 Raymond A. Clarke
Raymond A. Clarke Valsamma Eapen
Valsamma Eapen- 1Ingham Institute, School of Medicine, University of Western Sydney, Sydney, NSW, Australia
- 2School of Psychiatry, University of New South Wales & Academic Unit of Child Psychiatry, South West Sydney (AUCS), Liverpool Hospital, Sydney, NSW, Australia
Autism spectrum disorder (ASD) is characterized by a broad spectrum of behavioral deficits of unknown etiology. ASD associated mutations implicate numerous neurological pathways including a common association with the neurexin trans-synaptic connexus (NTSC) which regulates neuronal cell-adhesion, neuronal circuitry, and neurotransmission. Comparable DNA lesions affecting the NTSC, however, associate with a diversity of behavioral deficits within and without the autism spectrum including a very strong association with Tourette syndrome. The NTSC is comprised of numerous post-synaptic ligands competing for trans-synaptic connection with one of the many different neurexin receptors yet no apparent association exists between specific NTSC molecules/complexes and specific behavioral deficits. Together these findings indicate a fundamental role for NTSC-balance in stabilizing pre-behavioral control. Further molecular and clinical characterization and stratification of ASD and TS on the basis of NTSC status will help elucidate the molecular basis of behavior – and define how the NTSC functions in combination with other molecular determinates to strengthen behavioral control and specify behavioral deficits.
Introduction
Autism presents within a broad group of neurodevelopmental disabilities known as autism spectrum disorders (ASDs). ASDs are characterized by impaired social interaction and communication and by restricted interests and repetitive behaviors with high heritability estimates. Over 70% of individuals with autism present with intellectual disability (ID) and ~25% with epilepsy indicating overlapping etiologies in addition to secondary molecular determinates of behavior (Baird et al., 2006). There is currently no reliable biomarker, pathology, anatomical finding, or neuroimaging correlate that is specific for or predictive of ASD (Lord et al., 2000; Bauman and Kemper, 2005; Courchesne et al., 2007; Anagnostou and Taylor, 2011). Furthermore, precious little has been established regarding the precise neurological basis of ASD with many brain regions and circuits implicated (Bauman and Kemper, 2005; Courchesne et al., 2007; Amaral et al., 2008; Anagnostou and Taylor, 2011). Several competing hypotheses have been proposed to account for core behavioral deficits and ancillary symptomatic domains in ASD, but none have been widely accepted (Zoghbi, 2003; Belmonte et al., 2004; Courchesne et al., 2007; Geschwind and Levitt, 2007; Rubenstein, 2010). Genomic analyses indicate extreme genetic heterogeneity in ASD with a conservative estimation of between 380 and 820 loci implicated (Abrahams and Geschwind, 2008; O’Roak et al., 2012; Cristino et al., 2013), where many of the loci are associated with overlapping biological pathways (O’Roak et al., 2012; Cristino et al., 2013; Kenny et al., 2013; Yadav et al., 2013). Pathway overlap also extends to neuropsychiatric disorders with behavioral profiles outside the autism spectrum. In this respect, the neurexin trans-synaptic connexus (NTSC) (Clarke et al., 2012), which regulates development and maintenance of neuronal circuitry and neurotransmission is of particular relevance given its high mutation rate in ASD and other neuropsychiatric disorders (Clarke et al., 2012; O’Roak et al., 2012; Cristino et al., 2013; Kenny et al., 2013; Yadav et al., 2013).
Global genomic studies have identified numerous genes/variants and pathways implicated in the behavioral deficits associated with ASD. Cristino et al. (2013) used copy number variations (CNV) and SNP variant analyses to define 13 distinct protein modules involved in ASD including the NTSC. In addition, O’Roak et al. (2012) found high-density of mutations in the β-catenin and p53 signaling pathways consistent with the influence of both de novo and extremely rare inherited single nucleotide variations (SNVs) and CNVs contributing to the overall genetic risk. The wnt/β-catenin and Notch signaling pathways in neuronal development are also implicated commensurate with the importance of neuronal circuitry/boundaries and neurotransmission during development as intersecting determinates for ASD (Griswold et al., 2012; Kenny et al., 2013).
The neurexins (NRXNs) are one of the gene families most commonly mutated in ASD (Missler et al., 2003; Belloso et al., 2007; Gauthier et al., 2011; Clarke et al., 2012; Vaags et al., 2012). NRXNs are single-pass transmembrane proteins concentrated on the pre-synaptic side of the synapse which facilitate neuronal cell-adhesion through the formation of NRXN trans-synaptic cell-adhesion complexes which together comprise the NTSC (Laurén et al., 2003; Missler et al., 2003; Chen et al., 2006; Ko et al., 2009; Linhoff et al., 2009; Wright and Washbourne, 2011; Clarke et al., 2012). The extracellular domain of pre-synaptic NRXNs binds to one of a range of post-synaptic ligands including neuroligins (NLGNs), leucine-rich repeat transmembrane proteins (LRRTMs), or cerebellin precursor (CBLN) glutamate receptor delta (Glu/GRID) complexes (Figure 1) (Missler et al., 2003; Varoqueaux et al., 2006; Ko et al., 2009; Linhoff et al., 2009; Mondin et al., 2011; Wright and Washbourne, 2011; Clarke et al., 2012; Yasumura et al., 2012). Together the three alpha-NRXNs 1–3 are essential for survival and have a pivotal role in neurodevelopment and synaptic transmission where their roles partially overlap (Missler et al., 2003) and all have been implicated in ASD (Missler et al., 2003; Belloso et al., 2007; Wang et al., 2009; Sousa et al., 2010; Gauthier et al., 2011; Clarke et al., 2012; Vaags et al., 2012; Cristino et al., 2013; Jones et al., 2013; Kenny et al., 2013; Yadav et al., 2013). However, specific NTSC components do not associate with specific behavioral deficits in ASD. Moreover, many of the same NTSC gene families are found associated with other neuropsychiatric disorders outside the autism spectrum including Tourette syndrome (TS), Asperger syndrome, schizophrenia, and ID (Clarke et al., 2012). Neuroligin 4X (NLGN4X) is just one of the many NTSC single gene overlaps between TS, ASD, and ID. The first, a NLGN4X truncation mutation, was identified in a family comprising two affected brothers, one with autism and ID and the other with ASD–Asperger syndrome and normal intelligence (Jamain et al., 2003). Subsequently, a different NLGN4X truncating mutation was identified in a multigenerational pedigree with 13 affected males with either non-syndromic ID (10 individuals), ID with ASD, or ASD without ID (Laumonnier et al., 2004). In 2008, another familial NLGN4X truncating mutation was identified in two brothers with TS/motor tic, one with ASD and the other with attention deficit/hyperactivity disorder (ADHD) and a mother carrier with a learning disorder, anxiety, and depression (Lawson-Yuen et al., 2008). This latter NTSC association with TS and ADHD was just the first of many such associations which have emerged since between the NTSC and the divergent behavioral profiles of ASD and TS (Clarke et al., 2012).
