




- 1 Departments of Otolaryngology and Physiology/Neuroscience, Molecular Neurobiology Program, The Helen and Martin Kimmel Center for Biology and Medicine, Skirball Institute of Biomolecular Medicine, New York University School of Medicine, New York, NY, USA
- 2 Friedrich Miescher Institute for Biomedical Research, Basel, Switzerland
- 3 Brain Research Institute, University of Zurich, Zurich, Switzerland
- 4 Howard Hughes Medical Institute, University of California, Berkeley, CA, USA
- 5 Department of Molecular and Cell Biology, Division of Neurobiology, Helen Wills Neuroscience Institute, University of California, Berkeley, CA, USA
- 6 The John Curtin School of Medical Research, Australian National University, Canberra, ACT, Australia
While it has been appreciated for decades that synapse location in the dendritic tree has a powerful influence on signal processing in neurons, the role of dendritic synapse location on the induction of long-term synaptic plasticity has only recently been explored. Here, we review recent work revealing how learning rules for spike-timing-dependent plasticity (STDP) in cortical neurons vary with the spatial location of synaptic input. A common principle appears to be that proximal synapses show conventional STDP, whereas distal inputs undergo plasticity according to novel learning rules. One crucial factor determining location-dependent STDP is the backpropagating action potential, which tends to decrease in amplitude and increase in width as it propagates into the dendritic tree of cortical neurons. We discuss additional location-dependent mechanisms as well as the functional implications of heterogeneous learning rules at different dendritic locations for the organization of synaptic inputs.
Introduction
Cortical pyramidal neurons receive thousands of synaptic inputs distributed across an extensive dendritic tree. Rather than conducting synaptic events to the spike initiation zone unaltered, dendrites passively and actively shape the postsynaptic response to presynaptic input (Häusser and Mel, 2003; Stuart et al., 2008). Synaptic integration can be regulated by a number of dendritic phenomena, including cable filtering (Rall, 1964), activation and modulation of various ion channels (Migliore and Shepherd, 2002; Magee and Johnston, 2005; Nusser, 2009) and dendritic spike generation (Spruston, 2008). Because passive electrotonic propagation is generally weak, and active processes are often non-uniformly distributed throughout the dendritic arbor, one important determinant of the net strength of a given synapse is its dendritic location (Stuart and Spruston, 1998; Cash and Yuste, 1999; Magee and Cook, 2000; Segev and London, 2000; Häusser et al., 2001; Reyes, 2001; Tamás et al., 2002; Williams and Stuart, 2002).
Synaptic strength is not fixed, but can be altered by the pattern of neural activity (Buonomano and Merzenich, 1998; Feldman, 2009). In particular, repetitive pairing of pre- and postsynaptic action potentials (pre/post spike pairing) can induce persistent changes in synaptic strength depending on the temporal order and timing of pre/post pairing. Long-term potentiation (LTP) is induced at many glutamatergic synapses when presynaptic activity occurs just before postsynaptic spiking in the target cell (pre → post pairing). This timing can be viewed as causal, since the synaptic depolarization may contribute to eliciting the postsynaptic action potential. Conversely, long-term depression (LTD) is usually induced when the postsynaptic cell fires before the presynaptic input (post → pre pairing). These Hebbian forms of LTP and LTD are collectively known as spike-timing-dependent plasticity (STDP), because the sign and magnitude of changes in synaptic strength are dependent on the precise (millisecond) timing of pre/post spiking (Magee and Johnston, 1997; Markram et al., 1997; Bi and Poo, 1998; Debanne et al., 1998; Abbott and Nelson, 2000; Feldman, 2000; Song et al., 2000; Sjöström et al., 2001; Froemke and Dan, 2002; Kampa et al., 2006; Letzkus et al., 2006; Froemke et al., 2010).
Various forms of STDP have been observed in a variety of species, ranging from insects to humans. Despite the apparent generality of the STDP learning rule across synapses (Dan and Poo, 2006), there is considerable variability in the precise timing requirements for STDP induction, especially for LTD. Furthermore, several recent studies in cortical pyramidal neurons have revealed that the exact formulation of the temporal window for spike-timing-dependent LTD depends on dendritic location (Froemke et al., 2005; Letzkus et al., 2006; Sjöström and Häusser, 2006).
Here we review the dendritic factors that influence the induction of cortical STDP and set the timing requirements for associative synaptic plasticity. These include passive dendritic properties, action potential backpropagation, NMDA receptor (NMDAR) activation, and active processes such as dendritic spikes. We then discuss experimental and theoretical work on the selective targeting of synaptic inputs to different dendritic locations. Other related topics such as plasticity of dendritic excitability, branch formation and spine growth (Magee and Johnston, 2005; Sjöström et al., 2008; Holtmaat and Svoboda, 2009) will not be covered here. We focus on experiments in cortical brain slices, which have revealed basic differences in the size and shape of the STDP window at proximal and distal dendritic synapses. Spatial gradients for STDP along dendritic trees may be important for the development and functional organization of cortical synapses, the structuring of synaptic integration and information processing within dendritic sub-regions, and defining the receptive field properties of cortical neurons.
Location Dependence of STDP: Hebbian and Anti-Hebbian Learning Rules
Dendritic geometry, ion channels, and receptor distributions interact to control the local voltage at a given synapse, which as we discuss below is a dominant factor in determining the magnitude of long-term synaptic modifications. As action potential backpropagation and postsynaptic processing of excitatory postsynaptic potentials (EPSPs) are both spatially regulated in dendrites of cortical neurons, it has been proposed that the sign and magnitude of STDP will depend on the dendritic location of synaptic input (Sourdet and Debanne, 1999).
Three experimental studies have recently provided evidence for location-dependent differences in STDP learning rules in neocortical pyramidal neurons (Froemke et al., 2005; Letzkus et al., 2006; Sjöström and Häusser, 2006). Together, these experiments have shown that synapses proximal to the cell body, where backpropagating action potentials are large and narrow, express conventional STDP in which pre → post spike pairing induces LTP (for relatively short inter-spike intervals of ∼25 ms) and post → pre pairing leads to LTD (for inter-spike intervals of ∼50 ms). At synapses more distal from the soma, however, the timing requirements for pre/post pairing shift such that the magnitude, and eventually the sign, of synaptic modifications during STDP at distal synapses is profoundly different from that found at proximal inputs (Figure 1).
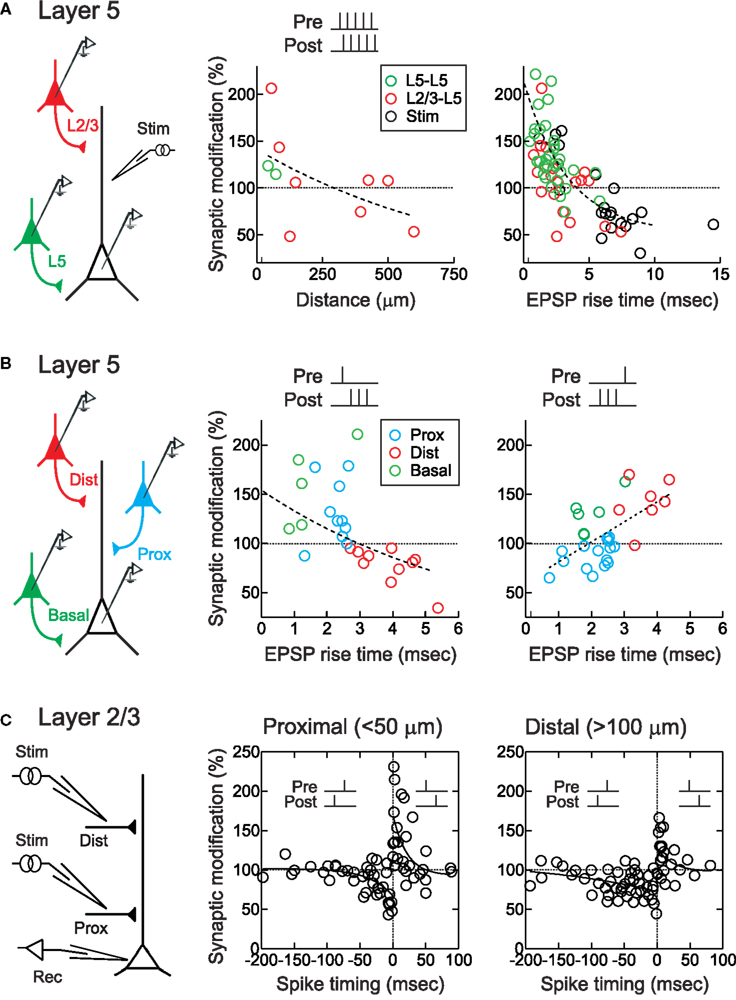
Figure 1. STDP timing rules depend on dendritic synapse location. (A) STDP of layer 2/3 (red) and layer 5 (green) EPSPs in layer 5 pyramidal neurons. EPSPs were evoked during paired recordings or by extracellular stimulation (“stim”, black). STDP was induced by repetitively pairing of five pre- and five postsynaptic action potentials/stimulations at +10 ms. Center, sign of synaptic modification was negatively correlated with distance of putative synaptic contact. Right, sign of synaptic modification was negatively correlated with EPSP rise time. From Sjöström and Häusser (2006). (B) STDP of proximal (blue) and distal (red) apical layer 2/3 inputs, and basal (green) layer 5 inputs, to layer 5 pyramidal neurons. Somatic EPSP rise time was used as a proxy for synaptic location, with fast EPSPs (<2.7 ms) indicating proximal EPSPs and slow rise times ( > 2.7 ms) indicating distal EPSPs. STDP induction consisted of repeated pairings of single EPSPs with high-frequency postsynaptic bursts (200 Hz). EPSP rise time was negatively correlated with the sign and magnitude of the change in synaptic strength for pre → post pairings (middle), but positively correlated with the sign and magnitude of changes in synaptic strength for post → pre pairings (right), leading to a switch to anti-Hebbian STDP for more distal inputs. From Kampa et al. (2006) and Letzkus et al. (2006). (C) STDP of proximal (<50 μm) and distal (>100 μm) inputs onto apical dendrites of layer 2/3 visual cortical pyramidal neurons. Post → pre pairing at −50 to −100 ms induced LTD distally (right) but not proximally (middle). From Froemke et al. (2005).
