 Aislinn D. Maguire
Aislinn D. Maguire John R. Bethea
John R. Bethea Bradley J. Kerr
Bradley J. Kerr- 1Neuroscience and Mental Health Institute, University of Alberta, Edmonton, AB, Canada
- 2Drexel University, Philadelphia, PA, United States
- 3Department of Pharmacology, University of Alberta, Edmonton, AB, Canada
- 4Department of Anesthesiology and Pain Medicine, University of Alberta, Edmonton, AB, Canada
Multiple Sclerosis (MS) is a debilitating autoimmune disease often accompanied by severe chronic pain. The most common type of pain in MS, called neuropathic pain, arises from disease processes affecting the peripheral and central nervous systems. It is incredibly difficult to study these processes in patients, so animal models such as experimental autoimmune encephalomyelitis (EAE) mice are used to dissect the complex mechanisms of neuropathic pain in MS. The pleiotropic cytokine tumor necrosis factor α (TNFα) is a critical factor mediating neuropathic pain identified by these animal studies. The TNF signaling pathway is complex, and can lead to cell death, inflammation, or survival. In complex diseases such as MS, signaling through the TNFR1 receptor tends to be pro-inflammation and death, whereas signaling through the TNFR2 receptor is pro-homeostatic. However, most TNFα-targeted therapies indiscriminately block both arms of the pathway, and thus are not therapeutic in MS. This review explores pain in MS, inflammatory TNF signaling, the link between the two, and how it could be exploited to develop more effective TNFα-targeting pain therapies.
MS and Its Mouse Models
MS Background
Multiple Sclerosis (MS) is an autoimmune disease characterized by aberrant immune cell activity leading to inflammation and demyelinating lesions of central nervous system (CNS) (1–3). It's causes, while not fully understood, are likely a combination of genetic and environmental factors (4). The MS disease course can follow multiple trajectories. Primary progressive disease worsens steadily from onset. Progressive relapsing disease increasingly worsens but with some relapsing and remitting characteristics, meaning there are periods where symptoms worsen, then improve again. Most patients have a biphasic disease course, wherein they initially present with a relapsing-remitting phenotype, but as the disease progresses there is a switch to the secondary progressive phenotype and disability continually worsens (2, 5). There are numerous symptoms and comorbidities associated with MS, which can affect sensory, motor, and cognitive modalities. One of the most debilitating ailments experienced by MS patients is chronic pain (6, 7).
Pain in MS
Pain is a common feature for many patients diagnosed with MS (6, 8). The pain MS patients may encounter includes chronic headache, sudden neck pain called Lhermitte's sign, trigeminal neuralgia, extremity pain and hypersensitivity due to neuropathy (central or peripheral) (9). Pain is one of the most devastating comorbidities of MS, significantly interfering with daily life and yet, there are few treatments available (9–11). This is likely because neuropathic pain (NP), chronic pain that is caused by injury or disease of the nervous system, underlies most pain in MS (12). Neuropathic pain cannot be treated with typical painkillers such as opioids or anti-inflammatory drugs but instead, is often treated with more non-specific drugs like anti-depressants or anti-convulsants which have severe side effects (9, 11, 13, 14). Treating neuropathic pain in autoimmune diseases is further complicated by a process called sensitization, which occurs in both the peripheral and central nervous system (PNS and CNS). Sensitization can involve both intra and inter-cellular changes that increase pain sensations and allow for the maintenance of pain regardless of disease progression or treatment (15). Studies in similar autoimmune diseases (namely Rheumatoid Arthritis) have demonstrated that pain is often not resolved by disease-modifying treatment, and must be studied and treated separately (16). To study the mechanisms of pain in MS, researchers have turned to animal models which exhibit comparable disease phenotypes in the PNS and CNS, and most importantly, pain.
Mouse Models of MS
There are several paradigms used to induce MS-like symptoms in laboratory animals. Although MS models have been developed in rats, non-human primates, and even zebrafish, the greatest diversity exists in mouse models (17). MS models are categorized into three main groups, though viral infection, self-antigens that become recognized by the immune system, or toxins that cause demyelination. Theiler's murine encephalomyelitis virus (TMEV) induced disease is the best example of a viral induced model of MS, while experimental autoimmune encephalomyelitis (EAE) represents the prototypical antigen induced disease model, and demyelination is induced by cuprizone or lysophosphatidyl choline (LPC) administration (Table 1).
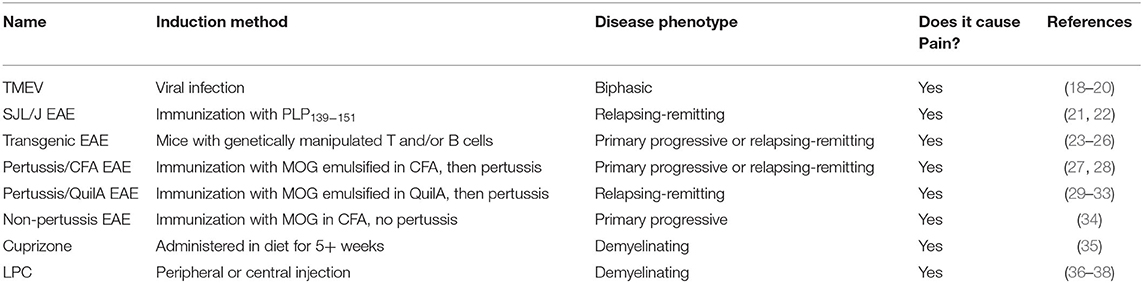
Table 1. Mouse models of Multiple Sclerosis.
Due to its induction method, mice infected with TMEV develop a biphasic disease phenotype that is useful for studying the viral contribution to MS (18, 19). EAE on the other hand, refers to a variety of ways to induce immune activation and demyelination that mimic MS pathophysiology. This is most often achieved by immunization with a myelin antigen. For example, EAE induced in Swiss Jim Lambert (SJL) mice with a fragment of proteolipid protein (PLP139−151) causes a relapsing-remitting disease phenotype (21). A primary progressive phenotype can be induced by immunization with myelin oligodendrocyte glycoprotein (MOG35−55) emulsified in an adjuvant such as CFA to trigger an immune response to myelin (27, 28, 39, 40). By modifying the concentration of MOG35−55 and the adjuvants used to induce EAE, a relapsing-remitting phenotype in C57Bl/6 mice can also be generated (29, 30, 41). MOG EAE immunization protocols are normally followed by injections with pertussis toxin to facilitate blood brain barrier breakdown (39), but this step can also be omitted (34). QuilA can also be used in place of Complete Freund's Adjuvant (CFA), the most used adjuvant in EAE models, to generate a relapsing-remitting phenotype (31).
Transgenic EAE is yet another method of mimicking MS in mice. T and/or B cells in these mice are genetically manipulated to react to MOG, and different strains have been developed to produce either a primary progressive or relapsing-remitting phenotype (23–26). Lastly, demyelination can be caused by either consumption of the copper chelator cuprizone which preferentially causes oligodendrocyte cell death (42, 43), or injection of LPC which integrates into membranes and disrupts myelin (44, 45). These models are useful to study demyelination separately from other MS disease processes. Although there are many ways to induce MS-like symptoms in mice, and each have their own strengths and weaknesses in modeling CNS lesions, demyelination, axonal damage, immune cell activation, they all produce pain (20, 22, 35).
Pain in MS Models
Animal models of MS have enabled researchers to study the mechanisms of chronic pain associated with the disease as the animals develop similar pain phenotypes to people with MS (46). Like MS patients, mice with EAE also exhibit cold and mechanical hypersensitivity, trigeminal neuralgia, and even sex differences in pain (9, 47, 48). Animals with TMEV and EAE exhibit hypersensitivity to painful and non-painful stimuli called, called hyperalgesia and allodynia, respectively (20, 46, 49). Interestingly, TMEV animals present with sex differences in pain, with females developing hypersensitivity more quickly than males (20). This sex difference is important as it allows researchers to better understand sex differences in human MS. In a foundational study of pain in EAE, animals exhibited heat-induced hyperalgesia not only when the disease was induced by immunization with a myelin peptide emulsified in CFA, but also when T cells from EAE mice were transferred to naïve mice (50). The cuprizone model has historically been studied less in the context of pain, but a recent study using electrical stimulation-induced paw withdrawal suggests there is a pain phenotype in cuprizone mice (35). LPC injection has also been associated with pain, but more commonly in the context of nerve or spinal cord injury (36–38).
One mechanism that may be responsible for some aspects of pain in MS animal models is immune cell activation and cytokine release, generating peripheral and central sensitization. Tumor necrosis factor alpha (TNFα) is of the most prominent pro-inflammatory cytokines present in MS and EAE, and it also has strong associations with many other chronic pain conditions (51–53).
TNFα Structure and Function
TNFα Signaling
TNFα is a pleiotropic cytokine with a multifaceted signaling pathway which can lead to cell death via either apoptosis or necrosis, or conversely, to survival and inflammation (Figure 1). TNFα originates in its transmembrane form (tmTNFα), produced by immune cells such as macrophages, monocytes, and lymphocytes (54, 55). Then it may be cleaved by TNFα converting enzyme (TACE) and released into its soluble form (sTNFα) (55). As the main determinant of TNFα isoform availability, TACE overactivity has been linked to inflammatory diseases (56). However, it is not currently a viable treatment target due to its similarity to other matrix metalloproteinases (57). There are two main subtypes of TNF receptors, TNFR1 and TNFR2. TNFR1 is expressed on most cell types and primarily mediates pro-inflammatory and pro-death signaling (58). It can bind both sTNFα and tmTNFα, but is preferentially activated by sTNFα (59, 60). TNFR2 is expressed mostly on immune cells and only associates with tmTNFα (61, 62). This receptor lacks a death domain and is associated with pro-survival and pro-homeostatic signaling (62). Interestingly, during this interaction, tmTNFα also transmits signals back into its host cell (55).
