 Keerthi Thirtamara Rajamani
Keerthi Thirtamara Rajamani Shlomo Wagner
Shlomo Wagner Valery Grinevich
Valery Grinevich Hala Harony-Nicolas
Hala Harony-Nicolas- 1The Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York City, NY, United States
- 2The Seaver Autism Center for Research and Treatment, Icahn School of Medicine at Mount Sinai, New York City, NY, United States
- 3Sagol Department of Neurobiology, Faculty of Natural Sciences, University of Haifa, Haifa, Israel
- 4Schaller Research Group on Neuropeptides at German Cancer Research Center (DKFZ), Central Institute of Mental Health and Cell Networks Cluster of Excellence, University of Heidelberg, Heidelberg, Germany
The neuropeptide oxytocin (OXT) is a crucial mediator of parturition and milk ejection and a major modulator of various social behaviors, including social recognition, aggression and parenting. In the past decade, there has been significant excitement around the possible use of OXT to treat behavioral deficits in neurodevelopmental disorders, including autism spectrum disorder (ASD). Yet, despite the fast move to clinical trials with OXT, little attention has been paid to the possibility that the OXT system in the brain is perturbed in these disorders and to what extent such perturbations may contribute to social behavior deficits. Large-scale whole-exome sequencing studies in subjects with ASD, along with biochemical and electrophysiological studies in animal models of the disorder, indicate several risk genes that play an essential role in brain synapses, suggesting that deficits in synaptic activity and plasticity underlie the pathophysiology in a considerable portion of these cases. OXT has been repeatedly shown, both in vitro and in vivo, to modify synaptic properties and plasticity and to modulate neural activity in circuits that regulate social behavior. Together, these findings led us to hypothesize that failure of the OXT system during early development, as a direct or indirect consequence of genetic mutations, may impact social behavior by altering synaptic activity and plasticity. In this article, we review the evidence that support our hypothesis.
Introduction
Behaviors are driven by diverse sets of functionally and anatomically connected brain regions that form brain circuits (Insel and Fernald, 2004; Goodson and Kabelik, 2009; Averbeck and Costa, 2017; Kohl et al., 2017; Roseberry and Kreitzer, 2017; Yang and Wang, 2017). Communications within brain circuits are not hard-wired but rather constantly adapting to the environment via neuromodulatory mechanisms. These mechanisms involve various neuromodulators, including neuropeptides, which exert their effect on neural ensembles to construct and modulate the circuit function and to shape a specific behavior (Marder, 2012; Nusbaum and Blitz, 2012). The last two decades have been enriched with studies exploring the behavioral effects of the pro-social oxytocin (OXT) neuropeptide (Heinrichs et al., 2003, 2009; Kirsch et al., 2005; Guastella et al., 2008a,b; Marsh et al., 2010; Guastella and MacLeod, 2012) and its mechanisms of action (Blume et al., 2008; Jurek et al., 2012; van den Burg et al., 2015). In parallel, several clinical and preclinical studies have focused on the therapeutic potential of OXT, mainly to treat social behavior deficits (Guastella and Hickie, 2016; Wagner and Harony-Nicolas, 2017). However, little attention has been paid to the possible implication of the OXT system in neurodevelopmental disorders and to whether perturbation in OXT may contribute to the social behavior phenotype. The objective of this review is to provide a framework for the role OXT plays in modulating synaptic plasticity and its implication in neurodevelopmental disorders. We begin by summarizing studies that examined the role of OXT in regulating synaptic plasticity underlying behavior. We then highlight studies that report specific alterations in the OXT system in rodent models of neurodevelopmental disorders. Finally, we explore the potential convergence between the OXT system and genes associated with neurodevelopmental disorders, focusing on the SHANK3 gene.
The Oxytocin System
OXT is a neuropeptide that is exclusively synthesized in neurons residing in the paraventricular (PVN), supraoptic (SON) and accessory nuclei (AN) of the hypothalamus. These nuclei harbor two major types of OXT-producing cells: magno- and parvocellular neurons (Althammer and Grinevich, 2017). Magnocellular OXT neurons project to the posterior pituitary for OXT release into systemic circulation and concomitantly send axonal collaterals to a large proportion of forebrain regions. Parvocellular (or preautonomic) OXT neurons differ in their projections to the midbrain, brainstem and spinal cord, thus controlling autonomic and metabolic processes as well as processing of nociceptive and non-nociceptive information (see reviews: (Althammer and Grinevich, 2017; Boll et al., 2017; Poisbeau et al., 2017)).
In rodents, OXT action is conveyed through the OXT receptor (OXTR), whose expression shows a significant overlap with axon terminals of OXT neurons within the brain (Grinevich et al., 2016; Marlin and Froemke, 2017). OXTR is a G-protein-coupled receptor that is capable of enhancing (Knobloch et al., 2012; Stoop, 2012) or reducing (Eliava et al., 2016) the excitability of neural cells via distinct mechanisms, including its dual coupling to Gq or Go/Gi proteins (Gravati et al., 2010; Busnelli et al., 2012). Among the intracellular signaling pathways activated by the OXTRs is the mitogen-activated protein kinase (MAPK) cascade (van den Burg and Neumann, 2011), whose role in regulating specific behaviors is now beginning to emerge. For example, it has been demonstrated that the OXT anxiolytic effect requires OXTR/MEK/ERK signaling (Blume et al., 2008; Jurek et al., 2012; van den Burg et al., 2015), and that this anxiolytic pathway strictly requires the influx of extra-cellular calcium through transient receptor potential vanilloid (TRPV) channels (van den Burg et al., 2015).
