 Yili Wu
Yili Wu Fei Hou3
Fei Hou3- 1Department of Psychiatry, Jining Medical University, Jining, China
- 2Collaborative Innovation Center for Birth Defect Research and Transformation of Shandong Province, Jining Medical University, Jining, China
- 3College of Science, Qufu Normal University, Jining, China
- 4Department of Biochemistry, Jining Medical University, Jining, China
Histone acetylation is a major mechanism of chromatin remodeling, contributing to epigenetic regulation of gene transcription. Histone deacetylases (HDACs) are involved in both physiological and pathological conditions by regulating the status of histone acetylation. Although histone deacetylase 4 (HDAC4), a member of the HDAC family, may lack HDAC activity, it is actively involved in regulating the transcription of genes involved in synaptic plasticity, neuronal survival, and neurodevelopment by interacting with transcription factors, signal transduction molecules and HDAC3, another member of the HDAC family. HDAC4 is highly expressed in brain and its homeostasis is crucial for the maintenance of cognitive function. Accumulated evidence shows that HDAC4 expression is dysregulated in several brain disorders, including neurodegenerative diseases and mental disorders. Moreover, cognitive impairment is a characteristic feature of these diseases. It indicates that aberrant HDAC4 expression plays a pivotal role in cognitive impairment of these disorders. This review aims to describe the current understanding of HDAC4’s role in the maintenance of cognitive function and its dysregulation in neurodegenerative diseases and mental disorders, discuss underlying molecular mechanisms, and provide an outlook into targeting HDAC4 as a potential therapeutic approach to rescue cognitive impairment in these diseases.
Introduction
Histone deacetylases (HDACs), accompanying with histone acetyltransferases (HATs), are implicated in chromatin remodeling and subsequent transcription regulation by controlling the status of histone acetylation. Histone acetylation makes chromatin conformation more relaxed, facilitating gene transcription, whereas histone deacetylation induces a condensed chromatin conformation repressing gene transcription. By controlling the status of histone acetylation, HDACs are involved in diverse physiological and pathological processes. Moreover, the function of HDACs is not limited to the histone deacetylation. Recent evidences suggest that HDACs may also contribute to the deacetylation of non-histone proteins (Lardenoije et al., 2015). In addition, HDACs also have deacetylase-independent functions, such as histone deacetylase 4 (HDAC4) (Lardenoije et al., 2015; Han et al., 2016).
HDAC4 is highly expressed in brain (Grozinger et al., 1999; Bolger and Yao, 2005; Darcy et al., 2010). It plays a key role in the maintenance of cognitive function and its alteration is associated with cognitive impairment in both age-related neurodegenerative diseases (e.g., Alzheimer’s disease, AD) and development-related mental disorders (e.g., autism). Therefore, the role of HDAC4 in cognitive function, its dysregulation in cognitive impairment-related neurodegenerative diseases and mental disorders, and underlying mechanisms are discussed in this review.
HDAC4 and HDACs
HDACs Classification
Eighteen human HDACs are identified and classed into four groups based on their homology to yeast HDACs (Didonna and Opal, 2015). Class I HDACs, consisting of HDAC1, 2, 3, and 8, are homologous to yeast RPD3 while class II HDACs have high identity to yeast HDA1, consisting of HDAC4, 5, 6, 7, 9, and 10. According to the protein structure and motif organization, class II HDACs are further divided into two subclasses, class IIa with HDAC4, 5, 7, and 9, and class IIb with HDAC6 and 10. Class III HDACs, named sirtuins, including SIRT1-7, are homologous to yeast SIR2. Compared with zinc-dependent HDACs of class I and class II, class III HDACs are nicotinamide-adenine-dinucleotide (NAD)-dependent. HDAC11 is the only member of Class IV, which is also a Zn-dependent HDAC.
The HDAC4 Gene and Protein
The human HDAC4 gene, located on chromosome 2q37.3, spans approximately 353,480 bp encoding HDAC4 protein with 1084 amino acids. HDAC4 shuttles between cytoplasm and nucleus depending on signal transduction-related phosphorylation status of HDAC4 (Mielcarek et al., 2013). Normally, phosphorylated HDAC4 retains in the cytoplasm, while dephosphorylated HDAC4 is imported into the nucleus (Nishino et al., 2008).
Histone deacetylase 4 protein consists of a long N-terminal domain and a highly conserved C-terminal catalytic domain. The deacetylase activity of HDAC4 is almost undetectable although it has a conserved C-terminal catalytic domain, which might be caused by a substitution of tyrosine to histidine in the enzyme active site (Lahm et al., 2007). However, HDAC4 does play an important role in the regulation of gene transcription via different ways (Figure 1). First, HDAC4 interacts with multiple transcriptional factors [e.g., myocyte enhancer 2 (MEF2), runt related transcription factor 2 (Runx2), serum response factor (SRF), heterochromatin protein 1(HP1), nuclear factor kappa B (NF-κB)] regulating gene transcription (Sando et al., 2012; Ronan et al., 2013). Although HDAC4 per se lacks deacetylase activity, it may be involved in histone deacetylation-mediated transcriptional regulation via interacting with HDAC3, another member of the HDAC family with deacetylase activity (Grozinger et al., 1999; Lee et al., 2015). For example, Lee et al. (2015) showed that HDAC4 is crucial for HDAC3-mediated deacetylation of mineralocorticoid receptor, which could be inhibited by class I HDAC inhibitor but not class II HDAC inhibitor, indicating that HDAC4 is implicated in protein deacetylation via the deacetylase activity of HDAC3. Moreover, the deacetylase activity of HDAC4 needs to be further investigated by multiple approaches as it is not convincing by the in vitro assay from one study (Lahm et al., 2007). As the nuclear localization of HDAC4 is regulated by its interaction with14-3-3, it is possible that the alteration of nuclear HDAC4 mediated by tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein (14-3-3) is involved in transcriptional regulation by its deacetylase activity (Nishino et al., 2008). A recent study suggests that HDAC4 may function to regulate protein SUMOylation via interacting with SUMO-conjugating enzyme Ubc9 (Ubc9), a SUMO E2-conjugating enzyme, contributing to memory formation (Figure 1) (Schwartz et al., 2016).