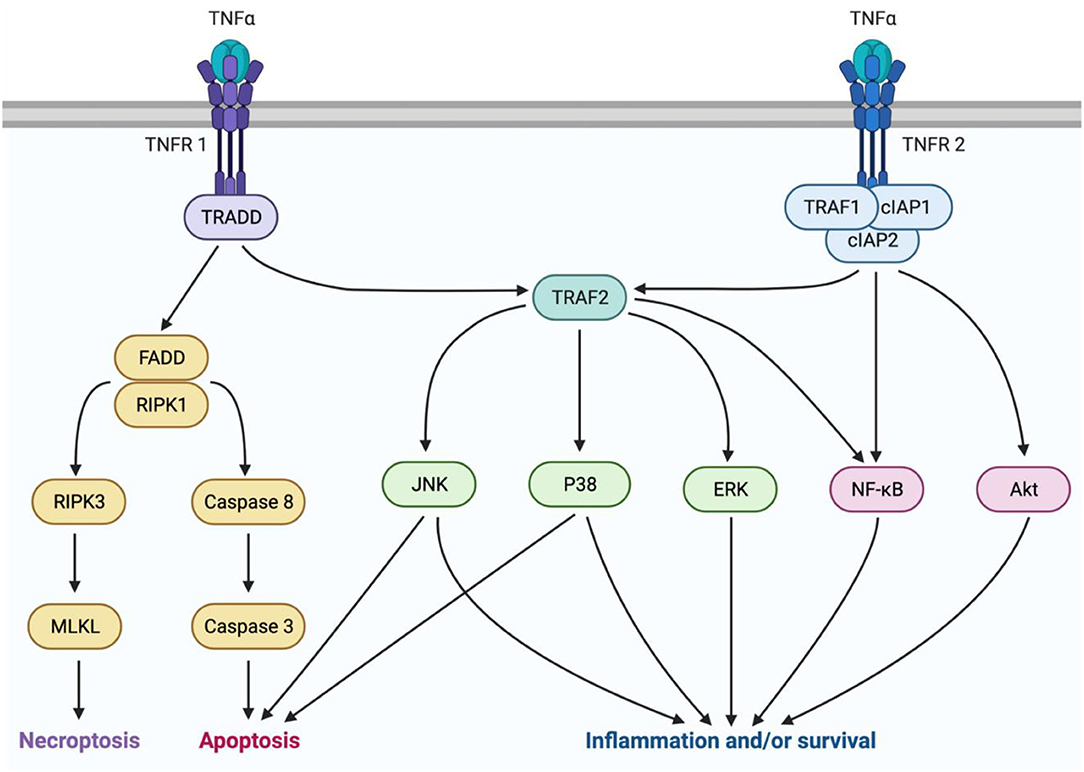
Figure 1. TNFα signaling is mediated by two isoforms of the cytokine and two receptor subtypes. sTNFα can interact with TNFR1 to mediate either pro-inflammatory or pro-death signaling. Pro-death signaling occurs through recruitment of FADD and RIPK1, which can either lead to necroptosis trough RIPK3 and MLKL activation, or apoptosis through caspase8 and caspase 3 activation. Pro-inflammatory signaling occurs through TRAF2 activation of P38, JNK, or ERK MAPKs, or NFκB. Alternatively, tmTNFα can interact with TNFR2 to mediate pro-homeostatic or pro-inflammatory signaling through TRAF1, cIAPs 1 and 2, and TRAF2. This pathway can be mediated by activation of MAPKs, NFκB, or Akt. This figure was made using BioRender.
sTNFα molecules act in a trimer and associate with three TNFR1 receptor subunits to activate the signaling complex by endocytosis into the cytoplasm (63). Next, Tumor necrosis factor receptor type 1-associated death domain protein (TRADD) associates with the receptor-ligand trimer. Further complex recruitment by TRADD then determines whether death or inflammation and survival will occur (63). Death signaling ensues if TRADD recruits fas-associated protein with death domain (FADD), and receptor-interacting serine/threonine-protein kinase (RIPK) 1 (64). Cell death occurs by apoptosis if initiator caspases 8 or 10 are recruited, or by necroptosis if RIPK3 and mixed lineage kinase domain-like pseudokinase (MLKL) are recruited (64). The pro-survival factor, TNF receptor-associated factor 2 (TRAF2) can prevent cell death by acting as an E3 ubiquitin ligase to target RIPK1 for degradation (65, 66). TRAF2 then initiates activation of the mitogen-activated protein kinases (MAPKs) P38, c-Jun-N-terminal kinase (JNK), and extracellular signal-regulate kinase (ERK), or the transcription factor nuclear factor kappa B (NFκB) (65). Pro-inflammatory signaling by these factors is a beneficial response to insults such as infection, but it can also be maladaptive, leading to pain (67, 68).
TNFR2 signaling also occurs in a trimeric fashion but rather interacts with tmTNFα, then recruits TRAF2 upon complex endocytosis. In addition, TNFR2 recruits TRAF1 and cellular inhibitors of apoptosis (cIAP1/2). This complex activates pro-survival signals through phosphatidylinositol 3-kinase (PI3K) and protein kinase B (Akt), and activates NFκB and JNK (69, 70). Although TNFR2 lacks a death domain, prolonged JNK activation by TNFR2 can lead to intrinsic apoptosis (65). Despite this ability to cause cell death, TNFR2 signaling is primarily pro-homeostatic and promotes many pro-survival activities including, cell proliferation, migration, and adhesion (71, 72).
TNFα and Pain
TNFα is involved in both central and peripheral mechanisms of chronic pain (73–75) (Figure 2). This has been demonstrated on a pre-clinical level in animal experiments which show that TNFα administration alone is sufficient to cause pain (76–78), and exogenous TNFα administered in animal models of inflammatory pain such as spinal nerve ligation (SNL) can exacerbate pain intensity and duration (79). In more complex animal models of neuropathic pain such as peripheral nerve injury (PNI), TNFα is elevated both centrally and peripherally, and TNF antagonists can be effective in relieving pain (80–84).
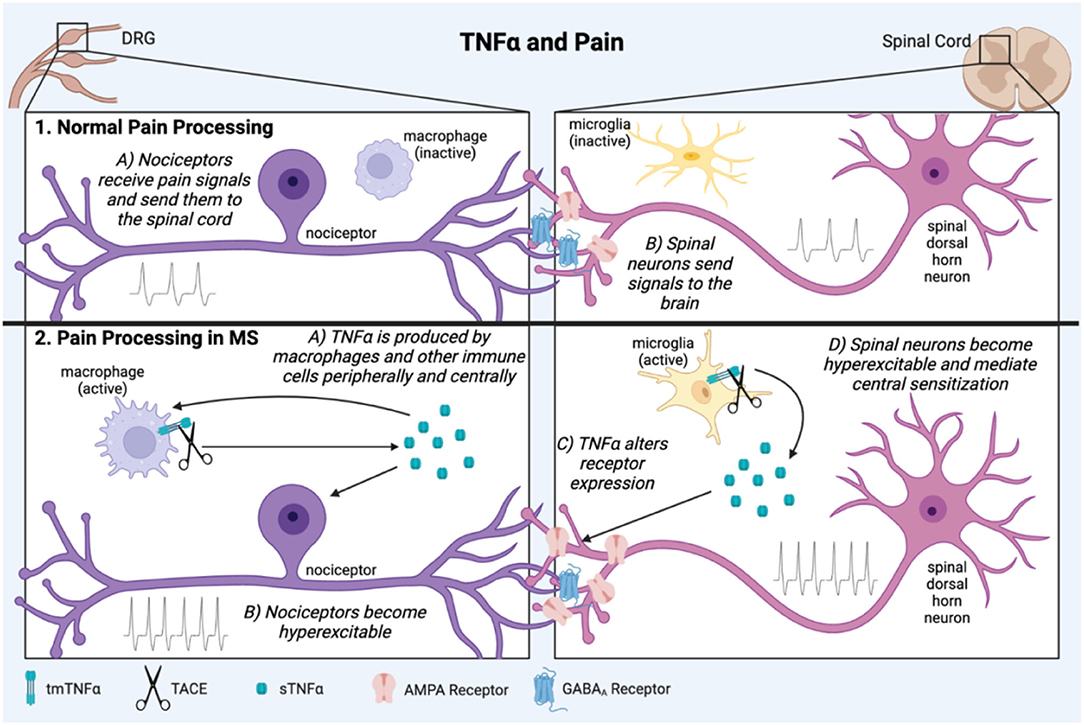
Figure 2. TNFα is involved in pathological pain processing in MS. In normal physiological pain processing, pain signals are sent from the periphery to the DRG, which relays information to the spinal cord then brain (1A,B). In neuropathic pain in MS, inflammation activates immune cells which secrete the pro-inflammatory cytokine, TNFα both peripherally and centrally (2A). It can then act back on the cells which produced it creating a positive feedback loop. TNFα contributes to sensitization of peripheral nociceptors and spinal dorsal horn neurons through mechanisms such as altered excitatory and inhibitory receptor expression (2B,C). These conditions lead to long term central pain sensitization of the brain and spinal cord (2D). This figure was made using BioRender.
