


- 1 Translational Neuroscience Program, Department of Psychiatry, University of Pittsburgh, Pittsburgh, PA, USA
- 2 Department of Neurology, University of Pittsburgh, Pittsburgh, PA, USA
- 3 VISN 4 Mental Illness Research, Education and Clinical Center, VA Pittsburgh Healthcare System, Pittsburgh, PA, USA
- 4 Department of Neuroscience, University of Pittsburgh, Pittsburgh, PA, USA
Converging lines of evidence indicate that schizophrenia is characterized by impairments of synaptic machinery within cerebral cortical circuits. Efforts to localize these alterations in brain tissue from subjects with schizophrenia have frequently been limited to the quantification of structures that are non-selectively identified (e.g., dendritic spines labeled in Golgi preparations, axon boutons labeled with synaptophysin), or to quantification of proteins using methods unable to resolve relevant cellular compartments. Multiple label fluorescence confocal microscopy represents a means to circumvent many of these limitations, by concurrently extracting information regarding the number, morphology, and relative protein content of synaptic structures. An important adaptation required for studies of human disease is coupling this approach to stereologic methods for systematic random sampling of relevant brain regions. In this review article we consider the application of multiple label fluorescence confocal microscopy to the mapping of synaptic alterations in subjects with schizophrenia and describe the application of a novel, readily automated, iterative intensity/morphological segmentation algorithm for the extraction of information regarding synaptic structure number, size, and relative protein level from tissue sections obtained using unbiased stereological principles of sampling. In this context, we provide examples of the examination of pre- and post-synaptic structures within excitatory and inhibitory circuits of the cerebral cortex.
Introduction
Evidence for Localized Impairments of Cerebral Cortical Circuits in Schizophrenia
Evidence from in vivo imaging and electrophysiologic studies, and from examination of postmortem tissue, indicate that schizophrenia is characterized by selective impairments of the synaptic machinery within cerebral cortical circuits. Though many brain regions may be affected, two regions in which there is strong convergence across these levels of inquiry are the dorsolateral prefrontal cortex (DLPFC) and primary auditory cortex (AI) (Lewis and Sweet, 2009 ).
Dorsolateral prefrontal cortex
Subjects with schizophrenia demonstrate impairments in working memory and executive functions referable to the DLPFC (Lewis and Sweet, 2009 ). Correlated with these deficits are reductions in underlying power of electroencephalographically recorded oscillations in the gamma (30–80 Hz) frequency range thought to be necessary for coordinated information processing within local cortical networks (Lewis et al., 2008 ). Parvalbumin-positive (PV+), fast-spiking GABA neurons (basket and chandelier cells) that provide synaptic input to the perisomatic region of pyramidal neurons are critical for the generation of gamma oscillations (Tukker et al., 2007 ; Cardin et al., 2009 ; Sohal et al., 2009 ). Postmortem studies of the DLPFC of subjects with schizophrenia have found disturbances in GABA neuron networks, in particular of PV+ neurons, which may contribute to impaired gamma oscillations in the DLPFC.
Lower mRNA levels of the 67 kDa isoform of glutamic acid decarboxylase (GAD67), the principal synthesizing enzyme for GABA, in the DLPFC is perhaps the most widely and consistently replicated pathological disturbance in schizophrenia (Torrey et al., 2005 ; Akbarian and Huang, 2006 ; Lewis and Sweet, 2009 ). At the cellular level, GAD67 mRNA is not detectable in ∼30% of GABA neurons in subjects with schizophrenia, but the remaining GABA neurons exhibit normal levels of GAD67 mRNA (Akbarian et al., 1995 ; Volk et al., 2000 ). Furthermore, levels of the mRNA for the GABA membrane transporter (GAT1), a protein responsible for re-uptake of released GABA into nerve terminals, is also decreased in schizophrenia (Ohnuma et al., 1999 ; Hashimoto et al., 2008b ), and this decrease is restricted to a similar minority of GABA neurons (Volk et al., 2001 ). These findings suggest that both the synthesis and re-uptake of GABA are lower in a subset of DLPFC neurons in schizophrenia. The affected neurons include the ∼25% of primate DLPFC GABA neurons that are PV+ (Condé et al., 1994 ) and exhibit fast-spiking firing properties (Zaitsev et al., 2005 ). In schizophrenia, the expression of PV mRNA is reduced (Hashimoto et al., 2003 ), although the number of PV+ neurons in the DLPFC appears to be unchanged (Woo et al., 1997 ; Hashimoto et al., 2003 ); in addition, approximately half of PV mRNA-containing neurons in subjects with schizophrenia lack detectable levels of GAD67 mRNA (Hashimoto et al., 2003 ).
Among PV+ neurons in subjects with schizophrenia, the chandelier class has lower GAT1 immunoreactivity in their characteristic axon boutons (cartridges) that target the axon initial segment of pyramidal neurons (Woo et al., 1998 ). In contrast, immunoreactivity for the GABA-A receptor α2 subunit, which is present in most GABA-A receptors in the axon initial segment of layer 2–3 pyramidal neurons (Nusser et al., 1996 ), is markedly increased in schizophrenia (Volk et al., 2002 ). Similar pre- and post-synaptic changes also appear to be present in the inputs of PV+ basket cells to the perisomatic region of pyramidal neurons. For example, the density of PV+ puncta, which likely represent the axon boutons of basket neurons (Erickson and Lewis, 2002 ), is reduced in DLPFC layers deep 3–4, but not in layer 2, of subjects with schizophrenia (Lewis et al., 2001 ), which parallels the laminar pattern of decreased PV mRNA expression in schizophrenia (Hashimoto et al., 2008a ). In the adult brain, PV+ basket cell inputs to pyramidal somata signal via GABA-A receptors containing α1 subunits. Interestingly, expression of the mRNA for this subunit is lower selectively in DLPFC layers 3–4 of schizophrenia subjects (Beneyto et al., 2009 ).
Primary auditory cortex
Primary auditory cortex offers another example of a cortical region in which convergent evidence implicates synaptic disruptions in the pathology of schizophrenia. In subjects with schizophrenia, the processing of sensory information within AI is impaired, manifest as the reduced ability to discriminate tones (Javitt et al., 1997 ). Tone discrimination deficits are evident even in the absence of an inter-tone interval (Javitt et al., 1997 ) and as such are unlikely to be due to impairments in working memory due to abnormalities of DLPFC as described above. Within subjects, impaired tone discrimination correlates with selective impairments in the ability to discriminate spoken emotion (prosody) (Leitman et al., 2005 ), and as such may underlie a core negative symptom in individuals with schizophrenia. Similarly, impaired tone discrimination may contribute to cognitive impairments, as it is correlated with deficits in phonemic processing and reading attainment in subjects with schizophrenia (Aspromonte et al., 2008 ). Related to these impairments in function of AI, individuals with schizophrenia also demonstrate impaired generation of early event-related potentials which localize to AI after auditory stimulus paradigms. These include deficits in the auditory N100 (Laurent et al., 1999 ; Shelley et al., 1999 ; O’Donnell et al., 2004 ; Ahveninen et al., 2006 ) and in mismatch negativity (MMN) (Javitt et al., 1995 ; Shelley et al., 1999 ; Ahveninen et al., 2006 ). MMN is of particular interest in that the degree of impairment in MMN and tone discrimination are correlated (Javitt et al., 2000 ), and both MMN and tone discrimination are likely to tap the same underlying intracortical mechanisms (Javitt et al., 1994 ).
Importantly for the design of postmortem studies of auditory cortex in subjects with schizophrenia, the source of MMN within the primate cortex is known. Intracortical recordings in monkey indicate that MMN arises in AI after the initial depolarizing thalamic volley (Javitt et al., 1994 , 1996 ). An initial component of MMN consists of increased multi-unit activity and field depolarization within deep layer 3 of AI. This is followed by a late component without multi-unit activity in which there is net depolarization within superficial layer 3, net inhibition in deep layer 3, or their combination (Javitt et al., 1994 ). This spread of activation through the supragranular AI is dependent on the intrinsic axon collaterals and their post-synaptic pyramidal cell targets within layer 3 (Mitani et al., 1985 ; Ojima et al., 1991 ; Wallace et al., 1991 ). The reduction in MMN in subjects with schizophrenia appears to represent an inability to generate maximum current flow in these circuits (Javitt et al., 1996 ). For example, reductions of MMN similar to those in subjects with schizophrenia can be modeled by infusing antagonists of N-methyl-d-aspartate (NMDA) excitatory glutamate receptors into the auditory cortex (Javitt et al., 1994 ), an observation paralleled by systemic administration of NMDA antagonists in normal humans (Umbricht et al., 2000 ).
Initial postmortem studies of AI in schizophrenia are convergent with the functional and electrophysiologic data. Thus, in deep layer 3 of AI, we observed reductions in mean pyramidal neuron somal volume (Sweet et al., 2004 ) and in the density of axon boutons (Sweet et al., 2007 ). More recently we have identified reductions in dendritic spine density in deep layer 3 of AI (Sweet et al., 2009 ). Dendritic spine and axon bouton densities correlated within subjects, suggesting excitatory synapse density is reduced in layer 3 of AI in subjects with schizophrenia (Sweet et al., 2009 ). Reductions in these excitatory synaptic components are unlikely to be restricted to AI, as reductions in dendritic spine density have also been observed in deep layer 3 in BA42 (Sweet et al., 2009 ) in deep layer 3 of DLPFC (Glantz and Lewis, 2000 ), layer 3 of PFC and temporal cortex (Garey et al., 1998 ), and in the hippocampal formation (Rosoklija et al., 2000 ).
Limitations of Existing Methods for Determination of Synaptic Pathology in Schizophrenia
Many of the above findings, while essential in establishing synaptic structural deficits within schizophrenia, have utilized methodologies that limit our understanding of these deficits. At the simplest level, studies using whole tissue extracts of protein, such as described for reductions in GAD67 (Guidotti et al., 2000 ), preclude identification of the synaptic structures specifically affected. Some findings that have identified alterations in specific structures, such as reduced bouton densities, have used a general marker such as synaptophysin, which labels nearly all boutons in cortex (Jahn et al., 1985 ; Navone et al., 1986 ), limiting our ability to identify whether deficits are selective among the subtypes of excitatory, inhibitory, and aminergic boutons which innervate the cortex. In cases where disease-related alterations have been found within a relatively specific synaptic structure, such as morphologically identified chandelier cell cartridges (summarized in Dorsolateral Prefrontal Cortex), the use of single-label approaches have restricted the ability to determine whether other proteins which may contribute to, or compensate for, synaptic dysfunction are concurrently altered within that compartment. In a similar vein, although the available evidence is consistent with correlated alterations in pre-and post-synaptic signaling in the PV+ basket cell neurons in schizophrenia, demonstration that these changes are present at the level of the synapse remains an important research objective that is not achievable using single-label approaches. These same considerations apply to studies of post-synaptic structural deficits in dendritic spines, within which the rapidly expanding knowledge base regarding signal transduction pathways allows the formulation of mechanistically important questions regarding whether proteins in specific pathways are altered in schizophrenia.