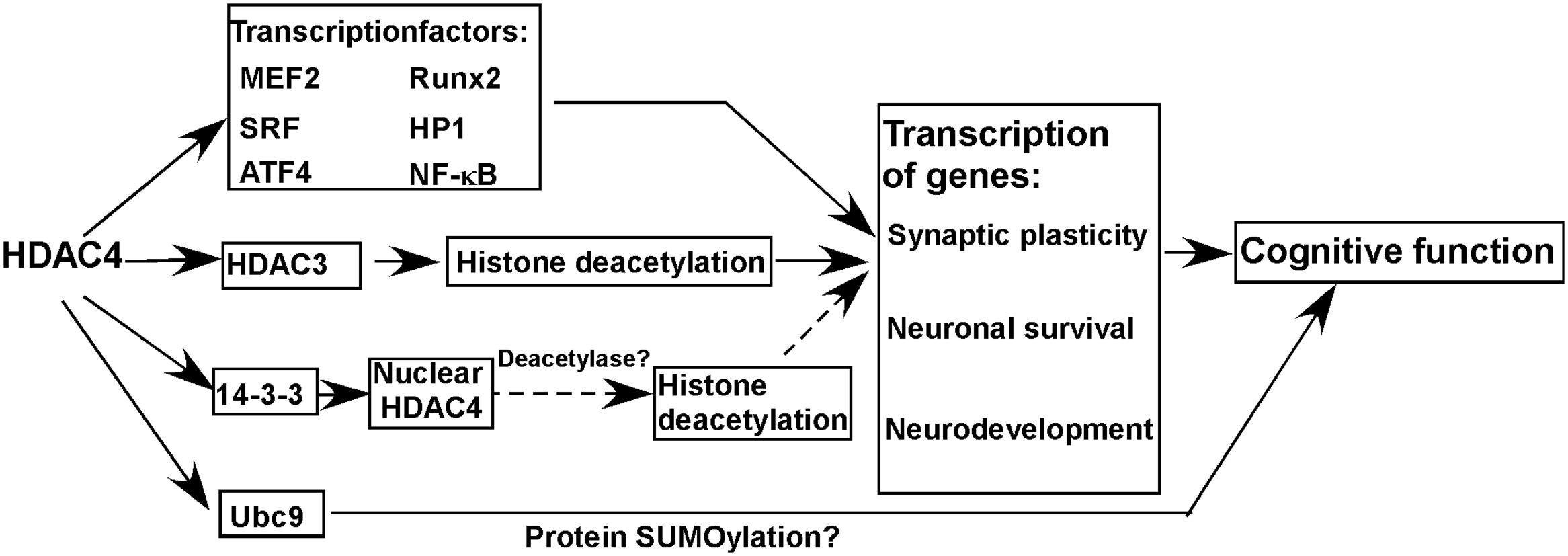
FIGURE 1. Histone deacetylase 4 (HDAC4) in cognitive function and molecular mechanisms. HDAC4 is a global regulator of the transcription of genes involved in synaptic plasticity, neuronal survival, and neurodevelopment by interacting with multiple proteins, which is essential for the maintenance of normal cognitive function. Moreover, HDAC4 may function to regulate protein SUMOylation via interacting with Ubc9 contributing to the maintenance of cognitive function. Solid line and dash line represent confirmed and possible mechanisms, respectively.
HDAC4 in Cognitive Function and Molecular Mechanisms
A growing body of evidence indicates that the homeostasis of HDAC4 is crucial for the maintenance of cognitive function by regulating genes involved in synaptic plasticity, neuronal survival and neurodevelopment (Figure 1) (Schwartz et al., 2016).
HDAC4 and Synaptic Plasticity
Histone deacetylase 4 interacts with multiple transcription factors (e.g., MEF2, Runx2, SRF, HP1), 14-3-3, HDAC3 etc. regulating the transcription of genes involved in synaptogenesis, synaptic plasticity and neurodevelopment, such as activity regulated cytoskeleton associated protein (Arc) and protocadherin (Pcdh10) (Figure 1) (Grozinger et al., 1999; Sando et al., 2012; Ronan et al., 2013; Rashid et al., 2014; Lee et al., 2015; Sharma et al., 2015; Krishna et al., 2016; Nott et al., 2016). First, nuclear HDAC4 represses the expression of constituents of synapses leading to the impairment of synaptic architecture and strength in mice (Sando et al., 2012). In addition, mice carrying a gain-of-function nuclear HDAC4 mutant exhibit deficits in neurotransmission, learning and memory (Sando et al., 2012). On the other hand, silencing HDAC4 expression does result in the impairment of synaptic plasticity, and learning and memory deficits in both mice and Drosophila (Kim et al., 2012; Fitzsimons et al., 2013). A proteomics analysis indicates that HDAC4 is a regulator of proteins involved in neuronal excitability and synaptic plasticity, which are differentially expressed in normal aging subjects and AD patients and associated with memory status (Neuner et al., 2016). A recent study showed that HDAC4 interacts with Ubc9 during memory formation, while the reduction of Ubc9 in adult brain of Drosophila impairs long-term memory, suggesting that the role of HDAC4 in memory formation may be associated with the regulation of protein SUMOylation (Figure 1) (Schwartz et al., 2016).
Above evidence suggests that HDAC4 homeostasis is crucial for the maintenance of synaptic plasticity and cognitive function, i.e., both HDAC4 elevation and reduction lead to cognitive deficits. It is not surprised that both up-regulation and down-regulation of HDAC4 impairs synaptic plasticity and memory function as previous studies have demonstrated that a number of molecules play a dual role in synaptic plasticity and memory function. For example, both overexpression and disruption of regulator of calcineurin 1 (RCAN1) leads to synaptic impairment and memory deficits in Drosophila and mice (Chang et al., 2003; Hoeffer et al., 2007; Chang and Min, 2009; Martin et al., 2012). Moreover, the bidirectional alterations of HDAC4 may differentially disrupt the balance between HDAC4 and its interacting partners leading to synaptic impairment and memory deficits as HDAC4 is implicated in multiple signaling pathways by interacting with many functional proteins (Figure 1). However, the underlying mechanisms need to be further investigated.