Various mechanisms for how TNFα causes pain have been investigated. TNFα produced in response to inflammation can increase excitatory synapse strength and decrease inhibitory synapse strength by altering AMPA and GABAA receptor surface expression on neurons (85). In the hippocampus this hyperexcitation leads to excitotoxicity and neuronal death (85, 86), but in the dorsal horn of the spinal cord it can cause either excitotoxic cell death or sensory sensitization and pain (87, 88). TNFα is also a well-characterized activator of microglia, leading to further secretion of inflammatory mediators (89, 90). This process has been implicated in spinal mechanisms of neuropathic pain (84, 91).
Whether TNFα leads to inflammation or cell death depends on its downstream signaling. For example, in male rats, SNL injury increased TNFα and P38 MAPK expression in the DRG and spinal cord, and inhibition of either TNFα or P38 was sufficient to reduce mechanical allodynia (92). While P38 and JNK MAPK signaling can be pro-inflammatory and pro-survival, they can also lead to intrinsic apoptosis through mitochondria (93). TNFα can also be involved in pain in a secondary manner. In an animal model of intervertebral disc degeneration, TNFα signaling contributed to disc degeneration by inducing apoptosis through caspase 3, and that subsequent disc degeneration caused pain (94).
The detrimental effects of TNF signaling in pain conditions are mediated primarily by TNFR1. Several anti-TNFR1 antibodies have been developed, as have inhibitors of sTNFα used to block TNFR1 signaling. An example of an sTNFα blocker, XPro1595, inhibited hyperalgesia in a CFA model of inflammatory pain (95) and in EAE (96). Additionally, after spinal cord injury, XPro1595 treatment increased TNFR2 expression (97). TNFR2 is considered pro-homeostatic, as evidenced by studies in which TNFR2 agonism has relieved pain after PNI (58, 98). Current evidence thus indicates that sTNFα signaling through TNFR1 is pathological in pain conditions, whereas tmTMFα signaling through TNFR2 is protective.
While animal models have the benefits of being well controlled for age, sex, environment, and the nature of illness or injury, the inherent variability in human populations complicates the study of pain and its treatment. However, TNFα is elevated is a number of painful conditions in humans including chemotherapy-induced neuropathic pain (CIPN) and rheumatoid arthritis (RA) (51). Non-specific TNFα antagonists have shown some effectiveness in relieving pain in RA, but not in all inflammatory pain conditions in which the cytokine might be involved, such as MS (99, 100).
TNFα in MS and EAE
TNF signaling may be involved in MS pathogenesis through several points of action. TNFα is elevated centrally in MS patients, and this is correlated with disease severity (101). However, it is incredibly difficult to study the precise mechanisms underlying TNF signaling in MS in humans. Much of the proposed actions of TNFα in MS have been discovered through study of animal models such as EAE. In both MS and its animal counterparts, the major pathological landmarks are immune cell infiltration into the CNS, and the development of demyelinating lesions which eventually lead to neuronal death. In EAE, TNFα transport is upregulated at the blood-brain barrier (BBB), as is TNFα expression by mast cells, which are involved in BBB breakdown (102). TNFα also promotes activation of T cells (103, 104), and is upregulated in demyelinating lesions in EAE where it is hypothesized to promote neuronal excitotoxicity and oligodendrocyte death (103, 105). Therefore, TNFα appears to be involved in immune cell activation and infiltration into the CNS, as well as demyelination and axonal injury. The receptor subtype employed in TNF signaling also shapes disease progression in MS and EAE. TNFR1 expression is correlated with disease progression, whereas TNFR2 promotes repair and remyelination through oligodendrocyte survival and differentiation (106–108) (Figure 3).
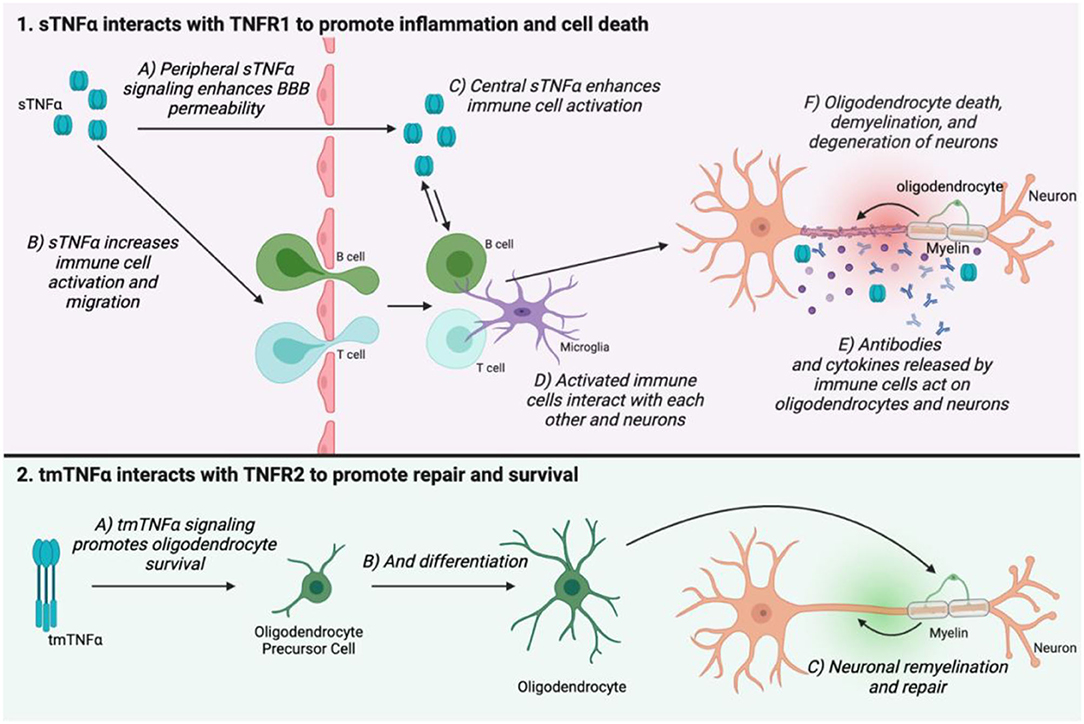
Figure 3. The two TNFα isoforms and their receptors have opposing actions on MS disease progression. sTNFα interacts with TNFR1 in the peripheral nervous system to enhance BBB permeability (1A), as well as increase immune cell activation and migration (1B). TNFα can migrate across the weakened BBB, but it is also produced in the central nervous system where it again promotes immune cell activation (1C,D). Immune cells secrete antibodies and cytokines which mediate oligodendrocyte death, demyelination, and neuronal damage (1E,F). tmTNFα interacts with TNFR2 to promote oligodendrocyte survival and differentiation. This helps to promote remyelination and repair (2C). This figure was made using BioRender.
While central TNF signaling is involved in disease progression in MS and EAE, peripheral TNFα elevation is a likely culprit for the development of chronic pain (52). Infiltration of TNFα-producing immune cells is evident peripherally in EAE (109). Macrophages, for example, both produce TNFα and are strongly affected by it (110). In a pro-inflammatory environment where sTNFα is the dominant isoform, TNFR1 signaling in macrophages can enhance their activation, resulting in a positive feedback loop (111). Primary pain sensing neurons, called nociceptors, express TNFR1 (112), and signaling through this receptor can cause nociceptors to become hyperexcitable, meaning they may be more likely to fire action potentials, and do so more intensely in response to painful stimuli (113, 114). These hyperexcitable nociceptors then signal to the spinal cord and brain, leading to central sensitization, which maintains pain chronically (115). This mechanism for pain has been proposed in other painful conditions like PNI and RA, where treating the peripheral causes of pain is ineffective once central sensitization has been established (115–117). It may also explain why in EAE, pain sensitivity to stimuli occurs early in the disease course, before full disease onset (118). While pain mechanisms in EAE have not been fully elucidated, understanding this model of peripheral and central sensitization will help inform future areas of study and potential treatments.
Targeting TNFα as an Analgesic Strategy
Anti-TNF Therapies
There are five non-specific TNFα inhibitors currently in clinical use (Table 2). Infliximab, Adalimimab, Golimumab, and Certolizumab are antibodies that target TNFα, and Etanercept is a soluble recombinant TNF receptor (119, 120). All of these drugs work by binding to and sequestering both the soluble and transmembrane forms of TNFα so they cannot interact with their receptors and initiate signaling cascades (55). Anti-TNF drugs can be beneficial in various types of arthritis, ankylosing sponditis, plaque psoriasis, Crohn's disease, and ulcerative colitis (121). However, there are severe side effects associated with TNF inhibition. TNFα is an important immune-mediator, and blocking its actions can be immunosuppressive, thereby increasing the risk of new infections as well as re-activation of dormant infections (122, 123). There is also evidence these drugs cause demyelination and liver damage (124, 125). The severe side effects of TNF inhibition may be due to blockade of the homeostatic functions TNFα, particularly through TNFR2 signaling. This indiscriminate blockade of TNFα may also help to explain why pain management is lacking with anti-TNF treatment (126–128).
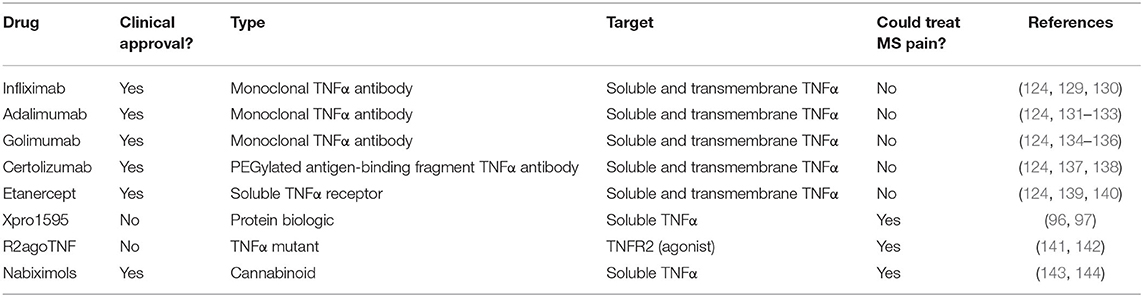
Table 2. TNFα-targeted therapies.
