellular neuronal antigens (classic paraneoplastic disorders) (47). The presence of these antibodies (such as anti-Hu, ant-Ri, and anti-Ma) is almost always associated with the presence of an underlying malignancy (Table 1). It is believed these disorders are caused by a misdirected immune response directed against tumor proteins (onconeural antigens) which are also expressed by neurons. But the auto-antibodies, which are aberrantly directed against intra-cellular neuronal antigens, have been shown in studies of live cultured neurons not to reach their intra-cellular targets and are not themselves pathogenic. They are useful, however, as bio-markers for these paraneoplastic disorders, which are caused mainly by T cell mediated pathology. This is supported by autopsy material which shows extensive infiltration of neurons by cytotoxic T cells, causing degeneration by perforin and granzyme B mediated mechanisms. Consequently patients with these paraneoplastic CNS disorders respond poorly to immunotherapeutic strategies involving removal of antibodies or antibody producing cells (Table 1); (2) Autoantibodies may be directed against neuronal cell surface and synaptic receptor proteins [(45), Table 1]. Cancer association in these disorders is less frequent (except in GABABR and AMPAR encephalitis where cancer association is more common). Several recently described encephalitides (such as NMDAR, LGI1, and CASPR2 encephalitis) have been shown to be secondary to the pathogenic effect of autoantibodies binding to extracellular epitopes of neuronal cell surface proteins (Table 1). Imunotherapy to remove or neutralize the effect of these pathogenic autoantibodies is effective. Similarly, autoantibodies to cell surface proteins at the neuromuscular junction (acetylcholine receptors and voltage gated calcium channels) are pathogenic and result in myasthenia gravis or Eaton-Lambert syndrome. Immunotherapy has been shown to be effective in these disorders.
In addition to these two categories (intracellular or cell surface antigenic expression) a more complicated pattern of expression is illustrated by glutamic acid decarboxylase (GAD) an intracellular enzyme whose physiological function is the decarboxylation of glutamate to gamma-aminobutyric acid (GABA) the main inhibitory transmitter within neurons. GAD 65 is able to associate with the plasma membrane and surge to the extracellular space. Thus, epitopes of GAD65 and also amphiphysin can be exposed to antibodies during synaptic fusion and reuptake. High titers of anti-GAD65 have been linked to stiff person spectrum disorders, cerebellar ataxia and limbic encephalitis (73).
Clinical Features Which Should Trigger Consideration of Autoimmune Diseases
The presentation of autoimmune encephalitis can be similar to encephalitis secondary to infections. Encephalitis is suggested by acute or subacute symptoms of decreased or altered level of consciousness, lethargy, short term memory loss, personality changes such as apathy, irritability, agitation and commonly, seizures.
The following features (developing over days to weeks not years), usually in patients with suspected encephalitis, should trigger consideration of autoimmune causes:
• After excluding infectious causes, acute or sub-acute development of cognitive decline, behavioral changes and seizures (typically intractable and resistant to anti-epileptic medications) should prompt consideration of autoimmune encephalitis. Seizures occur most commonly in autoimmune encephalitis with NMDAR, LGI1, and GABABR antibodies. When this clinical presentation is associated with bilateral medial temporal lobe abnormal signal on MRI FLAIR images, limbic encephalitis should be considered. Although limbic encephalitis can also be caused by CNS infections such as HSV or HHV6 encephalitis, when autoimmune in etiology the usual antibodies associated with limbic encephalitis are anti-Hu, Ma2, LGI1, GABAB, or AMPA receptor.
• Faciobrachial dystonic seizures characterized by brief (few seconds), lateralized (unilateral or bilaterally asynchronous) tonic contractions mainly affecting the upper limb and face often associated with hand dystonia. Episodes can recur numerous times a day and are considered pathognomic of anti-leucine-rich glioma inactivated 1 (LGI1) encephalitis (74).
• Prominent, recent, psychiatric manifestations including agitation, aggression and violence, visual or auditory hallucinations, delusions, disorganized or bizarre behavior, paranoia, disinhibition, apathy, mutism and catatonia; intolerance to neuroleptics may cause hyperthermia, rigidity, rhabdomyolysis or coma. Such features have most typically been described with NMDAR encephalitis (in up to 95% of patients) (75, 76).
• Cerebellar ataxia (pan cerebellar involvement) presenting subacutely (<3 months) as an isolated symptom or associated with limbic or brain stem encephalitis, myelitis or neuropathy. It is associated with multiple antibodies, such as anti-Hu (ANNA-1), mGluR1, CASPR2, GAD65, Ri (ANNA-2), Yo (PCA1), PCA2, CV2/CRMP5, or Sox1. Opsoclonus-myoclonus in an adult in isolation or together with cerebellar ataxia may be associated with anti-Ri antibodies. Decreased level of consciousness, bilateral external opthalmoplegia and ataxia developing over <4 weeks suggests Bickerstaff brainstem encephalitis and is associated with antibodies to anti-GQ1b antibodies.
• Sleep disorders such as abnormal sleep architecture with undifferentiated NREM sleep, REM sleep behavior disorder (RBD), obstructive sleep apnea and stridor, and agrypnia excitata (insomnia, motor, and autonomic hyperactivation). Various features of sleep disorders and RBD have been described in patients with antibodies to IgLON5, CASPR2, Ma2, and LGI1 (77). NMOSD with hypothalamic involvement can present with secondary narcolepsy.
▪ Movement disorders such as chorea, dyskinesias, dystonias, or myoclonus (associated with multiple antibodies including anti- NMDAR, CASPR2, LGI1,Ri, Hu, or CV2/CRMP5); and rarely parkinsonism (associated with anti-Ri, NMDAR, LGI1, and D2R) (78).
• Peripheral nerve hyperexcitability (neuromyotonia) presenting as spontaneous motor activity with cramps, myokimia or fasciculations which may be associated with encephalopathy (associated with anti-CASPR2 and LGI1 antibodies).
▪ Stiff person spectrum disorders presenting as fluctuating muscle spasms and rigidity and acquired hyperekplexia (startle response) is associated primarily with antibodies that target proteins expressed by inhibitory synapses such as glutamic acid decarboxylase 65 (GAD65), α1 subunit of the glycine receptor (GlyR) or amphiphysin (79–81).
• Autonomic disturbances such as hyperthermia, fluctuations of blood pressure, tachycardia, less commonly bradycardia requiring a temporary pacemaker, excessive salivation, hyperhidrosis, chronic gastro-intestinal pseudo-obstruction and central hypoventilation. These have typically been described in patients with anti- NMDAR and anti-Hu encephalitis.
• Sensory neuronopathies are a rare group of disorders secondary to degeneration of dorsal root ganglion presenting with asymmetric, multifocal, non-length dependent sensory loss, neuropathic pain and subacute sensory ataxia secondary to proprioceptive loss. This syndrome, in isolation or often together with encephalomyelitis, has been linked to anti-Hu, CV2/CRMP5 and amphiphysin antibodies in cancer patients.
• Multifocal encephalitic presentation (in children and young adults) characterized by multiple, large white matter and deep gray matter demyelinating lesions on brain MRI scans is suggestive of acute disseminated encephalomyelitis (ADEM). MOG antibodies may be present in serum in up to 50 per cent cases.
Unilateral or bilateral optic neuritis presenting as sudden visual loss may be associated with antibodies to AQP4 (NMO spectrum disease) or MOG (MOG antibody- associated disease) or multiple sclerosis (usually unilateral) (82) (Table 2). Myelitis presenting as rapid onset of paraparesis or quadriparesis may be associated with AQP4 antibodies (NMO spectrum disease); MOG antibodies (MOG antibody- associated disease) (83, 84); MS or ADEM.
Tests Done for Initial Evaluation (85, 86)
(1) CSF analysis in autoimmune encephalitis may be normal or non-specific with mild lymphocytic pleocytosis and elevation of CSF protein; (2) MRI brain studies in autoimmune encephalomyelitis may be normal or may show scattered white matter lesions on T2 weighted FLAIR images. Autoimmune limbic encephalitis is characterized by abnormal signal in bilateral medial temporal lobes; in NMO there may be swollen, enhancing optic nerves, longitudinally extensive (> 3 vertebral segments) spinal cord lesions with increased T2 signal with enhancement, and peri-aqueductal and dorsal medulla/pons lesions; (3) EEG findings are usually non-specific with focal or diffuse slowing with or without epileptogenic activity. In NMDAR encephalitis an unique EEG pattern of 20–30 Hz beta activity riding on rhythmic 1–3 Hz delta waves, called extreme delta brush has been described. In primary psychiatric or functional disorders MRI Brain, EEG and CSF analysis will be normal providing an useful initial assessment for the presence or absence of structural neurologic disease; (4) CT Chest, abdomen and pelvis with and without contrast should be done for tumor screening; consider pelvic/vaginal ultrasound for ovarian tumors and FDG-PET/CT scan for whole body tumor detection when clinical suspicion is high and other tests are non-revelatory; (5) PNS disorders are typically assessed by nerve conduction, repetitive stimulation and electromyographic (EMG) studies and creatine kinase (CK) estimation. Nerve or muscle biopsies may sometimes be useful.
