

- 1 Wolfson Institute for Biomedical Research, University College London, London, UK
- 2 The Neurosciences Institute, San Diego, CA, USA
Spike-timing-dependent plasticity (STDP) offers a powerful means of forming and modifying neural circuits. Experimental and theoretical studies have demonstrated its potential usefulness for functions as varied as cortical map development, sharpening of sensory receptive fields, working memory, and associative learning. Even so, it is unlikely that STDP works alone. Unless changes in synaptic strength are coordinated across multiple synapses and with other neuronal properties, it is difficult to maintain the stability and functionality of neural circuits. Moreover, there are certain features of early postnatal development (e.g., rapid changes in sensory input) that threaten neural circuit stability in ways that STDP may not be well placed to counter. These considerations have led researchers to investigate additional types of plasticity, complementary to STDP, that may serve to constrain synaptic weights and/or neuronal firing. These are collectively known as “homeostatic plasticity” and include schemes that control the total synaptic strength of a neuron, that modulate its intrinsic excitability as a function of average activity, or that make the ability of synapses to undergo Hebbian modification depend upon their history of use. In this article, we will review the experimental evidence for homeostatic forms of plasticity and consider how they might interact with STDP during development, and learning and memory.
Introduction
The typical academic’s day involves many long hours at one’s desk, reading the literature and writing articles. Picture yourself in London in January: it is cold, gray, and wet. Engrossed in reading, sitting at your desk for hours without exercise, you get chilled in your drafty office. What do you do? Simple: pull on a sweater, which you handily keep hanging on the back of your office door. But 20 min later, you realize that the sweater isn’t enough; you’re still cold. Again, no problem: just turn up the office radiator, and take a quick coffee break. Now you can work on, happy and comfortable. However, a few hours later, your office feels stuffy and warm, and you’re getting sleepy. Now what? Easy: you pull off your sweater, and once again comfortable, keep reading, hardly having noticed the interruption. What we’ve described above is a familiar experience, whether you live in London or Boston: you respond to the fluctuations in temperature in your surroundings.
Although important for brain function, maintenance of body temperature is not the type of homeostasis we will review in this article. The homeostatic plasticity mechanisms we will review concern a neuron’s activity, its synaptic drive, and its spiking output. This is a relatively young field, and evidence for these homeostatic plasticity mechanisms in the central nervous system (CNS) has been in the literature for just over a decade. Thus, many details of these mechanisms are still being elucidated.
What is the brain regulating with homeostatic plasticity mechanisms? There is now quite good evidence that homeostatic plasticity mechanisms regulate a neuron’s output – its firing rate – in such a way to keep it relatively stable. Why does the brain need such mechanisms? There are many changes the brain can undergo – for instance, Hebbian plasticity mechanisms and developmentally associated plasticity – that can alter a neuron’s input dramatically, pushing its output into an unstable regime. Homeostatic plasticity mechanisms counter things like this with the net result being a neuron whose activity is maintained in an optimal range.
To illustrate, take spike-timing-dependent plasticity (STDP). This is an attractive learning rule that seems to capture the essence of Donald Hebb’s postulate about the cellular basis of learning, namely that “cells that fire together wire together” (Hebb, 1949 ; Caporale and Dan, 2008 ). Though it appears that many forms of STDP exist at different synapse types (Abbott and Nelson, 2000 ; Caporale and Dan, 2008 ), in the archetypical form of STDP, spikes occurring in the presynaptic cell a few milliseconds before those in the postsynaptic cell trigger long-term potentiation (LTP), whereas the opposite temporal order results in long-term depression (LTD) (Markram et al., 1997 ; Bi and Poo, 1998 ; Debanne et al., 1998 ; Zhang et al., 1998 ). Encompassing both synaptic strengthening and synaptic weakening mechanisms, STDP may appear sufficient for neuronal stability, for instance, if that could be achieved through balanced and opposite changes in synaptic strength of different inputs. Although there may be cases when STDP can promote stability (discussed in Section “Balanced STDP”), there are also clear cases when it is insufficient. For instance, at high frequencies, STDP favors LTP regardless of precise spike timing (Sjostrom et al., 2001 ; Froemke et al., 2006 ). A regime in which LTP is favored would lack stability and move neurons out of their ideal functional regime (Tsodyks, 2002 ). To illustrate, picture two neurons that experience correlated pre- and postsynaptic firing at high frequencies, leading to LTP of the synaptic connection between them. Strengthening their synaptic connection will make it even more likely that they will experience correlated pre- and postsynaptic firing in the future, which would lead to yet more LTP. As the strength of the connection increases, the postsynaptic cell’s firing rate will slowly increase, since it is being driven more and more by this input. This will cause other presynaptic partners, whose activity has hitherto been uncorrelated with the postsynaptic cell’s activity, to become correlated with postsynaptic firing. This means that they will also undergo LTP, and their synapses will be strengthened as well. The postsynaptic cell’s firing will rapidly and dramatically increase, eventually rendering the neuron unable to transmit meaningful information, but allowing it to potentiate all its synapses, a positive feedback loop that is termed “runaway potentiation” (Figure 1 A). And the effect would spread: increasing firing rates would cause a cascade of runaway potentiation to travel from neuron to neuron, shifting the entire network’s activity into an unstable regime and possibly into pathology. And yet for the vast majority of healthy people, our brains never experience such extreme activity regimes, but keep their activity well within an optimal range. How do our brains achieve this feat?
When you feel cold, a range of homeostatic adaptations can occur: you may get “goose bumps,” shiver, or change your behavior or environment. In a similar vein, there are multiple forms of homeostatic plasticity that can work to stabilize a neuron’s output. We will discuss the major ones in this review, including synaptic scaling, homeostatic intrinsic plasticity, and metaplasticity, as well as stability mechanisms that are intrinsic to STDP. These different homeostatic mechanisms will intersect with Hebbian plasticity differently, and thus may have profoundly different effects on a neuron’s subsequent action within a circuit.
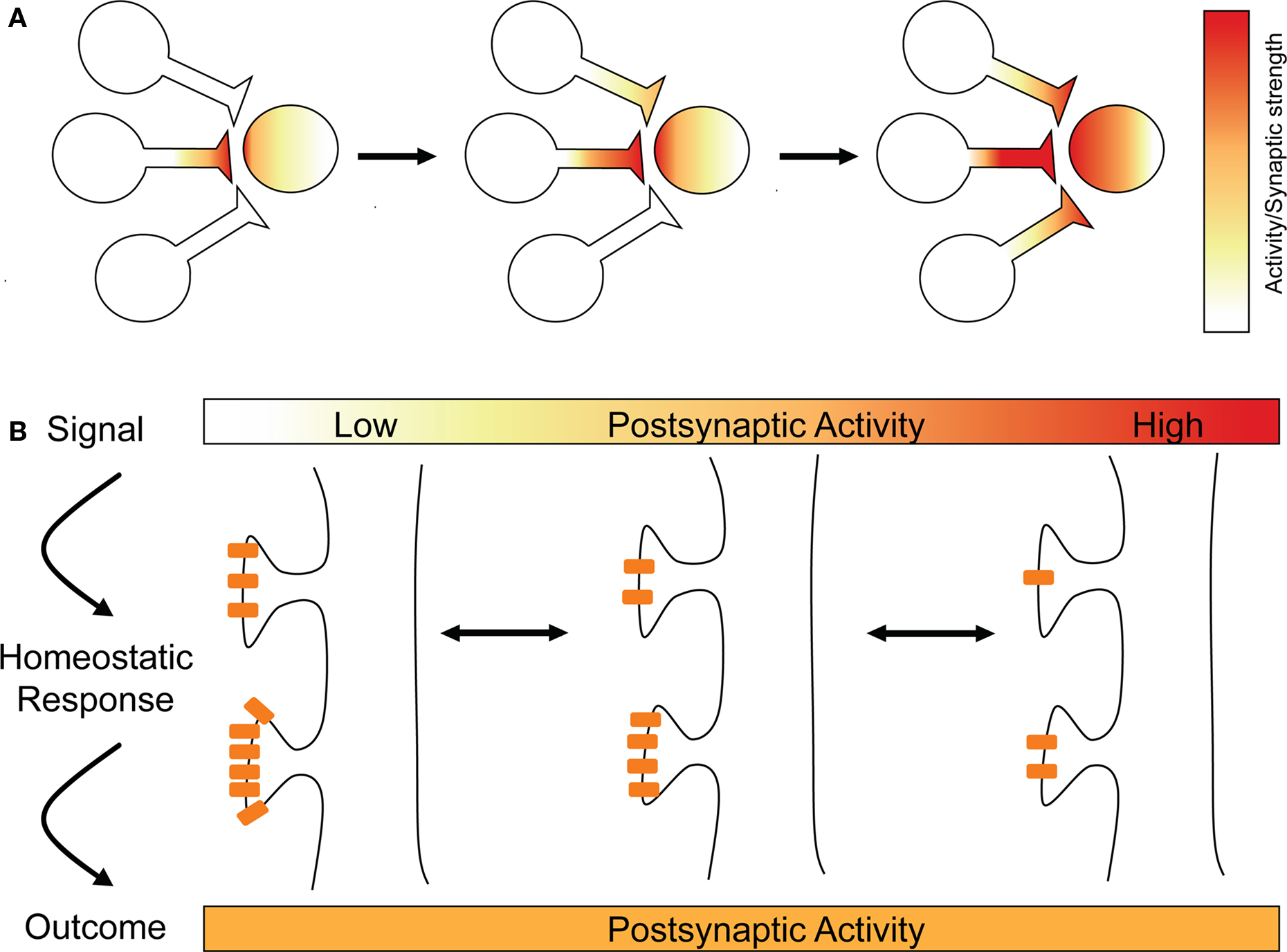
Figure 1. Runaway LTP and synaptic scaling. (A) Hebbian plasticity mechanisms can be unstable, as illustrated here with runaway potentiation. One synapse undergoes LTP (left), making it more likely that it will drive the postsynaptic cell, leading to more LTP (middle). As the postsynaptic cell’s firing increases, other synapses can now undergo LTP (right) that would not otherwise, leading to runaway potentiation and the loss of the postsynaptic neuron’s capacity to store information in its synaptic weights. (B). Long-term changes in postsynaptic activity is the signal for synaptic scaling, which acts homeostatically to bring the postsynaptic cell’s activity back to its optimal range. Synaptic scaling is illustrated by multiplicative changes in AMPA receptor content at synapses.
Synaptic Scaling
One adaptive mechanism possessed by many neurons to promote stability is synaptic scaling. Synaptic scaling appears to be a particularly parsimonious cellular stability mechanism. It directly regulates the strength of synapses – the same synapses that, undergoing synaptic plasticity like STDP, are likely to be among the sources of destabilization of a neuron’s firing rate. With scaling, a neuron can keep its synapses within some optimal size range, which might be energetically advantageous. Synaptic scaling has been widely observed in the brain: including in cortical (Turrigiano et al., 1998 ; Watt et al., 2000 ), hippocampal (Lissin et al., 1998 ; Burrone et al., 2002 ; Thiagarajan et al., 2005 ), and spinal neurons (O’Brien et al., 1998 ), with the result that synaptic scaling is one of the best-studied homeostatic plasticity mechanisms to date.
Basic Experimental Evidence
Synaptic scaling generally occurs on a much slower timescale than Hebbian plasticity mechanisms like STDP: from hours to days (Ibata et al., 2008 ). Much of the work to date on synaptic scaling has utilized primary cultured neurons as a model system: dissociated neurons from a mixed population of excitatory and inhibitory neurons in culture form synapses and develop spontaneous activity. Since these networks are grown on culture dishes, global activity levels can be easily modulated pharmacologically; additionally, it is straightforward to measure synaptic strengths either electrophysiologically or immunocytochemically in real time.
When global activity levels are lowered with either antagonists of excitatory synaptic transmission or tetrodotoxin (TTX), which blocks spiking activity, neurons scale their excitatory synapses up in strength (O’Brien et al., 1998 ; Turrigiano et al., 1998 ; Watt et al., 2000 ; Thiagarajan et al., 2005 ; Shepherd et al., 2006 ). This scaling takes the form of larger currents in response to spontaneous and/or evoked vesicle release (Figure 1 B). It is an adaptive response, meaning that its effect should lead the neuron to move its firing rate back to its functional regime, although neurons will not be able to compensate for the complete block of firing due to TTX. Synaptic scaling occurs gradually over hours to days (Turrigiano et al., 1998 ), but can be observed as early as 4 h after activity is blocked (Ibata et al., 2008 ). Synergistic, homeostatic changes at inhibitory synapses have also been observed (Kilman et al., 2002 ; Maffei et al., 2004 , 2006 ; Swanwick et al., 2006 ); however, this will focus on excitatory synapses.
As is true of temperature homeostasis, synaptic scaling is bidirectional, and neurons can respond to lowered neuronal activity as well as enhanced neuronal activity by altering the strength of their synapses. When inhibition mediated by the GABAA receptor is blocked in cultured neurons with drugs like bicuculline, this causes about a three-fold increase in the firing rates of excitatory neurons (Turrigiano et al., 1998 ). However, when this blockade is maintained for long time periods of 24–48 h, neurons slowly and progressively scale the strength of their synapses down, moving the firing rates of individual neurons back into their optimal range (Figure 1 B) (Lissin et al., 1998 ; Turrigiano et al., 1998 ). Similar effects on a neuron’s synapses are observed in spinal cultures when inhibition mediated by glycinergic neurons is blocked with strychnine (O’Brien et al., 1998 ).
One major difference between synaptic scaling and Hebbian forms of synaptic plasticity is that, in most experiments, the changes underlying synaptic scaling appear to occur across the entire population of a neuron’s synapses (Turrigiano et al., 1998 ; Harms and Craig, 2005 ; Thiagarajan et al., 2005 ; Ibata et al., 2008 ), rather than, as is normally true of LTP for instance, only a small fraction of a neuron’s inputs (Engert and Bonhoeffer, 1999 ). In this case, synaptic weights are scaled multiplicatively, which means that the relative differences in strength between any two inputs will be maintained (Figure 1 B). Functionally, this is important, since our current best understanding of processes like learning and memory in the brain suggest that memories are encoded in the relative differences in strength between inputs (Malenka and Nicoll, 1999 ; Neves et al., 2008 ).