HDAC4 and Apoptosis
Neuronal apoptosis is a major mechanism linking to cognitive deficits. In addition to synaptic plasticity, HDAC4 is also involved in neuronal apoptosis. For example, HDAC4 interacts with NF-κB repressing proapoptotic gene expression, and it also inhibit ER stress-induced apoptosis by interacting with activating transcription factor 4 (ATF4), a key transcriptional factor in ER stress response (Figure 1) (Zhang et al., 2014; Vallabhapurapu et al., 2015). Moreover, Majdzadeh et al. (2008) showed that HDAC4 overexpression protects mouse cerebellar granule neurons (CGNs) from apoptosis by inhibiting cyclin dependent kinase 1 (CDK1) activity. Consistently, upregulation of HDAC4 by a NMDAR antagonist protects mouse hippocampal neurons from naturally occurring neuronal death, whereas HDAC4 reduction promotes neuronal apoptosis during development (Chen and Cepko, 2009). Sando et al. (2012) further demonstrated that HDAC4-C-terminal is crucial for rescuing HDAC4 knockdown-induced cell death and reduction of synaptic strength in mouse brains. However, Bolger and Yao (2005) showed that increased expression of nuclear-localized HDAC4 promotes neuronal apoptosis in mouse CGNs, while down-regulation of HDAC4 protects neurons from stress-induced apoptosis. The conflicting results may be caused by different cell types and culture conditions. Majdzadeh et al. (2008) cultured CGNs for 4-5 days before transfection, while Bolger and Yao (2005) transfected CGNs immediately after cell isolation. Although CGNs were used in both studies, the maturation status of neurons when they were transfected may have significant effects on the conflict results. Moreover, short-term and long-term protein overexpression may have opposite effects. For example, a previous study showed that RCAN1 plays an opposite role in neuronal apoptosis at different culture stages, which may be associated with aging or maturation processes (Wu and Song, 2013).
HDAC4 and Brain Development
Histone deacetylase 4interacts with multiple transcriptional factors, repressing the transcription of genes involved in neurodevelopment (Figure 1) (Sando et al., 2012; Ronan et al., 2013). Moreover, HDAC4 may be implicated in neurodevelopment via interacting with HDAC3 which is necessary for brain development (Norwood et al., 2014). In human, both HDAC4 deletion and duplication lead to mental retardation and intelligence disability, suggesting that HDAC4 plays an important role in neurodevelopment which directly links to cognitive function (Shim et al., 2014).
Aberrant HDAC4 Expression/Localization in Neurodegenerative Diseases
Cognitive decline, in particular, learning and memory deficits, is a characteristic of neurodegenerative diseases [e.g., Alzheimer’s disease (AD), Huntington’s disease (HD), and Parkinson’s disease (PD)] which are associated with synaptic dysfunction and synaptic and neuronal loss. A large body of evidence indicates that aberrant HDAC4 expression and subcellular distribution may contribute to the cognitive decline in patients with neurodegenerative diseases (Table 1). First, increased HDAC4 expression was observed in prefrontal cortex of aged individuals, while aging is the major risk factor of neurodegenerative disorders (Sharma et al., 2008). In addition, a more recent study showed that HDAC4 is a global regulator of memory deficits with age (Neuner et al., 2016). Moreover, HDAC4 is involved in the regulation of SIRT1 which is implicated in both aging and memory process in rats (Sasaki et al., 2006; Sommer et al., 2006; Quintas et al., 2012; Han et al., 2016). Furthermore, as mentioned in the section 2.1, HDAC4 homeostasis is crucial for the maintenance of cognitive function, i.e., both HDAC4 elevation and reduction lead to cognitive deficits.
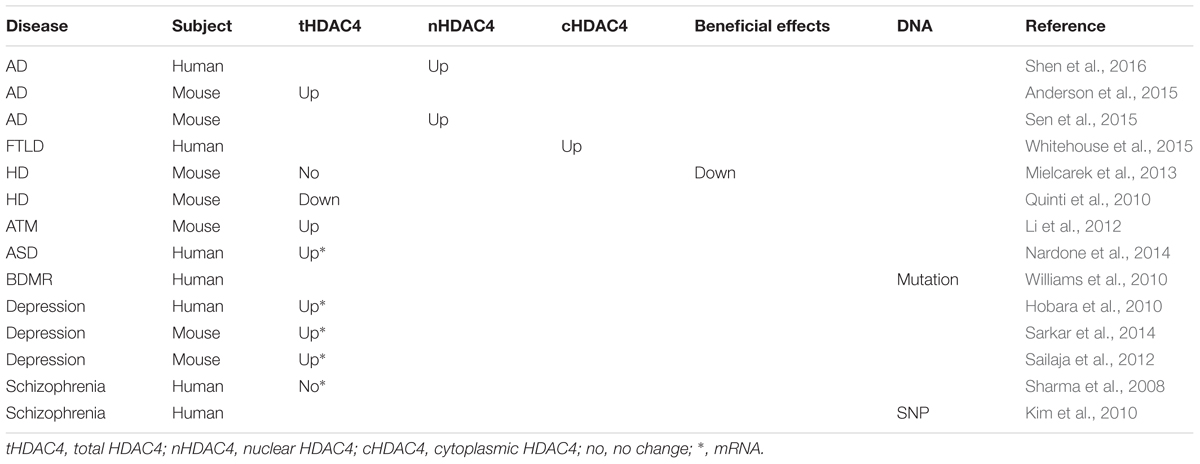
TABLE 1. The alteration of histone deacetylase 4 (HDAC4) in cognitive-related disorders.
HDAC4 in Alzheimer’s Disease
Alzheimer’s disease is the most common form of neurodegenerative disorders in the elderly leading to dementia. Progressive memory loss is the clinical characteristics of AD. Neuritic plaques, neurofibrillary tangles and neuronal loss are the neuropathological hallmarks of AD. Amyloid β (Aβ) and phosphorylated microtubule associated protein tau (Tau) are the major components of neuritic plaques and neurofibrillary tangles, respectively, while apoptosis is a major mechanism of neuronal loss (Wu et al., 2014).
The nuclear expression of HDAC4 is markedly increased in brains of AD patients, while the alteration of total HDAC4, including both cytoplasmic and nuclear HDAC4, is not conclusive (Shen et al., 2016). However, the expression of HDAC4 was significantly increased in AD model mice (Anderson et al., 2015). Moreover, ApoE4, the only confirmed genetic risk factor of late onset AD, increases nuclear HDAC4 levels compared with the ApoE3 in transgenic mice (Sen et al., 2015). It suggests that increased HDAC4 expression or its nuclear localization may contribute to learning and memory deficits in patients with AD.
HDAC4 in Frontotemporal Lobar Degeneration
Frontotemporal lobar degeneration (FTLD) is a heterogeneous neurodegenerative process resulting in frontotemporal dementia. Progressive difficulties in planning, organizing and language are the major characteristics of FTLD. The atrophy of frontal and temporal lobe and inclusions containing abnormal accumulation of Tau, TAR DNA binding protein (TDP-43) or FUS RNA bindind protein (FUS) are the characteristic pathological features of FTLD.
In FTLD patients, cytoplasmic HDAC4 is increased in granule cells of the dentate gyrus, while HDAC5, the other member of class IIa HDACs, is not altered, suggesting that HDAC4 may have a specific role in the pathology of FTLD (Whitehouse et al., 2015).
