 Boguslaw Lipinski
Boguslaw Lipinski Etheresia Pretorius
Etheresia Pretorius- 1Joslin Diabetes Center, Harvard Medical School, Boston, MA, USA
- 2Department of Physiology, Faculty of Health Sciences, University of Pretoria, Arcadia, South Africa
Amyloid hypothesis of Alzheimer’s disease (AD) has recently been challenged by the increasing evidence for the role of vascular and hemostatic components that impair oxygen delivery to the brain. One such component is fibrin clots, which, when they become resistant to thrombolysis, can cause chronic inflammation. It is not known, however, why some cerebral thrombi are resistant to the fibrinolytic degradation, whereas fibrin clots formed at the site of vessel wall injuries are completely, although gradually, removed to ensure proper wound healing. This phenomenon can now be explained in terms of the iron-induced free radicals that generate fibrin-like polymers remarkably resistant to the proteolytic degradation. It should be noted that similar insoluble deposits are present in AD brains in the form of aggregates with Abeta peptides that are resistant to fibrinolytic degradation. In addition, iron-induced fibrin fibers can irreversibly trap red blood cells (RBCs) and in this way obstruct oxygen delivery to the brain and induce chronic hypoxia that may contribute to AD. The RBC-fibrin aggregates can be disaggregated by magnesium ions and can also be prevented by certain polyphenols that are known to have beneficial effects in AD. In conclusion, we argue that AD can be prevented by: (1) limiting the dietary supply of trivalent iron contained in red and processed meat; (2) increasing the intake of chlorophyll-derived magnesium; and (3) consumption of foods rich in polyphenolic substances and certain aliphatic and aromatic unsaturated compounds. These dietary components are present in the Mediterranean diet known to be associated with the lower incidence of AD and other degenerative diseases.
Introduction
In this article we present a concept according to which the biologically most damaging hydroxyl radicals generated by free iron, and prevented by magnesium (Mg) ions, may cause neurodegenerative diseases. The primary target of the iron-induced free radicals is blood fibrinogen that becomes converted to fibrin-like material, which interferes with the effective delivery of oxygen by the altered red blood cells (RBCs) and consequently damages the brain’s neurological functions. We also review data on the protective role Mg, particularly in its ionized form. Mg has been shown to inhibit in vivo thrombosis (Ravn et al., 1997; Mussoni et al., 2001; Barbagallo et al., 2011). In this paper, the mechanism of its action will be discussed.
Efficient delivery of oxygen to tissues and organs is paramount for normal functioning of living organisms. It is well known that the brain is particularly susceptible to even short periods of hypoxia, and the chronic shortage of oxygen can cause irreversible neurologic consequences (Peers et al., 2007; Grammas et al., 2011). On the other hand it is generally believed that the prolonged ischemia followed by rapid reperfusion causes so-called “oxidative stress” believed to be preventable by the use of antioxidants (Nunomura et al., 2001). Yet potent antioxidants such as vitamin C and E have failed to provide health benefits in the degenerative diseases that are supposedly due to excessive blood oxygenation (Steinhubl, 2008). At the same time numerous natural substances endowed with oxidative properties (e.g., certain polyphenols) have been shown to provide protection against the damage caused by oxygen-centered free radicals (Summers, 2004; Lipinski, 2011). Therefore, in the current paper we present evidence for the neuroprotective effects of certain dietary components, such as Mg, polyphenols, aliphatic, and/or aromatic unsaturated natural substances.
Vascular and Hemostatic Factors
The prevailing amyloid hypothesis in Alzheimer’s disease (AD) has recently been challenged by several authors (Pimplikar, 2009; Skaper, 2012; Chetelat, 2013). This challenge is supported by the increasing evidence for the role of vascular components (Kovacic and Fuster, 2012; Diomedi and Misaggi, 2013; Sery et al., 2013), as well as hemostatic factors in neurological diseases and particularly in AD (Kalaria, 2003; Gupta et al., 2005; Kling et al., 2013). Thus, isoforms of fibrinogen gamma chain were identified among insoluble proteins in AD brains (Choi et al., 2002), and it was also shown that fibrin interacted with beta-amyloid protein (Merkle et al., 1996). Other researchers have demonstrated the perivascular leakage of fibrinogen around brain microvessels in AD and HIV-related neurological disorders (Fiala et al., 2002), as well as the presence of fibrin in CNS (Inoue et al., 1997). The existence of a protease-resistant complex of fibrinogen and albumin was also found in AD brains (Lipinski and Sajdel-Sulkowska, 2006). Moreover, Paul et al. (2007) documented that fibrin accelerates neurovascular damage in AD. In acute cerebral lesions of multiple sclerosis tissue plasminogen activator (tPA) was co-localized with fibrin(ogen) on large diameter axons, which finding was interpreted as an attempt to remove fibrin deposits and restore normal axon function (Gveric et al., 2001). Brain injury accompanied by cerebrovascular fibrin deposition was revealed in a hypertensive stroke model (Ninomia et al., 2000). Fibrin clearance and/or deposition was suggested to be a key regulatory mechanism for Schwann differentiation and nerve cell damage (Davalos et al., 2012) due to the altered fibrin structure. It is furthermore known that amyloid-beta delays fibrin clot lyses by altering fibrin structure and attenuating plasminogen binding to fibrin (Zamolodchikov and Strickland, 2012). Recently, Hultman et al. (2013) confirmed that fibrin(ogen) is deposited in the AD neurovasculature and interacts with beta-amyloid, resulting in increased formation of blood clots. Also, in postmortem investigations, an increased deposition of fibrin(ogen) was observed in AD cases compared with non-demented controls, and the authors suggested that a strong correlation exists between cerebral amyloid angiopathy severity and fibrin(ogen) deposition. There is therefore extensive evidence of the role of oxidative damage in the brain of patients with AD although the exact process is not fully understood. However, oxidative stress could be linked to alterations of amyloid-beta metabolism and beneficial effects of antioxidants against amyloid-beta toxicity or AD may be of great importance and are shown in many studies (Chakrabarti et al., 2013).
It is also of interest to note that high blood fibrinogen is associated with an increased risk of AD (van Oijen et al., 2005), and that antibodies to beta-amyloid peptide react with individual chains of fibrinogen (Stern et al., 1990). Moreover, amyloid fibrils were shown to contain fibrinogen antigen-reactive material (Ahn et al., 2010; Cortes-Canteli et al., 2010). It is of interest to note that AD is closely associated with atherosclerosis (Kovacic and Fuster, 2012; Yarchoan et al., 2012) in which blood fibrinogen levels are typically increased (Lipinska et al., 1976; van Oijen et al., 2005). Yet some people may develop a “pure” form of AD based strictly on the pathologic consequences of insoluble amyloid-beta without manifestation of the vascular disease as emphasized by Schneider et al. (2007).
Yet, despite intensive research, we still do not understand how fibrin is being formed in cerebral microcirculation, especially following episodes of hypoxia/ischemia. In contrast to coronary circulation, very few blood components are present in the extravascular brain space, as a result of a selective permeability of blood-brain-barrier (BBB). Perhaps it is not surprising that one of such components is tPA, which might serve as the first line of defense against fibrin formed from plasma leaked after damage to BBB (Gveric et al., 2001). On the other hand, it has been suggested that this activator in conjunction with plasmin may aid the clearance of Abeta peptide as shown in the brains of patients with AD (Melcher, 2004). Alternatively, plasmin formed in the cerebral extravascular space can be neurotoxic, by activating metaloproteinases that are known to degrade basement membrane matrix (Kaur et al., 2004).
Iron and Free Radicals
Several investigators discuss the involvement of two transition metals, iron and copper, in the pathogenesis of atherosclerosis and AD (Brewer, 2007; Barnham and Bush, 2008; Silvestri and Camaschella, 2008; Smith et al., 2010; Weinberg, 2010; Squitti, 2012; Zheng and Monnot, 2012). Out of these two redox metals, iron was shown to be particularly active in a number of degenerative diseases (Kiechl et al., 1997; Brewer, 2007; Ahluwalia et al., 2010; Depalma et al., 2010; Hahalis et al., 2011; Merono et al., 2011) including neurological disorders (Beard and Connor, 2003; Ke and Ming Qian, 2003; Thomas and Jankovic, 2004; Kell, 2010). It should be emphasized that free iron can participate in the formation reactive oxygen species (ROS) that, in turn, initiates so-called oxidative stress leading to AD (Casadesus et al., 2004; Castellani et al., 2012), as well as to those associated with aging (Szweda et al., 2002). The concept of labile iron pool was recently developed, which may explain the connection between iron and free-radical generation (Kruszewski, 2003; Breuer et al., 2008; Benarroch, 2009). However, despite the fact that the concept of oxidative stress has been generally accepted, there is no proof of the involvement of oxidation processes in degenerative diseases (Lipinski, 2011). On the other hand, the presence of biomolecules enriched in one or more atoms of oxygen indicates the involvement of a specific type of ROS – the hydroxyl radical. Hydroxyl radicals are the most biologically damaging species, particularly with respect to macromolecules such as proteins, nucleic acids, and carbohydrates (Cheeseman et al., 1988; Lipinski, 2011). We have recently documented that trivalent iron ion (Fe3+) reacts with the hydroxyl group of water to produce a powerful hydroxyl radical according to the following reaction:
This reaction can be considered as a special case of the Fenton reaction in which hydroxyl radicals are generated from divalent iron ion in the presence of hydrogen peroxide. The need for the oxidizing agent in the Fenton reaction led to a misleading concept that hydroxyl radicals are formed as a, result of the oxidation reaction. Whatever their origin, it should be emphasized that the most important neurological consequence of the action of hydroxyl radicals is their ability to convert soluble fibrinogen into an insoluble fibrin-like polymer (Lipinski and Pretorius, 2012). A characteristic feature of such a polymer is its resistance to the action of proteolytic enzymes that normally degrade fibrin(ogen) into smaller polypeptide fragments.
The damaging effect of hydroxyl radicals can be explained in terms of the following mechanism: the undesirable molecular interactions in blood proteins are prevented by holding their hydrophobic groups inside the interior of protein tridimensional structures stabilized by intra-molecular disulfide bonds. Once these bonds are broken, the polypeptide chains become unfolded with the consequent exposure of the hydrophobic domains, which form inter-molecular bonds and result in the formation of large a proteolytic enzymes, as is the case with human prion proteins (Das et al., 2010) and bacterial hydrophobins (Kwan et al., 2006). Moreover, in 2001, researchers suggested that free-radical-induced protein aggregates resistant to proteases are responsible for the amyloid formation (Squier, 2001). It is worth noting that one of the risk factors for AD is diabetes mellitus, in which excessive generation of free radicals was may play a major pathogenic role (Lipinski, 2001). This has recently resulted in raising intriguing questions regarding the common denominator in AD and diabetes (Craft, 2012; Adeghate et al., 2013; Vignini et al., 2013).
