 Lanjing Wang
Lanjing Wang Lijun Liu
Lijun Liu Yanhong Zhao1
Yanhong Zhao1 Min Chu
Min Chu Jijun Teng
Jijun Teng- 1Department of Neurology, The Affiliated Hospital of Qingdao University, Qingdao University, Qingdao, China
- 2Department of Neurology, Minhang Hospital, Fudan University, Qingdao, China
Objectives: Statins either barely affect or increase lipoprotein(a) [Lp(a)] levels. This study aimed to explore the factors correlated to the change of Lp(a) levels as well as the relationship between Lp(a) and the recurrent vascular events in statin-treated patients with first acute ischemic stroke (AIS).
Methods: Patients who were admitted to the hospital with first AIS from October 2018 to September 2020 were eligible for inclusion. Correlation between the change of Lp(a) levels and potential influencing factors was assessed by linear regression analysis. Cox proportional regression models were used to estimate the association between Lp(a) and recurrent vascular events including AIS, transient ischemic attack, myocardial infarction and coronary revascularization.
Results: In total, 303 patients, 69.6% males with mean age 64.26 ± 11.38 years, completed the follow-up. During the follow-up period, Lp(a) levels increased in 50.5% of statin-treated patients and the mean percent change of Lp(a) levels were 14.48% (95% CI 6.35–22.61%). Creatinine (β = 0.152, 95% CI 0.125–0.791, P = 0.007) and aspartate aminotransferase (AST) (β = 0.160, 95% CI 0.175–0.949, P = 0.005) were positively associated with the percent change of Lp(a) levels. During a median follow-up of 26 months, 66 (21.8%) patients had a recurrent vascular event. The median time period between AIS onset and vascular events recurrence was 9.5 months (IQR 2.0–16.3 months). The on-statin Lp(a) level ≥70 mg/dL (HR 2.539, 95% CI 1.076–5.990, P = 0.033) and the change of Lp(a) levels (HR 1.003, 95% CI 1.000–1.005, P = 0.033) were associated with the recurrent vascular events in statin-treated patients with first AIS. Furthermore, the on-statin Lp(a) levels ≥70 mg/dL (HR 3.612, 95% CI 1.018–12.815, P = 0.047) increased the risk of recurrent vascular events in patients with low-density lipoprotein cholesterol (LDL-C) levels < 1.8 mmol/L.
Conclusions: Lp(a) levels increased in half of statin-treated patients with first AIS. Creatinine and AST were positively associated with the percent change of Lp(a) levels. Lp(a) is a determinant of residual vascular risk and the change of Lp(a) is positively associated with the risk of recurrent vascular events in these patients.
Introduction
Hyperlipidemia is one of the most important causal risk factors for atherosclerotic cardiovascular disease (ASCVD) and stroke (1, 2). Treatment of hyperlipidemia is a vital aspect of the secondary prevention in these diseases (3). Statins can significantly reduce the low-density lipoprotein cholesterol (LDL-C) levels and are the most widely used lipid-lowering drugs (4). Even so, statin therapy has its limitations, such as increasing the risk of statin-associated myopathy and hepatotoxicity (5). Besides, statin therapy may increase lipoprotein(a) [Lp(a)] levels in patients with ASCVD (6).
Lp(a) is composed of a LDL-like particle in which apolipoprotein B100 is covalently linked by a disulfide bond to apolipoprotein(a) [apo(a)] (7). Regulated by the LPA gene encoding apo(a) and rarely affected by age, gender, or lifestyle, Lp(a) levels vary spectacularly (up to 1,000-fold) among individuals, and ~20% of the population have elevated Lp(a) levels (>30 mg/dL) (8–11). In the past long time, Lp(a) had been relatively neglected on account of no satisfactory therapeutic methods to reduce Lp(a) levels (12). However, there is ample evidence that elevated Lp(a) levels are significantly and independently associated with ASCVD and stroke, which may be attributes to the proatherogenic, proinflammatory, and potentially antifibrinolytic effects of Lp(a) (13–17). Moreover, the contribution of Lp(a) in cardiovascular disease (CVD) is not less than LDL-C (18). In recent years, the interest in Lp(a) has been reignited as a consequence of the evidence on its causality for CVD and the emergence of new targeted therapeutics including proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors, Lp(a) apheresis (LA), and RNA-targeted therapies which dramatically lower Lp(a) levels (19, 20).
Existing studies suggested that statin-treated individuals had the higher recurrent cardiovascular risk compared with those who did not taking statins when LDL-C levels were similar (21). Besides, the recurrent cardiovascular risk was positively correlated with the on-statin Lp (a) levels (22). The residual risk for cardiovascular events in patients receiving statin therapy may be mainly associated with Lp(a) (23). By comparison, evidence on residual risk in statin-treated patients with acute ischemic stroke (AIS) is insufficient. In this study, we aimed to explore the factors related to the change of Lp(a) levels as well as the association between the change of Lp(a) levels and recurrence of vascular events in statin-treated patients with first AIS.
Methods
Participants enrollment and baseline measurements
The present study was a single-center, prospective, observational cohort study conducted at the Affiliated Hospital of Qingdao University. Ethical approval was obtained from the Ethics Committee of the Affiliated Hospital of Qingdao University and informed consent was obtained from all patient or their family members.
Patients admitted to the hospital within 7 days of symptom onset with first AIS from October 2018 to September 2020, who did not take lipid-lowering drugs before onset and agreed to participate this study were consecutively screened for inclusion. Those with other serious medical diseases including renal failure, hepatic failure, the late stage of malignant tumor or with a life expectancy < 24 months were excluded. The diagnoses of AIS were confirmed based on clinical manifestations and signs in combination with cranial computerized tomography (CT) and/or magnetic resonance imaging (MRI). Two experienced neurologists assessed the stroke severity at admission using the National Institutes of Health Stroke Scale (NIHSS) and analyzed the stroke etiology according to the Trial of Org 10,172 in Acute Stroke Treatment (TOAST) criteria (24).
The following data of all participants were collected: age, gender, history of hypertension, diabetes mellitus, atrial fibrillation (AF), coronary heart disease (CHD), smoking and drinking, intravenous thrombolysis, intracranial or extracranial vascular stenosis, laboratory tests [i.e., Lp(a), triglyceride (TG), total cholesterol (TC), low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C), fasting blood glucose (FBG), urea nitrogen, uric acid, creatinine, aspartate aminotransferase (AST), alanine aminotransferase (ALT), prothrombin time (PT), activated partial thromboplastin time (APTT) and D-Dimer]. All blood samples tested in the laboratory were obtained in fasting state within 24 h after admission. Lp(a) levels were measured using immunoturbidimetric method by Olympus 2,700 (Olympus, Japan). On the basis of previous literature, Lp(a) levels of all participants were categorized into 5 groups: < 15, 15–30 mg/dL, 30–50, 50–70, and ≥70 mg/dL groups (25).