Oxytocin, Synaptic Plasticity and Behavior
Long-term potentiation (LTP) and long-term depression (LTD) are the most common forms of long-term synaptic plasticity. Both are long-lasting changes in synaptic strength induced by certain patterns of synaptic activity (Cooke and Bliss, 2006). LTP and LTD are considered as putative synaptic mechanisms that mediate learning and memory (Redondo and Morris, 2011). The effect of OXT on LTP was first demonstrated by Dubrovsky et al. (2002) in the rat hippocampus. The authors examined the effect of intracerebroventricular (ICV) administration of OXT (1 μg) on LTP induction in vivo using high-frequency tetanic stimulation in the dentate gyrus (DG) of anesthetized rats. They found that, in the presence of OXT, tetanic stimulation induced LTD rather than the expected LTP. We have reported a similar effect of OXT in the medial nucleus of the amygdala (MeA) in anesthetized rat. Specifically, we examined the effect of ICV OXT administration (1 μg) on synaptic plasticity induction by tetanic stimulation of the accessory olfactory bulb (AOB) (Gur et al., 2014) and demonstrated that OXT strongly augments LTD induction in the AOB-MeA pathway. The MeA is an essential component of the brain network that subserves social recognition memory (SRM) (Ferguson et al., 2002), a subtype of social memory that enables subjects to remember and distinguish individual conspecifics (Gheusi et al., 1994). In agreement with previous findings (Ferguson et al., 2001; Lukas et al., 2013), our study validated that OXT in the MeA is crucial for SRM and further suggested that the OXT mediated LTD in the AOB-MeA pathway is involved in consolidating long-term SRM (Gur et al., 2014). We have recently demonstrated that ICV administration of CRF-related peptide urocortin3 or 17β-estradiol 45 min before OXT administration induced LTP rather than LTD in the MeA in response to AOB stimulation, a result that suggests a bidirectional long-term plasticity in the AOB-MeA synaptic pathway (Frankiensztajn et al., 2018).
In 2003, Tomizawa et al. (2003) reported that OXT perfusion (1 μM) of mouse hippocampal slices enhanced the ability of subthreshold synaptic stimulation to induce long-lasting LTP (L-LTP) at Schaffer collateral-CA1 synapses. The authors also demonstrated that this induction was mediated by the activation of the MAPK cascade and phosphorylation of cyclic AMP-responsive element binding protein (CREB), suggesting that OXT induced neuronal plasticity in the hippocampus is transcription-dependent. In an attempt to correlate these findings with behavior, the authors demonstrated that ICV administration of OXT in virgin mice improved long-term spatial learning, a result that aligned with a previous discovery showing that spatial memory is enhanced during pregnancy, delivery and lactation, situations when OXT levels are substantially high (Kinsley et al., 1999). Similarly, Lin et al. (2012) demonstrated that endogenous OXT contributes to the maintenance of late but not early phase LTP, which was induced by subthreshold stimulation. Furthermore, they showed that the OXT-induced enhancement of LTP is OXTR dependent and involves an EGFR-mediated rapid and persistent increase in the local translation of an atypical protein kinase C (PKC) isoform, thus describing a mechanism for OXT-dependent LTP. Notably, stress is known to have a prolonged negative effect on memory and synaptic plasticity (Kim et al., 2015). In rats, an uncontrollable stress experience following unpredictable and unescapable shocks causes impairment in hippocampal-dependent memory tasks and leads to deficits in both LTP and LTD (Foy et al., 1987; Shors et al., 1989; Xu et al., 1997; Kim et al., 2001). The effect of OXT on stress-induced impairments in synaptic plasticity and cognition has been recently addressed in two separate studies by the same group (Lee et al., 2015; Park et al., 2017). These studies demonstrated that administration of intranasal OXT before or after the stress event could reverse the LTP and LTD deficits observed in hippocampal slices as well as improve spatial memory impairments by activating OXTRs and regulating ERK activity.
The nucleus accumbens (NAc) is a key component of the mesocorticolimbic dopamine reward circuit and is known to be a target for synaptic plasticity-associated changes induced by drugs of abuse (Luscher and Malenka, 2011). Dolen et al. (2013) have recently shown that within the NAc, a region that is also implicated in social reward, OXT acts as a social reinforcement signal, and blocking OXTRs in the NAc inhibits the establishment of a preference for social cues. Furthermore, they showed that bath application of OXT (1 μM) induced a presynaptic LTD in NAc medium spiny neurons caused by decreased presynaptic neurotransmitter release probability. Using viral genetic tools, they demonstrated that presynaptic OXTRs on serotonergic axon terminals, arriving from the dorsal raphe nucleus to the NAc, are required for social reward and OXT-induced LTD, which they found to be dependent on the coordinated activity of OXT and serotonin (Dolen et al., 2013).
The effect of OXT on synaptic plasticity has also been studied in mouse brain slices from other brain regions. In mouse slices of infralimbic medial prefrontal cortex, OXT (100 nM) produced a significant suppression of basal glutamatergic neurotransmission through reduction of presynaptic glutamate release and lead to conversion of the activity dependent LTD to LTP. This OXT-dependent conversion is NMDA receptor-dependent and requires synaptic insertion of calcium-permeable AMPA receptors (Ninan, 2011). In slices of the AOB, OXT (0.2–2 μM) facilitated the induction of a NMDA receptor-dependent LTP in reciprocal synapses of excitatory mitral cells on inhibitory interneurons, thus enhancing inhibition on the mitral cells (Fang et al., 2008). In slices of the left auditory cortex, OXT induced LTP and led to increased spike firing (Mitre et al., 2016). Finally, although out of the scope of the current review that focuses on OXT and synaptic plasticity in the context of behavior, it is important to note that the effect of OXT on synaptic plasticity has been also studied in the context of pain, where stimulation of OXT-PVN neurons or intrathecal OXT administration was shown to reduce or prevent LTP in spinal dorsal horn neurons and transiently interrupt the long-lasting LTP-mediated mechanical hyperalgesia (DeLaTorre et al., 2009).
Implications for Neurodevelopmental Disorders
Recent advances in genetic studies of autism spectrum disorder (ASD) and other neurodevelopmental disorder have implicated several risk genes that play an essential role in brain synapses (Xu et al., 2012; De Rubeis et al., 2014; Sanders et al., 2015), a finding that suggests that deficits in synaptic activity and plasticity may underlie the pathophysiology of these disorders in a considerable portion of the cases. Electrophysiological studies in animal models for ASD have supported this theory and have repeatedly shown that LTP and/or LTD are impaired in the vast majority of these models (Bhakar et al., 2012; Chung et al., 2012; Kirschstein, 2012; Harony-Nicolas et al., 2015, 2017; Till et al., 2015; Tian et al., 2017) (examples for are presented in Figure 1). The role that OXT plays in modulating synaptic plasticity led us to hypothesize that failure of the OXT system during early development may impact social behavior by altering synaptic plasticity in brain regions implicated in social behavior. Here we summarize several studies that propose that dysfunction in the OXT system early in life could account for the development of some of the social behavior symptoms.