Sjöström and Häusser (2006) and Letzkus et al. (2006) both observed a spatial gradient of STDP along the apical dendrites of layer 5 pyramidal neurons. For these experiments, EPSPs were evoked in postsynaptic layer 5 pyramidal neurons either by direct depolarization of a connected presynaptic layer 2/3 pyramidal neuron, or by focal stimulation of synaptic inputs at varying distances along the apical dendrite. Importantly, the rise time of the EPSP recorded at the soma was used as an indicator of the distance of synaptic inputs from the soma, allowing accurate determination of the dendritic location for a given synapse (Jack et al., 1971). For synapses close to the cell body (<100 μm), STDP induced by pre/post pairing was Hebbian: pre → post spike pairs induced LTP and post → pre pairing induced LTD (Figures 1A,B). But as dendritic distance increased, the amount of LTP induced by pre → post pairing gradually decreased to zero, and at more distal synaptic locations (>500 μm) pre → post pairing induced robust LTD. LTD at these distal synapses could be converted to LTP if postsynaptic action potentials were paired with dendritic depolarization (Sjöström and Häusser, 2006), which can boost action potential backpropagation (Larkum et al., 2001; Stuart and Häusser, 2001). Similarly, dendritic depolarization converting a single action potential to a high-frequency burst by generation of a dendritic calcium spike (Larkum et al., 1999b) could convert LTD to LTP (Letzkus et al., 2006). Consistent with a role of dendritic spikes in LTP induction, at distal synapses action potential bursts above the critical frequency for dendritic calcium spike generation (Larkum et al., 1999a) lead to LTP during post → pre pairing (Letzkus et al., 2006). A similar observation has been made for inputs onto basal dendrites of layer 5 pyramidal neurons (Kampa et al., 2006). This frequency dependence of STDP induction is reminiscent of earlier findings (Markram et al., 1997; Sjöström et al., 2001). The observation that post → pre pairing leads to LTD of proximal synapses, but can induce LTP at distal inputs when inputs were paired with high-frequency action potential bursts (Figure 1B), suggests a gradual shift of the timing requirements for STDP along the apical dendrite of layer 5 pyramidal neurons. Together with work in basal dendrites (Gordon et al., 2006; Kampa et al., 2006), these studies show that synapses located in the distal dendrites of layer 5 cortical pyramidal neurons express anti-Hebbian STDP timing rules, where LTD can be switched to LTP by boosting action potential backpropagation (Sjöström and Häusser, 2006), the generation of dendritic spikes (Kampa et al., 2006; Letzkus et al., 2006), or by pairing NMDA spikes with brain-derived neurotrophic factor (BDNF) application (Gordon et al., 2006).
Similarly, for lateral connections within layer 2/3 of developing visual cortex, the magnitude of STDP at more distal synapses (>100 μm from the soma) was found to be about half that of proximal synapses (<50 μm from the soma) (Froemke et al., 2005). In addition, the temporal window for spike-timing-dependent LTD of distal layer 2/3 synapses was much wider than that of proximal synapses (Figure 1C). As a result, during post → pre pairing between −50 and −100 ms LTD was induced at distal (Figure 1C, right) but not proximal inputs (Figure 1C, center).
Backpropagating Action Potentials and Dendritic Excitability
What factors govern the size and shape of the STDP time window at different dendritic sites? The amplitude and time course of the electrical events that cooperate to induce STDP – that is, EPSPs and postsynaptic action potentials – themselves depend on dendritic location. This implies that the local depolarization experienced by individual synapses during pre/post pairing will be affected not only by the temporal dynamics of pre- and postsynaptic spike trains, but also by the distance of synaptic inputs from the site of action potential initiation. Because the degree of postsynaptic depolarization is an important parameter controlling the induction of long-term synaptic plasticity (Zucker, 1999; Wespatat et al., 2004; Lisman and Spruston, 2005; Urakubo et al., 2008; Feldman, 2009), it is likely that the timing requirements for STDP will depend on the combined voltage change at the synapse when pre/post spikes are paired at different time intervals (Sjöström et al., 2001; Froemke et al., 2005; Sjöström and Häusser, 2006). Thus the basic learning rules for STDP are likely to be determined in part by the biophysical factors within dendrites that regulate action potential backpropagation and local EPSP amplitude and kinetics.
After being initiated in or near the axon initial segment (Coombs et al., 1957; Fuortes et al., 1957; Palmer and Stuart, 2006; Meeks and Mennerick, 2007; Shu et al., 2007; Schmidt-Hieber et al., 2008), action potentials backpropagate into apical and basal dendrites of cortical pyramidal neurons (Stuart and Sakmann, 1994; Antic, 2003; Kampa and Stuart, 2006; Nevian et al., 2007). Usually, these backpropagating action potentials become smaller in amplitude and broader in width with increasing dendritic distance from the soma (Figures 2A–D). This occurs due to passive current spread as described by cable theory (Rall, 1964). Neuronal cell membranes are leaky, giving rise to rapid attenuation of electrical signals along dendrites (Stuart and Spruston, 1998; Williams and Mitchell, 2008). In cerebellar Purkinje cells, for example, somatic action potentials decrease in size by 80% or more within the first 100 μm (Stuart and Häusser, 1994).
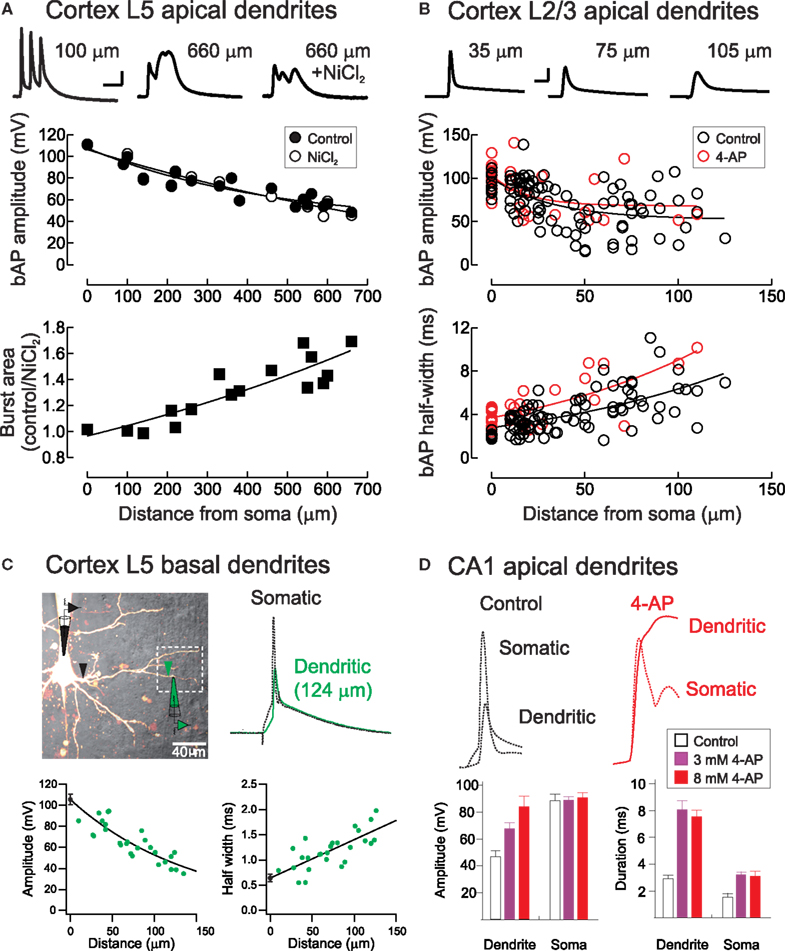
Figure 2. Backpropagating action potentials in pyramidal neuron dendrites. (A) Top, Voltage waveforms during a high-frequency (200 Hz) action potential burst at proximal and distal apical dendritic locations in a layer 5 pyramidal neuron before and after block of voltage-activated calcium channels with NiCl2 (0.1 mM, rat somatosensory cortex). Middle, the amplitude of single backpropagating action potentials (bAP) is unaffected by calcium channel block, whereas the area under action potential bursts is strongly Ca2+-dependent (bottom). Scale bar: 10 ms, 20 mV. From Letzkus et al. (2006). (B) Backpropagation of single action potentials in layer 2/3 pyramidal cells in rat visual cortex. Red symbols indicate experiments performed with the A-type K+ channel blocker 4-AP in the recording pipette. Scale bar, 5 ms, 40 mV. From Froemke et al. (2005). (C) Backpropagating action potentials in basal dendrites of layer 5 cortical pyramidal neurons. From Nevian et al. (2007). (D) Backpropagating action potentials in apical dendrites of hippocampal CA1 pyramidal neurons are enhanced in amplitude and duration by bath application of 4-AP. From Hoffman et al. (1997).
The dendrites of many central neurons, however, contain a variety of voltage-activated ion channels that support and regulate action potential backpropagation. As opposed to Purkinje cells, action potentials in neocortical and hippocampal pyramidal neurons decay in amplitude by less than 50% even several hundred μm from the cell body (Spruston et al., 1995; Stuart and Sakmann, 1994; Stuart and Spruston, 1998; Waters et al., 2003; Froemke et al., 2005). A relatively uniform distribution of dendritic Na+ channels in many neuronal cell types boosts action potentials as they propagate into dendrites, enhancing action potential backpropagation, which can be further amplified when paired with appropriately timed EPSPs (Magee and Johnston, 1995; Stuart and Häusser, 2001; Migliore and Shepherd, 2002). Nevertheless, backpropagating action potentials can fail to invade the most distal dendrites (Stuart and Häusser, 2001; Golding et al., 2002).
In addition to voltage-gated sodium channels, which enhance backpropagation, several other dendritic conductances have been found to exert dampening effects on dendritic excitation. A-type (Kv4) channels are fast-acting and inactivating K+ channels that counteract the depolarization produced by backpropagating action potentials. Blockade of dendritic A-type channels broadens dendritic EPSPs and backpropagating spikes (Figures 2B,D), suggesting that these channels help enforce spike-timing precision and reduce temporal summation of synaptic responses in dendrites (Hoffman et al., 1997; Froemke et al., 2006). Hyperpolarization-activated cyclic nucleotide-gated (HCN) channels can perform a similar normalizing function. Expressed at high levels in distal dendrites of hippocampal and cortical pyramidal neurons, HCN channels act to normalize both somatic EPSP time course and EPSP summation independent of the dendritic location of synaptic input (Magee, 2000; Williams and Stuart, 2000b). This effect depends on the total density of HCN channels, rather than their dendritic expression pattern (Angelo et al., 2007; Bullis et al., 2007).
Dendritic location profoundly impacts the amplitude and kinetics of synaptic responses as well as the characteristics of backpropagating action potentials. When measured in the dendrites close to the site of synaptic input, EPSPs evoked in distal dendrites are considerably larger (four-fold or more in amplitude) than EPSPs evoked more proximally (Magee and Cook, 2000; Williams and Stuart, 2002). For hippocampal CA1 pyramidal neurons, this scaling is due in part to a progressive increase in the number of AMPA receptors with dendritic distance from the soma (Smith et al., 2003). In contrast, synaptic conductance is relatively independent of dendritic location in layer 5 cortical pyramidal neurons (Williams and Stuart, 2002). For these cells, the smaller branch size of distal dendrites leads to a lower input capacitance, causing distal EPSPs to display larger amplitudes and faster kinetics than proximal inputs (Williams and Stuart, 2002; Nevian et al., 2007). In conjunction with the strong expression of HCN channels at distal sites, this leads to a brief temporal integration window for distal inputs (Williams and Stuart, 2002).