Follow-up and end points
The end points of this study included AIS, transient ischemic attack (TIA), myocardial infarction (MI) and coronary revascularization. Patients with recurrence of end points were followed up face-to-face when they were re-admitted. The follow-up time of these patients was calculated as the time from AIS onset to the end point events and their blood lipid levels were measured with the same detection method when end points occurred. In contrast, free-event patients, confirmed through telephone or face-to-face follow-up, were those who did not suffer from end points during a 2-year follow-up period and their blood lipid levels were assessed at the end of follow-up. The percent change of Lp(a) level was calculated as [the radio of follow-up Lp(a) level minus baseline Lp(a) level and baseline Lp(a) level]. Patients who lost to follow-up, declined to continue, discontinued statins or died due to conditions except end point events during follow-up were excluded from analysis.
Statistical analysis
Baseline data were described by summary statistics. We compared data between the free-event group and the end point group. The quantitative data were indicated by mean and standard deviation or median and interquartile range and compared by using Student's t-test or Mann-Whitney U-test depending on type of data distribution. The qualitative data was indicated by frequency and percentage, and compared by using Chi-squared test.
Wilcoxon text was used to compare the difference between baseline and follow-up blood lipid levels. Correlation between baseline Lp(a) levels and follow-up Lp(a) levels were assessed by Spearman correlation analysis. Univariate linear regression analysis was used to screen the influencing factors of the percent change of Lp(a) levels. Variables with a P-value of < 0.10 in univariate analysis were subsequently entered into a multivariable linear regression analysis to determine the independent factors associated with the percent change of Lp(a) levels in statin-treated patients with first AIS. A two-tailed P < 0.05 was considered statistically significant.
Cumulative survival free of recurrent vascular events during follow-up were assessed using Kaplan–Meier survival analysis followed by the log rank test. Univariate and multivariate Cox regression analysis were used to determine the associations between Lp(a) and recurrent vascular events. The multivariable Cox regression analysis were adjusted for the possible confounders, i.e., age, gender, hypertension, diabetes mellitus, AF, CHD, LDL-C, HDL-C, TC and TG. Three models were set up. Model 1 adjusted for age, gender. Model 2 adjusted for model 1 plus history of hypertension, diabetes, AF, and CHD. Model 3 adjusted for model 2 plus LDL-C, HDL-C, TC and TG. Results were expressed as hazard ratio (HR) with the corresponding 95% confidence interval (CI). A two-tailed P < 0.05 was considered statistically significant. All statistical analysis in this study was performed with Statistical Package for the Social Sciences (SPSS) software version 26.0 (SPSS, Inc., Chicago, IL, USA).
Results
Baseline characteristics
Three hundred and sixty two eligible patients were initially enrolled in this study among whom 303 cases completed the follow-up and were analyzed finally (nine patients were lost to follow-up, 18 patients declined to continue, 26 patients discontinued statins and six patients died due to conditions except end point events during follow-up) (Figure 1). All enrolled patients received Atorvastatin (10–40mg) or Rosuvastatin (5–20mg).
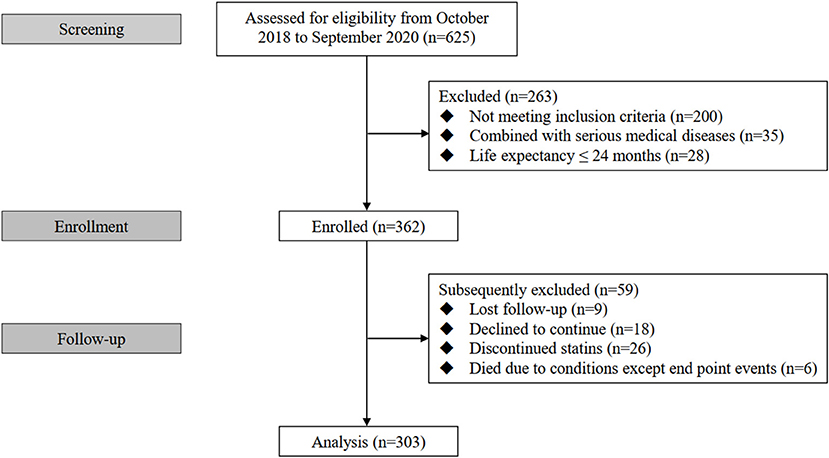
Figure 1. Flow diagram of the study.
Characteristics of the patients in the study cohort at baseline were presented in Table 1. Their mean age was 64.26 years and males accounted for 69.6%. With a median NIHSS of 4, 11.9% of patients received intravenous thrombolysis. The most common vascular risk factor in this study was hypertension (72.9%), followed by diabetes mellitus (37.6%). The etiological distribution according to the TOAST classification was large-artery atherosclerosis (LAA) 47.5%, cardioembolism (CE) 5.3%, small-artery occlusion (SAO) 27.4%, stroke of other determined etiology (SOE) 3.0% and stroke of undetermined etiology (SUE) 16.8%. The median baseline Lp(a) level was 18.90 mg/dL (IQR 11.20–31.00 mg/dL) and 79 (26.1%) patients had elevated Lp(a) levels (>30 mg/dL). In addition, 19 (6.3%), 43 (14.2%) and 23 (7.6%) patients had elevated TC levels (>6.2 mmol/L), TG levels (>2.3 mmol/L) and LDL-C levels (>4.1 mmol/L), respectively.