Anti-TNF therapies have mixed effectiveness in treating pain depending on the condition for which they are used. In a rat chronic constriction injury model of PNI, TNF inhibition reduced mechanical and thermal pain (99, 145). Pain reduction by anti-TNF drugs in PNI has been suggested to occur through an alteration of TNF receptor expression in the spinal cord (146). Anti-TNF treatment with either etanercept or infliximab decreased the TNFR1/TNFR2 ratio, and this correlated with better recovery (145). This finding conforms with the view of TNF receptors which considers TNFR1 to be involved in pathology/damage, and TNFR2 to be involved in repair and homeostasis (147, 148). Although it is unclear how exactly these anti-TNF drugs modulate receptor expression, these findings provide a strong rationale to further investigate TNF receptor modulation in neuropathic pain treatment, and there are currently drugs in development for this purpose (Table 2).
In another peripherally-driven chronic pain disorder, diabetic peripheral neuropathy (DPN), TNF inhibition blocked mechanical but not thermal pain in a rat model (99, 149). Anti-TNF treatment in this model has also improved signs of nerve degeneration associated with advancing DPN, restoring conduction velocity, myelination, myelin basic protein expression, as well as lamellar and axonal organization (150). Based on the findings in these two models, TNF inhibition can affect sensory neuron inflammation and degeneration, as well as spinal TNF receptor expression. It will be important for future studies in PNI and DPN models to address both peripheral and central mechanisms. This may provide a better understanding of how targeting TNF can prevent and/or relieve neuropathic pain.
Anti-TNF in MS: The Double-Edged Sword
Anti-TNF therapies are generally not only ineffective in treating MS, but they can also worsen disease severity. They are also known to increase the risk of developing MS in patients receiving anti-TNF treatment for other conditions (124, 151, 152). Studies using EAE have provided insight into why universal blockade of TNF signaling can be detrimental in the disease. Anti-TNF molecules sequester both soluble and transmembrane TNFα and block signaling through both TNFR1 and TNFR2 (55). In EAE, inhibition of soluble TNFα signaling through TNFR1 promotes remyelination and axon survival (96). However, transmembrane TNFα signaling through TNFR2 may be neuroprotective (153). TNFR2 signaling supports regulatory T cells (Tregs) (98, 154), and promotes remyelination through oligodendrocyte differentiation (106). Ultimately, while blocking all TNF signaling in MS and EAE can reduce its detrimental effects, it also reduces the beneficial aspects of TNFR2 signaling, leading to a net negative result for anti-TNF therapies in MS and EAE.
Drugs that are more specifically targeted to cell type, TNFα isoform, and TNF receptor type will be necessary to further explore TNF therapies for disease modification and pain treatment in MS (Table 2). For example, in EAE, treatment with a selective TNFR2 agonist reduced motor symptom and pain severity and improved various other hallmarks of the disease (155). Cannabinoids are another potential treatment for MS pain, as they are linked to a preferential reduction sTNFα production by both peripheral and central immune cells (156–158), and have shown promising analgesia in clinical trials (159, 160). Further investigation into similar receptor and/or isoform targeted drugs may make TNF therapy a viable option in MS pain treatment.
Gaps in Knowledge and Future Directions
TNFα mediates the development of neuropathic pain in many conditions. While indiscriminate TNF inhibition is effective in some human pain conditions and animal models, it is ineffective and can have deleterious consequences in MS. To develop therapies that effectively target TNF signaling to treat pain in MS we must first focus on developing a better understanding of the cell types, receptors, and downstream pathways involved both peripherally and centrally. Research in the EAE model has already led to the development of promising TNFR1 antagonists and TNFR2 agonists. Other components of the TNF pathway such MAPKs and NFκB may also provide points of intervention. These targeted therapies are the future of pain management in MS and other neuropathic pain conditions.
Author Contributions
AM, BK, and JB conceptualized the review. AM wrote the manuscript. BK and JB edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a Project Grant from the Canadian Institutes of Health Research (FRN-162434) and a Discovery Grant from the MS Society of Canada (EGID-3761).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Reich DS, Lucchinetti CF, Calabresi PA. Multiple Sclerosis. N Engl J Med. (2018) 378:169–80. doi: 10.1056/NEJMra1401483
2. Filippi M, Bar-Or A, Piehl F, Preziosa P, Solari A, Vukusic S, Rocca MA. Multiple sclerosis. Nat Rev Dis Primers. (2018) 4:43. doi: 10.1038/s41572-018-0041-4
3. Hemmer B, Kerschensteiner M, Korn T. Role of the innate and adaptive immune responses in the course of multiple sclerosis. Lancet Neurol. (2015) 14:406–19. doi: 10.1016/S1474-4422(14)70305-9
4. Olsson T, Barcellos LF, Alfredsson L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat Rev Neurol. (2017) 13:25–36. doi: 10.1038/nrneurol.2016.187
5. Lublin FD, Reingold SC, Cohen JA, Cutter GR, Sorensen PS, Thompson AJ, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. (2014) 83:278–86. doi: 10.1212/WNL.0000000000000560
6. Brochet B, Deloire MS, Ouallet JC, Salort E, Bonnet M, Jove J, et al. Pain and quality of life in the early stages after multiple sclerosis diagnosis:a 2-year longitudinal study. Clin J Pain. (2009) 25:211–7. doi: 10.1097/AJP.0b013e3181891347
7. Benson C, Kerr BJ. Pain and cognition in multiple sclerosis. Curr Top Behav Neurosci. (2014) 20:201–15. doi: 10.1007/7854_2014_309
8. O'Connor AB, Schwid SR, Herrmann DN, Markman JD, Dworkin RH. Pain associated with multiple sclerosis:systematic review and proposed classification. Pain. (2008) 137:96–111. doi: 10.1016/j.pain.2007.08.024
9. Iannitti T, Kerr BJ, Taylor BK. Mechanisms and pharmacology of neuropathic pain in multiple sclerosis. Curr Top Behav Neurosci. (2014) 20:75–97. doi: 10.1007/7854_2014_288
10. Solaro C, Trabucco E, Messmer Uccelli M. Pain and multiple sclerosis:pathophysiology and treatment Curr. Neurol Neurosci Rep. (2013) 13:320. doi: 10.1007/s11910-012-0320-5
11. Murphy KL, Bethea JR, Fischer R. Neuropathic pain in multiple sclerosis-current therapeutic intervention and future treatment perspectives. In: Zagon IS, McLaughlin PJ, editors. Multiple Sclerosis:Perspectives in Treatment and Pathogenesis Brisbane (AU). (2017). doi: 10.15586/codon.multiplesclerosis.2017.ch4
12. St. John Smith E. Advances in understanding nociception and neuropathic pain. J Neurol. (2018) 265:231–8. doi: 10.1007/s00415-017-8641-6
13. Bermejo PE, Oreja-Guevara C, Diez-Tejedor E. Pain in multiple sclerosis:prevalence, mechanisms, types and treatment. Rev Neurol. (2010) 50:101–8. doi: 10.33588/rn.5002.2008613