Given that distal inputs attenuate strongly on their way to the soma, additional mechanisms may be required for these events to influence axo-somatic synaptic integration and action potential generation. Dendritic spikes provide such a mechanism. Dendritic spikes are regenerative events which, depending on neuron type and dendritic location, can be mediated by voltage-gated Na+ and Ca2+ channels or by NMDARs (Häusser and Mel, 2003; Gulledge et al., 2005; Spruston, 2008). They can be evoked by correlated synaptic input (Williams and Stuart, 2002; Gasparini et al., 2004; Losonczy and Magee, 2006), by bursts of action potentials above a critical frequency (Figure 2A; Larkum et al., 1999a; Williams and Stuart, 2000a), or by coincidence of a distal EPSP with a backpropagating action potential within a narrow time window (Larkum et al., 1999b). As they lead to significant dendritic depolarization they act as an anti-Hebbian mechanism for LTP induction at distal inputs in cortical pyramidal neurons (Golding et al., 2002). Dendritic spikes can occur in isolation (Schiller et al., 1997; Stuart et al., 1997), or can forward-propagate to the soma where they typically elicit a burst of action potentials (Larkum et al., 1999b; Williams and Stuart, 1999), impacting on somatic spike-timing (Gasparini et al., 2004). Both local and global dendritic spikes can influence synaptic plasticity expressed at different dendritic locations (Sjöström et al., 2008).
NMDA Receptor Activation and STDP
At cortical layer 2/3 and layer 5 synapses, STDP at all dendritic locations requires activation of NMDARs, as synaptic modifications are prevented by application of the selective NMDAR antagonist APV (Froemke et al., 2005; Kampa et al., 2006; Letzkus et al., 2006). Furthermore, pairing action potentials with EPSPs has been shown to increase NMDAR activation. Block of NMDAR-mediated EPSPs by the activity-dependent NMDAR antagonist MK-801 was significantly greater when NMDAR EPSPs were paired with high-frequency (200 Hz) action potential bursts compared to NMDAR EPSPs evoked on their own (Figure 3A; Kampa et al., 2006). This activation of NMDARs by action potential bursts had similar timing requirements as STDP (Figure 3A), suggesting an important role of backpropagating action potentials and action potential bursts in enhancing NMDAR activation during STDP induction.
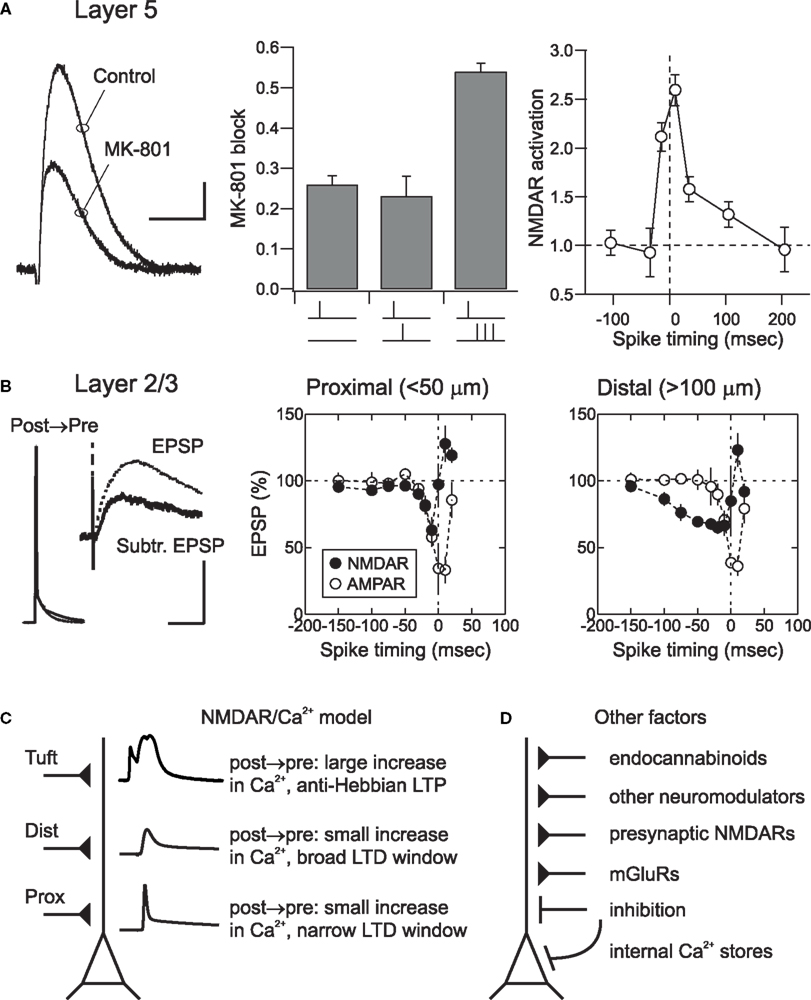
Figure 3. Spatial determinants and other mechanisms of cortical STDP. (A) NMDAR activation during pairing of EPSPs and action potentials in layer 5 neurons. Degree of NMDAR activation was measured using MK-801, an open channel blocker. MK801 block was defined as: 1 – EPSP(MK801)/EPSP(control). The greater the extent of NMDAR activation, the more channels will be blocked; as a result, the NMDAR component will become smaller over time. Scale bar: 0.5 mV, 20 ms (left). Pairing a presynaptic stimulus with a high-frequency (200 Hz) burst of three action potentials caused significant more NMDAR activation (i.e., a larger reduction in EPSP amplitude in the presence of MK-801) than pairing with single action potentials (center). The timing requirements for NMDAR activation (right) closely matched the STDP learning rule in these neurons. see Kampa et al. (2006) for methodological details. (B) During post → pre pairing in layer 2/3 neurons, NMDAR EPSPs were suppressed by postsynaptic action potentials. Scale bar: 50 or 2 mV, 100 ms (left). The time window for NMDAR EPSP suppression (filled circles) matched the time window for STDP induction at both proximal (center) and distal (right) synaptic locations. AMPA receptor mediated EPSPs seemed to be shunted by action potentials at short pairing intervals, but were otherwise unaffected by pairing (open circles). From Froemke et al. (2005). (C) The NMDAR/Ca2+ model of STDP. In this model, Ca2+ influx through postsynaptic NMDARs determines the degree of long-term synaptic modification. Depolarization from backpropagating action potentials relieves Mg2+ blockade of NMDARs, allowing postsynaptic influx of Ca2+. Lower levels of Ca2+ influx lead to LTD, whereas higher levels induce LTP. Conventional STDP is observed at proximal synapses. At more distal dendritic locations backpropagating action potentials are shorter and wider, increasing the time window of STDP. At the most distal dendritic locations, such as the apical tuft, action potentials interact locally with EPSPs to evoke dendritic Ca2+ spikes that induce anti-Hebbian LTP. (D) Other factors beyond Ca2+ influx through postsynaptic NMDARs also contribute to and control cortical STDP. These mechanisms may modulate postsynaptic excitability and spike generation, presynaptic release of neurotransmitter, or directly affect postsynaptic Ca2+ levels via release of Ca2+ from internal stores.
In layer 2/3, while pre → post pairing also directly increased the amplitude of NMDAR EPSPs during pairing, NMDAR EPSPs were strongly suppressed during post → pre pairing (Figure 3B). The location dependence of the spike-timing window for action potential-induced alteration of NMDAR EPSPs was similar to the location dependence of the STDP timing window. Enhancement of NMDAR EPSPs during pre → post pairing was almost certainly due to removal of the classical Mg2+ block (Mayer et al., 1984; Nowak et al., 1984), while NMDAR EPSP suppression by post → pre pairing required postsynaptic Ca2+ influx, suggesting that Ca2+-dependent NMDAR desensitization (Zorumski and Thio, 1992; Rosenmund et al., 1995; Tong et al., 1995; Kyrozis et al., 1996; Umemiya et al., 2001; Krupp et al., 2002) could be critical in setting the time window for LTD induction. EPSPs evoked by stimulation of layer 2/3 lateral connections in young visual cortex have strong NMDAR components even at hyperpolarized membrane potentials, apparently due to a relatively weak Mg2+ blockade (Kato and Yoshimura, 1993), similar to connections between layer 4 stellate cells (Fleidervish et al., 1998). This suggests that these connections might be highly dynamic and exquisitely sensitive to changes in pre- and postsynaptic activity patterns (Diamond et al., 1994; Das and Gilbert, 1995; Trachtenberg et al., 2000).
Analogously, Koester and Sakmann (1998) demonstrated that the precise timing of pre- and postsynaptic activity was directly related to the amplitude of Ca2+ signals in spines of layer 5 pyramidal cells. When the presynaptic EPSP was evoked first, Ca2+ signals in activated dendritic spines summed supralinearly, whereas when the postsynaptic cell fired first Ca2+ signals summed sublinearly.
These results support a causal relationship between modulation of NMDAR EPSPs and STDP induction in cortical pyramidal neurons. We therefore propose a model in which the extent of Ca2+ influx through NMDARs gated by the local depolarization due to backpropagating action potentials and dendritic spikes determines the sign and magnitude of plasticity (Figure 3C). While it has long been known that both NMDARs and postsynaptic Ca2+ influx are necessary for LTP and LTD induction at most central synapses (Malenka and Nicoll, 1999; Zucker, 1999), recent biophysical and biochemical models have quantitatively captured the dendritic location dependence of NMDAR activation and STDP. Simulations of the interactions between dendritic spikes and NMDAR kinetics recapitulated the progressive shift of the STDP learning rules with dendritic distance (Saudargiene et al., 2005; Letzkus et al., 2006), and could also account for anti- Hebbian LTP induced by short-interval post → pre pairing, depending on the rise time of the postsynaptic response (Saudargiene et al., 2004). Large-scale biochemical simulations of the mechanisms underlying long-term synaptic modification in layer 2/3 neurons revealed that the postsynaptic spike rapidly acts on NMDARs during post → pre pairing (Urakubo et al., 2008). This study revealed that, in addition to enzymatic action via calcineurin, it is likely that Ca2+-bound calmodulin allosterically regulates layer 2/3 NMDARs to induce LTD. Taken together, these experimental and theoretical studies suggest that Ca2+ influx through NMDARs is a dominant factor in STDP induction. During pre → post pairing high levels of Ca2+ influx lead to LTP, whereas during post → pre pairing low levels of Ca2+ influx through a desensitized NMDA channel induces LTD.
