 Lindsay Oberman
1
Lindsay Oberman
1 Fritz Ifert-Miller
1
Fritz Ifert-Miller
1 Umer Najib
1
Umer Najib
1 Shahid Bashir
1
Shahid Bashir
1 Ione Woollacott
2
Ione Woollacott
2 Joseph Gonzalez-Heydrich
3
Joseph Gonzalez-Heydrich
3 Jonathan Picker
3,4
Jonathan Picker
3,4 Alexander Rotenberg
1,5
Alexander Rotenberg
1,5 Alvaro Pascual-Leone
1*
Alvaro Pascual-Leone
1*
- 1 Department of Neurology, Berenson-Allen Center for Noninvasive Brain Stimulation, Beth Israel Deaconess Medical Center, Harvard University Medical School, Boston, MA, USA
- 2 Faculty of Medicine and Dentistry, University of Bristol, Bristol, UK
- 3 Department of Child and Adolescent Psychiatry, Children’s Hospital, Harvard Medical School, Boston, MA, USA
- 4 Division of Genetics, Children’s Hospital, Harvard Medical School, Boston, MA, USA
- 5 Department of Neurology, Children’s Hospital, Harvard Medical School, Boston, MA, USA
Fragile X Syndrome (FXS) is the most common heritable cause of intellectual disability. In vitro electrophysiologic data from mouse models of FXS suggest that loss of fragile X mental retardation protein affects intracortical excitability and synaptic plasticity. Specifically, the cortex appears hyperexcitable, and use-dependent long-term potentiation (LTP) and long-term depression (LTD) of synaptic strength are abnormal. Though animal models provide important information, FXS and other neurodevelopmental disorders are human diseases and as such translational research to evaluate cortical excitability and plasticity must be applied in the human. Transcranial magnetic stimulation paradigms have recently been developed to non-invasively investigate cortical excitability using paired pulse stimulation, as well as LTP- and LTD-like synaptic plasticity in response to theta burst stimulation (TBS) in vivo in the human. TBS applied on consecutive days can be used to measure metaplasticity (the ability of the synapse to undergo a second plastic change following a recent induction of plasticity). The current study investigated intracortical inhibition, plasticity and metaplasticity in full mutation females with FXS, participants with autism spectrum disorders (ASD), and neurotypical controls. Results suggest that intracortical inhibition is normal in participants with FXS, while plasticity and metaplasticity appear abnormal. ASD participants showed abnormalities in plasticity and metaplasticity, as well as heterogeneity in intracortical inhibition. Our findings highlight the utility of non-invasive neurophysiological measures to translate insights from animal models to humans with neurodevelopmental disorders, and thus provide direct confirmation of cortical dysfunction in patients with FXS and ASD.
Introduction
Fragile X syndrome (FXS), an X-linked disorder affecting 1 in 4000 males and at least 1 in 8000 females (O’Donnell and Warren, 2002 ), is the most common heritable cause of intellectual disability. Thirty percent of children with FXS are diagnosed with autism and 2–5% of autistic children have FXS (Kaufmann et al., 2004 ; Hagerman et al., 2005 ). FXS arises from a mutation in the X-chromosome gene FMR1, due to transcriptional silencing resulting from hypermethylation of an expansion of the CGG trinucleotide (Pfeiffer and Huber, 2009 ). The silencing of FMR1 transcription prevents or reduces translation of its protein product, fragile X mental retardation protein (FMRP) (Tassone et al., 1999 ). The cognitive behavioral phenotype of FXS includes general cognitive impairment (with moderate deficits in males and variable deficits in females that can range from as severely affected as males to asymptomatic), hyperarousal, hypersensitivity to sensory stimuli, anxiety, epilepsy, ADHD, and as mentioned above, characteristics of autism (Miller et al., 1999 ; Hagerman and Hagerman, 2002 ). Transgenic mouse and fly models, lack FMRP, and like affected humans, show an equivalent behavioral phenotype (Bakker, 1994 ; Musumeci et al., 2000 ; Nielsen et al., 2002 ; Spencer et al., 2005 ; Brennan et al., 2006 ).
Molecular Pathobiology of FXS
Fragile X mental retardation protein normally functions as a an inhibitory regulator of translation of metabotropic glutamate receptor 5 (mGluR5) (Laggerbauer et al., 2001 ; Li et al., 2001 ; Zhang et al., 2001 ; Bear et al., 2004 ). Thus the loss of FMRP results in excessive translation of mGluR5. mGluR5 plays an integral role in the establishment of new synaptic connections (Le Be and Markram, 2006 ) and thus is critical to stabilizing long-term depression (LTD) and inducing long-term potentiation (LTP) (Simonyi et al., 2005 ). The excessive mGluR5 activity caused by the lack of FMRP is apparent neuroanatomically with an increased density of long, thin aberrantly shaped dendritic spines in post mortem studies (Irwin et al., 2001 ) and animal models of fragile X (Irwin et al., 2002 ). This dysregulation caused by mGluR5 over activity is thought to lead to a number of characteristic FXS neurological and behavioral phenotypes, including epilepsy, cognitive impairment, developmental delay, and loss of motor coordination. Thus, if the mGluR receptor is allowed to activate without inhibition from FMRP, one would expect to see abnormalities in both cortical plasticity and excitability (see Bear et al., 2004 for a review). Indeed, in vitro data from Fmr1 knockout mouse FXS models show exaggerated LTD of excitatory synaptic strength with low frequency electrical stimulation (Huber et al., 2002 ; Koekkoek et al., 2005 ; Hou et al., 2006 ) and impaired LTP of excitatory synapses with high frequency electrical stimulation (Li et al., 2002 ; Zhao et al., 2005 ; Desai et al., 2006 ; Meredith et al., 2007 ; Wilson and Cox, 2007 ). The mouse model data also show decreased expression of the GABAA receptor in the cortex, hippocampus, diencephalon, and brainstem (see D’Hulst and Kooy, 2007 ) leading to an overall hyperexcitable brain due to lack of GABAA mediated intracortical inhibition.
Studying Cortical Excitability and Plasticity In Vivo in the Human
Though animal models provide an invaluable resource for understanding the neurophysiology of disease, it is often difficult to translate these insights into studies that can be done in patients with the disorders. In the human, the neurophysiological substrate may be the same as or different from the animal model, thus non-invasive neurophysiological studies using stimulation techniques in humans that are analogous to those used in basic physiologic experiments provide a means to validate findings reported in animal models. Here we propose that non-invasive methods can be used to evaluate cortical excitability and plasticity in vivo in patients with neurodevelopmental disorders. In doing so, we are able to show in patients with FXS that abnormalities in cortical plasticity exist, which are analogous to those reported in mouse models. Specifically, we used transcranial magnetic stimulation (TMS) to measure cortical excitability and plasticity in patients with FXS, individuals with autism spectrum disorder (ASD) without FXS, and healthy, neurotypical controls.