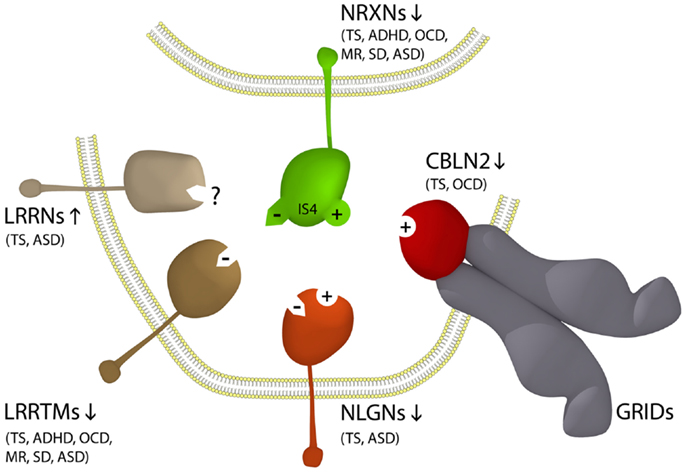
Figure 1. NTSC model for ASD and Tourette Syndrome (Clarke et al., 2012) implicates the full complement of known neurexin (NRXN – green) trans-synaptic cell-adhesion ligand gene families through multiple means of enquiry including the Neuroligin (NLGN – orange); Leucine-rich repeat transmembrane protein (LRRTM – brown); Cerebellin precursor (CBLN – grey); and glutamate receptor delta (GluD/GRID – red) gene families. The pre-synaptic NRXNs form trans-synaptic complexes with post-synaptic ligands NLGNs, LRRTMs, and CBLNs-GRIDs in the formation and/or maintenance of neuronal circuitry within the brain. Vertical arrows indicate putative pathogenic dose effects. Neurexin isoforms with (+) and without (−) the 30 amino acid insert at splice site 4 (IS4) dictate the different/competitive binding of NRXNs between ligands. Co-morbidities listed are those associated with the TS translocations and CNVs affecting the respective genes (Clarke et al., 2012).
Tourette syndrome (TS) is characterized by motor and vocal tics, with a pre-pubertal age of onset, a waxing and waning course, and improvement in symptoms in adulthood (Eapen and Crncec, 2009). Clinical and epidemiological studies point to a very high incidence of other childhood onset behavioral and developmental disorders including up to 60% with ADHD and up to 50% with obsessive–compulsive disorder (OCD). It has long been suggested that chronic tics and OCD within TS families are likely manifestations of the same underlying genetic etiology with gender-dependent differences in expression leading to male members of the family exhibiting more tic behaviors and the female members exhibiting OCD (Eapen et al., 1993). Furthermore, recent SNP association data suggests that OCD in the presence of TS/Chronic tics may have different underlying genetic susceptibility compared to OCD alone (Eapen et al., 1993).
In the fore mentioned affected families the different behavioral profiles appear to converge around the haploinsufficiency of NLGN4X as the common molecular deficit. The mutation, deletion, disruption and duplication of other NTSC components are also relatively common in ASD and consistent with dose effects (Sousa et al., 2010; Gilman et al., 2011; Sakai et al., 2011; Voineagu et al., 2011; O’Roak et al., 2012; Cristino et al., 2013). Association studies also show that many of the rare variants associated with ASD occur within NTSC genes including NRXN1-3, NRXN4/CNTNAP2, NLGN1, NLGN3, NLGN4X, NLGN4Y, LRRTM1, LRRTM2, GRID1 (Sudhof, 2008; Sousa et al., 2010; Gilman et al., 2011; Sakai et al., 2011; Voineagu et al., 2011; Clarke et al., 2012; O’Roak et al., 2012; Cristino et al., 2013) and genes encoding NTSC interacting proteins like SHANK1-3 (Cardno and Gottesman, 2000; O’Roak et al., 2011). Moreover, recent network analyses indicate synaptic transmission as the major protein hub within the ASD network and the only protein module with interactions with all other 12 major network modules including cell–cell adhesion (Cristino et al., 2013).