Other Potential Mechanisms for Location-Dependent STDP
In addition to backpropagating action potentials and postsynaptic NMDAR activation, a range of other mechanisms are known to be important for long-term synaptic plasticity in general and STDP in particular (Figure 3D). These factors include voltage-gated Ca2+ channels and Ca2+ spikes (Christie et al., 1997; Froemke et al., 2005; Kampa et al., 2006; Letzkus et al., 2006), metabotropic glutamate receptors (Nishiyama et al., 2000; Bender et al., 2006), endocannabinoid release (Sjöström et al., 2003; Sjöström and Häusser, 2006), BDNF signaling (Gordon et al., 2006), activation of other neuromodulators, G protein coupled receptors, and their downstream effectors (Lin et al., 2003; Froemke et al., 2007; Seol et al., 2007; Harvey et al., 2008), presynaptic NMDA autoreceptors (Sjöström et al., 2003; Corlew et al., 2007), Ca2+ release and/or sequestering by internal stores (Nishiyama et al., 2000) and disinhibition (Artola et al., 1990). Some of these systems are themselves expressed and regulated in a location-dependent manner, suggesting that processes and signaling mechanisms beyond direct activation and modulation of NMDARs is likely to control the local learning rules for synaptic modification at different inputs.
It remains unclear how these processes interact to ultimately control the induction of long-term synaptic modifications at cortical excitatory synapses. Some of these mechanisms can clearly influence dendritic excitability, such as adrenergic, cholinergic, or dopaminergic neuromodulation of K+ channel kinetics and downstream effects on action potential backpropagation (Hoffman and Johnston, 1999). Changes in dendritic excitability have been shown to alter the size and shape of backpropagating action potentials and synaptic events (Frick et al., 2004), which in turn affects how these events are integrated to produce STDP. In addition, other mechanisms, including metabotropic glutamate receptor and voltage-gated Ca2+ channel activation, might more directly affect internal Ca2+ levels and thus STDP, as previously observed for induction of STDP in hippocampus (Nishiyama et al., 2000) and cortex (Bender et al., 2006). Finally, release of retrograde messengers or activation of presynaptic autoreceptors might control the amount or kinetics of transmitter release (Sjöström et al., 2003), regulating the degree of postsynaptic NMDAR activation or inducing long-term changes in presynaptic release. Given this mechanistic diversity, future studies are required to determine how these mechanisms affect STDP in a location-dependent manner.
Recruitment of inhibitory inputs may also differentially affect STDP induction at different dendritic locations, particularly since several interneuron sub-populations target specific subcellular compartments of pyramidal neurons (Markram et al., 2004). Perisomatic inhibition provided by basket cells serves to inhibit action potential firing (Cobb et al., 1995; Pouille and Scanziani, 2001), but would allow synaptic modifications induced by dendritic spikes to occur. Conversely, several distinct classes of interneurons target the dendrites of pyramidal neurons in neocortex (Markram et al., 2004), providing a rich repertoire for location-dependent modulation of dendritic synapses. Activation of ionotropic GABA receptors in cortical pyramidal neurons can be both inhibitory or excitatory (Gulledge and Stuart, 2003). This suggests that, depending on dendritic location and timing relative to excitatory input, GABAergic synapses can either enhance or suppress action potential firing and STDP induction. In contrast, activation of dendritic GABAB receptors strongly inhibits the generation of dendritic spikes in the apical tuft of layer 5 pyramidal neurons, but leaves action potential backpropagation largely intact (Pérez-Garci et al., 2006). This may selectively inhibit STDP at distal apical synapses, which require dendritic calcium spikes for STDP (Letzkus et al., 2006), while leaving STDP at proximal inputs unaffected.
While we know very little about the engagement of these various inhibitory circuits during information processing in vivo, a well- understood example is provided by the disynaptic loop between layer 5 pyramidal neurons and dendrite-targeting Martinotti interneurons. Sensory stimulation has recently been shown to elicit calcium spikes in the apical tuft of layer 5 pyramids in vivo (Murayama et al., 2009). Associated high-frequency action potential bursts in turn activate Martinotti interneurons (Silberberg and Markram, 2007; Murayama et al., 2009), which inhibit subsequent dendritic calcium electrogenesis in surrounding pyramidal cells. In effect, this suggests that STDP induction in tuft inputs to one set of pyramidal neurons may render the same synapses in the other layer 5 implastic for a brief time window.
Although forms of STDP have been observed at many synapses, pre/post spike pairing is just one of several protocols for induction of long-term synaptic modification. Pairing single pre/post spikes at low frequency, even with dozens of repetitions over minutes, sometimes fails to induce significant changes in synaptic strength, particularly at unitary connections between cortical pyramidal neurons (Sjöström et al., 2001; Kampa et al., 2006; Letzkus et al., 2006; Froemke et al., 2010). Even pairing 5 Hz bursts of somatically-triggered spikes can fail to induce LTP in vitro (Markram et al., 1997; Sjöström et al., 2001), and pre/post spike pairing may be insufficient for induction of long-term synaptic modifications in vivo (Froemke et al., 2007; Jacob et al., 2007). For synapses where the predominant mechanism of long-term modification involves Ca2+ influx through postsynaptic NMDARs (Zucker, 1999; Urakubo et al., 2008), the local voltage change at the synapse is more important than somatic depolarization and spike generation (Lisman and Spruston, 2005). Thus while somatic spikes sometimes fail to invade distal dendrites and inhibitory inputs may prevent postsynaptic NMDAR activation, other processes such as dendritic calcium spikes (Schiller et al., 1997; Larkum et al., 1999b) or neuromodulation (Lin et al., 2003; Froemke et al., 2007) may be engaged to enable long-term synaptic modifications in the absence of somatic action potentials. For example, Golding et al. (2002) have shown that in hippocampal CA1 pyramidal neurons LTP of distal inputs can occur independently of somatic action potential backpropagation, and instead requires dendritic calcium spikes. LTD in layer 5 cortical pyramidal neurons can be induced by pairing presynaptic stimulation with subthreshold depolarization (Sjöström et al., 2004), a finding reminiscent of earlier work showing that the magnitude of postsynaptic depolarization determines the sign and magnitude of synaptic plasticity (Artola et al., 1990; Feldman et al., 1999; Zucker, 1999; Wespatat et al., 2004).
Thus pre/post spike pairing is sufficient to induce synaptic modification at many synapses, but the precise timing requirements, temporal ordering, and number of spikes required can be highly synapse specific. Furthermore, the exact timing rules for STDP at a given synapse are likely to be regulated by a large number of spatial and temporal phenomena. In the end, local depolarization and postsynaptic Ca2+ influx are the key factors underlying synaptic plasticity, independent of whether backpropagating action potentials are required or not, in a manner resonant with the classical BCM model (Sjöström and Nelson, 2002; Izhikevich and Desai, 2003).
Dendritic Organization of Synaptic Input
The recruitment of the different location-dependent plasticity mechanisms described above depends on the spatio-temporal activation pattern of synapses in the dendritic arbor. For this and other reasons, spatial organizing principles structuring input along the dendrites have recently received considerable attention. A landmark study by Petreanu et al. (2009) applied a novel technique to map the distribution of functional inputs to neocortical pyramidal neurons in barrel cortex. Using channelrhodopsin-2 to selectively activate various anatomical inputs (Figure 4A), they observed a hierarchical gradient of afferents on layer 3 pyramidal neurons, with bottom-up inputs impinging onto proximal dendritic locations and increasingly complex, more processed information arriving at progressively more distal sites (Figure 4B). A similar albeit more complex pattern was observed in layer 5B pyramidal neurons, where top-down inputs target both the basal dendritic domain and the apical tuft (Figure 4C). Since the rules of STDP induction depend on dendritic location, these input pathways are likely to display different timing requirements for potentiation and depression in response to postsynaptic firing. In response to uncorrelated firing, top-down inputs onto layer 3 neurons would be predicted to depress more than bottom-up synapses, possibly leading to an effective temporal sharpening of the top-down response (see below). In contrast, both bottom-up and top-down inputs to layer 5 pyramidal cells might be potentiated when activated after the initiation of a postsynaptic action potential, but only if top-down synapses are concomitantly active to transform the action potential into a burst by depolarizing the apical tuft (Larkum et al., 1999b).