Multiple Label Fluorescence Confocal Microscopy for Quantification of Synaptic Elements Within the Cerebral Cortex of Subjects with Schizophrenia
Overview
More effectively mapping the synaptic pathology within the cerebral cortex of subjects with schizophrenia would clearly benefit from an approach with the ability to detect small synaptic structures with high spatial resolution, with biochemical selectivity, and with the ability to assess the relative expression levels of multiple proteins within the identified structures. We review below the use of multiple label fluorescence confocal microscopy to meet these needs.
Technical Considerations
Confocal microscopic imaging
The emergence of confocal microscopy has markedly enhanced the simultaneous visualization of multiple markers of synaptic structures by fluorescent immunohistochemistry in tissue sections. Although it is ideal to use wide-field epifluorescence when performing quantitative multiple label fluorescent imaging (Swedlow et al., 2002 ; Murray et al., 2007 ), in tissue sections the high density of the synaptic structures of interest results in substantial secondary fluorescence (i.e., out of focus blur) that obscures the discrimination of fine features in epifluorescence systems. With that said, as a general rule of thumb wide-field epifluorescence microscopy combined with post-image-capture deconvolution algorithms can often be used to study synaptic architecture in sections that are <6 μm thick. However, even with these very thin sections, the presence of excess secondary fluorescence can overwhelm deconvolution algorithms (see Deconvolution and Image Segmentation) causing them to fail. In contrast, imaging with a confocal microscope substantially reduces secondary fluorescence, mitigating these concerns and improving discrimination of small, densely packed objects (Figure 1 ).
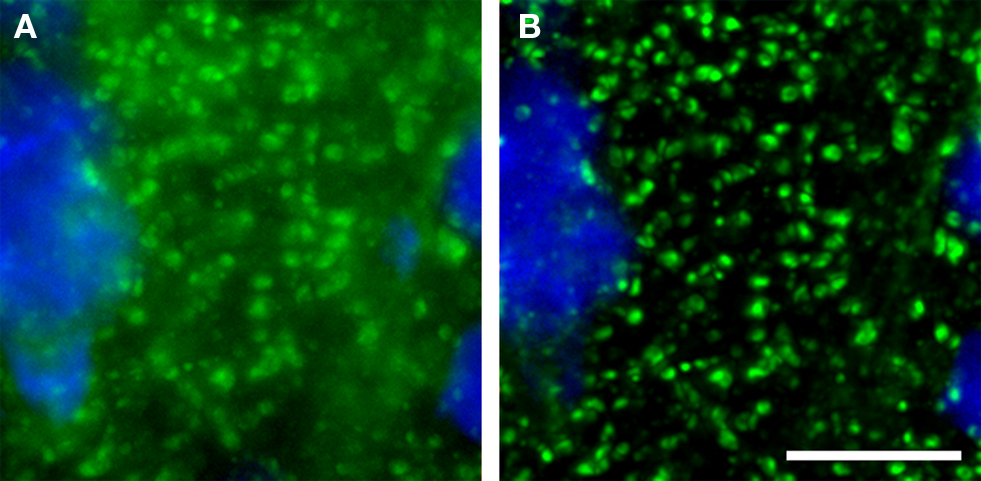
Figure 1. (A) Epifluorescence versus (B) confocal imaging of axon boutons labeled for GAD67 (green). A pyramidal cell stained for Nissl substance (blue) is also shown. The confocal image readily resolves small structures and markedly reduces out of plane fluorescence. Bar = 10 μm.
Two distinct approaches to fluorescent confocal microscopy that are widely used are the spinning disk confocal microscope (e.g., the Olympus DSU) and laser scanning confocal microscope (LSCM). Relative strengths of the spinning disk confocal microscope include having more photons reach the detector than in a typical LSCM setup (i.e., a pinhole of 1 Airy unit) (Sandison and Webb, 1994 ), providing greater fidelity of quantification of fluorescent intensity. Spinning disk confocals also use detectors [cooled charge-coupled device (CCD) camera] with higher quantum efficiency than the photomultiplier tubes use in LSCM (Sandison and Webb, 1994 ; Wang et al., 2005 ; Shaw, 2006 ; Murray et al., 2007 ). However, it is important to mention that the light path of spinning disk systems is notorious for attenuating both excitation and emission light. Thus, in experiments in which fluorescence signal intensities are low, e.g., live cell experiments, highly sensitive back-thinned electron multiplier CCD cameras may be needed. Back-thinned electron multiplier cameras offer greater sensitivity than a standard CCD camera, but at much higher purchase costs and offering lower resolution (μ/pixel). In our experience, it typically not necessary to use back-thinned electron multiplier CCD cameras to detect signal when studying synaptic organization in fixed tissue sections, providing the benefit of the higher resolution of small synaptic structures using a standard CCD camera (e.g., Hamamatsu ORCA-R2).Finally, spinning disk systems allow for rapid acquisition speed, with, as a benefit, greater throughput of image stacks.
The primary advantages of the LSCM include: greater resolution in the Z-axis (∼500 versus ∼700 nm for the DSU using a 60X 1.42 N.A. objective), which enhances the fidelity of object reconstructions in three-dimensions (3D) and thus improves volumetric data. Depending on the configuration, LSCMs offer greater excitation power above 600 nm (which is desirable as this spectrum has less autofluorescence in human tissue) that allows for efficient excitation of far red fluorochromes (e.g., Alexa 647). LSCM can also improve signal-to-background due to point illumination (Wang et al., 2005 ). In addition, LSCM scanning heads equipped for multi-spectral analysis can be very useful in dealing with tissue autofluorescence (Billinton and Knight, 2001 ), though spectral imaging and unmixing approaches for epifluorescence and spinning disk systems are becoming available (Levenson et al., 2008 ).
The comparison of spinning disk and LSCM is summarized in Table 1 . Knowledge of these tradeoffs can be used to select between microscopy approaches so as to enhance study design in imaging synaptic structures. For example, when maximal spatial resolution is required (e.g., see Figure 9 ) using a LSCM may prove most appropriate. When high throughput of multi-wavelength image stacks is needed (e.g., for stereologic sampling of a large cohort of subjects), spinning disk confocal microscopy is highly beneficial (Figure 4 ). Finally, because spinning disk confocal microscopes fall in between epifluorescence systems and LSCM with regard to both fluorescence quantification and the ability to discriminate densely packed small structures (especially when combined with deconvolution algorithms, see Deconvolution and Image Segmentation), they are often the ideal platform for studies in which both discrimination of structures for quantification of numbers and extraction of information on relative antigen expression are desired.
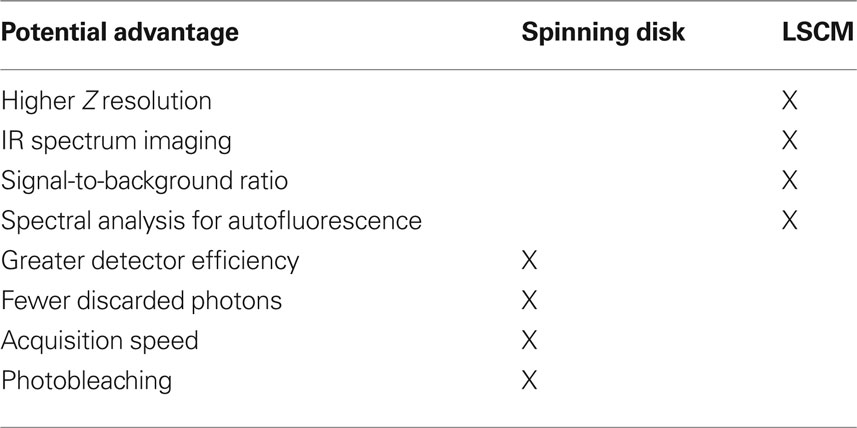
Table 1. Comparison of spinning disk confocal microscope and LSCM for human postmortem tissue studies.
Deconvolution and image segmentation
When using confocal microscopy in tissue sections, 2D images are collected at equidistant serial Z-axis positions and collated into 3D data sets. Prior to quantification of small synaptic structures, it is often desirable to further enhance the resolution of structures via processing with one of several deconvolution algorithms (Wallace et al., 2001 ) The choice of appropriate algorithm is at one level empirical, determined by a visual judgment of which algorithm provides the most robust discrimination and 3D representation of the labeled synaptic structures, and thus will best serve to estimate structure numbers. However, not all deconvolution approaches are created equally with regard to quantification of fluorescence intensity. Constrained iterative approaches that model the data as point sources, essentially placing scattered photons back at their origin, will allow quantification of fluorescence intensity. Other approaches which discard photons (e.g., nearest neighbor algorithms), cannot be used for quantification of fluorescence intensity (van der Voort and Strasters, 1995 ).
While manual counts of labeled structures of interest can be performed on a 3D image without further processing, the small size and large number of synaptic structures contained within a typical data set make it desirable to utilize automated approaches to identify structures of interest for quantification. Typically this is accomplished via threshold-based segmentation. In this process, a threshold value (either determined visually or by a thresholding algorithm) is applied to the fluorescence intensity histogram of the data set with the result that all pixels (or voxels) are reassigned a binary value according to whether they are below or above the threshold (which can be represented visually for example by assigning each value a separate color, e.g., black or blue, see Figure 2 A). The resulting image is referred to as the image mask. A group of adjacent pixels (or voxels) that are above threshold and are surrounded by pixels (or voxels) that are below threshold create an object mask. For a 3D data set, the number of such object masks provides the count of the underlying objects within the image stack. For each object mask, data such as mean and total fluorescence intensity in each wavelength detected, area (or volume), and centroid (or center of volume) are readily generated.

Figure 2. Iterative segmentation process for automated quantification of fluorescent structures (Fish et al., 2008 ). (A–C) Show the intensity histograms with the lower bounds for segmentation progressively migrating towards higher values, which results in fewer and fewer objects being masked (A′–C′). At each step, only mask objects within the selected size range are kept (A′′–C′′). After each iterative step the resulting masks (A′′–C′′) are combined. Even after only these 3 iterative steps, the combined mask shown in (D′′) already has excellent object representation [compare with the unmasked data in (D’)]. In practice any number of iterations can be chosen so as to ensure comprehensive masking of objects (see e.g., Figures 2 –5 ).
We recently described a novel approach to masking objects in 3D data sets which resulted in improved object masking in thick brain tissue sections (Fish et al., 2008 ). Rather than choosing a single set of intensity thresholds to create a binary selection mask, our approach used multiple iterations that systematically varied the intensity thresholds and combined the resultant masks via morphologic selection criteria (Figures 2 A–D). The use of varying intensity thresholds is particularly adapted for the selection of objects without uniform and/or high fluorescence intensities, which results when proteins differ in their abundance within synaptic structures, and can vary substantially over the course of development and as a result of disease states. The advantage of our approach over the use of a single threshold can be readily seen imaging a set of beads of uniform size, but of varying fluorescence intensity (Figures 3 A1–A3). The iterative method works similarly well for masking axon boutons labeled with an antibody to synaptophysin in primate cortex. Both bouton size and synaptophysin expression vary substantially in normal cortex, and yet are effectively masked to render accurate structure counts and represent with high fidelity structure shape (Figures 3 B1–B3).