However, some studies suggest that it is also possible for scaling-like processes to occur at a more local level, perhaps even at individual synapses (Sutton et al., 2006 ; Branco et al., 2008 ; Hou et al., 2008 ). Two points remain uncertain. First, there is the question of whether these local forms of scaling are the same as the global form that has typically been called “synaptic scaling.” In certain respects, the experiments supporting local scaling are difficult to reconcile with those supporting global scaling (e.g., compare Hou et al., 2008 , to Ehlers et al., 2007 ). And local scaling may be mediated by different physical mechanisms, sensitive to spontaneous rather than spike-evoked vesicle release (Sutton and Schuman, 2009 ). Second, it is not clear how local scaling coexists with Hebbian plasticity. The concern is that local scaling might counteract learning by erasing the relative differences in synaptic strength previously produced by LTP and LTD. A possible answer to this concern was offered by Rabinowitch and Segev (2008) . Noting that neighboring synapses on a single dendritic branch are not electrically isolated from each other, they argued that signals triggering homeostatic scaling at any one synapse would also affect synapses in close proximity. Multiplicative scaling would then still pertain, but the functional unit would be the dendritic compartment rather than the whole neuron. Rabinowitch and Segev further argued that local scaling of this type offers two distinct advantages: it prevents any one dendritic branch from dominating over the others and it allows for the selection of spatial patterns of inputs that are particularly efficacious in controlling neuronal output.
Although the culture preparation has proven to be ideally suited to characterizing synaptic scaling mechanisms, it is nonetheless somewhat removed from the intact animal. However, there is good evidence that synaptic scaling occurs in vivo (Desai et al., 2002 ; Maffei et al., 2004 ; Goel and Lee, 2007 ). In these studies, neurons of rodent visual cortex were deprived of normal activity for hours or days by either eyelid suture or intraocular TTX injection. Subsequent ex vivo recordings revealed that these manipulations produced homeostatic synaptic adaptations resulting in enhanced excitability in neurons of layers 2/3 and 4.
Detecting Activity
What is being regulated? Is it the firing rate of individual neurons, the levels of synaptic activation, or some property of the network as a whole? In principle all of these are possible – because the design of most of the experiments has not allowed for a clean separation of cellular, synaptic, and network parameters – but the first (or something closely related) seems most likely. Burrone and colleagues were able to address this question elegantly, by overexpressing a potassium channel (Kir2.1) to lower activity in a small fraction of neurons in a culture dish (Burrone and Murthy, 2003 ). They found that synaptic inputs onto a neuron were scaled up when the neuron’s firing rate was reduced, and that this synaptic scaling brought its firing rate back to near control levels. This result provided strong evidence that homeostatic synaptic scaling occurs at the level of individual neurons, and suggests that the strength of input that a neuron receives is titrated to keep its output stable. This study illustrates the complexity of homeostatic mechanisms, since changes in postsynaptic activity were precisely balanced by presynaptic changes in synaptic release dynamics. Furthermore, these changes happened only once synapses had been already established, and in fact opposite effects were observed when activity levels were reduced in younger cultures, when synapses were still forming. This is consistent with a recurring trend in the field of homeostatic plasticity: different mechanisms exist at different developmental stages.
What is the signal for synaptic scaling? Burrone’s work shows that a neuron’s own firing rate seems to be the key signal regulated, but what aspect of it is most important? Firing rate can be integrated over different time periods, for instance, and may look different in different parts of the neuron: its axon versus its dendrites, for instance. Furthermore, how a neuron reads this out, how it measures its own firing rate remains poorly understood. One recent study addressed the question of where activity is measured in a cell. By microperfusing TTX either onto a neuron’s soma or onto its primary apical dendrite, Ibata et al. (2008) demonstrated that somatic action potentials are required for synaptic scaling, and that blocking action potentials locally in the dendrite does not cause detectable alterations in synaptic strengths, either within the activity-blocked region or on dendritic regions outside of it. However, a study by Branco et al. (2008) shows that presynaptic release probability can be homeostatically controlled by depolarization of the local dendritic branch. How can we account for these apparently conflicting results? Perhaps synaptic scaling expressed through pre- and postsynaptic mechanisms are triggered by different signals that have different loci of origin.
One of the usual suspects when considering activity signals is internal calcium, an ion whose concentration is tightly regulated in neurons, and is known to play a critical role in many cellular processes. Indeed, Ibata et al. (2008) found that blocking somatic calcium transients had an effect on synaptic strengths similar to that of spiking activity. Furthermore, they found that activity deprivation caused decreased expression of the nuclear Ca2+-calmodulin-dependent kinase CaMKIV, and that this likely had its effect through protein transcription. Thiagarajan et al. (2005) have also found that homeostatic synaptic alterations are linked to reductions in calcium entry in the postsynaptic cell.
This would appear to make good intuitive sense: a neuron somehow integrates its calcium levels as a read-out for activity, leading to alterations in gene expression in the soma that is then directed to synapses. In this way, the same signal – however, with different location and integrated over a different timescale – could be responsible for both Hebbian synaptic plasticity (Amici et al., 2009 ) and synaptic scaling (Ibata et al., 2008 ). However elegant and satisfying this may be from a design perspective, it is probably an incomplete picture of synaptic plasticity. As is often the case in biological systems, the truth is somewhat more complicated. Indeed, there may be multiple signaling mechanisms involved in signaling in homeostatic plasticity. In particular, there is evidence that a number of molecules that are released into the extracellular space can affect scaling and may well mediate it in some cases (Rutherford et al., 1998 ; Stellwagen and Malenka, 2006 ; Aoto et al., 2008 ). These molecules can diffuse over distances spanning small numbers of neurons.
Perhaps the most surprising result of this sort in recent years was made by Stellwagen and Malenka (2006) , when they showed that a soluble released factor, a molecule called tumor necrosis factor alpha (TNFα) was required for synaptic scaling. What made this finding so surprising was that TNFα was released from glial cells rather than neurons. In hindsight, this finding makes sense: glial cells are abundant in the CNS, are intimately related to neurons, and have mechanisms in place to measure a neuron’s activity levels (Haydon, 2001 ). These experiments were performed in cultured neurons that are grown on beds of glial cells, a typical procedure for culturing neurons (Smith, 1998 ). However, it will be interesting to determine if in the intact neuron, it is a particular class of glial cells – perhaps one that preferentially associates with neuronal soma – that is responsible for mediating this signal for synaptic scaling. This work also suggests that in diseases that affect glial cells, pathogenesis may arise through feedback to synapses via this synaptic scaling mechanism (McCoy and Tansey, 2008 ).
Brain-derived neurotrophic factor (BDNF) is another soluble factor implicated in synaptic scaling (Rutherford et al., 1998 ). BDNF is produced in excitatory pyramidal neurons, and its expression and release depend on neuronal activity (Poo, 2001 ). Since soluble release factors participate in synaptic scaling, the relative physical locations of neurons and glia will partly determine whether neurons respond only to their own activity or also that of their neighbors. If a neuron is isolated and receives signals via BDNF in a mostly autocrine fashion, it is likely to respond to its own activity exclusively. Yet if the glial cells that insulated it from its neighbors were perturbed, it may begin to respond to more widespread changes in activity. Similarly, if glial cells sense a neuron’s activity levels and signal back to it by releasing TNFα, then the precise location and spread of those glial cells will be critical in determining just what activity is being sensed. Interestingly, BDNF is also implicated in LTP, where it is thought to be required for expression of the late, protein-dependent phase of LTP (Minichiello, 2009 ), or alternatively, synaptic tagging (Lu et al., 2008 ). How can a molecule be involved in two opposing processes: Hebbian and homeostatic plasticity? It could be that differences in release location or volume of BDNF determine whether homeostatic or Hebbian mechanisms are engaged. An alternative explanation is that BDNF is permissive for both processes, while other signals determine the type of plasticity expressed.
The usefulness of a cell autonomous signal, such as calcium, seems clear: it provides a straightforward means of implementing single-cell homeostasis (LeMasson et al., 1993 ). But why should there be cell non-autonomous (diffusable) signals as well? One idea is that they help to implement network-level homeostasis (Maffei and Fontanini, 2009 ). Single neurons obviously do not operate in isolation but as members of neural circuits. While it is possible, in principle, to maintain stable circuit behavior even without a coordinating signal between neurons (Renart et al., 2003 ), in practice, coordination might make for more robust regulation. Evidence that this is so comes from studies of the effects of sensory deprivation in visual cortex (Desai et al., 2002 ; Maffei et al., 2004 ; Maffei and Turrigiano, 2008 ). Rather than a simple upregulation of excitatory synapses, deprivation triggers a number of different homeostatic responses, which vary depending upon cortical layer, developmental age, cell identity, and the specific deprivation protocol employed. It has not actually been established that these responses are coordinated – much less what the coordinating signals are – but it is difficult picture how this system would work otherwise.
Expression Mechanisms
How are synapses altered during synaptic scaling? This has been one of the most well studied aspects of synaptic scaling to date, and has been reviewed elsewhere extensively (Turrigiano, 2008 ). Here we will limit ourselves to aspects of the question that intersect most directly with Hebbian plasticity.
Several groups have found evidence for postsynaptic changes in the densities of both AMPA receptors (Lissin et al., 1998 ; O’Brien et al., 1998 ; Turrigiano et al., 1998 ; Wierenga et al., 2005 ; Shepherd et al., 2006 ; Stellwagen and Malenka, 2006 ) and NMDA receptors (Rao and Craig, 1997 ; Watt et al., 2000 ; Mu et al., 2003 ). While there is broad agreement that AMPA receptor numbers are regulated, there are conflicting reports about the subunit composition of the affected receptors. Some researchers have observed changes in GluR2-containing receptors (O’Brien et al., 1998 ; Cingolani et al., 2008 ), others in GluR1-containing receptors (Ju et al., 2004 ; Thiagarajan et al., 2005 ), and still others in both (Wierenga et al., 2005 ).
AMPA and NMDA appear to be proportionally regulated by activity in cortical neurons (Watt et al., 2000 ). While we might expect that processes like LTP and LTD, which appear to affect AMPA receptors preferentially, would degrade this relationship (Lisman, 2003 ), this is apparently not always the case. Watt et al. (2004) demonstrated that, at neocortical synapses, LTP of AMPA currents, whether induced chemically or by an STDP protocol, is followed by LTP of NMDA currents on a slower timescale (hours versus minutes). Most importantly, the late NMDA potentiation is done in such a way that the original NMDA-to-AMPA ratio is regained (Watt et al., 2004 ). This highlights the importance of timescale in our understanding of how mechanisms like Hebbian and homeostatic plasticity interact.
In addition to postsynaptic changes, homeostatic presynaptic changes have also been observed (Murthy et al., 2001 ; Burrone et al., 2002 ; De Gois et al., 2005 ; Erickson et al., 2006 ; Wierenga et al., 2006 ; Branco et al., 2008 ). Some of the differences in expression location may reflect developmental differences: Wierenga and colleagues showed that in neurons in culture for less than 3 weeks, synaptic scaling is expressed purely postsynaptically, whereas in older cultures, both pre- and postsynaptic alterations contribute (Wierenga et al., 2006 ). This suggests that neurons may possess several different mechanisms for synaptic scaling, and that the precise expression mechanism utilized will depend on several factors, including developmental stage.
You may be forgiven if you are experiencing a sense of déjà vu at this moment; research into the expression mechanisms of synaptic scaling have interesting parallels with the LTP and LTD fields (Lisman, 2003 ; Duguid and Sjostrom, 2006 ), both in the “pre- versus postsynaptic” debate, and in the apparent resolution of this debate, that neurons may use either or both loci. Furthermore, many of the players in both the LTP and synaptic scaling fields are the same: calcium acts as the signal that is ultimately expressed as a change in AMPA receptors at synapses. Indeed, these are not the only similarities. Several other intracellular signaling molecules have been implicated in synaptic scaling that also may play a role in LTP or LTD, such as Arc (Rial Verde et al., 2006 ; Shepherd et al., 2006 ), polo-like kinase 2 (Plk2) and CDK5 (Seeburg et al., 2008 ), and CaMKIV (Ibata et al., 2008 ). Additionally, trans-synaptic signaling molecules like β3 Integrin (Cingolani et al., 2008 ) and MHC1 (Goddard et al., 2007 ) have been implicated in mediating synaptic scaling. How these signaling mechanisms interact remains to be determined.
Homeostatic Intrinsic Plasticity
The intrinsic electrical properties of neurons are determined largely by their expression of voltage- and calcium-gated ion channels. The diversity of these channels in mammalian central neurons is staggering: no fewer than 36 separate genes are required to account for the principal subunits of the potassium channels alone (Vacher et al., 2008 ). How the brain actually uses this diversity is among the most puzzling questions in neuroscience. It is made especially challenging by the fact that a neuron’s complement of ion channels is not simply dictated by a set of genetic instructions, nor is it hard-wired during early childhood. Rather, like its synapses, a neuron’s intrinsic electrical properties evolve throughout life, often under the influence of activity-dependent plasticity (Zhang and Linden, 2003 ; Frick and Johnston, 2005 ). Indeed, many of the same experimental protocols used to study synaptic plasticity (including STDP) also produce intrinsic plasticity.
Basic Experimental Evidence
The most straightforward way for changes in intrinsic properties to help neurons and circuits maintain appropriate levels of electrical activity is through overall shifts in cellular excitability. In particular, the gain and/or threshold of individual neurons might be adjusted to match whatever average synaptic input they receive. Consider an idealized f–I curve (Figure 2 A), which relates a neuron’s output (firing rate) to its input (synaptic current). If the average input is too low, the neuron will hardly ever fire, because of the spike threshold; if it is too high, the firing rate will saturate, because there is some physical limit on how fast a neuron can fire. Between the two extremes is a sensitive region, in which the neuron’s output really does reflect its input. A robust strategy for firing rate stability, when there are large or long-lasting fluctuations in average input, is to shift the position or slope of the f–I curve so that the sensitive region always corresponds well with the distribution of inputs. In this way, the neuron’s dynamic range can be preserved.