During TMS, small intracranial electrical currents are generated by a powerful fluctuating extracranial magnetic field. Measures of cortico-spinal excitability can then be obtained both at baseline and following a train of stimulation by peripherally measuring the motor-evoked potential in the muscle corresponding to the region of motor cortex that is stimulated. Specific TMS protocols have been introduced in an effort to investigate cortical excitability and plasticity mechanisms in the human brain. These protocols include adaptations of paired pulse stimulation (Kujirai et al., 1993 ) and theta burst stimulation (TBS) (Huang et al., 2005 ). Paired pulse stimulation can be delivered using different inter-pulse intervals to evaluate excitatory and inhibitory mechanisms and thus provide insights into cortical excitation/inhibition balance (E/I balance) (Kobayashi and Pascual-Leone, 2003 ). TBS can assess LTP-like and LTD-like plasticity by introducing a train of high frequency stimulation and then evaluating the cortical or cortical-spinal response to single-pulse TMS for a period of time following the plasticity-inducing train. Finally, we can evaluate metaplasticity by investigating the effect of a single session of TBS on a subsequent session of TBS carried out 24 h later (see Figure 1 ).
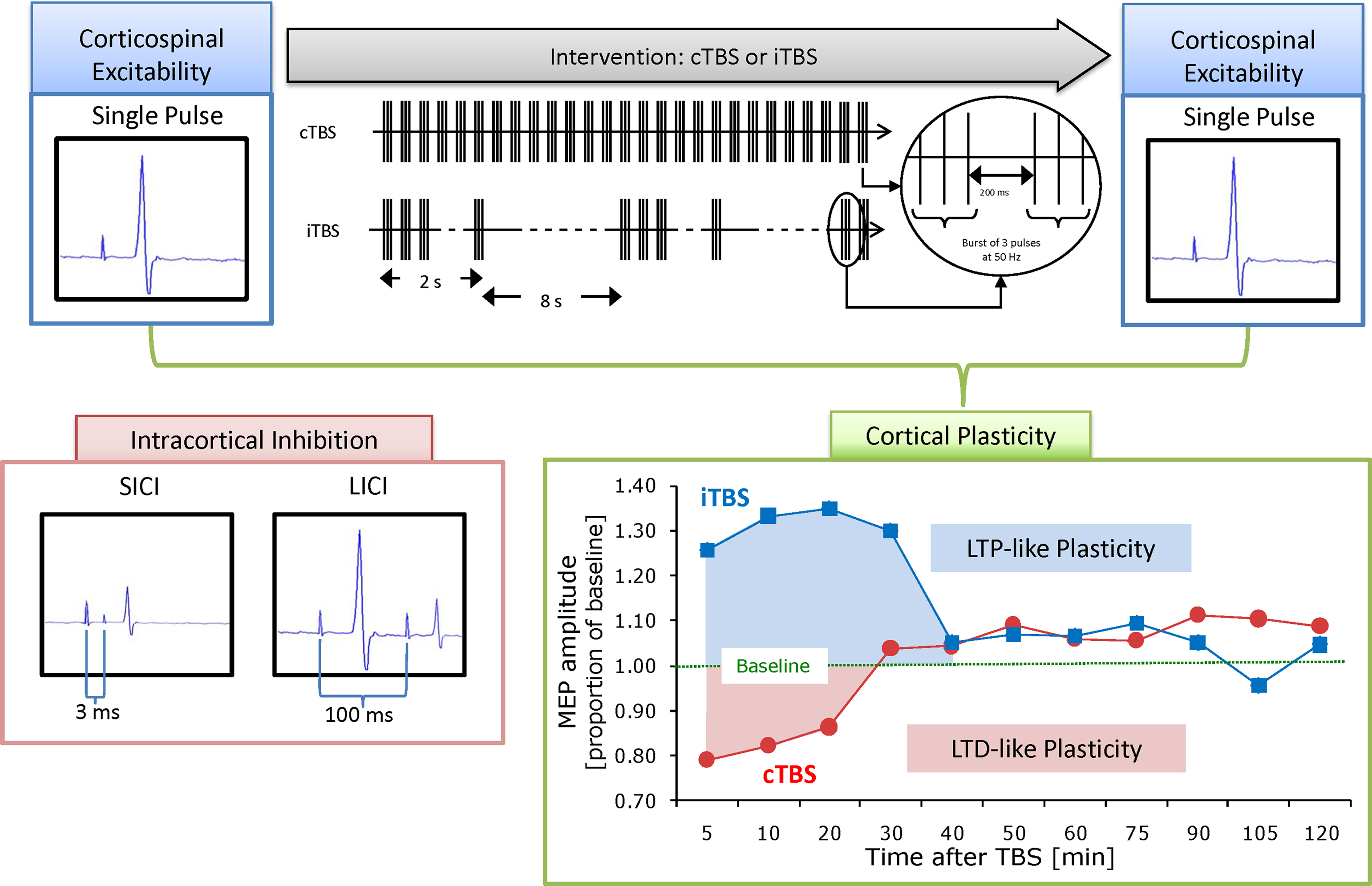
Figure 1. Schematic summary of applied methodology. Cortico-spinal excitability can be evaluated by comparing motor-evoked potentials (MEPs) recorded from the peripheral muscle in response to a single pulse to the primary motor cortex. These responses can then be obtained both at baseline and following continuous or intermittent theta burst stimulation (TBS), producing a measure of local cortical plasticity. TBS involves applying bursts of high frequency magnetic stimulation (three pulses at 50 Hz) repeated at intervals of 200 ms. After TBS is applied to the motor cortex in an intermittent fashion (iTBS), single pulse TMS-induced MEPs show increased amplitude for a period of 20–30 min, whereas continuous TBS (cTBS) leads to a suppression of the TMS-induced MEPs for approximately the same amount of time (Huang et al., 2005 ). Post-TBS enhancement (following iTBS) or suppression (after cTBS) of the cortical activity is considered an index of LTP and LTD-like induction of plasticity in the targeted brain area. Paired pulses to the primary motor cortex with either 3 or 100 ms interstimulus interval can be applied during a separate session to evaluate intracortical inhibition.
In the conventional paired pulse paradigm, two consecutive magnetic stimuli, a conditioning stimulus (CS) and a test stimulus (TS), are delivered through a TMS-coil over the motor cortex (Claus et al., 1992 ; Kujirai et al., 1993 ; Ziemann, 1999 ). An inter-pulse interval of 1–6 ms is thought to evaluate short interval intracortical inhibition (SICI). It is hypothesized that SICI is mediated by GABAergic intracortical inhibitory neurons and specifically by the GABAA receptor (Kujirai et al., 1993 ). Two suprathreshold pulses delivered at an inter-pulse interval of 50–200 ms is thought to evaluate long interval intracortical inhibition (LICI) and thought to reflect long-lasting cortical inhibition mediated by the GABAB receptor (Valls-Sole et al., 1992 ).