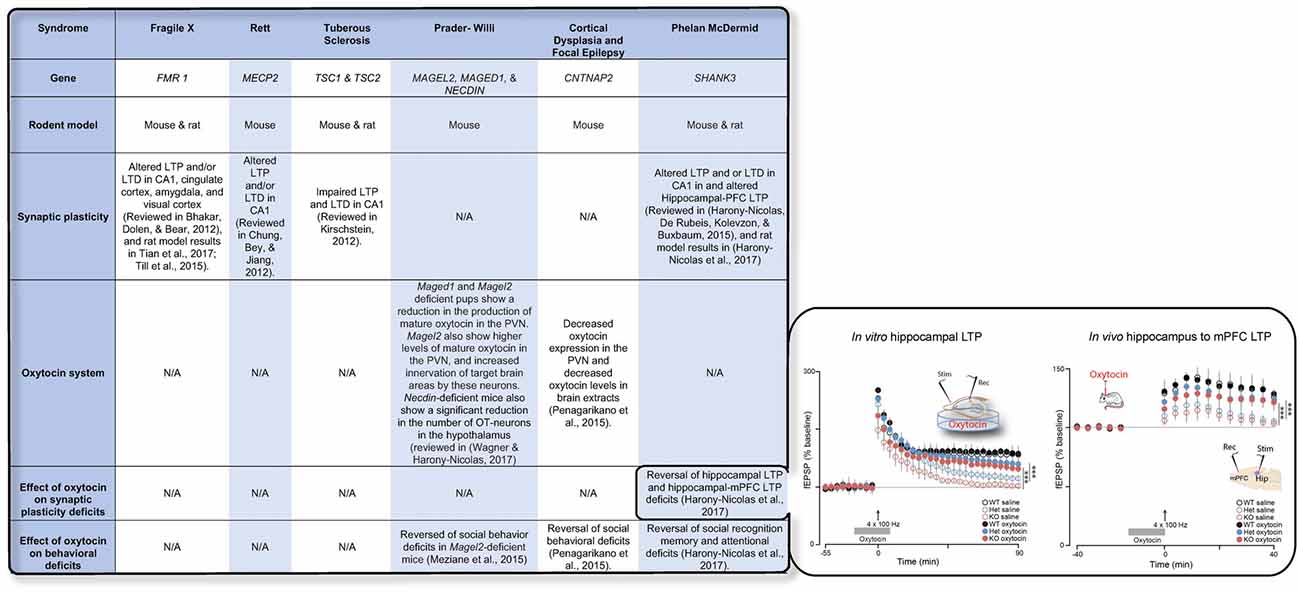
Figure 1. The table summarizes the available knowledge on synaptic plasticity deficits, alteration in the oxytocin (OXT) system, and the effect of OXT administration on behavioral and/or synaptic plasticity deficits in six syndromes associated with autism spectrum disorder (ASD). Inset shows the effect of OXT on synaptic plasticity deficits in the Shank3-deficient rat model. Reproduced from Figure 6, Harony-Nicolas et al., 2017; eLife, published under the Creative Commons Attribution 4.0 International Public License CC BY 4.0; (https://creativecommons.org/licenses/by/4.0/). FMR1, Fragile X mental retardation; MECP2, Methyl-CpG-binding protein 2; TSC1/2, Tuberous Sclerosis 1/2; MAGEL2, MAGE Family Member L2; MAGED1, MAGE Family Member D1; CNTNAP2, contactin associated protein like 2; SHANK3, SH3 And Multiple Ankyrin Repeat Domains 3; LTP, Long-term potentiation; LTD, Long-term depression; mPFC, medial prefrontal cortex; PVN, paraventricular nucleus.
Prader-Willi Syndrome
Prader-Willi Syndrome (PWS) is a rare multisystem neurodevelopmental disorder that presents with abnormal clinical features during development, starting with severe hypotonia and feeding difficulties in infants followed by unrelenting feelings of hunger and consequently excessive eating and obesity problems later in life (Angulo et al., 2015). Individuals with PWS also present with intellectual disability and some features of ASD (Bennett et al., 2015). PWS results from the lack of expression of the paternal allele of several contiguous genes including MKRN3, MAGEL2, MAGED1, NECDIN and SNURF-SNRPRN. Notably, subjects with PWS have a significantly decreased number of PVN-OXT neurons and decreased levels of circulating OXT (Swaab et al., 1995; Hæybye, 2004). These alterations are suggested to underlie the obesity phenotype in PWS patients (reviewed in Sabatier et al., 2013). Mouse models for PWS present with several phenotypes, some of which mimic those observed in subjects with PWS. Maged1-deficient mice develop progressive obesity, show impaired social interaction and social memory and display alerted sexual behavior, increased anxiety and self-grooming. Notably, in these mice, the synthesis of mature OXT in the brain is also significantly decreased, and restoring OXT levels via acute peripheral administration of OXT reverses the social memory deficits (Dombret et al., 2012). Similarly, the Magel2-deficient pups also show a significant reduction in the levels of mature OXT peptide in the brain. These abnormalities are accompanied by deficits in social and learning behaviors that are reversed following subcutaneous administration of OXT at an early postnatal stage (Schaller et al., 2010; Meziane et al., 2015). Notably, Magel2-deficient mice also exhibit feeding difficulties, a phenotype that mirrors those observed in patients with PWS. Finally, in the Necdin-deficient mouse model, the number of hypothalamic OXT-neurons is also significantly decreased (Muscatelli et al., 2000). Together, these studies suggest that the alterations in OXT system may underlie the social behavior deficits observed in subjects with PWS and that OXT treatment may be beneficial to treat these deficits. Similarly, a recent clinical study showed that intranasal application of OXT in subjects with PWS under age of 6 months improves feeding and social skills (Tauber et al., 2017).