Another factor has been suggested to be involved in the pathogenesis of AD, which is copper known to generate hydroxyl radicals in hypoxia. This transition metal as well as iron are present in normal brain tissue at normal concentrations, but are significantly increased in cerebrospinal fluid of AD patients (Multhaup, 1995) and in AD brains (Markesbery and Carney, 1999; Altamura and Muckenthaler, 2009). Moreover, transition metals have been implicated in the generation of free-radical and in the aggregation of amyloid protein in the brains of Alzheimer’s patients (Adlard and Bush, 2006). The role of iron in neurotoxicity was recently reviewed (Stankiewicz and Brass, 2009) and the iron overload in the early stages of life was suggested to induce cognitive impairment and the damage of the brain (de Lima et al., 2005). It is possible that under the reducing conditions of hypoxia, these metal ions generate hydroxyl radical with all its pathological consequences (Dajas-Bailador et al., 1998; Ciriolo et al., 2000). An imbalance between free-radical generation and scavenging was suggested to be one of the earliest pathological events in AD (Moreira et al., 2005).
Additional evidence for the involvement of hydroxyl radicals in the pathogenesis of neurodegenerative diseases comes from the experimental investigations showing the protective effect of a free-radical scavenger ferulic acid in mouse neuroblastoma 2a cells (Yan et al., 2012). Moreover, evidence for the involvement of free radicals in these diseases comes from experimental studies with the use of edaravone, a potent scavenger of hydroxyl radicals (Abe et al., 2004). On the basis of its scavenging properties, this compound was approved in Japan for the treatment of acute cerebral infarcts within 24 h of onset (Watanabe et al., 1996). Diabetic neuropathy was also shown to be prevented by edaravone in an animal experimental study (Saini et al., 2007). Perhaps, the best evidence for the connection between hypoxia and hydroxyl radical production was provided by showing the protective effect of edaravone in fetal lamb brain after umbilical cord occlusion (Nakajima et al., 2006).
Hemorheologic Disturbances
Although the hydroxyl radical is considered by mainstream scientists as an oxidant, it behaves as a reducing agent with respect to disulfide bridges in plasma proteins leading to the unfolding and scrambled refolding of the polypeptide chains. In this respect this type of free radicals is similar to a dithiol-reducing agent that when added to human whole blood causes rapid aggregation of RBC and an irreversible polymerization of the plasma proteins (Egyud and Lipinski, 1991). Thus, this in vitro phenomenon illustrates the pathologic consequences of the exposure of hydrophobic forces that are potentiated by antioxidants and prevented by the oxidizing agents (Pretorius et al., 2013a). For such a reason this concept offers an explanation for the failure of antioxidant therapies in the degenerative diseases – including the neurological disorders (Steinhubl, 2008).
At the same time numerous natural products that are not antioxidants were shown to provide protection against cardiovascular and neurological diseases. Particularly, a group of amphiphilic substances, known as “polyphenols” including resveratrol, epigallo-catechin-3-gallate (EGCG), certain anthocyanins, and flavonols has been shown to be effective scavengers of hydroxyl radicals (Lipinski, 2011). In addition, both EGCG and resveratrol were shown to cross BBB, a phenomenon that is not shared by other polyphenols (Mandel et al., 2006; Srividhya et al., 2008; Bieschke et al., 2010). However, most important biological function of polyphenols, is scavenging of hydroxyl radicals by means of aromatic hydroxylation. This reaction may explain the ability of EGCG to disrupt RBC-parafibrin aggregates, as previously reported by us (Pretorius et al., 2013b).
Continuous and unobstructed delivery of oxygen to the brain by the RBCs depends on their membrane fluidity and the state of aggregation that impairs the release of oxygen (Tateishi et al., 2001). Despite decades of intensive research it is not known what exactly causes RBC aggregation and disaggregation. Over 100 years ago Polish physician, Edmund Biernacki, documented that the phenomenon of erythrocyte sedimentation depends on the interaction with fibrinogen (Biernacki, 1897), the level of which in plasma is frequently elevated in degenerative and inflammatory diseases. Years later an argument was presented that it is not just fibrinogen itself but thrombin-induced soluble fibrin monomers that form bridges between individual cells and in this way increase their sedimentation and/or aggregation (Lipinski et al., 1969). The reason for this is that fibrin monomers are more hydrophobic than fibrinogen itself (van Oss, 1990) and thus can more readily interact with hydrophobic patches on RBC membranes. Thus, the dramatic increase of hydrophobicity of soluble parafibrin fibrils generated by hydroxyl radicals from fibrinogen makes them even more potent inducers of RBC aggregation. Whatever the mechanism, it is obvious that fibrinogen plays a critical role in this phenomenon, so the reduction of its reactivity seems to be of great potential in AD and in other neurodegenerative disorders. Davalos et al. (2012) have even suggested eliminating the effect of fibrinogen by the therapeutic defibrination, which is not a very realistic clinical practice.
Increased RBC sedimentation and/or their aggregation have been observed in the degenerative diseases such as atherosclerosis and inflammation, which are known to be associated with AD (Robinson et al., 1995; Andresdottir et al., 2003). Most recently it was shown that thrombosis mediated by RBC is potentiated by ferric chloride (Barr et al., 2013), which confirms our original observation on the role of iron in pathologic blood coagulation (Lipinski and Pretorius, 2012; Pretorius et al., 2013a,b). Other researchers have also emphasized the role of hemorheology in thrombosis and vascular diseases (Erikssen et al., 2000; Natali et al., 2003; Baskurt et al., 2004; Nagy et al., 2010). It is believed that the abnormalities in RBC obstruct oxygen delivery to the brain (Tateishi et al., 2001; Mohanty et al., 2008; Tripathy et al., 2013), which, in turn, causes hypoxia leading to chronic inflammation (Eltzschig and Carmeliet, 2011; Wyss-Coray and Rogers, 2012). Last but not least is the role of blood hemolysis that releases trivalent iron (Woollard et al., 2009) with all its pathologic consequences (Pretorius and Lipinski, 2013). It should be kept in mind that AD, especially when presented during the later stages of life, presents as a co-occurrence with vascular pathology, such as white matter disease. The question that now arises is exactly how damage due to hydroxyl radicals and typical vascular pathology that may develop independently, are interlinked. We suggest that the additional burden of the presence of hydroxyl radical damage might cause a faster progression of the disease.
Inflammation
A large body of data indicates that inflammation is a hallmark of AD and other neurodegenerative diseases (Tuppo and Arias, 2005; Paul et al., 2007; Ray and Lahiri, 2009; Davalos et al., 2012; Wyss-Coray and Rogers, 2012; Krstic and Knuesel, 2013). In view of a close relationship between inflammation and hypoxia (Eltzschig and Carmeliet, 2011) it is possible that the culprit is a persistent obstruction of cerebral blood flow caused by the chronic formation of iron-induced fibrin/RBC aggregate. In another scenario pathologic fibrin, being resistant to the proteolytic degradation, may present itself to the innate immune system as a foreign body. According to Mechnikov, inflammation is a protective adaptation response to a foreign body that activates macrophages in an attempt to eliminate it from a living organism (Palmblad, 2010). Whereas in most cases the powerful enzymatic machinery of macrophages eventually digests foreign pathogens, the specific hydrophobic properties of iron-induced material make it refractory to the proteolytic degradation, thus initiating a chronic state of inflammation.
The Protective Role of Magnesium
Mg is a cofactor in numerous enzymes and plays a critical role in many physiologic reactions, including the regulation of cell membrane stability (Woolf, 1961; Elin, 1994; Fawcett et al., 1999). Since Mg is not routinely measured in medical practice, its deficiency may remain undetected for a long period of time (Touyz, 2004; Assadi, 2010). Large epidemiological studies indicate that the inadequate dietary Mg intake and its low serum concentration are associated with insulin resistance, type 2 diabetes mellitus (Corica et al., 2006; Huang et al., 2012) and with cardiovascular diseases (Amighi et al., 2004; Adamopoulos et al., 2009). In addition, hypomagnesemia occurs frequently in association with hypertension (Houston, 2011) and is related to all-cause mortality (Reffelmann et al., 2011). It has also been argued that correcting hypomagnesia may enhance memory (Slutsky et al., 2010) and even prolong life (Rowe, 2012).
Mg deficiency is frequently encountered in critically ill patients (Limaye et al., 2011), particularly those with systemic inflammation (Mazur et al., 2007; Weglicki, 2012). Although Mg deficiency has been known for a long time to be correlated with thrombotic diseases, the mechanism of its hemostatic function is not well understood (Barbagallo et al., 2011). Total plasma Mg concentrations are remarkably constant in healthy subjects and do not substantially change with aging (Romani, 2013) but its ionized form, representing 55% of total Mg, has been shown to be decreased in cardiovascular disease (Kupetsky-Rincon and Uitto, 2012) and in AD as well (Durlach, 1990; Cilliler et al., 2007; Barbagallo et al., 2011).
Health beneficial effects of Mg can be explained by the results of our finding of its anticoagulant properties, as shown in Table 1.
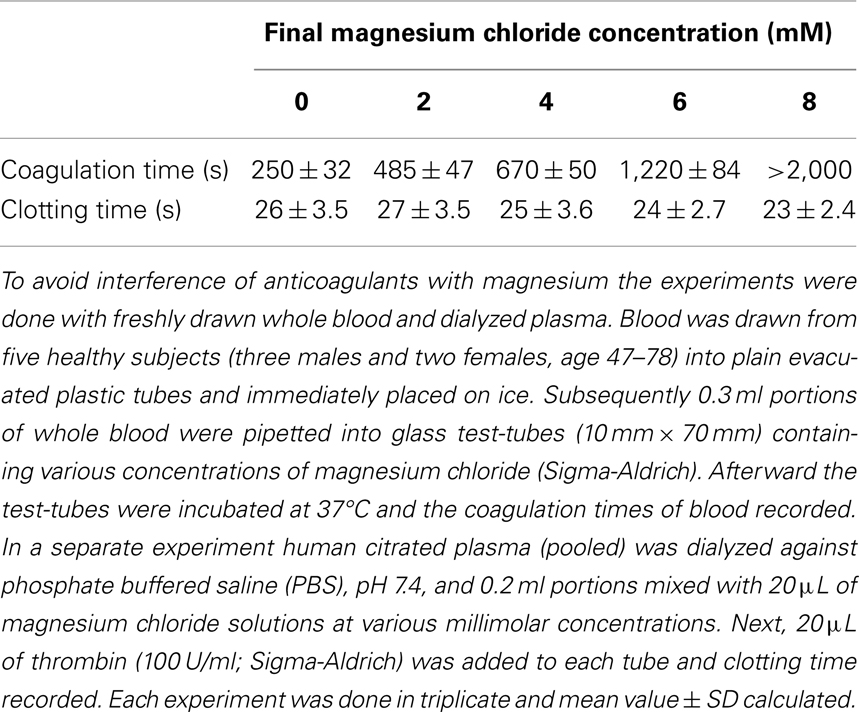
Table 1. Effect of magnesium chloride on spontaneous coagulation of whole blood and on thrombin-clotting times of plasma.