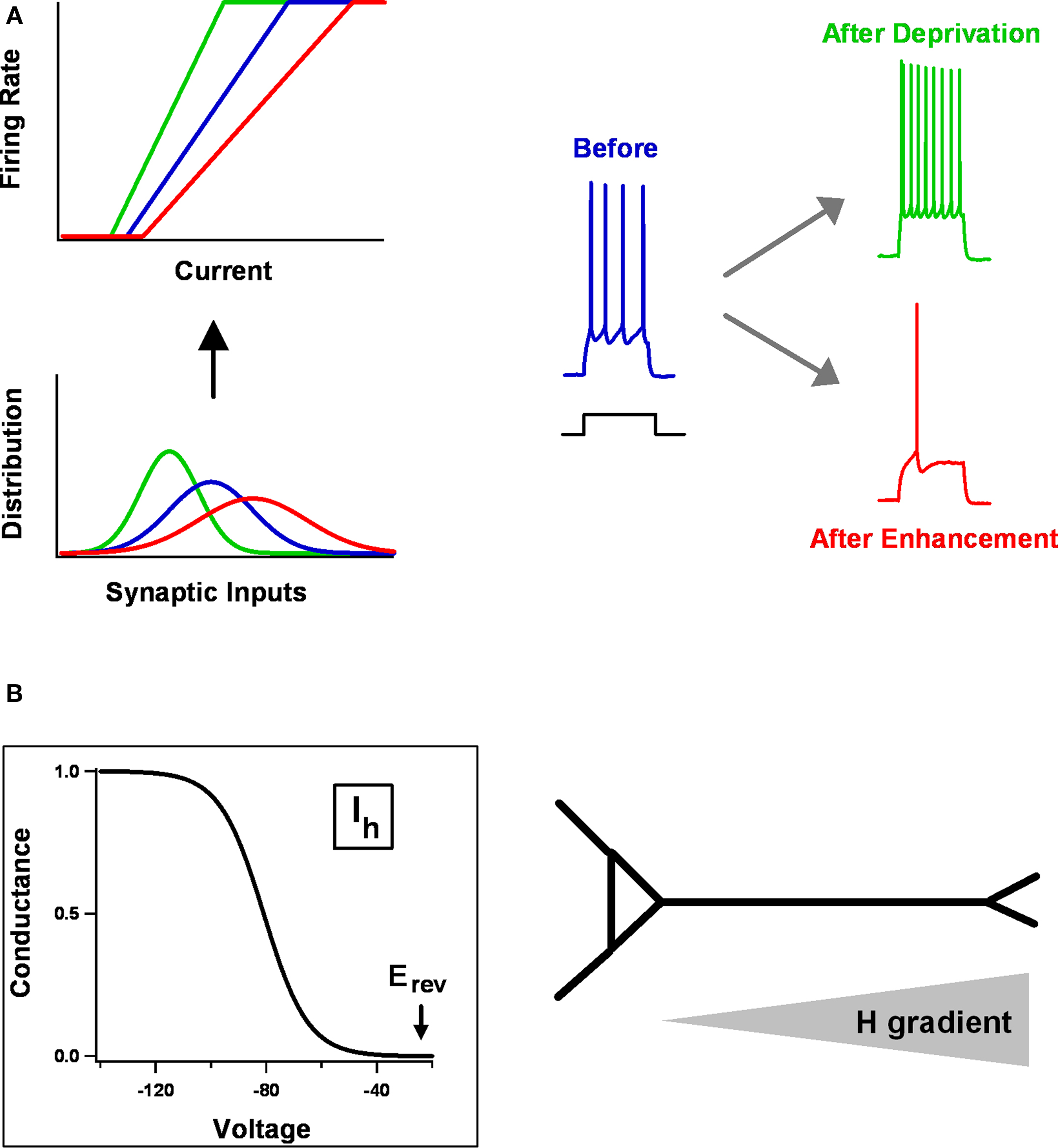
Figure 2. Homeostatic intrinsic plasticity. (A) A general way of adapting a neuron’s intrinsic excitability in response to changes in the mean or variance of synaptic inputs is by adjusting the gain (slope) or threshold (x-intercept) of its f–I curve. At top-left are stylized f–I curves, relating input current to output firing rate. These are matched to the three synaptic input distributions shown below. Experimentally what one observes is at right. After prolonged activity deprivation, neurons become hyper-excitable – for example, they fire more strongly in response to a given current step. After activity enhancement, they become hypo-excitable. (B) Distinctive properties of Ih make it interact with synaptic changes wrought by STDP. Ih activates in response to hyperpolarization, is partially active at resting potentials (≈ −60 mV), and its Erev sits at the base of the activation curve, making it a stabilizing force. In pyramidal neurons, HCN channels are expressed mostly in apical dendrites, with a density that increases as one moves distally.
This is a simple idea that nevertheless appears to hold (roughly) in a wide variety of real systems. Experimental evidence for the general strategy has been found in neocortex, hippocampus, striatum, and brainstem; in pyramidal neurons and interneurons; in mammalian brains and invertebrate brains; using tissue from juvenile animals and adult animals; after activity manipulation in cell cultures, slice cultures, and the intact brain (Turrigiano et al., 1994 ; Desai et al., 1999a ,b ; Aizenman et al., 2003 ; Nelson et al., 2003 ; Aptowicz et al., 2004 ; Gibson et al., 2006 ; Karmarkar and Buonomano, 2006 ; van Welie et al., 2006b ; Echegoyen et al., 2007 ; Pratt and Aizenman, 2007 ; Bartley et al., 2008 ; Maffei and Turrigiano, 2008 ; Azdad et al., 2009 ; Breton and Stuart, 2009 ; Mohapatra et al., 2009 ; Wilhelm et al., 2009 ; O’Leary et al., 2010 ). A typical experiment is that of Karmarkar and Buonomano (2006) , who examined the intrinsic excitability of CA1 pyramidal neurons in organotypic slice cultures. This preparation, like other culture systems, exhibits pronounced spontaneous spiking activity even in the absence of external stimuli. The activity is driven by the cultures’ dense excitatory and inhibitory connectivity. Karmarkar and Buonomano (2006) were therefore able, by applying pharmacological blockers of either inhibition or excitation, to drive average spontaneous firing rates up or down for a period of days. At the end of this period, they assayed the excitability of individual neurons by measuring the number of spikes fired in response to current injections of different amplitudes, and used the measurements to construct f–I curves. What they found was similar to the idealized case (Figure 2 A). Chronically reducing firing rates tilted the f–I curve up and to the left, whereas increasing them tilted it down and to the right. The changes in intrinsic excitability were accomplished without grossly deforming firing properties: individual spike waveforms were unaltered, as were the neurons’ electrophysiological identities (i.e., regular spiking neurons did not become fast spiking or rhythmically bursting). Instead, what was tuned by the activity manipulations was sensitivity: neurons responded to low (or high) activity conditions by becoming more (or less) sensitive to whatever input they received, as one would want for homeostatic regulation.
What is being regulated? Or more precisely, what signal controls whether neuronal excitability is increased or decreased? As is true of synaptic scaling, the answer appears to be calcium influx due to spike firing and depolarization (LeMasson et al., 1993 ). For mammalian central neurons, the argument rests on the observations that activity-dependent shifts in excitability can be produced by directly depolarizing neurons with externally-applied potassium (van Welie et al., 2004 ; O’Leary et al., 2010 ) or with puffs of glutamate (van Welie et al., 2004 ) and that homeostatic intrinsic plasticity can be prevented by blocking voltage-dependent calcium channels, other sources of calcium, and downstream calcium-related pathways (Fan et al., 2005 ; Frick and Johnston, 2005 ; Wu et al., 2008 ; O’Leary et al., 2010 ). Intracellular calcium levels track average firing rates quite well under physiological conditions. Using them as an “activity signal” would make for a robust regulatory mechanism, because each neuron could self-regulate. It would also allow for some kind of coordination between homeostatic intrinsic plasticity and both Hebbian and homeostatic forms of synaptic plasticity, as all would depend (somehow) on shared calcium signals. However, as is also true of synaptic scaling, there may be separate extracellular signaling pathways. In particular, manipulating BDNF levels and receptor activation affects the induction of intrinsic plasticity in culture (Desai et al., 1999a ). Whether BDNF actually mediates network-level homeostasis under physiological conditions is not known.
While the similarity of results obtained in different preparations and under different experimental conditions is remarkable, there are important differences. One is in the timescale of effects. In some cases, intrinsic excitability alters in response to activity only after hours or days have passed (Desai et al., 1999b ; Aptowicz et al., 2004 ; Gibson et al., 2006 ). In others, homeostatic regulation of excitability is apparent after only minutes (Nelson et al., 2003 ; Misonou et al., 2004 ; Fan et al., 2005 ; van Welie et al., 2006a ). The reason for the variability is uncertain. Part of it may simply be a consequence of differences in experimental techniques and methodology, but part of it probably reflects real differences in the underlying physiology. The experiments showing slow intrinsic plasticity were designed mainly to explore how neurons and neural circuits might respond to the stability challenges posed by postnatal development, which is an inherently slow process. On the other hand, the experiments showing fast intrinsic plasticity were motivated more by the need to counter the destabilizing effects of Hebbian plasticity and of temporary fluctuations in synaptic input. In fact, several actually employed standard LTP protocols in order to demonstrate homeostatic regulation of intrinsic excitability (Fan et al., 2005 ; Brager and Johnston, 2007 ; Campanac et al., 2008 ).
The other major difference between the various experimental results is in the specific ion channels regulated by homeostatic processes. In mammalian systems alone, depending on the activity manipulation employed and the type of neurons studied, the channels carrying one or more of the following currents might be altered: transient sodium, sustained potassium, M-type potassium, long-lasting calcium, hyperpolarization-activated cyclic nucleotide (HCN), and leak (Desai et al., 1999b ; van Welie et al., 2004 ; Fan et al., 2005 ; Gibson et al., 2006 ; Trasande and Ramirez, 2007 ; Wu et al., 2008 ; Breton and Stuart, 2009 ). Moreover, activity manipulation affects not only channel density, but also localization and gating characteristics. Consequently, while the idealized regulation of Figure 2 A (a simple shift of the f–I curve) is often true, the effects of homeostatic intrinsic plasticity can be quite complicated. In some cases, intrinsic plasticity involves not simply changes in gain or threshold but changes in spike frequency adaptation, afterpotentials, synaptic integration, local dendritic excitability, temporal firing patterns, and resonance characteristics (van Welie et al., 2004 ; Frick and Johnston, 2005 ; Trasande and Ramirez, 2007 ; Johnston and Narayanan, 2008 ). This diversity can be both good and bad, if the goal is neuronal and network stability. Some systems make excellent use of it. One of these is the crustacean stomatogastric ganglion (STG), a small circuit in which neurons have well-defined roles and stereotyped firing behavior (Marder and Bucher, 2007 ). Maintaining particular firing patterns (e.g., bursting or rhythmic activity) is crucial to circuit function; and the STG exhibits a broad range of homeostatic responses to perturbations that alter firing characteristics, adjusting inward and outward conductances in a coordinated way so as to restore function. But in other systems the diverse intrinsic changes can produce pathological states. Sensory deprivation of rat barrel cortex, by whisker trimming, downregulates HCN channel density in the dendrites of layer 5 pyramidal neurons (Breton and Stuart, 2009 ). This makes the dendrites more excitable, but it also increases the proportion of neurons that discharge strong bursts of action potentials. This might leave the system vulnerable to epileptic instability (Poolos, 2005 ). Indeed, such untoward effects of homeostatic plasticity have been hypothesized to contribute not only to epilepsy but to neuropsychiatric disorders (Ramocki and Zoghbi, 2008 ).
Relation to Synaptic Scaling
The experimental protocols used to induce homeostatic intrinsic plasticity have (with some exceptions) been identical to those used to induce synaptic scaling: chronic activity manipulation of cultured neurons and networks, sensory deprivation of cortex, pharmacological silencing of spiking activity in hippocampus. Even so, the relationship between the two types of homeostatic plasticity remains unclear. Why do they both exist? Are they expressed under the same circumstances? How do they work together? The answers to these questions are required if we are truly to understand how homeostatic regulation operates in real networks (Maffei and Fontanini, 2009 ).
An attractive idea is that synaptic scaling and homeostatic intrinsic plasticity operate in parallel as redundant regulatory mechanisms. Redundancy seems to be a ubiquitous design principle of biological systems (Tononi et al., 1999 ). Along these lines, a notable result is that of Maffei and Turrigiano (2008) . They studied the effects on the rodent monocular visual cortex of two forms of sensory deprivation: lid suture and intraocular injection of the sodium channel blocker TTX. Both disrupt normal visually-evoked drive to cortex, but whereas intraocular TTX eliminates all retinal activity, lid suture allows for the spontaneous firing of retinal ganglion cells. Maffei and Turrigiano found that layer 2/3 pyramidal neurons exhibited homeostatic responses to both types of deprivation, but that the nature of the responses differed. After lid suture, the intrinsic excitability of individual cells was upregulated; after intraocular TTX, the upregulation was instead that of excitatory synapses. Spontaneous retinal firing after lid suture is known to drive robust LTD in visual cortex (Rittenhouse et al., 1999 ). One explanation for the difference between intraocular TTX and lid suture is that, while synaptic scaling was able to produce a sufficient homeostatic response to the former, it was unable to overcome the strong LTD produced by the latter. Instead, a redundant homeostatic mechanism, intrinsic plasticity, was recruited.