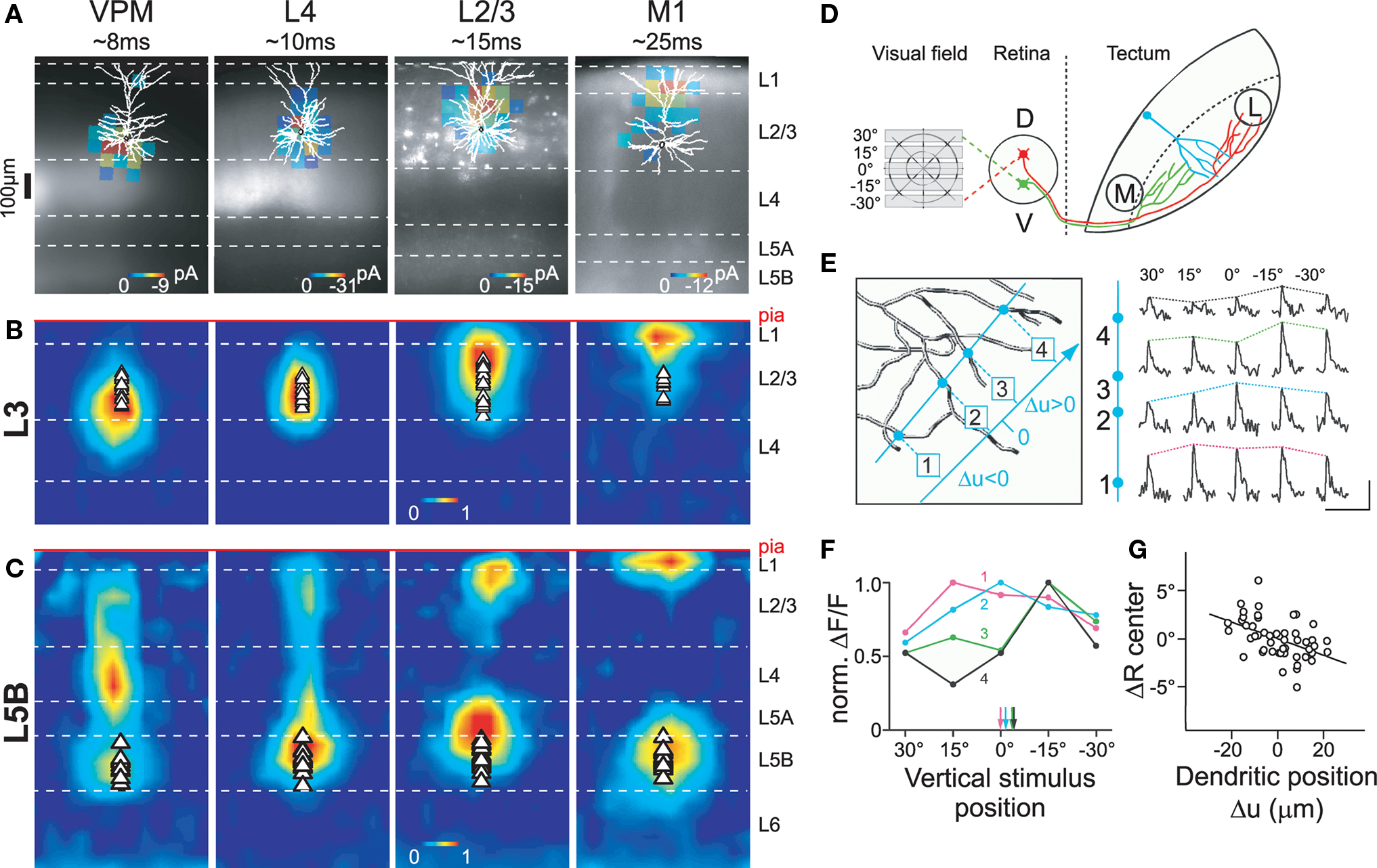
Figure 4. Dendritic compartmentalization of synaptic input. (A) Subcellular channelrhodopsin-2 assisted circuit mapping (sCRACM) was used to map the dendritic location of excitatory inputs from the ventral posterior medial nucleus (VPM), barrel cortex layer 4 and layer 2/3, and primary whisker motor cortex (M1) on layer 3 (A, B) and layer 5B (C) neurons in barrel cortex (for details see Petreanu et al., 2009). Example maps superimposed on reconstructed morphologies and fluorescent images of ChR2-expression. (B) Average sCRACM-derived input map of layer 3 pyramidal neurons, displaying a hierarchical gradient of organization: bottom-up input (VPM) impinges onto the basal dendritic domain. Progressively more processed information arrives at progressively further apical locations. (C) Average input map of layer 5B pyramidal neurons corrected for dendritic attenuation of postsynaptic currents. Note that L2/3 and M1 inputs impinge onto both the basal dendrites and the apical tuft. Individual maps were aligned at the pia. Triangles indicate soma position. (D) Retinal input to dendrites of tectal neurons was elicited by presenting horizontal bars at different positions within the visual field of Xenopus tadpoles. (E) Line scans (blue) across 4 dendritic branches of one tectal neuron filled with the calcium-sensitive dye OGB-1 (left) reveal differential responses of the branches to the same stimulus position. Scale bar, 50% ΔF/F, 5 s. (F) Plotting normalized responses against stimulus position suggests branch-specific retinotopic input tuning. Arrows indicate centers of mass for each branch. (G) The mean subtracted center of mass (ΔR center) correlates with the relative position of a branch in the tectum. Reproduced with permission from (Petreanu et al., 2009) (A–C) and (Bollmann and Engert, 2009) (D–G).
In addition, dendritic compartmentalization also controls which input pathways can potentially interact locally, for example, at the level of individual dendritic branches. Spatially clustered and simultaneously active synaptic activity is required for initiation of dendritic spikes (Schiller et al., 1997; Williams and Stuart, 2002; Losonczy and Magee, 2006), which is important for induction of some forms of STDP (Kampa et al., 2007), as well as branch-specific plasticity of excitability (Losonczy et al., 2008). For most inputs onto layer 3 neurons, these local effects are likely to be confined within single afferent pathways, whereas in layer 5B neurons between-pathway interactions are also possible (Figures 4B,C; Petreanu et al., 2009).
Within a given pathway, it has been hypothesized that synapses of similar information content are further clustered (Larkum and Nevian, 2008). The first direct corroboration of this hypothesis was found in tectal neurons of Xenopus tadpoles (Figures 4D–G; Bollmann and Engert, 2009). Visual stimulation revealed that inputs were organized topographically, where axons of neighboring retinal ganglion cells were clustered into distinct input domains in the dendritic arbor of tectal neurons. In contrast, Jia et al. (2010) recently observed in mouse primary visual cortex that inputs onto layer 2/3 neurons with similar orientation preference are distributed onto different dendritic branches. Thus, it is at present unclear whether clustering of inputs with similar information content onto the same dendritic branch is an important organizing principle or not, and future research is clearly required. Nonetheless, the organization of dendritic afferents characterized so far makes it possible that both STDP and dendritic-spike-dependent plasticity mechanisms may be engaged in vivo in a region-specific manner (Kampa et al., 2007; Letzkus et al., 2007). While clustering of co-active inputs would favor dendritic spikes as a mechanism for plasticity induction during learning, dispersion of co-active synapses in the dendritic arbor would make dendritic spikes less likely, and thus would favor backpropagating action potentials as the primary mechanism for the induction of long-term synaptic modifications. Despite a growing knowledge on the learning rules and underlying biophysics important for induction of synaptic plasticity, we still know very little about the temporal engagement of afferents during normal brain function. This information will be vital for a more complete understanding of the plasticity mechanisms governing synaptic transmission, perceptual learning, and memory formation.
Theoretical Considerations for Development and Information Processing
Synapses that exhibit STDP can perform a range of computations. These include sequence prediction (Abbott and Blum, 1996; Nowotny et al., 2003), memory storage (Roberts and Bell, 2002), principal component analysis (Oja, 1982; Abbott and Nelson, 2000), and temporal difference learning (Rao and Sejnowski, 2001). Simulations have shown that topographic maps and receptive field properties such as orientation tuning, ocular dominance, sound source localization, and velocity sensitivity can emerge when STDP is engaged as a developmental process (Gerstner et al., 1996; Song and Abbott, 2001; Tamosiunaite et al., 2007; Gandhi et al., 2008). Adult forms of plasticity, for example, stimulus-timing-dependent plasticity (Yao and Dan, 2001; Fu et al., 2002; Dahmen et al., 2008), recovery from retinal lesions (Young et al., 2007), and hippocampal phase procession (Mehta et al., 2002), have also been ascribed to forms of STDP.
In most of these theoretical studies, the STDP learning rule was assumed to be spatially and temporally homogeneous; that is, all synapses are assumed to have similar timing requirements for induction of LTP and LTD. Because experimental results demonstrate that STDP timing rules can depend on the dendritic location of synaptic inputs (Figure 1), and other studies document temporal modulation by spike bursts and high-frequency pre/post trains (Froemke et al., 2010), the assumption of uniformity for STDP time rules is probably only applicable for synapses located electrotonically close to the cell body and for pre- and postsynaptic spike trains at low frequencies (<10 Hz) that rarely contain bursts of two or more spikes. Furthermore, Goldberg et al. (2002) and Lisman and Spruston (2005) have raised several concerns about the importance of STDP as conventionally described, especially for synapses distal from the soma that have difficulty contributing to action potential generation in the absence of dendritic electrogenic mechanisms.
Spatial regulation of STDP helps alleviate some of these concerns. For example, synaptic weights in some neurons are scaled to normalize the effective strength of each input at the soma (Magee and Cook, 2000). As passive cable filtering of synaptic inputs would be expected to greatly reduce distal EPSP amplitudes, there may be mechanisms for adjusting synaptic efficacy, scaling the strength of synapses in proportion to their electrotonic distance from the spike initiation zone. Anti-Hebbian STDP is such a mechanism (Goldberg et al., 2002; Letzkus et al., 2006; Sjöström and Häusser, 2006). As formalized by Rumsey and Abbott (2004), Hebbian STDP promotes proximal synapses and reduces the amplitude of distal synapses, because all else being equal, proximally-located inputs have a greater impact on somatic spike generation. However, if distal synapses express anti-Hebbian STDP, such that post → pre pairing induces LTP, then distal synapses will eventually be strengthened until some equilibrium point has been reached (Figure 5A). In terms of synaptic organization, anti-Hebbian STDP provides a mechanism for retention of distal inputs, re-scaling synaptic strengths in proportion to electrotonic distance and preserving total excitatory drive. This can also be achieved with Hebbian STDP (Gidon and Segev, 2009) if formulated multiplicatively (Figure 5B, top) or if distal synapses have larger maximal peak conductances (Figure 5B, bottom), similar to reported experimental findings in CA1 hippocampal pyramidal cells (Magee and Cook, 2000).
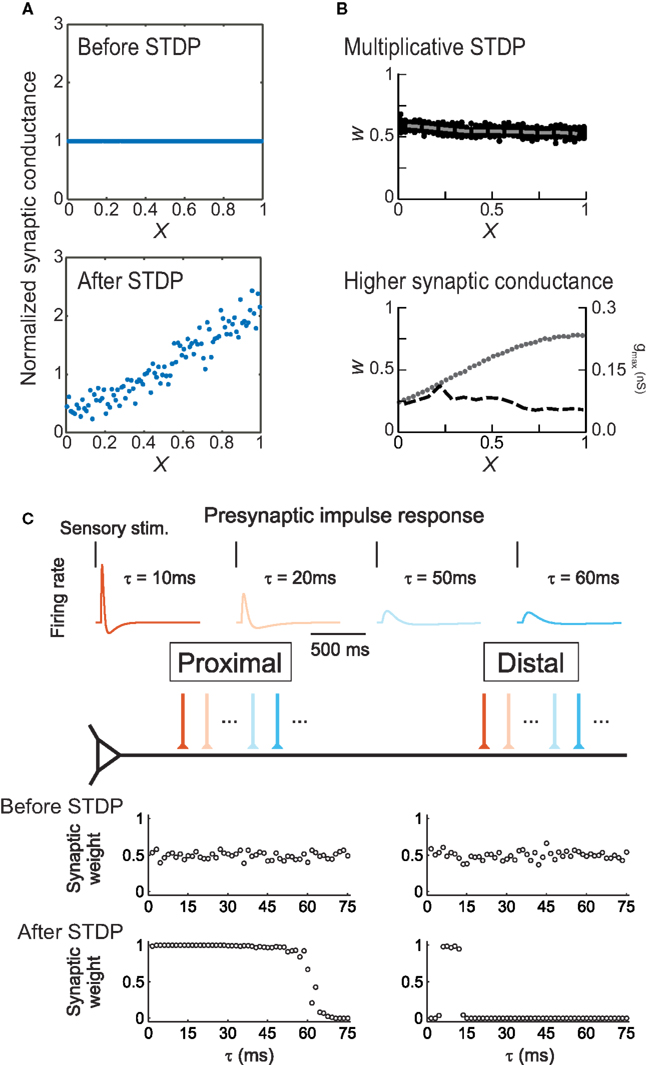
Figure 5. Modeling location-dependent STDP. (A) Anti-Hebbian STDP scales synaptic strength as a function of electrotonic distance from the soma (X). Top, distribution of synaptic weights before training. Bottom, equilibrium synaptic strength after training. From Rumsey and Abbott (2004). (B) Uniform synaptic strength (W) can be achieved by multiplicative, weight-dependent STDP (top) or if distal synapses have larger maximally-possible peak conductances gmax (bottom). Dashed lines indicate average synaptic strengths; dots indicate gmax. From Gidon and Segev (2009). (C) In layer 2/3 neurons, location-dependent STDP selects for transient distal inputs. Top, example presynaptic responses to brief sensory stimuli. Warmer colors indicate more transient responses (τ < 15 ms). Bottom, synaptic inputs before and after induction of STDP. From Froemke et al. (2005).