The recent identification of GRID1 associating with ASD (Cristino et al., 2013) and CBLN1 associating with autistic-like traits (Jones et al., 2013) extends the association between the NTSC and ASD and the molecular convergence between TS and ASD. GRID1 is an inter-synaptic ligand of the post-synaptic transmembrane protein CBLN2 found associated with TS (Clarke et al., 2012), that forms the tripartite NRXN–GRID1–CBLN2 trans-synaptic cell-adhesion complex (Matsuda and Yuzaki, 2011; Clarke et al., 2012). In fact all of the gene families encoding NTSC complexes, with the exception of the GRIDs, have been implicated in TS including the NRXN, NLGN, LRRTM, and CBLN gene families (Petek et al., 2001; Verkerk et al., 2003; Belloso et al., 2007; Lawson-Yuen et al., 2008; Sundaram et al., 2010; Patel et al., 2011; Clarke et al., 2012; Fernandez et al., 2012). Of the 11 novel TS gene disruptions, exonic deletions, and truncations reported to date that are either recurrent or familial, a total of 9 are associated with 1 of the 20 gene families encoding the NTSC [p = 5.5E – 26 (T-test)] (Petek et al., 2001; Verkerk et al., 2003; Belloso et al., 2007; Lawson-Yuen et al., 2008; Ercan-Sencicek et al., 2010; Sundaram et al., 2010; Patel et al., 2011; Clarke et al., 2012; Fernandez et al., 2012). As such, the NTSC emerges as a primary determinate for TS (Clarke et al., 2012) and thus by inference a primary determinate for that subset of ASDs with NTSC association. Moreover, as is the case with ASD the bulk of the NTSC mutations associated with TS to date are consistent with dose effects with no apparent correlation between any of the different receptors or ligands of the NTSC and specific behavioral deficits in or between ASD and TS. Rather, the stoichiometric balance between the various competitive NTSC ligands and receptors appears to play a protective gate-keeping role in behavioral control as outlined in the pathogenetic model (Figure 1) (Clarke et al., 2012).
The striking molecular convergence between TS and ASD at the NTSC may help explain epidemiological features shared between TS and ASD but not the behavioral divergence. TS and ASD are both conditions that begin during childhood (~1% of children affected) and both are more common in males than in females. The inheritance patterns of TS are also comparable with that of ASD. TS twin studies suggest a monozygotic to dizygotic concordance of up to 77% and family studies consistently demonstrate up to a 100-fold increase in the rates of TS in first-degree relatives comparable with the high heritability of the ASDs (O’Rourke et al., 2009). ASD is also over represented in TS, and clinically, symptoms such as obsessions, compulsive behaviors, involuntary movements (tics in TS and stereotypies in ASD), poor speech control, and echolalia are common in both conditions. Furthermore, the literature suggests that around 20–40% of individuals with ASD experience tics and over 50% of individuals with ID and ASD also exhibit tics (Kadesjo and Gillberg, 2000; O’Rourke et al., 2009). Such overlap in symptoms presumably stems from the interrelated neuronal circuitry involved in the final common pathways of behavioral expression (Eapen et al., 2013). However, the divergent behaviors seen in the two conditions with motor and vocal tics in TS, and impaired social interaction and communication and restricted interests seen in ASD presumably relate to secondary/auxillary molecular and/or environmental determinants impacting neuronal circuitry development/maintenance and/or transmission.
In addition to the prevalence of NTSC dose effects in ASD and TS, the competition for connections between NRXNs and their trans-synaptic ligands (Figure 1) further supports the requirement for NTSC-balance in behavioral control. This in turn provides insight into the behavioral role of molecules linked to the NTSC. For example, the SHANK proteins which function from the post-synaptic side of the NTSC are also commonly associated with ASD. The SHANK proteins mediate attachment of the intracellular PDZ-binding domains of NTSC receptor/ligand complexes, including NRXN–NLGN and NRXN–LRRTM (Clarke et al., 2012), to the local actin-based cytoskeleton within dendritic spines. Furthermore, in Purkinje cells, the post-synaptic clustering of SHANK2 with GRID2 appears dependent on the integrity of the tripartite NRXN–GRID2–CBLN1 trans-synaptic complex (Joo et al., 2011; Matsuda and Yuzaki, 2011; Jones et al., 2013). Another TS/ASD candidate gene of related interest to the SHANKs is synapse-associated protein 97 (SAP97) which encodes a scaffold-like protein located on the post-synaptic side of the synapse. Linkage analysis of a large TS pedigree identified the strongest linkage marker (D3S1311) within SAP97 (Verkerk et al., 2006) and a male individual with TS and ASD has been identified with duplication of the SAP97 gene locus (unpublished data), whereas micro-deletion of 3q inclusive of SAP97 is commonly associated with schizophrenia. SAP family proteins bind directly to NTSC complexes and to NMDA, AMP, and kainate receptors at the synapse (Rumbaugh et al., 2003) and membrane-diffusing AMPARs can be rapidly trapped at SAP90/PSD95 scaffolds assembled at nascent NTSC (NRXN–NLGN) adhesions (Mondin et al., 2011). Moreover, the TS candidate ZnT3 (Clarke et al., 2012) – a synaptic zinc transporter which controls concentrations of Zn2+ within post-synaptic vesicles – is of particular interest here given the concentration of Zn2+ ions within the post-synaptic density (PSD) is known to affect the recruitment of scaffolding proteins like SHANK2 and SHANK3 (Grabrucker et al., 2011).