Autoantibody Testing
Both serum and CSF should be tested for IgG autoantibodies. CSF should also be tested as CSF neuronal antibodies are more disease specific than serum antibodies and antibodies are only present in CSF in some cases. For example, in NMDAR encephalitis, no serum anti-NMDAR antibodies are found in ~15% of patients. It should be noted that conversely, in ~10% of patients of NMOSD and LGI1 encephalitis, antigen-specific autoantibodies can only be detected in serum, not in CSF (27). Auto-antibodies in the classic paraneoplastic disorders (e.g., anti-Hu, anti-Ri) are more specific or only detected in serum. Similarly only a minority of ant-MOG-seropositive patients have detectable antibodies in CSF (87).
Antibody screening methodology is usually indirect immunofluorescence or immunohistochemistry or radioimmunoassay but definitive confirmation may require a cell based assay or Western blot assay. For classic paraneoplastic disorders the first screen is by Western blot and cell based assays are often used from the outset if the clinical suspicion is high for autoimmune encephalitis. Most commercial laboratories test for panels of autoantibodies as similar presentations may underlie the effect of different auto-antibodies (Table 1). When a characteristic clinical presentation such as faciobrachial dystonic seizures are present a single antibody, LGI1 can be tested at the outset. Determination of immunoglobulin class is important as IgM or IgA antibody positivity may be seen in healthy controls and caution should be exercised before considering them clinically significant. Antibody positivity should be considered in the appropriate clinical context as low titers may be misleading. In NMDAR encephalitis high antibody titers correlate with a poor outcome (88), but this correlation is imperfect and antibody positivity may persist after recovery. Clinical assessment should therefore be used to guide treatment (89). Tests for CSF oligoclonal bands and CSF IgG index should be done as an indication of intra-thecal synthesis of antibodies.
Important Role of B Cells in Autoimmune Diseases of the Nervous System
Both T and B cells play important roles in the pathogenesis of various autoimmune inflammatory nervous system disorders (90). The predominant role of B cells in some of these disorders has recently been emphasized (90, 91). B cells and their secreted antibodies recognize distinct antigens through a rearranged B cell receptor (BCR). Naïve B cells arise from hematopoietic stem cells in the bone marrow and circulate in peripheral blood and lymphoid organs. Following recognition of their cognate antigen in germinal centers a subset may eventually become CD27+ memory B cells, which can further differentiate into CD38+ antibody secreting plasmablasts and long lived CD138+ plasma cells. In addition to antibody production, B cells can internalize proteins and present peptide fragments to-antigen-specific CD8+ and CD4+ T cells in association with MHC Class I and Class II molecules, thus modulating T cell function. Current evidence suggests that B cell antigen presentation plays an important role in T cell activation in MS (91). B cells also produce a number of pro-inflammatory cytokines lymphotoxin-α, interleukin 6 and tumor necrosis factor or anti-inflammatory cytokines such as IL-10 and IL-35, thus having the ability to either promote or suppress CNS inflammation.
Recent work has suggested that changes in B cell development in the bone marrow, secondary to altered B cell intrinsic signals can lead to an altered naïve B cell repertoire and the generation of autoantibody producing B cells (92). These signals consist of a complex interplay of BCR signaling and co-receptor signaling, mediated by B cell activating factor (BAFFR), CD40 and Toll-like receptors (TLRs). Such altered signaling programs (secondary to genetic variants associated with autoimmunity) could result in the production of a higher proportion of mature B cells which exhibit autoreactivity (92).
Illustrative CNS Autoimmune Disorders
Multiple Sclerosis
Although in MS the autoimmune response to unknown target antigens has traditionally been considered to be driven by T cells, the importance of B cells in pathophysiology has been highlighted by, (1) the detection of CSF oligoclonal bands, unique IgG fractions produced by an inthrathecal clonal B cell population which target ubiquitous self-antigens; (2) Detection of B cells in CNS lesions in MS, most abundantly in active lesions; (3) ectopic lymphoid follicle like structures containing CD20+ B cells, CD138+ plasma cells and follicular dendritic cells, which have been identified in the leptomeninges of patients with MS; (4) expanded populations of plasmablasts and plasma cells obtained from patients with MS, which target neurons, astrocytes and oligodendrocytes, although the target antigen is unknown; (5) the detection of CSF antibody reactivity against measles, rubella and varicella-zoster viral antigens (93). Collectively, it has been proposed that this data suggests that the humoral B cell response in MS may not be directed against a single antigen but may be directed against a wide array of self and non-self antigens which could differ from individual to individual (91). The broad range of targets may reflect epitope spreading and secondary immune reactions to CNS damage.
The remarkable efficacy of therapies which target CD20 in MS patients has confirmed the critical role of B cells in MS pathogenesis. Interestingly the clinical benefit of anti-CD20 B cell therapy in MS precedes reduction in total IgM and IgG levels, demonstrating that the effect is not due to a reduction in humoral immunity but may be secondary to impaired antigen presentation to T cells, or decrease in inflammatory cytokines such as IL-6 or TNF. Furthermore, the most effective therapies for MS involve the ablation or blocking the trafficking of B and T cells in the periphery, which results in a substantial reduction in new CNS lesions and relapses. This suggests that MS relapses are mediated by episodes of BBB breakdown with recruitment of peripheral lymphocytes and macrophages into the CNS (91). Thus, the lack of efficacy in some types of MS, such as in progressive MS, may be secondary to sequestration of the abnormal immune response behind an intact blood brain barrier.
NMO Spectrum Disease
NMOSD is an inflammatory demyelinating lesion primarily involving the optic nerves, spinal cord and brainstem. Anti–Aquaporin 4 (a membrane bound water channel expressed abundantly on astrocytic foot processes)-IgG is present in 70–90% of NMO patients. The pathogenicity of anti-AQP4 antibodies has been demonstrated both in vitro and in vivo and intracerebral injection of AQP4 IgG with complement results in demyelination resembling NMOSD. Serum titers of anti-AQP4 antibodies are 1,000-fold higher than in CSF and CSF oligoclonal bands, which are seen in only 15–30% of NMO patients usually disappear with disease progression, implying that B cell activation and the origin of the humoral immune response is outside the CNS. Activated B cells may then enter the CNS after BBB breakdown and cause pathology, as discussed earlier.
During normal early B cell development, tolerance is maintained by the removal of most self-reactive B cells. Anti-AQP4 IgG producing B cells seem to arise from defects in early B cell tolerance checkpoints, both centrally in the bone marrow and peripherally, resulting in an increased number of autoreactive B cells in the mature naiive B cell population (94). This reservoir of auto-reactive B cells can supply B cell clones which produce pathogenic anti-AQP4 antibodies after somatic hypermutations. This conclusion has been based on the observation that the unmutated precursors of B cells secreting anti-AQP4 antibodies do not bind to autoantigen, showing that anti-AQP4 specificity and the generation of pathogenic autoantibodies require affinity maturation and the acquisition of somatic hypermutations, a process regulated by T cells (94). Additionally, the activation of B cell clones may initially be triggered by an antigen other than AQP4, such as to a peptide from Clostridium perfringens, an indigenous intestinal bacterium (which has homology to an immunodominant epitope of AQP4).
Myelin Oligodendrocyte Glycoprotein—Antibody Associated Disease
MOG is produced by oligodendrocytes, the myelin forming cells of the CNS. It is expressed on the surface of oligodendrocytes and the external lamella of myelin sheaths. It has been of great interest historically in the study of MS as it had been shown to be the target antigen in an animal model of MS, experimental allergic encephalomyelitis (95). However, studies in MS showed that MOG antibodies were non-specific (96). It has only been recently with better antibody detection methods using cell based assays, which detect MOG in its native conformational state, that MOG-antibody has emerged as a biomarker for CNS inflammatory demyelinating disorders distinct from MS and NMOSD. A recent study has confirmed that the most reliable assays to detect MOG antibody are those using live cells expressing native full length human MOG (as some conformational epitopes are lost on fixation of MOG expressing cells) (64). Secondly, low positive samples are detected widely, including in healthy individuals, therefore an appropriate cutoff value to identify the clinically meaningful results is an area requiring further studies. Serum testing is recommended for detecting MOG-IgG as the reliability of CSF MOG-IgG is uncertain. Also oligoclonal bands are infrequently detected in CSF, unlike MS, implying the peripheral initiation of the autoimmune process.