Another attractive idea is that synaptic scaling and homeostatic intrinsic plasticity may be active at different developmental stages and may be engaged in a definite temporal order (Karmarkar and Buonomano, 2006 ; Echegoyen et al., 2007 ). Returning to the Karmarkar and Buonomano (2006) experiments described above: using the same recordings with which they examined plasticity of intrinsic excitability, they also examined plasticity of inhibition. They found that both were expressed in immature cultures, with changes in intrinsic excitability always appearing first, followed some time later by changes in inhibition. However, in mature cultures, only intrinsic plasticity was expressed. To the extent that development of organotypic slices seems to mimic that of hippocampus in vivo (De Simoni et al., 2003 ), this raises the possibility that intrinsic plasticity is the dominant homeostatic process of the adult brain. This notion is also hinted at by the work of Echegoyen et al. (2007) who subjected hippocampal networks in vivo to activity deprivation by sustained release of TTX via implanted polymer strips. These experiments showed that homeostatic intrinsic plasticity was robust in both juvenile and adult animals, but that effects on synaptic currents varied over development in ways that might not necessarily promote homeostasis. It may be that, in some cases, relying on homeostatic intrinsic plasticity is preferable to relying on synaptic scaling so as not to interfere with synaptically-stored information.
Perhaps the most attractive idea of all is that synaptic scaling and homeostatic intrinsic plasticity act synergistically. The term “homeostatic regulation” is a rather general one that can mean different things in different contexts. It can encompass tasks as distinct as controlling the firing rates of individual neurons or the activity of large networks, preserving uniform synaptic weight distributions (or at least ones in which weights do not cluster at extremes), and ensuring that the dynamic range of response properties matches that of inputs. Neither synaptic scaling nor homeostatic intrinsic plasticity, as complicated and flexible as they may be, seems well positioned to do all of these things by themselves. Lazar et al. (2009) explored this idea in a thought-provoking computational model. They endowed a recurrent neural network with three forms of plasticity – STDP, synaptic normalization (similar to scaling), and homeostatic intrinsic plasticity – and investigated its ability to learn spatio-temporal patterns in its inputs. Not only did the network outperform comparable, optimally tuned static networks, but both types of homeostatic plasticity were necessary to maintain healthy dynamics. Without synaptic normalization, the network exhibited seizure-like bursts of activity, even when driven by random inputs. Without intrinsic plasticity, many neurons in the network fired at aberrantly high rates, while others fell silent. Only when both were included was the network able to make efficient use of its resources.
Interaction with STDP
STDP is sensitive to several features of cellular excitability, including resting potential, firing frequency, and action potential backpropagation (reviewed by Sjostrom et al., 2008 ). We expect then that, rather than simply acting in parallel with synaptic plasticity, homeostatic intrinsic plasticity should be able to affect the induction of STDP itself. When a neuron’s average activity is low, increasing intrinsic excitability (by whatever means) should make subsequent synaptic potentiation more likely. Conversely, when average activity is high, decreasing intrinsic excitability should instead make subsequent synaptic depression more likely. This plasticity of plasticity, which has been termed “metaplasticity,” represents a way for plasticity of intrinsic properties to affect (indirectly) synaptic ones and a distinct method of homeostatic adaptation (Abraham, 2008 ). The idea has been treated most formally in the context of the Bienenstock, Cooper, and Munro (BCM) model of synaptic plasticity, which we discuss in the next section.
One class of the ion channels regulated by homeostatic plasticity, the HCN channels that carry the H current, display unique biophysical properties that make them especially suited to modulating STDP (Figure 2 B) (Atkinson and Williams, 2009 ; Biel et al., 2009 ). Unlike “normal” voltage-gated channels, HCN channels are activated by membrane hyperpolarization rather than depolarization, with a reversal potential that sits near the base of the activation curve. They do not display voltage-dependent inactivation, and they are partially open at typical resting potentials. In both hippocampal CA1 and neocortical layer 5 pyramidal neurons, HCN channels are highly expressed along the apical dendrite, with a channel density that gets progressively greater with distance from the soma. Together, these properties endow HCN channels with several important physiological roles (Narayanan and Johnston, 2008 ; Atkinson and Williams, 2009 ; Biel et al., 2009 ): they help determine resting potential and input resistance, shape the time course of synaptic potentials and how they summate, and control the spread of synaptic potentials along the dendritic tree.
All of these roles suggest ways in which plasticity of the H current might affect STDP and vice versa (Kampa et al., 2007 ; Shin and Chetkovich, 2007 ; Campanac and Debanne, 2008 ; Sjostrom et al., 2008 ). For the case of homeostatic regulation, most are as yet unexplored. However, there has been relevant work using traditional rate-based LTP and LTD protocols in hippocampal CA1 neurons. Fan et al. (2005) found that theta-burst pairing of Schaffer collateral inputs and postsynaptic firing not only resulted in robust LTP but also produced a general decrease in cellular excitability, which like LTP itself depended on NMDA activation and downstream calcium pathways. Brager and Johnston (2007) complemented this finding by showing that LTD evoked by low-frequency pairing was instead accompanied by a general increase in cellular excitability. In both cases, the intrinsic plasticity was mediated by up- or downregulation of the H current and the attendant effects on input resistance. At first glance, these two results might seem to contradict experiments showing that LTP (or LTD) evoked by STDP produces a localized increase (or decrease) in excitability (Campanac and Debanne, 2008 ). However, the discrepancy might be resolved by the observation that the magnitude of LTP controls whether the H current is up- or downregulated (Campanac et al., 2008 ). Strong LTP results in upregulation, weak LTP in downregulation. In other words, homeostatic intrinsic plasticity may only kick in when synapses are near saturation, which is when one would expect homeostatic regulation to be most necessary (Roth-Alpermann et al., 2006 ).
Metaplasticity and the BCM Model of Synaptic Plasticity
Most studies of homeostatic plasticity, whether synaptic or intrinsic, have treated its actions as essentially independent of those of Hebbian plasticity. That is, the general picture has been one of homeostatic plasticity operating in parallel with LTP and LTD, complementing them, but mediated by distinct biophysical mechanisms and operating without any specific coordination. But, as we have noted, this dichotomy between the two types of plasticity is not strictly necessary. Indeed, one might argue that, from the standpoint of network function, the most stable kind of homeostatic regulation is one that is embedded in the rules of Hebbian plasticity itself. One way of doing this is by making the capacity of synapses to undergo Hebbian modification depend upon their history of use or upon the history of neuronal activity, an idea called metaplasticity (Abraham, 2008 ). The most influential attempt to understand plasticity in this way was made by BCM nearly 30 years ago (Bienenstock et al., 1982 ).
BCM Rule
In the BCM formulation, individual excitatory synapses can undergo both potentiation and depression (Figure 3 A). Whether a synapse is strengthened or weakened by presynaptic activity depends upon whether postsynaptic activity is above or below a threshold. Synaptic input that drives postsynaptic firing to high levels results in an increase in synaptic strength, whereas input that produces only low levels of postsynaptic firing results in a decrease. The threshold firing rate – the crossover point between LTP and LTD – is itself a slow function of postsynaptic activity, moving so as to make LTP more likely when average activity is low and less likely when it is high. This sliding of the threshold can stabilize synaptic dynamics, if its dependence on activity is chosen appropriately. For example, a simple implementation of the BCM rule is:
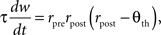
where w is the synaptic weight, τ is a time constant (a characteristic time for Hebbian modification), rpre and rpost are the presynaptic and postsynaptic firing rates, and Θth is the threshold. The condition for stability is that Θth grows faster than linearly with a running average of postsynaptic firing rate. The average is taken on a timescale slower than that of fluctuations in activity patterns but faster than that of changes in synaptic weight distribution. Other formulations of the BCM idea, with somewhat different properties, have also been proposed (Artola and Singer, 1993 ; Law and Cooper, 1994 ; Toyoizumi et al., 2005 ).
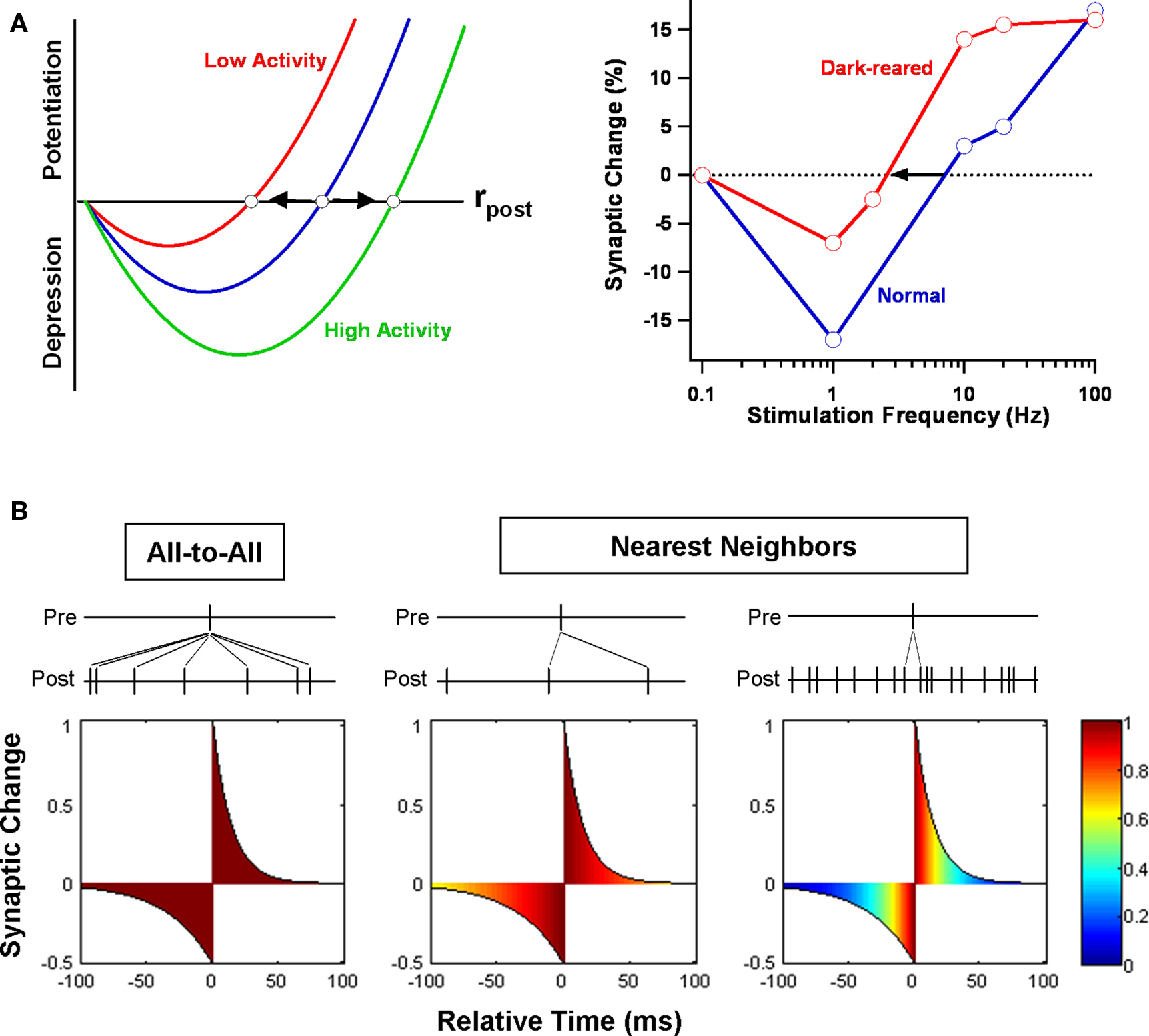
Figure 3. BCM model. (A) A standard implementation of the BCM rule is shown at left. Plasticity is a quadratic function of postsynaptic firing rate rpost. When rpost is below a specified threshold, presynaptic firing induces LTD; when above, LTP. Prolonged periods of low or high activity slide the position of the threshold bidirectionally. An experimental demonstration of the rule at rat visual cortical synapses is illustrated at right (Kirkwood et al., 1996 ). The threshold stimulation frequency separating LTD and LTP shifted left after several days of dark rearing. (B) Whether the BCM model and STDP are compatible depends on the STDP update rule employed (Izhikevich and Desai, 2003 ). If neurons are only weakly correlated, assuming that all combinations of pre- and postsynaptic spikes (all-to-all) contribute to plasticity means that the distribution of relative times will be uniform, as illustrated in the leftmost figure. (The color scale indicates the (normalized) fraction of contributing spike pairs for each relative time.) The integral of the STDP learning window is negative and only depression results in the all-to-all case. On the other hand, if only nearest-neighbor pairs contribute to plasticity, its sign depends on rpost. For low rates, the distribution of times is still mostly uniform, but for high rates, the distribution peaks near the origin. In the first case, net depression results, but in the second case, net potentiation results. Between the two rates sits a BCM-like threshold.
Numerous experimental tests of the BCM model have been conducted over the years – and it has stood up to them very well (Bear, 2003 ). Using rate-based protocols for induction of long-term plasticity, several different groups have confirmed the validity of key features of the BCM rule to synapses in both neocortex and hippocampus (Dudek and Bear, 1992 ; Kirkwood et al., 1996 ; Wang and Wagner, 1999 ; Abraham et al., 2001 ; Roth-Alpermann et al., 2006 ; Xu et al., 2009a ). Most importantly, they have demonstrated the existence of a threshold Θth that moves based on prior activity. To cite one example, when 2 days of dark rearing is used to decrease activity in rodent visual cortex, the threshold shifts so that LTP becomes easier to induce and LTD harder (Kirkwood et al., 1996 ; Philpot et al., 2001 ). More precisely, the minimum stimulation frequency necessary to produce LTP (at synapses connecting layer 4 to layer 2/3) is reduced, as is the amount of LTD produced by the smallest effective frequency (Figure 3 A). Brief re-exposure to light rapidly reverses the shift in the modification threshold, indicating that the effect is bidirectional. To cite another example, when granule cells of the dentate gyrus in awake rats are activated by strong stimulation of the medial perforant path, it becomes more difficult to potentiate synapses from the neighboring (but separate) lateral perforant path (Abraham et al., 2001 ). Moreover, a similar effect can be obtained by directly activating granule cells via antidromic stimulation of their mossy fiber axons. This is an important finding because it shows that sliding of the threshold is a postsynaptic, cell-wide property and not restricted to the synapses previously activated. It is also worth pointing out that, in just these two examples, the timescale of threshold modification varies between days, in the first case, to minutes, in the second.