HDAC4 in Huntington’s disease
Huntington’s disease is a common autosomal dominant neurodegenerative disease, which is caused by the expansion of polyglutamine repeats in huntingtin (HTT) protein, named as mutant HTT (mHTT) (MacDonald et al., 1993; Myers et al., 1993; Kremer et al., 1994). The characteristic clinical features are chorea, progressive cognitive decline, and psychiatric symptoms, while the cognitive problem is often the earliest symptom in patients with HD (Walker, 2007). mHTT impairs fast axonal transport, disrupts mitochondrial function and inflammatory response and promotes apoptosis, which may contribute to the cognitive decline (Szebenyi et al., 2003; Beal and Ferrante, 2004; Trushina et al., 2004).
Growing evidence suggests that increased HDAC4 is implicated in HD pathology, such that reducing HDAC4 expression has beneficial effects. First, overexpression of miR-22 has a protective effect on mHTT model cells, which may be mediated by HDAC4 reduction as HDAC4 is a target gene of miR-22 (Jovicic et al., 2013). Second, HDAC4 interacts with microtubule associated protein 1S (MAP1S) resulting in MAP1S destabilization and reduction, subsequently suppressing the clearance of mHTT aggregates and potentiating the toxicity of mHTT to cultured cells (Yue et al., 2015). Moreover, HDAC4 is associated with HTT in a polyglutamine-length-dependent manner and co-localized with cytoplasmic aggregates. However, reducing HDAC4 expression delays the formation of cytoplasmic aggregates, restores BDNF expression, and rescues synaptic dysfunction in HD mouse models (Mielcarek et al., 2013). In addition, suberoylanilide hydroxamic acid (SAHA) promotes HDAC4 degradation, suggesting that reducing HDAC4 expression may contribute to SAHA’s rescue effects on HD model mice via multiple HDAC4-associated pathways (Figure 1) (Mielcarek et al., 2011). However, SAHA is also an inhibitor of class I HDACs and HDAC6, suggesting that its rescue effects may also be mediated by inhibiting the deacetylase activity of class I HDACs and HDAC6. Although Quinti et al. (2010) showed that the reduction of HDAC4 is associated with the progression of HD in HD model mice, Mielcarek et al. (2013) did not observe the reduction in same HD model mice. However, they found that reducing HDAC4 expression has beneficial effects on HD mice (Mielcarek et al., 2013). As the alteration of HDAC4 in HD model mice remains inconclusive and it still lacks the evidence from HD patients, the alteration of HDAC4 in HD and its role in the pathology of HD need to be further investigated.
HDAC4 in Parkinson Disease
Parkinson disease is the second most common neurodegenerative disease in the elderly. In addition to tremor, rigidity, gait disturbances etc. motor dysfunctions, PD patients also have cognitive impairments (Jankovic, 2008). The major pathological hallmark of PD is the Lewy bodies which mainly consist of protein aggregates of a-synuclein, parkin, and ubiquitin (Jellinger, 2009). In addition, same pathological features were observed in patients with Lewy body (LB) dementia (Jellinger, 2009).
A couple of studies indicate that HDAC4 is associated with the pathology of PD. First, mutations in the Parkin gene cause early onset familial PD and the dysregualtion of parkin has also been observed in sporadic PD. Second, parkin controls the levels of sumoylated HDAC4 (Kirsh et al., 2002; Um et al., 2006). Moreover, HDAC4 co-localized with α-synuclein in the LB (Takahashi-Fujigasaki and Fujigasaki, 2006). In addition, paraquat, a widely used herbicide, implicated in the induction of the pathology of PD, reduces the expression of HDAC4 in culture cells (Song et al., 2011). Furthermore, previous studies showed that aberrant HDAC4 expression results in learning and memory deficits in both mice and Drosophila (Kim et al., 2012; Fitzsimons et al., 2013). Above evidence suggests that alteration of HDAC4 may contribute to cognitive decline in patients with PD. However, no direct evidence shows that HDAC4 is implicated in the pathology of PD.
HDAC4 in Ataxia-Telangiectasia
Ataxia-telangiectasia (A-T), a rare neurodegenerative disease, is caused by mutations in the ATM gene. A-T patients showed many premature aging components, characterized by difficulty in movement and coordination, and early cognitive impairment including learning and memory deficits (Vinck et al., 2011; Shiloh and Lederman, 2016).
In ATM deficient mice, nuclear HDAC4 is increased, which is mediated by the reduction of ATM-dependent phosphorylation of protein phosphatase 2A (PP2A). Reduced phosphorylation of PP2A results in increased HDAC4 dephosphorylation by enhancing PP2A-HDAC4 interaction (Li et al., 2012). HDAC4 dephosphorylation promotes its nuclear import and subsequent dysregulation of genes involved in synaptic plasticity, neuronal survival and neurodevelopment, which may contribute to the cognitive deficits in ATM mice. Consistently, reduced ATM accompanying with the increase of nuclear HDAC4 has been observed in brains of AD patients (Shen et al., 2016).
Aberrant HDAC4 Expression/Function in Mental Disorders
Many mental disorders, including autism spectrum disorders (ASDs), depression, and schizophrenia, are associated with neurodevelopment defects and cognitive impairment is a core feature of mental disorders (Ronan et al., 2013). Increased evidence indicates that aberrant HDAC4 expression or function plays an important role in cognitive deficits of mental disorders (Table 1).
HDAC4 in ASD and BMDR Syndrome
Autism spectrum disorder is characterized by the impairment of social and communication ability, as well as cognitive defects. Several lines of evidence suggests that dysregulation of HDAC4 is implicated in ASD (Pinto et al., 2014; Fisch et al., 2016). First, HDAC4 mRNA was significantly increased in autistic brains (Nardone et al., 2014). Moreover, ASD, intellectual disability, developmental delay etc. are the characteristics of Brachydactyly-mental-retardation (BDMR) syndrome which is caused by 2q37 microdeletion. Importantly, the HDAC4 gene is located in this small region (Doherty and Lacbawan, 1993–2016). A rare case of BMDR syndrome carries an inactive mutant of HDAC4, suggesting that HDAC4 deficiency may be the cause of BMDR syndrome (Doherty and Lacbawan, 1993–2016; Williams et al., 2010). Moreover, in patients with BMDR syndrome, HDAC4 modulates the severity of symptoms in a dosage dependent manner, which further confirms the role of HDAC4 in ASD and other BMDR features (Morris et al., 2012).