CNTNAP2
Missense heterozygous mutations in the contactin-associated protein-like 2 (CNTNAP2) are implicated in cortical dysplasia-focal epilepsy (CFDE) syndrome and are associated with epilepsy, seizures, attention-deficit hyperactivity disorder (ADHD) and ASD (Strauss et al., 2006; Elia et al., 2010; Mefford et al., 2010; Rodenas-Cuadrado et al., 2014). The CNTNAP2 gene encodes for the CASPR2 protein, which is a member of the neurexin superfamily. Presynaptic neurexins interact with members of the neuroligin family at the post synapse, and members of both protein families have been associated with ASD (Betancur et al., 2009). Similar to human subjects, mice with a Cntnap2 mutation also have epileptic seizures and show deficits in ASD-associated behaviors (Brunner et al., 2015; Peñagarikano et al., 2015). Notably, Cntnap2-KO mice exhibit a significant and specific reduction in the number of OXT expressing cells in the PVN as well as in OXT concentrations in brain extracts. Single intraperitoneal or intranasal application of OT in these mice is sufficient to transiently rescue their social behavior deficits. Interestingly, early postnatal sub-chronic intranasal application of OXT alleviates their social behavior deficits and restores PVN-OXT neurons and brain OXT concentrations to wild-type levels (Peñagarikano et al., 2015). Together, these findings suggest that the OXT system may be affected in individuals with CNTNAP2 mutations and that those individuals may particularly benefit from an early-life treatment with OXT.
Shank3
We have recently produced and validated the Shank3-deficient rat model, a novel transgenic rat model for ASD and intellectual disability that harbors a mutation in the Shank3 gene (Harony-Nicolas et al., 2017). Shank3 is a scaffolding protein at the postsynaptic density (PSD), which functions as a platform upon which other additional PSD proteins accrete (Grabrucker et al., 2011). In humans, deletions or mutations in the SHANK3 gene lead to Phelan-McDermid Syndrome (PMS) with approximately 80% meeting criteria for ASD (Soorya et al., 2013). Mouse models with Shank3 gene mutations display ASD-related behavioral phenotypes, altered synaptic transmission and changes in neural plasticity and synaptic morphology (Harony-Nicolas et al., 2015). In rats, we found that Shank3 mutations lead to deficits in attention, long- but not short-term SRM (Harony-Nicolas et al., 2017) and developmental social communication (Berg et al., 2018). We also found that these deficits are accompanied by impaired synaptic plasticity. Specifically, we showed that maintenance of LTP in the hippocampus (in vitro) and the hippocampal-PFC pathway (in vivo) is impaired in the Shank3-deficient rat. Finally, we demonstrated that acute ICV administration of OXT in these rats reversed both the behavioral and the in vitro and in vivo synaptic plasticity deficits (Harony-Nicolas et al., 2017). These findings were the first to report that OXT can reverse not only behavioral but also synaptic plasticity deficits in a genetic model for a neurodevelopmental disorder, suggesting that the reversal effect of OXT on synaptic plasticity, specifically LTP, may underlie its ameliorative effect on behavior. Further studies are needed to determine the effect of SHANK3 mutations on the OXT system in order to understand if perturbation in this system could explain some of the observed behavioral phenotypes and plasticity-related changes.
Concluding Remarks
Here, we provided an overview of the modulatory effects of OXT on synaptic activity that underlie diverse behaviors. We also reviewed findings from genetic rodent models of neurodevelopmental disorders that demonstrate alterations in the OXT system. Despite the major interest in the therapeutic potential of OXT to treat social behavior deficits, there is still a considerable gap in the knowledge about the plausible implication of the OXT system in the pathogenesis of neurodevelopmental disorders. To fill this gap, there is a need for future studies to investigate not only the effect of mutations associated with these disorders on the maturation of the OXT system during early development stages but also the integrity and functionality of this system during later postnatal windows and under different behavioral contexts. Many of the mutations associated with neurodevelopmental disorders reside in genes encoding for neural or synaptic proteins (Xu et al., 2012; De Rubeis et al., 2014; Sanders et al., 2015), suggesting that they may impact: (1) the development and maturation of OXT neurons; (2) the OXT projections; (3) the intrinsic properties of OXT neurons; and (4) the trafficking and release of OXT. An alteration in any of these elements could potentially lead to imbalanced OXT levels in the brain and/or a lack or diminished response by the OXT system to stimuli (e.g., stress or social stimuli). This consequence could affect synaptic activity and plasticity in target brain regions modulated by OXT and ultimately impair the behavioral response. The CD38−/− mouse model provides an example for how behavioral phenotypes can be attributed to deficits in OXT release. This model harbors a mutation in the CD38 gene that encodes for a transmembrane glycoprotein involved in OXT release. CD38−/− mice have reduced OXT plasma levels and increased number of large dense core vesicles (LDCVs), which package the OXT neuropeptide. These alterations are accompanied with impaired maternal nurturing and SRM, which can be rescued with OXT administration (Jin et al., 2007).
Our findings in the Shank3-deficient rat, where both the behavioral and the synaptic plasticity deficits are rescued with OXT administration (Harony-Nicolas et al., 2017), suggest that OXT delivery and/or release at target brain regions involved in social behavior may be impaired. Notably, OXT release is known to be reliant on rapid and transient depolymerization of actin filaments (Tobin et al., 2012), and Shank3-deficient mice show dysregulation of actin filaments via upregulation of cofilin, a known actin depolymerizing agent (Duffney et al., 2015). Based on these findings it is possible that mutations in the SHANK3 gene would impact the actin cytoskeleton in OXT neurons, the release of OXT, and consequently, synaptic plasticity and behaviors that are modulated by OXT. To address this theory, there is a need for future studies to assess the central and peripheral OXT levels in this model and OXT levels following behavioral (e.g., social or stress stimuli), drug-induced (e.g., CCK8) or chemogenetic (designer receptors exclusively activated by designer drug; DREADDS) activation of OXT-neurons. Moreover, given that OXT neurons project to several brain targets, it is important to elucidate whether the effect of OXT on modulating synaptic activity persists across all projection targets to influence a circuit and whether some of these target regions are more vulnerable to genetic insults than others.
Importantly, given the etiological and phenotypic heterogeneity in ASD and neurodevelopmental disorders, we do not expect impairment in the OXT system to explain the behavioral phenotypes of all individuals with the disorder. This highlights the need for future studies in additional genetic models of neurodevelopmental disorders to (1) identify mutations that pose a deleterious effect on the OXT system and therefore converge on a shared pathophysiology and (2) to define the mechanistic interplay between these mutations and the OXT system. Findings from these studies will inform targeted treatments in human individuals carrying these pathogenic mutations.
Author Contributions
KTR, SW, VG and HH-N contributed to the writing of the mini review.