Mapping STDP onto BCM
Since its introduction, the BCM rule has been widely used to model brain processes related to learning and experience-dependent development (Bear, 2003 ; Cooper et al., 2004 ; Abraham, 2008 ). Given its success, there has been considerable interest in understanding the relationship between BCM and STDP (Izhikevich and Desai, 2003 ; Burkitt et al., 2004 ; Pfister and Gerstner, 2006 ; Benuskova and Abraham, 2007 ). This is an aspect of a broader interest in understanding how traditional rate-based protocols for LTP and LTD are related to the newer timing-based protocols. If STDP rules can be mapped onto the BCM rule, the thinking goes, not only can a large body of experimental work be unified within a single conceptual framework, but network models based on STDP will inherit the useful competitive and stability properties that models based on BCM exhibit (Clopath et al., 2010 ). Efforts to combine STDP and BCM have typically fallen into one of two categories: phenomenology (reviewed by Morrison et al., 2008 ) or biophysics (Shouval et al., 2002 ; Yeung et al., 2004 ; Rubin et al., 2005 ).
The phenomenological efforts have sometimes adopted the strategy of beginning with a simple STDP rule and asking under what conditions it results in BCM-like plasticity. One builds up BCM from STDP by making the (not uncontested) assumption that spike timing is more fundamental to plasticity than spike rate (Lisman and Spruston, 2005 ). The simplest of these efforts is instructive, even if its applicability is limited (Izhikevich and Desai, 2003 ). Say that a neuron receives a large number of independent Poisson inputs and that each synapse evolves according to the rules of STDP. Is the end-result compatible with the BCM rule? The answer depends on how STDP is implemented (Figure 3 B). Under a standard implementation (Song et al., 2000 ), for each presynaptic spike, one sums the contributions from all preceding and all succeeding postsynaptic spikes. But if we do this, the distribution of relative times will be nearly flat. We end up sampling the whole STDP curve uniformly, and the net effect on synaptic weight is simply proportional to its integral. At most neocortical synapses, this is negative (Feldman, 2000 ), resulting in net depression regardless of postsynaptic firing rate. However, if we instead restrict STDP to nearest-neighbor pairs (Sjostrom et al., 2001 ), then, for high firing rates, relative times will mostly be short. We effectively sample the STDP curve only near the origin. The LTP maximum is larger than the LTD minimum, and we get potentiation. For low firing rates, longer relative times will also be common, so we sample more of the tails of the STDP curve. The LTD part decays more slowly than the LTP part, and we get depression. By varying the postsynaptic firing rate, we can obtain a function that mimics the BCM rule, with the threshold expressed in terms of the STDP parameters. One might then mimic sliding of the threshold by varying the parameters (Benuskova and Abraham, 2007 ).
Simple pair-based STDP rules, even if complemented with additional constraints, are not able to capture much of the phenomenology of STDP (Morrison et al., 2008 ). They do not, for example, account for the results of experiments in which the repetition frequency of pairs of spikes was varied or those in which spikes were paired, not singly, but in triplets or quadruplets. For this reason, more elaborate phenomenological models have been proposed, including ones that incorporate interactions between more than two spikes, postulate a hidden variable modulating efficacy, or allow synapses to exist in multiple states (Appleby and Elliott, 2005 ; Froemke et al., 2006 ; Pfister and Gerstner, 2006 ). A notable result is that of Clopath and colleagues (Clopath et al., 2010 ). In their model, synaptic changes elicited by presynaptic spikes depended not on postsynaptic spikes as such, but on postsynaptic membrane potential. Homeostasis was implemented by making the amount of LTD depend on a slow average of membrane potential. This approach not only accounted for much of the empirical STDP data, but allowed, in the case of Poisson inputs, for a good mapping to the BCM rule.
At the other end of the spectrum from phenomenology lies biophysics: perhaps more fundamental but also more difficult. The starting point for any biophysical description of plasticity is calcium (Castellani et al., 2001 ; Karmarkar and Buonomano, 2002 ; Shouval et al., 2002 ; Yeung et al., 2004 ; Rubin et al., 2005 ). The general approach models calcium influx and dynamics (e.g., buffering mechanisms), and relates these to variables known to affect synaptic transmission (e.g., activation of calcium-dependent enzymes). This approach is challenging for a couple of reasons: the biophysical workings of these processes are not fully understood, necessitating an element of phenomenology in every case; and there is evidence that the induction and the expression of synaptic plasticity have multiple loci, presynaptic as well as postsynaptic (Duguid and Sjostrom, 2006 ; Corlew et al., 2008 ). Even so, the work to date has been intriguing. For example, Shouval and colleagues built a model of plasticity that used calcium currents mediated by NMDA receptors as an associative signal (Shouval et al., 2002 ; Yeung et al., 2004 ). The resulting model neuron was both sensitive to temporal correlations in spike trains, as one expects from STDP, and able to respond selectively to a set of input rate patterns, as one expects from BCM. Stability was provided by a slow, homeostatic regulation of NMDA receptor levels. This seems compatible with the effects of synaptic scaling on NMDA conductance (Watt et al., 2000 ), but it is a bit difficult to reconcile with evidence that NMDA and AMPA currents are coregulated by LTP (Watt et al., 2004 ). We consider this issue next.
What Shifts the BCM Threshold?
The feature of the BCM rule that enables homeostatic regulation is the fact that the threshold between potentiation and depression shifts as a function of average postsynaptic activity. What might cause such a shift? Several answers to this question have been offered (Abraham, 2008 ).
The most compelling evidence involves activity-dependent regulation of NMDA receptor subunit composition (Cho et al., 2009 ; McCoy et al., 2009 ). NMDA receptors are heteromers consisting of two obligatory NR1 subunits and two regulatory subunits, usually a combination of NR2A and NR2B (Yashiro and Philpot, 2008 ). Early in life, NR2B levels are much higher than NR2A levels. However, as the brain matures, the ratio of NR2A to NR2B increases dramatically, as does the ratio of NMDA receptors containing NR2A to those containing NR2B. This increased NR2A/NR2B ratio has multiple effects on synaptic transmission and plasticity, because NR2B-containing receptors have slower channel kinetics and produce longer-lasting EPSPs; allow more calcium influx per EPSP; are more mobile; and interact more strongly with proteins important in the induction of LTP. For the BCM model, the important point is that the NR2A/NR2B ratio is sensitive to both sensory experience and activity-dependent plasticity. The leftward shift in the LTP/LTD threshold after dark rearing, described above, is accompanied by a decrease in the ratio; the rightward return after re-exposure to light is, conversely, accompanied by an increase (Quinlan et al., 1999 ; Philpot et al., 2001 , 2003 ). A causal relationship between changes in the ratio and sliding of the threshold is indicated by experiments on mutant mice in which the NR2A subunit was knocked out; these mice failed to show a threshold shift after dark rearing (Philpot et al., 2007 ). Furthermore, activity-dependent regulation of the NR2A/NR2B ratio is not limited to developmental timescales: at Schaffer collateral synapses onto CA1 pyramidal neurons, the ratio can be moved up or down by priming stimulations, with corresponding effects on LTP and LTD, after only minutes (Xu et al., 2009b ).
Might synaptic scaling of NMDA conductances (see Synaptic Scaling) contribute to sliding of the BCM threshold? It probably does. While both LTP and LTD require calcium influx through NMDA receptors, LTP is somewhat more sensitive to postsynaptic calcium than LTD, and in the developing neocortex LTD depends upon presynaptic rather than postsynaptic NMDA receptors (Sjostrom et al., 2003 ; Bender et al., 2006 ; Corlew et al., 2007 ). As a result, postsynaptic NMDA scaling might shift the balance between LTP and LTD, in a manner compatible with the BCM rule. However, NMDA scaling is unlikely to be the initial cause of threshold modification. NMDA and AMPA current amplitudes are coregulated by synaptic scaling in neocortical cultures, with no evidence that NMDA scaling precedes or triggers AMPA scaling (Watt et al., 2000 ). In fact, LTP of AMPA currents, whether induced pharmacologically or by STDP, is followed on a slower timescale by a corresponding increase in NMDA currents (Watt et al., 2004 ), which is the opposite of what the BCM model would suggest. Moreover, NMDA scaling does not affect the decay time constant of NMDA currents, as one would expect if the NR2A/NR2B ratio were being altered and as is observed in visual cortex after dark rearing (Watt et al., 2000 ; Philpot et al., 2001 ).
There are at least two other ways in which synaptic scaling and homeostatic intrinsic plasticity might affect the BCM threshold. These are in addition to and more direct than their general effects on average postsynaptic firing rate. One way is by regulating dendritic excitability. By this we mean such things as backpropagation of action potentials, generation of calcium bursts and dendritic spikes, and compartmentalization of the dendritic tree. All are believed to affect the induction of STDP at excitatory synapses (Kampa et al., 2007 ; Sjostrom et al., 2008 ), and all will be affected by the synaptic and intrinsic changes described in the previous two sections. The other way is simply by regulating average membrane conductance. Conductance is important to STDP because of its effect on EPSP kinetics (Fuenzalida et al., 2007 ). Intrinsic plasticity changes conductance more or less directly. Synaptic scaling does so through its effects on synaptic background activity, which is a ubiquitous feature of the brains of behaving animals and which, in pyramidal neurons, can increase average membrane conductance by as much as a factor of five (Destexhe et al., 2003 ). Crucially, postsynaptic conductance preferentially affects the LTP part of STDP; even high levels of conductance have only a minimal effect on timing-dependent LTD (Delgado et al., 2009 ). This asymmetry suggests that modulating conductance is an effective method of moving the threshold between LTP and LTD.
Balanced STDP
In the introduction, we noted that one motivation for studies of homeostatic plasticity is the concern that Hebbian networks are potentially unstable because of positive feedback loops. The concern is especially great when plasticity is based only on presynaptic–postsynaptic correlations. But for STDP at synapses that also express short-term plasticity, there may be ways, in some circumstances, to address this concern that do not require separate regulatory mechanisms. Here we describe two of them.
Redistribution of Synaptic Efficacy
Most experimental studies of STDP have quantified synaptic strength by measuring the postsynaptic response to a single presynaptic action potential or a single presynaptic stimulation. This is a useful metric because of its simplicity, but it neglects an important fact: neurons in the brains of behaving animals do not fire action potentials in isolation but as part of complex, irregular spike trains. Even among neocortical pyramidal cells, interspike intervals can be as short as 10 ms. This is easily short enough to engage non-Hebbian forms of short-term synaptic plasticity, including synaptic depression and facilitation, which can profoundly alter response properties (Zucker and Regehr, 2002 ).
In a landmark paper, Markram and Tsodyks (1996) demonstrated how the interaction between short- and long-term plasticity might result in changes in the content, rather than the gain, of neural signals. Using intracellular recordings, they examined synapses between individual layer 5 pyramidal cells from juvenile rats. These synapses exhibited considerable frequency-dependent synaptic depression, which is likely mediated by depletion of the pool of readily releasable vesicles at each synaptic site. Inducing LTP with paired burst activity increased EPSP amplitude in response to single presynaptic spikes. However, it did not alter the steady-state response to moderate- and high-frequency (>10 Hz) spike trains (Figure 4 A). Markram and Tsodyks argued that the LTP had been expressed presynaptically, as an increase in the probability of vesicle release. When the presynaptic neuron fired a train of spikes, this simply depleted the readily releasable pool more quickly, as reflected in an increased (post-pairing) rate of frequency-dependent depression, but it did not affect the size of the pool or any postsynaptic properties. In other words, LTP “redistributed” efficacy to the first spikes in the train, while having a minimal effect on the last ones. Describing quite what this means for a complex spike train, which might include a broad distribution of interspike intervals, is difficult, but it certainly suggests a useful means of imposing stability (Carpenter and Milenova, 2002 ). As rates increase, synapses become more depressed; the level at which they settle is unchanged by LTP.