In MOG-antibody associated disease preceding prodromal symptoms are commonly encountered and can include fever, malaise, cough and rhinorrhea (65). The majority of patients present with optic neuritis, transverse myelitis or acute disseminated encephalomyelitis (ADEM, which is most prevalent below the age of 20 years) (66). Brain stem or cerebellar involvement presenting with ataxia, facial palsy, diplopia or vertigo may be seen (97). A recent study has also reported the occurrence of overlapping central and peripheral nervous system involvement (acute inflammatory demyelinating neuropathy, myeloradiculitis, multifocal motor neuropathy or brachial neuritis) (98). Clinical course may be either monophasic or relapsing. In ADEM, high titers and persistent MOG-IgG positivity predict a higher risk of relapse (83). Optic neuritis can be unilateral or bilateral and is often associated with optic disc edema. MRI may show enhancement along the length of the optic nerve including the optic sheath. Bilateral anterior optic nerve enhancement without extension to the optic chiasm is typical (82). In patients presenting with myelitis on MRI, longitudinally extensive lesions occur in the majority, with involvement of the conus seen more commonly than in NMOSD. Spinal cord lesions on MRI typically involve the gray matter, distinguishing them from lesions in MS and NMOSD (84).
NMDAR Encephalitis
NMDAR encephalitis is the commonest autoimmune encephalitis, characterized by IgG auto-antibodies directed against the GluN1 subunit of the NMDAR (99). The disease shows a female predominance (female to male ratio of 8:2) with a median age at presentation of 21 years (range 1–85 years) and is commonly associated with ovarian teratoma. The most common presentation is abnormal behavior (visual or auditory hallucinations, acute schizoaffective disorder, depression, mania), cognitive dysfunction, seizures, movement disorders (oral, facial, lingual dyskinesias, chorea, athetosis, and dystonia), autonomic dysfunction or central hypoventilation. Most (80%) patients recover after immunotherapy and ovarian teratoma removal (if present). Diagnosis is confirmed by the presence of NMDAR IgG antibodies in cerebrospinal fluid. Brain biopsy or autopsy shows infiltrates of B cells, CD4+ T cells and deposits of IgG but little or no neuronal loss, providing an explanation for the significant recovery that can be achieved after early immunotherapy.
Multiple lines of evidence show that NMDAR antibodies in patients serum are pathogenic (100). In experiments involving cultured neurons, antibodies crosslink NMDARs, cause their internalization and impairment of synaptic plasticity (100). In animal experiments, infusion of patient CSF or IgG purified from patient CSF into the ventricular space of mice via intraventricular catheters leads to memory deficits, anhedonia and seizures (101–103). Furthermore, active immunization of mice with GluN1/GluN2B heteromers results in a fulminant encephalitis (104). The NMDAR antibodies produced cause a reduction of NMDAR and NMDAR currents in cultured neurons. Also transplacental transfer of NMDAR antibodies from pregnant patients to embryos results in neurological deficits in neonates (105, 106). Overall these findings support the role of NMDAR autoantibodies in disease causation as required by Witbesky's postulates.
Unlike NMOSD (where antibodies are much more readily detected in serum than in CSF), anti-NMDAR antibodies are more reliably detected in the CSF than in serum (both should be tested). Also, clonally expanded NMDAR-specific plasma cells have been detected in the CSF of patients with NMDAR encephalitis and B cells in the CSF of these patients can produce anti-NR1 antibodies. Taken together, in contrast to myasthenia gravis and NMOSD, in which antibody production occurs in the periphery, this data suggests that the NMDAR specific B cell response in anti-NMDAR encephalitis is compartmentalized in the CNS.
Checkpoint Inhibitor Therapy Induced Nervous System Autoimmune Disease
An uncommon but intriguing autoimmune disorder has recently been described (107). Inhibitory molecules (such as CTLA-4, PD-1, LAG-3, and TIM3) which are expressed on the surface of T and B lymphocytes inhibit and keep a check on excessive immune responses both normal and anti-self. Immune checkpoint inhibitors are a new treatment for cancer which is being used more commonly in recent years. They usually target programmed death 1 (PD-1) or cytotoxic T-lymphocyte antigen-4 (CTLA-4) expressed on T cells including Treg cells. Recently 4 patients were described who developed transverse myelitis, Guillain Barre syndrome or myasthenia gravis 4 weeks after medication (Pembrolizumab or Nivolumab and Iplimumab) administration. Conventional auto-antibodies associated with these conditions were absent but serum immunoglobulins from the myelitis and Guillain Barre syndrome patient showed a novel pattern of tissue reactivity, binding to rodent brain tissue or myelinating stem cell derived sensory neurons or rat primary Schwann cells. It was suggested that the short time between drug administration and disease onset implies that T and B cells which were circulating in a quiescent state, were disinhibited leading to the activation of pre-existing B cell reactivity to neural and neuromuscular proteins (107).
Illustrative PNS Autoimmune Disorders
Autoimmune disorders of the neuromuscular junction include myasthenia gravis (with auto-antibodies directed against the post-synaptic Acetylcholine receptor or muscle specific tyrosine kinase) or Lambert-Eaton syndrome with autoantibodies directed against pre-synaptic voltage gated calcium channels. The definitive demonstration of the autoimmune basis of myasthenia gravis is one of the classic examples of an autoimmune disease (108, 109).
Although not as definitive, considerable indirect evidence is supportive of an autoimmune basis for some disorders of the skeletal muscle and peripheral nerve.
Immune-Mediated Myopathies
Immune-mediate myopathies are a group of acquired muscle disorders which include dermatomyositis, necrotizing autoimmune myopathy, overlap myositis, and inclusion body myositis (Table 3) (69, 70). A good response to immunosuppressive therapy is seen in these disorders except in inclusion body myositis (see below). Dermatomyositis typically presents with proximal muscle weakness and a skin rash. Muscle biopsies show perifascicular atrophy with perimysial and perivascular infiltrates of CD4+ T cells, B cells and plasmacytoid dendritic cells with sarcolemmal upregulation of MHC1. The deposition of membrane attack complex on intramuscular capillaries is an early manifestation of dermatomysositis, but the primary cause of the microangiopathy remains unclear. About 70% of dermatomyositis patients have a specific autoantibody which are associated with unique clinical phenotypes (Table 3). For example the presence of anti-transcriptional factor 1α (TIF-1) or anti-nuclear matrix protein NXP2 antibodies are associated with a high risk of cancer. Patients with antibodies to melanoma differentiation associated gene 5 (MDA5) have severe cutaneous changes with skin ulcers and interstitial lung disease. Overlap myositis, typically represented by anti-synthetase syndrome, is a type of autoimmune myopathy associated with other connective tissue diseases. These patients have auto-antibodies to various aminoacyl tRNA synthetases (Table 3). Patients with anti-Jo antibodies have myopathy associated with interstitial lung disease, radial finger skin lesions called mechanic's hands and Raynaud's phenomenon. Immune mediated necrotizing myopathy is characterized by proximal muscle weakness with very creatine kinase levels and muscle biopsies showing necrosis and regeneration with minimal or no lymphocytic infiltration, upregulation of MHC 1 and membrane-attack complex deposition on non-necrotic fibers. About two-thirds of patients have autoantibodies to either signal recognition particle (anti-SRP) or 3-hydroxyl 3-methylglutaryl coenzyme-A reductase (HMGCR), an enzyme that catalyzes the rate limiting step in cholesterol synthesis. Antibodies to HMGCR are usually seen in patients who have been exposed to statins. Inclusion body myositis patients have evidence of CD8+ inflammatory infiltrates but also rimmed vacuoles, ragged red fibers indicating mitochondrial damage and abnormal protein aggregates on muscle biopsies suggesting both an inflammatory and a degenerative process. Auto-antibodies recognizing cytosolic 5'-nucleotidase 1 A are present in 30–60% of patients with sporadic inclusion body myositis. However, these are also found in 10–20% of patients with dermatomyositis, SLE and Sjogren's syndrome. The role of these autoantibodies in pathogenesis and in diagnosis requires further investigation.
Immune-Mediated Neuropathies
Immune mediated neuropathies typically include acute inflammatory demyelinating polyradiculoneuropathy (AIDP; Guillain Barre syndrome), chronic inflammatory demyelinating polyneuropathy (CIDP), multifocal motor neuropathy (MMN) and neuropathies associated with IgM monoclonal gammopathy of undetermined significance (MGUS) (Table 3) (71). It is believed that autoimmune peripheral nerve damage is mediated both by antibodies directed against myelin antigens and autoreactive T cells and macrophages that invade myelin sheath or axonal membranes. Autopsies from Guillain Barre syndrome patients have shown perivascular and endoneurial inflammatory infiltrates of T cells and macrophages throughout the nerves, roots or plexuses along with segmental demyelination mediated by macrophages (110). Autoantibodies to various gangliosides which are abundant in axons are found commonly in patients (in 80–90%) with the Miller-Fisher subtype (GQ1b, GT1a) or the axonal variant (GM1, GD1a) of Guillain-Barre syndrome but very infrequently in the most common demyelinating subtype. In 15–20% of CIDP patients auto-antibodies have been detected in serum to several proteins at the nodal or paranodal regions of peripheral nerves (such as CASPR1, contactin1, and NF155) which maintain axon-Schwann cell or axon-myelin binding. These autoantibodies could activate complement and macrophage mediated axonal degeneration, demyelination and disruption of nodal architecture.