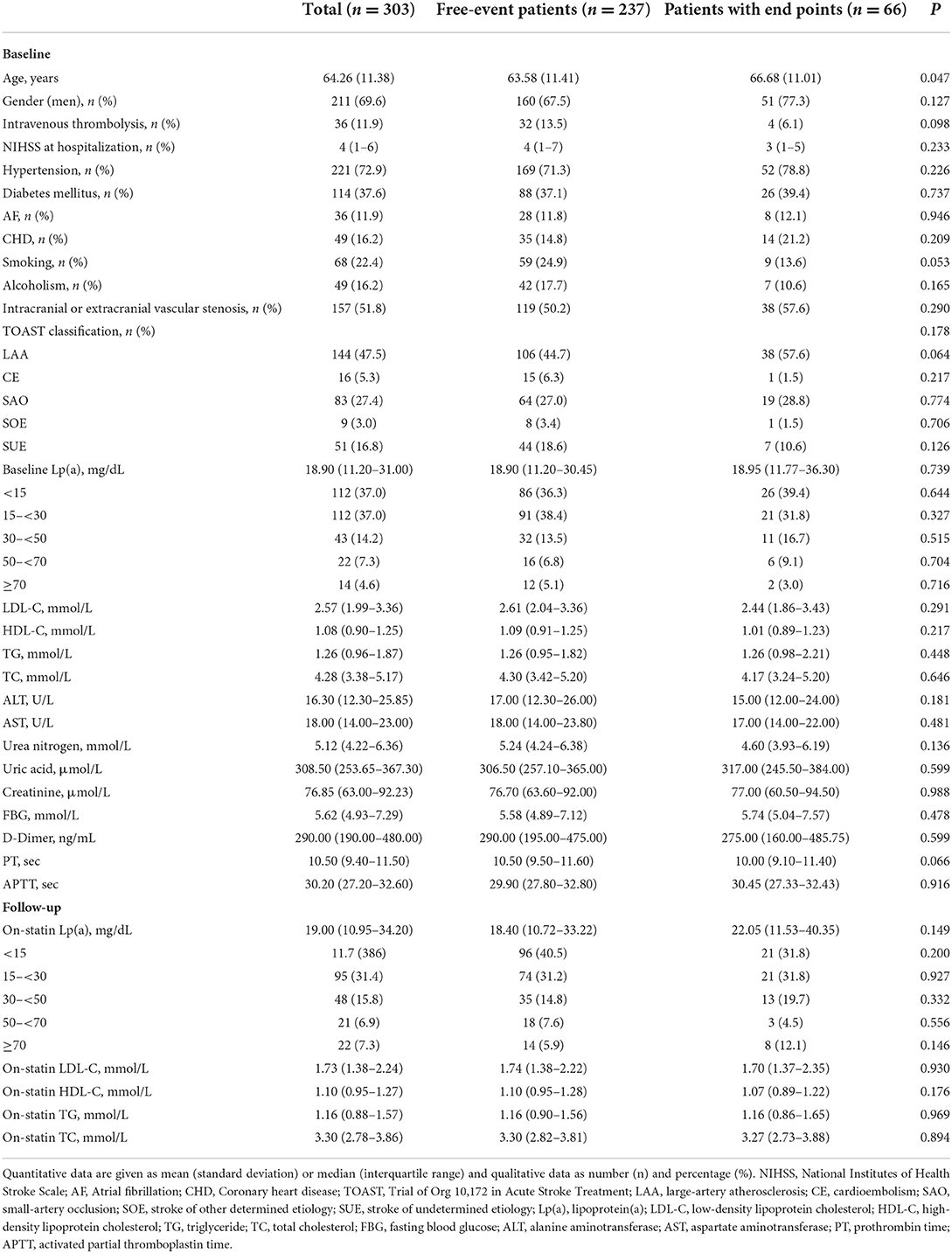
Table 1. Baseline and follow-up characteristics of patients in the study cohort.
Follow-up blood lipid levels
The overall median follow-up Lp(a) level was 19.00 mg/dL (IQR 10.95–34.20 mg/dL) and there was a strong correlation between baseline and follow-up Lp(a) levels (Spearman correlation rho: 0.824; P < 0.001) (Figure 2). During the follow-up period, 153 (50.5%) statin-treated patients' Lp(a) levels increased and the mean percent change of Lp(a) levels were 14.48% (95% CI 6.35–22.61%). The majority of the absolute percent change of Lp(a) levels were within 50% (Figure 3). There was a tendency for follow-up Lp(a) levels to be higher than baseline Lp(a) levels which could be obtained from the distribution of Lp(a) measurements in the two groups.
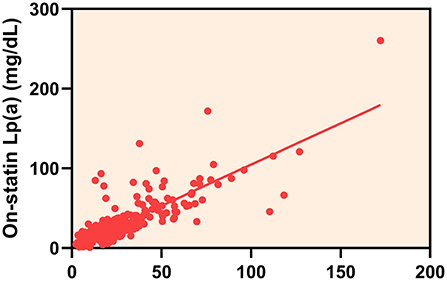
Figure 2. The correlation between baseline and on-statin lipoprotein(a) [Lp(a)] levels.
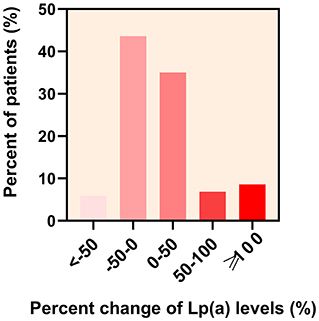
Figure 3. Distribution of the percent change of lipoprotein(a) [Lp(a)] levels in patients. The percent change of Lp(a) levels in statin-treated patients with first acute ischemic stroke at < −50,−50–0, 0–50, 50–100, and ≥100% was 5.9, 43.6, 35.0, 6.9, and 8.6%, respectively.
In addition, the LDL-C, TC and TG levels of patients decreased significantly after taking statins (2.57 vs. 1.73 mmol/L, P < 0.001; 4.28 vs. 3.30 mmol/L, P < 0.001; 1.26 vs. 1.16 mmol/L, P < 0.001). 162 (53.47%) statin-treated patients reached target LDL-C levels (< 1.8 mmol/L). The difference of HDL-C levels between baseline and follow-up were not significant (1.08 vs. 1.10 mmol/L, P = 0.192).
Factors correlated to the change of Lp(a) levels
The results of univariate linear regression analysis showed that the P-values of creatinine (β = 0.168, 95% CI 0.169–0.844, P = 0.003), AST (β = 0.161, 95% CI 0.173–0.961, P = 0.005), TG (β = 0.156, 95% CI 1.997–12.210, P = 0.007), TC (β = 0.107, 95% CI−0.283–10.325, P = 0.063), FBG (β = 0.104, 95% CI−0.265–6.333, P = 0.071) and APTT (β = −0.099, 95% CI−2.944–0.192, P = 0.085) were < 0.10 (Table 2). Therefore, these factors were entered into a multivariable linear regression analysis, which indicated that creatinine (β = 0.152, 95% CI 0.125–0.791, P = 0.007) and AST (β = 0.160, 95% CI 0.175–0.949, P = 0.005) were independently associated with the percent change of Lp(a) levels in statin-treated patients with first AIS (Table 3).