HDAC4 in Depressive Disorders
Depressive disorders are the most common mood disorder leading to disability, which is characterized by the presence of sad, empty, or irritable mood and cognitive impairment (Rock et al., 2014). Recent studies highly suggest that HDAC4 is implicated in the pathology of depressive disorders. First, aberrant expression of HDAC4 mRNA has been detected in patients with depression (Otsuki et al., 2012). Consistently, antidepressant reduces the recruitment of HDAC4 to the glial cell-derived neurotrophic factor (GDNF) promoter, consequently increasing the expression of GDNF which is reduced in patients with depression (Otsuki et al., 2012; Lin and Tseng, 2015). In patients with bipolar disorder (BPD), HDAC4 mRNA is significantly increased in a depressive state, while its expression is marked decreased in a remissive state (Hobara et al., 2010). In addition, HDAC4 mRNA is significantly increased in brains of forced-swim stress-induced- and postnatal fluoxetine-induced depression model mice (Sailaja et al., 2012; Sarkar et al., 2014). Intriguingly, adult fluoxetine application does not induce depression-like behavior in mice which is associated with unchanged HDAC4 expression. Ectopic overexpression of HDAC4 in hippocampus is sufficient to induce depression-like behavior in adult mice, indicating that HDAC4 elevation is the key to induce depression-like behavior (Sarkar et al., 2014). Furthermore, depression is a common feature in AD, which may be associated with the increase of HDAC4 expression in AD patients.
HDAC4 in Schizophrenia
Schizophrenia is a complex psychiatric disorder, characterized by impairments in behavior, thought, and emotion. Cognitive impairment is common in patients with schizophrenia, in particular, learning and memory deficits. A couple of evidence suggests that HDAC4 might be associated with the pathology of schizophrenia. First, one SNP (rs1063639) in the HDAC4 gene associates with the development of schizophrenia in a Korea population (Kim et al., 2010). Moreover, in patients with schizophrenia, HDAC4 mRNA is negatively associated with the expression of GAD67, a candidate gene of schizophrenia (Sharma et al., 2008). However, the exact role of HDAC4 in the cognitive deficits of schizophrenia needs to be further investigated.
HDAC4, A Specific Target for Cognitive Impairment
Growing evidence indicates that HDAC4 is a specific target for the treatment of cognitive impairment in multiple disorders, which is different from other HDACs. First, HDAC4 is highly enriched in brain compared with other HDACs. Second, HDAC4 has no or weak HDAC activity, suggesting that global HDAC inhibitors, targeting the catalytic sites of HDACs, may have no effect on HDAC4’s function (Sando et al., 2012). Consistently, HDAC4 has a different effect on cognitive function compared with other HDACs. For example, conditional deletion of HDAC4 leads to learning and memory deficits, while global HDACs inhibition or HDAC2 deficiency significantly improves learning and memory in mice (Vecsey et al., 2007; Guan et al., 2009; Kim et al., 2012). Moreover, the maintenance of HDAC4 homeostasis is crucial for the disease treatment as either increased or decreased HDAC4 expression is detrimental to the cognitive function. It suggests that HDAC4 is a potential target for the treatment of cognitive impairment. However, only one selective HDAC4 inhibitor, tasquinimod, is commercially available, and its effect on cognitive function has not been explored. Therefore, specific HDAC4 modulators should be developed and their roles in cognitive disorders need to be investigated.
Conclusion
Although HDAC4 belongs to the family of HDAC, its deacetylase activity is weak or undetectable. Thus, it remains elusive whether HDAC4 per se could repress gene transcription by its HDAC activity (Figure 1). However, HDAC4 could regulate the transcription of genes involved in synaptic plasticity, neuronal survival, and neurodevelopment by interacting with multiple proteins, which is essential for the maintenance of normal cognitive function (Figure 1). Moreover, HDAC4 may function to regulate protein SUMOylation via interacting with Ubc9 contributing to the maintenance of cognitive function (Figure 1). Moreover, aberrant expression of HDAC4 may be implicated in the cognitive impairment of neurodegenerative diseases and mental disorders. Therefore, HDAC4 is a potential therapeutic target to rescue cognitive deficits in above disorders.
Author Contributions
YW: Formulated the study, wrote the manuscript, and designed the figure. FH, XW: Formulated the study and wrote the manuscript. QK, XH, BB: Provided intellectual thoughts, revised the manuscript, and project leaders.
Funding
This work was supported by grants from the Natural Science Foundation of Shandong Province (No.ZR2013CM031 to QK), the Jining Science and Technology Program for Public Wellbeing (No.2014kjhm-10 to QK), the Jining Science and Technology Development Plan (No.2013jnwk75 to BB).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Anderson, K. W., Chen, J., Wang, M., Mast, N., Pikuleva, I. A., and Turko, I. V. (2015). Quantification of histone deacetylase isoforms in human frontal cortex, human retina, and mouse brain. PLoS ONE 10:e0126592. doi: 10.1371/journal.pone.0126592
Beal, M. F., and Ferrante, R. J. (2004). Experimental therapeutics in transgenic mouse models of Huntington’s disease. Nat. Rev. Neurosci. 5, 373–384. doi: 10.1038/nrn1386
Bolger, T. A., and Yao, T. P. (2005). Intracellular trafficking of histone deacetylase 4 regulates neuronal cell death. J. Neurosci. 25, 9544–9553. doi: 10.1523/JNEUROSCI.1826-05.2005
Chang, K. T., and Min, K. T. (2009). Upregulation of three Drosophila homologs of human chromosome 21 genes alters synaptic function: implications for Down syndrome. Proc. Natl. Acad. Sci. U.S.A. 106, 17117–17122. doi: 10.1073/pnas.0904397106
Chang, K. T., Shi, Y. J., and Min, K. T. (2003). The Drosophila homolog of Down’s syndrome critical region 1 gene regulates learning: implications for mental retardation. Proc. Natl. Acad. Sci. U.S.A. 100, 15794–15799. doi: 10.1073/pnas.2536696100
Chen, B., and Cepko, C. L. (2009). HDAC4 regulates neuronal survival in normal and diseased retinas. Science 323, 256–259. doi: 10.1126/science.1166226
Darcy, M. J., Calvin, K., Cavnar, K., and Ouimet, C. C. (2010). Regional and subcellular distribution of HDAC4 in mouse brain. J. Comp. Neurol. 518, 722–740. doi: 10.1002/cne.22241
Didonna, A., and Opal, P. (2015). The promise and perils of HDAC inhibitors in neurodegeneration. Ann. Clin. Transl. Neurol. 2, 79–101. doi: 10.1002/acn3.147
Doherty, E. S., and Lacbawan, F. L. (1993–2016). “2q37 Microdeletion syndrome,” in GeneReviews® [Internet], eds R. A. Pagon, M. P. Adam, H. H. Ardinger, S. E. Wallace, A. Amemiya, L. J. H. Bean, et al. (Seattle, WA: University of Washington).