As shown in Table 1, Mg progressively prolonged coagulation times until the samples became uncoagulable at 8 mM concentration. It is important to note that this novel effect can be observed only with native whole blood and is absent when tested with plasma clotted with thrombin. It has to be concluded, therefore, that the Mg ions interfere with the intrinsic activation of prothrombin. This is a very important fact that explains why even large doses of Mg administered to humans have never caused bleeding from the site of vascular injury, at which tissue factor-activated thrombin fulfilled its hemostatic function (Huntsman et al., 1960; Ravn et al., 1997; Ames et al., 1999; Mussoni et al., 2001). The dramatic influence of Mg on blood coagulation may explain its beneficial effects in numerous diseases described by several investigators (Ceremuzynski et al., 2000; Muir et al., 2004; Shechter, 2010; Barbagallo et al., 2011; Kupetsky-Rincon and Uitto, 2012; Muroi et al., 2012; Albrecht et al., 2013). An important fact described by us has to be emphasized – Mg anti-coagulated blood is hemostatically effective as a result of its preserved ability to form functional clots with thrombin that is physiologically generated by tissue factor at the site of vessel wall injury. Apparently, by contrast to coumarin-derived anticoagulants, Mg does not affect prothrombin biosynthesis, but inhibits its conversion to thrombin (Table 1). The role of Mg in hemostasis and in hemorheology is illustrated in Figure 1.
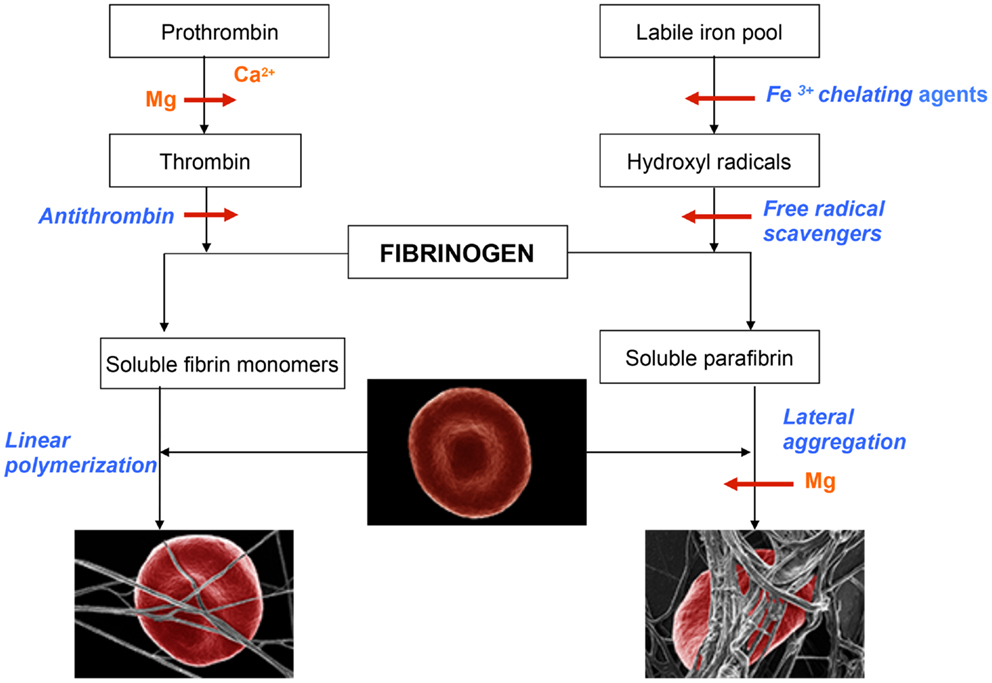
Figure 1. Schematic representation of events leading to the formation of normal blood clot (left panel) and pathological iron-induced parafibrin (right panel). Ca, calcium; Mg, magnesium; RBC, red blood cell.
In the normal process of blood coagulation prothrombin is activated to enzyme thrombin that, in turn, converts plasma fibrinogen into soluble fibrin monomer(s). Subsequently, these species undergo spontaneous linear polymerization, which leads to the formation of fibrin fibrils that mechanically trap RBCs (left panel). This phenomenon is very important for effective hemostasis and subsequent fibrinolysis, which is prerequisite for normal wound healing and the return of RBC into the circulation. By contrast to this physiological process, the pathological iron-induced blood coagulation involves polymerization and irreversible entrapment of RBCs (right panel). In this process hydrophobic soluble parafibrin interacts with the hydrophobic epitopes on RBC membranes, forming large aggregates that are resistant to the fibrinolytic degradation, and in this way prevents RBCs from effectively delivering oxygen to the brain.
Magnesium plays a dual role in hemostasis: first it delays and/or inhibits intravascular generation of fibrin monomers that might have been formed as the results of prothrombin activation; second, Mg ions prevent the interaction of soluble iron-induced parafibrin with RBC. Moreover, and even more importantly, preliminary studies have shown that Mg ions disrupt RBC-parafibrin aggregate as shown in Figure 2 and, in this way, allow erythrocytes to return to the circulation (in whole blood smears of six AD patients and noted in 100 RBCs per individual with and without added Mg). Figure 2 shows representative images for the six patients.
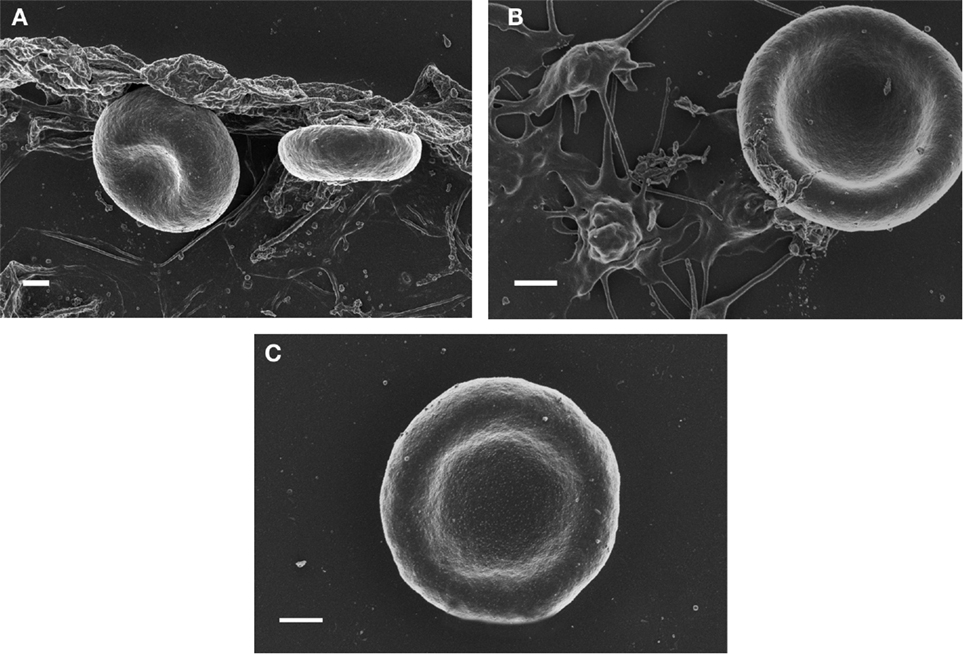
Figure 2. SEM micrographs of red blood cells from Alzheimer’s disease (A) RBC smear; (B) with 3 mM MgCl2; (C) with 30 mM MgCl2. Scale = 1 μm.
It should be emphasized that a substantial difference exists in the susceptibility to thrombolysis of these two types of clots. In normal blood the intravascular thrombi can be effectively, although only within a specific time window, eliminated with the administration of fibrinolytic therapy. By contrast, abnormal fibrin clots are remarkably resistant to the thrombolytic degradation, remain in the circulation and cause chronic inflammation. This pathologic reaction can be prevented by the chelation of free iron, scavenging of hydroxyl radicals, and/or administration of ionized Mg. Of note, Mg sulfate therapy was shown to be effective in women with eclampsia and pre-eclampsia (Duley et al., 2010; McDonald et al., 2012; Okereke et al., 2012). In addition, low dietary Mg intake was demonstrated to be associated with the risk of stroke (Larsson et al., 2012). Larger doses protect against neurological deficit after brain injury (McIntosh et al., 1989) and were shown to improve insulin sensitivity in non-diabetic subjects (Guerrero-Romero et al., 2004) and in type 2 DM patients (Rodriguez-Moran and Guerrero-Romero, 2003; Kim et al., 2010).
Other Neuroprotective Factors
It is well known that a healthy diet, rich in polyunsaturated fatty acids (PUFA) and polyphenols may have a positive effect on general health. In particular, the Mediterranean diet has long been known to have a positive effect on the health status of individuals, including that of AD patients (Scarmeas et al., 2006; Thaipisuttikul and Galvin, 2012). Countless papers show that the major components of this diet include mono- and PUFA (in the form of fish oil), minerals such as Mg, polyphenolic substances (red wine) and iron chelating agents contained in citrus fruits (Feart et al., 2010; Gu et al., 2010; Ayissi et al., 2013; Hu et al., 2013). All these substances and agents are known to scavenge hydroxyl radical by virtue of aromatic hydroxylation, as well as to chelate iron (Thaipisuttikul and Galvin, 2012). This specific combination of food products present in the Mediterranean diet results in the inhibition of the generation of parafibrin, which we suggest, contributes to the development and progression of AD. It should also be noted that these beneficial dietary effects may be important in prevention of cardiovascular disease (Lipinski and Pretorius, 2013).
Polyunsaturated Fatty Acids
The brain is known to contain elevated quantities of PUFA that paradoxically do not become oxidized by the high oxygen level present in the brain. By contrast, PUFAs have been shown to be neuroprotective, which can be explained in terms of their double bonds acting as scavengers of hydroxyl radicals by converting them to corresponding hydroxy-fatty acids (Czapski, 1984; Yavin et al., 2002; Song et al., 2004). A highly unsaturated eicosapentanoic acid was also shown to offer neuroprotection in the hippocampus of gamma-irradiated rats (Lonergan et al., 2002) and in other experimental models (Marszalek and Lodish, 2005). Concentrations of essential fatty acids in plasma and RBCs and brain tissue were found to be lower in patients with AD (Tully et al., 2003; Issa et al., 2006; Milte et al., 2009) and in the aging brain (Lukiw and Bazan, 2008). It is therefore not surprising that supplementation with fish oil in AD patients substantially improved their memory (Corrigan et al., 1991; Yehuda et al., 1996; Sinn et al., 2010). Moreover, consumption of fish was shown to be associated with a reduced risk of AD (Morris et al., 2003), which might explain the positive effect of the Mediterranean diet on cognitive functions and dementia (Vassallo and Scerri, 2013; Kesse-Guyot et al., 2013; King, 2013; Lourida et al., 2013). There are also other important nutrients in this type of diet, such as minerals (e.g., Mg in the form of chlorophyll contained in green vegetables) (Feart et al., 2012) and polyphenolic substances that can protect the brain against the pathogenic effect of iron overload.