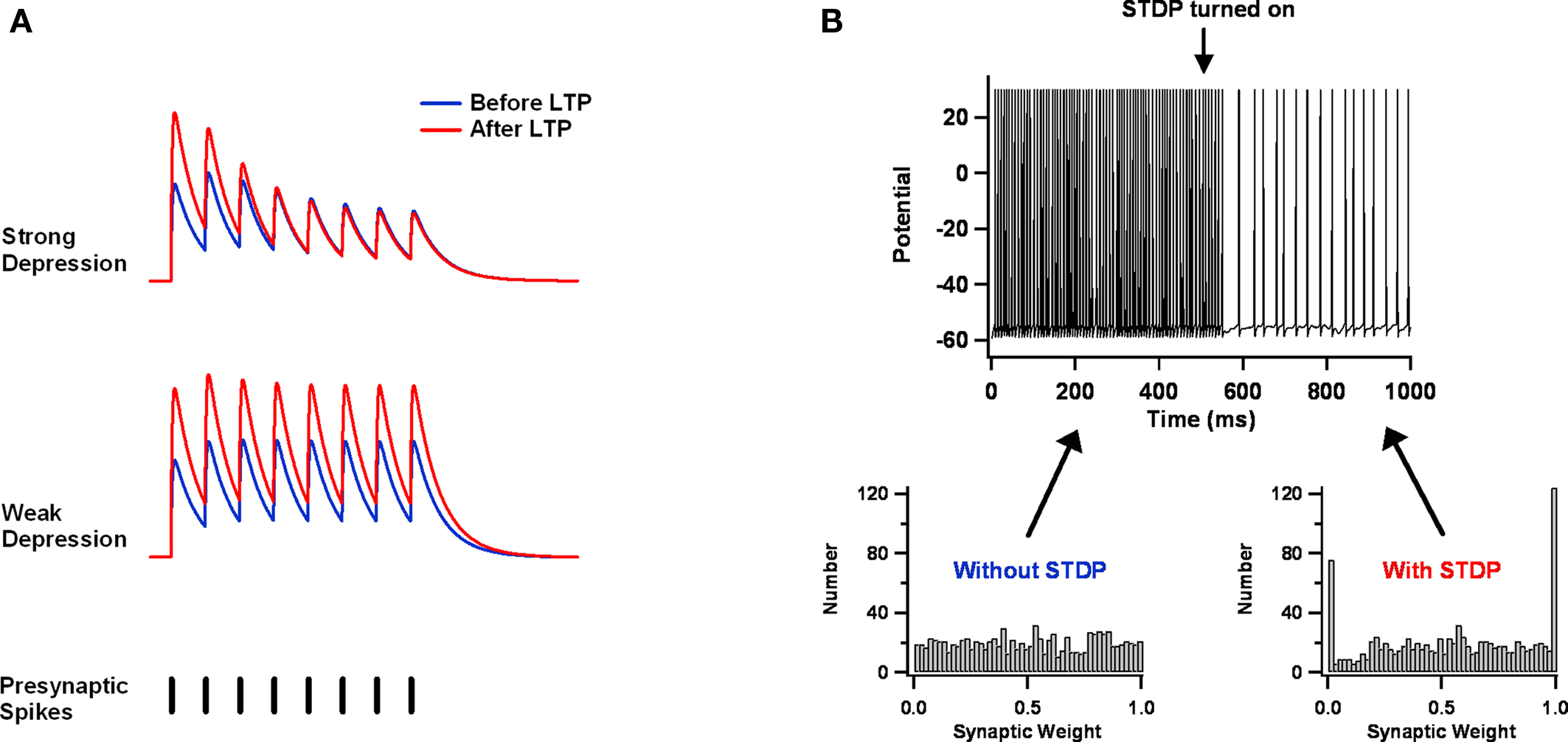
Figure 4. Balanced STDP. (A) Synaptic depression might balance STDP. At strongly depressing synapses, presynaptically expressed STDP increases the amplitude of the first potential in response to a train of presynaptic stimuli, but not the steady-state response. Weakly depressing synapses lack this normalization mechanism. The illustrations are derived from the phenomenological model of Tsodyks and Markram (1997) . (B) LTD might balance LTP. A leaky integrate and fire neuron receives a large number of excitatory and inhibitory Poisson inputs. Initially, the excitatory weight distribution is static and nearly uniform, and it results in aberrantly fast and regular firing. But once STDP is turned on, firing slows and becomes irregular. Excitatory weights move toward the two extremes. Parameters used here are similar to those of Song et al. (2000) .
How general a means of stabilization is this “redistribution of synaptic efficacy” phenomenon? The complementary effect (reduced short-term depression) has been observed after timing-dependent LTD in layer 5 neocortex (Sjostrom et al., 2003 ), and both effects have been observed at hippocampal CA1 synapses following certain protocols for LTP and LTD (Yasui et al., 2005 ). However, at some synapses, STDP appears to be mediated by postsynaptic changes in addition to, or instead of, presynaptic ones (Hardingham et al., 2007 ; Feldman, 2009 ). And many synapses either do not exhibit strong frequency-dependent depression or do so only early in development (Reyes and Sakmann, 1999 ; Zucker and Regehr, 2002 ). For these synapses, some other means of containing runaway potentiation, such as those we have described, would seem to be necessary.
LTP Versus LTD
In principle, purely Hebbian forms of plasticity might be able, by themselves, to regulate total synaptic drive, if the effects of LTP can be countered by those of LTD (and vice versa). This requires a rather delicate balance, which is difficult to maintain for rate-based plasticity. But STDP’s dependence on precise spike timing might allow for such a balance to be established and maintained.
In particular, this might happen if the STDP learning window is biased toward depression – that is, if the area over the LTD part of the curve is larger than the area under the LTP part (Feldman, 2000 ; Sjostrom et al., 2001 ). The basic argument is simple and intuitive (Abbott and Nelson, 2000 ). Imagine that a neuron receives excessively large synaptic input, which causes it to fire excessively fast. Each presynaptic spike will have only a weak effect on postsynaptic firing and is as likely to be preceded by postsynaptic spikes as it is to be followed by them. Both LTP and LTD will be induced as a result. If we assume that all spike pairings contribute to synaptic change (i.e., not just nearest neighbors), then the net effect will be depression of all synapses, because LTD dominates over LTP. As synapses get weaker, the firing rate will drop, until the only postsynaptic spikes are driven by chance clustering in the timing of presynaptic spikes. The synapses from these presynaptic neurons will grow stronger, because now their spikes will, on average, precede the postsynaptic ones. The end-result is a stable postsynaptic firing rate and a synaptic weight distribution in which some synapses are very strong and the others are very weak (assuming an additive STDP update rule; Figure 4 B) (Song et al., 2000 ).
As appealing as this argument is, simulation-based and analytical studies indicate that the approach has significant limitations (Song et al., 2000 ; van Rossum et al., 2000 ; Kempter et al., 2001 ; Rubin, 2001 ; Gutig et al., 2003 ; Morrison et al., 2007 ; Billings and van Rossum, 2009 ). Even for the simplified case of a single postsynaptic neuron receiving Poisson inputs, the final result depends quite a lot on the particular STDP implementation employed. Among the choices that matter: whether one adopts additive, multiplicative, or mixed update rules; how one treats multi-spike interactions; and whether one imposes hard bounds on the growth and decay of individual synapses. These choices determine the shape of the steady-state weight distribution, whether there is competition between synapses, and how stable synaptic weights are. Furthermore, some of the final results, like the bimodal weight distribution just described, contradict experimental data (Turrigiano et al., 1998 ; Song et al., 2005 ). Balancing LTP versus LTD should be useful for homeostasis, but present evidence suggests it is best complemented by additional mechanisms, such as lateral inhibition (Billings and van Rossum, 2009 ) or synaptic scaling (van Rossum et al., 2000 ).
Working Together
We have in this paper described multiple mechanisms for homeostatic regulation of firing rates. The list has been extensive while not being exhaustive. For example, we did not discuss anti-STDP (Rumsey and Abbott, 2006 ) or heterosynaptic LTP and LTD (Royer and Pare, 2003 ; Chistiakova and Volgushev, 2009 ), which are somewhat separate from STDP proper. An obvious question is: why are there so many mechanisms? The answer probably arises from a combination of things: (1) Somewhat different needs have been grouped together under the label of “homeostasis,” namely preventing runaway potentiation due to Hebbian synaptic change, and maintaining dynamic range during periods of intense synaptogenesis and neural growth. (2) The stability challenges faced by the nervous system differ between brain areas and at different developmental ages, perhaps necessitating different responses. (3) In complicated neural circuits, homeostatic regulation might require multiple processes acting on different timescales and in different ways. Given the complexity, what we most need now are investigations in which multiple forms of plasticity, both homeostatic and Hebbian, interact in defined systems. This is challenging but several studies of this sort have been done. To end this paper, we highlight a particularly illuminating one.
The rodent visual cortex is unusual in that one of its zones is strictly monocular, while the binocular cells in the other zone are interspersed and not segregated into patches based on eye preference. Mrsic-Flogel et al. (2007) exploited this organization to disentangle the effects of competition from those of activity deprivation. Using calcium imaging of individual cells, they quantified changes in response properties after short (2 days) and long (≥4 days) periods of monocular deprivation (MD). As expected, short MD durations decreased deprived-eye responses and long MD durations increased open-eye responses. But surprisingly, the longer MD durations also strengthened deprived-eye responses in neurons devoid of open-eye input; and binocular deprivation strengthened (not weakened) responsiveness to both eyes. Most strikingly, the bidirectional response adjustments in binocular cells after MD effectively preserved the net visual drive for each neuron. The most likely explanation for these results is that two distinct forces were at work: a Hebbian process (e.g., LTD of excitatory synapses) that weakened deprived-eye responses in binocular cells, and a non-Hebbian process that strengthened all responses. The first implemented competition between afferents from the two eyes; the second implemented homeostatic regulation in response to deprivation. Furthermore, the preservation of net visual drive suggests the two acted in complementary ways.
Thus, just as you have multiple mechanisms enabling you to cope with variations in temperature, ranging from the Sahara to the Arctic, a neuron is equipped with a range of different, and sometimes overlapping, homeostatic mechanisms that allow it to “maintain its cool” with its output, over a wide range of different input “environments”.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors’ work is supported by a Royal Society Dorothy Hodgkin Fellowship (Alanna J. Watt), and by the Neurosciences Research Foundation and the G. Harold and Leila Y. Mathers Charitable Foundation (Niraj S. Desai).
References
Abbott, L. F., and Nelson, S. B. (2000). Synaptic plasticity: taming the beast. Nat. Neurosci. 3(Suppl.), 1178–1183.
Abraham, W. C. (2008). Metaplasticity: tuning synapses and networks for plasticity. Nat. Rev. Neurosci. 9, 387.
Abraham, W. C., Mason-Parker, S. E., Bear, M. F., Webb, S., and Tate, W. P. (2001). Heterosynaptic metaplasticity in the hippocampus in vivo: a BCM-like modifiable threshold for LTP. Proc. Natl. Acad. Sci. U.S.A. 98, 10924–10929.
Aizenman, C. D., Akerman, C. J., Jensen, K. R., and Cline, H. T. (2003). Visually driven regulation of intrinsic neuronal excitability improves stimulus detection in vivo. Neuron 39, 831–842.
Amici, M., Doherty, A., Jo, J., Jane, D., Cho, K., Collingridge, G., and Dargan, S. (2009). Neuronal calcium sensors and synaptic plasticity. Biochem. Soc. Trans. 37, 1359–1363.
Aoto, J., Nam, C. I., Poon, M. M., Ting, P., and Chen, L. (2008). Synaptic signaling by all-trans retinoic acid in homeostatic synaptic plasticity. Neuron 60, 308–320.
Appleby, P. A., and Elliott, T. (2005). Synaptic and temporal ensemble interpretation of spike-timing-dependent plasticity. Neural Comput. 17, 2316–2336.
Aptowicz, C. O., Kunkler, P. E., and Kraig, R. P. (2004). Homeostatic plasticity in hippocampal slice cultures involves changes in voltage-gated Na+ channel expression. Brain Res. 998, 155–163.
Artola, A., and Singer, W. (1993). Long-term depression of excitatory synaptic transmission and its relationship to long-term potentiation. Trends Neurosci. 16, 480–487.
Atkinson, S. E., and Williams, S. R. (2009). Postnatal development of dendritic synaptic integration in rat neocortical pyramidal neurons. J. Neurophysiol. 102, 735–751.
Azdad, K., Chavez, M., Don Bischop, P., Wetzelaer, P., Marescau, B., De Deyn, P. P., Gall, D., and Schiffmann, S. N. (2009). Homeostatic plasticity of striatal neurons intrinsic excitability following dopamine depletion. PLoS ONE 4, e6908. doi: 10.1371/journal.pone.0006908.
Bartley, A. F., Huang, Z. J., Huber, K. M., and Gibson, J. R. (2008). Differential activity-dependent, homeostatic plasticity of two neocortical inhibitory circuits. J. Neurophysiol. 100, 1983–1994.
Bear, M. F. (2003). Bidirectional synaptic plasticity: from theory to reality. Philos. Trans. R. Soc. Lond., B, Biol. Sci. 358, 649–655.
Bender, V. A., Bender, K. J., Brasier, D. J., and Feldman, D. E. (2006). Two coincidence detectors for spike timing-dependent plasticity in somatosensory cortex. J. Neurosci. 26, 4166–4177.
Benuskova, L., and Abraham, W. C. (2007). STDP rule endowed with the BCM sliding threshold accounts for hippocampal heterosynaptic plasticity. J. Comput. Neurosci. 22, 129–133.
Bi, G. Q., and Poo, M. M. (1998). Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J. Neurosci. 18, 10464–10472.
Biel, M., Wahl-Schott, C., Michalakis, S., and Zong, X. (2009). Hyperpolarization-activated cation channels: from genes to function. Physiol. Rev. 89, 847–885.
Bienenstock, E. L., Cooper, L. N., and Munro, P. W. (1982). Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. J. Neurosci. 2, 32–48.
Billings, G., and van Rossum, M. C. (2009). Memory retention and spike-timing-dependent plasticity. J. Neurophysiol. 101, 2775–2788.
Brager, D. H., and Johnston, D. (2007). Plasticity of intrinsic excitability during long-term depression is mediated through mGluR-dependent changes in I(h) in hippocampal CA1 pyramidal neurons. J. Neurosci. 27, 13926–13937.
Branco, T., Staras, K., Darcy, K. J., and Goda, Y. (2008). Local dendritic activity sets release probability at hippocampal synapses. Neuron 59, 475–485.
Breton, J. D., and Stuart, G. J. (2009). Loss of sensory input increases the intrinsic excitability of layer 5 pyramidal neurons in rat barrel cortex. J. Physiol. 587, 5107–5119.
Burkitt, A. N., Meffin, H., and Grayden, D. B. (2004). Spike-timing-dependent plasticity: the relationship to rate-based learning for models with weight dynamics determined by a stable fixed point. Neural Comput. 16, 885–940.
Burrone, J., and Murthy, V. N. (2003). Synaptic gain control and homeostasis. Curr. Opin. Neurobiol. 13, 560–567.
Burrone, J., O’Byrne, M., and Murthy, V. N. (2002). Multiple forms of synaptic plasticity triggered by selective suppression of activity in individual neurons. Nature 420, 414–418.
Campanac, E., Daoudal, G., Ankri, N., and Debanne, D. (2008). Downregulation of dendritic I(h) in CA1 pyramidal neurons after LTP. J. Neurosci. 28, 8635–8643.
Campanac, E., and Debanne, D. (2008). Spike timing-dependent plasticity: a learning rule for dendritic integration in rat CA1 pyramidal neurons. J. Physiol. 586, 779–793.
Caporale, N., and Dan, Y. (2008). Spike timing-dependent plasticity: a Hebbian learning rule. Annu. Rev. Neurosci. 31, 25–46.
Carpenter, G. A., and Milenova, B. L. (2002). Redistribution of synaptic efficacy supports stable pattern learning in neural networks. Neural Comput. 14, 873–888.
