

- Department of Integrative Neurophysiology, Center for Neurogenomics and Cognitive Research, Neuroscience Campus Amsterdam, VU University, Amsterdam, Netherlands
Development of cognitive function requires the formation and refinement of synaptic networks of neurons in the brain. Morphological abnormalities of synaptic spines occur throughout the brain in a wide variety of syndromic and non-syndromic disorders of mental retardation (MR). In both neurons from human post-mortem tissue and mouse models of retardation, the changes observed in synaptic spine and dendritic morphology can be subtle, in the range of 10–20% alterations for spine protrusion length and density. Functionally, synapses in hippocampus and cortex show deficits in long-term potentiation (LTP) and long-term depression (LTD) in an array of neurodevelopmental disorders including Down’s, Angelman, Fragile X and Rett syndrome. Recent studies have shown that in principle the machinery for synaptic plasticity is in place in these synapses, but that significant alterations in spike-timing-dependent plasticity (STDP) induction rules exist in cortical synaptic pathways of Fragile X MR syndrome. In this model, the threshold for inducing timing-dependent long-term potentiation (tLTP) is increased in these synapses. Increased postsynaptic activity can overcome this threshold and induce normal levels of tLTP. In this review, we bring together recent studies investigating STDP in neurodevelopmental learning disorders using Fragile X syndrome as a model and we argue that alterations in dendritic excitability underlie deficits seen in STDP. Known and candidate dendritic mechanisms that may underlie the plasticity deficits are discussed. Studying STDP in monogenic MR syndromes with clear deficits in information processing at the cognitive level also provides the field with an opportunity to make direct links between cognition and processing rules at the synapse during development.
Introduction: Mental Retardation, Synapse Pathology and Plasticity
The formation and refinement of synaptic connections in the brain is crucial for development of cognitive function. Changing synaptic strength is widely believed to be a cellular mechanism underlying storage of information in the brain (Martin et al., 2000 ). Mental retardation (MR) is characterized by deficits in learning and memory with significantly low IQ levels (<70) and often accompanied by impaired social communication and autistic traits. Over the last decade, traditional methods for studying synaptic plasticity in mouse models for MR syndromes have revealed alterations in long-term potentiation (LTP) and long-term depression (LTD) using tetanic stimulation protocols or chemical induction methods. Deficits in LTP using these paradigms are reported in hippocampal and cortical synapses in an array of syndromes including Down’s (Siarey et al., 1997 ), Angelman (Jiang et al., 1998 ; Weeber et al., 2003 ), Fragile X (Li et al., 2002 ) and Rett syndrome (Asaka et al., 2006 ; Moretti et al., 2006 ). A number of impairments in intracellular signaling pathways are implicated in these differing forms of retardation, which often have co-morbidity for autistic traits and epilepsy. However, this variety of signaling deficits all lead to changes in synaptic plasticity and show abnormalities in dendrite and synaptic structures in the brain.
Synapse and Dendrite Structural Abnormalities
There are many signaling pathways implicated in the underlying causes of MR but the majority have direct interactions with Rho, Rac and Ras small GTPases (Ramakers, 2002 ; Newey et al., 2005 ). These proteins are all key regulators of the actin and microtubule cytoskeleton and affect both the structure and function of dendrites and synapses. A morphological characteristic common to many different forms of syndromic and non-syndromic MR is an altered filopodia-to-spine ratio and abnormal distribution of dendritic protrusions in both humans and mouse models for MR (Ramakers, 2002 ). The majority of excitatory synapses in the brain are made on to these dendritic protrusions. Over the course of normal development, long, thin filopodia commonly found in immature networks are replaced in later stages by shorter, more stubby spines (Harris, 1999 ). Spine abnormalities in MR are seen in many different brain regions and during both early and late stages of postnatal development (Purpura, 1974 ). For example, in the Fmr1-KO mouse model of Fragile X syndrome, more immature filopodial protrusions are observed along with an increased protrusion density along the dendrite (Irwin et al., 2000 ) and are seen from the end of the first postnatal week (Nimchinsky et al., 2001 ). In Rett syndrome, differing spine distribution patterns are reported in mouse models but the overall consensus is of a decrease in spine density at symptomatic stages, in line with findings in human post-mortem tissue and methyl CpG binding protein 2 (MeCP2) knockdown in cell cultures (Chapleau et al., 2009 ). Manipulation of expression levels of Rho and Rac GTPases in cultured neurons also leads to clear changes in spine morphology. Expression of a constitutively active form of Rac in mice leads to increased spine density and reduction in spine size (Luo et al., 1996 ; Tashiro et al., 2000 ). In contrast, expression of constitutively active Rho led to dramatic decreases in spine density, reduction in spine length and in some cases an absence of spines on pyramidal neurons (Tashiro et al., 2000 ).
Abnormalities of dendrite morphology in the brain are also a feature common to many different forms of MR (Kaufmann and Moser, 2000 ; Nadif Kasri and Van Aelst, 2008 ). The distribution of dendritic branching patterns shows abnormalities in cortical neurons in genetic forms of MR, such as Rett and Fragile X syndromes, (Kaufmann and Moser, 2000 ; Galvez et al., 2003 ; Restivo et al., 2005 ). Dendritic morphology is significantly altered following alteration of Rac and Rho expression levels, with Rac and Cdc42 increasing the growth and branching of neurites whereas Rho may act to negatively regulate dendritic growth (Threadgill et al., 1997 ; Negishi and Katoh, 2005 ). Thus, Rac and Rho both play a role in spine and dendrite morphology, with Rac promoting protrusion and branch growth and Rho having a regulatory role in dendrite and spine formation and maintenance. In the Fmr1-KO mouse model, Fragile X Mental Retardation Protein (FMRP) interacts directly with two cytoplastic proteins located in synaptosome extracts, CYFIP1/2 (Schenck et al., 2001 ), with CYFIP1 interacting with Rac1 small Rho GTPase (Kobayashi et al., 1998 ). Mutations in regulators and effectors of Rho GTPases have been associated with MR (Nadif Kasri and Van Aelst, 2008 ). Thus, effects upon spine dysmorphogenesis single gene MR disorders are likely to arise via effects mediated by interactions with pathways such as Rho and Rac signaling cascades.