NTSC Relation to Neurological Pathologies in ASD and TS
The neuronal cell-adhesion complexes of the NTSC promote synapse formation and/or maintenance bi-directionally in the glutamatergic and GABA-ergic nervous system. As such, NTSC-imbalance will translate as an imbalance in neuronal connectivity through changes in synapse patterning and transmission (Missler et al., 2003; Varoqueaux et al., 2006; Ko et al., 2009; Linhoff et al., 2009; Clarke et al., 2012). Loss of CBLN2, as reported in TS (Clarke et al., 2012), is associated with reduced mediation of inhibitory synaptogenesis (Yasumura et al., 2012). This however, appears in opposition with the reduced number of excitatory synapses associated with the downregulation of NLGN4X or the LRRTMs in TS (Figure 1) (Ko et al., 2009; Wright and Washbourne, 2011), albeit the recurrent loss/disruption of NRXN1 in TS and ASD infers loss of both excitatory and inhibitory synaptic connections. Together these findings further reinforce the importance of a balanced NTSC repertoire rather than “specific complexes” as the basis of NTSC related behavioral disorders.
Synaptic homeostasis depends on the balance between the strength of excitation, inhibition, and the intrinsic excitability of the neuronal circuitry. Evidence suggests that the balance between excitation and inhibition is tightly regulated with even small changes affecting neuronal firing (Atallah and Scanziani, 2009; Pouille et al., 2009). When this balance is perturbed, mechanisms come into play to restore synaptic homeostasis by modifying the balance between excitatory and inhibitory inputs or the application of intrinsic mechanisms to modify the balance of inward and outward voltage-dependent current (Gainey et al., 2009). Synapses are formed even when αNRXN l is deleted from the mouse genome, however, this compromises synaptic transmission (Missler et al., 2003). The pre-synaptic co-assembly of Ca2+ channels with the secretory apparatus is a prerequisite for the release of neurotransmitters like glutamate and this channel function is impaired in αNRXN1 knockout mice with consequent reductions in neurotransmitter release (Missler et al., 2003). The NTSC trans-synaptic connections NRXN–NLGN and NRXN–LRRTM are both sensitive to extracellular Ca2+ concentrations which appear to trigger post-synaptic differentiation and control the balance of inhibitory GABA-ergic and excitatory glutamatergic inputs. Glutamate, the main excitatory neurotransmitter in the vertebrate brain, has a major role in cortico-striatal-thalamo-cortical circuits and several lines of evidence support the role of glutamate in TS including: the TS association of glutamate receptors that are localized in the cellular membranes of both neurons and glia; the recognized extensive interaction between glutamate and dopamine systems; results of familial genetic studies; and data from neurochemical analyses of post-mortem brain samples (Felling and Singer, 2011; Clarke et al., 2012). Interestingly, LRRTM1 null mice have altered distribution of the excitatory pre-synaptic vesicular glutamate transporter VGLUT1 (Ko et al., 2009; Linhoff et al., 2009). Furthermore, loss of excitatory synaptic connections results in a hypo-glutamatergic state that is consistent with a loss in the synaptic weight, which is an all important factor for the circuit strength required in language development (Matsuda and Yuzaki, 2011).
Neural Circuitry as a Function of Synaptic Pruning and Boundary Formation
Synaptic pruning plays an important role during maturation of the brain by limiting neural circuitry, and neural circuitry within specific brain regions is implicated in behavioral control. As such the integrity of neural/brain boundaries may be a factor in neuropsychiatric disorders. In this respect, it is most interesting to note that both ASD and TS have been associated with leucine-rich repeat neuronal (LRRN) type I transmembrane protein genes. LRRN3 is localized within the genomic region most commonly duplicated in ASD (Kroisel et al., 2001; Maestrini et al., 2010; Pagnamenta et al., 2010). LRRN3 is also nested in an antisense orientation within the IMMP2L gene recurrently disrupted in TS and ASD (Clarke et al., 2012). Moreover, the nearest gene relation to LRRN3, LRRN1, has been duplicated in ASD (Davis et al., 2009). These associations suggest increased dose of LRRN1 and LRRN3 maybe pathogenic for ASD and TS. Little is known about the function of LRRN3, however, LRRN1 is known to have a key role in regional boundary formation within the brain (Chen et al., 2006; Tossell et al., 2011). Studies in the developing chick demonstrate that the midbrain–hindbrain boundary (MHB) is established through the down regulation of Lrrn1 by Fgf8 on the posterior side of the future boundary (Tossell et al., 2011), thereby creating a differential cellular affinity between the two compartments likely to involve an as yet unspecified extracellular binding partner for Lrrn1. Lrrn1 in turn regulates the expression of the Lunatic Fringe gene which modulates Notch signaling to complete MHB formation. Over-expression of Lrrn1 disrupts the MHB with mixing of cells between compartments (Tossell et al., 2011). For further insight into this association see (Clarke et al., 2012).
Auxillary Molecular and Environmental Determinants Specify Behavioral Deficits
Imbalance in the NTSC appears to be sufficient for but not definitive in specifying the nature of behavioral pathogenesis. Moreover, recent evidence suggests that numerous gene variants combine with environmental and physiological factors to specify behavioral deficits. For example, the sex-specific imprinting of NRXN4/CNTNAP2, CTNNA3, and LRRTM1 is known to alter the expression of these genes and their parent-of-origin phenotypic inheritance patterns (Oudejans et al., 2004; Francks et al., 2007). Thus, a particular phenotypic co-morbidity may present based on the type and level of involvement of the different NTSC neurotransmitter pathways in combination with secondary determinates that mediate or modulate NTSC pathways during neurodevelopment whereas an early environmental insult could specify an alternate behavioral deficit/neural outcome (Herbert, 2010) including effects associated with prematurity, perinatal trauma, hypoxia, injury oxidative stress, inflammations, infections and autoimmunity, neural and psychosocial stressors, gender effects, etc. (Eapen, 2011). Gender-specific differences exist in the topographic segregation and functionality of GABA-A systems in the substantia nigra, moreover, circulating testosterone is essential for the development of the substantia nigra region in the neonatal period and to a lesser extent for final maturation in the peripubertal period (Veliskova and Moshe, 2001). In this regard, a role for testosterone has been suggested in the extreme male brain hypothesis in ASD (Baron-Cohen, 2002). Similar mechanisms may affect the TS genes leading to gender-dependent difference in phenotypic expression – with male members of TS families exhibiting more tic behaviors and female members more OCD (Eapen et al., 1993, 1997). Thus, an NTSC related imbalance that impacts development of different neuronal regions and circuitry maybe further specified by secondary genetic and/or environmental events (Eapen et al., 2014). The penetrance of the different co-morbidities may also be related to gender, gene dose effects, or the timing of events when different brain regions are being formed, thus resulting in different clinical phenotypes (Eapen et al., 2013).