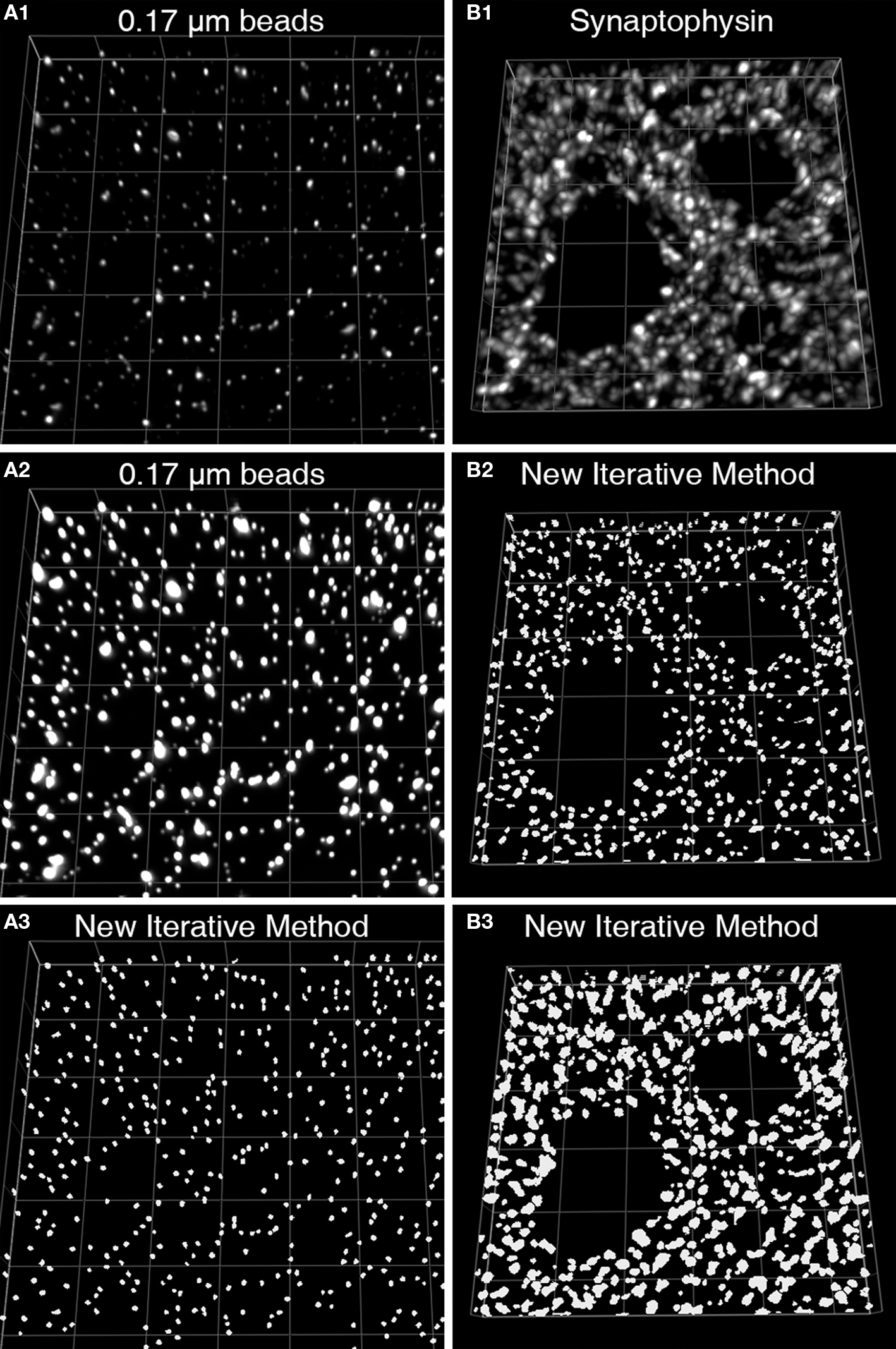
Figure 3. Iterative segmentation effectively masks objects of varying fluorescent intensity (Fish et al., 2008 ). (A1) An image projection of the deconvolved data set, consisting of 11 z-planes. (A2) The same image is displayed using different intensity levels so that all the beads in the image stack can be easily discerned. (A3) The set of mask objects derived from the same data set using our iterative segmentation approach, note the low variability in size of the mask objects, reflecting the uniform size of the imaged beads. (B1) An image projection of a monkey DLPFC section labeled for synaptophysin after deconvolution. This data set underwent iterative segmentation with objects selected by size using a range of 0.0125–0.1 μm3 (B2) or a range of 0.0125–0.5 μm3 (B3). For each size range the final mask contained a relatively uniform set of object masks that approached the upper size limit of the selection criteria. While the smaller size selection provides greater resolution of individual labeled boutons, mask objects made with the larger size range cover the underlying bouton with greater fidelity.
Needless to say when applying this approach to mask synaptic structures in tissue sections, care must be taken to ensure a high fidelity of agreement between the masked objects and the structures of interest. Both over- and under-representation of structures may occur. On the one hand, inadequate antibody penetration may lead to failure to sufficiently label and mask structures deeper within sections. Alternatively, if not accounted for, non-specific binding of the antibody or tissue autofluorescence may be detected and masked by the algorithm. Finally, if the antibodies used are directed against a protein within a localized domain of a synaptic structure, the masked object may represent that domain without masking the entire synaptic structure. For example, using an antibody against a post-synaptic density protein may result in masking the post-synaptic density within a dendritic spine, leaving much of the spine itself unmasked. Such a situation could also lead to counting two masks in a single spine if it contained two post-synaptic densities sufficiently spaced so as to be resolved by the microscope system.
Other technical issues
z-Axis sampling interval. For 3D deconvolution microscopy, it is imperative that the z-sampling frequency is sufficient to adequately represent objects of interest in the image stack. The Nyquist sampling theorem explains exactly how great the sampling density (in both x–y and the spacing of z planes) has to be to record all information from a sample. The theorem is based on the realization that one must sample at a rate greater than twice the maximum frequency in the signal in order to reconstruct the original from the sampled version, otherwise aliasing occurs. The theorem takes into account the numerical aperture of the objective, mounting/immersion media, excitation and emission wavelengths, and the number of excitation photons. There are several online Nyquist rate calculators. As a rule of thumb, for confocal microscopy imaging sampling distances along the z-axis can be up to 1.7 times the Nyquist ones. Note that oversampling is harmless unless it results in photobleaching. However, it increases capture and computational time, and image storage needs.
Resolution. The resolution of light microscopes is significantly lower in the z-axis than in the x and y dimensions. Resolution, r can be estimated for x and y axes as (Born and Wolf, 1999 ):
rx,y = 0.61λ/NA
where λ is the wavelength of the emitted light and NA is the numerical aperture of the objective. In contrast, resolution in the z-axis is estimated as (Born and Wolf, 1999 ):
rz = 2λη/NA2
where η is the refractive index of the mounting/immersion media (where the mounting and immersion media are assumed to have the same refractive index, and the refractive index of the cover slip is greater than or equal to that of the mounting media). Note that the above equations are for epifluorescence microscopy wherein the objective also serves as the condenser.
Thus for imaging an Alexa488 fluorophore (emission 519) using a 60X Oil 1.42NA objective with immersion oil of refractive index 1.51, x- and y-axis resolution is 223 nm, while z-axis resolution is 777 nm. For these parameters the Nyquist sampling for the z-axis equals 123 nm. Thus, the spacing between optical z planes should be ≤209 nm.
Fluorescence intensity measurements. When using fluorescent protein recombinants (e.g., GFP), the relationship between fluorescence intensity and antigen concentration is likely to be linear. In contrast, when using fluorescence immunocytochemistry to label proteins in tissue sections, the absolute relationship of fluorescence intensity to protein abundance can only be determined by conducting rigorous control experiments, and thus linearity of this relationship should not be presumed. With that said, because CCD and photomultiplier tube detection systems are linear across a broad range of fluorescence intensities (e.g., see Fish et al., 2008 ; Figure 4 ) quantitative fluorescence microscopy has been used in numerous studies to measure relative changes in protein levels (Sugiyama et al., 2005 ; Glynn and McAllister, 2006 ; Fish and Krucker, 2008 ; Aridor and Fish, 2009 ). In a system where the expression of a protein is only affected in a subpopulation of cells (e.g., as described in Dorsolateral Prefrontal Cortex for GAD67 expression in PV+ interneurons in schizophrenia) quantitative fluorescence immunohistochemistry will be more informative than techniques (e.g., Western Blot) that utilize the mixture of cells in tissue as a whole.