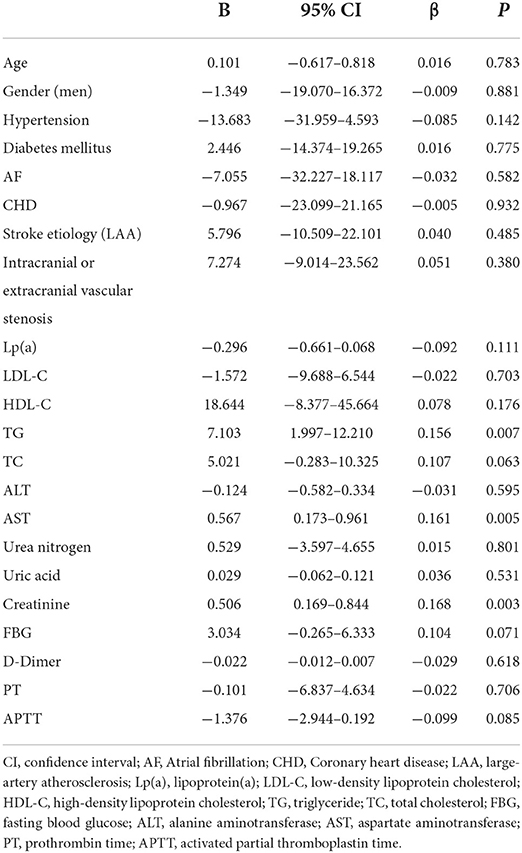
Table 2. Univariate linear regression analysis on the association between baseline factors and the percent change of lipoprotein(a) levels.
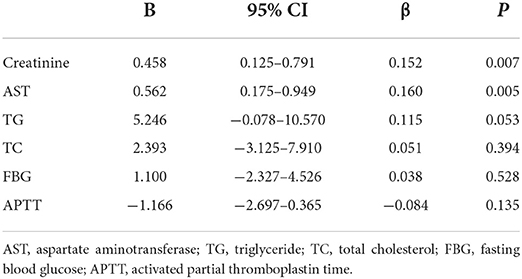
Table 3. Multivariable linear regression analysis on the association between baseline factors and the percent change of lipoprotein(a) levels.
Lp(a) and end point events
During a median follow-up of 26 months, there were a total of 66 recurrent vascular events (61 AIS, 1 TIA, 2 MI and 2 coronary revascularizations). The median time period between AIS onset and vascular events recurrence was 9.5 months (IQR 2.0–16.3 months). Individuals with recurrent vascular events were older than those without events (63.58 vs. 66.68 years, P = 0.047). No significant differences were observed in other baseline characteristics (Table 1).
Among patients with Lp(a) levels < 15 mg/dL, 21 of 117 patients (17.9%) experienced a recurrent vascular event at a median time of 11 months (IQR 7.8–17.0 months), whereas in patients with Lp(a) levels ≥ 70 mg/dL, 8 of 22 patients (36.4%) experienced an event at a median time of 11 months (IQR 3.0–17.5 months) (log rank P = 0.037) (Figure 4).
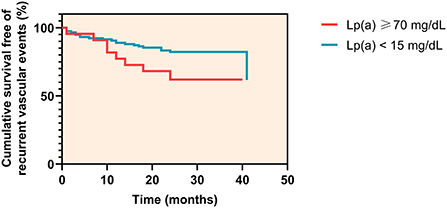
Figure 4. Kaplan-Meier curves for cumulative survival free of recurrent vascular events.
The association between baseline Lp(a) levels and recurrent vascular events was not significant in univariate or multivariate Cox regression analysis. On-statin Lp(a) level ≥70 mg/dL significantly increased the risk for recurrent vascular events after adjustment for relevant confounding factors (HR 2.539, 95% CI 1.076–5.990, P = 0.033). The increased percent change of Lp(a) levels tended to increase the risk for recurrent vascular events (HR 1.003, 95% CI 1.000–1.005, P = 0.033) (Table 4). In patients with LDL-C levels < 1.8 mmol/L, on-statin Lp(a) levels ≥70 mg/dL also increased the risk for recurrent vascular events after adjustment for relevant confounding factors (HR 3.612, 95% CI 1.018–12.815, P = 0.047) (Table 5). In multivariate Cox regression analysis, age was not independently associated with recurrent vascular events.
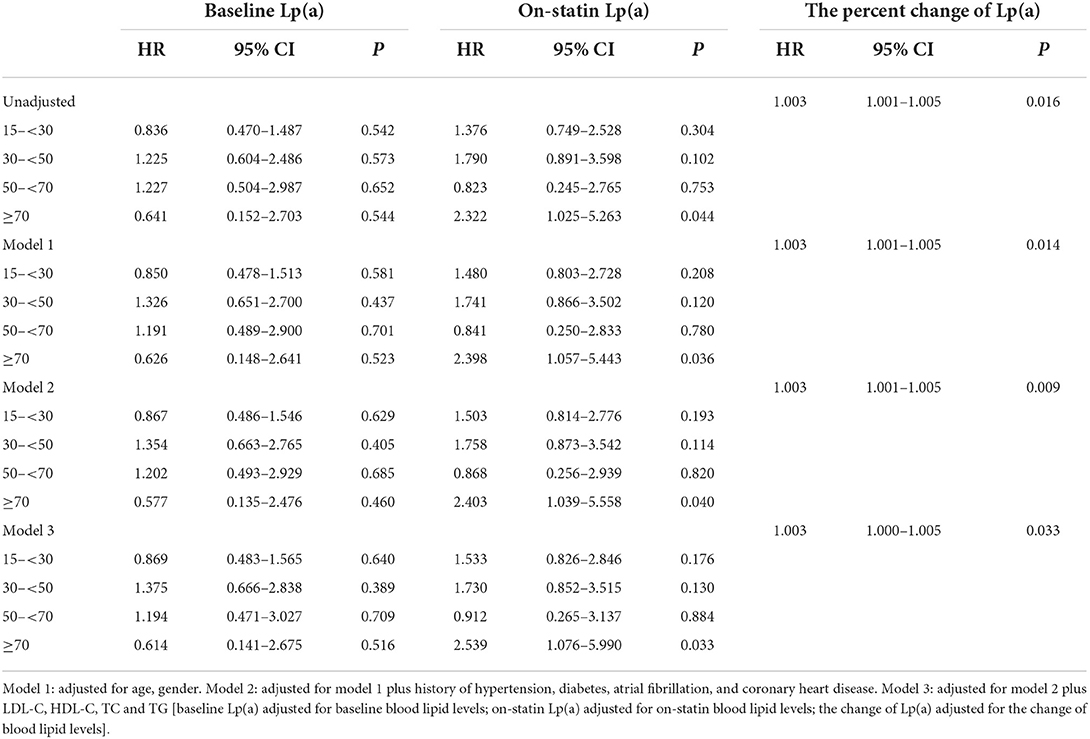
Table 4. Multivariate Cox regression analysis on the association between lipoprotein(a) levels and recurrent vascular events.