Conclusion
The NTSC provides an invaluable window into the molecular basis of behavior. The role of NTSC-balance as a gate keeper of behavioral control provides a firm basis for more in depth molecular and clinical characterization and stratification of behavioral disorders. To this end, NTSC’s common association with ASD and Tourette syndrome provides the ideal starting point for molecular and clinical comparisons between select ASD and TS families.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abrahams, B. S., and Geschwind, D. H. (2008). Advances in autism genetics: on the threshold of a new neurobiology. Nat. Rev. Genet. 9, 341. doi: 10.1038/nrg2346
Amaral, D. G., Schumann, C. M., and Nordahl, C. W. (2008). Neuroanatomy of autism. Trends Neurosci. 31, 137–145. doi:10.1016/j.tins.2007.12.005
Anagnostou, E., and Taylor, M. J. (2011). Review of neuroimaging in autism spectrum disorders: what have we learned and where we go from here. Mol. Autism 2, 4. doi:10.1186/2040-2392-2-4
Atallah, B. V., and Scanziani, M. (2009). Instantaneous modulation of gamma oscillation frequency by balancing excitation with inhibition. Neuron 62, 566–577. doi:10.1016/j.neuron.2009.04.027
Baird, G., Simonoff, E., Pickles, A., Chandler, S., Loucas, T., Meldrum, D., et al. (2006). Prevalence of disorders of the autism spectrum in a population cohort of children in South Thames: the special needs and autism project (SNAP). Lancet 368, 210–215. doi:10.1016/S0140-6736(06)69041-7
Bauman, M. L., and Kemper, T. L. (2005). Neuroanatomic observations of the brain in autism: a review and future directions. Int. J. Dev. Neurosci. 23, 183–187. doi:10.1016/j.ijdevneu.2004.09.006
Belloso, J. M., Bache, I., Guitart, M., Caballin, M. R., Halgren, C., Kirchhoff, M., et al. (2007). Disruption of the CNTNAP2 gene in a t(7;15) translocation family without symptoms of Gilles de la Tourette syndrome. Eur. J. Hum. Genet. 15, 711–713. doi:10.1038/sj.ejhg.5201824
Belmonte, M. K., Allen, G., Beckel-Mitchener, A., Boulanger, L. M., Carper, R. A., and Webb, S. J. (2004). Autism and abnormal development of brain connectivity. J. Neurosci. 24, 9228–9231. doi:10.1523/JNEUROSCI.3340-04.2004
Cardno, A. G., and Gottesman, I. I. (2000). Twin studies of schizophrenia: from bow-and-arrow concordances to star wars Mx and functional genomics. Am. J. Med. Genet. 97, 12–17. doi:10.1002/(SICI)1096-8628(200021)97:1<12::AID-AJMG3>3.0.CO;2-U
Chen, Y., Aulia, S., Li, L., and Tang, B. L. (2006). AMIGO and friends: an emerging family of brain-enriched, neuronal growth modulating, type I transmembrane proteins with leucine-rich repeats (LRR) and cell adhesion molecule motifs. Brain Res. Rev. 51, 265–274. doi:10.1016/j.brainresrev.2005.11.005
Clarke, R. A., Lee, S., and Eapen, V. (2012). Pathogenetic model for Tourette syndrome delineates overlap with related neurodevelopmental disorders including Autism. Transl. Psychiatry 2, e163. doi:10.1038/tp.2012.75
Courchesne, E., Pierce, K., Schumann, C. M., Redcay, E., Buckwalter, J. A., Kennedy, D. P., et al. (2007). Mapping early brain development in autism. Neuron 56, 399–413. doi:10.1016/j.neuron.2007.10.016
Cristino, A. S., Williams, S. M., Hawi, Z., An, J. Y., Bellgrove, M. A., Schwartz, C. E., et al. (2013). Neurodevelopmental and neuropsychiatric disorders represent an interconnected molecular system. Mol. Psychiatry. doi:10.1038/mp.2013.16. [Epub ahead of print].