Castellani, G. C., Quinlan, E. M., Cooper, L. N., and Shouval, H. Z. (2001). A biophysical model of bidirectional synaptic plasticity: dependence on AMPA and NMDA receptors. Proc. Natl. Acad. Sci. U.S.A. 98, 12772–12777.
Chistiakova, M., and Volgushev, M. (2009). Heterosynaptic plasticity in the neocortex. Exp. Brain Res. 199, 377–390.
Cho, K. K., Khibnik, L., Philpot, B. D., and Bear, M. F. (2009). The ratio of NR2A/B NMDA receptor subunits determines the qualities of ocular dominance plasticity in visual cortex. Proc. Natl. Acad. Sci. U.S.A. 106, 5377–5382.
Cingolani, L. A., Thalhammer, A., Yu, L. M., Catalano, M., Ramos, T., Colicos, M. A., and Goda, Y. (2008). Activity-dependent regulation of synaptic AMPA receptor composition and abundance by beta3 integrins. Neuron 58, 749–762.
Clopath, C., Busing, L., Vasilaki, E., and Gerstner, W. (2010). Connectivity reflects coding: a model of voltage-based STDP with homeostasis. Nat. Neurosci. 13, 344–352.
Cooper, L. N., Intrator, N., Blais, B. S., and Shouval, H. Z. (2004). Theory of Cortical Plasticity. River Edge, NJ: World Scientific.
Corlew, R., Brasier, D. J., Feldman, D. E., and Philpot, B. D. (2008). Presynaptic NMDA receptors: newly appreciated roles in cortical synaptic function and plasticity. Neuroscientist 14, 609–625.
Corlew, R., Wang, Y., Ghermazien, H., Erisir, A., and Philpot, B. D. (2007). Developmental switch in the contribution of presynaptic and postsynaptic NMDA receptors to long-term depression. J. Neurosci. 27, 9835–9845.
Debanne, D., Gahwiler, B. H., and Thompson, S. M. (1998). Long-term synaptic plasticity between pairs of individual CA3 pyramidal cells in rat hippocampal slice cultures. J. Physiol. 507, 237–247.
De Gois, S., Schafer, M. K., Defamie, N., Chen, C., Ricci, A., Weihe, E., Varoqui, H., and Erickson, J. D. (2005). Homeostatic scaling of vesicular glutamate and GABA transporter expression in rat neocortical circuits. J. Neurosci. 25, 7121–7133.
Delgado, J. Y., Gomez, J. F., and Desai, N. S. (2009). In vivo-like conductance levels control the induction of spike-timing dependent plasticity. Soc. Neurosci. Abstr. 35, 41.8.
Desai, N. S., Cudmore, R. H., Nelson, S. B., and Turrigiano, G. G. (2002). Critical periods for experience-dependent synaptic scaling in visual cortex. Nat. Neurosci. 5, 783–789.
Desai, N. S., Rutherford, L. C., and Turrigiano, G. G. (1999a). BDNF regulates the intrinsic excitability of cortical neurons. Learn. Mem. 6, 284–291.
Desai, N. S., Rutherford, L. C., and Turrigiano, G. G. (1999b). Plasticity in the intrinsic excitability of cortical pyramidal neurons. Nat. Neurosci. 2, 515–520.
De Simoni, A., Griesinger, C. B., and Edwards, F. A. (2003). Development of rat CA1 neurones in acute versus organotypic slices: role of experience in synaptic morphology and activity. J. Physiol. 550, 135–147.
Destexhe, A., Rudolph, M., and Pare, D. (2003). The high-conductance state of neocortical neurons in vivo. Nat. Rev. Neurosci. 4, 739–751.
Dudek, S. M., and Bear, M. F. (1992). Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-D-aspartate receptor blockade. Proc. Natl. Acad. Sci. U.S.A. 89, 4363–4367.
Duguid, I., and Sjostrom, P. J. (2006). Novel presynaptic mechanisms for coincidence detection in synaptic plasticity. Curr. Opin. Neurobiol. 16, 312–322.
Echegoyen, J., Neu, A., Graber, K. D., and Soltesz, I. (2007). Homeostatic plasticity studied using in vivo hippocampal activity-blockade: synaptic scaling, intrinsic plasticity and age-dependence. PLoS ONE 2, e700. doi: 10.1371/journal.pone.0000700.
Ehlers, M. D., Heine, M., Groc, L., Lee, M. C., and Choquet, D. (2007). Diffusional trapping of GluR1 AMPA receptors by input-specific synaptic activity. Neuron 54, 447–460.
Engert, F., and Bonhoeffer, T. (1999). Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature 399, 66–70.
Erickson, J. D., De Gois, S., Varoqui, H., Schafer, M. K., and Weihe, E. (2006). Activity-dependent regulation of vesicular glutamate and GABA transporters: a means to scale quantal size. Neurochem. Int. 48, 643–649.
Fan, Y., Fricker, D., Brager, D. H., Chen, X., Lu, H. C., Chitwood, R. A., and Johnston, D. (2005). Activity-dependent decrease of excitability in rat hippocampal neurons through increases in I(h). Nat. Neurosci. 8, 1542–1551.
Feldman, D. E. (2009). Synaptic mechanisms for plasticity in neocortex. Annu. Rev. Neurosci. 32, 33–55.
Frick, A., and Johnston, D. (2005). Plasticity of dendritic excitability. J. Neurobiol. 64, 100–115.
Froemke, R. C., Tsay, I. A., Raad, M., Long, J. D., and Dan, Y. (2006). Contribution of individual spikes in burst-induced long-term synaptic modification. J. Neurophysiol. 95, 1620–1629.
Fuenzalida, M., Fernandez de Sevilla, D., and Buno, W. (2007). Changes of the EPSP waveform regulate the temporal window for spike-timing-dependent plasticity. J. Neurosci. 27, 11940–11948.
Gibson, J. R., Bartley, A. F., and Huber, K. M. (2006). Role for the subthreshold currents ILeak and IH in the homeostatic control of excitability in neocortical somatostatin-positive inhibitory neurons. J. Neurophysiol. 96, 420–432.
Goddard, C. A., Butts, D. A., and Shatz, C. J. (2007). Regulation of CNS synapses by neuronal MHC class I. Proc. Natl. Acad. Sci. U.S.A. 104, 6828–6833.
Goel, A., and Lee, H. K. (2007). Persistence of experience-induced homeostatic synaptic plasticity through adulthood in superficial layers of mouse visual cortex. J. Neurosci. 27, 6692–6700.
Gutig, R., Aharonov, R., Rotter, S., and Sompolinsky, H. (2003). Learning input correlations through nonlinear temporally asymmetric Hebbian plasticity. J. Neurosci. 23, 3697–3714.
Hardingham, N. R., Hardingham, G. E., Fox, K. D., and Jack, J. J. (2007). Presynaptic efficacy directs normalization of synaptic strength in layer 2/3 rat neocortex after paired activity. J. Neurophysiol. 97, 2965–2975.
Harms, K. J., and Craig, A. M. (2005). Synapse composition and organization following chronic activity blockade in cultured hippocampal neurons. J. Comp. Neurol. 490, 72–84.
Hebb, D. O. (1949). The Organization of Behavior: A Neuropsychological Theory. New York, NY: John Wiley and Sons.
Hou, Q., Zhang, D., Jarzylo, L., Huganir, R. L., and Man, H. Y. (2008). Homeostatic regulation of AMPA receptor expression at single hippocampal synapses. Proc. Natl. Acad. Sci. U.S.A. 105, 775–780.
Ibata, K., Sun, Q., and Turrigiano, G. G. (2008). Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron 57, 819–826.
Johnston, D., and Narayanan, R. (2008). Active dendrites: colorful wings of the mysterious butterflies. Trends Neurosci. 31, 309–316.
Ju, W., Morishita, W., Tsui, J., Gaietta, G., Deerinck, T. J., Adams, S. R., Garner, C. C., Tsien, R. Y., Ellisman, M. H., and Malenka, R. C. (2004). Activity-dependent regulation of dendritic synthesis and trafficking of AMPA receptors. Nat. Neurosci. 7, 244–253.
Kampa, B. M., Letzkus, J. J., and Stuart, G. J. (2007). Dendritic mechanisms controlling spike-timing-dependent synaptic plasticity. Trends Neurosci. 30, 456–463.
Karmarkar, U. R., and Buonomano, D. V. (2002). A model of spike-timing dependent plasticity: one or two coincidence detectors? J. Neurophysiol. 88, 507–513.
Karmarkar, U. R., and Buonomano, D. V. (2006). Different forms of homeostatic plasticity are engaged with distinct temporal profiles. Eur. J. Neurosci. 23, 1575–1584.
Kempter, R., Gerstner, W., and van Hemmen, J. L. (2001). Intrinsic stabilization of output rates by spike-based Hebbian learning. Neural Comput. 13, 2709–2741.
Kilman, V., van Rossum, M. C., and Turrigiano, G. G. (2002). Activity deprivation reduces miniature IPSC amplitude by decreasing the number of postsynaptic GABA(A) receptors clustered at neocortical synapses. J. Neurosci. 22, 1328–1337.
Kirkwood, A., Rioult, M. C., and Bear, M. F. (1996). Experience-dependent modification of synaptic plasticity in visual cortex. Nature 381, 526–528.
Law, C. C., and Cooper, L. N. (1994). Formation of receptive fields in realistic visual environments according to the Bienenstock, Cooper, and Munro (BCM) theory. Proc. Natl. Acad. Sci. U.S.A. 91, 7797–7801.
Lazar, A., Pipa, G., and Triesch, J. (2009). SORN: a self-organizing recurrent neural network. Front. Comput. Neurosci. 3:23. doi: 10.3389/neuro.10.023.2009.
LeMasson, G., Marder, E., and Abbott, L. F. (1993). Activity-dependent regulation of conductances in model neurons. Science 259, 1915–1917.
Lisman, J. (2003). Long-term potentiation: outstanding questions and attempted synthesis. Philos. Trans. R. Soc. Lond., B, Biol. Sci. 358, 829–842.
Lisman, J., and Spruston, N. (2005). Postsynaptic depolarization requirements for LTP and LTD: a critique of spike timing-dependent plasticity. Nat. Neurosci. 8, 839–841.
Lissin, D. V., Gomperts, S. N., Carroll, R. C., Christine, C. W., Kalman, D., Kitamura, M., Hardy, S., Nicoll, R. A., Malenka, R. C., and von Zastrow, M. (1998). Activity differentially regulates the surface expression of synaptic AMPA and NMDA glutamate receptors. Proc. Natl. Acad. Sci. U.S.A. 95, 7097–7102.
Lu, Y., Christian, K., and Lu, B. (2008). BDNF: a key regulator for protein synthesis-dependent LTP and long-term memory? Neurobiol. Learn. Mem. 89, 312–323.
Maffei, A., and Fontanini, A. (2009). Network homeostasis: a matter of coordination. Curr. Opin. Neurobiol. 19, 168–173.
Maffei, A., Nataraj, K., Nelson, S. B., and Turrigiano, G. G. (2006). Potentiation of cortical inhibition by visual deprivation. Nature 443, 81–84.
Maffei, A., Nelson, S. B., and Turrigiano, G. G. (2004). Selective reconfiguration of layer 4 visual cortical circuitry by visual deprivation. Nat. Neurosci. 7, 1353–1359.
Maffei, A., and Turrigiano, G. G. (2008). Multiple modes of network homeostasis in visual cortical layer 2/3. J. Neurosci. 28, 4377–4384.
Malenka, R. C., and Nicoll, R. A. (1999). Long-term potentiation – a decade of progress? Science 285, 1870–1874.
Marder, E., and Bucher, D. (2007). Understanding circuit dynamics using the stomatogastric nervous system of lobsters and crabs. Annu. Rev. Physiol. 69, 291–316.
Markram, H., Lubke, J., Frotscher, M., and Sakmann, B. (1997). Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science 275, 213–215.
Markram, H., and Tsodyks, M. (1996). Redistribution of synaptic efficacy between neocortical pyramidal neurons. Nature 382, 807–810.
McCoy, M. K., and Tansey, M. G. (2008). TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. J. Neuroinflammation 5, 45.
McCoy, P. A., Huang, H. S., and Philpot, B. D. (2009). Advances in understanding visual cortex plasticity. Curr. Opin. Neurobiol. 19, 298–304.
Minichiello, L. (2009). TrkB signalling pathways in LTP and learning. Nat. Rev. Neurosci. 10, 850–860.
Misonou, H., Mohapatra, D. P., Park, E. W., Leung, V., Zhen, D., Misonou, K., Anderson, A. E., and Trimmer, J. S. (2004). Regulation of ion channel localization and phosphorylation by neuronal activity. Nat. Neurosci. 7, 711–718.
Mohapatra, D. P., Misonou, H., Pan, S. J., Held, J. E., Surmeier, D. J., and Trimmer, J. S. (2009). Regulation of intrinsic excitability in hippocampal neurons by activity-dependent modulation of the KV2.1 potassium channel. Channels (Austin) 3, 46–56.
Morrison, A., Aertsen, A., and Diesmann, M. (2007). Spike-timing-dependent plasticity in balanced random networks. Neural Comput. 19, 1437–1467.
Morrison, A., Diesmann, M., and Gerstner, W. (2008). Phenomenological models of synaptic plasticity based on spike timing. Biol. Cybern. 98, 459–478.
Mrsic-Flogel, T. D., Hofer, S. B., Ohki, K., Reid, R. C., Bonhoeffer, T., and Hubener, M. (2007). Homeostatic regulation of eye-specific responses in visual cortex during ocular dominance plasticity. Neuron 54, 961–972.
Mu, Y., Otsuka, T., Horton, A. C., Scott, D. B., and Ehlers, M. D. (2003). Activity-dependent mRNA splicing controls ER export and synaptic delivery of NMDA receptors. Neuron 40, 581–594.
Murthy, V. N., Schikorski, T., Stevens, C. F., and Zhu, Y. (2001). Inactivity produces increases in neurotransmitter release and synapse size. Neuron 32, 673–682.