Polyphenols
Phenolic compounds and/or polyphenols constitute an important group of compounds occurring in plants, comprising at least 8,000 different known structures – including simple phenols, phenolic acids, coumarins and isocoumarins, naphthoquinones, xanthones, stilbenes, flavonoids, and lignins (Rossi et al., 2008; Dudonne et al., 2009). These natural substances exhibit a wide range of biological effects including antibacterial-, anti-inflammatory-, antiallergic-, antifungal-, antithrombotic-, and vasodilatory actions (Lin, 2011; Angeloni et al., 2012; Kumar et al., 2012). More importantly the substances are endowed with a capacity to scavenge hydroxyl radicals (Zielonka et al., 2003; Lipinski, 2011), which might explain their neuroprotective properties (Bieschke et al., 2010; Wang et al., 2012).
Only very few polyphenols can cross BBB, the most active in this respect being EGCG (Mandel et al., 2006; Meng et al., 2010; Palhano et al., 2013; Srividhya and Kalaiselvi, 2013). Other polyphenolic substances, such as curcumin (Baum and Ng, 2004; Ringman et al., 2005; Zhao et al., 2012) have to be first metabolized to small molecular-weight benzoic acid derivatives that are easily absorbable and can enter the brain circulation after crossing BBB (Kahle et al., 2007; Wang et al., 2012). These metabolites are known to scavenge hydroxyl radical (⋅OH) by virtue of their addition to double bonds with the formation of a corresponding hydroxyl derivative:
It should be emphasized, however, that in the case of polyphenols, only those with available ortho-position in their phenolic rings will effectively scavenge hydroxyl radicals. A classic example of such a mechanism, is the aromatic hydroxylation of salicylates this constitutes a principle for the quantitative determination of hydroxyl radicals in vivo (Grootveld and Halliwell, 1986; Ueno et al., 2006). Anti-inflammatory effects of aspirin and salicylates (Berk et al., 2013), as well as other small molecular-weight phenolic substances including melatonin (Srinivasan et al., 2005; Galano, 2011), ferulic, chlorogenic, and coumaric acids (Srinivasan et al., 2007), are very likely due to their ability to scavenge hydroxyl radicals by means of aromatic hydroxylation.
Conclusion
In the present paper we argue that the neurological disturbances in AD can be explained in terms of the hemostatic and hemorheologic effects of free iron in the cerebral circulation. It is known that the iron pool increases with age because no mechanism exists for its elimination from the human body. At a certain critical concentration trivalent iron ions will react with water to give rise to the biologically most reactive hydroxyl radicals that, in turn, unfold and randomly refold fibrinogen polypeptide chains, which results in the formation of a fibrin-like polymer (parafibrin). Such a polymer, by contrast to a thrombin-induced fibril, is remarkably resistant to degradation by the fibrinolytic enzyme system. In addition, soluble parafibrin interacts with hydrophobic patches on RBC membranes, forming huge aggregates that obstruct cerebral blood flow and thus impair oxygen delivery to the brain. In addition, parafibrin deposits residing for longer times in the brain tissue may assume the role of a foreign body, which in turn induces the state of chronic inflammation. Therefore, according to our hypothesis, AD is associated with chronic hypoxia caused by the gradual accumulation of parafibrin that is generated by a non-enzymatic pathway and is totally resistant to the proteolytic degradation. By contrast, fibrin clots formed in the brain by the action of thrombin, as in vascular dementia, are eventually removed by the powerful fibrinolytic enzyme system and may not be found in “pure” AD. We suggest that parafibrin, which is responsible for chronic brain hypoxia, is generated by the mechanisms other that the activation of blood coagulation. Therefore, vascular risk factors are not detectable in patients with pure AD (shown in Figure 1). In order to stop the vicious cycle it is necessary to break this pathologic chain of events by: (1) limiting the dietary supply of trivalent iron; (2) supplying sufficient quantities of natural iron chelating substances (e.g., certain polyphenols); (3) increasing the consumption of omega-3 fatty acids (e.g., fish oil) and other unsaturated aliphatic and/or aromatic substances that will scavenge hydroxyl radicals; and (4) increase the intake of Mg. It should be noted that these conditions are already met in the so-called Mediterranean diet, which is known to be associated with lower incidence of cardiovascular and AD (Lourida et al., 2013). Animal models of AD are frequently used to study the mechanisms underlying AD pathogenesis, genetic interactions with genes of interest, and environmental risk factors that cause sporadic AD, as well as to test the therapeutic effects of AD drug-candidates on neuropathology and cognitive function (Lee and Han, 2013). An important next step with regard to this hypothesis would be to investigate using, for example, animal models, the effect of Mg and iron chelating, and/or hydroxyl radical scavengers.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
We would like to thank Janette Bester for technical preparation of Alzheimer’s Disease erythrocytes.
References
Abe, S., Kirima, K., Tsuchiya, K., Okamoto, M., Hasegawa, T., Houchi, H., et al. (2004). The reaction rate of edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one (MCI-186)) with hydroxyl radical. Chem. Pharm. Bull. 52, 186–191. doi: 10.1248/cpb.52.186
Adamopoulos, C., Pitt, B., Sui, X., Love, T. E., Zannad, F., and Ahmed, A. (2009). Low serum magnesium and cardiovascular mortality in chronic heart failure: a propensity-matched study. Int. J. Cardiol. 136, 270–277. doi:10.1016/j.ijcard.2008.05.006
Adeghate, E., Donath, T., and Adem, A. (2013). Alzheimer disease and diabetes mellitus: do they have anything in common? Curr. Alzheimer Res. 10, 609–617. doi:10.2174/15672050113109990009
Adlard, P. A., and Bush, A. I. (2006). Metals and Alzheimer’s disease. J. Alzheimers Dis. 10, 145–163.
Ahluwalia, N., Genoux, A., Ferrieres, J., Perret, B., Carayol, M., Drouet, L., et al. (2010). Iron status is associated with carotid atherosclerotic plaques in middle-aged adults. J. Nutr. 140, 812–816. doi:10.3945/jn.109.110353
Ahn, H. J., Zamolodchikov, D., Cortes-Canteli, M., Norris, E. H., Glickman, J. F., and Strickland, S. (2010). Alzheimer’s disease peptide beta-amyloid interacts with fibrinogen and induces its oligomerization. Proc. Natl. Acad. Sci. U.S.A. 107, 21812–21817. doi:10.1073/pnas.1010373107
Albrecht, E., Kirkham, K. R., Liu, S. S., and Brull, R. (2013). Peri-operative intravenous administration of magnesium sulphate and postoperative pain: a meta-analysis. Anaesthesia 68, 79–90. doi:10.1111/j.1365-2044.2012.07335.x
Altamura, S., and Muckenthaler, M. U. (2009). Iron toxicity in diseases of aging: Alzheimer’s disease, Parkinson’s disease and atherosclerosis. J. Alzheimers Dis. 16, 879–895. doi:10.3233/JAD-2009-1010
Ames, W. A., Mcdonnell, N., and Potter, D. (1999). The effect of ionised magnesium on coagulation using thromboelastography. Anaesthesia 54, 999–1001. doi:10.1046/j.1365-2044.1999.00647.x
Amighi, J., Sabeti, S., Schlager, O., Mlekusch, W., Exner, M., Lalouschek, W., et al. (2004). Low serum magnesium predicts neurological events in patients with advanced atherosclerosis. Stroke 35, 22–27. doi:10.1161/01.STR.0000105928.95124.1F
Andresdottir, M. B., Sigfusson, N., Sigvaldason, H., and Gudnason, V. (2003). Erythrocyte sedimentation rate, an independent predictor of coronary heart disease in men and women: the Reykjavik Study. Am. J. Epidemiol. 158, 844–851. doi:10.1093/aje/kwg222
Angeloni, C., Pirola, L., Vauzour, D., and Maraldi, T. (2012). Dietary polyphenols and their effects on cell biochemistry and pathophysiology. Oxid. Med. Cell. Longev. 2012, 583901. doi:10.1155/2012/583901
Assadi, F. (2010). Hypomagnesemia: an evidence-based approach to clinical cases. Iran J. Kidney Dis. 4, 13–19.
Ayissi, V. B., Ebrahimi, A., and Schluesenner, H. (2013). Epigenetic effects of natural polyphenols: a focus on SIRT1-mediated mechanisms. Mol. Nutr. Food Res. doi:10.1002/mnfr.201300195
Barbagallo, M., Belvedere, M., Di Bella, G., and Dominguez, L. J. (2011). Altered ionized magnesium levels in mild-to-moderate Alzheimer’s disease. Magnes. Res. 24, S115–S121. doi:10.1684/mrh.2011.0287
Barnham, K. J., and Bush, A. I. (2008). Metals in Alzheimer’s and Parkinson’s diseases. Curr. Opin. Chem. Biol. 12, 222–228. doi:10.1016/j.cbpa.2008.02.019
Barr, J. D., Chauhan, A. K., Schaeffer, G. V., Hansen, J. K., and Motto, D. G. (2013). Red blood cells mediate the onset of thrombosis in the ferric chloride murine model. Blood 121, 3733–3741. doi:10.1182/blood-2012-11-468983
Baskurt, O. K., Yalcin, O., and Meiselman, H. J. (2004). Hemorheology and vascular control mechanisms. Clin. Hemorheol. Microcirc. 30, 169–178.
Baum, L., and Ng, A. (2004). Curcumin interaction with copper and iron suggests one possible mechanism of action in Alzheimer’s disease animal models. J. Alzheimers Dis. 6, 367–377; discussion 443–369.
Beard, J. L., and Connor, J. R. (2003). Iron status and neural functioning. Annu. Rev. Nutr. 23, 41–58. doi:10.1146/annurev.nutr.23.020102.075739
Benarroch, E. E. (2009). Brain iron homeostasis and neurodegenerative disease. Neurology 72, 1436–1440. doi:10.1212/WNL.0b013e3181a26b30
Berk, M., Dean, O., Drexhage, H., Mcneil, J. J., Moylan, S., Oneil, A., et al. (2013). Aspirin: a review of its neurobiological properties and therapeutic potential for mental illness. BMC Med. 11:74. doi:10.1186/1741-7015-11-74
Biernacki, E. (1897). Samoistna sedymentacja krwi, jako naukowa i praktyczno-kliniczna metoda badania (spontaneous sedimentation of red blood cells in clinical practice). Gazeta Lekarska 17, 962–964.