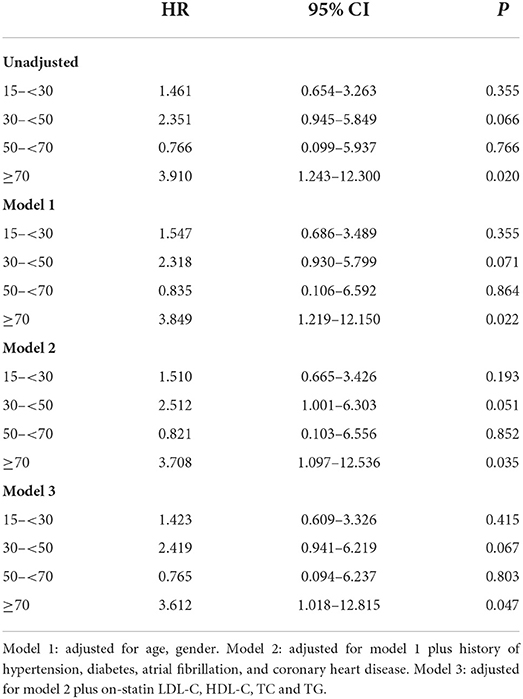
Table 5. Multivariate Cox regression analysis on the association between on-statin lipoprotein(a) levels and recurrent vascular events in patients with LDL-C levels < 1.8 mmol/L.
Discussion
To the best of our knowledge, this is the first study investigating the association between the change of Lp(a) with recurrent vascular events in a cohort of statin-treated patients with first AIS. Our results indicated that Lp(a) levels increased in half of statin-treated patients and the on-statin Lp(a) level ≥70 mg/d instead of the baseline Lp(a) level at admission was significantly associated with the recurrent vascular events. The relationship exists even the LDL-C levels are very low, suggesting that patients had residual vascular risk even while taking statins and Lp(a) is a determinant of the residual vascular risk. Moreover, the change of Lp(a) levels was positively associated with the risk of recurrent vascular events.
Statin therapy has minimal effect on Lp(a) even increases Lp(a) levels, which may be partly attributed to that LDL receptor does not play a major role in the clearance of Lp(a) (26). A meta-analysis showed that statins increase Lp(a) levels by 8.5–19.6% from baseline (6). In line with this study, we observed a 14.48% increasement in Lp(a) levels from baseline during statin therapy. Previous studies established that Lp(a) levels of individuals with low molecular weight apo (a) ( ≤ 22 kringle IV repeats) phenotype or baseline Lp(a) levels ≥70 nmol/L (≈34 mg/dL) were more likely to increase after taking statins (27, 28). And high-intensity statin therapy was more remarkably associated with increased Lp(a) levels in patients with CVD (29). Based on this situation, it is extremely necessary to explore which factors are associated with the change of Lp (a) levels in statin-treated AIS patients and whether the change of Lp (a) is associated with the recurrence risk for vascular events in these patients.
The underlying mechanisms by which statins affect circulating Lp(a) levels remain unclear. Statins elevate the expression of LPA mRNA as well as the synthesis and secretion of apo(a) protein in HepG2 cells, which may result in the increase of Lp(a) levels (6). Moreover, statins activate the expression of PCSK9 genes and increase PCSK9 levels, which then enhance Lp(a) production (30–32). Deficiently, there are few studies on the factors associated with the change of Lp(a) in statin-treated patients. The existing literature have provided some evidence that non-genetic factors can affect Lp(a) levels to a limited extent, such as kidney impairment, liver disease, inflammation, weight reduction or high saturated fat intake (33–38). In our study, we discovered that creatinine and AST were positively correlated with the change of Lp(a) levels despite these two factors can only explain the change to a small extent. The circulating Lp(a) levels are determined by the velocity of synthesis (39). The liver is responsible for the synthesis and catabolism of Lp(a) (40, 41). Therefore, liver dysfunction impairs Lp(a) metabolism (42). Available researches have validated that severe chronic liver diseases can lead to the decrease of Lp(a) synthesis so that the circulating Lp(a) levels can be decreased (36, 43). However, Lp(a) levels may be increased in some patients with hepatocarcinoma (44). Further studies are needed to validate the relationship between liver function and Lp(a) levels. Kidney may be involved in Lp(a) catabolism and the clearance of Lp(a) from circulation (45, 46). Therefore, renal impairment may block Lp(a) catabolism and result in the increase of circulating Lp(a) levels (47). Trinder et al. (28) discovered that there was a weak negative correlation between the percent change of Lp(a) and the estimated glomerular filtration rate. Large sample trials are warranted to investigate the factors associated with the influences of statins on Lp(a) levels.
Elevated Lp(a) levels are significantly and independently associated with the risk of ASCVD and Lp(a) has been confirmed as a momentous determinant of residual risk for cardiovascular events in statin-treated patients (13, 14, 23). But so far, few studies have investigated the residual vascular risks for AIS patients with elevated Lp(a) levels. Lange et al. (48) firstly reported that elevated Lp(a) levels contributed to the risk of recurrent vascular events after the first ischemic stroke in a 12-month follow-up study. Hong et al. (49) discovered that elevated Lp(a) levels were associated with early stroke recurrence in patients with AIS. The positive association between Lp(a) and the recurrence risk of cerebrovascular events was found in patients who were either < 60 years or had evident LAA stroke etiology (50). However, there is still a lack of studies on the relationship between the on-statin Lp(a) levels or the change of Lp(a) levels and the recurrence risk of vascular events in patients with AIS in a secondary prevention setting. Nestel et al. (51) claimed that increased Lp(a) levels after using statins 1 year were associated with future CVD events in patients with CHD. A latest study assessed the relationship between change of Lp(a) and risk of coronary artery disease and found the two mentioned above are not correlated (28). In this study, we discovered that the risk for recurrent vascular events is significantly higher in the patients whose on-statin Lp(a) levels ≥ 70 mg/dL and the change of Lp(a) was independently associated with the risk for recurrent vascular events in statin-treated patients with AIS, even in patients with LDL-C lower than 1.8 mmol/L. Furthermore, the risk associated with elevated Lp(a) increased in the context of very low LDL-C levels. Therefore, Lp(a) is a residual risk determinant in statin-treated patients with AIS.