Davis, L. K., Meyer, K. J., Rudd, D. S., Librant, A. L., Epping, E. A., Sheffield, V. C., et al. (2009). Novel copy number variants in children with autism and additional developmental anomalies. J. Neurodev. Disord. 1, 292–301. doi:10.1007/s11689-009-9013-z
Eapen, V. (2011). Genetic basis of autism: is there a way forward? Curr. Opin. Psychiatry 24, 226–236. doi:10.1097/YCO.0b013e328345927e
Eapen, V., and Crncec, R. (2009). Tourette syndrome in children and adolescents: special considerations. J. Psychosom. Res. 67, 525–532. doi:10.1016/j.jpsychores.2009.08.003
Eapen, V., Crncec, R., and Walter, A. (2013). Exploring links between genotypes, phenotypes, and clinical predictors of response to early intensive behavioral intervention in autism spectrum disorder. Front. Hum. Neurosci. 7:567. doi:10.3389/fnhum.2013.00567
Eapen, V., Ward, P., and Clarke, R. (2014). Clonidine in Tourette syndrome and sensorimotor gating. Psychiatry Res. 215, 494–496. doi:10.1016/j.psychres.2013.10.009
Eapen, V., Pauls, D. L., and Robertson, M. M. (1993). Evidence for autosomal dominant transmission in Tourette’s syndrome. United Kingdom cohort study. Br. J. Psychiatry 162, 593–596. doi:10.1192/bjp.162.5.593
Eapen, V., Robertson, M. M., Alsobrook, J. P. II, and Pauls, D. L. (1997). Obsessive compulsive symptoms in Gilles de la Tourette syndrome and obsessive compulsive disorder: differences by diagnosis and family history. Am. J. Med. Genet. 74, 432–438. doi:10.1002/(SICI)1096-8628(19970725)74:4<432::AID-AJMG15>3.0.CO;2-J
Ercan-Sencicek, A. G., Stillman, A. A., Ghosh, A. K., Bilguvar, K., O’Roak, B. J., Mason, C. E., et al. (2010). L-histidine decarboxylase and Tourette’s syndrome. N. Engl. J. Med. 362, 1901–1908. doi:10.1056/NEJMoa0907006
Felling, R. J., and Singer, H. S. (2011). Neurobiology of Tourette syndrome: current status and need for further investigation. J. Neurosci. 31, 12387–12395. doi:10.1523/JNEUROSCI.0150-11.2011
Fernandez, T. V., Sanders, S. J., Yurkiewicz, I. R., Ercan-Sencicek, A. G., Kim, Y. S., Fishman, D. O., et al. (2012). Rare copy number variants in tourette syndrome disrupt genes in histaminergic pathways and overlap with autism. Biol. Psychiatry 71, 392–402. doi:10.1016/j.biopsych.2011.09.034
Francks, C., Maegawa, S., Laurén, J., Abrahams, B. S., Velayos-Baeza, A., Medland, S. E., et al. (2007). LRRTM1 on chromosome 2p12 is a maternally suppressed gene that is associated paternally with handedness and schizophrenia. Mol. Psychiatry 12, 1129–1139.
Gainey, M. A., Hurvitz-Wolff, J. R., Lambo, M. E., and Turrigiano, G. G. (2009). Synaptic scaling requires the GluR2 subunit of the AMPA receptor. J. Neurosci. 29, 6479–6489. doi:10.1523/JNEUROSCI.3753-08.2009
Gauthier, J., Siddiqui, T. J., Huashan, P., Yokomaku, D., Hamdan, F. F., Champagne, N., et al. (2011). Truncating mutations in NRXN2 and NRXN1 in autism spectrum disorders and schizophrenia. Hum. Genet. 130, 563–573. doi:10.1007/s00439-011-0975-z
Geschwind, D. H., and Levitt, P. (2007). Autism spectrum disorders: developmental disconnection syndromes. Curr. Opin. Neurobiol. 17, 103–111. doi:10.1016/j.conb.2007.01.009
Gilman, S. R., Iossifov, I., Levy, D., Ronemus, M., Wigler, M., and Vitkup, D. (2011). Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron 70, 898–907. doi:10.1016/j.neuron.2011.05.021
Grabrucker, A. M., Knight, M. J., Proepper, C., Bockmann, J., Joubert, M., Rowan, M., et al. (2011). Concerted action of zinc and ProSAP/Shank in synaptogenesis and synapse maturation. EMBO J. 30, 569–581. doi:10.1038/emboj.2010.336
Griswold, A. J., Ma, D., Cukier, H. N., Nations, L. D., Schmidt, M. A., Chung, R. H., et al. (2012). Evaluation of copy number variations reveals novel candidate genes in autism spectrum disorder-associated pathways. Hum. Mol. Genet. 21, 3513–3523. doi:10.1093/hmg/dds164
Herbert, M. R. (2010). Contributions of the environment and environmentally vulnerable physiology to autism spectrum disorders. Curr. Opin. Neurol. 23, 103–110. doi:10.1097/WCO.0b013e328336a01f
Jamain, S., Quach, H., Betancur, C., Råstam, M., Colineaux, C., Gillberg, I. C., et al. (2003). Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 34, 27–29. doi:10.1038/ng1136
Jones, R. M., Cadby, G., Melton, P. E., Abraham, L. J., Whitehouse, A. J., and Moses, E. K. (2013). Genome-wide association study of autistic-like traits in a general population study of young adults. Front. Hum. Neurosci. 7:658. doi:10.3389/fnhum.2013.00658
Joo, J. Y., Lee, S. J., Uemura, T., Yoshida, T., Yasumura, M., Watanabe, M., et al. (2011). Differential interactions of cerebellin precursor protein (Cbln) subtypes and neurexin variants for synapse formation of cortical neurons. Biochem. Biophys. Res. Commun. 406, 627–632. doi:10.1016/j.bbrc.2011.02.108
Kadesjo, B., and Gillberg, C. (2000). Tourette’s disorder: epidemiology and comorbidity in primary school children. J. Am. Acad. Child Adolesc. Psychiatry 39, 548–555. doi:10.1097/00004583-200005000-00007
Kenny, E. M., Cormican, P., Furlong, S., Heron, E., Kenny, G., Fahey, C., et al. (2013). Excess of rare novel loss-of-function variants in synaptic genes in schizophrenia and autism spectrum disorders. Mol. Psychiatry. doi:10.1038/mp.2013.127. [Epub ahead of print].