Bieschke, J., Russ, J., Friedrich, R. P., Ehrnhoefer, D. E., Wobst, H., Neugebauer, K., et al. (2010). EGCG remodels mature alpha-synuclein and amyloid-beta fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. U.S.A. 107, 7710–7715. doi:10.1073/pnas.0910723107
Breuer, W., Shvartsman, M., and Cabantchik, Z. I. (2008). Intracellular labile iron. Int. J. Biochem. Cell Biol. 40, 350–354. doi:10.1016/j.biocel.2007.03.010
Brewer, G. J. (2007). Iron and copper toxicity in diseases of aging, particularly atherosclerosis and Alzheimer’s disease. Exp. Biol. Med. (Maywood) 232, 323–335.
Casadesus, G., Smith, M. A., Zhu, X., Aliev, G., Cash, A. D., Honda, K., et al. (2004). Alzheimer disease: evidence for a central pathogenic role of iron-mediated reactive oxygen species. J. Alzheimers Dis. 6, 165–169.
Castellani, R. J., Moreira, P. I., Perry, G., and Zhu, X. (2012). The role of iron as a mediator of oxidative stress in Alzheimer disease. Biofactors 38, 133–138. doi:10.1002/biof.1010
Ceremuzynski, L., Gebalska, J., Wolk, R., and Makowska, E. (2000). Hypomagnesemia in heart failure with ventricular arrhythmias. Beneficial effects of magnesium supplementation. J. Intern. Med. 247, 78–86. doi:10.1046/j.1365-2796.2000.00585.x
Chakrabarti, S., Sinha, M., Thakurta, I. G., Banerjee, P., and Chattopadhyay, M. (2013). Oxidative stress and amyloid beta toxicity in Alzheimer’s disease: intervention in a complex relationship by antioxidants. Curr. Med. Chem.
Cheeseman, K. H., Beavis, A., and Esterbauer, H. (1988). Hydroxyl-radical-induced iron-catalysed degradation of 2-deoxyribose. Quantitative determination of malondialdehyde. Biochem. J. 252, 649–653.
Chetelat, G. (2013). Alzheimer disease: Abeta-independent processes-rethinking preclinical AD. Nat. Rev. Neurol. 9, 123–124. doi:10.1038/nrneurol.2013.21
Choi, J., Malakowsky, C. A., Talent, J. M., Conrad, C. C., and Gracy, R. W. (2002). Identification of oxidized plasma proteins in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 293, 1566–1570. doi:10.1016/S0006-291X(02)00420-5
Cilliler, A. E., Ozturk, S., and Ozbakir, S. (2007). Serum magnesium level and clinical deterioration in Alzheimer’s disease. Gerontology 53, 419–422. doi:10.1159/000110873
Ciriolo, M. R., De Martino, A., Lafavia, E., Rossi, L., Carri, M. T., and Rotilio, G. (2000). Cu,Zn-superoxide dismutase-dependent apoptosis induced by nitric oxide in neuronal cells. J. Biol. Chem. 275, 5065–5072. doi:10.1074/jbc.275.7.5065
Corica, F., Corsonello, A., Ientile, R., Cucinotta, D., Di Benedetto, A., Perticone, F., et al. (2006). Serum ionized magnesium levels in relation to metabolic syndrome in type 2 diabetic patients. J. Am. Coll. Nutr. 25, 210–215. doi:10.1080/07315724.2006.10719534
Corrigan, F. M., Van Rhijn, A., and Horrobin, D. F. (1991). Essential fatty acids in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 640, 250–252.
Cortes-Canteli, M., Paul, J., Norris, E. H., Bronstein, R., Ahn, H. J., Zamolodchikov, D., et al. (2010). Fibrinogen and beta-amyloid association alters thrombosis and fibrinolysis: a possible contributing factor to Alzheimer’s disease. Neuron 66, 695–709. doi:10.1016/j.neuron.2010.05.014
Craft, S. (2012). Alzheimer disease: insulin resistance and AD – extending the translational path. Nat. Rev. Neurol. 8, 360–362. doi:10.1038/nrneurol.2012.112
Dajas-Bailador, F. A., Martinez-Borges, A., Costa, G., Abin, J. A., Martignoni, E., Nappi, G., et al. (1998). Hydroxyl radical production in the substantia nigra after 6-hydroxydopamine and hypoxia-reoxygenation. Brain Res. 813, 18–25. doi:10.1016/S0006-8993(98)00989-5
Das, D., Luo, X., Singh, A., Gu, Y., Ghosh, S., Mukhopadhyay, C. K., et al. (2010). Paradoxical role of prion protein aggregates in redox-iron induced toxicity. PLoS ONE 5:e11420. doi:10.1371/journal.pone.0011420
Davalos, D., Ryu, J. K., Merlini, M., Baeten, K. M., Le Moan, N., Petersen, M. A., et al. (2012). Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat. Commun. 3, 1227. doi:10.1038/ncomms2230
de Lima, M. N., Polydoro, M., Laranja, D. C., Bonatto, F., Bromberg, E., Moreira, J. C., et al. (2005). Recognition memory impairment and brain oxidative stress induced by postnatal iron administration. Eur. J. Neurosci. 21, 2521–2528. doi:10.1111/j.1460-9568.2005.04083.x
Depalma, R. G., Hayes, V. W., Chow, B. K., Shamayeva, G., May, P. E., and Zacharski, L. R. (2010). Ferritin levels, inflammatory biomarkers, and mortality in peripheral arterial disease: a substudy of the Iron (Fe) and Atherosclerosis Study (FeAST) Trial. J. Vasc. Surg. 51, 1498–1503. doi:10.1016/j.jvs.2009.12.068
Diomedi, M., and Misaggi, G. (2013). Vascular contribution to Alzheimer disease: predictors of rapid progression. CNS Neurol. Disord. Drug Targets 12, 532–537.
Dudonne, S., Vitrac, X., Coutiere, P., Woillez, M., and Merillon, J. M. (2009). Comparative study of antioxidant properties and total phenolic content of 30 plant extracts of industrial interest using DPPH, ABTS, FRAP, SOD, and ORAC assays. J. Agric. Food Chem. 57, 1768–1774. doi:10.1021/jf803011r
Duley, L., Matar, H. E., Almerie, M. Q., and Hall, D. R. (2010). Alternative magnesium sulphate regimens for women with pre-eclampsia and eclampsia. Cochrane Database Syst. Rev. (8):CD007388. doi:10.1002/14651858.CD007388.pub2
Durlach, J. (1990). Magnesium depletion and pathogenesis of Alzheimer’s disease. Magnes. Res. 3, 217–218.
Egyud, L. G., and Lipinski, B. (1991). Significance of fibrin formation and dissolution in the pathogenesis and treatment of cancer. Med. Hypotheses 36, 336–340. doi:10.1016/0306-9877(91)90006-K
Elin, R. J. (1994). Magnesium: the fifth but forgotten electrolyte. Am. J. Clin. Pathol. 102, 616–622.
Eltzschig, H. K., and Carmeliet, P. (2011). Hypoxia and inflammation. N. Engl. J. Med. 364, 656–665. doi:10.1056/NEJMra0910283
Erikssen, G., Liestol, K., Bjornholt, J. V., Stormorken, H., Thaulow, E., and Erikssen, J. (2000). Erythrocyte sedimentation rate: a possible marker of atherosclerosis and a strong predictor of coronary heart disease mortality. Eur. Heart J. 21, 1614–1620. doi:10.1053/euhj.2000.2148
Fawcett, W. J., Haxby, E. J., and Male, D. A. (1999). Magnesium: physiology and pharmacology. Br. J. Anaesth. 83, 302–320. doi:10.1093/bja/83.2.302
Feart, C., Alles, B., Merle, B., Samieri, C., and Barberger-Gateau, P. (2012). Adherence to a Mediterranean diet and energy, macro-, and micronutrient intakes in older persons. J. Physiol. Biochem. 68, 691–700. doi:10.1007/s13105-012-0190-y
Feart, C., Samieri, C., and Barberger-Gateau, P. (2010). Mediterranean diet and cognitive function in older adults. Curr. Opin. Clin. Nutr. Metab. Care 13, 14–18. doi:10.1097/MCO.0b013e3283331fe4
Fiala, M., Liu, Q. N., Sayre, J., Pop, V., Brahmandam, V., Graves, M. C., et al. (2002). Cyclooxygenase-2-positive macrophages infiltrate the Alzheimer’s disease brain and damage the blood-brain barrier. Eur. J. Clin. Invest. 32, 360–371. doi:10.1046/j.1365-2362.2002.00994.x
Galano, A. (2011). On the direct scavenging activity of melatonin towards hydroxyl and a series of peroxyl radicals. Phys. Chem. Chem. Phys. 13, 7178–7188. doi:10.1039/c0cp02801k
Grammas, P., Tripathy, D., Sanchez, A., Yin, X., and Luo, J. (2011). Brain microvasculature and hypoxia-related proteins in Alzheimer’s disease. Int. J. Clin. Exp. Pathol. 4, 616–627.
Grootveld, M., and Halliwell, B. (1986). Aromatic hydroxylation as a potential measure of hydroxyl-radical formation in vivo. Identification of hydroxylated derivatives of salicylate in human body fluids. Biochem. J. 237, 499–504.