Paraneoplastic neuropathies are commonly sensory neuronopathies secondary to dorsal root ganglion pathology associated with anti-Hu, CRMP5 or amphiphysin antibodies. Peripheral nerve hyperexcitability and neuropathic pain may be seen with CASPR2 antibodies. Enteric autonomic involvement with severe disturbances of gastrointestinal mobility has been described with anti-Hu antibodies. Treatment of these disorders usually involves plasmapheresis, IVIG, and/or immunosuppression.
Therapy
Current therapies for CNS and PNS autoimmune diseases and mechanism of action of various treatment modalities are detailed in Table 4.

Table 4. Therapy of autoimmune diseases of the nervous system.
Management of Autoimmune Encephalitis
Best practice recommendations for the management of autoimmune encephalitis have recently been made (111). If cancer is present it should be treated promptly as early treatment is associated with better prognosis. For example ovarian teratoma removal in NMDAR encephalitis is associated with expedited recovery (112). Typically patients are given immunosuppressive therapy which should also be started as early as possible as this is associated with a better prognosis (112). First line immunotherapy includes high dose intravenous methylprednisolone for 5 days. This is usually followed by plasmapheresis (5–7 exchanges) and/or intravenous immunoglobulins (IVIG). Second line therapy for non-responders may include IV Rituximab and Cyclophosphamide. The IL6 blocker tocilizumab may be beneficial in special circumstances such as refractory status epilepticus in these patients. In more severe cases 3–6 months of Cyclophosphamide may be added. Cyclophosphamide is often considered in the classic paraneoplastic disorders given its effectiveness in depleting T cells. Bortezomib, a proteasome inhibitor which targets long lived auto-reactive plasma cells can be a useful adjunct to standard immunotherapy in treatment-refractory patients (113).
The extent of recovery varies according to the type of encephalitis. In a large study of 577 patients with NMDAR encephalitis, after immunotherapy, 53% improved within 4 weeks and 81% improved by 2 years (112). Although 47% failed first line immunotherapy, second line immunotherapy was still found to be effective. Outcomes were found to continue to improve for up to 18 months after symptom onset (112). In LGI1 encephalitis, a study of 103 patients showed a striking time-sensitive response to early immunotherapy, with 51% showing cessation of faciobrachial dystonic seizures within 4 weeks of starting treatment (in contrast to only 10% when anti-epileptic drugs alone were used) (114). Early cessation of faciobrachial dystonic seizures with immunotherapy prevented the development of cognitive impairment (all patients were also shown to have IgG4-LGI1 antibodies but those with cognitive impairment had a higher proportion of complement fixing IgG1 antibodies (114, 115).
Lower response rates are seen in encephalitis associated with antibodies to AMPAR and GABABR, which are often associated with cancer. In general patients with antibodies to intracellular antigens (classic paraneoplastic disorders which are predominantly T cell mediated) have a worse prognosis after immunotherapy, compared to those with antibodies to cell surface or synaptic antigens. The frequency of relapses in encephalitides associated with antibodies against NMDAR, AMPAR, LGI1, or CASPR2 can range from 12 to 35%. Long term immunosuppression with Azathioprine or mycophenolate mofetil can be given to prevent relapses although the evidence for benefit is not conclusive.
Treatment of NMOSD
An acute attack of NMO is treated with high dose corticosteroids (1 gm IV methylprednisolone for 5 days). Most patients should also receive early treatment with 5–7 plasma exchanges. Maintenance therapy is very important as NMOSD is a relapsing disorder. The most commonly used medications for maintenance are oral prednisone along with the steroid sparing drugs azathioprine, mycophenolate mofetil, methotrexate or rituximab. MOG-antibody associated disorders are treated in a similar manner.
New treatment options are becoming available for the treatment of NMOSD. AQP4-IgG1 antibodies are known to trigger the complement cascade, leading to inflammatory CNS damage. Eculizumab a terminal complement (C5) inhibitor was shown in a phase 3 trial to reduce the risk of relapses in NMO by 94% compared to placebo (116). Benefit was maintained long term (median 2.5 years) (117). Another strategy has been to use IL6 receptor blocking monoclonal antibodies such as satralizumab and tocilizumab. Disruption of IL6 signaling might affect disease by reducing AQP4 antibody production, inhibition of pro-inflammatory T cell differentiation and lowering of blood-brain barrier permeability. These agents reduce the relapse rate by about 60%.
B Cell Depleting Therapy
The most significant advance in the treatment of MS (and to some extent other nervous system autoimmune diseases) has been the advent of highly efficacious monoclonal antibodies which deplete B cells (118, 119). Anti- CD 20 monoclonal antibodies rapidly deplete circulating B lineage cells which express CD20 (a cell surface marker expressed on pre-B, mature and memory B cells but not on stem cells, pro-B cells, plasmablasts or plasma cells). The anti-CD20 antibodies currently being used are Rituximab, a chimeric mouse human monoclonal antibody; Ocrelizumab, a humanized monoclonal antibody, and Ofatumumab a fully human antibody. These therapies lead to rapid and almost complete depletion of circulating B cells (but less so in lymph nodes) for ~1 year after a single course, by antibody dependent and complement dependent cytotoxicity. Since auto-antibody levels are not markedly reduced by Rituximab, the major mechanism of action is believed to be the depletion of naïve and memory B cells, impaired antigen presentation by B cells and decreased release of inflammatory cytokines such as tumor necrosis factorα and lymphotoxin and increase in anti-inflammatory IL-10. Although intrathecal concentration of Rituximab after systemic administration is <0.1% of plasma levels, CSF B cells decrease dramatically after therapy, implying that peripheral depletion of B cells leads to decreased CNS migration. However, CD20+B cells still persist in meninges and CNS parenchyma after treatment (120), indicating the possibility of relapses. Also, since profound B cell depletion may lead to increased risk of new infections, reactivation of latent infections such as JC virus, tuberculosis and hepatitis or malignancies, longer term safety issues should be considered.
Research Into Novel Targeted Therapies for Autoimmune Diseases
Most current treatment choices for autoimmune diseases (Table 4), although effective, involve broad immunosuppression, with its concurrent risk of increased infection or malignancy. An ideal treatment for autoimmune diseases would eliminate auto-reactive immune cells but spare protective immunity.
Cell Therapy With Immunosuppressive Treg Cells
Treg cells are a small subset, 5–7%, of CD4+ cells (CD4+, CD25+, FOXP3+, and CD127low), which maintain immune homeostasis by suppressing excessive immune activation and inflammatory damage (121, 122). Several pre-clinical and early clinical trials with polyclonal Treg cells are underway. However, Treg therapy which only targets antigen-specific autoreactive T cells is more potent and has less risk of generalized immunosuppression than using polyclonal Treg cells (123). Strategies to generate antigen-specific Treg cells are: (i) engineering the T cell receptor (TCR) by transducing TCR variable (V) genes from a patient's antigen-specific, auto-reactive T cell clone into Treg cells; (ii) introducing a chimeric antigen receptor (CAR), comprising an extracellular auto-antigenic peptide-MHC molecule linked to a TCR signaling domain into Treg cells. Treg cells engineered in these ways recognize and are activated by MHC-peptide on antigen presenting cells or by pathogenic auto-reactive T cells and mediate localized immunosuppression. For example, myelin basic protein specific Treg cells with an engineered T cell receptor (124) or a CAR (125) have been shown to ameliorate EAE in mice.
Nanoparticles Coated With Peptide-MHC for Antigen-Specific Immunosuppression
In another successful strategy in animals, nanoparticles coated with peptide fragments from myelin oligodendrocyte protein, bound to MHC Class II proteins were injected into EAE mice (126). On receiving this signal, autoreactive CD4+ T cells with receptors for that peptide (in the absence of co-stimulatory signals from antigen presenting cells) expanded and differentiated into an immunosuppressive subset of regulatory T cells, instead of inducing an immune response against the self-protein. This led to further recruitment of auto-reactive B cells as regulatory cells, suppression of antigen presentation and even restoration of strength in paralytic mice (126).
CAR T Cell Antigen-Specific Immunosuppression
Significant advances in cancer treatment have involved the use of chimeric antigen T cell receptors (CAR-T cells), which are engineered T cell receptors with a single chain extracellular antibody molecule, capable of binding to a specific antigen, and destroying cancer cells expressing that antigen (127). A similar approach for the treatment of autoimmunity involves the generation of engineered T cells which bind specifically and only to auto-reactive B cells and destroys them. This has been validated in a mouse model of an autoimmune disease, pemphigus, caused by B cell production of auto-antibodies to Desmoglein3. T cells with chimeric CAR-T receptors expressing extracellular Desmoglein 3, fused to the zeta chain of the T cell receptor CD3 signaling domain (CD3 ζ), specifically destroyed only anti-Desmoglein3 producing B cells and ameliorated disease in mice (128). Such an approach may be feasible against autantibody-mediated disease where the target antigen is known, such as Aquaporin4 in NMO.