The association between Lp(a) and residual risk of ASCVD makes Lp(a) a novel therapeutic target in ASCVD. Ongoing clinical trials provided some promising results that PCSK9 inhibitors, LA, and RNA-targeted therapies may be available Lp(a) lowering therapies (20). Existing studies have confirmed that lowering Lp(a) level by 80 mg/dL might reduce the risk of CHD by approximately 18 to 20% (52). A study reported that reducing Lp (a) level by 63% with LA achieved a 94% reduction in the major adverse cardiovascular events over a mean treatment period of 4 years (53). RNA-targeted therapies such as pelacarsen, olpasiran, and SLN360 which are currently investigated in trials have shown to lower Lp(a) levels by 90% (54). Decrease of Lp(a) levels by 50 mg/dL in 5 years in individuals with preexisting CVD may reduce the recurrence risk of CVD by 20% (55). The clinical benefits may be directly proportional to the absolute decrease of Lp (a) levels (52). Therefore, patients with higher baseline Lp(a) levels had greater reductions in the incidence of CVD events receiving Lp(a) lowering therapy (52, 56). Whether reducing Lp(a) levels in patients preexisting ischemic stroke can reduce the risk of recurrent vascular events deserves further study. And we should consider both patients' Lp(a) levels and their absolute risk of having a vascular event to determine whether they were likely to benefit from Lp(a)-lowering therapy (57).
Our study has some limitations. First, our study was a single center study, therefore, the sample size was relatively small. Second, a part of free-event patients was followed up by telephone, which might miss some end point events, especially TIA. Third, dietary habits and physical activities in the patients were not taken into account which might alter results of the study. In addition, it could not be ignored that the times of reaching the end point were different in patients, the measurement times of on-statin Lp(a) levels were different, so the total durations of statin use were different, which might cause certain errors in the results. The relationship between the change of Lp(a) and the recurrence of vascular events still needs to be confirmed by large-scale prospective studies. This study may provide evidence for the application of Lp(a) lowering therapy in AIS patients with elevated Lp(a) levels. Further prospective studies are warranted to evaluate the effect of Lp(a) lowering therapy on the recurrence of vascular events for patients with elevated Lp(a) levels and to identify those might benefit from Lp(a) lowering therapy.
Conclusions
In the patients receiving statin therapy after first AIS, creatinine and AST were positively associated with the percent change of Lp(a) levels. Lp(a) is a determinant of residual vascular risk and the change of Lp(a) is positively associated with the risk of recurrent vascular events. For AIS patients with elevated Lp(a) levels, Lp(a) lowering therapy may be desirable. With the emergence of new lipid-lowering drugs, drugs that reduce LDL-C levels and do not increase Lp(a) levels can be considered in future treatment.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
Study conception and design: JT. Acquisition of data: LW, YZ, and MC. Statistical analysis and drafting the manuscript: LW. Critical revision of the article for important intellectual content: LL and JT. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hussain I, Patni N, Garg A. Lipodystrophies, dyslipidaemias and atherosclerotic cardiovascular disease. Pathology. (2019) 51:202–12. doi: 10.1016/j.pathol.2018.11.004
2. Alloubani A, Nimer R, Samara R. Relationship between hyperlipidemia, cardiovascular disease and stroke: a systematic review. Curr Cardiol Rev. (2021) 17:e051121189015. doi: 10.2174/1573403X16999201210200342
3. Tsai H, Kim J, Jouvent E, Gurol M. Updates on prevention of hemorrhagic and lacunar strokes. J Stroke. (2018) 20:167–79. doi: 10.5853/jos.2018.00787
4. Reith C, Armitage J. Management of residual risk after statin therapy. Atherosclerosis. (2016) 245:161–70. doi: 10.1016/j.atherosclerosis.2015.12.018
5. Bellosta S, Corsini A. Statin drug interactions and related adverse reactions: an update. Expert Opin Drug Saf. (2018) 17:25–37. doi: 10.1080/14740338.2018.1394455
6. Tsimikas S, Gordts P, Nora C, Yeang C, Witztum J. Statin therapy increases lipoprotein(a) levels. Eur Heart J. (2020) 41:2275–84. doi: 10.1093/eurheartj/ehz310
7. Schmidt K, Noureen A, Kronenberg F, Utermann G. Structure, function, and genetics of lipoprotein (a). J Lipid Res. (2016) 57:1339–59. doi: 10.1194/jlr.R067314
8. Trinder M, Uddin M, Finneran P, Aragam K, Natarajan P. Clinical utility of lipoprotein(a) and LPA genetic risk score in risk prediction of incident atherosclerotic cardiovascular disease. JAMA Cardiol. (2020) 6:1–9. doi: 10.1001/jamacardio.2020.5398
9. Kronenberg F. Human genetics and the causal role of lipoprotein(a) for various diseases. Cardiovasc Drug Ther. (2016) 30:87–100. doi: 10.1007/s10557-016-6648-3
10. Durlach V, Bonnefont-Rousselot D, Boccara F, Varret M, Di-Filippo Charcosset M, Cariou B, et al. Lipoprotein(a): pathophysiology, measurement, indication and treatment in cardiovascular disease. a consensus statement from the nouvelle société francophone d'athérosclérose (NSFA). Arch Cardiovasc Dis. (2021) 114:828–47. doi: 10.1016/j.acvd.2021.10.009
11. Swerdlow D, Rider D, Yavari A, Wikström Lindholm M, Campion G, Nissen S. Treatment and prevention of lipoprotein(a)-mediated cardiovascular disease: the emerging potential of RNA interference therapeutics. Cardiovasc Res. (2022) 118:1218–31. doi: 10.1093/cvr/cvab100