Gu, Y., Luchsinger, J. A., Stern, Y., and Scarmeas, N. (2010). Mediterranean diet, inflammatory and metabolic biomarkers, and risk of Alzheimer’s disease. J. Alzheimers Dis. 22, 483–492. doi:10.3233/JAD-2010-100897
Guerrero-Romero, F., Tamez-Perez, H. E., Gonzalez-Gonzalez, G., Salinas-Martinez, A. M., Montes-Villarreal, J., Trevino-Ortiz, J. H., et al. (2004). Oral magnesium supplementation improves insulin sensitivity in non-diabetic subjects with insulin resistance. A double-blind placebo-controlled randomized trial. Diabetes Metab. 30, 253–258. doi:10.1016/S1262-3636(07)70116-7
Gupta, A., Watkins, A., Thomas, P., Majer, R., Habubi, N., Morris, G., et al. (2005). Coagulation and inflammatory markers in Alzheimer’s and vascular dementia. Int. J. Clin. Pract. 59, 52–57. doi:10.1111/j.1742-1241.2004.00143.x
Gveric, D., Hanemaaijer, R., Newcombe, J., Van Lent, N. A., Sier, C. F., and Cuzner, M. L. (2001). Plasminogen activators in multiple sclerosis lesions: implications for the inflammatory response and axonal damage. Brain 124, 1978–1988. doi:10.1093/brain/124.10.1978
Hahalis, G., Kalogeropoulos, A., Terzis, G., Tselepis, A. D., Kourakli, A., Mylona, P., et al. (2011). Premature atherosclerosis in non-transfusion-dependent beta-thalassemia intermedia. Cardiology 118, 159–163. doi:10.1159/000327997
Houston, M. (2011). The role of magnesium in hypertension and cardiovascular disease. J. Clin. Hypertens (Greenwich) 13, 843–847. doi:10.1111/j.1751-7176.2011.00538.x
Hu, N., Yu, J. T., Tan, L., Wang, Y. L., Sun, L., and Tan, L. (2013). Nutrition and the risk of Alzheimer’s disease. Biomed. Res. Int. 2013, 524820. doi:10.1155/2013/524820
Huang, J. H., Lu, Y. F., Cheng, F. C., Lee, J. N., and Tsai, L. C. (2012). Correlation of magnesium intake with metabolic parameters, depression and physical activity in elderly type 2 diabetes patients: a cross-sectional study. Nutr. J. 11, 41. doi:10.1186/1475-2891-11-41
Hultman, K., Strickland, S., and Norris, E. H. (2013). The APOE varepsilon4/varepsilon4 genotype potentiates vascular fibrin(ogen) deposition in amyloid-laden vessels in the brains of Alzheimer’s disease patients. J. Cereb. Blood Flow Metab. 33, 1251–1258. doi:10.1038/jcbfm.2013.76
Huntsman, R. G., Hurn, B. A., and Lehmann, H. (1960). Observations on the effect of magnesium on blood coagulation. J. Clin. Pathol. 13, 99–101. doi:10.1136/jcp.13.2.99
Inoue, A., Koh, C. S., Yamazaki, M., Yanagisawa, N., Ishihara, Y., and Kim, B. S. (1997). Fibrin deposition in the central nervous system correlates with the degree of Theiler’s murine encephalomyelitis virus-induced demyelinating disease. J. Neuroimmunol. 77, 185–194. doi:10.1016/S0165-5728(97)00072-6
Issa, A. M., Mojica, W. A., Morton, S. C., Traina, S., Newberry, S. J., Hilton, L. G., et al. (2006). The efficacy of omega-3 fatty acids on cognitive function in aging and dementia: a systematic review. Dement. Geriatr. Cogn. Disord. 21, 88–96. doi:10.1159/000090224
Kahle, K., Huemmer, W., Kempf, M., Scheppach, W., Erk, T., and Richling, E. (2007). Polyphenols are intensively metabolized in the human gastrointestinal tract after apple juice consumption. J. Agric. Food Chem. 55, 10605–10614. doi:10.1021/jf071942r
Kalaria, R. N. (2003). Vascular factors in Alzheimer’s disease. Int. Psychogeriatr. 15(Suppl. 1), 47–52. doi:10.1017/S1041610203008950
Kaur, J., Zhao, Z., Klein, G. M., Lo, E. H., and Buchan, A. M. (2004). The neurotoxicity of tissue plasminogen activator? J. Cereb. Blood Flow Metab. 24, 945–963. doi:10.1097/01.WCB.0000137868.50767.E8
Ke, Y., and Ming Qian, Z. (2003). Iron misregulation in the brain: a primary cause of neurodegenerative disorders. Lancet Neurol. 2, 246–253. doi:10.1016/S1474-4422(03)00353-3
Kell, D. B. (2010). Towards a unifying, systems biology understanding of large-scale cellular death and destruction caused by poorly liganded iron: Parkinson’s, Huntington’s, Alzheimer’s, prions, bactericides, chemical toxicology and others as examples. Arch. Toxicol. 84, 825–889. doi:10.1007/s00204-010-0577-x
Kesse-Guyot, E., Andreeva, V. A., Lassale, C., Ferry, M., Jeandel, C., Hercberg, S., et al. (2013). Mediterranean diet and cognitive function: a French study. Am. J. Clin. Nutr. 97, 369–376. doi:10.3945/ajcn.112.047993
Kiechl, S., Willeit, J., Egger, G., Poewe, W., and Oberhollenzer, F. (1997). Body iron stores and the risk of carotid atherosclerosis: prospective results from the Bruneck study. Circulation 96, 3300–3307. doi:10.1161/01.CIR.96.10.3300
Kim, D. J., Xun, P., Liu, K., Loria, C., Yokota, K., Jacobs, D. R. Jr., et al. (2010). Magnesium intake in relation to systemic inflammation, insulin resistance, and the incidence of diabetes. Diabetes Care 33, 2604–2610. doi:10.2337/dc10-0994
King, A. (2013). Prevention: the benefits of a Mediterranean diet. Nat. Rev. Cardiol. 10, 239. doi:10.1038/nrcardio.2013.36
Kling, M. A., Trojanowski, J. Q., Wolk, D. A., Lee, V. M. A., and Arnold, S. E. (2013). Vascular disease and dementias: paradigm shifts to drive research in new directions. Alzheimers Dement. 9, 76–92. doi:10.1016/j.jalz.2012.02.007
Kovacic, J. C., and Fuster, V. (2012). Atherosclerotic risk factors, vascular cognitive impairment, and Alzheimer disease. Mt. Sinai J. Med. 79, 664–673. doi:10.1002/msj.21347
Krstic, D., and Knuesel, I. (2013). Deciphering the mechanism underlying late-onsetalzheimer disease. Nat. Rev. Neurol. 9, 25–34. doi:10.1038/nrneurol.2012.236
Kruszewski, M. (2003). Labile iron pool: the main determinant of cellular response to oxidative stress. Mutat. Res. 531, 81–92. doi:10.1016/j.mrfmmm.2003.08.004
Kumar, S., Okello, E. J., and Harris, J. R. (2012). Experimental inhibition of fibrillogenesis and neurotoxicity by amyloid-beta (Abeta) and other disease-related peptides/proteins by plant extracts and herbal compounds. Subcell. Biochem. 65, 295–326. doi:10.1007/978-94-007-5416-4_13
Kupetsky-Rincon, E. A., and Uitto, J. (2012). Magnesium: novel applications in cardiovascular disease – a review of the literature. Ann. Nutr. Metab. 61, 102–110. doi:10.1159/000339380
Kwan, A. H., Winefield, R. D., Sunde, M., Matthews, J. M., Haverkamp, R. G., Templeton, M. D., et al. (2006). Structural basis for rodlet assembly in fungal hydrophobins. Proc. Natl. Acad. Sci. U.S.A. 103, 3621–3626. doi:10.1073/pnas.0505704103
Larsson, S. C., Orsini, N., and Wolk, A. (2012). Dietary magnesium intake and risk of stroke: a meta-analysis of prospective studies. Am. J. Clin. Nutr. 95, 362–366. doi:10.3945/ajcn.111.022376
Lee, J. E., and Han, P. L. (2013). An update of animal models of Alzheimer disease with a reevaluation of plaque depositions. Exp. Neurobiol. 22, 84–95. doi:10.5607/en.2013.22.2.84
Limaye, C. S., Londhey, V. A., Nadkart, M. Y., and Borges, N. E. (2011). Hypomagnesemia in critically ill medical patients. J. Assoc. Physicians India 59, 19–22.
Lin, B. (2011). Polyphenols and neuroprotection against ischemia and neurodegeneration. Mini Rev. Med. Chem. 11, 1222–1238. doi:10.2174/138955711804586784
Lipinska, I., Lipinski, B., Gurewich, V., and Hoffmann, K. D. (1976). Fibrinogen heterogeneity in cancer, in occlusive vascular disease, and after surgical procedures. Am. J. Clin. Pathol. 66, 958–966.
Lipinski, B. (2001). Pathophysiology of oxidative stress in diabetes mellitus. J. Diabetes Complicat. 15, 203–210. doi:10.1016/S1056-8727(01)00143-X
Lipinski, B. (2011). Hydroxyl radical and its scavengers in health and disease. Oxid. Med. Cell. Longev. 2011, 809696. doi:10.1155/2011/809696
Lipinski, B., and Pretorius, E. (2012). Novel pathway of ironinduced blood coagulation: implications for diabetes mellitus and its complications. Pol. Arch. Med. Wewn. 122, 115–122.
Lipinski, B., and Pretorius, E. (2013). Iron-induced fibrin in cardiovascular disease. Curr. Neurovasc. Res. 10, 269–274. doi:10.2174/15672026113109990016
Lipinski, B., and Sajdel-Sulkowska, E. M. (2006). New insight into Alzheimer disease: demonstration of fibrin(ogen)-serum albumin insoluble deposits in brain tissue. Alzheimer Dis. Assoc. Disord. 20, 323–326. doi:10.1097/01.wad.0000213844.21001.a2
Lipinski, B., Worowski, K., Mysliwiec, M., and Farbiszewski, R. (1969). Erythrocyte sedimentation and soluble fibrin monomer complexes. Thromb. Diath. Haemorrh. 21, 196–202.