Fisch, G. S., Falk, R. E., Carey, J. C., Imitola, J., Sederberg, M., Caravalho, K. S., et al. (2016). Deletion 2q37 syndrome: cognitive-behavioral trajectories and autistic features related to breakpoint and deletion size. Am. J. Med. Genet. A 170, 2282–2291. doi: 10.1002/ajmg.a.37782
Fitzsimons, H. L., Schwartz, S., Given, F. M., and Scott, M. J. (2013). The histone deacetylase HDAC4 regulates long-term memory in Drosophila. PLoS ONE 8:e83903. doi: 10.1371/journal.pone.0083903
Grozinger, C. M., Hassig, C. A., and Schreiber, S. L. (1999). Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc. Natl. Acad. Sci. U.S.A. 96, 4868–4873. doi: 10.1073/pnas.96.9.4868
Guan, J. S., Haggarty, S. J., Giacometti, E., Dannenberg, J. H., Joseph, N., Gao, J., et al. (2009). HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459, 55–60. doi: 10.1038/nature07925
Han, X., Niu, J., Zhao, Y., Kong, Q., Tong, T., and Han, L. (2016). HDAC4 stabilizes SIRT1 via sumoylation SIRT1 to delay cellular senescence. Clin. Exp. Pharmacol. Physiol. 43, 41–46. doi: 10.1111/1440-1681.12496
Hobara, T., Uchida, S., Otsuki, K., Matsubara, T., Funato, H., Matsuo, K., et al. (2010). Altered gene expression of histone deacetylases in mood disorder patients. J. Psychiatr. Res. 44, 263–270. doi: 10.1016/j.jpsychires.2009.08.015
Hoeffer, C. A., Dey, A., Sachan, N., Wong, H., Patterson, R. J., Shelton, J. M., et al. (2007). The Down syndrome critical region protein RCAN1 regulates long-term potentiation and memory via inhibition of phosphatase signaling. J. Neurosci. 27, 13161–13172. doi: 10.1523/JNEUROSCI.3974-07.2007
Jankovic, J. (2008). Parkinson’s disease: clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 79, 368–376. doi: 10.1136/jnnp.2007.131045
Jellinger, K. A. (2009). Significance of brain lesions in Parkinson disease dementia and Lewy body dementia. Front. Neurol. Neurosci. 24:114–125. doi: 10.1159/000197890
Jovicic, A., Zaldivar Jolissaint, J. F., Moser, R., Silva Santos Mde, F., and Luthi-Carter, R. (2013). MicroRNA-22 (miR-22) overexpression is neuroprotective via general anti-apoptotic effects and may also target specific Huntington’s disease-related mechanisms. PLoS ONE 8:e54222. doi: 10.1371/journal.pone.0054222
Kim, M. S., Akhtar, M. W., Adachi, M., Mahgoub, M., Bassel-Duby, R., Kavalali, E. T., et al. (2012). An essential role for histone deacetylase 4 in synaptic plasticity and memory formation. J. Neurosci. 32, 10879–10886. doi: 10.1523/JNEUROSCI.2089-12.2012
Kim, T., Park, J. K., Kim, H. J., Chung, J. H., and Kim, J. W. (2010). Association of histone deacetylase genes with schizophrenia in Korean population. Psychiatry Res. 178, 266–269. doi: 10.1016/j.psychres.2009.05.007
Kirsh, O., Seeler, J. S., Pichler, A., Gast, A., Muller, S., Miska, E., et al. (2002). The SUMO E3 ligase RanBP2 promotes modification of the HDAC4 deacetylase. EMBO J. 21, 2682–2691. doi: 10.1093/emboj/21.11.2682
Kremer, B., Goldberg, P., Andrew, S. E., Theilmann, J., Telenius, H., Zeisler, J., et al. (1994). A worldwide study of the Huntington’s disease mutation. The sensitivity and specificity of measuring CAG repeats. N. Engl. J. Med. 330, 1401–1406.
Krishna, K., Behnisch, T., and Sajikumar, S. (2016). Inhibition of histone deacetylase 3 restores amyloid-beta oligomer-induced plasticity deficit in hippocampal CA1 pyramidal neurons. J. Alzheimers Dis. 51, 783–791. doi: 10.3233/JAD-150838
Lahm, A., Paolini, C., Pallaoro, M., Nardi, M. C., Jones, P., Neddermann, P., et al. (2007). Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc. Natl. Acad. Sci. U.S.A. 104, 17335–17340. doi: 10.1073/pnas.0706487104
Lardenoije, R., Iatrou, A., Kenis, G., Kompotis, K., Steinbusch, H. W., Mastroeni, D., et al. (2015). The epigenetics of aging and neurodegeneration. Prog. Neurobiol. 131, 21–64. doi: 10.1016/j.pneurobio.2015.05.002
Lee, H. A., Song, M. J., Seok, Y. M., Kang, S. H., Kim, S. Y., and Kim, I. (2015). Histone deacetylase 3 and 4 complex stimulates the transcriptional activity of the mineralocorticoid receptor. PLoS ONE 10:e0136801. doi: 10.1371/journal.pone.0136801
Li, J., Chen, J., Ricupero, C. L., Hart, R. P., Schwartz, M. S., Kusnecov, A., et al. (2012). Nuclear accumulation of HDAC4 in ATM deficiency promotes neurodegeneration in ataxia telangiectasia. Nat. Med. 18, 783–790. doi: 10.1038/nm.2709
Lin, P. Y., and Tseng, P. T. (2015). Decreased glial cell line-derived neurotrophic factor levels in patients with depression: a meta-analytic study. J. Psychiatr. Res. 63, 20–27. doi: 10.1016/j.jpsychires.2015.02.004
MacDonald, M. E., Strobel, S. A., Draths, K. M., Wales, J. L., and Dervan, P. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72, 971–983.