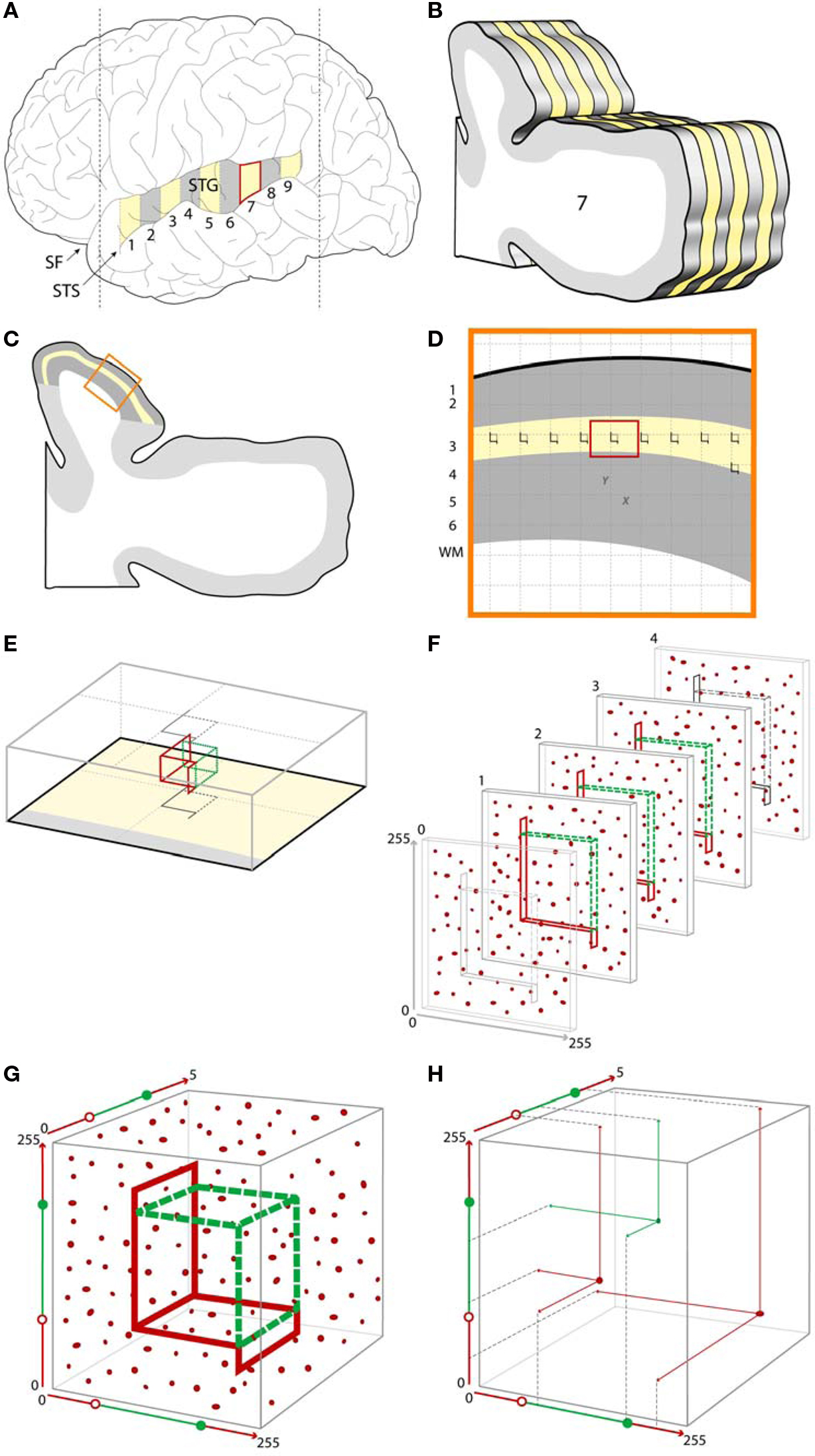
Figure 4. Example of a typical stereologic sampling scheme incorporating confocal microscopy with post-processing object masking. (A). A schematic of a human brain, showing the superior temporal gyrus (STG) which can be sampled systematic uniformly random, shown here as a series of numbered blocks from which every other block (yellow) is selected for further processing. (B) Each of the selected blocks is further subsampled into a systematic random series of sections for microscopy [shown here for one of the blocks, with every other section in this example selected for further processing (yellow)]. (C) Within each sampled section the region of interest is identified (e.g., shown here as the yellow outline of deep layer 3 within the larger cytoarchitectonically defined Primary Auditory Cortex area of interest shown in dark gray). (D) An enlarged view of a portion of the region of interest showing a systematic random sampling grid which has been placed over each sampled section. Sites for microscopic sampling are identified by each grid intersection with the region of interest. (E) An enlarged view of a single sampling site, showing the optical disector located within the z-axis of the tissue section. The gray outlines represent the projections of the disector unto the superficial and deep surfaces of the tissue section. (F) A series of 2D confocal images at a fixed distance apart in the z-axis (see Other Technical Issues for a discussion of appropriate z-axis sampling distances) are collected at each sampling site. Immunoreactive synaptic structures are shown as red puncta. (G) The series of images are combined to form a 3D data set from each site, resulting in a sampled cube with x and y dimensions represented here in pixels and the z dimension as plane number. Within this data set, the disector can be conceived of as defined by a set of values in 3D. (H) Using the masking approach described above, a center of volume, that is a single point defined by x, y, z coordinates, can be assigned for each immunoreactive puncta. Puncta with centers of volume falling within the boundaries of the optical disector are sampled for quantification (shown here in green), while those puncta with centers of volume falling outside the disector, or touching one of the exclusion lines (red) are not selected for quantification. SF, Sylvian fissure; STS, superior temporal sulcus.
Photobleaching and choice of fluorophores. Photobleaching refers to the loss of fluorescence that can occur as a result of fluorophores cycling through emission and excitation. As a result, the extent of photobleaching will be proportional to the intensity of the excitation light and the duration of exposure. When it occurs, photobleaching can make it difficult to detect the target protein and, if non-uniform, can bias quantification of fluorescence intensity. Photobleaching tends to be more severe in live cell imaging compared to imaging fixed samples because the antifade reagents mixed in mounting media more effectively protect samples than those mixed into cell media (Lichtman and Conchello, 2005 ). Photobleaching may also be mitigated by using newer generation fluorescence dyes (e.g., the Alexa series dyes) instead of traditional dyes (e.g., FITC), which are destroyed after significantly fewer emission-excitation cycles. Photobleaching may be nearly eliminated altogether through the use of other fluorescent labeling approaches such as quantum dots (Pathak et al., 2006 ).
Incorporation of Stereologic Sampling in Mapping of Synaptic Pathology
The application of stereologic methods for sampling of relevant brain regions has emerged as the standard for studies quantifying anatomical structures (Saper, 1996 ). Thus, any microscopic methods used to map synaptic pathology in disease needs to be readily integrated into a stereologic sampling scheme. A comprehensive review of stereologic principles and approaches is beyond the current manuscript (for more thorough treatments see Howard and Reed, 1998 ; Evans et al., 2004 ). At its core, stereology is about unbiased estimation of 3D parameters of objects using sampling strategies that represent the full population of structures and geometric probes for determination of 3D properties. For sampling, the key principle is that each structure of interest (e.g., each presynaptic bouton within the region of interest) should have a known (typically equal) probability of being sampled. For probes the key principle is to interact with the obtained (often 2D) sample to obtain simple geometric data (e.g., hits by points) from which the wanted 3D property can directly be estimated.
For the assessment of synaptic pathology, the necessary sampling can be conceptualized as occurring in two stages. First is the sampling of the tissue needed to generate a set of confocal image stacks (Figures 4 A–D). Second is sampling within the Z-axis of stacks (Figure 4 F). Finally, an appropriate probe or probes may be applied (Figures 4 G,H). We will briefly review several key steps at each stage.
Sampling tissue
While a number of sampling strategies are available (Evans et al., 2004 ), use of a systematic, uniformly random sampling approach is often chosen. The use of systematic sampling (e.g., every 10th section) is efficient and ensures robust coverage of the region of interest, so that the resulting sample is likely to account for variability within the region of interest. Uniformly random speaks to the final sample of structures of interest having been selected with equal probability from among the complete set of structures of interest. In the above example of selecting every 10th section from a region of interest, the starting section would be uniformly random selected from among sections 1–10 (e.g., using a random number generator), with every 10th section thereafter systematically selected.
A typical systematic, uniformly random sampling schema in studies of human postmortem tissue is often stratified, with systematic uniformly random sampling occurring at each of several levels of tissue processing. As illustrated in Figures 4 A,B, a systematic uniformly random set of blocks from the gyrus of interest is first selected, and then each selected block is exhaustively sectioned, with a systematic uniformly random set of those sections selected for further study.
Implementation of such an approach requires one to be able to define the boundaries of the region of interest. This may be relatively straightforward (e.g., whole brain). However, especially within the human brain in which there is substantial diversity of gyrification, the methodologic challenges in defining the boundaries of structures such as an individual gyrus (Kwon et al., 1999 ) or a cytoarchitectonic region (Rademacher et al., 2001 ; Sweet et al., 2005 ) should not be underestimated and must be guarded against. Methodologic variability in the delineation of the regional boundaries will inflate the variability of final estimates of synaptic structures within the region (Sweet et al., 2005 ), potentially reducing the power to detect differences between groups of interest.
In practice, two common scenarios for systematic uniformly random sampling a region of interest occur. When the region of interest can be identified by its gross structure (e.g., whole brain, cerebral cortex, superior frontal gyrus), the grossly identified structure is sampled in the manner illustrated in Figures 4 A,B. Often the region of interest can only be determined via microscopy. In this case, it is important that the grossly determined region known to completely contain the region of interest is first identified and sampled as in Figures 4 A,B. Within each of the selected sections, the boundaries of the region of interest are then identified microscopically and delineated (Figure 4 C), often with the assistance of interactive software. In both approaches the region of interest can then again be sampled systematic uniformly random in the x and y dimensions of the microscopic slide (Figure 4 D). This is usually accomplished via stereology software programs controlling a motorized microscope stage. A grid of fixed size is generated by the software and randomly positioned with respect to the region of interest. At each point of intersection of the grid with the region, a confocal stack can then be acquired (Figure 4 F).
Probes for image stacks
The first step in quantifying features of synaptic structures is the unbiased selection of these structures within the stack. In most cases this requires a means of selecting equally among structures irrespective of their size. The stereology probe used to make this selection is the disector (Sterio, 1984 ) or more typically the optical disector (Gundersen, 1986 ; Dorph-Petersen et al., 2001 ), consisting of an unbiased counting frame located within an optical plane of a thick section and at least one subsequent optical plane in the section, shown in Figures 4 E,F. In each plane, any structure falling within the counting frame, or in contact with either of the two green inclusion sides, but not the two red exclusion lines and their extensions, and not in contact with the subsequent plane, is counted.
Several important prerequisites apply when using this approach with confocal image stacks in thick sections. The counting frame must be surrounded by guard zones in x, y, and z dimensions (Figures 4 E,F). Guard zones in the z-axis should typically be similar in size to the corresponding dimensions of the structures being quantified, to avoid the possibility that one object may appear and be counted in two adjacent sections, and may need to be empirically adjusted to avoid loss of objects at the surface due to cutting artifacts (Andersen and Gundersen, 1999 ; Dorph-Petersen et al., 2009 ). In the case where deconvolution algorithms are used (see Deconvolution and Image Segmentation), guard zones also prevent quantification of the stack edges where these algorithms often fail. It is also imperative that no object be able to “hide” between the z-planes. As described in Section “Other Technical Issues”, when the z-axis is oversampled to allow for 3D reconstruction and deconvolution, this concern is automatically mitigated.
The above approach assigns what in essence is a unique associated point for each structure (its top) which is independent of object size. Counting these unique points can be accomplished in differing ways. Confocal image 3D data sets can be read into appropriate stereologic software, and the optical disector can then be applied (Gundersen, 1986 ; Dorph-Petersen et al., 2001 ). In this approach, the investigator scrolls through successive z-planes and manually defines the unique associated point of each structure as its first point of appearance in focus, using the disector counting rules described above. Alternatively, when using the approach described in Section “Deconvolution and Image Segmentation” to generate mask objects in 3D, a unique X, Y, Z coordinate for each masked structure, for example its center of volume, is readily generated by the image analysis software, and can serve as the unique associated point to be used to select it for quantification from the data file according to equations defining the unbiased counting frame in 3D. Figures 4 G,H demonstrates this “virtual” implementation of an optical disector probe which samples a set of objects using unbiased principles, from which data on object number, or object properties (e.g., fluorescence intensity) can be extracted.