While paired pulse TMS is capable of evaluating intracortical inhibition, TBS provides a non-invasive index of LTP-like and LTD-like cortical plasticity in the human brain (Huang et al., 2005 ). Theta burst TMS protocols lead to changes in cortical activity that last well beyond the duration of the TMS application, show a time course consistent with that found with LTP, and LTD in slice preparations, and are also modulated by glutamatergic and GABAergic mediators (Huang et al., 2007 ; Stagg et al., 2009 ). The propensity of a given synapse to exhibit LTP or LTD can be altered based on its recent history. This “plasticity of plasticity” is referred to as metaplasticity and entails a change in the state of the synapse that alters its ability to make subsequent plastic changes. Metaplasticity involves many of the same mechanisms as conventional LTP and LTD (including activation of metabotropic glutamate receptors and changes in intrinsic excitability of the post-synaptic neuron) and is thought to serve a homeostatic function to ensure that plasticity is kept within a normal working range (Abraham, 2008 ).
Evidence for Abnormal Plasticity in ASD
In a recent study, we used TBS to investigate cortical plasticity of the motor system in 25 individuals with ASDs. Results suggest that the change in cortical excitability following TBS lasts approximately 200–300% longer in the ASD group, compared with a matched control group. While the typical response to TBS lasts approximately 25–30 min, the average time to return to baseline in the ASD group was 80–90 min. Importantly, this abnormal prolongation of the plastic response to TBS is induced by both, continuous and intermittent TBS (cTBS and iTBS) thus suggesting aberrant plasticity in both, LTD- and LTP-like mechanisms. Furthermore, the same hyperplastic state is present in all 25 individuals with ASD, suggesting a consistent dysfunction despite likely genetic variability. These findings provide novel insights into the pathophysiology of ASD, consistent with recent results of animal models (Markram et al., 2007 ) that might become a biomarker for ASD, and ultimately offer a target for novel therapeutic interventions.
The Current Study
The current study aimed to evaluate intracortical inhibition, plasticity, and metaplasticity in individuals with FXS, and individuals with ASD. We assessed intracortical inhibition through paired pulse stimulation, cortical plasticity through response to TBS, and metaplasticity through response to TBS given 24 h after an initial TBS session, in individuals with FXS, ASD, and neurotypical controls. Based on data from animal models of FXS, and on prior data in our laboratory, we hypothesized that intracortical inhibition, plasticity, and metaplasticity might be abnormal in individuals with FXS and ASD, as compared to typically developing control participants. In addition to new insights into the pathophysiology of FXS and ASD in humans, our results demonstrate the utility of non-invasive measures to characterize E/I balance and cortical plasticity in translational neuroscience to non-invasively extend the findings in animal models to human populations with neurodevelopment disorders.
Materials and Methods
Participants
We studied two full mutation females with FXS (age 16 and 33), five individuals with ASD (three males and two females; ages 26, 35, 44, 45, 54) and five neurotypical controls (two males and three females; ages 22, 27, 41, 49, and 54). The two individuals with FXS participated in both the paired pulse and plasticity/metaplasticity sessions, four of the five individuals with ASD participated in both sessions while one only participated in the paired pulse session, and two neurotypical controls participated in both sessions while the other three participated in only the paired pulse session. All participants were right handed as assessed by revised Edinburgh Handedness Inventory (Oldfield, 1971 ; Schachter, 1994 ). Participants were recruited through the National Fragile X Society, FRAXA, local community advertisement, and local Asperger’s Associations and clinics. Both participants with FXS had a diagnosis with molecular confirmation of full mutation status, and all participants in the ASD group met DSM-IV-TR criteria for Asperger’s syndrome. All participants in all groups scored within the normal range on the Weschler Abbreviated Scale of Intelligence. The participants in the typically developing group had no neurological or psychological disorder.
All participants underwent a neurological exam to assess strength, tone, fine, and gross motor skills, involuntary movements, and gait, to ensure normal motor functioning. All participants signed an informed consent form. The project was reviewed and approved by the local institutional review board.
Stimulation and Recording
For both the paired pulse and TBS sessions TMS induced motor-evoked potentials (MEPs) were recorded with Ag–AgCl EMG surface electrodes placed over the right first dorsal interosseus (FDI) muscle of the participant’s right hand. Raw signals were amplified and band-pass-filtered between 20 and 2000 Hz. EMG signals were digitized and sampled at a rate of 5000 Hz. TMS was delivered using a hand-held figure-eight coil attached to the Nexstim stimulator (for paired pulse paradigms, Nexstim Ltd, Helsinki, Finland)) and a Magstim Super Rapid stimulator (for TBS paradigms, Magstim Company Ltd, Spring Gardens, Wales, UK). The coil was placed tangentially to the scalp with the handle pointing posteriorly for all stimulation.
All stimulation was given over the hand area of the left motor cortex and individually localized for each participant based on the optimal position for eliciting MEPs in the right FDI. Cortico-spinal excitability was assessed by measuring peak-to-peak amplitude of MEPs in the contralateral FDI muscle in response to TMS pulses.
Resting motor threshold (RMT) was determined as the minimum single pulse intensity required to produce an MEP of greater than 50 μV on more than 5 out of 10 consecutive trials from the contralateral FDI muscle while the subject was at rest. Active motor threshold (AMT) was determined as the minimum single pulse intensity required to produce an MEP of greater than 200 μV on more than 5 out of 10 consecutive trials from the contralateral FDI muscle while the subject contracted the muscle at approximately 20% of the maximal voluntary contraction.
In order to precisely target the stimulation site and keep the brain target constant throughout the paired pulse session, we used the Nexstim frameless stereotaxic system. (Nexstim Ltd, Helsinki, Finland). For the TBS sessions, we used a Brainsight frameless stereotactic system (Brainsight, Rogue Inc, Montreal, Canada). Both systems allow for online synchronization of a specific targeted brain region on an MRI with the online position of the subject and coil at a given moment and achieve a spatial resolution in the order of a fraction of a millimeter. In both cases the location of stimulation was kept constant through online feedback throughout the experimental session. These systems ensure that the exact same brain region is targeted on each trial and eliminate variability in the response as a result of changes in coil position.
Experimental Design
Participants were seated in a comfortable chair for the duration of the study. Paired pulse and TBS sessions were conducted on separate days with a minimum of 1 week between these sessions. TBS sessions were conducted on two consecutive days (see Figure 2 ) in order to simultaneously evaluate plasticity and metaplasticity processes. Resulting in three experimental sessions (one paired pulse session and two TBS sessions).