Majdzadeh, N., Wang, L., Morrison, B. E., Bassel-Duby, R., Olson, E. N., and D’Mello, S. R. (2008). HDAC4 inhibits cell-cycle progression and protects neurons from cell death. Dev. Neurobiol. 68, 1076–1092. doi: 10.1002/dneu.20637
Martin, K. R., Corlett, A., Dubach, D., Mustafa, T., Coleman, H. A., Parkington, H. C., et al. (2012). Over-expression of RCAN1 causes Down syndrome-like hippocampal deficits that alter learning and memory. Hum. Mol. Genet. 21, 3025–3041. doi: 10.1093/hmg/dds134
Mielcarek, M., Benn, C. L., Franklin, S. A., Smith, D. L., Woodman, B., Marks, P. A., et al. (2011). SAHA decreases HDAC 2 and 4 levels in vivo and improves molecular phenotypes in the R6/2 mouse model of Huntington’s disease. PLoS ONE 6:e27746. doi: 10.1371/journal.pone.0027746
Mielcarek, M., Landles, C., Weiss, A., Bradaia, A., Seredenina, T., Inuabasi, L., et al. (2013). HDAC4 reduction: a novel therapeutic strategy to target cytoplasmic huntingtin and ameliorate neurodegeneration. PLoS Biol. 11:e1001717. doi: 10.1371/journal.pbio.1001717
Morris, B., Etoubleau, C., Bourthoumieu, S., Reynaud-Perrine, S., Laroche, C., Lebbar, A., et al. (2012). Dose dependent expression of HDAC4 causes variable expressivity in a novel inherited case of brachydactyly mental retardation syndrome. Am. J. Med. Genet. A 158A, 2015–2020. doi: 10.1002/ajmg.a.35463
Myers, R. H., MacDonald, M. E., Koroshetz, W. J., Duyao, M. P., Ambrose, C. M., Taylor, S. A., et al. (1993). De novo expansion of a (CAG)n repeat in sporadic Huntington’s disease. Nat. Genet. 5, 168–173. doi: 10.1038/ng1093-168
Nardone, S., Sams, D. S., Reuveni, E., Getselter, D., Oron, O., Karpuj, M., et al. (2014). DNA methylation analysis of the autistic brain reveals multiple dysregulated biological pathways. Transl. Psychiatry 4:e433. doi: 10.1038/tp.2014.70
Neuner, S. M., Wilmott, L. A., Hoffmann, B. R., Mozhui, K., and Kaczorowski, C. C. (2016). Hippocampal proteomics defines pathways associated with memory decline and resilience in ‘normal’ aging and Alzheimer’s disease mouse models. Behav. Brain Res. doi: 10.1016/j.bbr.2016.06.002 [Epub ahead of print].
Nishino, T. G., Miyazaki, M., Hoshino, H., Miwa, Y., Horinouchi, S., and Yoshida, M. (2008). 14-3-3 regulates the nuclear import of class IIa histone deacetylases. Biochem. Biophys. Res. Commun. 377, 852–856. doi: 10.1016/j.bbrc.2008.10.079
Norwood, J., Franklin, J. M., Sharma, D., and D’Mello, S. R. (2014). Histone deacetylase 3 is necessary for proper brain development. J. Biol. Chem. 289, 34569–34582. doi: 10.1074/jbc.M114.576397
Nott, A., Cheng, J., Gao, F., Lin, Y. T., Gjoneska, E., Ko, T., et al. (2016). Histone deacetylase 3 associates with MeCP2 to regulate FOXO and social behavior. Nat. Neurosci. doi: 10.1038/nn.4347 [Epub ahead of print].
Otsuki, K., Uchida, S., Hobara, T., Yamagata, H., and Watanabe, Y. (2012). [Epigenetic regulation in depression]. Nihon Shinkei Seishin Yakurigaku Zasshi 32, 181–186.
Pinto, D., Delaby, E., Merico, D., Barbosa, M., Merikangas, A., Klei, L., et al. (2014). Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 94, 677–694. doi: 10.1016/j.ajhg.2014.03.018
Quintas, A., de Solis, A. J., Diez-Guerra, F. J., Carrascosa, J. M., and Bogonez, E. (2012). Age-associated decrease of SIRT1 expression in rat hippocampus: prevention by late onset caloric restriction. Exp. Gerontol. 47, 198–201. doi: 10.1016/j.exger.2011.11.010
Quinti, L., Chopra, V., Rotili, D., Valente, S., Amore, A., Franci, G., et al. (2010). Evaluation of histone deacetylases as drug targets in Huntington’s disease models. Study of HDACs in brain tissues from R6/2 and CAG140 knock-in HD mouse models and human patients and in a neuronal HD cell model. PLoS Curr. 2:RRN1172. doi: 10.1371/currents.RRN1172
Rashid, A. J., Cole, C. J., and Josselyn, S. A. (2014). Emerging roles for MEF2 transcription factors in memory. Genes Brain Behav. 13, 118–125. doi: 10.1111/gbb.12058
Rock, P. L., Roiser, J. P., Riedel, W. J., and Blackwell, A. D. (2014). Cognitive impairment in depression: a systematic review and meta-analysis. Psychol. Med. 44, 2029–2040. doi: 10.1017/S0033291713002535
Ronan, J. L., Wu, W., and Crabtree, G. R. (2013). From neural development to cognition: unexpected roles for chromatin. Nat. Rev. Genet. 14, 347–359. doi: 10.1038/nrg3413
Sailaja, B. S., Cohen-Carmon, D., Zimmerman, G., Soreq, H., and Meshorer, E. (2012). Stress-induced epigenetic transcriptional memory of acetylcholinesterase by HDAC4. Proc. Natl. Acad. Sci. U.S.A. 109, E3687–E3695. doi: 10.1073/pnas.1209990110
Sando, R. III, Gounko, N., Pieraut, S., Liao, L., Yates, J. III, and Maximov, A. (2012). HDAC4 governs a transcriptional program essential for synaptic plasticity and memory. Cell 151, 821–834. doi: 10.1016/j.cell.2012.09.037
Sarkar, A., Chachra, P., Kennedy, P., Pena, C. J., Desouza, L. A., Nestler, E. J., et al. (2014). Hippocampal HDAC4 contributes to postnatal fluoxetine-evoked depression-like behavior. Neuropsychopharmacology 39, 2221–2232. doi: 10.1038/npp.2014.73
Sasaki, T., Maier, B., Bartke, A., and Scrable, H. (2006). Progressive loss of SIRT1 with cell cycle withdrawal. Aging Cell 5, 413–422. doi: 10.1111/j.1474-9726.2006.00235.x
Schwartz, S., Truglio, M., Scott, M. J., and Fitzsimons, H. L. (2016). Long-term memory in Drosophila is influenced by the histone deacetylase HDAC4 interacting with the SUMO-conjugating enzyme Ubc9. Genetics 203, 1249–1264. doi: 10.1534/genetics.115.183194
Sen, A., Nelson, T. J., and Alkon, D. L. (2015). ApoE4 and abeta oligomers reduce BDNF expression via HDAC nuclear translocation. J. Neurosci. 35, 7538–7551. doi: 10.1523/JNEUROSCI.0260-15.2015
Sharma, M., Shetty, M. S., Arumugam, T. V., and Sajikumar, S. (2015). Histone deacetylase 3 inhibition re-establishes synaptic tagging and capture in aging through the activation of nuclear factor kappa B. Sci. Rep. 5:16616. doi: 10.1038/srep16616
Sharma, R. P., Grayson, D. R., and Gavin, D. P. (2008). Histone deactylase 1 expression is increased in the prefrontal cortex of schizophrenia subjects: analysis of the national brain databank microarray collection. Schizophr. Res. 98, 111–117. doi: 10.1016/j.schres.2007.09.020
Shen, X., Chen, J., Li, J., Kofler, J., and Herrup, K. (2016). Neurons in vulnerable regions of the Alzheimer’s disease brain display reduced ATM signaling. eNeuro 3, 1–18. doi: 10.1523/ENEURO.0124-15.2016
Shiloh, Y., and Lederman, H. M. (2016). Ataxia-telangiectasia (A-T): an emerging dimension of premature ageing. Ageing Res. Rev. doi: 10.1016/j.arr.2016.05.002 [Epub ahead of print].