Lonergan, P. E., Martin, D. S., Horrobin, D. F., and Lynch, M. A. (2002). Neuroprotective effect of eicosapentaenoic acid in hippocampus of rats exposed to gamma-irradiation. J. Biol. Chem. 277, 20804–20811. doi:10.1074/jbc.M202387200
Lourida, I., Soni, M., Thompson-Coon, J., Purandare, N., Lang, I. A., Ukoumunne, O. C., et al. (2013). Mediterranean diet, cognitive function, and dementia: a systematic review. Epidemiology 24, 479–489. doi:10.1097/EDE.0b013e3182944410
Lukiw, W. J., and Bazan, N. G. (2008). Docosahexaenoic acid and the aging brain. J. Nutr. 138, 2510–2514. doi:10.3945/jn.108.096016
Mandel, S., Amit, T., Reznichenko, L., Weinreb, O., and Youdim, M. B. (2006). Green tea catechins as brain-permeable, natural iron chelators-antioxidants for the treatment of neurodegenerative disorders. Mol. Nutr. Food Res. 50, 229–234. doi:10.1002/mnfr.200500156
Markesbery, W. R., and Carney, J. M. (1999). Oxidative alterations in Alzheimer’s disease. Brain Pathol. 9, 133–146. doi:10.1111/j.1750-3639.1999.tb00215.x
Marszalek, J. R., and Lodish, H. F. (2005). Docosahexaenoic acid, fatty acid-interacting proteins, and neuronal function: breastmilk and fish are good for you. Annu. Rev. Cell Dev. Biol. 21, 633–657. doi:10.1146/annurev.cellbio.21.122303.120624
Mazur, A., Maier, J. A., Rock, E., Gueux, E., Nowacki, W., and Rayssiguier, Y. (2007). Magnesium and the inflammatory response: potential physiopathological implications. Arch. Biochem. Biophys. 458, 48–56. doi:10.1016/j.abb.2006.03.031
McDonald, S. D., Lutsiv, O., Dzaja, N., and Duley, L. (2012). A systematic review of maternal and infant outcomes following magnesium sulfate for pre-eclampsia/eclampsia in real-world use. Int. J. Gynaecol. Obstet. 118, 90–96. doi:10.1016/j.ijgo.2012.01.028
McIntosh, T. K., Vink, R., Yamakami, I., and Faden, A. I. (1989). Magnesium protects against neurological deficit after brain injury. Brain Res. 482, 252–260. doi:10.1016/0006-8993(89)91188-8
Melcher, K. (2004). New chemical crosslinking methods for the identification of transient protein-protein interactions with multiprotein complexes. Curr. Protein Pept. Sci. 5, 287–296. doi:10.2174/1389203043379701
Meng, F., Abedini, A., Plesner, A., Verchere, C. B., and Raleigh, D. P. (2010). The flavanol (-)-epigallocatechin 3-gallate inhibits amyloid formation by islet amyloid polypeptide, disaggregates amyloid fibrils, and protects cultured cells against IAPP-induced toxicity. Biochemistry 49, 8127–8133. doi:10.1021/bi100939a
Merkle, D. L., Cheng, C. H., Castellino, F. J., and Chibber, B. A. (1996). Modulation of fibrin assembly and polymerization by the beta-amyloid of Alzheimer’s disease. Blood Coagul. Fibrinolysis 7, 650–658. doi:10.1097/00001721-199609000-00011
Merono, T., Rosso, L. G., Sorroche, P., Boero, L., Arbelbide, J., and Brites, F. (2011). High risk of cardiovascular disease in iron overload patients. Eur. J. Clin. Invest. 41, 479–486. doi:10.1111/j.1365-2362.2010.02429.x
Milte, C. M., Sinn, N., and Howe, P. R. (2009). Polyunsaturated fatty acid status in attention deficit hyperactivity disorder, depression, and Alzheimer’s disease: towards an omega-3 index for mental health? Nutr. Rev. 67, 573–590. doi:10.1111/j.1753-4887.2009.00229.x
Mohanty, J. G., Eckley, D. M., Williamson, J. D., Launer, L. J., and Rifkind, J. M. (2008). Do red blood cell-beta-amyloid interactions alter oxygen delivery in Alzheimer’s disease? Adv. Exp. Med. Biol. 614, 29–35. doi:10.1007/978-0-387-74911-2_4
Moreira, P. I., Honda, K., Liu, Q., Santos, M. S., Oliveira, C. R., Aliev, G., et al. (2005). Oxidative stress: the old enemy in Alzheimer’s disease pathophysiology. Curr. Alzheimer Res. 2, 403–408. doi:10.2174/156720505774330537
Morris, M. C., Evans, D. A., Bienias, J. L., Tangney, C. C., Bennett, D. A., Wilson, R. S., et al. (2003). Consumption of fish and n-3 fatty acids and risk of incident Alzheimer disease. Arch. Neurol. 60, 940–946. doi:10.1001/archneur.60.7.940
Muir, K. W., Lees, K. R., Ford, I., and Davis, S. (2004). Magnesium for acute stroke (Intravenous Magnesium Efficacy in Stroke trial): randomised controlled trial. Lancet 363, 439–445. doi:10.1016/S0140-6736(04)15490-1
Multhaup, K. S. (1995). Aging, source, and decision criteria: when false fame errors do and do not occur. Psychol. Aging 10, 492–497. doi:10.1037/0882-7974.10.3.492
Muroi, C., Burkhardt, J. K., Hugelshofer, M., Seule, M., Mishima, K., and Keller, E. (2012). Magnesium and the inflammatory response: potential pathophysiological implications in the management of patients with aneurysmal subarachnoid hemorrhage? Magnes. Res. 25, 64–71.
Mussoni, L., Sironi, L., Tedeschi, L., Calvio, A. M., Colli, S., and Tremoli, E. (2001). Magnesium inhibits arterial thrombi after vascular injury in rat: in vivo impairment of coagulation. Thromb. Haemost. 86, 1292–1295.
Nagy, E., Eaton, J. W., Jeney, V., Soares, M. P., Varga, Z., Galajda, Z., et al. (2010). Red cells, hemoglobin, heme, iron, and atherogenesis. Arterioscler. Thromb. Vasc. Biol. 30, 1347–1353. doi:10.1161/ATVBAHA.110.206433
Nakajima, Y., Masaoka, N., Hayakawa, Y., Watanabe, M., Nagaishi, M., Sakata, H., et al. (2006). The production of hydroxyl radicals in the fetal lamb brain resulting from occlusion of the umbilical circulation and the transplacental effect of MCI-186 to inhibit hydroxyl radical production. Pediatr. Res. 59, 216–220. doi:10.1203/01.pdr.0000196373.52086.c2
Natali, A., L’Abbate, A., and Ferrannini, E. (2003). Erythrocyte sedimentation rate, coronary atherosclerosis, and cardiac mortality. Eur. Heart J. 24, 639–648. doi:10.1016/S0195-668X(02)00741-8
Ninomia, T., Wang, L., Kumar, S. R., Kim, A., and Zlokovic, B. V. (2000). Brain injury and cerebrovascular fibrin deposition correlate with reduced antithrombotic brain capillary functions in a hypertensive stroke model. J. Cereb. Blood Flow Metab. 20, 998–1009. doi:10.1097/00004647-200006000-00012
Nunomura, A., Perry, G., Aliev, G., Hirai, K., Takeda, A., Balraj, E. K., et al. (2001). Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 60, 759–767.
Okereke, E., Ahonsi, B., Tukur, J., Ishaku, S. M., and Oginni, A. B. (2012). Benefits of using magnesium sulphate (MgSO(4)) for eclampsia management and maternal mortality reduction: lessons from Kano State in Northern Nigeria. BMC Res. Notes 5:421. doi:10.1186/1756-0500-5-421
Palhano, F. L., Lee, J., Grimster, N. P., and Kelly, J. W. (2013). Toward the molecular mechanism(s) by which EGCG treatment remodels mature amyloid fibrils. J. Am. Chem. Soc. 135, 7503–7510. doi:10.1021/ja3115696
Palmblad, J. (2010). The acute inflammatory reaction: the legacy of Mechnikov. J. Intern. Med. 268, 35–37.
Paul, J., Strickland, S., and Melchor, J. P. (2007). Fibrin deposition accelerates neurovascular damage and neuroinflammation in mouse models of Alzheimer’s disease. J. Exp. Med. 204, 1999–2008. doi:10.1084/jem.20070304
Peers, C., Pearson, H. A., and Boyle, J. P. (2007). Hypoxia and Alzheimer’s disease. Essays Biochem. 43, 153–164. doi:10.1042/BSE0430153
Pimplikar, S. W. (2009). Reassessing the amyloid cascade hypothesis of Alzheimer’s disease. Int. J. Biochem. Cell Biol. 41, 1261–1268. doi:10.1016/j.biocel.2008.12.015
Pretorius, E., Bester, J., Vermeulen, N., and Lipinski, B. (2013a). Oxidation inhibits iron-induced blood coagulation. Curr. Drug Targets 14, 13–19. doi:10.2174/138945013804806541
Pretorius, E., Vermeulen, N., Bester, J., Lipinski, B., and Kell, D. B. (2013b). A novel method for assessing the role of iron and its functional chelation in fibrin fibril formation: the use of scanning electron microscopy. Toxicol. Mech. Methods 23, 352–359. doi:10.3109/15376516.2012.762082
Pretorius, E., and Lipinski, B. (2013). Iron alters red blood cell morphology. Blood 121, 9. doi:10.1182/blood-2012-09-454793
Ravn, H. B., Kristensen, S. D., Hjortdal, V. E., Thygesen, K., and Husted, S. E. (1997). Early administration of intravenous magnesium inhibits arterial thrombus formation. Arterioscler. Thromb. Vasc. Biol. 17, 3620–3625. doi:10.1161/01.ATV.17.12.3620
Ray, B., and Lahiri, D. K. (2009). Neuroinflammation in Alzheimer’s disease: different molecular targets and potential therapeutic agents including curcumin. Curr. Opin. Pharmacol. 9, 434–444. doi:10.1016/j.coph.2009.06.012
Reffelmann, T., Ittermann, T., Dorr, M., Volzke, H., Reinthaler, M., Petersmann, A., et al. (2011). Low serum magnesium concentrations predict cardiovascular and all-cause mortality. Atherosclerosis 219, 280–284. doi:10.1016/j.atherosclerosis.2011.05.038
Ringman, J. M., Frautschy, S. A., Cole, G. M., Masterman, D. L., and Cummings, J. L. (2005). A potential role of the curry spice curcumin in Alzheimer’s disease. Curr. Alzheimer Res. 2, 131–136. doi:10.2174/1567205053585882
Robinson, D., Bucci, J., Fenn, H., Miller, T., Tinklenberg, J., and Yesavage, J. A. (1995). Erythrocyte sedimentation rate in Alzheimer’s dementia. J. Am. Geriatr. Soc. 43, 1177–1178.