Examples of Using Multiple Label Fluorescence Confocal Microscopy for Mapping Synaptic Pathology
Mapping GABA alterations in DLPFC
As summarized in Section “Dorsolateral Prefrontal Cortex”, multiple lines of evidence indicate abnormalities of the PV+ class of GABA neurons in the DLPFC of subjects with schizophrenia. Importantly, the PV+ GABA neurons comprise two functionally and morphologically distinct subgroups: chandelier neurons whose axon boutons form distinctive arrays (cartridges) surrounding the axon initial segment of pyramidal cells and basket cells, whose boutons surround neuron cell bodies (Figure 5 A). Because both chandelier cell cartridge boutons and PV+ basket cell boutons can be readily masked using iterative segmentation (Figures 5 B–D) quantitative comparisons of relative fluorescence of proteins (e.g., GAD67) in these PV+ GABA bouton types in normal individuals, and in subjects with schizophrenia, can be accomplished. Moreover, within the PV+ basket cell subtype, boutons may target either pyramidal cell bodies, or the cell bodies of other inhibitory neurons. These can be distinguished via multiple label immunofluorescence (Figure 6 ), and the pre- and post-synaptic components masked for quantification of number and/or relative fluorescence intensity.

Figure 5. Parvalbumin-positive inhibitory boutons. (A) Projection images of deconvolved confocal image stacks (13 z-planes taken 0.22 μm apart) labeled for PV in Cryostat sections (40 μm). Arrowheads indicate a chandelier cell cartridge. An unlabeled pyramidal cell (P) is surrounded by PV+ presumptive basket cell boutons. (B,C) The entire PV cartridge was masked (B) followed by the masking of puncta that make up the cartridge using a modification of our iterative segmentation methodology (C). (D) Shows the mask objects remaining after using our masking method followed by the subtraction of mask objects that colocalized with the mask in (B). The objects thus represent the PV+ boutons not located within cartridges. Bar = 10 μm.
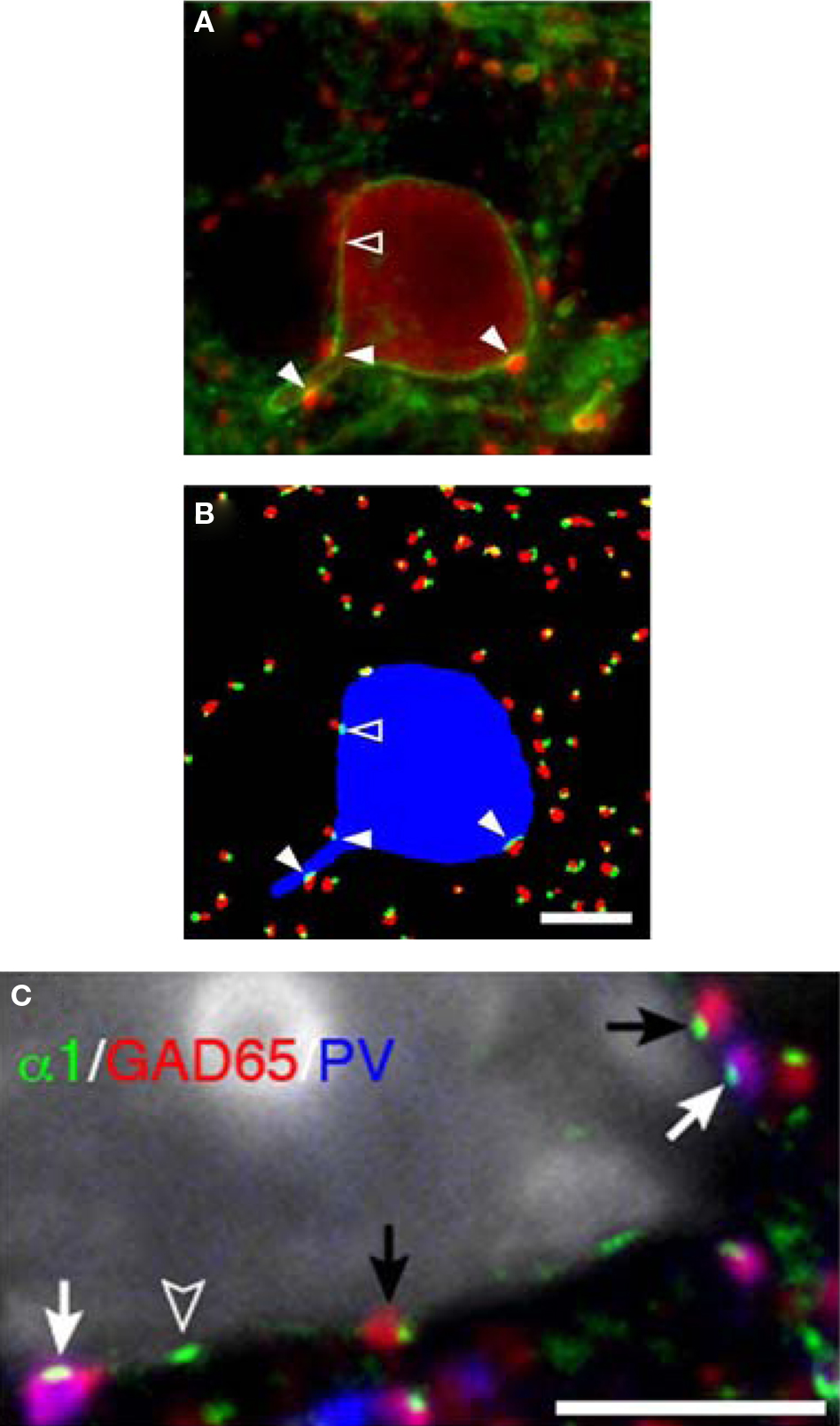
Figure 6. Parvalbumin-positive basket cell boutons apposing GABAA α1 subunits in the plasma membrane of neurons. (A) In a single confocal z-plane GABAA α1 (green) is seen in the plasma membrane of a PV (red) neuron in a tissue section. (B) Color segmentation masks of the image in (A) which represents the intracellular PV (blue), PV boutons (red) and GABAA α1 clusters (green). The different masks were made using different parameters for our iterative approach. Only the red and green object masks that partially overlap, and thus correspond to PV puncta and α1 clusters that are directly apposed, are shown. The arrowheads point to α1 clusters that are within the PV neuron and are directly apposed by PV puncta. Note that in (A) several PV puncta appear to juxtapose the soma near the open arrowhead. However, in this z-plane only one is directly apposed to an α1 cluster (open arrowhead). (C) A single confocal z-plane of a tissue section was labeled for GABAA α1 subunit (green), GAD65 (red), PV (blue), and Nissl substance (gray). Note GAD65 and PV dual-labeled (purple) boutons (white arrows) and GAD65 labeled boutons (black arrows) in apposition to GABAA α1 subunits in the plasma membrane of a pyramidal cell. The open arrowhead indicates an α1 cluster for which the apposed bouton is below the plane of focus. All Bars = 5 μm.
Mapping bouton reductions in AI
The prior observations of reduced bouton density in AI were based on identification of boutons using immunoreactivity for synaptophysin, a non-specific marker of most bouton types in cortex. Identifying whether these reductions reflect fewer boutons of intracortical or thalamocortical origin, and the extent to which excitatory and inhibitory boutons are reduced, is of fundamental importance in mapping the circuits involved in schizophrenia pathology. We have developed a multiple label fluorescent microscopic approach using antibodies directed against vesicular glutamate transporter 1 (VGlut1) which in rodent cortex has been shown to selectively label intracortical excitatory boutons, and VGlut2, which selectively labels thalamocortical boutons (Kaneko and Fujiyama, 2002 ). An example of a confocal micrograph in AI of a human subject labeled for VGlut1 and VGlut 2 and showing the predicted distribution of labeled boutons is shown in Figure 7 . A higher power example from another subject is shown in Figure 8 , triple labeled for VGlut1, VGlut2, and synaptophysin. It is readily apparent that VGlut1 and VGlut2 each label presynaptic boutons, indicated by the colocalization of each with synaptophysin (Figures 8 A–D). An example of how the iterative segmentation procedure to generate object masks can be utilized to extract information on relative protein expression within boutons is shown in Figure 8 E, for object masks created using VGlut1. It can be seen that this approach readily confirms both the lack of co-expressed VGlut2 within these boutons, and the range of synaptophysin relative expression within boutons. It is anticipated that this approach can be to map the expression of other presynaptic proteins which impact signaling characteristics within select populations of excitatory boutons in subjects with schizophrenia (Santos et al., 2009 ).
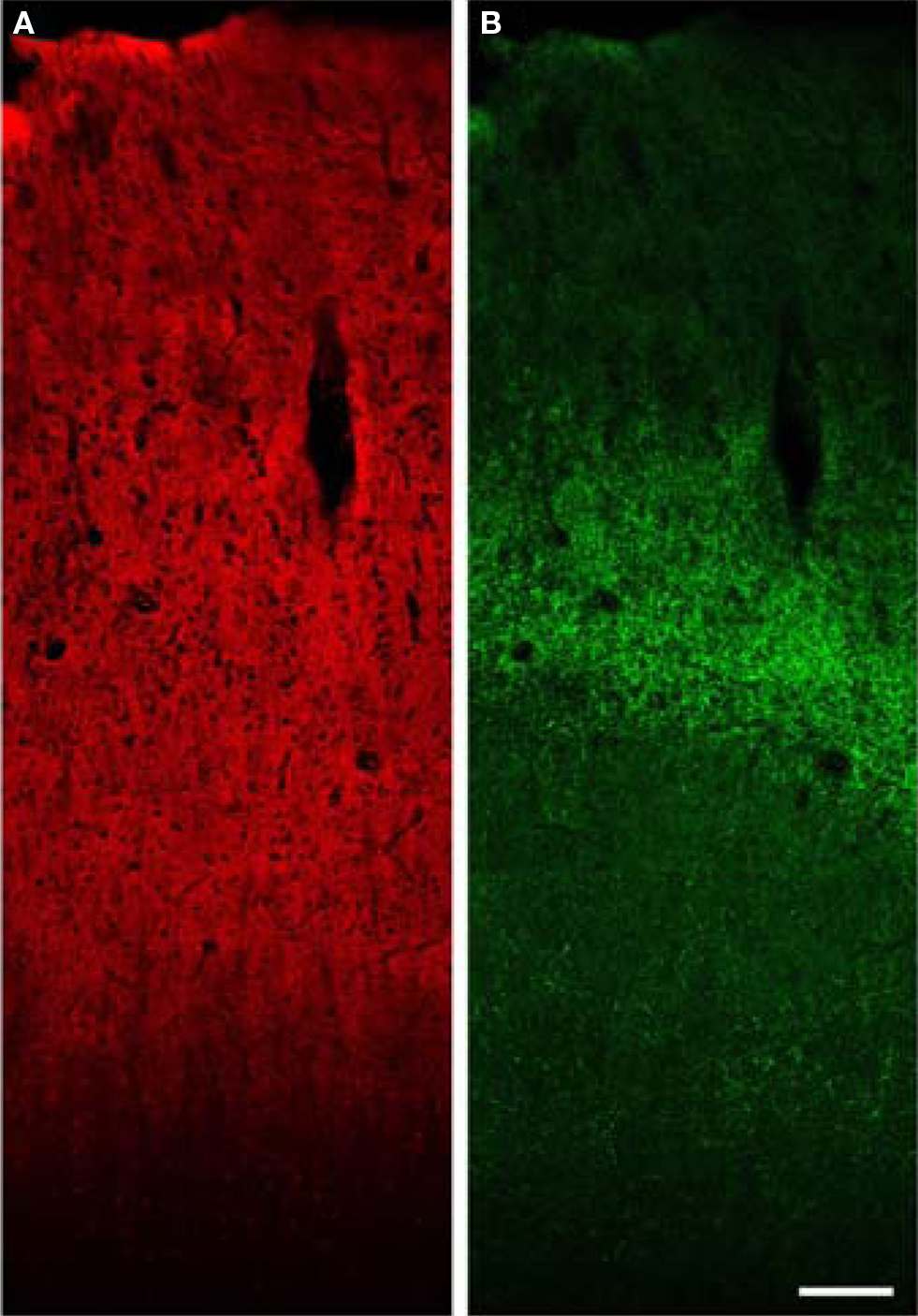
Figure 7. Low power confocal XY montage of human primary auditory cortex. (A) Immunoreactivity for VGlut1 demonstrates intense label of the entire gray matter, consistent with localization in boutons of cortical origin. (B) VGlut2 labels boutons in the thalamic termination zone in layer 4 and deep layer 3. Bar = 150 μm.