The direct influence of spine morphology on spike-timing- dependent plasticity (STDP) has not been widely studied, although the effects of spine shape upon biochemical and electrical compartmentalization of synaptic input and AP backpropagation are beginning to be understood (see later section on ‘Dendritic Excitability’). Geometry of the postsynaptic spine is correlated with functional AMPA receptor expression, with thin long-necked protrusions showing little or no sensitivity to synaptic glutamate and thicker spines of greater volume showing large glutamate-induced synaptic currents (Matsuzaki et al., 2001 ). Thus the greater proportion of longer protrusions reported in Fragile X syndrome, particularly in early cortical development (Nimchinsky et al., 2001 ) may account for a higher proportion of silent synapses observed in sensory cortex (Harlow et al., 2010 ). Changes in dendritic spines and dendritic branching underlying MR syndromes can be small, in the region of 10–20% in cortex and hippocampus. These small structural changes cause only modest or no significant effects on the somatic-recordings of overall baseline measures of quantal synaptic function in MR models (Jiang et al., 1998 ; Braun and Segal, 2000 ; Desai et al., 2006 ; Pfeiffer and Huber, 2007 ; Best et al., 2008 ). However, these structural changes and the intracellular signaling mediated across the synaptic junctions have significant effects upon the synaptic plasticity in cortical and hippocampal circuits. In the Fmr1-KO mouse, an impairment in Ras-dependent trafficking of AMPA receptor subunits underlies a decrease in the level of LTP following a pairing protocol in cultured slices (Hu et al., 2008 ). By enhancing Ras signaling directly or via manipulation of the PI3K-PKB pathway, the deficits in paired LTP could be overcome, thus demonstrating a direct signaling cascade link between factors affecting both spine morphology and synapse plasticity.
STDP in Mouse Models for Inherited Mental Retardation
Activity-dependent alterations in synapse strength are key factors in refining neural circuits and learning based on experience. Compromised synaptic plasticity is likely to contribute to deficits underlying cognitive impairments in many MR syndromes. In the last decades, it has become clear that synapse strength can be modified depending on the millisecond timing of action potential firing with the sign of synaptic plasticity depending on the spike order of presynaptic and postsynaptic neurons (Levy and Steward, 1983 ; Gustafsson et al., 1987 ; Bell et al., 1997 ; Magee and Johnston, 1997 ; Markram et al., 1997 ). By varying the timing and order of pre- and postsynaptic spiking, it was found that critical time windows exist for synaptic modification on the order of tens of milliseconds (Bi and Poo, 1998 , 2001 ). Recently, it was found that induction rules for timing-dependent LTP (tLTP) and LTD (tLTD) are different in two mouse models for the X-linked syndromes of Rett and Fragile X. These syndromes, although caused by different distinct mutations on the X chromosome, have an overlap of phenotypic features such as MR and co-morbidity of autistic traits and epilepsy in some cases.
Postsynaptic Threshold for STDP
Spike-timing-dependent plasticity protocols with timing- dependent stimulation of pre- and postsynaptic activity using single postsynaptic action potentials were tested in the Fmr1-KO mouse, a model for Fragile X syndrome (Bakker et al., 1994 ). Fragile X syndrome, resulting from mutations in the Fmr1 gene on the X chromosome (Jin and Warren, 2000 ), is accompanied by significantly reduced or absent levels of FMRP which plays a key role in regulation of mRNA translation at or near dendritic spines (Weiler et al., 1997 ; Zalfa et al., 2003 ). In sensory and prefrontal cortex, there is a deficit in tLTP in sensory and prefrontal cortex from 2- to 3-week-old Fmr1-KO mice (Desai et al., 2006 ; Meredith et al., 2007 ). A similar deficit in tLTP was also observed in prefrontal cortex of older Fmr1-KO mice (Meredith et al., 2007 ). In hippocampal cultures and slices, a timing-dependent pairing protocol also showed a reduction in LTP in Fmr1-KO mice (Hu et al., 2008 ). Thus in both hippocampal and cortical tissue, timing-dependent stimulation with single postsynaptic action potentials revealed tLTP deficits in the Fmr1-KO mouse.
Absence of LTP could imply that the cellular machinery to support plasticity is not present in the dendrites and spines. Fragile X Mental Retardation Protein acts as a local regulator of mRNA translation and affects a number of cytoskeletal and synaptic proteins, many of which could have deleterious effects on plasticity. However, it was possible to overcome the deficits in tLTP by enhancement of postsynaptic activity or intracellular signaling factors. In 2-week-old cortex, increasing the level of postsynaptic stimulation from single action potentials to postsynaptic bursting activity in vitro rescued the tLTP deficit in Fmr1-KO mice (Meredith et al., 2007 ). Boosting postsynaptic activity via environmental enrichment stimulation for 2 months prior to recording also improved tLTP in old prefrontal cortex of Fmr1-KO mice (Meredith et al., 2007 ). Thus at both developmental ages, increased postsynaptic stimulation in STDP paradigms was able to demonstrate that cortical synapses in Fmr1-KO mice could potentiate but that stronger activity was necessary to rescue the synaptic plasticity impairment. Furthermore, the mechanisms capable of supporting potentiation are still present in the synapse but require a higher threshold for activation (see Figure 1 ). These findings echo data using traditional synaptic plasticity paradigms in Fmr1-KO mice: no significant differences in field LTP were reported initially in the Fmr1-KO mouse model following tetanic stimulation in hippocampus (Godfraind et al., 1996 ; Paradee et al., 1999 ; Li et al., 2002 ). However, reduced LTP induction was later observed with weak but not strong theta-burst stimulation in hippocampus (Lauterborn et al., 2007 ), suggesting that use of strong stimulation to induce LTP can mask some of the subtle changes in plasticity and overcome ‘threshold’ limitations that may be present in more physiological stimulation patterns in the retardation syndrome.
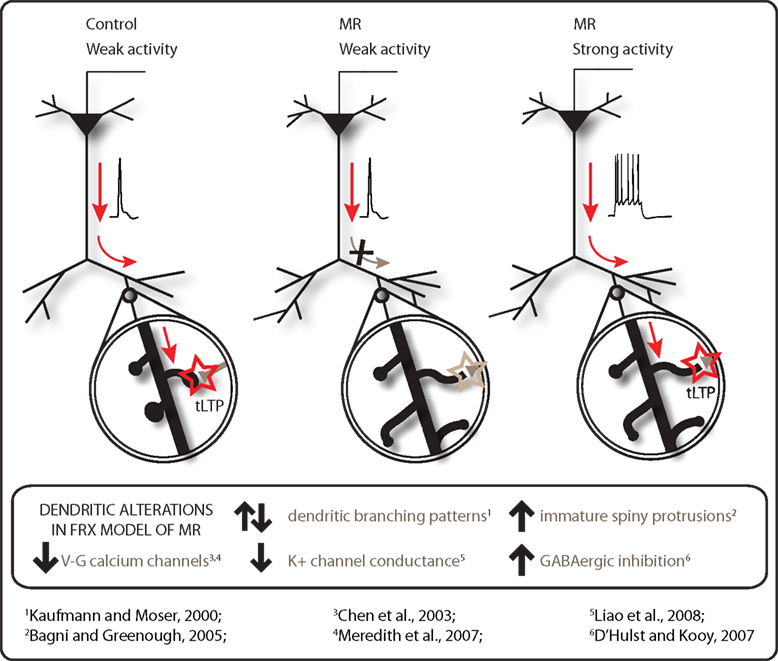
Figure 1. Modulation of dendritic excitability in mental retardation (MR). tLTP can be induced in pyramidal neurons with single backpropagating APs during a timing-dependent paradigm in control cortex but not in that from a mouse model for Fragile X syndrome. Instead, the postsynaptic threshold for tLTP is shifted in Fragile X MR synapses, requiring a burst of postsynaptic APs during the STDP protocol to induce synaptic potentiation (data from Meredith et al., 2007 ). Abnormalities of dendritic branching patterns and spiny protrusion density and maturity are observed in many forms of MR. A number of candidate mechanisms including down-regulation of calcium channels, increased GABAergic inhibition and alteration of K+ conductance affecting function of dendrites and spines may underlie this tLTP deficit to reduce the dendritic excitability in the brain in MR syndromes.