Ko, J., Fuccillo, M. V., Malenka, R. C., and Südhof, T. C. (2009). LRRTM2 functions as a neurexin ligand in promoting excitatory synapse formation. Neuron 64, 791–798. doi:10.1016/j.neuron.2009.12.012
Kroisel, P. M., Petek, E., Emberger, W., Windpassinger, C., Wladika, W., and Wagner, K. (2001). Candidate region for Gilles de la Tourette syndrome at 7q31. Am. J. Med. Genet. 101, 259–261. doi:10.1002/1096-8628(20010701)101:3<259::AID-AJMG1374>3.0.CO;2-
Laumonnier, F., Bonnet-Brilhault, F., Gomot, M., Blanc, R., David, A., Moizard, M. P., et al. (2004). X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am. J. Hum. Genet. 74, 552–557. doi:10.1086/382137
Laurén, J., Airaksinen, M. S., Saarma, M., and Timmusk, T. (2003). A novel gene family encoding leucine-rich repeat transmembrane proteins differentially expressed in the nervous system. Genomics 81, 411–421.
Lawson-Yuen, A., Saldivar, J. S., Sommer, S., and Picker, J. (2008). Familial deletion within NLGN4 associated with autism and Tourette syndrome. Eur. J. Hum. Genet. 16, 614–618. doi:10.1038/sj.ejhg.5202006
Linhoff, M. W., Laurén, J., Cassidy, R. M., Dobie, F. A., Takahashi, H., Nygaard, H. B., et al. (2009). An unbiased expression screen for synaptogenic proteins identifies the LRRTM protein family as synaptic organizers. Neuron 61, 734–749. doi:10.1016/j.neuron.2009.01.017
Lord, C., Cook, E. H., Leventhal, B. L., and Amaral, D. G. (2000). Autism spectrum disorders. Neuron 28, 355–363. doi:10.1016/S0896-6273(00)00115-X
Maestrini, E., Pagnamenta, A. T., Lamb, J. A., Bacchelli, E., Sykes, N. H., Sousa, I., et al. (2010). High-density SNP association study and copy number variation analysis of the AUTS1 and AUTS5 loci implicate the IMMP2L-DOCK4 gene region in autism susceptibility. Mol. Psychiatry 15, 954–968. doi:10.1038/mp.2009.34
Matsuda, K., and Yuzaki, M. (2011). Cbln family proteins promote synapse formation by regulating distinct neurexin signaling pathways in various brain regions. Eur. J. Neurosci. 33, 1447–1461. doi:10.1111/j.1460-9568.2011.07638.x
Missler, M., Zhang, W., Rohlmann, A., Kattenstroth, G., Hammer, R. E., Gottmann, K., et al. (2003). Alpha-neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature 423, 939–948. doi:10.1038/nature01755
Mondin, M., Labrousse, V., Hosy, E., Heine, M., Tessier, B., Levet, F., et al. (2011). Neurexin-neuroligin adhesions capture surface-diffusing AMPA receptors through PSD-95 scaffolds. J. Neurosci. 31, 13500–13515. doi:10.1523/JNEUROSCI.6439-10.2011
O’Roak, B. J., Deriziotis, P., Lee, C., Vives, L., Schwartz, J. J., Girirajan, S., et al. (2011). Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 43, 585–589. doi:10.1038/ng.835
O’Roak, B. J., Vives, L., Girirajan, S., Karakoc, E., Krumm, N., Coe, B. P., et al. (2012). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–U136. doi:10.1038/nature10989
O’Rourke, J. A., Scharf, J. M., Yu, D., and Pauls, D. L. (2009). The genetics of Tourette syndrome: a review. J. Psychosom. Res. 67, 533–545. doi:10.1016/j.jpsychores.2009.06.006
Oudejans, C. B., Mulders, J., Lachmeijer, A. M., van Dijk, M., Könst, A. A., Westerman, B. A., et al. (2004). The parent-of-origin effect of 10q22 in pre-eclamptic females coincides with two regions clustered for genes with down-regulated expression in androgenetic placentas. Mol. Hum. Reprod. 10, 589–598. doi:10.1093/molehr/gah080
Pagnamenta, A. T., Bacchelli, E., de Jonge, M. V., Mirza, G., Scerri, T. S., Minopoli, F., et al. (2010). Characterization of a family with rare deletions in CNTNAP5 and DOCK4 suggests novel risk loci for autism and dyslexia. Biol. Psychiatry 68, 320–328. doi:10.1016/j.biopsych.2010.02.002
Patel, C., Cooper-Charles, L., McMullan, D. J., Walker, J. M., Davison, V., and Morton, J. (2011). Translocation breakpoint at 7q31 associated with tics: further evidence for IMMP2L as a candidate gene for Tourette syndrome. Eur. J. Hum. Genet. 19, 634–639. doi:10.1038/ejhg.2010.238
Petek, E., Windpassinger, C., Vincent, J. B., Cheung, J., Boright, A. P., Scherer, S. W., et al. (2001). Disruption of a novel gene (IMMP2L) by a breakpoint in 7q31 associated with Tourette syndrome. Am. J. Hum. Genet. 68, 848–858. doi:10.1086/319523
Pouille, F., Marin-Burgin, A., Adesnik, H., Atallah, B. V., and Scanziani, M. (2009). Input normalization by global feedforward inhibition expands cortical dynamic range. Nat. Neurosci. 12, 1577–1585. doi:10.1038/nn.2441
Rubenstein, J. L. (2010). Three hypotheses for developmental defects that may underlie some forms of autism spectrum disorder. Curr. Opin. Neurol. 23, 118–123. doi:10.1097/WCO.0b013e328336eb13
Rumbaugh, G., Sia, G. M., Garner, C. C., and Huganir, R. L. (2003). Synapse-associated protein-97 isoform-specific regulation of surface AMPA receptors and synaptic function in cultured neurons. J. Neurosci. 23, 4567–4576.