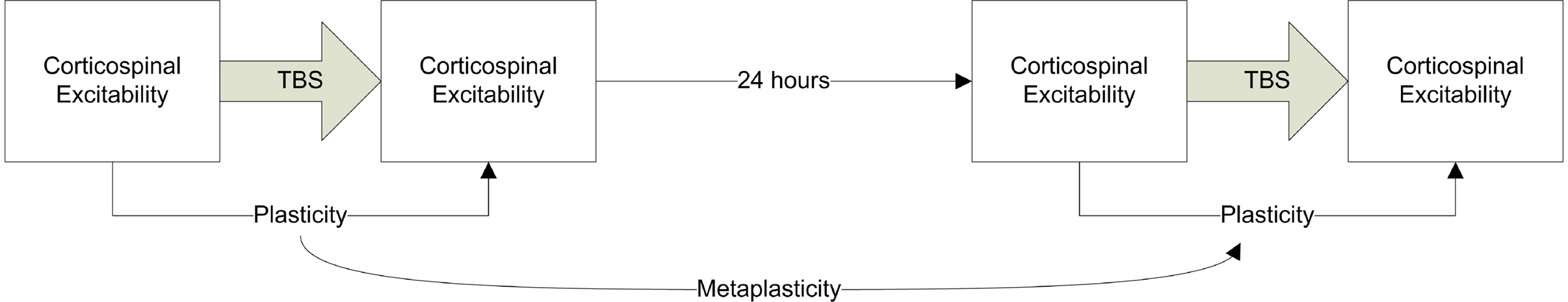
Figure 2. Schematic summary of the TBS experimental design. Cortico-spinal excitability was assessed using single pulse TMS. TBS was applied either as cTBS or iTBS to assess LTD- and LTP-like plasticity respectively. This procedure was then repeated 24 h later. The difference between the MEPs at baseline and following TBS can be used as an index of plasticity (LTD- or LTP-like depending on whether c or iTBS was applied) while the plasticity measure on day 1 compared to day 2 provides an index of metaplasticity.
Intracortical inhibition
Paired pulse paradigms were delivered at two inter-pulse intervals: 3 ms to elicit SICI and 100 ms to elicit LICI. Each pair was delivered at a rate of approximately 0.1 Hz (a random jitter of ±1 s was introduced to avoid any train effects). For SICI the first pulse was delivered at 80% of RMT (subthreshold) while the second pulse was delivered at 120% of RMT (suprathreshold). LICI was evaluated using two pulses delivered at 120% of RMT. For each subject an average SICI and LICI response was determined by calculating the mean of 20 MEPs each. These paired stimulations were then compared to the mean of 20 MEPs in response to single pulses at 120% RMT, to create a ratio indicating the degree of suppression in the MEP during SICI and LICI as compared to single pulses. The order of SICI, LICI, and single pulses was counterbalanced between subjects to control for order effects.
Cortical plasticity and metaplasticity
Theta burst stimulation was applied using the parameters introduced by Huang et al., (2005) (three pulses at 50 Hz at an intensity of 80% of AMT at 200 ms intervals (5 Hz), Figure 1 ). Two patterns of TBS stimulation were applied on consecutive days: intermittent theta burst stimulation (iTBS), shown to cause facilitation of the post-stimulation MEP, and continuous theta burst stimulation (cTBS), shown to cause suppression of the post-stimulation MEP (Huang et al., 2005 ). These sessions were counterbalanced such that cTBS was sometimes applied on day 1 and iTBS on day 2, while other times iTBS was applied on day 1 and cTBS on day 2. In the iTBS paradigm participants received a 2-s train of TBS repeated every 10-s for a total of 190 s (600 pulses), while in the cTBS paradigm they received a 47-s train of uninterrupted TBS (600 pulses).
To establish a baseline prior to TBS, three batches of 10 MEPs were recorded and measured in response to stimulation at a rate of approximately 0.1 Hz (a random jitter of ±1 s was introduced to avoid any train effects). Following TBS, batches of MEPs to 10 single pulses also at a rate of approximately 0.1 Hz were measured at 5, 10, 20, 30, 40, 50, 60, 75, 90, 105, and 120 min following TBS, to track changes in amplitude over time. MEP amplitude at a given time point was defined as the mean amplitude of the 10 MEPs to single TMS pulses recorded in a given 2 min time window.
As an index of the duration of the TBS-induced modulation of cortical response (index of cortical plasticity), we defined for each participant the time point at which the average MEP amplitude fell within the 95% confidence interval of the baseline amplitude and did not return to outside that interval on subsequent time point measures. MEP amplitudes were standardized forming a ratio of MEP amplitudes following TBS relative to average baseline MEP amplitude for each individual. Metaplasticity was indexed as the difference in response to cTBS or iTBS on the second day as compared to the first day.
Results
Intracortical Inhibition
All participants tolerated the TMS without any side-effects or complications. Figure 3 shows the average response to SICI and LICI for all individuals. All neurotypical control participants showed the expected suppression to both SICI and LICI. In both individuals with FXS SICI shows an approximately 63% suppression from baseline, and matches the findings in neurotypical controls. Both FXS individuals also show a large degree (92%) reduction in the MEP amplitude to the second stimuli in the LICI paired pulse paradigm. Again these findings match those in the neurotypical controls. On the other hand, findings in the ASD participants revealed a heterogeneity in the sample, with one individual showing an enhancement during the SICI paradigm and a different individual showing enhancement during the LICI paradigm.