12. Tada H, Takamura M, Kawashiri M. Lipoprotein(a) as an old and new causal risk factor of atherosclerotic cardiovascular disease. J Atheroscler Thromb. (2019) 26:583–91. doi: 10.5551/jat.RV17034
13. Vavuranakis M, Jones S, Cardoso R, Gerstenblith G, Leucker T. The role of Lipoprotein(a) in cardiovascular disease: current concepts and future perspectives. Hellenic J Cardiol. (2020) 61:398–403. doi: 10.1016/j.hjc.2020.09.016
14. Mehta A, Vasquez N, Ayers C, Patel J, Hooda A, Khera A, et al. Independent association of lipoprotein(a) and coronary artery calcification with atherosclerotic cardiovascular risk. J Am Coll Cardiol. (2022) 79:757–68. doi: 10.1016/j.jacc.2021.11.058
15. Arora P, Kalra R, Callas P, Alexander K, Zakai N, Wadley V, et al. Lipoprotein(a) and risk of ischemic stroke in the regards study. Arterioscler Thromb Vasc Biol. (2019) 39:810–8. doi: 10.1161/ATVBAHA.118.311857
16. Tsimikas S. A test in context: lipoprotein(a): diagnosis, prognosis, controversies, and emerging therapies. J Am Coll Cardiol. (2017) 69:692–711. doi: 10.1016/j.jacc.2016.11.042
17. Tziomalos K, Athyros V, Wierzbicki A, Mikhailidis D. Lipoprotein a: where are we now? Curr Opin Cardiol. (2009) 24:351–7. doi: 10.1097/HCO.0b013e32832ac21a
18. Langsted A, Nordestgaard B. Lipoprotein(a): is it more, less or equal to LDL as a causal factor for cardiovascular disease and mortality? Curr Opin Lipidol. (2020) 31:125–31. doi: 10.1097/MOL.0000000000000681
19. Tokgozoglu L, Orringer C, Ginsberg H, Catapano A. The year in cardiovascular medicine 2021: dyslipidaemia. Eur Heart J. (2022) 43:807–17. doi: 10.1093/eurheartj/ehab875
20. Kosmas C, Sourlas A, Mallarkey G, Silverio D, Ynoa D, Montan P, et al. Therapeutic management of hyperlipoproteinemia (a). Drugs Context. (2019) 8:212609. doi: 10.7573/dic.212609
21. Lieb W, Enserro D, Larson M, Vasan R. Residual cardiovascular risk in individuals on lipid-lowering treatment: quantifying absolute and relative risk in the community. Open Heart. (2018) 5:e000722. doi: 10.1136/openhrt-2017-000722
22. Willeit P, Ridker P, Nestel P, Simes J, Tonkin A, Pedersen T, et al. Baseline and on-statin treatment lipoprotein(a) levels for prediction of cardiovascular events: individual patient-data meta-analysis of statin outcome trials. Lancet. (2018) 392:1311–20. doi: 10.1016/S0140-6736(18)31652-0
23. Khera A, Everett B, Caulfield M, Hantash F, Wohlgemuth J, Ridker P, et al. Lipoprotein(a) concentrations, rosuvastatin therapy, and residual vascular risk: an analysis from the JUPITER Trial (justification for the use of statins in prevention: an intervention trial evaluating rosuvastatin). Circulation. (2014) 129:635–42. doi: 10.1161/CIRCULATIONAHA.113.004406
24. Adams H, Bendixen B, Kappelle L, Biller J, Love B, Gordon D, et al. Classification of subtype of acute ischemic stroke. definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in acute stroke treatment. Stroke. (1993) 24:35–41. doi: 10.1161/01.STR.24.1.35
25. Wong N, Zhao Y, Sung J, Browne A. Relation of first and total recurrent atherosclerotic cardiovascular disease events to increased lipoprotein(a) levels among statin treated adults with cardiovascular disease. Am J Cardiol. (2021) 145:12–7. doi: 10.1016/j.amjcard.2020.12.075
26. Saleheen D, Haycock P, Zhao W, Rasheed A, Taleb A, Imran A, et al. Apolipoprotein(a) isoform size, lipoprotein(a) concentration, and coronary artery disease: a mendelian randomisation analysis. Lancet Diabetes Endocrinol. (2017) 5:524–33. doi: 10.1016/S2213-8587(17)30088-8
27. Yahya R, Berk K, Verhoeven A, Bos S, van der Zee L, Touw J, et al. Statin treatment increases lipoprotein(a) levels in subjects with low molecular weight apolipoprotein(a) phenotype. Atherosclerosis. (2019) 289:201–5. doi: 10.1016/j.atherosclerosis.2019.07.001
28. Trinder M, Paruchuri K, Haidermota S, Bernardo R, Zekavat S, Gilliland T, et al. Repeat measures of lipoprotein(a) molar concentration and cardiovascular risk. J Am Coll Cardiol. (2022) 79:617–28. doi: 10.1016/j.jacc.2021.11.055
29. de Boer L, Oorthuys A, Wiegman A, Langendam M, Kroon J, Spijker R, et al. Statin therapy and lipoprotein(a) levels: a systematic review and meta-analysis. Eur J Prev Cardiol. (2021). doi: 10.1016/j.atherosclerosis.2022.06.202
30. Guo Y, Liu J, Xu R, Zhu C, Wu N, Jiang L, et al. Short-term impact of low-dose atorvastatin on serum proprotein convertase subtilisin/kexin type 9. Clin Drug Investig. (2013) 33:877–83. doi: 10.1007/s40261-013-0129-2
31. Awan Z, Seidah N, MacFadyen J, Benjannet S, Chasman D, Ridker P, et al. Rosuvastatin, proprotein convertase subtilisin/kexin type 9 concentrations, and LDL cholesterol response: the JUPITER trial. Clin Chem. (2012) 58:183–9. doi: 10.1373/clinchem.2011.172932
32. Villard E, Thedrez A, Blankenstein J, Croyal M, Tran T, Poirier B, et al. PCSK9 modulates the secretion but not the cellular uptake of lipoprotein(a) ex vivo: an effect blunted by alirocumab. JACC Basic Transl Sci. (2016) 1:419–27. doi: 10.1016/j.jacbts.2016.06.006
33. Faghihnia N, Tsimikas S, Miller E, Witztum J, Krauss R. Changes in lipoprotein(a), oxidized phospholipids, and LDL subclasses with a low-fat high-carbohydrate diet. J Lipid Res. (2010) 51:3324–30. doi: 10.1194/jlr.M005769
34. Kronenberg F, Kuen E, Ritz E, Junker R, König P, Kraatz G, et al. Lipoprotein(a) serum concentrations and apolipoprotein(a) phenotypes in mild and moderate renal failure. J Am Soc Nephrol. (2000) 11:105–15. doi: 10.1681/ASN.V111105