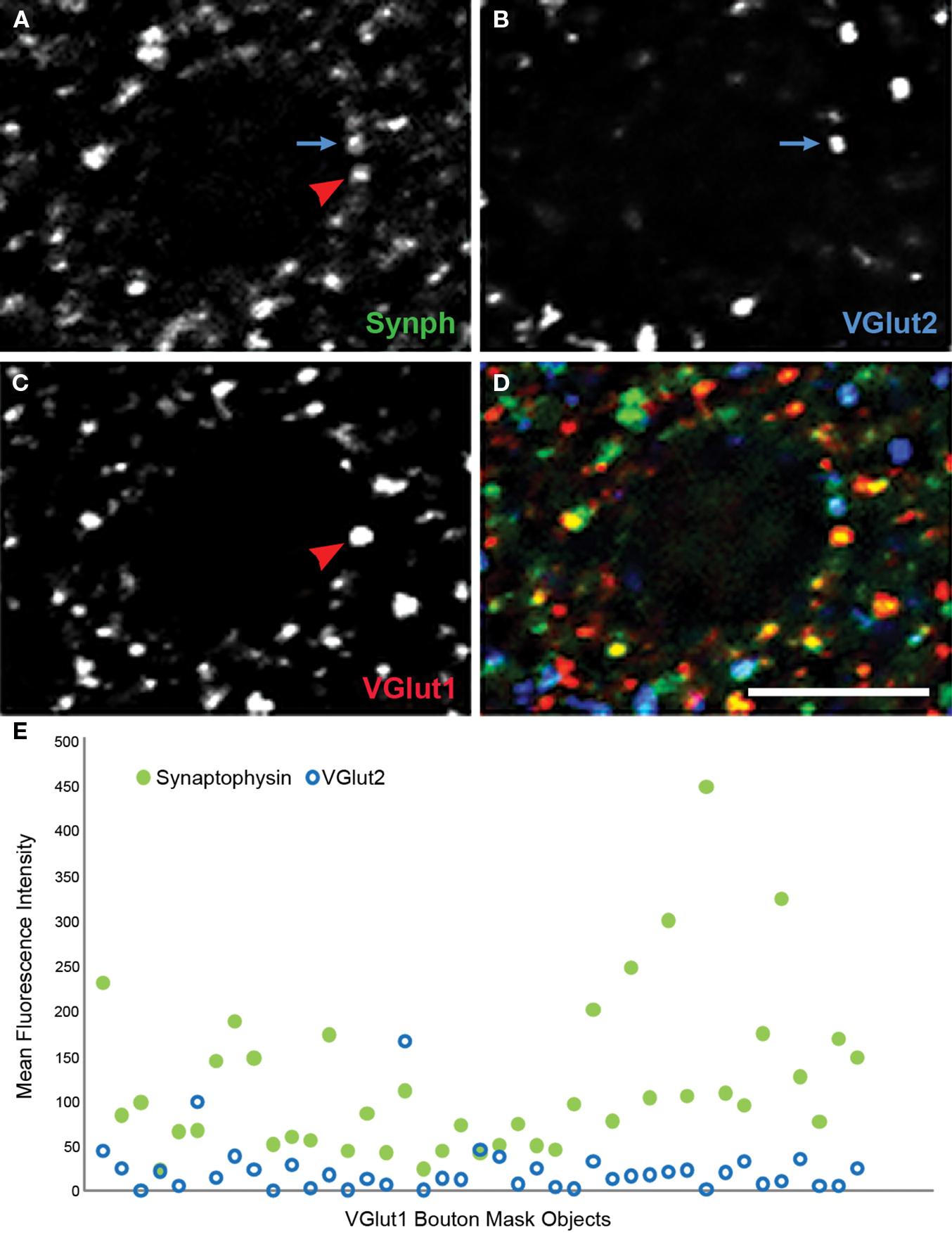
Figure 8. Human primary auditory cortex tissue labeled for synaptophysin (Synph, green), VGlut2 (blue), and VGlut1 (red). (A–D) Arrows demonstrate colocalization between synaptophysin and either VGlut1 or VGlut2. (E) VGlut2, and synaptophysin fluorescence intensities extracted from mask objects of the VGlut1-immunoreactive boutons in the micrograph shown in (A)–(D). It can be seen that quantitative data reflecting the degree of colocalization are readily extracted. Bar = 5 μm.
Mapping dendritic spine plasticity
Even in adult animals, spines are dynamic structures, with continuing generation and retraction of a proportion of spines on cortical pyramidal neurons (Trachtenberg et al., 2002 ). Experience-dependent signals can lead to spine enlargement and stabilization, or conversely shrinkage and retraction, which are dependent on F-actin stabilization or destabilization, respectively (Okamoto et al., 2004 ). The signaling cascades converging on bidirectional spine plasticity are complex and interacting, comprised of many different proteins (Calabrese et al., 2006 ). Two such spine-specific proteins, spinophilin and synaptopodin, have opposing effects on spine structural plasticity (Feng et al., 2000 ; Ryan et al., 2005 ; Vlachos et al., 2009 ). Figure 9 demonstrates labeling of dendritic spines using antibodies to these proteins, and the F-actin binding toxin, phalloidin. Despite their close approximation within spines, the separate compartments containing each label can be resolved using LSCM. This imaging approach has the additional advantage of readily controllable power of excitation, allowing the detection of low abundance post-synaptic proteins within these small structures.
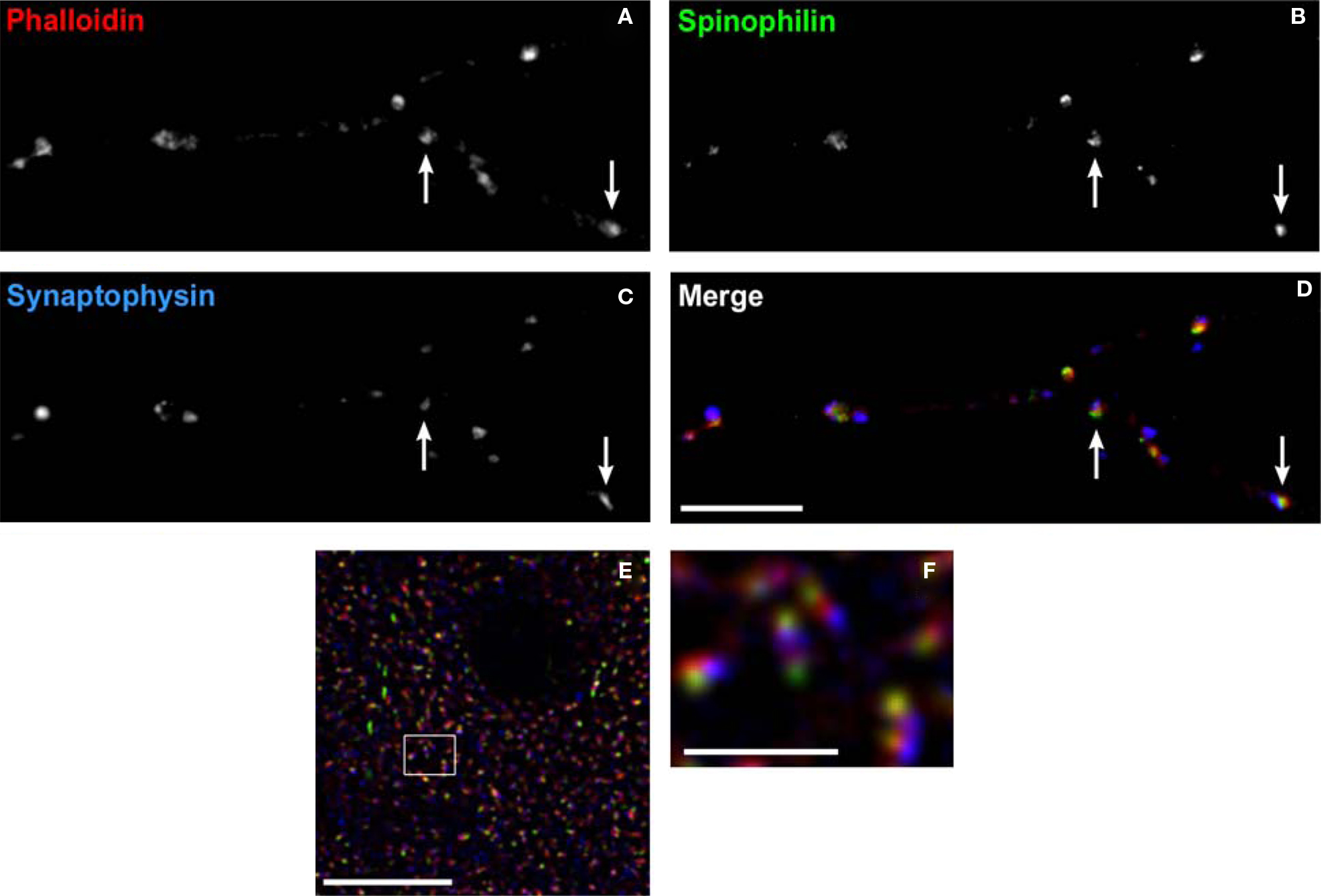
Figure 9. Labeling dendritic spines. (A–D) Spine labeling by anti-spinophilin antibody in a mature primary neuronal culture. Spines labeled with the F-actin binding toxin, phalloidin (red) can be seen to colocalize spinophilin (green). Axon boutons labeled by antibody to synaptophysin (blue) appose some of these spines (arrows). (E) Laser scanning confocal micrograph showing colocalization of the F-actin binding toxin phalloidin (red), immunoreactivity to synaptopodin (green), and to spinophilin (blue) within dendritic spines in monkey cortex. (F) Higher power view of (E) Note the ability to resolve spine subcompartments, with phalloidin in the spine head, synaptopodin at the spine apparatus, and spinophilin at the post-synaptic density. Bar in (A), (E) = 10 μm, in bar (F) = 2 μm.