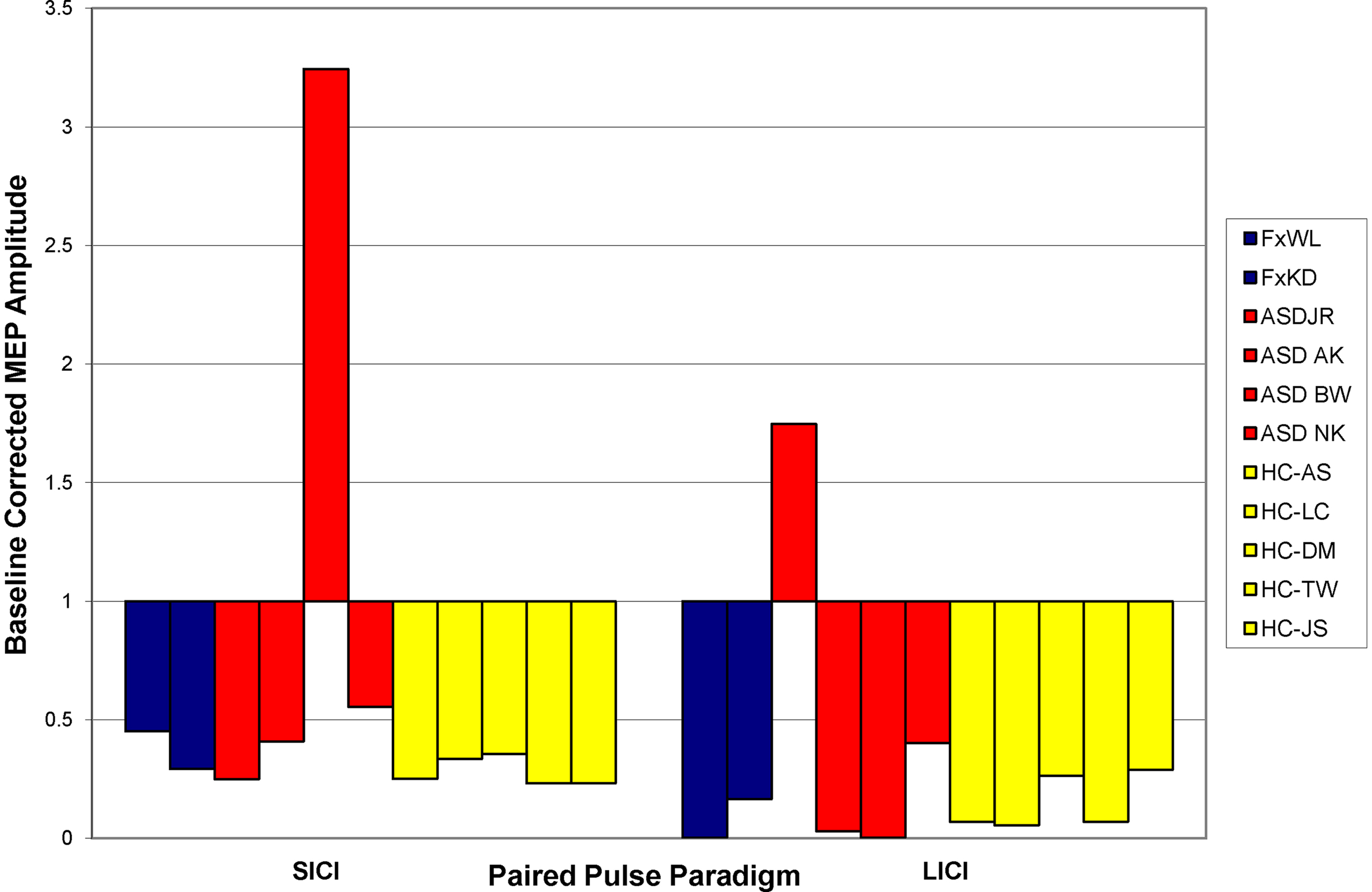
Figure 3. Baseline responses to SICI and LICI paradigms. Bars indicate the average degree of suppression of the MEP as compared to baseline single pulse measures. Blue bars represent patients with FXS; red bars, participants with ASD; and yellow bars, healthy control participants. A value of less than one indicates suppression while a value of greater than one indicates enhancement.
Cortical Plasticity and Metaplasticity
The neurotypical individuals showed the expected suppression to cTBS and enhancement to iTBS on day 1, lasting approximately 30 min (replicating previous reports, Huang et al., 2005 ). They also showed no effect of the previous day’s stimulation when given the opposite paradigm on day 2, indicating that metaplasticity mechanisms had been restored to baseline levels by 24 h (see Figure 4 A).
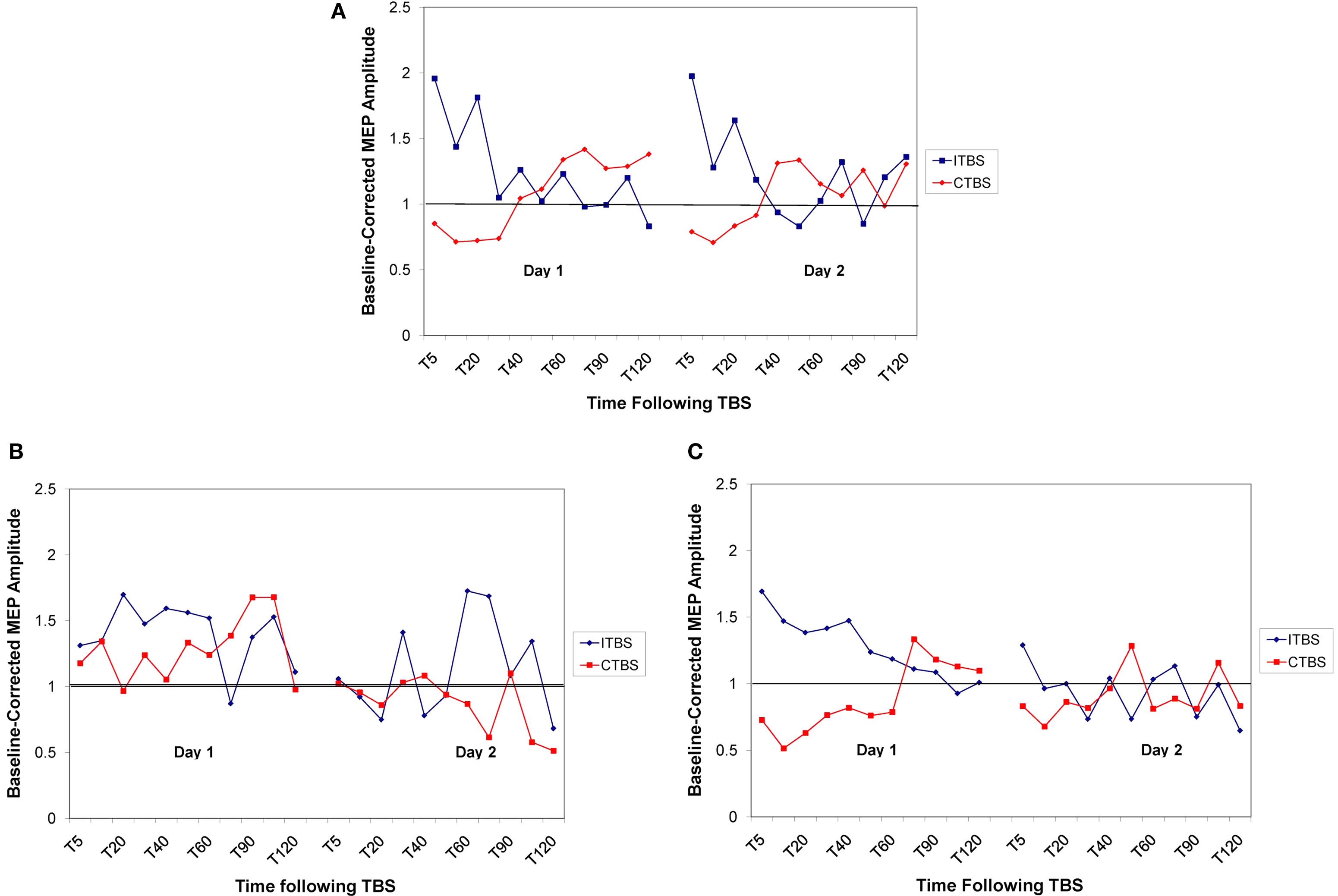
Figure 4. Baseline corrected MEP amplitude following cTBS (in red) and iTBS (in blue) on day 1 and day 2 for the control group (A), FXS group (B), and ASD group (C). Values are represented as proportion of baseline amplitude with a line at 1.0 (representing baseline amplitude).