The idea of a higher threshold for induction of plasticity is not unique to Fragile X syndrome and is emerging as a common factor from data in other mouse models. In the UBE3A-KO mouse model for Angelman syndrome, a similar higher threshold level for induction of plasticity using tetanic and theta-burst stimulation paradigms is observed in neocortex and in hippocampus (Jiang et al., 1998 ; Weeber et al., 2003 ; Yashiro et al., 2009 ). Deficits in the Ts65Dn mouse model for Down’s syndrome observed with theta-burst stimulation can also be overcome with stronger tetanic paradigms or by unblocking inhibition (Kleschevnikov et al., 2004 ; Costa and Grybko, 2005 ). A modifiable threshold level to describe the balance of potentiation and depression in synapses is described in the Bienenstock, Cooper, Munro (BCM) model (Bienenstock et al., 1982 ). Here, the threshold for modification is a non-linear function of postsynaptic activity in the synapses with low activity levels leading to depression and higher rates leading to potentiation. The application of this theory to STDP protocols is not straightforward but has been applied to experimental data using triplet spiking rules and a burst-firing paradigm with differing postsynaptic activity levels (Sjostrom et al., 2001 ; Morrison et al., 2008 ). In Fmr1-KO mouse model, no evidence exists for a shift in BCM threshold at layer 2/3 cortical synapses, with tLTD and tLTP resulting from low and high respective frequency of postsynaptic bursts during pairing in a manner similar to WT synapses (Supplementary data in Meredith et al., 2007 ).
Despite significant effects upon tLTP, timing-dependent synaptic depression (tLTD) appears normal in both sensory and prefrontal cortex of Fmr1-KO mice mouse (Desai et al., 2006 ; Meredith et al., 2007 ). The reasons for this lack of change in cortical synaptic depression in the Fmr1-KO mouse model are not currently known. In hippocampus and cerebellum, plasticity mechanisms for chemically-induced mGluR-mediated synaptic depression in the Fmr1-KO mouse reveal an enhanced LTD (Huber et al., 2002 ; Koekkoek et al., 2005 ). These findings led to the theory of altered signaling via group 1 mGluRs in Fragile X syndrome (Bear et al., 2004 ). tLTD protocols can be considered distinct from mGluR-dependent forms of synaptic depression, relying on presynaptic rather than postsynaptic NMDA receptors for induction (Rodriguez-Moreno and Paulsen, 2008 ; Banerjee et al., 2009 ) and not being heavily-dependent upon protein synthesis. Further investigations into these distinct synaptic depression mechanisms in cortex, hippocampus and cerebellum may provide mechanisms to explain the absence of effects upon tLTD in the Fmr1-KO mouse to-date.
Neurodevelopmental Onset of Plasticity
Many forms of autism and MR are classified as neurodevelopmental cognitive disorders with noticeable onset of phenotypic symptoms and immature dendritic spine morphology occurring during early postnatal stages (Zoghbi, 2003 ). Rett syndrome is a neurodevelopmental disorder typified by autistic behaviors, movement impairments and MR and caused by mutations in the X-linked MECP2 gene, which acts to regulation transcription (Nan et al., 1997 ; Amir et al., 1999 ). A MECP2-null mouse model was created that develops symptoms reminiscent of Rett syndrome with a delayed phenotypic onset similar to the human condition (Chen et al., 2001 ; Guy et al., 2001 ). Early onset of spine pathologies in MR raises the question of whether abnormalities are causal for changes in synaptic plasticity for example, or arise as a consequence of changes in synaptic processing (Fiala et al., 2002 ). The MECP2 mouse model gives the possibility to investigate the relationship between plasticity, synapse morphology and behavioral abnormalities.
At pre-symptomatic stages of two postnatal weeks old, MECP2 mutant mice show no alterations in tLTP paradigms with single postsynaptic action potentials in layer 5 of sensory cortex. At 4 weeks old, when onset of symptoms first occurs in MECP2 null mice, tLTP is also unimpaired, despite decreases in synaptic connectivity, weakened excitatory synapse strength and alteration in excitatory/inhibitory balance (Dani et al., 2005 ; Dani and Nelson, 2009 ). Impairments in LTP, induced with tetanic or theta-burst stimulation, are only seen in older symptomatic mice in both hippocampus and cortex (Asaka et al., 2006 ; Moretti et al., 2006 ). This suggests that deficits in synaptic plasticity seen after the onset of behavioral abnormalities, occur as a consequence of baseline synaptic function and connectivity impairments (Guy et al., 2007 ). Thus, alterations in mechanisms that underlie functional processing and integration of signaling in both dendrites and spines are likely candidates for a role in MR pathophysiology regulating STDP.
Dendritic Mechanisms Affecting Local Membrane Excitability
Dendritic excitability plays a key role in understanding the mechanisms of timing-dependent synaptic plasticity in the brain. Structural alterations, as documented in MR syndromes, will have profound effects upon distribution of ion channels in dendritic membranes and resultant effects on active and passive processing properties of signaling in the dendrites. During tLTP paradigms, backpropagation of a single action potential depolarizes the dendritic membrane, opening voltage-gated calcium channels on both the dendritic shaft (Callaway and Ross, 1995 ) and spines (Koester and Sakmann, 1998 ). The action potential does not always propagate throughout the entire dendritic tree and can follow a distant-dependent profile with lack of signal at distal dendritic regions (Jaffe et al., 1992 ). The degree of backpropagation into the dendrites depends critically upon dendritic morphology, including dendritic diameter and branching patterns (Vetter et al., 2001 ). Failure of signal propagation can also occur after dendritic branching points (Spruston et al., 1995 ). In Fragile X syndrome, alterations in basal dendritic branching patterns have been observed in both visual and somatosensory cortex of adult Fmr1-KO mice on a C57BL/6 background (Galvez et al., 2003 ; Restivo et al., 2005 ). Dendritic excitability may also be affected via alterations in axonal arborization patterns, with more spatially diffuse aborization seen during a restricted developmental period in superficial barrel cortex layers of the Fmr1-KO mouse (Bureau et al., 2008 ).