Funding
The Seaver Foundation, The Ministry of Science, Technology and Space of Israel (Grant #3-12068), Chica and Heinz Schaller Research Foundation, Deutsche Forschungsgemeinschaft (DFG) within the Collaborative Research Center (SFB) 1134 and 1158, SNSF-DFG grant GR 3619/8-1, ANR-DFG grant GR 3619/7-1, Fritz Thyssen Research grant 10.16.2.018MN and Human Frontier Science Program grant RGP0019/2015, National Institute of Mental Health R01-MH101584.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Althammer, F., and Grinevich, V. (2017). Diversity of oxytocin neurons: beyond magno- and parvocellular cell types? J. Neuroendocrinol. doi: 10.1111/jne.12549 [Epub ahead of print].
Angulo, M. A., Butler, M. G., and Cataletto, M. E. (2015). Prader-Willi syndrome: a review of clinical, genetic, and endocrine findings. J. Endocrinol. Invest. 38, 1249–1263. doi: 10.1007/s40618-015-0312-9
Averbeck, B. B., and Costa, V. D. (2017). Motivational neural circuits underlying reinforcement learning. Nat. Neurosci. 20, 505–512. doi: 10.1038/nn.4506
Bennett, J. A., Germani, T., Haqq, A. M., and Zwaigenbaum, L. (2015). Autism spectrum disorder in Prader-Willi syndrome: a systematic review. Am. J. Med. Genet. A 167A, 2936–2944. doi: 10.1002/ajmg.a.37286
Berg, E. L., Copping, N. A., Rivera, J. K., Pride, M. C., Careaga, M., Bauman, M. D., et al. (2018). Developmental social communication deficits in the Shank3 rat model of phelan-mcdermid syndrome and autism spectrum disorder. Autism Res. 11, 587–601. doi: 10.1002/aur.1925
Betancur, C., Sakurai, T., and Buxbaum, J. D. (2009). The emerging role of synaptic cell-adhesion pathways in the pathogenesis of autism spectrum disorders. Trends Neurosci. 32, 402–412. doi: 10.1016/j.tins.2009.04.003
Bhakar, A. L., Dölen, G., and Bear, M. F. (2012). The pathophysiology of fragile X (and what it teaches us about synapses). Annu. Rev. Neurosci. 35, 417–443. doi: 10.1146/annurev-neuro-060909-153138
Blume, A., Bosch, O. J., Miklos, S., Torner, L., Wales, L., Waldherr, M., et al. (2008). Oxytocin reduces anxiety via ERK1/2 activation: local effect within the rat hypothalamic paraventricular nucleus. Eur. J. Neurosci. 27, 1947–1956. doi: 10.1111/j.1460-9568.2008.06184.x
Boll, S., Almeida de Minas, A. C., Raftogianni, A., Herpertz, S. C., and Grinevich, V. (2017). Oxytocin and pain perception: from animal models to human research. Neuroscience doi: 10.1016/j.neuroscience.2017.09.041 [Epub ahead of print].
Brunner, D., Kabitzke, P., He, D., Cox, K., Thiede, L., Hanania, T., et al. (2015). Comprehensive analysis of the 16p11.2 deletion and null Cntnap2 mouse models of autism spectrum disorder. PLoS One 10:e0134572. doi: 10.1371/journal.pone.0134572
Busnelli, M., Saulière, A., Manning, M., Bouvier, M., Galés, C., and Chini, B. (2012). Functional selective oxytocin-derived agonists discriminate between individual G protein family subtypes. J. Biol. Chem. 287, 3617–3629. doi: 10.1074/jbc.M111.277178
Chung, L., Bey, A. L., and Jiang, Y. H. (2012). Synaptic plasticity in mouse models of autism spectrum disorders. Korean J. Physiol. Pharmacol. 16, 369–378. doi: 10.4196/kjpp.2012.16.6.369
Cooke, S. F., and Bliss, T. V. (2006). Plasticity in the human central nervous system. Brain 129, 1659–1673. doi: 10.1093/brain/awl082
De Rubeis, S., He, X., Goldberg, A. P., Poultney, C. S., Samocha, K., Cicek, A. E., et al. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. doi: 10.1038/nature13772
DeLaTorre, S., Rojas-Piloni, G., Martínez-Lorenzana, G., Rodríguez-Jiménez, J., Villanueva, L., and Condes-Lara, M. (2009). Paraventricular oxytocinergic hypothalamic prevention or interruption of long-term potentiation in dorsal horn nociceptive neurons: electrophysiological and behavioral evidence. Pain 144, 320–328. doi: 10.1016/j.pain.2009.05.002
Dolen, G., Darvishzadeh, A., Huang, K. W., and Malenka, R. C. (2013). Social reward requires coordinated activity of nucleus accumbens oxytocin and serotonin. Nature 501, 179–184. doi: 10.1038/nature12518
Dombret, C., Nguyen, T., Schakman, O., Michaud, J. L., Hardin-Pouzet, H., Bertrand, M. J., et al. (2012). Loss of Maged1 results in obesity, deficits of social interactions, impaired sexual behavior and severe alteration of mature oxytocin production in the hypothalamus. Hum. Mol. Genet. 21, 4703–4717. doi: 10.1093/hmg/dds310
Dubrovsky, B., Harris, J., Gijsbers, K., and Tatarinov, A. (2002). Oxytocin induces long-term depression on the rat dentate gyrus: possible ATPase and ectoprotein kinase mediation. Brain Res. Bull. 58, 141–147. doi: 10.1016/s0361-9230(01)00748-1
Duffney, L. J., Zhong, P., Wei, J., Matas, E., Cheng, J., Qin, L., et al. (2015). Autism-like deficits in shank3-deficient mice are rescued by targeting actin regulators. Cell Rep. 11, 1400–1413. doi: 10.1016/j.celrep.2015.04.064
Elia, J., Gai, X., Xie, H. M., Perin, J. C., Geiger, E., Glessner, J. T., et al. (2010). Rare structural variants found in attention-deficit hyperactivity disorder are preferentially associated with neurodevelopmental genes. Mol. Psychiatry 15, 637–646. doi: 10.1038/mp.2009.57