The FXS participants, on day 1, showed no discernable response to the cTBS paradigm while showing an enhanced duration of response to the iTBS paradigm. The lack of response to cTBS was also present on day 2 when the participants had received iTBS on day 1. The enhanced response to iTBS that was present on day 1, however, was completely absent on day 2 when the participants had received cTBS on day 1, indicating an enhancement in metaplasticity (see Figure 4 B).
In the ASD sample, we replicated our previous finding of hyperplasticity in both the cTBS and iTBS paradigms on day 1. However, the response was strongly attenuated on day 2 in both paradigms when the opposite paradigm was applied on day 1, indicating an enhancement in both plasticity and metaplasticity in these individuals (see Figure 4 C).
Discussion
Our results demonstrate the utility of non-invasively assessing intracortical inhibition and cortical plasticity in vivo in patients with neurodevelopmental disorders. In doing so, we have introduced a paradigm capable of translating the findings from animal models of these disorders that can be applied across a wide range of ages and is not subject to participant’s performance or behavioral variability. Overall, our findings were consistent with animal models showing abnormalities in plasticity and metaplasticity in both the FXS and ASD groups. The intracortical inhibition measures reveal heterogeneity in ASD, as might be expected based on the behavioral heterogeneity of the population.
In both FXs participants the lack of response to the cTBS, enhanced response to the iTBS and complete blocking of the iTBS response on day 2 are indicative of abnormalities in both plasticity and metaplasticity. Larger scale studies involving more participants as well as both males and females are necessary to further elucidate the extent of this apparent dysfunction. However, these results are certainly consistent with findings in animal models. The animal models predict abnormalities in both LTP and LTD based on overactivity of the mGLUR5 receptor.
On the other hand the normal SICI and LICI findings are surprising. SICI and LICI are both measures of intracortical inhibition thought to primarily reveal GABAergic mechanisms. Animal models of FXS demonstrate a cortical GABAergic dysfunction and thus our findings in humans are unexpected. Of course, it is possible that the assumption that SICI and LICI reveal GABAergic activity may be overly simplistic and incorrect. SICI is a complex phenomenon and recent studies have revealed different mechanisms involved including excitation at certain ISIs and CSs. To measure pure GABAergic inhibitory system, ISI between 3–5 ms should be used so that SICI will not be contaminated and does not overlap with ICF (Hanajima et al., 2003 ; Vucic et al., 2009 ). Thus, our choice of ISI 3 ms was proper, as in earlier studies (Boroojerdi et al., 2000 ; Hammond et al., 2004 ). Some studies have proposed that ISIs between 2.1 and 2.6 ms to be most effective (Fisher et al., 2002 ; Ilic et al., 2002 ; Orth et al., 2003 ; Roshan et al., 2003 ; Vucic et al., 2009 ), but these ISIs are in the range of concurrent facilitation (Ziemann et al., 1998 ). All of these ISIs are based on a neurotypical brain, thus it is possible that in an individual with FXS, these ISIs are not appropriate to evaluate purely GABAergic functioning. Furthermore, we have only studied two FXS patients thus far here, making these findings very preliminary. The results warrant further replication, including the study of areas other than the motor cortex by combining TMS with electroencephalography (EEG). The typical response of the FXS patients to SICI and LICI could also be due to the fact that both our patients were females with full mutation FXS. Though they likely do not have as much FMRP as a neurotypical individual, the one unaffected X chromosome may be producing enough FMRP to compensate for the affected chromosome. Both participants were largely clinically unaffected, showing no signs of epilepsy or intellectual disability. Ultimately, it is also possible that the underlying neurophysiological mechanisms that have been predicted based on animal models may not translate to the human disease.
In the ASD sample we replicated our previous findings, with the four individuals with ASD displaying hyperplasticity in both the cTBS and iTBS paradigms on day 1. Additionally, we extended our findings, showing that metaplasticity is also affected in this population. The neurotypical individuals showed no “carry-over” from day 1 to day 2 in their response to cTBS and iTBS. However, in the four individuals with ASD, if they received cTBS on day 1, their response to iTBS on day 2 was attenuated. Similarly, if they received iTBS on day 1, their response to cTBS on day 2 was also attenuated. This enhanced metaplasticity indicates that the synapse was still being affected from the previous day’s TBS session altering its ability to make subsequent plastic changes. The LICI and SICI results highlighted heterogeneity in the sample, suggesting that some individuals with ASD may show abnormalities in the GABAergic system while others may not. Again, this is very preliminary and larger studies with individuals across the autism spectrum are necessary to evaluate the degree of heterogeneity in cortical excitability in this population.
Certainly, more studies with larger samples are necessary to fully appreciate the extent to which our findings generalize. In addition, combination of TMS with electroencephalography (EEG), rather than MEP electromyographic measures, will allow assessment of cortical reactivity, and plasticity in cortical regions outside of the motor cortex. If our preliminary findings are replicated, such TMS paradigms might prove to be valuable tools to translate the findings in animal models to humans. Our results raise the possibility that aberrant plasticity and metaplasticity might lead to alterations in development leading to the clinical manifestations of FXS and ASD. If these results are replicated, therapeutic interventions aimed at regulating plasticity and metaplasticity may be worth pursuing. Importantly, our TMS paradigms will enable monitoring and assessment of the efficacy of such interventions and possibly serve as valuable physiologic biomarkers. Treatments and interventions utilizing non-invasive brain stimulation or pharmacological agents that reverse aberrant plasticity and metaplasticity, may functionally rescue the affected individuals, the consequential neuropathological abnormalities might be minimized or even prevented, and the behavioral symptoms that define these disorders might be reversed or even prevented.