In modeling studies, the role of spine morphology on dendritic excitability and AP backpropagation is shown to be influential, where the inactivation threshold of sodium channels along the dendrite and potentially in the spines can regulate signal failure in the distal dendritic regions (Tsay and Yuste, 2002 ). In proximal dendrites of cortical layer 5 pyramidal neurons, AP backpropagation reliably transmitted to large spines without loss of voltage (Palmer and Stuart, 2009 ). However, the propagation into spines on smaller, more distal dendrites or into the filopodial protrusions with differing spine neck diameters and resistance more commonly seen along dendrites in MR syndromes, could well differ. Thus, alterations in dendritic morphology and cytoskeleton in retardation syndromes are likely to affect the efficacy of backpropagation into the dendritic tree and have consequences upon integrative signaling processes in dendritic compartments.
Calcium Transient Failures
Induction of LTP of excitatory synapses is critically dependent on increases in postsynaptic calcium signaling during both tetanic induction or STDP stimulation paradigms (Lynch et al., 1983 ; Nevian and Sakmann, 2006 ). In pyramidal neurons of prefrontal cortex from Fmr1-KO mice, action potential-mediated calcium transients in both dendrites and postsynaptic spines were more prone to failures than those in control neurons (Meredith et al., 2007 ). Active properties of dendrites are strongly shaped by the density, localization and properties of voltage-gated calcium channels distributed in the membrane (Magee and Johnston, 2005 ). Dendrites and spines express a variety of different voltage-gated calcium channels, localized to specific regions of the dendritic tree and spine location on the dendrite (Magee and Johnston, 1995 ; Bloodgood and Sabatini, 2007 ). In Fmr1-KO neurons in prefrontal cortex, calcium transients were less sensitive to L-type calcium channel antagonists, that could explain the decrease in calcium transients in these dendritic protrusions (Meredith et al., 2007 ). L-type calcium channel protein levels were also found to be down-regulated in frontal cortex in these KO mice (Chen et al., 2003 ). L-type calcium channels are found in both dendrites and spines and contribute to both resting calcium levels (Magee et al., 1996 ), excitability of the dendrite in the hippocampus (Moyer et al., 1992 ) and coupling of postsynaptic calcium influx activity to gene transcription (Murphy et al., 1991 ). Blocking L-type calcium channels in prefrontal cortex of wild-type mice prevents tLTP (Meredith et al., 2007 ). Thus, alterations in L-type channels in dendritic spines are one indicator of an impairment in activity-coupled plasticity mechanisms mediated via calcium transients in neuronal dendrites in Fragile X syndrome.
K+ Channels and Dendrite Excitability
Dendritic excitability is also modulated by alterations in the distribution and modulatory state of sodium and potassium channels in the dendritic membrane. Modeling studies demonstrate severe attenuation of the spread of backpropagation via subtle alterations in the distribution and density of sodium and potassium channels along the dendrite (Golding et al., 2001 ). In the peripheral nervous system, such ‘channelopathies’ affect dendritic excitability and mediate severe neuromuscular and cell signaling disorders. In temporal lobe epilepsy, a channelopathy involving A-type potassium channels directly modulates dendritic excitability via transcriptional and post-translational mechanisms (Bernard et al., 2004 ). The prevalence of epilepsy increases greatly with severe intellectual disability in many forms of MR. In both human and mouse forms of Fragile X syndrome, an increased incidence of epileptic activity is reported (Musumeci et al., 1999 ; Chen and Toth, 2001 ). It is not currently known whether significant alterations in ion channel distributions occur in dendrites in models of MR.
Levels of protein expression of specific potassium channels revealed significant up-regulation in subunits of the G-protein coupled inwardly-rectifying K+ channel in hippocampus and frontal cortex of the Ts65Dn mouse model for Down’s syndrome (Chie et al., 2006 ). Proteomics analysis of cortical synaptosomes from Fmr1-KO mice revealed a down-regulation in a set of proteins governing membrane excitability, including a large conductance, calcium-activated BK potassium channel subunit, Kcnma1α (Liao et al., 2008 ). BK ion channels are activated by membrane depolarization and increases in intracellular calcium concentration, resulting in hyperpolarization and decreased local dendritic excitability (Faber and Sah, 2003 ).
It is interesting to speculate that the increased threshold for tLTP in dendrites of Fmr1-KO neurons point towards regional dendritic impairments in excitability that are coupled to an interplay between local calcium transients, voltage-activated ion channels and synaptic stimulation. In prefrontal cortex, tLTP is blocked by application of the group 1 mGluR antagonist MPEP (Meredith et al., 2007 ) but the mechanisms governing this blockade are not known. The perisynaptic location of group 1 mGluRs (Baude et al., 1993 ) make them ideally placed postsynaptically to regulate dendritic channels governing excitability. Stimulation of group 1 mGluRs on hippocampal dendrites causes depolarization and firing of CA1 pyramidal cells, mediated by both voltage-gated calcium channels and inhibition of differing potassium currents (Anwyl, 1999 ). Although no changes in group 1 mGluR5 protein expression alone are found in Fmr1-KO mice, the alteration in linkage with postsynaptic density proteins such as Homer family may have resultant effects on other dendritically-located postsynaptic ion channels to mediate an alteration in excitability (Giuffrida et al., 2005 ).