Sakai, Y., Shaw, C. A., Dawson, B. C., Dugas, D. V., Al-Mohtaseb, Z., Hill, D. E., et al. (2011). Protein interactome reveals converging molecular pathways among autism disorders. Sci. Transl. Med. 3, 86ra49. doi:10.1126/scitranslmed.3002166
Sousa, I., Clark, T. G., Holt, R., Pagnamenta, A. T., Mulder, E. J., Minderaa, R. B., et al. (2010). Polymorphisms in leucine-rich repeat genes are associated with autism spectrum disorder susceptibility in populations of European ancestry. Mol. Autism 1, 7. doi:10.1186/2040-2392-1-7
Sudhof, T. C. (2008). Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455, 903–911. doi:10.1038/nature07456
Sundaram, S. K., Huq, A. M., Wilson, B. J., and Chugani, H. T. (2010). Tourette syndrome is associated with recurrent exonic copy number variants. Neurology 74, 1583–1590. doi:10.1212/WNL.0b013e3181e0f147
Tossell, K., Andreae, L. C., Cudmore, C., Lang, E., Muthukrishnan, U., Lumsden, A., et al. (2011). Lrrn1 is required for formation of the midbrain-hindbrain boundary and organiser through regulation of affinity differences between midbrain and hindbrain cells in chick. Dev. Biol. 352, 341–352. doi:10.1016/j.ydbio.2011.02.002
Vaags, A. K., Lionel, A. C., Sato, D., Goodenberger, M., Stein, Q. P., Curran, S., et al. (2012). Rare deletions at the neurexin 3 locus in autism spectrum disorder. Am. J. Hum. Genet. 90, 133–141. doi:10.1016/j.ajhg.2011.11.025
Varoqueaux, F., Aramuni, G., Rawson, R. L., Mohrmann, R., Missler, M., Gottmann, K., et al. (2006). Neuroligins determine synapse maturation and function. Neuron 51, 741–754. doi:10.1016/j.neuron.2006.09.003
Veliskova, J., and Moshe, S. L. (2001). Sexual dimorphism and developmental regulation of substantia nigra function. Ann. Neurol. 50, 596–601. doi:10.1002/ana.1248
Verkerk, A. J., Cath, D. C., van der Linde, H. C., Both, J., Heutink, P., Breedveld, G., et al. (2006). Genetic and clinical analysis of a large Dutch Gilles de la Tourette family. Mol. Psychiatry 11, 954–964. doi:10.1038/sj.mp.4001877
Verkerk, A. J., Mathews, C. A., Joosse, M., Eussen, B. H., Heutink, P., Oostra, B. A., et al. (2003). CNTNAP2 is disrupted in a family with Gilles de la Tourette syndrome and obsessive compulsive disorder. Genomics 82, 1–9. doi:10.1016/S0888-7543(03)00097-1
Voineagu, I., Wang, X., Johnston, P., Lowe, J. K., Tian, Y., Horvath, S., et al. (2011). Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474, 380–384. doi:10.1038/nature10110
Wang, K., Zhang, H., Ma, D., Bucan, M., Glessner, J. T., Abrahams, B. S., et al. (2009). Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature 459, 528–533. doi:10.1038/nature07999
Wright, G. J., and Washbourne, P. (2011). Neurexins, Neuroligins and LRRTMs: synaptic adhesion getting fishy. J. Neurochem. 117, 765–778. doi:10.1111/j.1471-4159.2010.07141.x
Yadav, R., Hillman, B. G., Gupta, S. C., Suryavanshi, P., Bhatt, J. M., Pavuluri, R., et al. (2013). Deletion of glutamate delta-1 receptor in mouse leads to enhanced working memory and deficit in fear conditioning. PLoS ONE 8:e60785. doi:10.1371/journal.pone.0060785
Yasumura, M., Yoshida, T., Lee, S. J., Uemura, T., Joo, J. Y., and Mishina, M. (2012). Glutamate receptor delta 1 induces preferentially inhibitory presynaptic differentiation of cortical neurons by interacting with neurexins through cerebellin precursor protein subtypes. J. Neurochem. 121, 705–716. doi:10.1111/j.1471-4159.2011.07631.x
Keywords: neurexin, NLGN, LRRTM, CBLN, GRID, LRRN, Autism, Tourette
Citation: Clarke RA and Eapen V (2014) Balance within the neurexin trans-synaptic connexus stabilizes behavioral control. Front. Hum. Neurosci. 8:52. doi: 10.3389/fnhum.2014.00052
Received: 17 June 2013; Accepted: 23 January 2014;
Published online: 27 February 2014.
Edited by:
Rudi Crncec, South Western Sydney Local Health District, AustraliaReviewed by:
Alexandre Santos Cristino, The University of Queensland, AustraliaAndrea Kathleen Vaags, Alberta Children’s Hospital, Canada
Copyright: © 2014 Clarke and Eapen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Raymond A. Clarke, Ingham Institute, School of Medicine, University of Western Sydney, 1 Campbell Street, Liverpool, Sydney, NSW 2170, Australia e-mail:cmF5bW9uZC5jbGFya2VAdXdzLmVkdS5hdQ==