35. Lin J, Reilly M, Terembula K, Wilson F. Plasma lipoprotein(a) levels are associated with mild renal impairment in type 2 diabetics independent of albuminuria. PLoS ONE. (2014) 9:e114397. doi: 10.1371/journal.pone.0114397
36. Meroni M, Longo M, Lombardi R, Paolini E, Macchi C, Corsini A, et al. Low lipoprotein(a) levels predict hepatic fibrosis in patients with nonalcoholic fatty liver disease. Hepatol Commun. (2022) 6:535–49. doi: 10.1002/hep4.1830
37. Reyes-Soffer G, Westerterp M. Beyond lipoprotein(a) plasma measurements: lipoprotein(a) and inflammation. Pharmacol Res. (2021) 169:105689. doi: 10.1016/j.phrs.2021.105689
38. Brandstätter A, Lingenhel A, Zwiauer K, Strobl W, Kronenberg F. Decrease of Lp(a) during weight reduction in obese children is modified by the apo(a) kringle-IV copy number variation. Int J Obes. (2009) 33:1136–42. doi: 10.1038/ijo.2009.144
39. Krempler F, Kostner G, Bolzano K, Sandhofer F. Turnover of lipoprotein (a) in man. J Clin Invest. (1980) 65:1483–90. doi: 10.1172/JCI109813
40. Cain W, Millar J, Himebauch A, Tietge U, Maugeais C, Usher D, et al. Lipoprotein [a] is cleared from the plasma primarily by the liver in a process mediated by apolipoprotein [a]. J Lipid Res. (2005) 46:2681–91. doi: 10.1194/jlr.M500249-JLR200
41. Kronenberg F, Utermann G. Lipoprotein(a): resurrected by genetics. J Intern Med. (2013) 273:6–30. doi: 10.1111/j.1365-2796.2012.02592.x
42. Jiang J, Nilsson-Ehle P, Xu N. Influence of liver cancer on lipid and lipoprotein metabolism. Lipids Health Dis. (2006) 5:4. doi: 10.1186/1476-511X-5-4
43. Malaguarnera M, Giugno I, Trovato B, Panebianco M, Restuccia N, Ruello P. Lipoprotein(a) in cirrhosis. a new index of liver functions? Curr Med Res Opin. (1996) 13:479–85. doi: 10.1185/03007999609115228
44. Basili S, Andreozzi P, Vieri M, Maurelli M, Cara D, Cordova C, et al. Lipoprotein (a) serum levels in patients with hepatocarcinoma. Clin Chim Acta. (1997) 262:53–60. doi: 10.1016/S0009-8981(97)06533-9
45. Rosas S, Joffe M, Wolfe M, Brayman K, Rader D. Effects of renal replacement therapy on plasma lipoprotein(a) levels. Am J Nephrol. (2008) 28:361–5. doi: 10.1159/000112225
46. Kronenberg F, Trenkwalder E, Lingenhel A, Friedrich G, Lhotta K, Schober M, et al. Renovascular arteriovenous differences in Lp[a] plasma concentrations suggest removal of Lp[a] from the renal circulation. J Lipid Res. (1997) 38:1755–63. doi: 10.1016/S0022-2275(20)37150-9
47. Kronenberg F. Causes and consequences of lipoprotein(a) abnormalities in kidney disease. Clin Exp Nephrol. (2014) 18:234–7. doi: 10.1007/s10157-013-0875-8
48. Lange K, Nave A, Liman T, Grittner U, Endres M, Ebinger M. Lipoprotein(a) levels and recurrent vascular events after first ischemic stroke. Stroke. (2017) 48:36–42. doi: 10.1161/STROKEAHA.116.014436
49. Hong X, Wu D, Lu J, Zheng Y, Tu W, Yan J. Lipoprotein (a) as a predictor of early stroke recurrence in acute ischemic stroke. Mol Neurobiol. (2018) 55:718–26. doi: 10.1007/s12035-016-0346-9
50. Arnold M, Schweizer J, Nakas C, Schütz V, Westphal L, Inauen C, et al. Lipoprotein(a) is associated with large artery atherosclerosis stroke aetiology and stroke recurrence among patients below the age of 60 years: results from the BIOSIGNAL study. Eur Heart J. (2021) 42:2186–96. doi: 10.1093/eurheartj/ehab081
51. Nestel P, Barnes E, Tonkin A, Simes J, Fournier M, White H, et al. Plasma lipoprotein(a) concentration predicts future coronary and cardiovascular events in patients with stable coronary heart disease. Arterioscler Thromb Vasc Biol. (2013) 33:2902–8. doi: 10.1161/ATVBAHA.113.302479
52. Burgess S, Ference B, Staley J, Freitag D, Mason A, Nielsen S, et al. Association of LPA variants with risk of coronary disease and the implications for lipoprotein(a)-lowering therapies: a mendelian randomization analysis. JAMA Cardiol. (2018) 3:619–27. doi: 10.1001/jamacardio.2018.1470
53. Moriarty P, Gray J, Gorby L. Lipoprotein apheresis for lipoprotein(a) and cardiovascular disease. J Clin Lipidol. (2019) 13:894–900. doi: 10.1016/j.jacl.2019.09.010
54. Nurmohamed N, Kraaijenhof J, Stroes E. Lp(a): a new pathway to target? Curr Atheroscler Rep. (2022). doi: 10.1007/s11883-022-01060-4
55. Madsen C, Kamstrup P, Langsted A, Varbo A, Nordestgaard B. Lipoprotein(a)-lowering by 50 mg/dL (105 nmol/L) may be needed to reduce cardiovascular disease 20% in secondary prevention: a population-based study. Arterioscler Thromb Vasc Biol. (2020) 40:255–66. doi: 10.1161/ATVBAHA.119.312951
56. O'Donoghue M, Fazio S, Giugliano R, Stroes E, Kanevsky E, Gouni-Berthold I, et al. Lipoprotein(a), PCSK9 inhibition, and cardiovascular risk. Circulation. (2019) 139:1483–92. doi: 10.1161/CIRCULATIONAHA.118.037184
Keywords: lipoprotein(a), stroke, statin, recurrence, vascular risk
Citation: Wang L, Liu L, Zhao Y, Chu M and Teng J (2022) Lipoprotein(a) and residual vascular risk in statin-treated patients with first acute ischemic stroke: A prospective cohort study. Front. Neurol. 13:1004264. doi: 10.3389/fneur.2022.1004264
Received: 27 July 2022; Accepted: 17 October 2022;
Published: 03 November 2022.
Edited by:
Linxin Li, University of Oxford, United KingdomReviewed by:
Patricia De La Riva, Donostia University Hospital, SpainAndrea Zini, IRCCS Institute of Neurological Sciences of Bologna (ISNB), Italy
Copyright © 2022 Wang, Liu, Zhao, Chu and Teng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jijun Teng, drtengjijun@163.com