Eliava, M., Melchior, M., Knobloch-Bollmann, H. S., Wahis, J., da Silva Gouveia, M., Tang, Y., et al. (2016). A new population of parvocellular oxytocin neurons controlling magnocellular neuron activity and inflammatory pain processing. Neuron 89, 1291–1304. doi: 10.1016/j.neuron.2016.01.041
Fang, L. Y., Quan, R. D., and Kaba, H. (2008). Oxytocin facilitates the induction of long-term potentiation in the accessory olfactory bulb. Neurosci. Lett. 438, 133–137. doi: 10.1016/j.neulet.2007.12.070
Ferguson, J. N., Aldag, J. M., Insel, T. R., and Young, L. J. (2001). Oxytocin in the medial amygdala is essential for social recognition in the mouse. J. Neurosci. 21, 8278–8285. doi: 10.1523/JNEUROSCI.21-20-08278.2001
Ferguson, J. N., Young, L. J., and Insel, T. R. (2002). The neuroendocrine basis of social recognition. Front. Neuroendocrinol. 23, 200–224. doi: 10.1006/frne.2002.0229
Foy, M. R., Stanton, M. E., Levine, S., and Thompson, R. F. (1987). Behavioral stress impairs long-term potentiation in rodent hippocampus. Behav. Neural Biol. 48, 138–149. doi: 10.1016/s0163-1047(87)90664-9
Frankiensztajn, L. M., Gur-Pollack, R., and Wagner, S. (2018). A combinatorial modulation of synaptic plasticity in the rat medial amygdala by oxytocin, urocortin3 and estrogen. Psychoneuroendocrinology 92, 95–102. doi: 10.1016/j.psyneuen.2018.04.006
Gheusi, G., Bluthe, R. M., Goodall, G., and Dantzer, R. (1994). Social and individual recognition in rodents: methodological aspects and neurobiological bases. Behav. Processes 33, 59–87. doi: 10.1016/0376-6357(94)90060-4
Goodson, J. L., and Kabelik, D. (2009). Dynamic limbic networks and social diversity in vertebrates: from neural context to neuromodulatory patterning. Front. Neuroendocrinol. 30, 429–441. doi: 10.1016/j.yfrne.2009.05.007
Grabrucker, A. M., Schmeisser, M. J., Schoen, M., and Boeckers, T. M. (2011). Postsynaptic ProSAP/Shank scaffolds in the cross-hair of synaptopathies. Trends Cell Biol. 21, 594–603. doi: 10.1016/j.tcb.2011.07.003
Gravati, M., Busnelli, M., Bulgheroni, E., Reversi, A., Spaiardi, P., Parenti, M., et al. (2010). Dual modulation of inward rectifier potassium currents in olfactory neuronal cells by promiscuous G protein coupling of the oxytocin receptor. J. Neurochem. 114, 1424–1435. doi: 10.1111/j.1471-4159.2010.06861.x
Grinevich, V., Knobloch-Bollmann, H. S., Eliava, M., Busnelli, M., and Chini, B. (2016). Assembling the puzzle: pathways of oxytocin signaling in the brain. Biol. Psychiatry 79, 155–164. doi: 10.1016/j.biopsych.2015.04.013
Guastella, A. J., and Hickie, I. B. (2016). Oxytocin treatment, circuitry, and autism: a critical review of the literature placing oxytocin into the autism context. Biol. Psychiatry 79, 234–242. doi: 10.1016/j.biopsych.2015.06.028
Guastella, A. J., and MacLeod, C. (2012). A critical review of the influence of oxytocin nasal spray on social cognition in humans: evidence and future directions. Horm. Behav. 61, 410–418. doi: 10.1016/j.yhbeh.2012.01.002
Guastella, A. J., Mitchell, P. B., and Dadds, M. R. (2008a). Oxytocin increases gaze to the eye region of human faces. Biol. Psychiatry 63, 3–5. doi: 10.1016/j.biopsych.2007.06.026
Guastella, A. J., Mitchell, P. B., and Mathews, F. (2008b). Oxytocin enhances the encoding of positive social memories in humans. Biol. Psychiatry 64, 256–258. doi: 10.1016/j.biopsych.2008.02.008
Gur, R., Tendler, A., and Wagner, S. (2014). Long-term social recognition memory is mediated by oxytocin-dependent synaptic plasticity in the medial amygdala. Biol. Psychiatry 76, 377–386. doi: 10.1016/j.biopsych.2014.03.022
Harony-Nicolas, H., De Rubeis, S., Kolevzon, A., and Buxbaum, J. D. (2015). Phelan McDermid syndrome: from genetic discoveries to animal models and treatment. J. Child Neurol. 30, 1861–1870. doi: 10.1177/0883073815600872
Harony-Nicolas, H., Kay, M., Hoffmann, J. D., Klein, M. E., Bozdagi-Gunal, O., Riad, M., et al. (2017). Oxytocin improves behavioral and electrophysiological deficits in a novel Shank3-deficient rat. Elife 6:e18904. doi: 10.7554/eLife.18904
Heinrichs, M., Baumgartner, T., Kirschbaum, C., and Ehlert, U. (2003). Social support and oxytocin interact to suppress cortisol and subjective responses to psychosocial stress. Biol. Psychiatry 54, 1389–1398. doi: 10.1016/s0006-3223(03)00465-7
Heinrichs, M., von Dawans, B., and Domes, G. (2009). Oxytocin, vasopressin, and human social behavior. Front. Neuroendocrinol. 30, 548–557. doi: 10.1016/j.yfrne.2009.05.005
Höybye, C. (2004). Endocrine and metabolic aspects of adult Prader-Willi syndrome with special emphasis on the effect of growth hormone treatment. Growth Horm. IGF Res. 14, 1–15. doi: 10.1016/j.ghir.2003.09.003
Insel, T. R., and Fernald, R. D. (2004). How the brain processes social information: searching for the social brain. Annu. Rev. Neurosci. 27, 697–722. doi: 10.1146/annurev.neuro.27.070203.144148
Jin, D., Liu, H. X., Hirai, H., Torashima, T., Nagai, T., Lopatina, O., et al. (2007). CD38 is critical for social behaviour by regulating oxytocin secretion. Nature 446, 41–45. doi: 10.1038/nature05526