Inhibitory Dendritic Tone
In addition to alterations in voltage-activated ion channels, another key player affecting synaptic plasticity is the role of GABAergic inhibition. In many different forms of MR, there are abnormalities in the balance of excitation to inhibition in the brain (Kleschevnikov et al., 2004 ; Dani et al., 2005 ; Gibson et al., 2008 ). GABAergic inhibition has significant effects on synaptic plasticity. Blocking GABAergic inhibition via GABA-A receptors reveals tLTP in mature hippocampal neurons (Meredith et al., 2003 ; Campanac and Debanne, 2008 ) and also rescues LTP induction in a mouse model for Down’s syndrome (Kleschevnikov et al., 2004 ). At the dendritic level, interneurons target highly specific compartments of dendrites of pyramidal neurons (Klausberger and Somogyi, 2008 ). Alterations in inhibitory tone in dendritic branches would have significant impact upon integration and plasticity at sites away from the cell soma. Backpropagation of an AP along the apical dendrite is under inhibitory regulation, suggesting that attenuation or even failure of signaling could be due to increased inhibitory tone (Tsubokawa and Ross, 1996 ; Meredith and Groen, 2010 ). It is not known whether inhibitory mechanisms in dendrites are altered in MR. In Fragile X syndrome, decreased expression of mRNA and protein levels of a number of GABA-A subunits are reported in the Fmr1-KO mouse (El Idrissi et al., 2005 ; D’Hulst et al., 2006 ). Furthermore, GABAergic inhibition at the network level is altered, as seen by prolonged epileptoform activity and altered connectivity in both hippocampus and cortex (Chuang et al., 2005 ; Gibson et al., 2008 ). The disinhibition leading to epileptogenesis in the Fmr1-KO model is sensitive to excitation via group 1 metabotropic receptors, since the antagonist MPEP suppresses epileptiform discharges in hippocampus (Chuang et al., 2005 ). Group 1 mGluRs are also present on inhibitory interneurons (Lopez-Bendito et al., 2002 ). Their direct activation may also regulate dendritic excitability of pyramidal neurons via alterations in inhibitory tone upon dendritic compartments. Dissection of the alterations in GABAergic inhibition, the role of specific interneurons involved and their activation patterns may elucidate many of the impairments in synaptic plasticity and network processing observed in the Fragile X retardation model. This delicate balance of inhibition in the brain may be an important feature common to many neurodevelopmental cognitive disorders (Rubenstein and Merzenich, 2003 ).
Summary
In summary, this review highlights timing-based cortical plasticity in the Fragile X syndrome model of MR and proposes that alterations in dendritic excitability underlie deficits seen in STDP. Subtle alterations in dendritic and spine morphology can have dramatic effects on efficacy of calcium signaling throughout the dendritic tree, with downstream consequences on calcium-activated intracellular signaling cascades. In Fragile X syndrome, FMRP interacts directly with many other proteins playing key roles in synapse morphology and plasticity, including Rho GTPase family members and excitatory mGluRs. Morphological abnormalities can also alter the distribution of ion channels, in particular K+ conductances that play a critical role in dendritic excitability. Finally, the alteration in inhibition/excitation balance observed from measurements at neuronal soma is likely to arise from significant imbalances in dendritic compartments and as a consequence, alter local dendritic depolarization necessary for STDP. Further investigation into these local dendritic mechanisms will give new insight into the deficits in dendrites and spiny protrusions that mediate deficits in these cognitive disorders.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Related research in the department is funded by FRAXA Research Organisation, Fondation Jérôme Lejeune, Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO) to HDM and RMM.
References
Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188.
Anwyl, R. (1999). Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res. Brain Res. Rev. 29, 83–120.
Asaka, Y., Jugloff, D. G., Zhang, L., Eubanks, J. H., and Fitzsimonds, R. M. (2006). Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol. Dis. 21, 217–227.
Bagni, C., and Greenough, W. T. (2005). From mRNP trafficking to spine dysmorphogenesis: the roots of fragile X syndrome. Nat. Rev. Neurosci. 6, 376–387.
Bakker, E. C., Verheij, C., Willemsen, R., van der Helm, R., Oerlemans, F., Vermey, M., Bygraved, A., Hoogeveen, A. T., Oostra, B. A., Reyniers, E., De Boule, K., D’Hooge, R., Cras, P., van Velzen, D., Nagels, G., Martin, J. J., De Deyn, P. P., Darby, J. K., Willems, P. J. (1994). Fmr1 knockout mice: a model to study fragile X mental retardation. The Dutch-Belgian Fragile X Consortium. Cell 78, 23–33.
Banerjee, A., Meredith, R. M., Rodriguez-Moreno, A., Mierau, S. B., Auberson, Y. P., and Paulsen, O. (2009). Double dissociation of spike timing-dependent potentiation and depression by subunit-preferring NMDA receptor antagonists in mouse barrel cortex. Cereb. Cortex 19, 2959–2969.
Baude, A., Nusser, Z., Roberts, J. D., Mulvihill, E., McIlhinney, R. A., and Somogyi, P. (1993). The metabotropic glutamate receptor (mGluR1 alpha) is concentrated at perisynaptic membrane of neuronal subpopulations as detected by immunogold reaction. Neuron 11, 771–787.
Bear, M. F., Huber, K. M., and Warren, S. T. (2004). The mGluR theory of fragile X mental retardation. Trends Neurosci. 27, 370–377.
Bell, C. C., Han, V. Z., Sugawara, Y., and Grant, K. (1997). Synaptic plasticity in a cerebellum-like structure depends on temporal order. Nature 387, 278–281.
Bernard, C., Anderson, A., Becker, A., Poolos, N. P., Beck, H., and Johnston, D. (2004). Acquired dendritic channelopathy in temporal lobe epilepsy. Science 305, 532–535.
Best, T. K., Cho-Clark, M., Siarey, R. J., and Galdzicki, Z. (2008). Speeding of miniature excitatory post-synaptic currents in Ts65Dn cultured hippocampal neurons. Neurosci. Lett. 438, 356–361.
Bi, G., and Poo, M. (2001). Synaptic modification by correlated activity: Hebb’s postulate revisited. Annu. Rev. Neurosci. 24, 139–166.
Bi, G. Q., and Poo, M. M. (1998). Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J. Neurosci. 18, 10464–10472.
Bienenstock, E. L., Cooper, L. N., and Munro, P. W. (1982). Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. J. Neurosci. 2, 32–48.
Bloodgood, B. L., and Sabatini, B. L. (2007). Ca(2+) signaling in dendritic spines. Curr. Opin. Neurobiol. 17, 345–351.
Braun, K., and Segal, M. (2000). FMRP involvement in formation of synapses among cultured hippocampal neurons. Cereb. Cortex 10, 1045–1052.
Bureau, I., Shepherd, G. M., and Svoboda, K. (2008). Circuit and plasticity defects in the developing somatosensory cortex of FMR1 knock-out mice. J. Neurosci. 28, 5178–5188.
Callaway, J. C., and Ross, W. N. (1995). Frequency-dependent propagation of sodium action potentials in dendrites of hippocampal CA1 pyramidal neurons. J. Neurophysiol. 74, 1395–1403.
Campanac, E., and Debanne, D. (2008). Spike timing-dependent plasticity: a learning rule for dendritic integration in rat CA1 pyramidal neurons. J. Physiol. (Lond.) 586, 779–793.
Chapleau, C. A., Calfa, G. D., Lane, M. C., Albertson, A. J., Larimore, J. L., Kudo, S., Armstrong, D. L., Percy, A. K., and Pozzo-Miller, L. (2009). Dendritic spine pathologies in hippocampal pyramidal neurons from Rett syndrome brain and after expression of Rett-associated MECP2 mutations. Neurobiol. Dis. 35, 219–233.