Spatial gradients of STDP also increase the computational capacity of cortical pyramidal neurons. Previous simulations showed that LTD induced by post → pre spike pairs preferentially weakens synaptic inputs with long response latencies during competitive Hebbian synaptic modification (Song and Abbott, 2001). Dendritic regulation of STDP may therefore lead to differential selectivity of inputs along the apical dendrite. However, theoretical work from Mel and colleagues has demonstrated that additional computational power can only be harvested if synaptic inputs carrying different signals are clustered into distinct regions of the dendritic tree (Mel et al., 1998; Archie and Mel, 2000; Häusser and Mel, 2003). Thus, the location dependence of STDP not only demonstrates dendritic inhomogeneities in the properties governing activity-dependent synaptic modification, but also points to their potential role in the formation of branch-specific synaptic inputs; a feature that can greatly enrich the repertoire of dendritic information processing (Losonczy et al., 2008; Makara et al., 2009).
This idea was tested using a simple integrate-and-fire model neuron (Figure 5C), in which proximal and distal dendrites exhibited location-dependent STDP using presynaptic spike trains that were either transient and phasic or more prolonged and sustained, as observed in vivo (Baddeley et al., 1997). After training, inputs with relatively transient responses were strengthened and those with sustained responses were weakened at both distal and proximal locations. However, distal synapses were much more selective, retaining only those synaptic inputs that fired extremely transiently in response to sensory stimuli; these inputs must act synchronously to bring the postsynaptic cell to threshold, then quickly adapt so as to minimize the amount of depression caused by post → pre spiking in the postsynaptic cell. This is a direct consequence of the competitive advantage conferred to the proximal inputs over the distal inputs, by the difference in STDP. These simulations demonstrate that STDP may lead to functional differentiation of the capacity of distal and proximal dendrites to process signals with distinct temporal characteristics. Due to selection of more transient inputs, the distal dendrite may be specialized for processing the precise timing of sensory signals. These characteristics for distal inputs are precisely what have been found to be required for the initiation of distal dendritic Ca2+ spikes, which represent an effective way for distal inputs to influence neuronal output (Williams and Stuart, 2002). Moreover, variation in the statistical properties of inputs along the apical dendrite allows for topographic organization and coordination of receptive field properties such as temporal modulation sensitivity (Atencio and Schreiner, 2010).
Conclusion
There is a large literature on the dendritic factors that influence synaptic integration and action potential backpropagation in pyramidal neurons. These studies have demonstrated that action potentials and EPSPs are not the same size and shape in different regions of the dendritic tree, due to differences in dendritic geometry and passive properties, as well as differences in the dendritic distributions of neurotransmitter receptors, voltage-gated ion channels, Ca2+ buffers and stores, intracellular signaling molecules and mRNAs (Spruston, 2008; Stuart et al., 2008). Given the possible permutations of these factors, all of which may be crucial for long-term synaptic plasticity (Sanes and Lichtman, 1999), it is perhaps not surprising that STDP learning rules are different at different dendritic locations onto the same postsynaptic neuron, as previous hypothesized (Sourdet and Debanne, 1999).
In contrast to our more detailed understanding of dendritic integration, location dependence of STDP has to date only been investigated experimentally in a few studies (Froemke et al., 2005; Letzkus et al., 2006; Sjöström and Häusser, 2006). These data suggest that in general, proximal synapses undergo STDP according to conventional learning rules, while inputs onto distal dendrites display novel STDP induction requirements. A dominant factor shaping local learning rules is the decremental nature of action potential backpropagation (Stuart and Sakmann, 1994; Svoboda et al., 1999; Waters et al., 2003), causing distal synapses to experience a smaller local depolarization during STDP induction than proximal inputs. All forms of location-dependent STDP characterized so far employ NMDARs as coincidence detectors of synaptic activation and the backpropagating action potential (Froemke et al., 2005; Letzkus et al., 2006). However, STDP is known to depend on a variety of other signaling pathways, and future research will be needed to uncover their involvement in location-dependent STDP. Different input pathways have recently been shown to segregate onto distinct dendritic domains (Petreanu et al., 2009), suggesting that location-dependent forms of STDP may provide a mechanism for the regulation of dendritic inputs carrying different sensory signals.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abbott, L. F., and Blum, K. I. (1996). Functional significance of long-term potentiation for sequence learning and prediction. Cereb. Cortex 6, 406–416.
Abbott, L. F., and Nelson, S. B. (2000). Synaptic plasticity: taming the beast. Nat. Neurosci. 3, 1178–1183.
Angelo, K., London, M., Christensen, S. R., and Häusser, M. (2007). Local and global effects of Ih distribution in dendrites of mammalian neurons. J. Neurosci. 27, 8643–8653.
Antic, S. D. (2003). Action potentials in basal and oblique dendrites of rat neocortical neurons. J. Physiol. 550, 35–50.
Archie, K. A., and Mel, B. W. (2000). A model for intradendritic computation of binocular disparity. Nat. Neurosci. 3, 54–63.
Artola, A., Bröcher, S., and Singer, W. (1990). Different voltage-dependent thresholds for inducing long-term depression and long-term potentiation in slices of rat visual cortex. Nature 347, 69–72.
Atencio, C. A., and Schreiner, C. E. (2010). Laminar diversity of dynamic sound processing in cat primary auditory cortex. J. Neurophysiol. 103, 192–205.
Baddeley, R., Abbott, L. F., Booth, M. C., Sengpiel, F., Freeman, T., Wakeman, E. A., and Rolls, E. T. (1997). Responses of neurons in primary and inferior temporal visual cortices to natural scenes. Proc. Biol. Sci. 264, 1775–1783.
Bender, V. A., Bender, K. J., Brasier, D. J., and Feldman, D. E. (2006). Two coincidence detectors for spike timing-dependent plasticity in somatosensory cortex. J. Neurosci. 26, 4166–4177.
Bi, G.-Q., and Poo, M.-M. (1998). Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J. Neurosci. 18, 10464–10472.
Bollmann, J. H., and Engert, F. (2009). Subcellular topography of visually driven dendritic activity in the vertebrate visual system. Neuron 61, 895–905.
Bullis, J. B., Jones, T. D., and Poolos, N. P. (2007). Reversed somatodendritic Ih gradient in a class of rat hippocampal neurons with pyramidal morphology. J. Physiol. 579, 431–443.
Buonomano, D. V., and Merzenich, M. M. (1998). Cortical plasticity: from synapses to maps. Annu. Rev. Neurosci. 21, 149–186.
Cash, S., and Yuste, R. (1999). Linear summation of excitatory inputs by CA1 neurons. Neuron 22, 383–394.
Christie, B. R., Schexnayder, L. K., and Johnston, D. (1997). Contribution of voltage-gated Ca2+ channels to homosynaptic long-term depression in the CA1 region in vitro. J. Neurophysiol. 77, 1651–1655.
Cobb, S. R., Buhl, E. H., Halasy, K., Paulsen, O., and Somogyi, P. (1995). Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature 378, 75–78.
Coombs, J. S., Curtis, D. R., and Eccles, J. C. (1957). The interpretation of spike potentials of motoneurones. J. Physiol. 139, 198–231.
Corlew, R., Wang, Y., Ghermazien, H., Erisir, A., and Philpot, B. D. (2007). Developmental switch in the contribution of presynaptic and postsynaptic NMDA receptors to long-term depression. J. Neurosci. 27, 9835–9845.
Dahmen, J. C., Hartley, D. E., and King, A. J. (2008). Stimulus-timing-dependent plasticity of cortical frequency processing. J. Neurosci. 28, 13629–13639.
Dan, Y., and Poo, M.-M. (2006). Spike timing-dependent plasticity: from synapse to perception. Physiol. Rev. 86, 1033–1048.
Das, A., and Gilbert, C. D. (1995). Long-range horizontal connections and their role in cortical reorganization revealed by optical recording of cat primary visual cortex. Nature 375, 780–784.
Debanne, D., Gahwiler, B. H., and Thompson, S. M. (1998). Long-term synaptic plasticity between pairs of individual CA3 pyramidal cells in rat hippocampal slice cultures. J. Physiol. 507, 237–247.
Diamond, M. E., Huang, W., and Ebner, F. F. (1994). Laminar comparison of somatosensory cortical plasticity. Science 265, 1885–1888.
Feldman, D. E. (2000). Timing-based LTP and LTD at vertical inputs to layer II/III pyramidal cells in rat barrel cortex. Neuron 27, 45–56.
Feldman, D. E. (2009). Synaptic mechanisms for plasticity in neocortex. Annu. Rev. Neurosci. 32, 33–55.
Feldman, D. E., Nicoll, R. A., and Malenka, R. C. (1999). Synaptic plasticity at thalamocortical synapses in developing rat somatosensory cortex: LTP, LTD, and silent synapses. J. Neurobiol. 41, 92–101.
Fleidervish, I. A., Binshtok, A. M., and Gutnick, M. J. (1998). Functionally distinct NMDA receptors mediate horizontal connectivity within layer 4 of mouse barrel cortex. Neuron 21, 1055–1065.
Frick, A., Magee, J., and Johnston, D. (2004). LTP is accompanied by an enhanced local excitability of pyramidal neuron dendrites. Nat. Neurosci. 7, 126–135.
Froemke, R. C., and Dan, Y. (2002). Spike-timing-dependent synaptic modification induced by natural spike trains. Nature 416, 433–438.
Froemke, R. C., Debanne, D., and Bi, G.-Q. (2010). Temporal modulation of spike-timing-dependent plasticity. Front. Syn. Neurosci. 2:19. doi:10.3389/fnsyn.2010.00019.
Froemke, R. C., Merzenich, M. M., and Schreiner, C. E. (2007). A synaptic memory trace for cortical receptive field plasticity. Nature 450, 425–429.
Froemke, R. C., Poo, M.-M., and Dan, Y. (2005). Spike-timing-dependent plasticity depends on dendritic location. Nature 434, 221–225.
Froemke, R. C., Tsay, I. A., Raad, M., Long, J. D., and Dan, Y. (2006). Contribution of individual spikes in burst-induced long-term synaptic modification. J. Neurophysiol. 95, 1620–1629.
Fu, Y.-X., Djupsund, K., Gao, H., Hayden, B., Shen, K., and Dan, Y. (2002). Temporal specificity in the cortical plasticity of visual space representation. Science 296, 1999–2003.