14. Sansone RA, Sansone LA. Pain, pain, go away:antidepressants and pain management. Psychiatry. (2008) 5:16–19.
15. Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. (2009) 139:267–84. doi: 10.1016/j.cell.2009.09.028
16. Cao Y, Fan D, Yin Y. Pain mechanism in rheumatoid arthritis:from cytokines to central sensitization. Mediators Inflamm. (2020). (2020:2076328. doi: 10.1155/2020/2076328
17. Burrows DJ, McGown A, Jain SA, De Felice M, Ramesh TM, Sharrack B, et al. Animal models of multiple sclerosis:From rodents to zebrafish. Mult Scler. (2019) 25:306–24. doi: 10.1177/1352458518805246
18. Tsunoda I, Fujinami RS. Neuropathogenesis of Theiler's murine encephalomyelitis virus infection, an animal model for multiple sclerosis. J Neuroimmune Pharmacol. (2010) 5:355–69. doi: 10.1007/s11481-009-9179-x
19. Oleszak EL, Chang JR, Friedman H, Katsetos CD, Platsoucas CD. Theiler's virus infection:a model for multiple sclerosis. Clin Microbiol Rev. (2004) 17:174–207. doi: 10.1128/CMR.17.1.174-207.2004
20. Lynch JL, Gallus NJ, Ericson ME, Beitz AJ. Analysis of nociception, sex and peripheral nerve innervation in the TMEV animal model of multiple sclerosis. Pain. (2008) 136:293–304. doi: 10.1016/j.pain.2007.07.007
21. McRae BL, Kennedy MK, Tan LJ, Dal Canto MC, Picha KS, Miller SD. Induction of active and adoptive relapsing experimental autoimmune encephalomyelitis (EAE) using an encephalitogenic epitope of proteolipid protein. J Neuroimmunol. (1992) 38:229–40. doi: 10.1016/0165-5728(92)90016-E
22. Lu J, Kurejova M, Wirotanseng LN, Linker RA, Kuner R, Tappe-Theodor A. Pain in experimental autoimmune encephalitis:a comparative study between different mouse models. J Neuroinflammation. (2012) 9:233. doi: 10.1186/1742-2094-9-233
23. Krishnamoorthy G, Lassmann H, Wekerle H, Holz A. Spontaneous opticospinal encephalomyelitis in a double-transgenic mouse model of autoimmune T cell/B cell cooperation. J Clin Invest. (2006) 116:2385–92. doi: 10.1172/JCI28330
24. Litzenburger T, Fassler R, Bauer J, Lassmann H, Linington C, Wekerle H, et al. B lymphocytes producing demyelinating autoantibodies:development and function in gene-targeted transgenic mice. J Exp Med. (1998) 188:169–80. doi: 10.1084/jem.188.1.169
25. Pollinger B, Krishnamoorthy G, Berer K, Lassmann H, Bosl MR, Dunn R, et al. Spontaneous relapsing-remitting EAE in the SJL/J mouse:MOG-reactive transgenic T cells recruit endogenous MOG-specific B cells. J Exp Med. (2009) 206:1303–16. doi: 10.1084/jem.20090299
26. Anderson AC, Chandwaskar R, Lee DH, Sullivan JM, Solomon A, Rodriguez-Manzanet R, et al. A transgenic model of central nervous system autoimmunity mediated by CD4+ and CD8+ T and B cells. J Immunol. (2012) 188:2084–92. doi: 10.4049/jimmunol.1102186
27. Mendel I, Kerlero de Rosbo N, Ben-Nun A. A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2b mice: fine specificity and T cell receptor V beta expression of encephalitogenic T cells. Eur J Immunol. (1995) 25:1951–9. doi: 10.1002/eji.1830250723
28. Giralt M, Molinero A, Hidalgo J. Active Induction of Experimental Autoimmune Encephalomyelitis (EAE) with MOG35-55 in the Mouse. Meth Mol Biol. (2018) 1791:227–32. doi: 10.1007/978-1-4939-7862-5_17
29. Segal JP, Bannerman CA, Silva JR, Haird CM, Baharnoori M, Gilron I, et al. Chronic mechanical hypersensitivity in experimental autoimmune encephalomyelitis is regulated by disease severity and neuroinflammation. Brain Behav Immun. (2020) 89:314–25. doi: 10.1016/j.bbi.2020.07.010
30. Olechowski CJ, Truong JJ, Kerr BJ. Neuropathic pain behaviours in a chronic-relapsing model of experimental autoimmune encephalomyelitis (EAE). Pain. (2009) 141:156–64. doi: 10.1016/j.pain.2008.11.002
31. Peiris M, Monteith GR, Roberts-Thomson SJ, Cabot PJ. A model of experimental autoimmune encephalomyelitis (EAE) in C57BL/6 mice for the characterisation of intervention therapies. J Neurosci Methods. (2007) 163:245–54. doi: 10.1016/j.jneumeth.2007.03.013
32. Flebbe LM, Braley-Mullen H. Immunopotentiation by SGP and Quil A. II. Identification of responding cell populations. Cell Immunol. (1986) 99:128–39. doi: 10.1016/0008-8749(86)90222-4
33. Khan N, Woodruff TM, Smith MT. Establishment and characterization of an optimized mouse model of multiple sclerosis-induced neuropathic pain using behavioral, pharmacologic, histologic and immunohistochemical methods. Pharmacol Biochem Behav. (2014) 126:13–27. doi: 10.1016/j.pbb.2014.09.003
34. Murphy KL, Fischer R, Swanson KA, Bhatt IJ, Oakley L, Smeyne R, et al. Synaptic alterations and immune response are sexually dimorphic in a non-pertussis toxin model of experimental autoimmune encephalomyelitis. Exp Neurol. (2020) 323:113061. doi: 10.1016/j.expneurol.2019.113061
35. Tsukahara R, Yamamoto S, Yoshikawa K, Gotoh M, Tsukahara T, Neyama H, et al. LPA5 signaling is involved in multiple sclerosis-mediated neuropathic pain in the cuprizone mouse model. J Pharmacol Sci. (2018) 136:93–6. doi: 10.1016/j.jphs.2018.01.001
36. Inoue M, Xie W, Matsushita Y, Chun J, Aoki J, Ueda H. Lysophosphatidylcholine induces neuropathic pain through an action of autotaxin to generate lysophosphatidic acid. Neuroscience. (2008) 152:296–8. doi: 10.1016/j.neuroscience.2007.12.041
37. Wang HY, Tsai YJ, Chen SH, Lin CT, Lue JH. Lysophosphatidylcholine causes neuropathic pain via the increase of neuronal nitric oxide synthase in the dorsal root ganglion and cuneate nucleus. Pharmacol Biochem Behav. (2013) 106:47–56. doi: 10.1016/j.pbb.2013.03.002
38. Uranbileg B, Ito N, Kurano M, Saigusa D, Saito R, Uruno A, et al. Alteration of the lysophosphatidic acid and its precursor lysophosphatidylcholine levels in spinal cord stenosis: a study using a rat cauda equina compression model. Sci Rep. (2019) 9:16578. doi: 10.1038/s41598-019-52999-5
39. Schreiner B, Heppner FL, Becher B. Modeling multiple sclerosis in laboratory animals. Semin Immunopathol. (2009) 31:479–95. doi: 10.1007/s00281-009-0181-4
40. Bittner S, Afzali AM, Wiendl H, Meuth SG. Myelin oligodendrocyte glycoprotein (MOG35-55) induced experimental autoimmune encephalomyelitis (EAE) in C57BL/6 mice. J Vis Exp. (2014) 86:51275. doi: 10.3791/51275
41. Berard JL, Wolak K, Fournier S, David S. Characterization of relapsing-remitting and chronic forms of experimental autoimmune encephalomyelitis in C57BL/6 mice. Glia. (2010) 58:434–45. doi: 10.1002/glia.20935
42. Torkildsen O, Brunborg LA, Myhr KM, Bo L. The cuprizone model for demyelination. Acta Neurol Scand Suppl. (2008) 188:72–6. doi: 10.1111/j.1600-0404.2008.01036.x
43. Zhan J, Mann T, Joost S, Behrangi N, Frank M, Kipp M. The Cuprizone Model:Dos and Do Nots. Cells. (2020) 9:843. doi: 10.3390/cells9040843
44. Ayanwuyi L, Tokarska N, McLean NA, Johnston JM, Verge VMK. Brief electrical nerve stimulation enhances intrinsic repair capacity of the focally demyelinated central nervous system. Neural Regen Res. (2022) 17:1042–50. doi: 10.4103/1673-5374.324848
45. Plemel JR, Michaels NJ, Weishaupt N, Caprariello AV, Keough MB, Rogers JA, et al. Mechanisms of lysophosphatidylcholine-induced demyelination: a primary lipid disrupting myelinopathy. Glia. (2018) 66:327–47. doi: 10.1002/glia.23245
46. Tian DH, Perera CJ, Moalem-Taylor G. Neuropathic pain in animal models of nervous system autoimmune diseases. Mediators Inflamm. (2013) 2013:298326. doi: 10.1155/2013/298326
47. Thorburn KC, Paylor JW, Webber CA, Winship IR, Kerr BJ. Facial hypersensitivity and trigeminal pathology in mice with experimental autoimmune encephalomyelitis. Pain. (2016) 157:627–42. doi: 10.1097/j.pain.0000000000000409
48. Rahn EJ, Iannitti T, Donahue RR, Taylor BK. Sex differences in a mouse model of multiple sclerosis:neuropathic pain behavior in females but not males and protection from neurological deficits during proestrus. Biol Sex Differ. (2014) 5:4. doi: 10.1186/2042-6410-5-4