Conclusion
Schizophrenia is a disorder that appears to be associated with both structural and biochemical evidence of synaptic pathology. It is likely that important therapeutic developments will derive from a more complete understanding of the nature of this pathology. Reaching this next level of understanding will clearly benefit from approaches able to map pathology at the level of expressed proteins within selectively identified cortical circuits. We have summarized, above, one approach able to address this need: multiple label fluorescence confocal microscopy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by USPHS grants MH 071533 and MH 084053. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Mental Health or the National Institutes of Health. The authors would like to thank Mrs. Mary Brady and Caitlin Moyer for assistance with the figures, and Dr. Karl-Anton Dorph-Petersen for his comments on the manuscript. The authors gratefully acknowledge the efforts of the research staff of the Translational Neuroscience Program and the Conte Center for Neuroscience of Mental Disorders at the University of Pittsburgh.
References
Ahveninen, J., Jääskeläinen, I. P., Osipova, D., Huttunen, M. O., Ilmoniemi, R. J., Kaprio, J., Lönnqvist, J., Manninen, M., Pakarinen, S., Therman, S., Näätänen, R., and Cannon, T. D. (2006). Inherited auditory-cortical dysfunction in twin pairs discordant for schizophrenia. Biol. Psychiatry 60, 612–620.
Akbarian, S., and Huang, H. S. (2006). Molecular and cellular mechanisms of altered GAD1/GAD67 expression in schizophrenia and related disorders. Brain Res. Rev. 52, 293–304.
Akbarian, S., Kim, J. J., Potkin, S. G., Hagman, J. O., Tafazzoli, A., Bunney, W. E. Jr., and Jones, E. G. (1995). Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch. Gen. Psychiatry 52, 258–266.
Andersen, B. B., and Gundersen, H. J. G. (1999). Pronounced loss of cell nuclei and anisotropic deformation of thick sections. J. Microsc. 196, 69–73.
Aridor, M., and Fish, K. N. (2009). Selective targeting of ER exit sites supports axon development. Traffic 10, 1669–1684.
Aspromonte, J., Saccente, E., Ziwich, R., Javitt, D. C., and Revheim, N. (2008). Sounding it out: auditory processing deficits and reading impairment in schizophrenia. Biol. Psychiatry 63, 248S–249S.
Beneyto, M., Abbott, A., Hashimoto, T., and Lewis, D. A. (2009). Expression of cortical GABAA receptor subunit transcripts in schizophrenia: altered inhibitory neurotransmission in specific cortical circuits. Schizophr. Bulletin 35, 224.
Billinton, N., and Knight, A. W. (2001). Seeing the wood through the trees: a review of techniques for distinguishing green fluorescent protein from endogenous autofluorescence. Anal. Biochem. 291, 175–197.
Born, M., and Wolf, E. (1999). Principles of Optics: Electromagnetic Theory of Propagation, Interference and Diffraction of Light, 7th Edn. Cambridge: Cambridge University Press.
Calabrese, B., Wilson, M. S., and Halpain, S. (2006). Development and regulation of dendritic spine synapses. Physiology (Bethesda) 21, 38–47.
Cardin, J. A., Carlen, M., Meletis, K., Knoblich, U., Zhang, F., Deisseroth, K., Tsai, L. H., and Moore, C. I. (2009). Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature 459, 663–667.
Condé, F., Lund, J. S., Jacobowitz, D. M., Baimbridge, K. G., and Lewis, D. A. (1994). Local circuit neurons immunoreactive for calretinin, calbindin D-28k, or parvalbumin in monkey prefrontal cortex: distribution and morphology. J. Comp. Neurol. 341, 95–116.
Dorph-Petersen, K.-A., Caric, D., Saghafi, R., Zhang, W., Sampson, A. R., and Lewis, D. A. (2009). Volume and neuron number of the lateral geniculate nucleus in schizophrenia and mood disorders. Acta Neuropathol. 117, 369–384.
Dorph-Petersen, K.-A., Nyengaard, J. R., and Gundersen, H. J. G. (2001). Tissue shrinkage and unbiased stereological estimation of particle number and size. J. Microsc. 204, 232–246.
Erickson, S. L., and Lewis, D. A. (2002). Postnatal development of parvalbumin- and GABA transporter-immunoreactive axon terminals in monkey prefrontal cortex. J. Comp. Neurol. 448, 186–202.
Evans, S. M., Janson, A. M., and Nyengaard, J. R. (2004). Quantitative Methods in Neuroscience: A Neroanatomical Approach. Oxford: Oxford University Press.
Feng, J., Yan, Z., Ferreira, A., Tomizawa, K., Liauw, J. A., Zhuo, M., Allen, P. B., Ouimet, C. C., and Greengard, P. (2000). Spinophilin regulates the formation and function of dendritic spines. Proc. Natl. Acad. Sci. U.S.A. 97, 9287–9292.
Fish, K. N., and Krucker, T. (2008). Functional consequences of hippocampal neuronal ectopia in the apolipoprotein E receptor-2 knockout mouse. Neurobiol. Dis. 32, 391–401.
Fish, K. N., Sweet, R. A., Deo, A. J., and Lewis, D. A. (2008). An automated segmentation methodology for quantifying immunoreactive puncta number and fluorescence intensity in tissue sections. Brain Res. 1240, 62–72.
Garey, L. J., Ong, W. Y., Patel, T. S., Kanani, M., Davis, A., Mortimer, A. M. Barnes, T. R. E., and Hirsch, S. R. (1998). Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. J. Neurol. Neurosurg. Psychiatr. 65, 446–453.
Glantz, L. A., and Lewis, D. A. (2000). Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch. Gen. Psychiatry 57, 65–73.
Glynn, M. W., and McAllister, A. K. (2006). Immunocytochemistry and quantification of protein colocalization in cultured neurons. Nat. Protoc. 1, 1287–1296.
Guidotti, A., Auta, J., Davis, J. M., Di-Giorgi-Gerevini, V., Dwivedi, Y., Grayson, D. R., Impagnatiello, F., Pandey, G., Pesold, C., Sharma, R., Uzunov, D., and Costa, E. (2000). Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch. Gen. Psychiatry 57, 1061–1069.
Gundersen, H. J. G. (1986). Stereology of arbitrary particles. A review of unbiased number and size estimators and the presentation of some new ones, in memory of William R. Thompson. J. Microsc. 143 (Pt 1), 3–45.
Hashimoto, T., Arion, D., Unger, T., Maldonado-Aviles, J. G., Morris, H. M., Volk, D. W., Mirnics, K., and Lewis, D. A. (2008a). Alterations in GABA-related transcriptome in the dorsolateral prefrontal cortex of subjects with schizophrenia. Mol. Psychiatry 13, 147–161.
Hashimoto, T., Bazmi, H. H., Mirnics, K., Wu, Q., Sampson, A. R., and Lewis, D. A. (2008b). Conserved regional patterns of GABA-related transcript expression in the neocortex of subjects with schizophrenia. Am. J. Psychiatry 165, 479–489.
Hashimoto, T., Volk, D. W., Eggan, S. M., Mirnics, K., Pierri, J. N., Sun, Z., Sampson, A. R., and Lewis, D. A. (2003). Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J. Neurosci. 23, 6315–6326.
Howard, C. V., and Reed, M. G. (1998). Unbiased Stereology. Three-Dimensional Measurement in Microscopy. New York: Springer-Verlag.
Jahn, R., Schiebler, W., Ouimet, C., and Greengard, P. (1985). A 38,000-dalton membrane protein (p38) present in synaptic vesicles. Proc. Natl. Acad. Sci. U.S.A. 82, 4137–4141.
Javitt, D. C., Doneshka, P., Grochowski, S., and Ritter, W. (1995). Impaired mismatch negativity generation reflects widespread dysfunction of working memory in schizophrenia. Arch. Gen. Psychiatry 52, 550–558.
Javitt, D. C., Shelley, A.-M., and Ritter, W. (2000). Associated deficits in mismatch negativity generation and tone matching in schizophrenia. Clin. Neurophysiol. 111, 1733–1737.
Javitt, D. C., Steinschneider, M., Schroeder, C. E., and Arezzo, J. C. (1996). Role of cortical N-methyl-d-aspartate receptors in auditory sensory memory and mismatch negativity generation: implications for schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 93, 11962–11967.
Javitt, D. C., Steinschneider, M., Schroeder, C. E., Vaughan, H. G. Jr., and Arezzo, J. C. (1994). Detection of stimulus deviance within primate primary auditory cortex: intracortical mechanisms of mismatch negativity (MMN) generation. Brain Res. 667, 192–200.
Javitt, D. C., Strous, R. D., Grochowski, S., Ritter, W., and Cowan, N. (1997). Impaired precision, but normal retention, of auditory sensory (“echoic”) memory information in schizophrenia. J. Abnorm. Psychol. 106, 315–324.
Kaneko, T., and Fujiyama, F. (2002). Complementary distribution of vesicular glutamate transporters in the central nervous system. Neurosci. Res. 42, 243–250.
Kwon, J. S., McCarley, R. W., Hirayasu, Y., Anderson, J. E., Fischer, I. A., Kikinis, R., Jolesz, F. A., and Shenton, M. E. (1999). Left planum temporale volume reduction in schizophrenia. Arch. Gen. Psychiatry 56, 142–148.
Laurent, A., Garcia-Larréa, L., d’Amato, T., Bosson, J.-L., Saoud, M., Marie-Cardine, M., Maugière, F., and Dalery, J. (1999). Auditory event-related potentials and clinical scores in unmedicated schizophrenic patients. Psychiatry Res. 86, 229–238.
Leitman, D. I., Foxe, J. J., Butler, P. D., Saperstein, A., Revheim, N., and Javitt, D. C. (2005). Sensory contributions to impaired prosodic processing in schizophrenia. Biol. Psychiatry 58, 56–61.
Levenson, R. M., Lynch, D. T., Kobayashi, H., Backer, J. M., and Backer, M. V. (2008). Multiplexing with multispectral imaging: from mice to microscopy. ILAR J. 49, 78–88.
Lewis, D. A., Cho, R. Y., Carter, C. S., Eklund, K., Forster, S., Kelly, M. A., and Montrose, D. (2008). Subunit-selective modulation of GABA type A receptor neurotransmission and cognition in schizophrenia. Am. J. Psychiatry 165, 1585–1593.