Positive Allosteric Modulation as a Therapeutic Strategy
NMDAR antibodies binding in patients result in NMDAR loss and hypofunction. Preliminary work suggests that allosteric binding of compounds such as synthetic oxysterols to extracellular regulatory sites of the NMDAR (distinct from the agonist binding and channel permeating sites) can restore NMDAR function in cultured rat hippocampal neurons and prevent the pathogenic effects of NMDAR auto-antibodies in a mouse model, suggesting a potential new strategy to counteract abnormal NMDAR function (129).
Although promising in in vitro or animal studies, the ability to translate these innovative approaches to treat human diseases, safely and effectively, is a subject of ongoing research.
Conclusion
Recent research has outlined a broad range of autoimmune disorders of both the central and peripheral nervous system, which may be triggered by diverse factors in genetically susceptible individuals. Several new disorders such as NMOSD, MOG-antibody associated disease and the various encephalitides caused by antibodies to synaptic receptors in the CNS have been defined, permitting rapid diagnosis and treatment. The antigenic target of this autoimmune attack has been well-defined in many of these disorders, such as in myasthenia gravis, NMDA receptor encephalitis and NMOSD but is uncertain in others such as multiple sclerosis and Guillain Barre syndrome. The autoimmune attack and pathology may be predominantly B cell and auto-antibody mediated, such as in myasthenia gravis and NMOSD or predominantly T cell mediated as in the classic paraneoplastic disorders. Current therapy encompasses eliminating pathogenic auto-antibodies by plasma exchange and broad immunosuppression. Novel research avenues being pursued raise hopes for the possibility of targeted immunotherapy in the future.
Author Contributions
The entire article, figures and tables have been conceived and written by SB.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I acknowledge support from the Department of Neurology, SUNY Downstate College of Medicine, New York.
References
1. Hayter SM, Cook MC. Updated assessment of the prevalence, spectrum and case definition of autoimmune disease. Autoimmun Rev. (2012) 11:754–65. doi: 10.1016/j.autrev.2012.02.001
2. Theofilopoulos AN, Kono DH, Baccala R. The multiple pathways to autoimmunity. Nat Immunol. (2017) 18:716–24. doi: 10.1038/ni.3731
3. Willer CJ, Dyment DA, Risch NJ, Sadovnick AD, Ebers GC. Canadian collaborative study group. Twin concordance and sibling recurrence rates in multiple sclerosis. Proc Nat Acad Sci USA. (2003) 100:12877–82. doi: 10.1073/pnas.1932604100
4. Avidan N, Le Panse R, Berrih-Aknin S, Miller A. Genetic basis of myasthenia gravis- a comprehensive review. J Autoimmun. (2014) 52:146–53. doi: 10.1016/j.jaut.2013.12.001
5. Cooper GS, Stroehla BC. The epidemiology of autoimmune diseases. Autoimmun Rev. (2003) 2:119–125. doi: 10.1016/S1568-9972(03)00006-5
6. Flanagan EP, Cabre P, Weinshenker BG, St Sauver J, Jacobson DJ, Majed M, et al. Epidemiology of aquaporin-4 autoimmunity and neuromyelitis optica spectrum Ann Neurol. (2016) 79:775–83. doi: 10.1002/ana.24617
7. Libert C, Dejager D, Pinheiro I. The X chromosome in immune functions: when a chromosome makes the difference. Nature. (2010) 10:594–604. doi: 10.1038/nri2815
8. Klein SL, Flanagan KL. Sex differences in immune responses. Nat Rev Immunol. (2016) 16:626–38. doi: 10.1038/nri.2016.90
9. Dendrou CA, Peterson J, Rossjohn J, Fugger L. HLA variation and disease. Nat Rev Immunol. (2018) 18:325–38. doi: 10.1038/nri.2017.143
10. Hahn M, Nicholson MJ, Wucherpfennig KW. Unconventional topology of self peptide-major histocompatibility complex binding by a human autoimmune T cell receptor. Nat Immunol. (2005) 6:490–6. doi: 10.1038/ni1187
11. van Sonderen A, Roelen DL, Stoop JA, Verdik RM, Haasnoot GW, Thijs RD, et al. Anti-LGI1 encephalitis is strongly associated with HLA-DR7 and HLA-DRB4. Ann Neurol. (2017) 81:193–8. doi: 10.1002/ana.24858
12. Gaig C, Guadalupe E, Daura X, Ezquerra M, Fernandez-Santiago R, Palou E, et al. HLA and microtubule associated protein tau H1 haplotype associations in anti-IgLON5 disease. Neurol Neuroimmunol Neuroinflamm. (2019) 6:e605. doi: 10.1212/NXI.0000000000000605
13. Binks S, Varley J, Lee W, Makuch M, Elliott K, Gelfand JM, et al. Distinct HLA associations of LGI1 and CASPR2-antibody diseases. Brain. (2018) 141:2263–71. doi: 10.1093/brain/awy109
14. Hong Y, Li HF, Romi F, Skeiego, Gilhus NE. HLA and MUSK-positive myasthenia gravis: a systematic review and meta-analysis. Acta Neurol Scand. (2018) 138:219–26. doi: 10.1111/ane.12951
15. Maniaol AH, Elsais A, Lorentzen AR, Viken MK, Sæther H, Flåm ST, et al. Late onset myasthenia gravis is associated with HLA DRB*15:01 in the Norwegian population. PLoS ONE. (2012) 7:e36603. doi: 10.1371/journal.pone.0036603
16. Estrada K, Whelan CW, Zhao F, Bronson P, Handsaker RE, Sun C, et al. A whole-genome sequence study identifies genetic risk factors for neuromyelitis optica. Nat Commun. (2018) 9:1929. doi: 10.1038/s41467-018-04332-3
17. Cerosaletti K, Schneider A, Schwedhelm K, Frank I, Tatum M, Wei S, et al. Multiple autoimmune-associated variants confer decreased IL-2R signaling in CD4+CD25hi T cells of Type 1 diabetic and multiple sclerosis patients. PLoS ONE. (2013) 8:e83811. doi: 10.1371/journal.pone.0083811
18. Giraud M, Taubert R, Vandiedonck C, Ke X, Levi-Strauss M, Pagani F, et al. An IRF8-binding promoter variant and AIRE control CHRNA1 promiscuous expression in thymus. Nature. (2007) 448:934–7. doi: 10.1038/nature06066
19. Costapas C. International multiple sclerosis genetics consortium. Low-frequency and rare-coding variation contributes to multiple sclerosis risk. Cell. (2018) 175:1679–87.e7. doi: 10.1016/j.cell.2018.09.049
20. Pruss H. Postviral autoimmune encephalitis: manifestations in children and adults. Neurology. (2006) 67:652–9. doi: 10.1097/WCO.0000000000000445
21. ‘t Hart BA, Hintzen RQ, Laman JD. Multiple sclerosis-a response-to-damage model. Trends Mol Med. (2016) 22:1012–24. doi: 10.1016/j.molmed.2016.10.007
22. Oldstone MB. Molecular mimicry and immune-mediated diseases. Faseb J. (1998) 12:1255–65. doi: 10.1096/fasebj.12.13.1255
23. Wilson DB, Wilson DH, Schroeder K, Schroder K, Pinilla C, Blondelle S, et al. Specificity and degeneracy of T cells. Mol Immunol. (2004) 40:1047–55. doi: 10.1016/j.molimm.2003.11.022
24. Wooldridge L, Ekeruche-Makinde J, van den Berg HA, Skowera A, Miles JJ, Tan MP, et al. A single autoimmune T cell receptor recognizes more than a million different peptides. J Biol Chem. (2012) 287:1168–77. doi: 10.1074/jbc.M111.289488
25. Enouz S, Carrie L, Merkler D, Bevan MJ, Zehn D. Autoreactive T cells bypass negative selection and respond to self-antigen stimulation during infection. J Exp Med. (2012) 209:1769–79. doi: 10.1084/jem.20120905
26. Ang CW, Jacobs BC, Laman JD. The Guillain-Barre syndrome: a true case of molecular mimicry. Trends Immunol. (2004) 25:61–6. doi: 10.1016/j.it.2003.12.004
27. Sun B, Ramberger M, O'Connor KC, Bashford-Rogers RJM, Irani S. The B cell immunobiology that underlies CNS autoantibody-mediated diseases. Nat Rev Neurol. (2020) 16:481–92. doi: 10.1038/s41582-020-0381-z
28. Armangue T, Spatola M, Vlagea A, Mattozzi S, Cárceles-Cordon M, Martinez-Heras E, et al. Frequency, symptoms, risk factors, and outcomes of autoimmune encephalitis after herpes simplex encephalitis: a prospective observational study and retrospective analysis. Lancet Neurol. (2018) 17:760–72. doi: 10.1016/S1474-4422(18)30244-8
29. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. (2015) 523:337–41. doi: 10.1038/nature14432
30. de Vos AF, van Meurs M, Brok HP, Boven LA, Hintzen RQ, van der Valk P, et al. Transfer of central nervous system autoantigens and presentation in secondary lymphoid organs. J Immunol. (2002) 169:5415–23. doi: 10.4049/jimmunol.169.10.5415
31. Di Zenzo G, Di Lullio G, Corti D, Calabresi V, Sinistro A, Vanzetta F, et al. Pemphigus autoantibodies generated through somatic mutations target the desmoglein-3 cis interface. J Clin Invest. (2012) 122:3781–90. doi: 10.1172/JCI64413
32. Mietzner B, Tsuiji M, Scheid J, Velinzon K, Tiller T, Abraham K, et al. Autoreactive IgG memory antibodies in patients with systemic lupus erythematosus arise from nonreactive and polyreactive precursors. Proc Natl Acad Sci USA. (2008) 105:9277–32. doi: 10.1073/pnas.0803644105