49. Khan N, Smith MT. Multiple sclerosis-induced neuropathic pain:pharmacological management and pathophysiological insights from rodent EAE models. Inflammopharmacology. (2014) 22:1–22. doi: 10.1007/s10787-013-0195-3
50. Aicher SA, Silverman MB, Winkler CW, Bebo BF Jr. Hyperalgesia in an animal model of multiple sclerosis. Pain. (2004) 110:560–70. doi: 10.1016/j.pain.2004.03.025
51. Leung L, Cahill CM. TNF-alpha and neuropathic pain—a review. J Neuroinflammation. (2010) 7:27. doi: 10.1186/1742-2094-7-27
52. Ribeiro CM, Oliveira SR, Alfieri DF, Flauzino T, Kaimen-Maciel DR, Simao ANC, et al. Tumor necrosis factor alpha (TNF-alpha) and its soluble receptors are associated with disability, disability progression and clinical forms of multiple sclerosis. Inflamm Res. (2019) 68:1049–59. doi: 10.1007/s00011-019-01286-0
53. Madsen PM, Desu HL, de Rivero Vaccari JP, Florimon Y, Ellman DG, Keane RW, et al. Oligodendrocytes modulate the immune-inflammatory response in EAE via TNFR2 signaling. Brain Behav Immun. (2020) 84:132–46. doi: 10.1016/j.bbi.2019.11.017
54. Idriss HT, Naismith JH. TNF alpha and the TNF receptor superfamily:structure-function relationship(s). Microsc Res Tech. (2000) 50:184–95. doi: 10.1002/1097-0029(20000801)50:3<184::AID-JEMT2>3.0.CO;2-H
55. Horiuchi T, Mitoma H, Harashima S, Tsukamoto H, Shimoda T. Transmembrane TNF-alpha:structure, function and interaction with anti-TNF agents. Rheumatology. (2010) 49:1215–28. doi: 10.1093/rheumatology/keq031
56. Yu Y, Cao Y, Bell B, Chen X, Weiss RM, Felder RB, et al. Brain TACE (tumor necrosis factor-alpha-converting enzyme) contributes to sympathetic excitation in heart failure rats. Hypertension. (2019) 74:63–72. doi: 10.1161/HYPERTENSIONAHA.119.12651
57. Bahia MS, Silakari O. Tumor necrosis factor alpha converting enzyme:an encouraging target for various inflammatory disorders. Chem Biol Drug Des. (2010) 75:415–43. doi: 10.1111/j.1747-0285.2010.00950.x
58. Atretkhany KN, Gogoleva VS, Drutskaya MS, Nedospasov SA. Distinct modes of TNF signaling through its two receptors in health and disease. J Leukoc Biol. (2020) 107:893–905. doi: 10.1002/JLB.2MR0120-510R
59. Grell M, Wajant H, Zimmermann G, Scheurich P. The type 1 receptor (CD120a) is the high-affinity receptor for soluble tumor necrosis factor. Proc Natl Acad Sci USA. (1998) 95:570–75. doi: 10.1073/pnas.95.2.570
60. Rossol M, Meusch U, Pierer M, Kaltenhauser S, Hantzschel H, Hauschildt S, et al. Interaction between transmembrane TNF and TNFR1/2 mediates the activation of monocytes by contact with T cells. J Immunol. (2007) 179:4239–48. doi: 10.4049/jimmunol.179.6.4239
61. Grell M, Douni E, Wajant H, Lohden M, Clauss M, Maxeiner B, et al. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell. (1995) 83:793–802. doi: 10.1016/0092-8674(95)90192-2
62. Rauert H, Wicovsky A, Muller N, Siegmund D, Spindler V, Waschke J, et al. Membrane tumor necrosis factor (TNF) induces p100 processing via TNF receptor-2 (TNFR2). J Biol Chem. (2010) 285:7394–404. doi: 10.1074/jbc.M109.037341
63. Tsao DH, McDonagh T, Telliez JB, Hsu S, Malakian K, Xu GY, et al. Solution structure of N-TRADD and characterization of the interaction of N-TRADD and C-TRAF2, a key step in the TNFR1 signaling pathway. Mol Cell. (2000) 5:1051–7. doi: 10.1016/S1097-2765(00)80270-1
64. Liu Y, Liu T, Lei T, Zhang D, Du S, Girani L, et al. RIP1/RIP3-regulated necroptosis as a target for multifaceted disease therapy (Review). Int J Mol Med. (2019) 44:771–86. doi: 10.3892/ijmm.2019.4244
65. Borghi A, Verstrepen L, Beyaert R. TRAF2 multitasking in TNF receptor-induced signaling to NF-kappaB, MAP kinases and cell death. Biochem Pharmacol. (2016) 116:1–10. doi: 10.1016/j.bcp.2016.03.009
66. Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell. (2008) 30:689–700. doi: 10.1016/j.molcel.2008.05.014
67. Chen Y, Chen X, Yu J, Xu X, Wei X, Gu X, Xu Z, et al. JAB1 is involved in neuropathic pain by regulating JNK and NF-kappaB activation after chronic constriction injury. Neurochem Res. (2016) 41:1119–29. doi: 10.1007/s11064-015-1802-z
68. Obata K, Noguchi K. MAPK activation in nociceptive neurons and pain hypersensitivity. Life Sci. (2004) 74:2643–53. doi: 10.1016/j.lfs.2004.01.007
69. Borghi A, Haegman M, Fischer R, Carpentier I, Bertrand MJM, Libert C, Beyaert R, et al. The E3 ubiquitin ligases HOIP and cIAP1 are recruited to the TNFR2 signaling complex and mediate TNFR2-induced canonical NF-kappaB signaling. Biochem Pharmacol. (2018) 153:292–8. doi: 10.1016/j.bcp.2018.01.039
70. Jupp OJ, McFarlane SM, Anderson HM, Littlejohn AF, Mohamed AA, MacKay RH, MacEwan DJ, et al. Type II tumour necrosis factor-alpha receptor (TNFR2) activates c-Jun N-terminal kinase (JNK) but not mitogen-activated protein kinase (MAPK) or p38 MAPK pathways. Biochem J. (2001) 359:525–35. doi: 10.1042/bj3590525
71. Yang S, Wang J, Brand DD, Zheng SG. Role of TNF-TNF Receptor 2 Signal in Regulatory T Cells and Its Therapeutic Implications. Front Immunol. (2018) 9:784. doi: 10.3389/fimmu.2018.00784
72. Zhang R, Xu Y, Ekman N, Wu Z, Wu J, Alitalo K, Min W. Etk/Bmx transactivates vascular endothelial growth factor 2 and recruits phosphatidylinositol 3-kinase to mediate the tumor necrosis factor-induced angiogenic pathway. J Biol Chem. (2003) 278:51267–76. doi: 10.1074/jbc.M310678200
73. Wei XH, Na XD, Liao GJ, Chen QY, Cui Y, Chen FY Li YY, Liu XG, et al. The up-regulation of IL-6 in DRG and spinal dorsal horn contributes to neuropathic pain following L5 ventral root transection. Exp Neurol. (2013) 241:159–68. doi: 10.1016/j.expneurol.2012.12.007
74. Wu Y, Na X, Zang Y, Cui Y, Xin W, Pang R, Liu X, et al. Upregulation of tumor necrosis factor-alpha in nucleus accumbens attenuates morphine-induced rewarding in a neuropathic pain model. Biochem Biophys Res Commun. (2014) 449:502–7. doi: 10.1016/j.bbrc.2014.05.025
75. Genevay S, Finckh A, Payer M, Mezin F, Tessitore E, Gabay C, et al. Elevated levels of tumor necrosis factor-alpha in periradicular fat tissue in patients with radiculopathy from herniated disc. Spine. (2008) 33:2041–6. doi: 10.1097/BRS.0b013e318183bb86
76. Sorkin LS, Doom CM. Epineurial application of TNF elicits an acute mechanical hyperalgesia in the awake rat. J Peripher Nerv Syst. (2000) 5:96–100. doi: 10.1046/j.1529-8027.2000.00012.x
77. Wagner R, Myers RR. Endoneurial injection of TNF-alpha produces neuropathic pain behaviors. Neuroreport. (1996) 7:2897–901. doi: 10.1097/00001756-199611250-00018
78. Schafers M, Sorkin LS, Sommer C. Intramuscular injection of tumor necrosis factor-alpha induces muscle hyperalgesia in rats. Pain. (2003) 104:579–88. doi: 10.1016/S0304-3959(03)00115-5
79. Schafers M, Lee DH, Brors D, Yaksh TL, Sorkin LS. Increased sensitivity of injured and adjacent uninjured rat primary sensory neurons to exogenous tumor necrosis factor-alpha after spinal nerve ligation. J Neurosci. (2003) 23:3028–38. doi: 10.1523/JNEUROSCI.23-07-03028.2003
80. Sommer C, Schmidt C, George A. Hyperalgesia in experimental neuropathy is dependent on the TNF receptor 1. Exp Neurol. (1998) 151:138–42. doi: 10.1006/exnr.1998.6797
81. Shubayev VI, Myers RR. Upregulation and interaction of TNFalpha and gelatinases A and B in painful peripheral nerve injury. Brain Res. (2000) 855:83–9. doi: 10.1016/S0006-8993(99)02321-5
82. Jancalek R, Dubovy P, Svizenska I, Klusakova I. Bilateral changes of TNF-alpha and IL-10 protein in the lumbar and cervical dorsal root ganglia following a unilateral chronic constriction injury of the sciatic nerve. J Neuroinflammation. (2010) 7:11. doi: 10.1186/1742-2094-7-11
83. Sacerdote P, Franchi S, Trovato AE, Valsecchi AE, Panerai AE, Colleoni M. Transient early expression of TNF-alpha in sciatic nerve and dorsal root ganglia in a mouse model of painful peripheral neuropathy. Neurosci Lett. (2008) 436:210–3. doi: 10.1016/j.neulet.2008.03.023
84. Liu Y, Zhou LJ, Wang J, Li D, Ren WJ, Peng J, et al. TNF-alpha Differentially regulates synaptic plasticity in the hippocampus and spinal cord by microglia-dependent mechanisms after peripheral nerve injury. J Neurosci. (2017) 37:871–81. doi: 10.1523/JNEUROSCI.2235-16.2016
85. Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci. (2005) 25:3219–28. doi: 10.1523/JNEUROSCI.4486-04.2005
86. Leonoudakis D, Zhao P, Beattie EC. Rapid tumor necrosis factor alpha-induced exocytosis of glutamate receptor 2-lacking AMPA receptors to extrasynaptic plasma membrane potentiates excitotoxicity. J Neurosci. (2008) 28:2119–30. doi: 10.1523/JNEUROSCI.5159-07.2008
87. Wigerblad G, Huie JR, Yin HZ, Leinders M, Pritchard RA, Koehrn FJ, et al. Inflammation-induced GluA1 trafficking and membrane insertion of Ca(2+) permeable AMPA receptors in dorsal horn neurons is dependent on spinal tumor necrosis factor, PI3 kinase and protein kinase A. Exp Neurol. (2017) 293:144–58. doi: 10.1016/j.expneurol.2017.04.004
88. Ferguson AR, Christensen RN, Gensel JC, Miller BA, Sun F, Beattie EC, Beattie MS, et al. Cell death after spinal cord injury is exacerbated by rapid TNF alpha-induced trafficking of GluR2-lacking AMPARs to the plasma membrane. J Neurosci. (2008) 28:11391–400. doi: 10.1523/JNEUROSCI.3708-08.2008
89. Hoogland IC, Houbolt C, van Westerloo DJ, van Gool WA, van de Beek D. Systemic inflammation and microglial activation:systematic review of animal experiments. J Neuroinflammation. (2015) 12:114. doi: 10.1186/s12974-015-0332-6
90. Lively S, Schlichter LC. Microglia Responses to Pro-inflammatory Stimuli (LPS, IFNgamma+TNFalpha) and Reprogramming by Resolving Cytokines (IL-4, IL-10). Front Cell Neurosci. (2018) 12:215. doi: 10.3389/fncel.2018.00215
91. Zhao H, Alam A, Chen Q. M AE, Pal A, Eguchi S, Wu L and Ma D. The role of microglia in the pathobiology of neuropathic pain development:what do we know? Br J Anaesth. (2017) 118:504–16. doi: 10.1093/bja/aex006
92. Schafers M, Svensson CI, Sommer C, Sorkin LS. Tumor necrosis factor-alpha induces mechanical allodynia after spinal nerve ligation by activation of p38 MAPK in primary sensory neurons. J Neurosci. (2003) 23:2517–21. doi: 10.1523/JNEUROSCI.23-07-02517.2003
93. Taylor CA, Zheng Q, Liu Z, Thompson JE. Role of p38 and JNK MAPK signaling pathways and tumor suppressor p53 on induction of apoptosis in response to Ad-eIF5A1 in A549 lung cancer cells. Mol Cancer. (2013) 12:35. doi: 10.1186/1476-4598-12-35
94. Qiu X, Zhuang M, Lu Z, Liu Z, Cheng D, Zhu C, Liu J. RIPK1 suppresses apoptosis mediated by TNF and caspase-3 in intervertebral discs. J Transl Med. (2019) 17:135. doi: 10.1186/s12967-019-1886-3
95. Lis K, Grygorowicz T, Cudna A, Szymkowski DE, Balkowiec-Iskra E. Inhibition of TNF reduces mechanical orofacial hyperalgesia induced by Complete Freund's Adjuvant by a TRPV1-dependent mechanism in mice. Pharmacol Rep. (2017) 69:1380–5. doi: 10.1016/j.pharep.2017.05.013
96. Brambilla R, Ashbaugh JJ, Magliozzi R, Dellarole A, Karmally S, Szymkowski DE, Bethea JR. Inhibition of soluble tumour necrosis factor is therapeutic in experimental autoimmune encephalomyelitis and promotes axon preservation and remyelination. Brain. (2011) 134:2736–54. doi: 10.1093/brain/awr199
97. Novrup HG, Bracchi-Ricard V, Ellman DG, Ricard J, Jain A, Runko E, et al. Central but not systemic administration of XPro1595 is therapeutic following moderate spinal cord injury in mice. J Neuroinflammation. (2014) 11:159. doi: 10.1186/s12974-014-0159-6
98. Fischer R, Sendetski M, Del Rivero T, Martinez GF, Bracchi-Ricard V, Swanson KA, et al. TNFR2 promotes Treg-mediated recovery from neuropathic pain across sexes. Proc Natl Acad Sci USA. (2019) 116:17045–50. doi: 10.1073/pnas.1902091116
99. Hung AL, Lim M, Doshi TL. Targeting cytokines for treatment of neuropathic pain. Scand J Pain. (2017) 17:287–93. doi: 10.1016/j.sjpain.2017.08.002
100. Radner H, Aletaha D. Anti-TNF in rheumatoid arthritis:an overview. Wien Med Wochenschr. (2015) 165:3–9. doi: 10.1007/s10354-015-0344-y
101. Sharief MK, Hentges R. Association between tumor necrosis factor-alpha and disease progression in patients with multiple sclerosis. N Engl J Med. (1991) 325:467–72. doi: 10.1056/NEJM199108153250704
102. Sayed BA, Christy AL, Walker ME, Brown MA. Meningeal mast cells affect early T cell central nervous system infiltration and blood-brain barrier integrity through TNF:a role for neutrophil recruitment? J Immunol. (2010) 184:6891–900. doi: 10.4049/jimmunol.1000126
103. Fresegna D, Bullitta S, Musella A, Rizzo FR, De Vito F, Guadalupi L, et al. Re-examining the role of TNF in MS pathogenesis and therapy. Cells. (2020) 9:2290 doi: 10.3390/cells9102290
104. Mehta AK, Gracias DT, Croft M. TNF activity and T cells. Cytokine. (2018) 101:14–8. doi: 10.1016/j.cyto.2016.08.003
105. Selmaj K, Raine CS, Cannella B, Brosnan CF. Identification of lymphotoxin and tumor necrosis factor in multiple sclerosis lesions. J Clin Invest. (1991) 87:949–54. doi: 10.1172/JCI115102
106. Madsen PM, Motti D, Karmally S, Szymkowski DE, Lambertsen KL, Bethea JR, et al. Oligodendroglial TNFR2 Mediates Membrane TNF-Dependent Repair in Experimental Autoimmune Encephalomyelitis by Promoting Oligodendrocyte Differentiation and Remyelination. J Neurosci. (2016) 36:5128–43. doi: 10.1523/JNEUROSCI.0211-16.2016
107. Magliozzi R, Howell OW, Durrenberger P, Arico E, James R, Cruciani C, et al. Meningeal inflammation changes the balance of TNF signalling in cortical grey matter in multiple sclerosis. J Neuroinflammation. (2019) 16:259. doi: 10.1186/s12974-019-1650-x
108. Kassiotis G, Kollias G. Uncoupling the proinflammatory from the immunosuppressive properties of tumor necrosis factor (TNF) at the p55 TNF receptor level:implications for pathogenesis and therapy of autoimmune demyelination. J Exp Med. (2001) 193:427–34. doi: 10.1084/jem.193.4.427
109. Yousuf MS, Noh MC, Friedman TN, Zubkow K, Johnson JC, Tenorio G, et al. Sensory neurons of the dorsal root ganglia become hyperexcitable in a T-cell-mediated MOG-EAE model of multiple sclerosis. eNeuro. (2019) 6:ENEURO.0024-19.2019. doi: 10.1523/ENEURO.0024-19.2019
110. Wajant H, Siegmund D. TNFR1 and TNFR2 in the control of the life and death balance of macrophages. Front Cell Dev Biol. (2019) 7:91. doi: 10.3389/fcell.2019.00091
111. Parameswaran N, Patial S. Tumor necrosis factor-alpha signaling in macrophages. Crit Rev Eukaryot Gene Expr. (2010) 20:87–103. doi: 10.1615/CritRevEukarGeneExpr.v20.i2.10
112. Wheeler MA, Heffner DL, Kim S, Espy SM, Spano AJ, Cleland CL, et al. TNF-alpha/TNFR1 signaling is required for the development and function of primary nociceptors. Neuron. (2014) 82:587–602. doi: 10.1016/j.neuron.2014.04.009
113. Gangadharan V, Kuner R. Pain hypersensitivity mechanisms at a glance. Dis Model Mech. (2013) 6:889–95. doi: 10.1242/dmm.011502
114. Julius D, Basbaum AI. Molecular mechanisms of nociception. Nature. (2001) 413:203–10. doi: 10.1038/35093019
115. Latremoliere A, Woolf CJ. Central sensitization:a generator of pain hypersensitivity by central neural plasticity. J Pain. (2009) 10:895–926. doi: 10.1016/j.jpain.2009.06.012
116. Ten Klooster PM, de Graaf N, Vonkeman HE. Association between pain phenotype and disease activity in rheumatoid arthritis patients:a non-interventional, longitudinal cohort study. Arthritis Res Ther. (2019) 21:257. doi: 10.1186/s13075-019-2042-4
117. Sunder RA, Toshniwal G, Dureja GP. Ketamine as an adjuvant in sympathetic blocks for management of central sensitization following peripheral nerve injury. J Brachial Plex Peripher Nerve Inj. (2008) 3:22. doi: 10.1186/1749-7221-3-22
118. Duffy SS, Perera CJ, Makker PG, Lees JG, Carrive P, Moalem-Taylor G. Peripheral and central neuroinflammatory changes and pain behaviors in an animal model of multiple sclerosis. Front Immunol. (2016) 7:369. doi: 10.3389/fimmu.2016.00369
119. Tumor Necrosis Factor Antagonists. LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. (2012). Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases.