Lewis, D. A., Cruz, D. A., Melchitzky, D. S., and Pierri, J. N. (2001). Lamina-specific deficits in parvalbumin-immunoreactive varicosities in the prefrontal cortex of subjects with schizophrenia: evidence for fewer projections from the thalamus. Am. J. Psychiatry 158, 1411–1422.
Lewis, D. A., and Sweet, R. A. (2009). Schizophrenia from a neural circuitry perspective: advancing toward rational pharmacological therapies. J. Clin. Invest. 119, 706–716.
Mitani, A., Shimokouchi, M., Itoh, K., Nomura, S., Kudo, M., and Mizuno, N. (1985). Morphology and laminar organization of electrophysiologically identified neurons in the primary auditory cortex in the cat. J. Comp. Neurol. 235, 430–447.
Murray, J. M., Appleton, P. L., Swedlow, J. R., and Waters, J. C. (2007). Evaluating performance in three-dimensional fluorescence microscopy. J. Microsc. 228, 390–405.
Navone, F., Jahn, R., Di Gioia, G., Stukenbrok, H., Greengard, P., and De Camilli, P. (1986). Protein p38: an integral membrane protein specific for small vesicles of neurons and neuroendocrine cells. J. Cell Biol. 103, 2511–2527.
Nusser, Z., Sieghart, W., Benke, D., Fritschy, J.-M., and Somogyi, P. (1996). Differential synaptic localization of two major γ-aminobutyric acid type A receptor α subunits on hippocampal pyramidal cells. Proc. Natl. Acad. Sci. U.S.A. 93, 11939–11944.
O’Donnell, B. F., Vohs, J. L., Hetrick, W. P., Carroll, C. A., and Shekhar, A. (2004). Auditory event-related potential abnormalities in bipolar disorder and schizophrenia. Int. J. Psychophysiol. 53, 45–55.
Ohnuma, T., Augood, S. J., Arai, H., McKenna, P. J., and Emson, P. C. (1999). Measurement of GABAergic parameters in the prefrontal cortex in schizophrenia: focus on GABA content, GABAA receptor α-1 subunit messenger RNA and human GABA transporter-1 (HGAT-1) messenger RNA expression. Neuroscience 93, 441–448.
Ojima, H., Honda, C. N., and Jones, E. G. (1991). Patterns of axon collateralization of identified supragranular pyramidal neurons in the cat auditory cortex. Cereb. Cortex 1, 80–94.
Okamoto, K., Nagai, T., Miyawaki, A., and Hayashi, Y. (2004). Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat. Neurosci. 7, 1104–1112.
Pathak, S., Cao, E., Davidson, M. C., Jin, S., and Silva, G. A. (2006). Quantum dot applications to neuroscience: new tools for probing neurons and glia. J. Neurosci. 26, 1893–1895.
Rademacher, J., Morosan, P., Schormann, T., Schleicher, A., Werner, C., Freund, H. J., and Zilles, K. (2001). Probabilistic mapping and volume measurement of human primary auditory cortex. Neuroimage 13, 669–683.
Rosoklija, G., Toomayan, G., Ellis, S. P., Keilp, J., Mann, J. J., Latov, N., Hays, A. P., and Dwork, A. J. (2000). Structural abnormalities of subicular dendrites in subjects with schizophrenia and mood disorders. Arch. Gen. Psychiatry 57, 349–356.
Ryan, X. P., Alldritt, J., Svenningsson, P., Allen, P. B., Wu, G. Y., Nairn, A. C., and Greengard, P. (2005). The Rho-specific GEF Lfc interacts with neurabin and spinophilin to regulate dendritic spine morphology. Neuron 47, 85–100.
Sandison, D. R., and Webb, W. W. (1994). Background rejection and signal-to-noise optimization in confocal and alternative fluorescence microscopes. Appl. Opt. 33, 603–615.
Santos, M. S., Li, H., and Voglmaier, S. M. (2009). Synaptic vesicle protein trafficking at the glutamate synapse. Neuroscience 158, 189–203.
Saper, C. B. (1996). Any way you cut it: a new journal policy for the use of unbiased counting methods. J. Comp. Neurol. 364, 5.
Shaw, P. J. (2006). “Comparison of widefield/deconvolution and confocal microscopy for three-dimensional imaging.” in Handbook of Biological Confocal Microscopy, ed. J. B. Pawley (New York: Springer), 453–467.
Shelley, A.-M., Silipo, G., and Javitt, D. C. (1999). Diminished responsiveness of ERPs in schizophrenic subjects to changes in auditory stimulation parameters: implications for theories of cortical dysfunction. Schizophr. Res. 37, 65–79.
Sohal, V. S., Zhang, F., Yizhar, O., and Deisseroth, K. (2009). Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature 459, 698–702.
Sterio, D. C. (1984). The unbiased estimation of number and sizes of arbitrary particles using the disector. J. Microsc. 134 (Pt 2), 127–136.
Sugiyama, Y., Kawabata, I., Sobue, K., and Okabe, S. (2005). Determination of absolute protein numbers in single synapses by a GFP-based calibration technique. Nat. Methods 2, 677–684.
Swedlow, J. R., Hu, K., Andrews, P. D., Roos, D. S., and Murray, J. M. (2002). Measuring tubulin content in Toxoplasma gondii: a comparison of laser-scanning confocal and wide-field fluorescence microscopy. Proc. Natl. Acad. Sci. U.S.A. 99, 2014–2019.
Sweet, R. A., Bergen, S. E., Sun, Z., Marcsisin, M. J., Sampson, A. R., and Lewis, D. A. (2007). Anatomical evidence of impaired feedforward auditory processing in schizophrenia. Biol. Psychiatry 61, 854–864.
Sweet, R. A., Bergen, S. E., Sun, Z., Sampson, A. R., Pierri, J. N., and Lewis, D. A. (2004). Pyramidal cell size reduction in schizophrenia: evidence for involvement of auditory feedforward circuits. Biol. Psychiatry 55, 1128–1137.
Sweet, R. A., Dorph-Petersen, K.-A., and Lewis, D. A. (2005). Mapping auditory core, lateral belt, and parabelt cortices in the human superior temporal gyrus. J. Comp. Neurol. 491, 270–289.
Sweet, R. A., Henteleff, R. A., Zhang, W., Sampson, A. R., and Lewis, D. A. (2009). Reduced dendritic spine density in auditory cortex of subjects with schizophrenia. Neuropsychopharmacology 34, 374–389.
Torrey, E. F., Barci, B. M., Webster, M. J., Bartko, J. J., Meador-Woodruff, J. H., and Knable, M. B. (2005). Neurochemical markers for schizophrenia, bipolar disorder, and major depression in postmortem brains. Biol. Psychiatry 57, 252–260.
Trachtenberg, J. T., Chen, B. E., Knott, G. W., Feng, G., Sanes, J. R., Welker, E. and Svoboda, K. (2002). Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature 420, 788–794.
Tukker, J. J., Fuentealba, P., Hartwich, K., Somogyi, P., and Klausberger, T. (2007). Cell type-specific tuning of hippocampal interneuron firing during gamma oscillations in vivo. J. Neurosci. 27, 8184–8189.
Umbricht, D., Schmid, L., Koller, R., Vollenweider, F. X., Hell, D., and Javitt, D. C. (2000). Ketamine-induced deficits in auditory and visual context-dependent processing in healthy volunteers: implications for models of cognitive deficits in schizophrenia. Arch. Gen. Psychiatry 57, 1139–1147.
van der Voort, H., and Strasters, K. (1995). Restoration of confocal images for quantitative image analysis. J. Microsc. 178, 165–181.
Vlachos, A., Korkotian, E., Schonfeld, E., Copanaki, E., Deller, T., and Segal, M. (2009). Synaptopodin regulates plasticity of dendritic spines in hippocampal neurons. J. Neurosci. 29, 1017–1033.
Volk, D. W., Austin, M. C., Pierri, J. N., Sampson, A. R., and Lewis, D. A. (2000). Decreased glutamic acid decarboxylase67 messenger RNA expression in a subset of prefrontal cortical gamma-aminobutyric acid neurons in subjects with schizophrenia. Arch. Gen. Psychiatry 57, 237–245.
Volk, D. W., Austin, M. C., Pierri, J. N., Sampson, A. R., and Lewis, D. A. (2001). GABA transporter-1 mRNA in the prefrontal cortex in schizophrenia: decreased expression in a subset of neurons. Am. J. Psychiatry 158, 256–265.
Volk, D. W., Pierri, J. N., Fritschy, J.-M., Auh, S., Sampson, A. R., and Lewis, D. A. (2002). Reciprocal alterations in pre- and postsynaptic inhibitory markers at chandelier cell inputs to pyramidal neurons in schizophrenia. Cereb. Cortex 12, 1063–1070.
Wallace, M. N., Kitzes, L. M., and Jones, E. G. (1991). Intrinsic inter- and intralaminar connections and their relationship to the tonotopic map in cat primary auditory cortex. Exp. Brain Res. 86, 527–544.
Wallace, W., Schaefer, L. H., and Swedlow, J. R. (2001). A workingperson’s guide to deconvolution in light microscopy. BioTechniques 31: 1076-8, 1080, 1082.
Wang, E., Babbey, C. M., and Dunn, K. W. (2005). Performance comparison between the high-speed Yokogawa spinning disc confocal system and single-point scanning confocal systems. J. Microsc. 218, 148–159.
Woo, T.-U., Miller, J. L., and Lewis, D. A. (1997). Schizophrenia and the parvalbumin-containing class of cortical local circuit neurons. Am. J. Psychiatry 154, 1013–1015.
Woo, T.-U., Whitehead, R. E., Melchitzky, D. S., and Lewis, D. A. (1998). A subclass of prefrontal gamma-aminobutyric acid axon terminals are selectively altered in schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 95, 5341–5346.
Keywords: schizophrenia, synapse, cerebral cortex, postmortem, immunofluorescence
Citation: Sweet RA, Fish KN and Lewis DA (2010) Mapping synaptic pathology within cerebral cortical circuits in subjects with schizophrenia. Front. Hum. Neurosci. 4:44. doi: 10.3389/fnhum.2010.00044
Received: 13 August 2009;
Paper pending published: 19 November 2009;
Accepted: 26 April 2010;
Published online: 23 June 2010
Edited by:
Andreas Jeromin, Banyan Biomarkers, USAReviewed by:
Daniel A. Peterson, Rosalind Franklin University of Medicine and Science, USAPatrik Krieger, Karolinska Institutet, Sweden
Copyright: © 2010 Sweet, Fish and Lewis. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Robert A. Sweet, Biomedical Science Tower, Rm W-1645, 3811 O’Hara Street, Pittsburgh, PA 15213-2593, USA. e-mail:c3dlZXRyYUB1cG1jLmVkdQ==