Jurek, B., Slattery, D. A., Maloumby, R., Hillerer, K., Koszinowski, S., Neumann, I. D., et al. (2012). Differential contribution of hypothalamic MAPK activity to anxiety-like behaviour in virgin and lactating rats. PLoS One 7:e37060. doi: 10.1371/journal.pone.0037060
Kim, J. J., Lee, H. J., Han, J. S., and Packard, M. G. (2001). Amygdala is critical for stress-induced modulation of hippocampal long-term potentiation and learning. J. Neurosci. 21, 5222–5228. doi: 10.1523/JNEUROSCI.21-14-05222.2001
Kim, E. J., Pellman, B., and Kim, J. J. (2015). Stress effects on the hippocampus: a critical review. Learn. Mem. 22, 411–416. doi: 10.1101/lm.037291.114
Kinsley, C. H., Madonia, L., Gifford, G. W., Tureski, K., Griffin, G. R., Lowry, C., et al. (1999). Motherhood improves learning and memory. Nature 402, 137–138. doi: 10.1038/45957
Kirsch, P., Esslinger, C., Chen, Q., Mier, D., Lis, S., Siddhanti, S., et al. (2005). Oxytocin modulates neural circuitry for social cognition and fear in humans. J. Neurosci. 25, 11489–11493. doi: 10.1523/JNEUROSCI.3984-05.2005
Kirschstein, T. (2012). Synaptic plasticity and learning in animal models of tuberous sclerosis complex. Neural Plast. 2012:279834. doi: 10.1155/2012/279834
Knobloch, H. S., Charlet, A., Hoffmann, L. C., Eliava, M., Khrulev, S., Cetin, A. H., et al. (2012). Evoked axonal oxytocin release in the central amygdala attenuates fear response. Neuron 73, 553–566. doi: 10.1016/j.neuron.2011.11.030
Kohl, J., Autry, A. E., and Dulac, C. (2017). The neurobiology of parenting: a neural circuit perspective. Bioessays 39, 1–11. doi: 10.1002/bies.201600159
Lee, S. Y., Park, S. H., Chung, C., Kim, J. J., Choi, S. Y., and Han, J. S. (2015). Oxytocin protects hippocampal memory and plasticity from uncontrollable stress. Sci. Rep. 5:18540. doi: 10.1038/srep18540
Lin, Y. T., Huang, C. C., and Hsu, K. S. (2012). Oxytocin promotes long-term potentiation by enhancing epidermal growth factor receptor-mediated local translation of protein kinase Mζ. J. Neurosci. 32, 15476–15488. doi: 10.1523/JNEUROSCI.2429-12.2012
Lukas, M., Toth, I., Veenema, A. H., and Neumann, I. D. (2013). Oxytocin mediates rodent social memory within the lateral septum and the medial amygdala depending on the relevance of the social stimulus: male juvenile versus female adult conspecifics. Psychoneuroendocrinology 38, 916–926. doi: 10.1016/j.psyneuen.2012.09.018
Luscher, C., and Malenka, R. C. (2011). Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron 69, 650–663. doi: 10.1016/j.neuron.2011.01.017
Marder, E. (2012). Neuromodulation of neuronal circuits: back to the future. Neuron 76, 1–11. doi: 10.1016/j.neuron.2012.09.010
Marlin, B. J., and Froemke, R. C. (2017). Oxytocin modulation of neural circuits for social behavior. Dev. Neurobiol. 77, 169–189. doi: 10.1002/dneu.22452
Marsh, A. A., Yu, H. H., Pine, D. S., and Blair, R. J. (2010). Oxytocin improves specific recognition of positive facial expressions. Psychopharmacology 209, 225–232. doi: 10.1007/s00213-010-1780-4
Mefford, H. C., Muhle, H., Ostertag, P., von Spiczak, S., Buysse, K., Baker, C., et al. (2010). Genome-wide copy number variation in epilepsy: novel susceptibility loci in idiopathic generalized and focal epilepsies. PLoS Genet. 6:e1000962. doi: 10.1371/journal.pgen.1000962
Meziane, H., Schaller, F., Bauer, S., Villard, C., Matarazzo, V., Riet, F., et al. (2015). An early postnatal oxytocin treatment prevents social and learning deficits in adult mice deficient for Magel2, a gene involved in prader-willi syndrome and autism. Biol. Psychiatry 78, 85–94. doi: 10.1016/j.biopsych.2014.11.010
Mitre, M., Marlin, B. J., Schiavo, J. K., Morina, E., Norden, S. E., Hackett, T. A., et al. (2016). A distributed network for social cognition enriched for oxytocin receptors. J. Neurosci. 36, 2517–2535. doi: 10.1523/JNEUROSCI.2409-15.2016
Muscatelli, F., Abrous, D. N., Massacrier, A., Boccaccio, I., Le Moal, M., Cau, P., et al. (2000). Disruption of the mouse Necdin gene results in hypothalamic and behavioral alterations reminiscent of the human Prader-Willi syndrome. Hum. Mol. Genet. 9, 3101–3110. doi: 10.1093/hmg/9.20.3101
Ninan, I. (2011). Oxytocin suppresses basal glutamatergic transmission but facilitates activity-dependent synaptic potentiation in the medial prefrontal cortex. J. Neurochem. 119, 324–331. doi: 10.1111/j.1471-4159.2011.07430.x
Nusbaum, M. P., and Blitz, D. M. (2012). Neuropeptide modulation of microcircuits. Curr. Opin. Neurobiol. 22, 592–601. doi: 10.1016/j.conb.2012.01.003
Park, S. H., Kim, Y. J., Park, J. C., Han, J. S., and Choi, S. Y. (2017). Intranasal oxytocin following uncontrollable stress blocks impairments in hippocampal plasticity and recognition memory in stressed rats. Int. J. Neuropsychopharmacol. 20, 861–866. doi: 10.1093/ijnp/pyx061
Peñagarikano, O., Lázaro, M. T., Lu, X. H., Gordon, A., Dong, H., Lam, H. A., et al. (2015). Exogenous and evoked oxytocin restores social behavior in the Cntnap2 mouse model of autism. Sci. Transl. Med. 7:271ra278. doi: 10.1126/scitranslmed.3010257
Poisbeau, P., Grinevich, V., and Charlet, A. (2017). Oxytocin signaling in pain: cellular, circuit, system, and behavioral levels. Curr. Top. Behav. Neurosci. doi: 10.1007/7854_2017_14 [Epub ahead of print].