Chen, L., and Toth, M. (2001). Fragile X mice develop sensory hyperreactivity to auditory stimuli. Neuroscience 103, 1043–1050.
Chen, L., Yun, S. W., Seto, J., Liu, W., and Toth, M. (2003). The fragile X mental retardation protein binds and regulates a novel class of mRNAs containing U rich target sequences. Neuroscience 120, 1005–1017.
Chen, R. Z., Akbarian, S., Tudor, M., and Jaenisch, R. (2001). Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 27, 327–331.
Chie, H., David, M. J., Jassir, W., Rosemary, C. B., Tyler, K. B., Richard, J. S., and Zygmunt, G. (2006). Abnormal expression of the G-protein-activated inwardly rectifying potassium channel 2 (GIRK2) in hippocampus, frontal cortex, and substantia nigra of Ts65Dn mouse: a model of Down syndrome. J. Comp. Neurol. 494, 815–833.
Chuang, S. C., Zhao, W., Bauchwitz, R., Yan, Q., Bianchi, R., and Wong, R. K. (2005). Prolonged epileptiform discharges induced by altered group I metabotropic glutamate receptor-mediated synaptic responses in hippocampal slices of a fragile X mouse model. J. Neurosci. 25, 8048–8055.
Costa, A. C., and Grybko, M. J. (2005). Deficits in hippocampal CA1 LTP induced by TBS but not HFS in the Ts65Dn mouse: a model of Down syndrome. Neurosci. Lett. 382, 317–322.
D’Hulst, C., De Geest, N., Reeve, S. P., Van Dam, D., De Deyn, P. P., Hassan, B. A., and Kooy, R. F. (2006). Decreased expression of the GABA(A) receptor in fragile X syndrome. Brain Res. 1121, 238–245.
D’Hulst, C., and Kooy, R. F. (2007). The GABAA receptor: a novel target for treatment of fragile X? Trends in Neurosci. 30:425-431.
Dani, V. S., Chang, Q., Maffei, A., Turrigiano, G. G., Jaenisch, R., and Nelson, S. B. (2005). Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 102, 12560–12565.
Dani, V. S., and Nelson, S. B. (2009). Intact long-term potentiation but reduced connectivity between neocortical layer 5 pyramidal neurons in a mouse model of Rett syndrome. J. Neurosci. 29, 11263–11270.
Desai, N. S., Casimiro, T. M., Gruber, S. M., and Vanderklish, P. W. (2006). Early postnatal plasticity in neocortex of Fmr1 knockout mice. J. Neurophysiol. 96, 1734–1745.
El Idrissi, A., Ding, X. H., Scalia, J., Trenkner, E., Brown, W. T., and Dobkin, C. (2005). Decreased GABA(A) receptor expression in the seizure-prone fragile X mouse. Neurosci. Lett. 377, 141–146.
Faber, E. S. L., and Sah, P. (2003). Calcium-activated potassium channels: multiple contributions to neuronal function. Neuroscientist. 9, 181–194.
Fiala, J. C., Spacek, J., and Harris, K. M. (2002). Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res. Brain Res. Rev. 39, 29–54.
Galvez, R., Gopal, A. R., and Greenough, W. T. (2003). Somatosensory cortical barrel dendritic abnormalities in a mouse model of the fragile X mental retardation syndrome. Brain Res. 971, 83–89.
Gibson, J. R., Bartley, A. F., Hays, S. A., and Huber, K. M. (2008). Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. J. Neurophysiol. 100, 2615–2626.
Giuffrida, R., Musumeci, S., D’Antoni, S., Bonaccorso, C. M., Giuffrida-Stella, A. M., Oostra, B. A., and Catania, M. V. (2005). A reduced number of metabotropic glutamate subtype 5 receptors are associated with constitutive homer proteins in a mouse model of fragile X syndrome. J. Neurosci. 25, 8908–8916. doi: 10.1523/JNEUROSCI.0932-05.2005.
Godfraind, J. M., Reyniers, E., De Boulle, K., D’Hooge, R., De Deyn, P. P., Bakker, C. E., Oostra, B. A., Kooy, R. F., and Willems, P. J. (1996). Long-term potentiation in the hippocampus of fragile X knockout mice. Am. J. Med. Genet. 64, 246–251.
Golding, N. L., Kath, W. L., and Spruston, N. (2001). Dichotomy of action- potential backpropagation in CA1 pyramidal neuron dendrites. J. Neurophysiol. 86, 2998–3010.
Gustafsson, B., Wigstrom, H., Abraham, W. C., and Huang, Y. Y. (1987). Long-term potentiation in the hippocampus using depolarizing current pulses as the conditioning stimulus to single volley synaptic potentials. J. Neurosci. 7, 774–780.
Guy, J., Gan, J., Selfridge, J., Cobb, S., and Bird, A. (2007). Reversal of neurological defects in a mouse model of Rett syndrome. Science 315, 1143–1147. doi: 10.1126/science.1138389.
Guy, J., Hendrich, B., Holmes, M., Martin, J. E., and Bird, A. (2001). A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 27, 322–326.
Harlow, E. G., Till, S. M., Russell, T. A., Wijetunge, L. S., Kind, P., and Contractor, A. (2010). Critical period plasticity is disrupted in the barrel cortex of FMR1 knockout mice. Neuron 65, 385–398.
Harris, K. M. (1999). Structure, development, and plasticity of dendritic spines. Curr. Opin. Neurobiol. 9, 343–348.
Hu, H., Qin, Y., Bochorishvili, G., Zhu, Y., van Aelst, L., and Zhu, J. J. (2008). Ras signaling mechanisms underlying impaired GluR1-dependent plasticity associated with fragile X syndrome. J. Neurosci. 28, 7847–7862.
Huber, K. M., Gallagher, S. M., Warren, S. T., and Bear, M. F. (2002). Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. U.S.A. 99, 7746–7750.
Irwin, S. A., Galvez, R., and Greenough, W. T. (2000). Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb. Cortex 10, 1038–1044.
Jaffe, D. B., Johnston, D., Lasser-Ross, N., Lisman, J. E., Miyakawa, H., and Ross, W. N. (1992). The spread of Na+ spikes determines the pattern of dendritic Ca2+ entry into hippocampal neurons. Nature 357, 244–246.
Jiang, Y. H., Armstrong, D., Albrecht, U., Atkins, C. M., Noebels, J. L., Eichele, G., Sweatt, J. D., and Beaudet, A. L. (1998). Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron 21, 799–811.
Jin, P., and Warren, S. T. (2000). Understanding the molecular basis of fragile X syndrome. Hum. Mol. Genet. 9, 901–908.