Fuortes, M. G., Frank, K., and Becker, M. C. (1957). Steps in the production of motoneuron spikes. J. Gen. Physiol. 40, 735–752.
Gandhi, S. P., Yanagawa, Y., and Stryker, M. P. (2008). Delayed plasticity of inhibitory neurons in developing visual cortex. Proc. Natl. Acad. Sci. U.S.A. 105, 16797–16802.
Gasparini, S., Migliore, M., and Magee, J. C. (2004). On the initiation and propagation of dendritic spikes in CA1 pyramidal neurons. J. Neurosci. 24, 11046–11056.
Gerstner, W., Kempter, R., van Hemmen, J. L., and Wagner, H. (1996). A neuronal learning rule for sub- millisecond temporal coding. Nature 383, 76–81.
Gidon, A., and Segev, I. (2009). Spike-timing-dependent synaptic plasticity and synaptic democracy in dendrites. J. Neurophysiol. 101, 3226–3234.
Goldberg, J., Holthoff, K., and Yuste, R. (2002). A problem with Hebb and local spikes. Trends Neurosci. 25, 433–435.
Golding, N. L., Staff, N. P., and Spruston, N. (2002). Dendritic spikes as a mechanism for cooperative long-term potentiation. Nature 418, 326–331.
Gordon, U., Polsky, A., and Schiller, J. (2006). Plasticity compartments in basal dendrites of neocortical pyramidal neurons. J. Neurosci. 26, 12717–12726.
Gulledge, A. T., Kampa, B. M., and Stuart, G. J. (2005). Synaptic integration in dendritic trees. J. Neurobiol. 64, 75–90.
Gulledge, A. T., and Stuart, G. J. (2003). Excitatory actions of GABA in the cortex. Neuron 37, 299–309.
Harvey, C. D., Yasuda, R., Zhong, H., and Svoboda, K. (2008). The spread of Ras activity triggered by activation of a single dendritic spine. Science 321, 136–140.
Häusser, M., Major, G., and Stuart, G. J. (2001). Differential shunting of EPSPs by action potentials. Science 291, 138–141.
Hoffman, D. A., and Johnston, D. (1999). Neuromodulation of dendritic action potentials. J. Neurophysiol. 81, 408–411.
Hoffman, D. A., Magee, J. C., Colbert, C. M., and Johnston, D. (1997). K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature 387, 869–875.
Holtmaat, A., and Svoboda, K. (2009). Experience-dependent structural synaptic plasticity in the mammalian brain. Nat. Rev. Neurosci. 10, 647–658.
Jack, J. J., Miller, S., Porter, R., and Redman, S. J. (1971). The time course of minimal excitatory post-synaptic potentials evoked in spinal motoneurones by group Ia afferent fibres. J. Physiol. 215, 353–380.
Jacob, V., Brasier, D. J., Erchova, I., Feldman, D., and Shulz, D. E. (2007). Spike timing-dependent synaptic depression in the in vivo barrel cortex of the rat. J. Neurosci. 27, 1271–1284.
Jia, H., Rochefort, N. L., Chen, X., and Konnerth, A. (2010). Dendritic organization of sensory input to cortical neurons in vivo. Nature 464, 1307–1312.
Kampa, B. M., Letzkus, J. J., and Stuart, G. J. (2006). Requirement of dendritic calcium spikes for induction of spike-timing-dependent synaptic plasticity. J. Physiol. 574, 283–290.
Kampa, B. M., Letzkus, J. J., and Stuart, G. J. (2007). Dendritic mechanisms controlling spike-timing-dependent synaptic plasticity. Trends Neurosci. 30, 456–463.
Kampa, B. M., and Stuart, G. J. (2006). Calcium spikes in basal dendrites of layer 5 pyramidal neurons during action potential bursts. J. Neurosci. 26, 7424–7432.
Kato, N., and Yoshimura, H. (1993). Reduced Mg2+ block of N-methyl-D-aspartate receptor-mediated synaptic potentials in developing visual cortex. Proc. Natl. Acad. Sci. U.S.A. 90, 7114–7118.
Koester, H. J., and Sakmann, B. (1998). Calcium dynamics in single spines during coincident pre- and postsynaptic activity depend on relative timing of back-propagating action potentials and subthreshold excitatory postsynaptic potentials. Proc. Natl. Acad. Sci. U.S.A. 95, 9596–9601.
Krupp, J. J., Vissel, B., Thomas, C. G., Heinemann, S. F., and Westbrook, G. L. (2002). Calcineurin acts via the C-terminus of NR2A to modulate desensitization of NMDA receptors. Neuropharmacology 42, 593–602.
Kyrozis, A., Albuquerque, C., Gu, J., and MacDermott, A. B. (1996). Ca2+-dependent inactivation of NMDA receptors: fast kinetics and high Ca2+ sensitivity in rat dorsal horn neurons. J. Physiol. 495, 449–463.
Larkum, M. E., Kaiser, K. M., and Sakmann, B. (1999a). Calcium electrogenesis in distal apical dendrites of layer 5 pyramidal cells at a critical frequency of back-propagating action potentials. Proc. Natl. Acad. Sci. U.S.A. 96, 14600–14604.
Larkum, M. E., Zhu, J. J., and Sakmann, B. (1999b). A new cellular mechanism for coupling inputs arriving at different cortical layers. Nature 398, 338–341.
Larkum, M. E., Zhu, J. J., and Sakmann, B. (2001). Dendritic mechanisms underlying the coupling of the dendritic with the axonal action potential initiation zone of adult rat layer 5 pyramidal neurons. J. Physiol. 533, 447–466.
Larkum, M. E., and Nevian, T. (2008). Synaptic clustering by dendritic signalling mechanisms. Curr. Opin. Neurobiol. 18, 321–331.
Letzkus, J. J., Kampa, B. M., and Stuart, G. J. (2006). Learning rules for spike timing-dependent plasticity depend on dendritic synapse location. J. Neurosci. 26, 10420–10429.
Letzkus, J. J., Kampa, B. M., and Stuart, G. J. (2007). Does spike timing- dependent synaptic plasticity underlie memory formation? Clin. Exp. Pharmacol. Physiol. 34, 1070–1076.
Lin, Y. W., Min, M. Y., Chiu, T. H., and Yang, H. W. (2003). Enhancement of associative long-term potentiation by activation of beta-adrenergic receptors at CA1 synapses in rat hippocampal slices. J. Neurosci. 23, 4173–4181.
Lisman, J., and Spruston, N. (2005). Postsynaptic depolarization requirements for LTP and LTD: a critique of spike-timing-dependent plasticity. Nat. Neurosci. 8, 839–841.
Losonczy, A., and Magee, J. C. (2006). Integrative properties of radial oblique dendrites in hippocampal CA1 pyramidal neurons. Neuron 50, 291–307.
Losonczy, A., Makara, J. K., and Magee, J. C. (2008). Compartmentalized dendritic plasticity and input feature storage in neurons. Nature 452, 436–441.
Magee, J. C. (2000). Dendritic integration of excitatory synaptic input. Nat. Rev. Neurosci. 1, 181–190.
Magee, J. C., and Cook, E. P. (2000). Somatic EPSP amplitude is independent of synapse location in hippocampal pyramidal neurons. Nat. Neurosci. 3, 895–903.
Magee, J. C., and Johnston, D. (1995). Synaptic activation of voltage-gated channels in the dendrites of hippocampal pyramidal neurons. Science 268, 301–304.
Magee, J.C., and Johnston, D. (1997). A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science 275, 209–213.
Magee, J. C., and Johnston, D. (2005). Plasticity of dendritic function. Curr. Opin. Neurobiol. 15, 334–342.
Makara, J. K., Losonczy, A., Wen, Q., and Magee, J. C. (2009). Experience-dependent compartmentalized dendritic plasticity in rat hippocampal CA1 pyramidal neurons. Nat. Neurosci. 12, 1485–1487.
Malenka, R. C., and Nicoll, R. A. (1999). Long-term potentiation- a decade of progress? Science 285, 1870–1874.
Markram, H., Lübke, J., Frotscher, M., and Sakmann, B. (1997). Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science 275, 213–215.
Markram, H., Toledo-Rodriguez, M., Wang, Y., Gupta, A., Silberberg, G., and Wu, C. (2004). Interneurons of the neocortical inhibitory system. Nat. Rev. Neurosci. 5, 793–807.
Mayer, M. L., Westbrook, G. L., and Guthrie, P. B. (1984). Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature 309, 261–263.
Meeks, J. P., and Mennerick, S. (2007). Action potential initiation and propagation in CA3 pyramidal axons. J. Neurophysiol. 97, 3460–3472.
Mehta, M. R., Lee, A. K., and Wilson, M. A. (2002). Role of experience and oscillations in transforming a rate code into a temporal code. Nature 417, 741–746.
Mel, B. W., Ruderman, D. L., and Archie, K. A. (1998). Translation-invariant orientation tuning in visual “complex” cells could derive from intradendritic computations. J. Neurosci. 18, 4325–4334.
Migliore, M., and Shepherd, G. M. (2002). Emerging rules for the distributions of active dendritic conductances. Nat. Rev. Neurosci. 3, 362–270.
Murayama, M., Pérez-Garci, E., Nevian, T., Bock, T., Senn, W., and Larkum, M. E. (2009). Dendritic encoding of sensory stimuli controlled by deep cortical interneurons. Nature 457, 1137–1141.
Nevian, T. Larkum, M. E., Polsky, A., and Schiller, J. (2007). Properties of basal dendrites of layer 5 pyramidal neurons: a direct patch-clamp recording study. Nat. Neurosci. 10, 206–214.
Nishiyama, M., Hong, K., Mikoshiba, K., Poo, M. M., and Kato, K. (2000). Calcium stores regulate the polarity and input specificity of synaptic modification. Nature 408, 584–588.
Nowak, L., Bregestovski, P., Ascher, P., Herbet, P., and Prochiantz, A. (1984). Magnesium gates glutamate-activated channels in mouse central neurones. Nature 307, 462–465.
Nowotny, T., Rabinovich, M. I., and Abarbanel, H. D. (2003). Spatial representation of temporal information through spike-timing-dependent plasticity. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 68, 011908.
Nusser, Z. (2009). Variability in the subcellular distribution of ion channels increases neuronal diversity. Trends Neurosci. 32, 267–274.
Oja, E. (1982). A simplified neuron model as a principal component analyzer. J. Math. Biol. 15, 267–273.
Palmer, L.M., and Stuart, G.J. (2006). Site of action potential initiation in layer 5 pyramidal neurons. J. Neurosci. 26, 1854–1863.
Pérez-Garci, E., Gassmann, M., Bettler, B., and Larkum, M. E. (2006). The GABAB1b isoform mediates long- lasting inhibition of dendritic Ca2+ spikes in layer 5 somatosensory pyramidal neurons. Neuron 50, 603–616.