Shim, S. H., Shim, J. S., Min, K., Lee, H. S., Park, J. E., Park, S. H., et al. (2014). Siblings with opposite chromosome constitutions, dup(2q)/del(7q) and del(2q)/dup(7q). Gene 534, 100–106. doi: 10.1016/j.gene.2013.09.093
Sommer, M., Poliak, N., Upadhyay, S., Ratovitski, E., Nelkin, B. D., Donehower, L. A., et al. (2006). DeltaNp63alpha overexpression induces downregulation of Sirt1 and an accelerated aging phenotype in the mouse. Cell Cycle 5, 2005–2011. doi: 10.4161/cc.5.17.3194
Song, C., Kanthasamy, A., Jin, H., Anantharam, V., and Kanthasamy, A. G. (2011). Paraquat induces epigenetic changes by promoting histone acetylation in cell culture models of dopaminergic degeneration. Neurotoxicology 32, 586–595. doi: 10.1016/j.neuro.2011.05.018
Szebenyi, G., Morfini, G. A., Babcock, A., Gould, M., Selkoe, K., Stenoien, D. L., et al. (2003). Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport. Neuron 40, 41–52. doi: 10.1016/S0896-6273(03)00569-5
Takahashi-Fujigasaki, J., and Fujigasaki, H. (2006). Histone deacetylase (HDAC) 4 involvement in both Lewy and Marinesco bodies. Neuropathol. Appl. Neurobiol. 32, 562–566. doi: 10.1111/j.1365-2990.2006.00733.x
Trushina, E., Dyer, R. B., Badger, J. D. II, Ure, D., Eide, L., Tran, D. D., et al. (2004). Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol. Cell. Biol. 24, 8195–8209. doi: 10.1128/MCB.24.18.8195-8209.2004
Um, J. W., Min, D. S., Rhim, H., Kim, J., Paik, S. R., and Chung, K. C. (2006). Parkin ubiquitinates and promotes the degradation of RanBP2. J. Biol. Chem. 281, 3595–3603. doi: 10.1074/jbc.M504994200
Vallabhapurapu, S. D., Noothi, S. K., Pullum, D. A., Lawrie, C. H., Pallapati, R., Potluri, V., et al. (2015). Transcriptional repression by the HDAC4-RelB-p52 complex regulates multiple myeloma survival and growth. Nat. Commun. 6, 8428. doi: 10.1038/ncomms9428
Vecsey, C. G., Hawk, J. D., Lattal, K. M., Stein, J. M., Fabian, S. A., Attner, M. A., et al. (2007). Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J. Neurosci. 27, 6128–6140. doi: 10.1523/JNEUROSCI.0296-07.2007
Vinck, A., Verhagen, M. M., Gerven, M., de Groot, I. J., Weemaes, C. M., Maassen, B. A., et al. (2011). Cognitive and speech-language performance in children with ataxia telangiectasia. Dev. Neurorehabil. 14, 315–322. doi: 10.3109/17518423.2011.603368
Whitehouse, A., Doherty, K., Yeh, H. H., Robinson, A. C., Rollinson, S., Pickering-Brown, S., et al. (2015). Histone deacetylases (HDACs) in frontotemporal lobar degeneration. Neuropathol. Appl. Neurobiol. 41, 245–257. doi: 10.1111/nan.12153
Williams, S. R., Aldred, M. A., Der Kaloustian, V. M., Halal, F., Gowans, G., McLeod, D. R., et al. (2010). Haploinsufficiency of HDAC4 causes brachydactyly mental retardation syndrome, with brachydactyly type E, developmental delays, and behavioral problems. Am. J. Hum. Genet. 87, 219–228. doi: 10.1016/j.ajhg.2010.07.011
Wu, Y., Ly, P. T., and Song, W. (2014). Aberrant expression of RCAN1 in Alzheimer’s pathogenesis: a new molecular mechanism and a novel drug target. Mol. Neurobiol. 50, 1085–1097. doi: 10.1007/s12035-014-8704-y
Wu, Y., and Song, W. (2013). Regulation of RCAN1 translation and its role in oxidative stress-induced apoptosis. FASEB J. 27, 208–221. doi: 10.1096/fj.12-213124
Yue, F., Li, W., Zou, J., Chen, Q., Xu, G., Huang, H., et al. (2015). Blocking the association of HDAC4 with MAP1S accelerates autophagy clearance of mutant Huntingtin. Aging (Albany N. Y.) 7, 839–853. doi: 10.18632/aging.100818
Keywords: HDAC4, cognitive function, cognitive impairment, neurodegenerative diseases, mental disorders
Citation: Wu Y, Hou F, Wang X, Kong Q, Han X and Bai B (2016) Aberrant Expression of Histone Deacetylases 4 in Cognitive Disorders: Molecular Mechanisms and a Potential Target. Front. Mol. Neurosci. 9:114. doi: 10.3389/fnmol.2016.00114
Received: 12 August 2016; Accepted: 17 October 2016;
Published: 01 November 2016.
Edited by:
Andrew Harkin, Trinity College Dublin, IrelandReviewed by:
Elizabeth A. Thomas, Scripps Research Institute, USAShuchi Mittal, Harvard Medical School, USA
Copyright © 2016 Wu, Hou, Wang, Kong, Han and Bai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qingsheng Kong, cWluZ3NoZW5na29uZzIwMTZAMTI2LmNvbQ==; am55eHlrcXNAMTYzLmNvbQ== Xiaolin Han, c2Rqbmh4bGxAc29odS5jb20= Bo Bai, YmJhaUBtYWlsLmpubWMuZWR1LmNu