Kaufmann, W. E., and Moser, H. W. (2000). Dendritic anomalies in disorders associated with mental retardation. Cereb. Cortex 10, 981–991.
Klausberger, T., and Somogyi, P. (2008). Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science 321, 53–57.
Kleschevnikov, A. M., Belichenko, P. V., Villar, A. J., Epstein, C. J., Malenka, R. C., and Mobley, W. C. (2004). Hippocampal long-term potentiation suppressed by increased inhibition in the Ts65Dn mouse, a genetic model of down syndrome. J. Neurosci. 24, 8153–8160.
Kobayashi, K., Kuroda, S., Fukata, M., Nakamura, T., Nagase, T., Nomura, N., Matsuura, Y., Yoshida-Kubomura, N., Iwamatsu, A., and Kaibuchi, K. (1998). p140Sra-1 (specifically Rac1-associated protein) is a novel specific target for Rac1 small GTPase. J. Biol. Chem. 273, 291–295.
Koekkoek, S. K., Yamaguchi, K., Milojkovic, B. A., Dortland, B. R., Ruigrok, T. J., Maex, R., De Graaf, W., Smit, A. E., VanderWerf, F., Bakker, C. E., Willemsen, R., Ikeda, T., Kakizawa, S., Onodera, K., Nelson, D. L., Mientjes, E., Joosten, M., De Schutter, E., Oostra, B. A., Ito, M., De Zeeuw, C. I. (2005). Deletion of FMR1 in Purkinje cells enhances parallel fiber LTD, enlarges spines, and attenuates cerebellar eyelid conditioning in Fragile X syndrome. Neuron 47, 339–352.
Koester, H. J., and Sakmann, B. (1998). Calcium dynamics in single spines during coincident pre- and postsynaptic activity depend on relative timing of back-propagating action potentials and subthreshold excitatory postsynaptic potentials. Proc. Natl. Acad. Sci. U.S.A. 95, 9596–9601.
Lauterborn, J. C., Rex, C. S., Kramar, E., Chen, L. Y., Pandyarajan, V., Lynch, G., and Gall, C. M. (2007). Brain-derived neurotrophic factor rescues synaptic plasticity in a mouse model of fragile x syndrome. J. Neurosci. 27, 10685–10694.
Levy, W. B., and Steward, O. (1983). Temporal contiguity requirements for long-term associative potentiation/depression in the hippocampus. Neuroscience 8, 791–797.
Li, J., Pelletier, M. R., Perez Velazquez, J. L., and Carlen, P. L. (2002). Reduced cortical synaptic plasticity and GluR1 expression associated with fragile X mental retardation protein deficiency. Mol. Cell. Neurosci. 19, 138–151.
Liao, L., Park, S. K., Xu, T., Vanderklish, P., and Yates, J. R., 3rd. (2008). Quantitative proteomic analysis of primary neurons reveals diverse changes in synaptic protein content in fmr1 knockout mice. Proc. Natl. Acad. Sci. U.S.A. 105, 15281–15286.
Lopez-Bendito, G., Shigemoto, R., Fairen, A., and Lujan, R. (2002). Differential distribution of group I metabotropic glutamate receptors during rat cortical development. Cereb. Cortex 12, 625–638.
Luo, L., Hensch, T. K., Ackerman, L., Barbel, S., Jan, L. Y., and Jan, Y. N. (1996). Differential effects of the Rac GTPase on Purkinje cell axons and dendritic trunks and spines. Nature 379, 837–840.
Lynch, G., Larson, J., Kelso, S., Barrionuevo, G., and Schottler, F. (1983). Intracellular injections of EGTA block induction of hippocampal long-term potentiation. Nature 305, 719–721.
Magee, J. C., Avery, R. B., Christie, B. R., and Johnston, D. (1996). Dihydropyridine-sensitive, voltage-gated Ca2+ channels contribute to the resting intracellular Ca2+ concentration of hippocampal CA1 pyramidal neurons. J. Neurophysiol. 76, 3460–3470.
Magee, J. C., and Johnston, D. (1995). Characterization of single voltage-gated Na+ and Ca2+ channels in apical dendrites of rat CA1 pyramidal neurons. J. Physiol. (Lond.) 487, 67–90.
Magee, J. C., and Johnston, D. (1997). A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science 275, 209–213.
Magee, J. C., and Johnston, D. (2005). Plasticity of dendritic function. Curr. Opin. Neurobiol. 15, 334–342.
Markram, H., Lubke, J., Frotscher, M., and Sakmann, B. (1997). Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science 275, 213–215.
Martin, S. J., Grimwood, P. D., and Morris, R. G. M. (2000). Synaptic plasticity and memory: an evaluation of the hypothesis. Annu. Rev. Neurosci. 23, 649–711.
Matsuzaki, M., Ellis-Davies, G. C., Nemoto, T., Miyashita, Y., Iino, M., and Kasai, H. (2001). Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat. Neurosci. 4, 1086–1092.
Meredith, R. M., Floyer-Lea, A. M., and Paulsen, O. (2003). Maturation of long-term potentiation induction rules in rodent hippocampus: role of GABAergic inhibition. J. Neurosci. 23, 11142–11146.
Meredith, R. M., and Groen, M. R. (2010). Inhibition of action potential backpropagation during postnatal development of the hippocampus. J. Neurophysiol. 103, 2313.
Meredith, R. M., Holmgren, C. D., Weidum, M., Burnashev, N., and Mansvelder, H. D. (2007). Increased threshold for spike-timing-dependent plasticity is caused by unreliable calcium signaling in mice lacking fragile X gene FMR1. Neuron 54, 627–638.
Moretti, P., Levenson, J. M., Battaglia, F., Atkinson, R., Teague, R., Antalffy, B., Armstrong, D., Arancio, O., Sweatt, J. D., and Zoghbi, H. Y. (2006). Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J. Neurosci. 26, 319–327.
Morrison, A., Diesmann, M., and Gerstner, W. (2008). Phenomenological models of synaptic plasticity based on spike timing. Biol. Cybern. 98, 459–478.
Moyer, J. R., Jr., Thompson, L. T., Black, J. P., and Disterhoft, J. F. (1992). Nimodipine increases excitability of rabbit CA1 pyramidal neurons in an age- and concentration- dependent manner. J. Neurophysiol. 68, 2100–2109.
Murphy, T. H., Worley, P. F., and Baraban, J. M. (1991). L-type voltage-sensitive calcium channels mediate synaptic activation of immediate early genes. Neuron 7, 625–635.
Musumeci, S. A., Hagerman, R. J., Ferri, R., Bosco, P., Dalla Bernardina, B., Tassinari, C. A., De Sarro, G. B., and Elia, M. (1999). Epilepsy and EEG findings in males with fragile X syndrome. Epilepsia 40, 1092–1099.