Narayanan, R., and Johnston, D. (2008). The h channel mediates location dependence and plasticity of intrinsic phase response in rat hippocampal neurons. J. Neurosci. 28, 5846–5860.
Nelson, A. B., Krispel, C. M., Sekirnjak, C., and du Lac, S. (2003). Long-lasting increases in intrinsic excitability triggered by inhibition. Neuron 40, 609–620.
Neves, G., Cooke, S. F., and Bliss, T. V. (2008). Synaptic plasticity, memory and the hippocampus: a neural network approach to causality. Nat. Rev. Neurosci. 9, 65–75.
O’Brien, R. J., Kamboj, S., Ehlers, M. D., Rosen, K. R., Fischbach, G. D., and Huganir, R. L. (1998). Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron 21, 1067–1078.
O’Leary, T., van Rossum, M. C., and Wyllie, D. J. (2010). Homeostasis of intrinsic excitability in hippocampal neurones: dynamics and mechanism of the response to chronic depolarization. J. Physiol. 588, 157–170.
Pfister, J. P., and Gerstner, W. (2006). Triplets of spikes in a model of spike timing-dependent plasticity. J. Neurosci. 26, 9673–9682.
Philpot, B. D., Cho, K. K., and Bear, M. F. (2007). Obligatory role of NR2A for metaplasticity in visual cortex. Neuron 53, 495–502.
Philpot, B. D., Espinosa, J. S., and Bear, M. F. (2003). Evidence for altered NMDA receptor function as a basis for metaplasticity in visual cortex. J. Neurosci. 23, 5583–5588.
Philpot, B. D., Sekhar, A. K., Shouval, H. Z., and Bear, M. F. (2001). Visual experience and deprivation bidirectionally modify the composition and function of NMDA receptors in visual cortex. Neuron 29, 157–169.
Poolos, N. P. (2005). The h-channel: a potential channelopathy in epilepsy? Epilepsy Behav. 7, 51–56.
Pratt, K. G., and Aizenman, C. D. (2007). Homeostatic regulation of intrinsic excitability and synaptic transmission in a developing visual circuit. J. Neurosci. 27, 8268–8277.
Quinlan, E. M., Philpot, B. D., Huganir, R. L., and Bear, M. F. (1999). Rapid, experience-dependent expression of synaptic NMDA receptors in visual cortex in vivo. Nat. Neurosci. 2, 352–357.
Rabinowitch, I., and Segev, I. (2008). Two opposing plasticity mechanisms pulling a single synapse. Trends Neurosci. 31, 377–383.
Ramocki, M. B., and Zoghbi, H. Y. (2008). Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature 455, 912–918.
Rao, A., and Craig, A. M. (1997). Activity regulates the synaptic localization of the NMDA receptor in hippocampal neurons. Neuron 19, 801–812.
Renart, A., Song, P., and Wang, X. J. (2003). Robust spatial working memory through homeostatic synaptic scaling in heterogeneous cortical networks. Neuron 38, 473–485.
Reyes, A., and Sakmann, B. (1999). Developmental switch in the short-term modification of unitary EPSPs evoked in layer 2/3 and layer 5 pyramidal neurons of rat neocortex. J. Neurosci. 19, 3827–3835.
Rial Verde, E. M., Lee-Osbourne, J., Worley, P. F., Malinow, R., and Cline, H. T. (2006). Increased expression of the immediate-early gene arc/arg3.1 reduces AMPA receptor-mediated synaptic transmission. Neuron 52, 461–474.
Rittenhouse, C. D., Shouval, H. Z., Paradiso, M. A., and Bear, M. F. (1999). Monocular deprivation induces homosynaptic long-term depression in visual cortex. Nature 397, 347–350.
Roth-Alpermann, C., Morris, R. G., Korte, M., and Bonhoeffer, T. (2006). Homeostatic shutdown of long-term potentiation in the adult hippocampus. Proc. Natl. Acad. Sci. U.S.A. 103, 11039–11044.
Royer, S., and Pare, D. (2003). Conservation of total synaptic weight through balanced synaptic depression and potentiation. Nature 422, 518–522.
Rubin, J. E. (2001). Steady states in an iterative model for multiplicative spike-timing-dependent plasticity. Network 12, 131–140.
Rubin, J. E., Gerkin, R. C., Bi, G. Q., and Chow, C. C. (2005). Calcium time course as a signal for spike-timing-dependent plasticity. J. Neurophysiol. 93, 2600–2613.
Rumsey, C. C., and Abbott, L. F. (2006). Synaptic democracy in active dendrites. J. Neurophysiol. 96, 2307–2318.
Rutherford, L. C., Nelson, S. B., and Turrigiano, G. G. (1998). BDNF has opposite effects on the quantal amplitude of pyramidal neuron and interneuron excitatory synapses. Neuron 21, 521–530.
Seeburg, D. P., Feliu-Mojer, M., Gaiottino, J., Pak, D. T., and Sheng, M. (2008). Critical role of CDK5 and Polo-like kinase 2 in homeostatic synaptic plasticity during elevated activity. Neuron 58, 571–583.
Shepherd, J. D., Rumbaugh, G., Wu, J., Chowdhury, S., Plath, N., Kuhl, D., Huganir, R. L., and Worley, P. F. (2006). Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron 52, 475–484.
Shin, M., and Chetkovich, D. M. (2007). Activity-dependent regulation of h channel distribution in hippocampal CA1 pyramidal neurons. J. Biol. Chem. 282, 33168–33180.
Shouval, H. Z., Bear, M. F., and Cooper, L. N. (2002). A unified model of NMDA receptor-dependent bidirectional synaptic plasticity. Proc. Natl. Acad. Sci. U.S.A. 99, 10831–10836.
Sjostrom, P. J., Rancz, E. A., Roth, A., and Hausser, M. (2008). Dendritic excitability and synaptic plasticity. Physiol. Rev. 88, 769–840.
Sjostrom, P. J., Turrigiano, G. G., and Nelson, S. B. (2001). Rate, timing, and cooperativity jointly determine cortical synaptic plasticity. Neuron 32, 1149–1164.
Sjostrom, P. J., Turrigiano, G. G., and Nelson, S. B. (2003). Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron 39, 641–654.
Song, S., Miller, K. D., and Abbott, L. F. (2000). Competitive Hebbian learning through spike-timing-dependent synaptic plasticity. Nat. Neurosci. 3, 919–926.
Song, S., Sjostrom, P. J., Reigl, M., Nelson, S., and Chklovskii, D. B. (2005). Highly nonrandom features of synaptic connectivity in local cortical circuits. PLoS Biol. 3, e68. doi: 10.1371/journal.pbio.0030068.
Stellwagen, D., and Malenka, R. C. (2006). Synaptic scaling mediated by glial TNF-alpha. Nature 440, 1054–1059.
Sutton, M. A., Ito, H. T., Cressy, P., Kempf, C., Woo, J. C., and Schuman, E. M. (2006). Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell 125, 785–799.
Sutton, M. A., and Schuman, E. M. (2009). Partitioning the synaptic landscape: distinct microdomains for spontaneous and spike-triggered neurotransmission. Sci. Signal. 2, pe19.
Swanwick, C. C., Murthy, N. R., and Kapur, J. (2006). Activity-dependent scaling of GABAergic synapse strength is regulated by brain-derived neurotrophic factor. Mol. Cell. Neurosci. 31, 481–492.
Thiagarajan, T. C., Lindskog, M., and Tsien, R. W. (2005). Adaptation to synaptic inactivity in hippocampal neurons. Neuron 47, 725–737.
Tononi, G., Sporns, O., and Edelman, G. M. (1999). Measures of degeneracy and redundancy in biological networks. Proc. Natl. Acad. Sci. U.S.A. 96, 3257–3262.
Toyoizumi, T., Pfister, J. P., Aihara, K., and Gerstner, W. (2005). Generalized Bienenstock–Cooper–Munro rule for spiking neurons that maximizes information transmission. Proc. Natl. Acad. Sci. U.S.A. 102, 5239–5244.
Trasande, C. A., and Ramirez, J. M. (2007). Activity deprivation leads to seizures in hippocampal slice cultures: is epilepsy the consequence of homeostatic plasticity? J. Clin. Neurophysiol. 24, 154–164.
Tsodyks, M. (2002). Spike-timing-dependent synaptic plasticity – the long road towards understanding neuronal mechanisms of learning and memory. Trends Neurosci. 25, 599–600.
Tsodyks, M. V., and Markram, H. (1997). The neural code between neocortical pyramidal neurons depends on neurotransmitter release probability. Proc. Natl. Acad. Sci. U.S.A. 94, 719–723.
Turrigiano, G., Abbott, L. F., and Marder, E. (1994). Activity-dependent changes in the intrinsic properties of cultured neurons. Science 264, 974–977.
Turrigiano, G. G. (2008). The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135, 422–435.
Turrigiano, G. G., Leslie, K. R., Desai, N. S., Rutherford, L. C., and Nelson, S. B. (1998). Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391, 892–896.
Vacher, H., Mohapatra, D. P., and Trimmer, J. S. (2008). Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiol. Rev. 88, 1407–1447.
van Rossum, M. C., Bi, G. Q., and Turrigiano, G. G. (2000). Stable Hebbian learning from spike timing-dependent plasticity. J. Neurosci. 20, 8812–8821.
van Welie, I., Remme, M. W., van Hooft, J. A., and Wadman, W. J. (2006a). Different levels of Ih determine distinct temporal integration in bursting and regular-spiking neurons in rat subiculum. J. Physiol. 576, 203–214.
van Welie, I., van Hooft, J. A., and Wadman, W. J. (2006b). Background activity regulates excitability of rat hippocampal CA1 pyramidal neurons by adaptation of a K+ conductance. J. Neurophysiol. 95, 2007–2012.
van Welie, I., van Hooft, J. A., and Wadman, W. J. (2004). Homeostatic scaling of neuronal excitability by synaptic modulation of somatic hyperpolarization-activated Ih channels. Proc. Natl. Acad. Sci. U.S.A. 101, 5123–5128.
Wang, H., and Wagner, J. J. (1999). Priming-induced shift in synaptic plasticity in the rat hippocampus. J. Neurophysiol. 82, 2024–2028.
Watt, A. J., Sjostrom, P. J., Hausser, M., Nelson, S. B., and Turrigiano, G. G. (2004). A proportional but slower NMDA potentiation follows AMPA potentiation in LTP. Nat. Neurosci. 7, 518–524.
Watt, A. J., van Rossum, M. C., MacLeod, K. M., Nelson, S. B., and Turrigiano, G. G. (2000). Activity coregulates quantal AMPA and NMDA currents at neocortical synapses. Neuron 26, 659–670.
Wierenga, C. J., Ibata, K., and Turrigiano, G. G. (2005). Postsynaptic expression of homeostatic plasticity at neocortical synapses. J. Neurosci. 25, 2895–2905.
Wierenga, C. J., Walsh, M. F., and Turrigiano, G. G. (2006). Temporal regulation of the expression locus of homeostatic plasticity. J. Neurophysiol. 96, 2127–2133.
Wilhelm, J. C., Rich, M. M., and Wenner, P. (2009). Compensatory changes in cellular excitability, not synaptic scaling, contribute to homeostatic recovery of embryonic network activity. Proc. Natl. Acad. Sci. U.S.A. 106, 6760–6765.
Wu, W. W., Chan, C. S., Surmeier, D. J., and Disterhoft, J. F. (2008). Coupling of L-type Ca2+ channels to KV7/KCNQ channels creates a novel, activity- dependent, homeostatic intrinsic plasticity. J. Neurophysiol. 100, 1897–1908.
Xu, T. X., Sotnikova, T. D., Liang, C., Zhang, J., Jung, J. U., Spealman, R. D., Gainetdinov, R. R., and Yao, W. D. (2009a). Hyperdopaminergic tone erodes prefrontal long-term potential via a D2 receptor-operated protein phosphatase gate. J. Neurosci. 29, 14086–14099.
Xu, Z., Chen, R. Q., Gu, Q. H., Yan, J. Z., Wang, S. H., Liu, S. Y., and Lu, W. (2009b). Metaplastic regulation of long-term potentiation/long-term depression threshold by activity-dependent changes of NR2A/NR2B ratio. J. Neurosci. 29, 8764–8773.
Yashiro, K., and Philpot, B. D. (2008). Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology 55, 1081–1094.
Yasui, T., Fujisawa, S., Tsukamoto, M., Matsuki, N., and Ikegaya, Y. (2005). Dynamic synapses as archives of synaptic history: state-dependent redistribution of synaptic efficacy in the rat hippocampal CA1. J. Physiol. 566, 143–160.
Yeung, L. C., Shouval, H. Z., Blais, B. S., and Cooper, L. N. (2004). Synaptic homeostasis and input selectivity follow from a calcium-dependent plasticity model. Proc. Natl. Acad. Sci. U.S.A. 101, 14943–14948.
Zhang, L. I., Tao, H. W., Holt, C. E., Harris, W. A., and Poo, M. (1998). A critical window for cooperation and competition among developing retinotectal synapses. Nature 395, 37–44.
Zhang, W., and Linden, D. J. (2003). The other side of the engram: experience-driven changes in neuronal intrinsic excitability. Nat. Rev. Neurosci. 4, 885–900.
Keywords: homeostatic plasticity, synaptic scaling, intrinsic plasticity, STDP, BCM, LTP, LTD, stability
Citation: Watt AJ and Desai NS (2010) Homeostatic plasticity and STDP: keeping a neuron’s cool in a fluctuating world. Front. Syn. Neurosci. 2:5. doi: 10.3389/fnsyn.2010.00005
Received: 18 February 2010;
Paper pending published: 23 April 2010;
Accepted: 17 May 2010;
Published online: 07 June 2010
Edited by:
Henry Markram, Ecole Polytechnique Federale de Lausanne, SwitzerlandReviewed by:
Gina Turrigiano, Brandeis University, USACopyright: © 2010 Watt and Desai. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Niraj S. Desai, The Neurosciences Institute, 10640 John Jay Hopkins Drive, San Diego, CA 92121, USA. e-mail:ZGVzYWlAbnNpLmVkdQ==