33. Lanzavecchia A. Dissecting human antibody responses: useful, basic and surprising findings. EMBO Mol Med. (2018) 10:e8879. doi: 10.15252/emmm.201808879
34. Lutz HU, Binder CJ, Kaveri S. Naturally occurring auto-antibodies in homeostasis and disease. Trends Immunol. (2009) 30:43–51. doi: 10.1016/j.it.2008.10.002
35. Avrameas S, Alexopoulos H, Moutsopoulos HM. Natural autoantibodies: an undersung hero of the immune system and autoimmune disorders—a point of view. Front Immunol. (2018) 9:1320. doi: 10.3389/fimmu.2018.01320
36. Wekerle H. Brain autoimmunity and intestinal microbiota: 100 trillion game changers. Trends Immunol. (2017) 38:483–97. doi: 10.1016/j.it.2017.03.008
37. Goverman J, Woods A, Larson L, Weiner LP, Hood L, Zaller DM, et al. Transgenic mice express a myelin basic protein- specific T cell receptor develop spontaneous autoimmunity. Cell. (1993) 72:551–60. doi: 10.1016/0092-8674(93)90074-Z
38. Berer K, Gerdes LA, Cekanaviciute E, Jia X, Xiao L, Xia Z, et al. Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc Natl Acad Sci USA. (2017) 114:10719–24. doi: 10.1073/pnas.1711233114
39. Cekanaviciute E, Yoo BB, Runia TF, Debelius JW, Singh S, Nelson CA, et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc Natl Acad Sci USA. (2017) 114:10713–8. doi: 10.1073/pnas.1711235114
40. Planas R, Santos R, Tomas-Ojer P, Lutterotti A, Faigle W, Schaeren-Wiemers N, et al. GDP-L-fucose synthetase is a CD4+T cell-specific autoantigen in DRB3*02:02 patients with multiple sclerosis. Sci Transl Med. (2018) 10:eaat4301. doi: 10.1126/scitranslmed.aat4301
41. Sabatino JJ, Zamvil SS. T cells take aim at a ubiquitous autoantigen in multiple sclerosis. Sci Transl Med. (2018) 10:eaau8826. doi: 10.1126/scitranslmed.aau8826
42. Horai R, Zarate-Blades CR, Dillenburg-Pilla P, Chen J, Kielczewski JL, Silver PB, et al. Microbiota-dependent activation of an autoreactive T cell receptor provokes autoimmunity in an immunologically privileged site. Immunity. (2015) 43:343–53. doi: 10.1016/j.immuni.2015.07.014
43. Brandle SM, Obermeier B, Senel M, Mentele R, Khademi M, Olsson T, et al. Distinct oligoclonal band antibodies in multiple sclerosis recognize ubiquitous self-proteins. Proc Natl Acad Sci USA. (2016) 113:7864–9. doi: 10.1073/pnas.1522730113
44. Dalmau J, Graus F. Antibody mediated encephalitis. New Eng J Med. (2018) 378:840–51. doi: 10.1056/NEJMra1708712
45. Crisp SJ, Kullmann DM, Vincent A. Autoimmune synaptopathies. Nat Rev Neurosci. (2016) 17:103–17. doi: 10.1038/nrn.2015.27
46. Dalmau J, Geis C, Graus F. Autoantibodies to synaptic receptors and neuronal cell surface proteins in autoimmune disease of the central nervous system. Physiol Rev. (2017) 97:839–87. doi: 10.1152/physrev.00010.2016
47. Mckeon A, Pittock SJ. Paraneoplastic encephalomyelopathies: pathology and mechanisms. Acta Neuropathol. (2011) 122:381–400. doi: 10.1007/s00401-011-0876-1
48. Bauer J, Bien CG. Neuropathology of autoimmune encephalitides. Handbook Clin Neurol. (2016) 133:107–20. doi: 10.1016/B978-0-444-63432-0.00007-4
49. Giannoccaro MP, Wright SK, Vincent A. In vivo mechanisms of antibody mediated neurological disorders; animal models and potential implications. Front Neurol. (2020) 10:1394. doi: 10.3389/fneur.2019.01394
50. Rosenfeld MR, Dalmau J. Paraneoplastic neurologic syndrome. Neurol Clin. (2018) 36:675–85. doi: 10.1016/j.ncl.2018.04.015
51. Dubey D, Wilson MR, Clarkson B, Giannini C, Gandhi M, Cheville J, et al. Expanded clinical phenotype, oncological associations and immunopathologic insights of paraneoplastic Kelch-like protein-11. JAMA Neurol. (2020) 77:1420–9. doi: 10.1001/jamaneurol.2020.2231
52. Dalmau J, Tuzun E, Wu H, Rossi JE, Voloschin A, Baehring JM, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. (2007) 61:25–36. doi: 10.1002/ana.21050
53. Makuch M, Wilson R, Al-Diwani A, Varley J, Kienzler AK, Taylor J, et al. N-Methyl-D-Aspartate receptor antibody production from germinal center reactions: Therapeutic implications. Ann Neurol. (2018) 83:553–61. doi: 10.1002/ana.25173
54. Graus F, Dalmau J. Paraneoplastic neurological syndromes in the era of immune-checkpoint inhibitors. Nat Rev Clin Oncol. (2019) 16:535–48. doi: 10.1038/s41571-019-0194-4
55. Small M, Treilleux I, Couillault C, Pissaloux D, Picard G, Paindavoine S, et al. Genetic alterations and tumor immune attack in Yo paraneoplastic cerebellar degeneration. Acta Neuropathol. (2018) 135:569–79. doi: 10.1007/s00401-017-1802-y
56. Miclea A, Bagnoud M, Chan A, Hoepner R. A brief review of the effects of Vitamin D on multiple sclerosis. Front Immunol. (2020) 11:781. doi: 10.3389/fimmu.2020.00781
57. Bellan M, Andreoli L, Mele C, Rigamonti C, Piantoni S, De Benedittis C, et al. Pathophysiological role and therapeutic implications of Vitamin D in autoimmunity: Focus on chronic autoimmune diseases. Nutrients. (2020) 12:789. doi: 10.3390/nu12030789
58. Munger KL, Levin LI, Hollis BW, Howard NS, Ascherio A. Serum 25-hydroxyvitamin D levels and risk of multiple sclerosis. JAMA. (2006) 296:2832–8. doi: 10.1001/jama.296.23.2832
59. Gianfrancesco MA, Stridh P, Shao X, Rhead B, Graves JS, Chitnis T, et al. Genetic risk factors for pediatric-onset multiple sclerosis. Mult Scler. (2018) 14:1825–34. doi: 10.1177/1352458517733551
60. Fragoso YD, Adoni T, Alves-Leon SV, Arruda WO, Brooks JB, Cal HS, et al. No correlation was observed between Vitamin D levels and disability of patients with multiplw sclerosis between latitudes of 18 and 30 South. Arq Neuropsiquiatr. (2017) 75:3–8. doi: 10.1590/0004-282x20160173
61. Jagannath VA, Fillipini G, Pietrantonj C, Asokan GV, Robak EW, Whamond L, et al. Vitamin D for the management of multiple sclerosis. Cochrane Database Syst Rev. (2018) 9:CD008422. doi: 10.1002/14651858.CD008422.pub3
62. Lodygin D, Hermann M, Schweingruber N, Flügel-Koch C, Watanabe T, Schlosser C, et al. βsynuclein-reactive T cells induce autoimmune CNS greay matter degeneration. Nature. (2019) 566:503–8. doi: 10.1038/s41586-019-0964-2
63. Latorre D, Kallweit U, Armentani E, Foglierini M, Mele F, Cassotta A, et al. T cells in patients with narcolepsy target self-antigens of hypocretin neurons. Nature. (2018) 562:63–8. doi: 10.1038/s41586-018-0540-1
64. Reindl M, Schanda K, Woodhall M, Tea F, Ramanathan S, Sagen J, et al. International multicenter examination of MOG antibody assays. Neurol Neuroimmunol Neuroinflamm. (2020) 7:e674. doi: 10.1212/NXI.0000000000000674
65. Flanagan EP. Neuromyelitis optica spectrum disorder and other non-multiple sclerosis central nervous system inflammatory diseases. Continuum. (2019) 25:815–44. doi: 10.1212/CON.0000000000000742
66. Jurynczyk M, Messina S, Woodhall MR, Raza N, Everett R, Roca-Fernandez A, et al. Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain. (2017) 140:3128–38. doi: 10.1093/brain/awx276
67. Andrews SP, Lim MD, Thomas E, Scammell MD. The trouble with Tribbles: do antibodies against TRIB2 cause narcolepsy. Sleep. (2010) 33:875–8. doi: 10.1093/sleep/33.7.857
68. Flanagan EP, Hinson SR, Lennon VA, Fang B, Aksamit AJ, Morris PP, et al. Glial fibrillary acidic protein immunoglobulin G as biomarker of autoimmune astrocytopathy: analysis of 102 patients. Ann Neurol. (2017) 81:298–309. doi: 10.1002/ana.24881
69. Selva-O'Callaghan A, Pinal-Fernandez I, Trallero-Araguas E, Milisenda JC, Grau-Junyent JC, Mammen AL. Classification and management of adult inflammatory myopathies. Lancet Neurol. (2018) 17:816–28. doi: 10.1016/S1474-4422(18)30254-0
70. Milone M. Diagnosis and management of immune-mediated myopathies. Mayo Clin Proc. (2017) 92:826–37. doi: 10.1016/j.mayocp.2016.12.025
71. Querol L, Devaux J, Rojas-Garcia R, Illa I. Autoantibodies in chronic inflammatory neuropathies: diagnostic and therapeutic implications. Nat Rev Neurol. (2017) 13:533–47. doi: 10.1038/nrneurol.2017.84
72. Bien CG, Vincent A, Barnett MH, Becker AJ, Blumcke I, Graus F, et al. Immunopathology of autoantibody-associated encephalitides: clues for pathogenesis. Brain. (2012) 135:1622–38. doi: 10.1093/brain/aws082
73. Budharan A, Sechi E, Flanagan EP, Dubey D, Zekeridou A, Shah SS, et al. Clinical spectrum of high-titre GAD antibodies. J Neurol Neurosurg Psychiatry. (2021) 1−10. doi: 10.1136/jnnp-2020-325275. [Epub ahead of print].