120. Mitoma H, Horiuchi T, Tsukamoto H, Ueda N. Molecular mechanisms of action of anti-TNF-alpha agents—Comparison among therapeutic TNF-alpha antagonists. Cytokine. (2018) 101:56–63. doi: 10.1016/j.cyto.2016.08.014
121. Maxwell LJ, Zochling J, Boonen A, Singh JA, Veras MM, Tanjong Ghogomu E, et al. TNF-alpha inhibitors for ankylosing spondylitis. Cochrane Database Syst Rev. (2015) 4:CD005468. doi: 10.1002/14651858.CD005468.pub2
122. Minozzi S, Bonovas S, Lytras T, Pecoraro V, Gonzalez-Lorenzo M, Bastiampillai AJ, et al. Risk of infections using anti-TNF agents in rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis:a systematic review and meta-analysis. Expert Opin Drug Saf. (2016) 15:11–34. doi: 10.1080/14740338.2016.1240783
123. Downey C. Serious infection during etanercept, infliximab and adalimumab therapy for rheumatoid arthritis:A literature review. Int J Rheum Dis. (2016) 19:536–50. doi: 10.1111/1756-185X.12659
124. Kemanetzoglou E, Andreadou E. CNS demyelination with TNF-alpha blockers. Curr Neurol Neurosci Rep. (2017) 17:36. doi: 10.1007/s11910-017-0742-1
125. French JB, Bonacini M, Ghabril M, Foureau D, Bonkovsky HL. Hepatotoxicity associated with the use of anti-TNF-alpha agents. Drug Saf. (2016) 39:199–208. doi: 10.1007/s40264-015-0366-9
126. Ma X, Xu S. TNF inhibitor therapy for rheumatoid arthritis. Biomed Rep. (2013) 1:177–84. doi: 10.3892/br.2012.42
127. Pimentel DC, El Abd O, Benyamin RM, Buehler AM, Leite VF, Mazloomdoost D, et al. Anti-tumor necrosis factor antagonists in the treatment of low back pain and radiculopathy:a systematic review and meta-analysis. Pain Physician. (2014) 17:E27–44. doi: 10.36076/ppj.2014/17/E27
128. Lu D, Song H, Shi G. Anti-TNF-alpha treatment for pelvic pain associated with endometriosis. Cochrane Database Syst Rev. (2013) 3:CD008088. doi: 10.1002/14651858.CD008088.pub3
129. Hemperly A, Vande Casteele N. Clinical pharmacokinetics and pharmacodynamics of infliximab in the treatment of inflammatory bowel disease. Clin Pharmacokinet. (2018) 57:929–42. doi: 10.1007/s40262-017-0627-0
130. Guo C, Wu K, Liang X, Liang Y, Li R. Infliximab clinically treating ulcerative colitis: a systematic review and meta-analysis. Pharmacol Res. (2019) 148:104455. doi: 10.1016/j.phrs.2019.104455
131. Cvetkovic RS, Scott LJ. Adalimumab:a review of its use in adult patients with rheumatoid arthritis. BioDrugs. (2006) 20:293–311. doi: 10.2165/00063030-200620050-00005
132. Menter A, Thaci D, Wu JJ, Abramovits W, Kerdel F, Arikan D, et al. Long-term safety and effectiveness of adalimumab for moderate to severe psoriasis: results from 7-year interim analysis of the ESPRIT registry. Dermatol Ther. (2017) 7:365–81. doi: 10.1007/s13555-017-0198-x
133. Scheinfeld N. Adalimumab:a review of side effects. Expert Opin Drug Saf. (2005) 4:637–41. doi: 10.1517/14740338.4.4.637
136. Maillart E, Papeix C, Mellerio C, Bertrand A, Lubetzki C, Louapre C. Extensive and severe CNS demyelination associated with golimumab therapy. J Neurol. (2016) 263:1869–71. doi: 10.1007/s00415-016-8238-5
137. Esposito M, Carubbi F, Giunta A, Alunno A, Giacomelli R, Fargnoli MC. Certolizumab pegol for the treatment of psoriatic arthritis and plaque psoriasis. Expert Rev Clin Immunol. (2020) 16:119–28. doi: 10.1080/1744666X.2020.1713754
139. Burness CB, Duggan ST. Etanercept (SB4): a review in autoimmune inflammatory diseases. BioDrugs. (2016) 30:371–8. doi: 10.1007/s40259-016-0188-z
140. Gomez-Gallego M, Meca-Lallana J, Fernandez-Barreiro A. Multiple sclerosis onset during etanercept treatment. Eur Neurol. (2008) 59:91–3. doi: 10.1159/000109576
141. Inoue M, Yamashita K, Tsuji Y, Miki M, Amano S, Okumura T, et al. Characterization of a TNFR2-selective agonistic TNF-alpha mutant and its derivatives as an optimal regulatory T cell expander. J Immunol. (2021) 206:1740–51. doi: 10.4049/jimmunol.2000871
142. Duffy SS, Keating BA, Moalem-Taylor G. Adoptive transfer of regulatory T cells as a promising immunotherapy for the treatment of multiple sclerosis. Front Neurosci. (2019) 13:1107. doi: 10.3389/fnins.2019.01107
143. Jones E, Vlachou S. A critical review of the role of the cannabinoid compounds delta(9)-tetrahydrocannabinol (delta(9)-THC) and cannabidiol (CBD) and their combination in multiple sclerosis treatment. Molecules. (2020) 25:4930. doi: 10.3390/molecules25214930
144. Feliu A, Moreno-Martet M, Mecha M, Carrillo-Salinas FJ, de Lago E, Fernandez-Ruiz Jet al. A Sativex((R))-like combination of phytocannabinoids as a disease-modifying therapy in a viral model of multiple sclerosis. Br J Pharmacol. (2015) 172:3579–95. doi: 10.1111/bph.13159
145. Andrade P, Hoogland G, Del Rosario JS, Steinbusch HW, Visser-Vandewalle V, Daemen MA. Tumor necrosis factor-alpha inhibitors alleviation of experimentally induced neuropathic pain is associated with modulation of TNF receptor expression. J Neurosci Res. (2014) 92:1490–8. doi: 10.1002/jnr.23432
146. Andrade P, Visser-Vandewalle V, Del Rosario JS, Daemen MA, Buurman WA, Steinbusch HW, Hoogland G. The thalidomide analgesic effect is associated with differential TNF-alpha receptor expression in the dorsal horn of the spinal cord as studied in a rat model of neuropathic pain. Brain Res. (2012) 1450:24–32. doi: 10.1016/j.brainres.2012.02.033
147. Medler J, Wajant H. Tumor necrosis factor receptor-2 (TNFR2):an overview of an emerging drug target. Expert Opin Ther Targets. (2019) 23:295–307. doi: 10.1080/14728222.2019.1586886
148. Bradley JR. TNF-mediated inflammatory disease. J Pathol. (2008) 214:149–60. doi: 10.1002/path.2287
149. Dogrul A, Gul H, Yesilyurt O, Ulas UH, Yildiz O. Systemic and spinal administration of etanercept, a tumor necrosis factor alpha inhibitor, blocks tactile allodynia in diabetic mice. Acta Diabetol. (2011) 48:135–42. doi: 10.1007/s00592-010-0237-x
150. Shi X, Chen Y, Nadeem L, Xu G. Beneficial effect of TNF-alpha inhibition on diabetic peripheral neuropathy. J Neuroinflammation. (2013) 10:69. doi: 10.1186/1742-2094-10-69
151. Batoulis H, Recks MS, Holland FO, Thomalla F, Williams RO, Kuerten S. Blockade of tumour necrosis factor-alpha in experimental autoimmune encephalomyelitis reveals differential effects on the antigen-specific immune response and central nervous system histopathology. Clin Exp Immunol. (2014) 175:41–8. doi: 10.1111/cei.12209
152. Titelbaum DS, Degenhardt A, Kinkel RP. Anti-tumor necrosis factor alpha-associated multiple sclerosis. AJNR Am J Neuroradiol. (2005) 26:1548–50.
153. Taoufik E, Tseveleki V, Chu SY, Tselios T, Karin M, Lassmann H, et al. Transmembrane tumour necrosis factor is neuroprotective and regulates experimental autoimmune encephalomyelitis via neuronal nuclear factor-kappaB. Brain. (2011) 134:2722–35. doi: 10.1093/brain/awr203
154. Chen X, Wu X, Zhou Q, Howard OM, Netea MG, Oppenheim JJ. TNFR2 is critical for the stabilization of the CD4+Foxp3+ regulatory T. cell phenotype in the inflammatory environment. J Immunol. (2013). 190:1076–84. doi: 10.4049/jimmunol.1202659
155. Fischer R, Padutsch T, Bracchi-Ricard V, Murphy KL, Martinez GF, Delguercio N, et al. Exogenous activation of tumor necrosis factor receptor 2 promotes recovery from sensory and motor disease in a model of multiple sclerosis. Brain Behav Immun. (2019) 81:247–59. doi: 10.1016/j.bbi.2019.06.021
156. Robinson ES, Alves P, Bashir MM, Zeidi M, Feng, Werth VP. Cannabinoid reduces inflammatory cytokines, tumor necrosis factor-alpha, and type I interferons in dermatomyositis in vitro. J Invest Dermatol. (2017) 137:2445–7. doi: 10.1016/j.jid.2017.05.035
157. Nagarkatti P, Pandey R, Rieder SA, Hegde VL, Nagarkatti M. Cannabinoids as novel anti-inflammatory drugs. Future Med Chem. (2009) 1:1333–49. doi: 10.4155/fmc.09.93
158. Zheng ZM, Specter SC. Delta-9-tetrahydrocannabinol suppresses tumor necrosis factor alpha maturation and secretion but not its transcription in mouse macrophages. Int J Immunopharmacol. (1996) 18:53–68. doi: 10.1016/0192-0561(95)00107-7
159. Zajicek JP, Apostu VI. Role of cannabinoids in multiple sclerosis. CNS Drugs. (2011) 25:187–201. doi: 10.2165/11539000-000000000-00000
Keywords: pain, cytokine, TNF-α, EAE (experimental autoimmune encephalomyelitis), NFkapapB, MAP kinase (MAPK), autoimmune disease
Citation: Maguire AD, Bethea JR and Kerr BJ (2021) TNFα in MS and Its Animal Models: Implications for Chronic Pain in the Disease. Front. Neurol. 12:780876. doi: 10.3389/fneur.2021.780876
Received: 21 September 2021; Accepted: 15 November 2021;
Published: 06 December 2021.
Edited by:
Gila Moalem-Taylor, University of New South Wales, AustraliaReviewed by:
Andrew David Gaudet, University of Texas at Austin, United StatesKate Lykke Lambertsen, University of Southern Denmark, Denmark
Copyright © 2021 Maguire, Bethea and Kerr. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bradley J. Kerr, bradley.kerr@ualberta.ca