Conflict of Interest Statement
We declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work on this study was supported by grants from the National Center for Research Resources: Harvard-Thorndike General Clinical Research Center at BIDMC (NCRR MO1 RR01032) and Harvard Clinical and Translational Science Center (UL1 RR025758); NIH grant K24 RR018875 and a grant from the Nancy Lurie Marks Family Foundation to Alvaro Pascual-Leone. Lindsay Oberman was supported by NIH fellowship F32MH080493 and 1KL2RR025757-01. Ione Woollacott was supported by the Wellcome Trust [grant number 090692/Z/09/Z]. The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the Nancy Lurie Marks Family Foundation, National Center for Research Resources or the National Institutes of Health.
References
Abraham, W. C. (2008). Metaplasticity: tuning synapses and networks for plasticity. Nat. Rev. Neurosci. 9, 387.
Bakker, D. B. (1994). Fmr1 knockout mice: a model to study fragile X mental retardation. The Dutch-Belgian Fragile X Consortium. Cell 78, 23–33.
Bear, M. F., Huber, K. M., and Warren, S. T. (2004). The mGluR theory of fragile X mental retardation. Trends Neurosci. 27, 370–377.
Boroojerdi, B., Kopylev, L., Battaglia, F., Facchini, S., Ziemann, U., Muellbacher, W., and Cohen, L. G. (2000). Reproducibility of intracortical inhibition and facilitation using the paired-pulse paradigm. Muscle Nerve 23, 1594–1597.
Brennan, F. X., Albeck, D. S., and Paylor, R. (2006). Fmr1 knockout mice are impaired in a leverpress escape/avoidance task. Genes Brain Behav. 5, 467–471.
Claus, D., Weis, M., Jahnke, U., Plewe, A., and Brunholzl, C. (1992). Corticospinal conduction studied with magnetic double stimulation in the intact human. J. Neurol. Sci. 111, 180–188.
D’Hulst, C., and Kooy, R. F. (2007). The GABAA receptor: a novel target for treatment of fragile X? Trends Neurosci. 30, 425–431.
Desai, N. S., Casimiro, T. M., Gruber, S. M., and Vanderklish, P. W. (2006). Early postnatal plasticity in neocortex of Fmr1 knockout mice. J. Neurophysiol. 96, 1734–1745.
Fisher, R. J., Nakamura, Y., Bestmann, S., Rothwell, J. C., and Bostock, H. (2002). Two phases of intracortical inhibition revealed by transcranial magnetic threshold tracking. Exp. Brain Res. 143, 240–248.
Hagerman, R. J., and Hagerman, P. J. (2002). The fragile X premutation: into the phenotypic fold. Curr. Opin. Genet. Dev. 12, 278–283.
Hagerman, R. J., Ono, M. Y., and Hagerman, P. J. (2005). Recent advances in fragile X: a model for autism and neurodegeneration. Curr. Opin. Psychiatry 18, 490–496.
Hammond, G., Faulkner, D., Byrnes, M., Mastaglia, F., and Thickbroom, G. (2004). Transcranial magnetic stimulation reveals asymmetrical efficacy of intracortical circuits in primary motor cortex. Exp. Brain Res. 155, 19–23.
Hanajima, R., Furubayashi, T., Iwata, N. K., Shiio, Y., Okabe, S., Kanazawa, I., and Ugawa, Y. (2003). Further evidence to support different mechanisms underlying intracortical inhibition of the motor cortex. Exp. Brain Res. 151, 427–434.
Hou, L., Antion, M. D., Hu, D., Spencer, C. M., Paylor, R., and Klann, E. (2006). Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron 51, 441–454.
Huang, Y. Z., Chen, R. S., Rothwell, J. C., and Wen, H. Y. (2007). The after-effect of human theta burst stimulation is NMDA receptor dependent. Clin. Neurophysiol. 118, 1028–1032.
Huang, Y. Z., Edwards, M. J., Rounis, E., Bhatia, K. P., and Rothwell, J. C. (2005). Theta burst stimulation of the human motor cortex. Neuron 45, 201–206.
Huber, K. M., Gallagher, S. M., Warren, S. T., and Bear, M. F. (2002). Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. U.S.A. 99, 7746–7750.
Ilic, T. V., Meintzschel, F., Cleff, U., Ruge, D., Kessler, K. R., and Ziemann, U. (2002). Short-interval paired-pulse inhibition and facilitation of human motor cortex: the dimension of stimulus intensity. J. Physiol. (Lond.) 545, 153–167.
Irwin, S. A., Idupulapati, M., Gilbert, M. E., Harris, J. B., Chakravarti, A. B., Rogers, E. J., Crisostomo, R. A., Larsen, B. P., Mehta, A., Alcantara, C. J., Patel, B., Swain, R. A., Weiler, I. J., Oostra, B. A., and Greenough, W. T. (2002). Dendritic spine and dendritic field characteristics of layer V pyramidal neurons in the visual cortex of fragile-X knockout mice. Am. J. Med. Genet. 111, 140–146.
Irwin, S. A., Patel, B., Idupulapati, M., Harris, J. B., Crisostomo, R. A., Larsen, B. P., Kooy, F., Willems, P. J., Cras, P., Kozlowski, P. B., Swain, R. A., Weiler, I. J., and Greenough, W. T. (2001). Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am. J. Med. Genet. 98, 161–167.
Kaufmann, W. E., Cortell, R., Kau, A. S., Bukelis, I., Tierney, E., Gray, R. M., Cox, C., Capone, G. T., and Stanard, P. (2004). Autism spectrum disorder in fragile X syndrome: communication, social interaction, and specific behaviors. Am. J. Med. Genet. A 129A, 225–234.
Kobayashi, M., and Pascual-Leone, A. (2003). Transcranial magnetic stimulation in neurology. Lancet Neurol. 2, 145–156.
Koekkoek, S. K., Yamaguchi, K., Milojkovic, B. A., Dortland, B. R., Ruigrok, T. J., Maex, R., De Graaf, W., Smit, A. E., VanderWerf, F., Bakker, C. E., Willemsen, R., Ikeda, T., Kakizawa, S., Onodera, K., Nelson, D. L., Mientjes, E., Joosten, M., De Schutter, E., Oostra, B. A., Ito, M., and De Zeeuw, C. I. (2005). Deletion of FMR1 in Purkinje cells enhances parallel fiber LTD, enlarges spines, and attenuates cerebellar eyelid conditioning in Fragile X syndrome. Neuron 47, 339–352.
Kujirai, T., Caramia, M. D., Rothwell, J. C., Day, B. L., Thompson, P. D., Ferbert, A., Wroe, S., Asselman, P., and Marsden, C. D. (1993). Corticocortical inhibition in human motor cortex. J. Physiol. (Lond.) 471, 501–519.
Laggerbauer, B., Ostareck, D., Keidel, E. M., Ostareck-Lederer, A., and Fischer, U. (2001). Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum. Mol. Genet. 10, 329–338.