Nadif Kasri, N., and Van Aelst, L. (2008). Rho-linked genes and neurological disorders. Pflugers Arch. 455, 787–797.
Nan, X., Campoy, F. J., and Bird, A. (1997). MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell 88, 471–481.
Negishi, M., and Katoh, H. (2005). Rho family GTPases and dendrite plasticity. Neuroscientist. 11, 187–191.
Nevian, T., and Sakmann, B. (2006). Spine Ca2+ signaling in spike-timing-dependent plasticity. J. Neurosci. 26, 11001–11013.
Newey, S. E., Velamoor, V., Govek, E. E., and Van Aelst, L. (2005). Rho GTPases, dendritic structure, and mental retardation. J. Neurobiol. 64, 58–74.
Nimchinsky, E. A., Oberlander, A. M., and Svoboda, K. (2001). Abnormal development of dendritic spines in FMR1 knock-out mice. J. Neurosci. 21, 5139–5146.
Palmer, L. M., and Stuart, G. J. (2009). Membrane potential changes in dendritic spines during action potentials and synaptic input. J. Neurosci. 29, 6897–6903.
Paradee, W., Melikian, H. E., Rasmussen, D. L., Kenneson, A., Conn, P. J., and Warren, S. T. (1999). Fragile X mouse: strain effects of knockout phenotype and evidence suggesting deficient amygdala function. Neuroscience 94, 185–192.
Pfeiffer, B. E., and Huber, K. M. (2007). Fragile X mental retardation protein induces synapse loss through acute postsynaptic translational regulation. J. Neurosci. 27, 3120–3130. doi: 10.1523/JNEUROSCI.0054-07.2007.
Ramakers, G. J. (2002). Rho proteins, mental retardation and the cellular basis of cognition. Trends Neurosci. 25, 191–199.
Restivo, L., Ferrari, F., Passino, E., Sgobio, C., Bock, J., Oostra, B. A., Bagni, C., and Ammassari-Teule, M. (2005). Enriched environment promotes behavioral and morphological recovery in a mouse model for the fragile X syndrome. Proc. Natl. Acad. Sci. U.S.A. 102, 11557–11562.
Rodriguez-Moreno, A., and Paulsen, O. (2008). Spike timing-dependent long-term depression requires presynaptic NMDA receptors. Nat. Neurosci. 11, 744–745.
Rubenstein, J. L., and Merzenich, M. M. (2003). Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2, 255–267.
Schenck, A., Bardoni, B., Moro, A., Bagni, C., and Mandel, J.-L. (2001). A highly conserved protein family interacting with the fragile X mental retardation protein (FMRP) and displaying selective interactions with FMRP-related proteins FXR1P and FXR2P. Proc. Natl. Acad. Sci. U.S.A. 98, 8844–8849.
Siarey, R. J., Stoll, J., Rapoport, S. I., and Galdzicki, Z. (1997). Altered long-term potentiation in the young and old Ts65Dn mouse, a model for Down syndrome. Neuropharmacology 36, 1549–1554.
Sjostrom, P. J., Turrigiano, G. G., and Nelson, S. B. (2001). Rate, timing, and cooperativity jointly determine cortical synaptic plasticity. Neuron 32, 1149–1164.
Spruston, N., Schiller, Y., Stuart, G., and Sakmann, B. (1995). Activity-dependent action potential invasion and calcium influx into hippocampal CA1 dendrites. Science 268, 297–300.
Tashiro, A., Minden, A., and Yuste, R. (2000). Regulation of dendritic spine morphology by the Rho family of small GTPases: antagonistic roles of Rac and Rho. Cereb. Cortex 10, 927–938.
Threadgill, R., Bobb, K., and Ghosh, A. (1997). Regulation of dendritic growth and remodeling by Rho, Rac, and Cdc42. Neuron 19, 625–634.
Tsay, D., and Yuste, R. (2002). Role of dendritic spines in action potential backpropagation: a numerical simulation study. J. Neurophysiol. 88, 2834–2845.
Tsubokawa, H., and Ross, W. N. (1996). IPSPs modulate spike backpropagation and associated [Ca2+]i changes in the dendrites of hippocampal CA1 pyramidal neurons. J. Neurophysiol. 76, 2896–2906.
Vetter, P., Roth, A., and Hausser, M. (2001). Propagation of action potentials in dendrites depends on dendritic morphology. J. Neurophysiol. 85, 926–937.
Weeber, E. J., Jiang, Y.-H., Elgersma, Y., Varga, A. W., Carrasquillo, Y., Brown, S. E., Christian, J. M., Mirnikjoo, B., Silva, A., Beaudet, A. L., and Sweatt, J. D. (2003). Derangements of hippocampal calcium/calmodulin-dependent protein kinase II in a mouse model for Angelman mental retardation syndrome. J. Neurosci. 23, 2634–2644.
Weiler, I. J., Irwin, S. A., Klintsova, A. Y., Spencer, C. M., Brazelton, A. D., Miyashiro, K., Comery, T. A., Patel, B., Eberwine, J., and Greenough, W. T. (1997). Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc. Natl. Acad. Sci. U.S.A. 94, 5395–5400.
Yashiro, K., Riday, T. T., Condon, K. H., Roberts, A. C., Bernardo, D. R., Prakash, R., Weinberg, R. J., Ehlers, M. D., and Philpot, B. D. (2009). Ube3a is required for experience-dependent maturation of the neocortex. Nat. Neurosci. 12, 777–783.
Zalfa, F., Giorgi, M., Primerano, B., Moro, A., Di Penta, A., Reis, S., Oostra, B., and Bagni, C. (2003). The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell 112, 317–327.
Keywords: STDP, mental retardation, Fragile X syndrome, LTP, dendritic excitability, calcium, spine, dendrite
Citation: Meredith RM and Mansvelder HD (2010) STDP and mental retardation: dysregulation of dendritic excitability in Fragile X syndrome. Front. Syn. Neurosci. 2:10. doi: 10.3389/fnsyn.2010.00010
Received: 01 February 2010;
Paper pending published: 20 February 2010;
Accepted: 17 May 2010;
Published online: 10 June 2010
Edited by:
Per Jesper Sjöström, University College London, UKReviewed by:
Niraj S. Desai, The Neurosciences Institute, USAGül Dölen, Massachusetts Institute of Technology, USA
Copyright: © 2010 Meredith and Mansvelder. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Rhiannon M. Meredith, Department of Integrative Neurophysiology, Center for Neurogenomics and Cognitive Research, Neuroscience Campus Amsterdam, VU University, De Boelelaan 1085, 1081 HV Amsterdam, Netherlands. e-mail:cmhpYW5ub24ubWVyZWRpdGhAY25jci52dS5ubA==