Redondo, R. L., and Morris, R. G. (2011). Making memories last: the synaptic tagging and capture hypothesis. Nat. Rev. Neurosci. 12, 17–30. doi: 10.1038/nrn2963
Rodenas-Cuadrado, P., Ho, J., and Vernes, S. C. (2014). Shining a light on CNTNAP2: complex functions to complex disorders. Eur. J. Hum. Genet. 22, 171–178. doi: 10.1038/ejhg.2013.100
Roseberry, T., and Kreitzer, A. (2017). Neural circuitry for behavioural arrest. Philos. Trans. R. Soc. Lond. B Biol. Sci. 372:20160197. doi: 10.1098/rstb.2016.0197
Sabatier, N., Leng, G., and Menzies, J. (2013). Oxytocin, feeding, and satiety. Front. Endocrinol. 4:35. doi: 10.3389/fendo.2013.00035
Sanders, S. J., He, X., Willsey, A. J., Ercan-Sencicek, A. G., Samocha, K. E., Cicek, A. E., et al. (2015). Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 87, 1215–1233. doi: 10.1016/j.neuron.2015.09.016
Schaller, F., Watrin, F., Sturny, R., Massacrier, A., Szepetowski, P., and Muscatelli, F. (2010). A single postnatal injection of oxytocin rescues the lethal feeding behaviour in mouse newborns deficient for the imprinted Magel2 gene. Hum. Mol. Genet. 19, 4895–4905. doi: 10.1093/hmg/ddq424
Shors, T. J., Seib, T. B., Levine, S., and Thompson, R. F. (1989). Inescapable versus escapable shock modulates long-term potentiation in the rat hippocampus. Science 244, 224–226. doi: 10.1126/science.2704997
Soorya, L., Kolevzon, A., Zweifach, J., Lim, T., Dobry, Y., Schwartz, L., et al. (2013). Prospective investigation of autism and genotype-phenotype correlations in 22q13 deletion syndrome and SHANK3 deficiency. Mol. Autism 4:18. doi: 10.1186/2040-2392-4-18
Stoop, R. (2012). Neuromodulation by oxytocin and vasopressin. Neuron 76, 142–159. doi: 10.1016/j.neuron.2012.09.025
Strauss, K. A., Puffenberger, E. G., Huentelman, M. J., Gottlieb, S., Dobrin, S. E., Parod, J. M., et al. (2006). Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N. Engl. J. Med. 354, 1370–1377. doi: 10.1056/nejmoa052773
Swaab, D. F., Purba, J. S., and Hofman, M. A. (1995). Alterations in the hypothalamic paraventricular nucleus and its oxytocin neurons (putative satiety cells) in Prader-Willi syndrome: a study of five cases. J. Clin. Endocrinol. Metab. 80, 573–579. doi: 10.1210/jcem.80.2.7852523
Tauber, M., Boulanouar, K., Diene, G., Çabal-Berthoumieu, S., Ehlinger, V., Fichaux-Bourin, P., et al. (2017). The use of oxytocin to improve feeding and social skills in infants with prader-willi syndrome. Pediatrics 139:e20162976. doi: 10.1542/peds.2016-2976
Tian, Y., Yang, C., Shang, S., Cai, Y., Deng, X., Zhang, J., et al. (2017). Loss of FMRP impaired hippocampal long-term plasticity and spatial learning in rats. Front. Mol. Neurosci. 10:269. doi: 10.3389/fnmol.2017.00269
Till, S. M., Asiminas, A., Jackson, A. D., Katsanevaki, D., Barnes, S. A., Osterweil, E. K., et al. (2015). Conserved hippocampal cellular pathophysiology but distinct behavioural deficits in a new rat model of FXS. Hum. Mol. Genet. 24, 5977–5984. doi: 10.1093/hmg/ddv299
Tobin, V., Leng, G., and Ludwig, M. (2012). The involvement of actin, calcium channels and exocytosis proteins in somato-dendritic oxytocin and vasopressin release. Front. Physiol. 3:261. doi: 10.3389/fphys.2012.00261
Tomizawa, K., Iga, N., Lu, Y. F., Moriwaki, A., Matsushita, M., Li, S. T., et al. (2003). Oxytocin improves long-lasting spatial memory during motherhood through MAP kinase cascade. Nat. Neurosci. 6, 384–390. doi: 10.1038/nn1023
van den Burg, E. H., and Neumann, I. D. (2011). Bridging the gap between GPCR activation and behaviour: oxytocin and prolactin signalling in the hypothalamus. J. Mol. Neurosci. 43, 200–208. doi: 10.1007/s12031-010-9452-8
van den Burg, E. H., Stindl, J., Grund, T., Neumann, I. D., and Strauss, O. (2015). Oxytocin stimulates extracellular Ca2+ influx through TRPV2 channels in hypothalamic neurons to exert its anxiolytic effects. Neuropsychopharmacology 40, 2938–2947. doi: 10.1038/npp.2015.147
Wagner, S., and Harony-Nicolas, H. (2017). Oxytocin and animal models for autism spectrum disorder. Curr. Top. Behav. Neurosci. doi: 10.1007/7854_2017_15 [Epub ahead of print].
Xu, L., Anwyl, R., and Rowan, M. J. (1997). Behavioural stress facilitates the induction of long-term depression in the hippocampus. Nature 387, 497–500. doi: 10.1038/387497a0
Xu, L. M., Li, J. R., Huang, Y., Zhao, M., Tang, X., and Wei, L. (2012). AutismKB: an evidence-based knowledgebase of autism genetics. Nucleic Acids Res. 40, D1016–D1022. doi: 10.1093/nar/gkr1145
Keywords: oxytocin, synaptic plasticity, neurodevelopmental disorder, autism spectrum disorder (ASD), animal models for ASD
Citation: Thirtamara Rajamani K, Wagner S, Grinevich V and Harony-Nicolas H (2018) Oxytocin as a Modulator of Synaptic Plasticity: Implications for Neurodevelopmental Disorders. Front. Synaptic Neurosci. 10:17. doi: 10.3389/fnsyn.2018.00017
Received: 28 March 2018; Accepted: 25 May 2018;
Published: 19 June 2018.
Edited by:
Maria Elisa Calcagnotto, Universidade Federal do Rio Grande do Sul (UFRGS), BrazilReviewed by:
Robert C. Froemke, New York University, United StatesZhiping P. Pang, Rutgers University, United States
Copyright © 2018 Thirtamara Rajamani, Wagner, Grinevich and Harony-Nicolas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hala Harony-Nicolas, aGFsYS5oYXJvbnktbmljb2xhc0Btc3NtLmVkdQ==