Le Be, J. V., and Markram, H. (2006). Spontaneous and evoked synaptic rewiring in the neonatal neocortex. Proc. Natl. Acad. Sci. U.S.A. 103, 13214–13219.
Li, J., Pelletier, M. R., Perez Velazquez, J. L., and Carlen, P. L. (2002). Reduced cortical synaptic plasticity and GluR1 expression associated with fragile X mental retardation protein deficiency. Mol. Cell. Neurosci. 19, 138–151.
Li, Z., Zhang, Y., Ku, L., Wilkinson, K. D., Warren, S. T., and Feng, Y. (2001). The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res. 29, 2276–2283.
Markram, H., Rinaldi, T., and Markram, K. (2007). The intense world syndrome – an alternative hypothesis for autism. Front. Neurosci. 1, 77–96.
Meredith, R. M., Holmgren, C. D., Weidum, M., Burnashev, N., and Mansvelder, H. D. (2007). Increased threshold for spike-timing-dependent plasticity is caused by unreliable calcium signaling in mice lacking fragile X gene FMR1. Neuron 54, 627–638.
Miller, L. J., McIntosh, D. N., McGrath, J., Shyu, V., Lampe, M., Taylor, A. K., Tassone, F., Neitzel, K., Stackhouse, T., and Hagerman, R. J. (1999). Electrodermal responses to sensory stimuli in individuals with fragile X syndrome: a preliminary report. Am. J. Med. Genet. 83, 268–279.
Musumeci, S. A., Bosco, P., Calabrese, G., Bakker, C., De Sarro, G. B., Elia, M., Ferri, R., and Oostra, B. A. (2000). Audiogenic seizures susceptibility in transgenic mice with fragile X syndrome. Epilepsia 41, 19–23.
Nielsen, D. M., Derber, W. J., McClellan, D. A., and Crnic, L. S. (2002). Alterations in the auditory startle response in Fmr1 targeted mutant mouse models of fragile X syndrome. Brain Res. 927, 8–17.
O’Donnell, W. T., and Warren, S. T. (2002). A decade of molecular studies of fragile X syndrome. Annu. Rev. Neurosci. 25, 315–338.
Oldfield, R. C. (1971). The assessment and analysis of handedness: the Edinburgh inventory. Neuropsychologia 9, 97–113.
Orth, M., Snijders, A. H., and Rothwell, J. C. (2003). The variability of intracortical inhibition and facilitation. Clin. Neurophysiol. 114, 2362–2369.
Pfeiffer, B. E., and Huber, K. M. (2009). The state of synapses in fragile X syndrome. Neuroscientist 15, 549–567.
Roshan, L., Paradiso, G. O., and Chen, R. (2003). Two phases of short-interval intracortical inhibition. Exp. Brain Res. 151, 330–337.
Schachter, S. C. (1994). Ambilaterality: definition from handedness preference questionnaires and potential significance. Int. J. Neurosci. 77, 47–51.
Simonyi, A., Schachtman, T. R., and Christofferson, G. R. (2005). The role of metabotropic glutamate receptor 5 in learning and memory processes. Drug News Perspect. 18, 353–361.
Spencer, C. M., Alekseyenko, O., Serysheva, E., Yuva-Paylor, L. A., and Paylor, R. (2005). Altered anxiety-related and social behaviors in the Fmr1 knockout mouse model of fragile X syndrome. Genes Brain Behav. 4, 420–430.
Stagg, C. J., Wylezinska, M., Matthews, P. M., Johansen-Berg, H., Jezzard, P., Rothwell, J. C., and Bestmann, S. (2009). Neurochemical effects of theta burst stimulation as assessed by magnetic resonance spectroscopy. J. Neurophysiol. 101, 2872–2877.
Tassone, F., Hagerman, R. J., Ikle, D. N., Dyer, P. N., Lampe, M., Willemsen, R., Oostra, B. A., and Taylor, A. K. (1999). FMRP expression as a potential prognostic indicator in fragile X syndrome. Am. J. Med. Genet. 84, 250–261.
Valls-Sole, J., Pascual-Leone, A., Wassermann, E. M., and Hallett, M. (1992). Human motor evoked responses to paired transcranial magnetic stimuli. Electroencephalogr. Clin. Neurophysiol. 85, 355–364.
Vucic, S., Cheah, B. C., Krishnan, A. V., Burke, D., and Kiernan, M. C. (2009). The effects of alterations in conditioning stimulus intensity on short interval intracortical inhibition. Brain Res. 1273, 39–47.
Wilson, B. M., and Cox, C. L. (2007). Absence of metabotropic glutamate receptor-mediated plasticity in the neocortex of fragile X mice. Proc. Natl. Acad. Sci. U.S.A. 104, 2454–2459.
Zhang, Y. Q., Bailey, A. M., Matthies, H. J., Renden, R. B., Smith, M. A., Speese, S. D., Rubin, G. M., and Broadie, K. (2001). Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell 107, 591–603.
Zhao, M. G., Toyoda, H., Ko, S. W., Ding, H. K., Wu, L. J., and Zhuo, M. (2005). Deficits in trace fear memory and long-term potentiation in a mouse model for fragile X syndrome. J. Neurosci. 25, 7385–7392.
Ziemann, U. (1999). Intracortical inhibition and facilitation in the conventional paired TMS paradigm. Electroencephalogr. Clin. Neurophysiol. Suppl. 51, 127–136.
Keywords: plasticity, excitability, fragile X syndrome, autism spectrum disorders, transcranial magnetic stimulation, theta burst stimulation, paired pulse stimulation
Citation: Oberman L, Ifert-Miller F, Najib U, Bashir S, Woollacott I, Gonzalez-Heydrich J, Picker J, Rotenberg A and Pascual-Leone A (2010) Transcranial magnetic stimulation provides means to assess cortical plasticity and excitability in humans with fragile X syndrome and autism spectrum disorder. Front. Syn. Neurosci. 2:26. doi: 10.3389/fnsyn.2010.00026
Received: 03 March 2010;
Paper pending published: 27 April 2010;
Accepted: 10 June 2010;
Published online: 28 June 2010
Edited by:
Susana Cohen-Cory, University of California, USAReviewed by:
Henry Markram, Ecole Polytechnique Federale de Lausanne, SwitzerlandPaul Fitzgerald, Monash University, Australia
Copyright: © 2010 Oberman, Ifert-Miller, Najib, Bashir, Woollacott, Gonzalez-Heydrich, Picker, Rotenberg and Pascual-Leone. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Alvaro Pascual-Leone, Department of Neurology, Berenson-Allen Center for Noninvasive Brain Stimulation, Beth Israel Deaconess Medical Center,330 Brookline Avenue, KS-158, Boston, MA 02215, USA. e-mail:YXBsZW9uZUBiaWRtYy5oYXJ2YXJkLmVkdQ==