Petreanu, L., Mao, T., Sternson, S. M., and Svoboda, K. (2009). The subcellular organization of neocortical excitatory connections. Nature 457, 1142–1145.
Pouille, F., and Scanziani, M. (2001). Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science 293, 1159–1163.
Rall, W. (1964). “Theoretical significance of dendritic trees for neuronal input-output relations,” in Neural Theory and Modeling, ed R. F. Reiss (Palo Alto, CA: Stanford University Press), 73–97.
Rao, R. P. N., and Sejnowski, T. J. (2001). Spike-timing-dependent Hebbian plasticity as temporal difference learning. Neural. Comput. 13, 2221–2237.
Reyes, A. (2001). Influence of dendritic conductances on the input-output properties of neurons. Annu. Rev. Neurosci. 24, 653–675.
Roberts, P. D., and Bell, C. C. (2002). Spike timing dependent plasticity in biological systems. Biol. Cybern. 87, 392–403.
Rosenmund, C., Feltz, A., and Westbrook, G. L. (1995). Calcium-dependent inactivation of synaptic NMDA receptors in hippocampal neurons. J. Neurophysiol. 73, 427–430.
Rumsey, C. C., and Abbott, L. F. (2004). Equalization of synaptic efficacy by activity- and timing-dependent synaptic plasticity. J. Neurophysiol. 91, 2273–2280.
Sanes, J. R., and Lichtman, J. W. (1999). Can molecules explain long-term potentiation? Nat. Neurosci. 2, 597–604.
Saudargiene, A., Porr., B., and Wörgötter, F. (2004). How the shape of pre- and postsynaptic signals can influence STDP: a biophysical model. Neural. Comput. 16, 595–625.
Saudargiene, A., Porr., B., and Wörgötter, F. (2005). Local learning rules: predicted influence of dendritic location on synaptic modification in spike- timing-dependent plasticity. Biol. Cybern. 92, 128–138.
Schiller, J., Schiller, Y., Stuart, G., and Sakmann, B. (1997). Calcium action potentials restricted to distal apical dendrites of rat neocortical pyramidal neurons. J. Physiol. 505, 605–616.
Schmidt-Hieber, C., Jonas, P., and Bischofberger, J. (2008). Action potential initiation and propagation in hippocampal mossy fibre axons. J. Physiol. 586, 1849–1857.
Segev, I., and London, M. (2000). Untangling dendrites with quantitative models. Science 290, 744–750.
Seol, G. H., Ziburkus, J., Huang, S., Song, L., Kim, I. T., Takamiya, K., Huganir, R. L., Lee, H. -K., and Kirkwood, A. (2007). Neuromodulators control the polarity of spike-timing- dependent synaptic plasticity. Neuron 20, 919–929.
Shu, Y., Duque, A., Yu, Y., Haider, B., and McCormick, D. A. (2007). Properties of action-potential initiation in neocortical pyramidal cells: evidence from whole cell axon recordings. J. Neurophysiol. 97, 746–760.
Silberberg, G., and Markram, H. (2007). Disynaptic inhibition between neocortical pyramidal cells mediated by Martinotti cells. Neuron 53, 735–746.
Sjöström, P. J., and Häusser, M. (2006). A cooperative switch determines the sign of synaptic plasticity in distal dendrites of neocortical pyramidal neurons. Neuron 51, 227–238.
Sjöström, P. J., and Nelson, S. B. (2002). Spike timing, calcium signals, and synaptic plasticity. Curr. Opin. Neurobiol. 12, 305–314.
Sjöström, P. J., Rancz, E. A., Roth, A., and Häusser, M. (2008). Dendritic excitability and synaptic plasticity. Physiol. Rev. 88, 769–840.
Sjöström, P. J., Turrigiano, G. G., and Nelson, S. B. (2001). Rate, timing, and cooperativity jointly determine cortical synaptic plasticity. Neuron 32, 1149–1164.
Sjöström, P. J., Turrigiano, G. G., and Nelson, S. B. (2003). Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron 39, 641–654.
Sjöström, P. J., Turrigiano, G. G., and Nelson, S. B. (2004). Endocannabinoid-dependent neocortical layer-5 LTD in the absence of postsynaptic spiking. J. Neurophysiol. 92, 3338–3343.
Smith, M. A., Ellis-Davies, G. C. R., and Magee, J. C. (2003). Mechanism of the distance-dependent scaling of Schaffer collateral synapses in rat CA1 pyramidal neurons. J. Physiol. 548, 245–258.
Song, S., and Abbott, L. F. (2001). Column and map development and cortical re-mapping through spike timing-dependent synaptic plasticity. Neuron 32, 339–350.
Song, S., Miller, K. D., and Abbott, L. F. (2000). Competitive Hebbian learning through spike-timing-dependent synaptic plasticity. Nat. Neurosci. 3, 919–926.
Sourdet, V., and Debanne, D. (1999). The role of dendritic filtering in associative long-term synaptic plasticity. Learn. Mem. 6, 422–447.
Spruston, N. (2008). Pyramidal neurons: dendritic structure and synaptic integration. Nat. Rev. Neurosci. 9, 206–221.
Spruston, N., Schiller, Y., Stuart, G., and Sakmann, B. (1995). Activity-dependent action potential invasion and calcium influx into hippocampal CA1 dendrites. Science 268, 297–300.
Stuart, G. J., and Häusser, M. (1994). Initiation and spread of sodium action potentials in cerebellar Purkinje cells. Neuron 13, 703–712.
Stuart, G. J., and Häusser, M. (2001). Dendritic coincidence detection of EPSPs and action potentials. Nat. Neurosci. 4, 63–71.
Stuart, G. J., and Sakmann, B. (1994). Active propagation of somatic action potentials into neocortical pyramidal cell dendrites. Nature 367, 69–72.
Stuart, G., Schiller, J., and Sakmann, B. (1997). Action potential initiation and propagation in rat neocortical pyramidal neurons. J. Physiol. 505, 617–632.
Stuart, G. J., and Spruston, N. (1998). Determinants of voltage attenuation in neocortical pyramidal neuron dendrites. J. Neurosci. 18, 3501–3510.
Svoboda, K., Helmchen, F., Denk, W., and Tank, D. W. (1999). Spread of dendritic excitation in layer 2/3 pyramidal neurons in rat barrel cortex in vivo. Nat. Neurosci. 2, 65–73.
Tamás, G., Szabadics, J., and Somogyi, P. (2002). Cell type- and subcellular position-dependent summation of unitary postsynaptic potentials in neocortical neurons. J. Neurosci. 22, 740–747.
Tamosiunaite, M., Porr, B., and Wörgötter, F. (2007). Developing velocity sensitivity in a model neuron by local synaptic plasticity. Biol. Cybern. 96, 507–518.
Tong, G., Shepherd, D., and Jahr, C. E. (1995). Synaptic desensitization of NMDA receptors by calcineurin. Science 267, 1510–1512.
Trachtenberg, J. T., Trepel, C., and Stryker, M. P. (2000). Rapid extragranular plasticity in the absence of thalamocortical plasticity in the developing primary visual cortex. Science 287, 2029–2032.
Umemiya, M., Chen, N., Raymond, L. A., and Murphy, T. H. (2001). A calcium-dependent feedback mechanism participates in shaping single NMDA miniature EPSCs. J. Neurosci. 21, 1–9.
Urakubo, H., Honda, M., Froemke, R. C., and Kuroda, S. (2008). Requirement of an allosteric kinetics of NMDA receptors for spike timing-dependent plasticity. J. Neurosci. 28, 3310–3323.
Waters, J., Larkum, M., Sakmann, B., and Helmchen, F. (2003). Supralinear Ca2+ influx into dendritic tufts of layer 2/3 neocortical pyramidal neurons in vitro and in vivo. J. Neurosci. 23, 8558–8567.
Wespatat, V., Tennigkeit, F., and Singer, W. (2004). Phase sensitivity of synaptic modifications in oscillating cells of rat visual cortex. J. Neurosci. 24, 9067–9075.
Williams, S. R., and Mitchell, S. J. (2008). Direct measurement of somatic voltage clamp errors in central neurons. Nat. Neurosci. 11, 790–798.
Williams, S. R., and Stuart, G. J. (1999). Mechanisms and consequences of action potential burst firing in rat neocortical pyramidal neurons. J. Physiol. 521, 467–482.
Williams, S. R., and Stuart, G. J. (2000a). Backpropagation of physiological spike trains in neocortical pyramidal neurons: implications for temporal coding in dendrites. J. Neurosci. 20, 8238–8246.
Williams, S. R., and Stuart, G. J. (2000b). Site independence of EPSP time course is mediated by dendritic Ih in neocortical pyramidal neurons. J. Neurophysiol. 83, 3177–3182.
Williams, S. R., and Stuart, G. J. (2002). Dependence of EPSP efficacy on synapse location in neocortical pyramidal neurons. Science 295, 1907–1910.
Yao, H., and Dan, Y. (2001). Stimulus timing-dependent plasticity in cortical processing of orientation. Neuron 32, 315–323.
Young, J. M., Waleszczyk, W. J., Wang, C., Calford, M. B., Dreher, B., and Obermayer, K. (2007). Cortical reorganization consistent with spike timing- but not correlation-dependent plasticity. Nat. Neurosci. 10, 887–895.
Zorumski, C. F., and Thio, L. L. (1992). Properties of vertebrate glutamate receptors: calcium mobilization and desensitization. Prog. Neurobiol. 39, 295–336.
Keywords: cortex, dendrites, LTD, LTP, NMDA receptors, spikes, STDP, synaptic plasticity
Citation: Froemke RC, Letzkus JJ, Kampa BM, Hang GB, Stuart GJ (2010) Dendritic synapse location and neocortical spike-timing-dependent plasticity. Front. Syn. Neurosci. 2:29. doi: 10.3389/fnsyn.2010.00029
Received: 02 February 2010;
Paper pending published: 23 February 2010;
Accepted: 27 June 2010;
Published online: 21 July 2010
Edited by:
Per Jesper Sjöström, University College London, UKReviewed by:
Attila Losonczy, Howard Hughes Medical Institute, USAIthai Rabinowitch, Medical Research Council Laboratory of Molecular Biology, UK
Dalton J. Surmeier, Northwestern University, USA
Copyright: © 2010 Froemke, Letzkus, Kampa, Hang and Stuart. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Robert C. Froemke, Molecular Neurobiology Program, Departments of Otolaryngology, Physiology and Neuroscience, The Helen and Martin Kimmel Center for Biology and Medicine, Skirball Institute of Biomolecular Medicine, New York University School of Medicine, New York, NY 10016, USA. e-mail:cm9iZXJ0LmZyb2Vta2VAbWVkLm55dS5lZHU=