Rodriguez-Moran, M., and Guerrero-Romero, F. (2003). Oral magnesium supplementation improves insulin sensitivity and metabolic control in type 2 diabetic subjects: a randomized double-blind controlled trial. Diabetes Care 26, 1147–1152. doi:10.2337/diacare.26.4.1147
Romani, A. M. (2013). Magnesium homeostasis in mammalian cells. Met. Ions Life Sci. 12, 69–118. doi:10.1007/978-94-007-5561-1_4
Rossi, L., Mazzitelli, S., Arciello, M., Capo, C. R., and Rotilio, G. (2008). Benefits from dietary polyphenols for brain aging and Alzheimer’s disease. Neurochem. Res. 33, 2390–2400. doi:10.1007/s11064-008-9696-7
Rowe, W. J. (2012). Correcting magnesium deficiencies may prolong life. Clin. Interv. Aging 7, 51–54. doi:10.2147/CIA.S28768
Saini, A. K., Kumar, H. S. A., and Sharma, S. S. (2007). Preventive and curative effect of edaravone on nerve functions and oxidative stress in experimental diabetic neuropathy. Eur. J. Pharmacol. 568, 164–172. doi:10.1016/j.ejphar.2007.04.016
Scarmeas, N., Stern, Y., Mayeux, R., and Luchsinger, J. A. (2006). Mediterranean diet, Alzheimer disease, and vascular mediation. Arch. Neurol. 63, 1709–1717. doi:10.1001/archneur.63.12.noc60109
Schneider, J. A., Arvanitakis, Z., Bang, W., and Bennett, D. A. (2007). Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 69, 2197–2204. doi:10.1212/01.wnl.0000271090.28148.24
Sery, O., Povova, J., Misek, I., Pesak, L., and Janout, V. (2013). Molecular mechanisms of neuropathological changes in Alzheimer’s disease: a review. Folia Neuropathol. 51, 1–9. doi:10.5114/fn.2013.34190
Silvestri, L., and Camaschella, C. (2008). A potential pathogenetic role of iron in Alzheimer’s disease. J. Cell. Mol. Med. 12, 1548–1550. doi:10.1111/j.1582-4934.2008.00356.x
Sinn, N., Milte, C., and Howe, P. R. (2010). Oiling the brain: a review of randomized controlled trials of omega-3 fatty acids in psychopathology across the lifespan. Nutrients 2, 128–170. doi:10.3390/nu2020128
Skaper, S. D. (2012). Alzheimer’s disease and amyloid: culprit or coincidence? Int. Rev. Neurobiol. 102, 277–316. doi:10.1016/B978-0-12-386986-9.00011-9
Slutsky, I., Abumaria, N., Wu, L. J., Huang, C., Zhang, L., Li, B., et al. (2010). Enhancement of learning and memory by elevating brain magnesium. Neuron 65, 165–177. doi:10.1016/j.neuron.2009.12.026
Smith, M. A., Zhu, X., Tabaton, M., Liu, G., Mckeel, D. W. Jr., Cohen, M. L., et al. (2010). Increased iron and free radical generation in preclinical Alzheimer disease and mild cognitive impairment. J. Alzheimers Dis. 19, 363–372. doi:10.3233/JAD-2010-1239
Song, C., Leonard, B. E., and Horrobin, D. F. (2004). Dietary ethyl-eicosapentaenoic acid but not soybean oil reverses central interleukin-1-induced changes in behavior, corticosterone and immune response in rats. Stress 7, 43–54. doi:10.1080/10253890410001667188
Squier, T. C. (2001). Oxidative stress and protein aggregation during biological aging. Exp. Gerontol. 36, 1539–1550. doi:10.1016/S0531-5565(01)00139-5
Squitti, R. (2012). Metals in Alzheimer’s disease: a systemic perspective. Front. Biosci. 17:451–472. doi:10.2741/3938
Srinivasan, M., Sudheer, A. R., and Menon, V. P. (2007). Ferulic acid: therapeutic potential through its antioxidant property. J. Clin. Biochem. Nutr. 40, 92–100. doi:10.3164/jcbn.40.92
Srinivasan, V., Pandi-Perumal, S. R., Maestroni, G. J., Esquifino, A. I., Hardeland, R., and Cardinali, D. P. (2005). Role of melatonin in neurodegenerative diseases. Neurotox. Res. 7, 293–318. doi:10.1007/BF03033887
Srividhya, R., Jyothilakshmi, V., Arulmathi, K., Senthilkumaran, V., and Kalaiselvi, P. (2008). Attenuation of senescence-induced oxidative exacerbations in aged rat brain by (-)-epigallocatechin-3-gallate. Int. J. Dev. Neurosci. 26, 217–223. doi:10.1016/j.ijdevneu.2007.12.003
Srividhya, R., and Kalaiselvi, P. (2013). Neuroprotective potential of epigallo catechin-3-gallate in PC-12 cells. Neurochem. Res. 38, 486–493. doi:10.1007/s11064-012-0940-9
Stankiewicz, J. M., and Brass, S. D. (2009). Role of iron in neurotoxicity: a cause for concern in the elderly? Curr. Opin. Clin. Nutr. Metab. Care 12, 22–29. doi:10.1097/MCO.0b013e32831ba07c
Steinhubl, S. R. (2008). Why have antioxidants failed in clinical trials? Am. J. Cardiol. 101, 14D–19D. doi:10.1016/j.amjcard.2008.02.003
Stern, R. A., Trojanowski, J. Q., and Lee, V. M. (1990). Antibodies to the beta-amyloid peptide cross-react with conformational epitopes in human fibrinogen subunits from peripheral blood. FEBS Lett. 264, 43–47. doi:10.1016/0014-5793(90)80760-G
Summers, W. K. (2004). Alzheimer’s disease, oxidative injury, and cytokines. J. Alzheimers Dis. 6, 651–657; discussion 673–681.
Szweda, P. A., Friguet, B., and Szweda, L. I. (2002). Proteolysis, free radicals, and aging. Free Radic. Biol. Med. 33, 29–36. doi:10.1016/S0891-5849(02)00837-7
Tateishi, N., Suzuki, Y., Cicha, I., and Maeda, N. (2001). O(2) release from erythrocytes flowing in a narrow O(2)-permeable tube: effects of erythrocyte aggregation. Am. J. Physiol. Heart Circ. Physiol. 281, H448–H456.
Thaipisuttikul, P., and Galvin, J. E. (2012). Use of medical foods and nutritional approaches in the treatment of Alzheimer’s disease. Clin. Pract. (Lond.) 9, 199–209. doi:10.2217/cpr.12.3
Thomas, M., and Jankovic, J. (2004). Neurodegenerative disease and iron storage in the brain. Curr. Opin. Neurol. 17, 437–442. doi:10.1097/01.wco.0000137534.61244.d1
Tripathy, D., Sanchez, A., Yin, X., Luo, J., Martinez, J., and Grammas, P. (2013). Thrombin, a mediator of cerebrovascular inflammation in AD and hypoxia. Front. Aging Neurosci. 5:19. doi:10.3389/fnagi.2013.00019
Tully, A. M., Roche, H. M., Doyle, R., Fallon, C., Bruce, I., Lawlor, B., et al. (2003). Low serum cholesteryl ester-docosahexaenoic acid levels in Alzheimer’s disease: a case-control study. Br. J. Nutr. 89, 483–489. doi:10.1079/BJN2002804
Tuppo, E. E., and Arias, H. R. (2005). The role of inflammation in Alzheimer’s disease. Int. J. Biochem. Cell Biol. 37, 289–305. doi:10.1016/j.biocel.2004.07.009
Ueno, S., Kashimoto, T., Susa, N., Wada, K., Ito, N., Takeda-Homma, S., et al. (2006). Estimation of hydroxyl radical generation by salicylate hydroxylation method in multiple organs of mice exposed to whole-body X-ray irradiation. Free Radic. Res. 40, 944–951. doi:10.1080/10715760600817963
van Oijen, M., Witteman, J. C., Hofman, A., Koudstaal, P. J., and Breteler, M. M. (2005). Fibrinogen is associated with an increased risk of Alzheimer disease and vascular dementia. Stroke 36, 2637–2641. doi:10.1161/01.STR.0000189721.31432.26
van Oss, C. J. (1990). Surface properties of fibrinogen and fibrin. J. Protein Chem. 9, 487–491. doi:10.1007/BF01024625
Vassallo, N., and Scerri, C. (2013). Mediterranean diet and dementia of the Alzheimer type. Curr. Aging Sci. 6, 150–162.
Vignini, A., Giulietti, A., Nanetti, L., Raffaelli, F., Giusti, L., Mazzanti, L., et al. (2013). Alzheimer’s disease and diabetes: new insights and unifying therapies. Curr. Diabetes Rev. 9, 218–227. doi:10.2174/1573399811309030003
Wang, J., Ferruzzi, M. G., Ho, L., Blount, J., Janle, E. M., Gong, B., et al. (2012). Brain-targeted proanthocyanidin metabolites for Alzheimer’s disease treatment. J. Neurosci. 32, 5144–5150. doi:10.1523/JNEUROSCI.6437-11.2012
Watanabe, M., Suga, T., Yoshioka, K., Doi, M., Chiba, M., and Ogawa, A. (1996). [A case of acute occlusion of left middle cerebral artery due to an embolus treated successfully with percutaneous transluminal angioplasty]. No Shinkei Geka 24, 1027–1032.
Weglicki, W. B. (2012). Hypomagnesemia and inflammation: clinical and basic aspects. Annu. Rev. Nutr. 32, 55–71. doi:10.1146/annurev-nutr-071811-150656
Woolf, N. (1961). The distribution of fibrin within the aortic intima. An immunohistochemical study. Am. J. Pathol. 39, 521–532.
Woollard, K. J., Sturgeon, S., Chin-Dusting, J. P., Salem, H. H., and Jackson, S. P. (2009). Erythrocyte hemolysis and hemoglobin oxidation promote ferric chloride-induced vascular injury. J. Biol. Chem. 284, 13110–13118. doi:10.1074/jbc.M809095200
Wyss-Coray, T., and Rogers, J. (2012). Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb. Perspect. Med. 2, a006346. doi:10.1101/cshperspect.a006346
Yan, Y., Gong, K., Ma, T., Zhang, L., Zhao, N., Zhang, X., et al. (2012). Protective effect of edaravone against Alzheimer’s disease-relevant insults in neuroblastoma N2a cells. Neurosci. Lett. 531, 160–165. doi:10.1016/j.neulet.2012.10.043
Yarchoan, M., Xie, S. X., Kling, M. A., Toledo, J. B., Wolk, D. A., Lee, E. B., et al. (2012). Cerebrovascular atherosclerosis correlates with Alzheimer pathology in neurodegenerative dementias. Brain 135, 3749–3756. doi:10.1093/brain/aws271
Yavin, E., Brand, A., and Green, P. (2002). Docosahexaenoic acid abundance in the brain: a biodevice to combat oxidative stress. Nutr. Neurosci. 5, 149–157. doi:10.1080/10284150290003159
Yehuda, S., Rabinovtz, S., Carasso, R. L., and Mostofsky, D. I. (1996). Essential fatty acids preparation (SR-3) improves Alzheimer’s patients quality of life. Int. J. Neurosci. 87, 141–149. doi:10.3109/00207459609070833
Zamolodchikov, D., and Strickland, S. (2012). Abeta delays fibrin clot lysis by altering fibrin structure and attenuating plasminogen binding to fibrin. Blood 119, 3342–3351. doi:10.1182/blood-2011-11-389668
Zhao, L. N., Chiu, S. W., Benoit, J., Chew, L. Y., and Mu, Y. (2012). The effect of curcumin on the stability of Abeta dimers. J. Phys. Chem. B 116, 7428–7435. doi:10.1021/jp3034209
Zheng, W., and Monnot, A. D. (2012). Regulation of brain iron and copper homeostasis by brain barrier systems: implication in neurodegenerative diseases. Pharmacol. Ther. 133, 177–188. doi:10.1016/j.pharmthera.2011.10.006
Keywords: Alzheimer’s disease, iron, fibrin, red blood cells, magnesium
Citation: Lipinski B and Pretorius E (2013) The role of iron-induced fibrin in the pathogenesis of Alzheimer’s disease and the protective role of magnesium. Front. Hum. Neurosci. 7:735. doi: 10.3389/fnhum.2013.00735
Received: 18 June 2013; Accepted: 14 October 2013;
Published online: 29 October 2013.
Edited by:
Srikantan S. Nagarajan, University of California San Francisco, USAReviewed by:
Keith Vossel, University of California San Francisco, USAKamalini Gayathree Ranasinghe, University of California San Francisco, USA
Copyright: © 2013 Lipinski and Pretorius. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Etheresia Pretorius, Department of Physiology, Faculty of Health Sciences, University of Pretoria, Private Bag x323, Arcadia 0007, South Africa e-mail:cmVzaWEucHJldG9yaXVzQHVwLmFjLnph