74. Morano A, Fanella M, Giallandro T, Di Bonaventura C. Faciobrachial dystonic seizures: the borderland between epilepsy and movement disorders. Movem Disord Clin Practice. (2020) 7:228–9. doi: 10.1002/mdc3.12884
75. Pollack TA, Lennox BR, Muller S, Nicholson TR. Autoimmune psychosis: an international consensus on an approach to the diagnosis and management of psychosis of suspected origin. Lancet Psychiatry. (2020) 7:93–108. doi: 10.1016/S2215-0366(19)30290-1
76. Masdeu JC, Dalmau J, Berman KF. NMDA receptor internalization by autoantibodies: A reversible mechanism underlying psychosis? Trends Neurosci. (2016) 39:300–10. doi: 10.1016/j.tins.2016.02.006
77. Sabater L, Gaig C, Gelpi E, Bataller L, Lewerenz J, Torres-Vega E, et al. A novel NREM and REM parasomnia with sleep breathing disorder associated with antibodies against IGLON5: a case series, characterization of the antigen and post- mortem studies. Lancet Neurol. (2014) 13:575–86. doi: 10.1016/S1474-4422(14)70051-1
78. Balint B, Vincent A, Meinck H-M, Irani SR, Bhatia KP. Movement disorders with neuronal antibodies: asyndromic approach, genetic parallels and pathophysiology. Brain. (2018) 141:13–36. doi: 10.1093/brain/awx189
79. Crisp S, Dixon CL, Jacobson L, et Chabrol E, Irani SR, Leite MI, et al. Glycine receptor autoantibodies disrupt inhibitory neurotransmission. Brain. (2019) 142:3398–410. doi: 10.1093/brain/awz297
80. Crisp S, Balint B, Vincent A. Redefining progressive encephalomyelitis with rigidity and myoclonus after the discovery of antibodies to glycine receptors. Curr Opin Neurol. (2017) 30:310–6. doi: 10.1097/WCO.0000000000000450
81. Martinez-Hernandez E, Arino H, McKeon A, Lizuka T, Titulaer MJ, Simabukuro MM, et al. Clinical and immunologic investigations in patients with stiff-person spectrum disorder. JAMA Neurol. (2016) 73:714–20. doi: 10.1001/jamaneurol.2016.0133
82. Chen J, Pittock SJ, Flanagan EP, Lennon VA, Bhatti MT. Optic neuritis in the era of biomarkers. Survey Opthalmol. (2020) 65:12–7. doi: 10.1016/j.survophthal.2019.08.001
83. Lopez-Chiriboga AS, Majed M, Fryer J, Dubey D, McKeon A, Flanagan EP, et al. Association of MOG-IgG serostatus with relapse after acute disseminated encephalomyelitis and proposed criterion for MOG-IgG-associated disorders. JAMA Neurol. (2018) 75:1355–63. doi: 10.1001/jamaneurol.2018.1814
84. Dubey D, Pittock SJ, Flanagan EP. Clinical, radiographic and prognostic features of myelitis associated with myelin oligodendrocyte glycoprotein autoantibody. JAMA Neurol. (2019) 76:301–9. doi: 10.1001/jamaneurol.2018.4053
85. Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. (2016) 15:391–404. doi: 10.1016/S1474-4422(15)00401-9
86. Cellucci T, Van Mater H, Graus F, Muscal E, Gallentine W, Klein-Gitelman MS, et al. Clinical approach to the diagnosis of autoimmune encephalitis in the pediatric patient. Neurol Neuroimmunol Neuroinflamm. (2020) 7:e663. doi: 10.1212/NXI.0000000000000663
87. Jarius S, Paul F, Atkas O, Dale RC, de Seze J, Franciotta D, et al. MOG encephalomyelitis: international recommendation on diagnosis and testing. J Neuroinflammation. (2018) 89:1388–99. doi: 10.1186/s12974-018-1144-2
88. Gresa-Arribas N, Titulaer MJ, Torrents A, Aguilar E, McCracken L, Leypoldt F, et al. Antibody titres at diagnosis and during follow up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol. (2014) 13:167–77. doi: 10.1016/S1474-4422(13)70282-5
89. Dalmau J, Armangue T, Planaguma J, Radosevic M, Mannara F, Leypoldt F, et al. An update on anti-NMDA receptor encephalitis for neurologists and psychiatrists: mechanism and models. Lancet Neurol. (2019) 18:1045–57. doi: 10.1016/S1474-4422(19)30244-3
90. Weissert R. Adaptive immunity is the key to the understanding of autoimmune and paraneoplastic inflammatory central nervous system disorders. Front Immunol. (2017) 8:336. doi: 10.3389/fimmu.2017.00336
91. R Sabatino JJ, Probstel AK, Zamvil SS. B cells in autoimmune and neurodegenerative central nervous system diseases. Nat Rev Neuroscience. (2019) 20:728–44. doi: 10.1038/s41583-019-0233-2
92. Rawlings DJ, Metzier G, Wray-Dutra M, Jackson SW. Altered B cell signaling in autoimmunity. Nat Rev Immunol. (2017) 17:421–36. doi: 10.1038/nri.2017.24
93. Jarius S, Eichhorn P, Franciotta D, Petereit HF, Akman-Demir G, Wick M, et al. The MRZ reaction as a highly specific marker of multiple sclerosis; re-evaluation and structured review of the literature. J Neurol. (2017) 264:453–64. doi: 10.1007/s00415-016-8360-4
94. Cotzomi E, Stathopoulos P, Lee CS, Ritchie AM, Soltys JN, Delmotte FR, et al. Early B cell tolerance defects in neuromyelitis optica favor anti-AQP4 autoantibody production. Brain. (2019) 142:1598–615. doi: 10.1093/brain/awz106
95. von Büdingen HC, Tanuma N, Villoslada P, Villoslada P, Ouallet JC, Hauser SL, et al. Immune responses against the myelin/oligodendrocyte glycoprotein in experimental autoimmune demyelination. J Clin Immunol. (2001) 21:155–70. doi: 10.1023/A:1011031014433
96. Karni A, Bakimer-Kleiner R, Abramsky O, Ben-Nun A. Elevated levels of antibody to myelin oligodendrocyte glycoprotein is not specific for patients with multiple sclerosis. Arch Neurol. (1999) 56:311–5. doi: 10.1001/archneur.56.3.311
97. Banks SA, Morris PP, Chen JJ, Pittock SJ, Sechi E, Kunchok A, et al. Brainstem and cerebellar involvement in MOG0IgG-associated disorder versus aquaporin-4-IgG and MS. J Neurol Neurosurg Psychiatry. (2020) 1–7. doi: 10.1136/jnnp-2020-325121. [Epub ahead of print].
98. Rinaldi S, Davies A, Fehmi J, Wang J, Hardy TA, Barnett MH, et al. Overlapping central and peripheral nervous system syndromes in MOG-antibody associated disorders. Neurol Neuroimmunol Neuroinflamm. (2021) 8:e924. doi: 10.1212/NXI.0000000000000924
99. Kreye J, Wenke NK, Chayka M, Leubner J, Murugan R, Maier N, et al. Human cerebrospinal fluid monoclonal N-methyl-D-aspartate receptor autoantibodies are sufficient for encephalitis pathogenesis. Brain. (2016) 139:2641–52. doi: 10.1093/brain/aww208
100. Hughes EG, Peng X, Gleichman AJ, Lai M, Zhou L, Tsou R, et al. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J Neurosci. (2010) 28:5866–75. doi: 10.1523/JNEUROSCI.0167-10.2010
101. Planaguma J, Leypoldt F, Mannara F, Gutiérrez-Cuesta J, Martín-García E, Aguilar E, et al. Human N-methyl D-aspartate receptor antibodies alter memory and behavior in mice. Brain. (2015) 138:94–105. doi: 10.1093/brain/awu310
102. Taraschenko O, Fox HS, Pittock S, Zekeridou A, Gafurova M, Eldridge E, et al. A mouse model of seizures in anti-N-methyl-D-aspartate receptor encephalitis. Epilepsia. (2019) 60:452–63. doi: 10.1111/epi.14662
103. Wright S, Hashemi K, Stasiak L, Bartram J, Lang B, Vincent A, et al. Epileptogenic effects of NMDAR antibodies in a passive transfer mouse model. Brain. (2015) 138:3159–67. doi: 10.1093/brain/awv257
104. Jones BE, Tovar KR, Goehring A, Jalali-Yazdi F, Okada NJ, Gouaux E, et al. Autoimmune receptor encephalitis in mice induced by active immunization with conformationally stabilized holoreceptors. Sci Transl Med. (2019) 11:eeaaw0044. doi: 10.1126/scitranslmed.aaw0044
105. Shi YC, Chen XJ, Zhang HM, Wang Z, Du DY. Anti-N-Methyl-D-Aspartate receptor encephalitis during pregnancy: clinical analysis of reported cases. Taiwan J Obstet Gynaecol. (2017) 56:315–9. doi: 10.1016/j.tjog.2017.04.009
106. Kumar MA, Jain A, Dechant VE, Saito T, Rafael T, Aizawa H, et al. Anti-N-methyl –D-aspartate receptor encephalitis during pregnancy. JAMA Neurol. (2010) 67:884–87. doi: 10.1001/archneurol.2010.133
107. Wilson R, Menassa DA, Davies AJ, Michael S, Hester J, Kuker W, et al. Seronegative antibody-mediated neurology after immune checkpoint inhibitors. Ann Clin Transl Neurol. (2018) 5:640–5. doi: 10.1002/acn3.547
108. Toyka KV, Brachman DB, Pestronk A, Kao I. Myasthenia gravis: passive transfer from man to mouse. Science. (1975) 190:397–9. doi: 10.1126/science.1179220
109. Patrick J, Lindstrom J. Autoimmune response to acetylcholine receptor. Science. (1973) 180:871–2. doi: 10.1126/science.180.4088.871
110. Yuki N, Hartung H-P. Guillain- Barre syndrome. New Eng J Med. (2012) 366:2294–304. doi: 10.1056/NEJMra1114525
111. Abboud H, Probasco JC, Irani S, Ances B, Benavides DR, Bradshaw M, et al. Autoimmune encephalitis: proposed best practice recommendations for diagnosis and acute management. J Neurol Neurosurg Psychiatry. (2021). doi: 10.1136/jnnp-2020-325300. [Epub ahead of print].
112. Titulaer MJ, McCracken L, Gabilondo I, Armangué T, Glaser C, Iizuka T, et al. Treatment and prognostic factors for long-term outcome in patients with anti-N-Methyl-D-Aspartate (NMDA) receptor encephalitis: a cohort study. Lancet Neurol. (2013) 12:157–65. doi: 10.1016/S1474-4422(12)70310-1
113. Scheibe F, Pruss H, Mengel AA, Kohler S, Nümann A, Köhnlein M, et al. Bortezomib for treatment of therapy-refractory anti-NMDA receptor encephalitis. Neurology. (2017) 88:1–5. doi: 10.1212/WNL.0000000000003536
114. Thompson J, Bi M, Murchison AG, Makuch M, Bien CG, Chu K, et al. The importance of early immunotherapy in patients with faciobrachial dystonic seizures. Brain. (2018) 141:348–56. doi: 10.1093/brain/awx323
115. Irani SR, Stagg CJ, Schott JM, Rosenthal CR, Schneider SA, Pettingill P, et al. Faciobrachial dystonic seizures: the influence of immunotherapy on seizure control and prevention of cognitive impairment in a broadening phenotype. Brain. (2013) 136:3151–62. doi: 10.1093/brain/awt212
116. Pittock SJ, Berthele A, Fujihara K, Kim HJ, Levy M, Palace J, et al. Eculizumab in aquaporin-4-positive neuromyelitis optica spectrum disorder. New Eng J Med. (2019) 381:614–25. doi: 10.1056/NEJMoa1900866
117. Wingerchuk DM, Fujihara K, Palace J, Berthele A, Levy M, Kim HJ, et al. Long-term safety and efficacy of eculizumab in aquaporin-4 IgG-positive NMOSD. Ann Neurol. (2021) 1–11. doi: 10.1002/ana.26049. [Epub ahead of print].
118. Greenfield AL, Hauser SL. B cell therapy for Multiple sclerosis; entering an era. Ann Neurol. (2018) 83:13–26. doi: 10.1002/ana.25119
119. Sabatino JJ, Wilson MR, Calabresi PA, Hauser SL, Schneck JP, Zamvil S. Anti CD20 therapy depletes activated myelin specific CD8+ T cells in multiple sclerosis. Proc Natl Acad Sci USA. (2019) 116:25800–7. doi: 10.1073/pnas.1915309116
120. Esfandi S, Salimian S, Corboy J, Alvarez E. Persistent B lymphocytes in multiple sclerosis plaques after Rituximab treatment (P5.341). Neurology. (2019) 88(Suppl. 16).
121. Raffin C, Vo LT, Bluestone JA. Treg based cell therapies: challenges and perspectives. Nat Rev Immunol. (2020) 20:158–72. doi: 10.1038/s41577-019-0232-6
122. Ominguez-Villar M, Hafler DA. Regulatory T cells in autoimmune disease. Nat Immunol. (2018) 19:665–73. doi: 10.1038/s41590-018-0120-4
123. Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, et al. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med. (2004) 199:1455–65. doi: 10.1084/jem.20040139
124. Kim YC, Zhang A, Yoon J, Culp WE, Lees JR, Wucherpfennig KW, et al. Engineered MBP-specific human Tregs ameliorate MOG-induced EAE through IL-2 triggered inhibition of effector T cells. J Autoimmunity. (2018) 92:77–86. doi: 10.1016/j.jaut.2018.05.003
125. Mekala DJ, Geiger TL. Immunotherapy of autoimmune encephalomyelitis with redirected CD4+ CD25+ T lymphocytes. Blood. (2005) 105:2090–2. doi: 10.1182/blood-2004-09-3579
126. Clemente-Casares X, Blanco J, Ambalavanan P, Yamanouchi J, Singha S, Fandos C, et al. Expanding antigen-specific regulatory networks to treat autoimmunity. Nature. (2016) 530:434–40. doi: 10.1038/nature16962
127. June CH, O'Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. (2018) 359:1361–5. doi: 10.1126/science.aar6711
128. Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science. (2016) 353:179–84. doi: 10.1126/science.aaf6756
Keywords: autoimmunity, nervous system, clinical, therapy, pathophysiology
Citation: Bhagavati S (2021) Autoimmune Disorders of the Nervous System: Pathophysiology, Clinical Features, and Therapy. Front. Neurol. 12:664664. doi: 10.3389/fneur.2021.664664
Received: 05 February 2021; Accepted: 19 March 2021;
Published: 14 April 2021.
Edited by:
Robert Weissert, University of Regensburg, GermanyReviewed by:
Sudarshini Ramanathan, The University of Sydney, AustraliaHarry Alexopoulos, National and Kapodistrian University of Athens, Greece
Copyright © 2021 Bhagavati. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Satyakam Bhagavati, sbhagavati@downstate.edu