 Antonio Heras-Garvin
Antonio Heras-Garvin Nadia Stefanova
Nadia Stefanova- Division of Neurobiology, Department of Neurology, Medical University of Innsbruck, Innsbruck, Austria
Since its discovery 30 years ago, α-synuclein (α-syn) has been one of the most studied proteins in the field of neuroscience. Dozens of groups worldwide have tried to reveal not only its role in the CNS but also in other organs. α-syn has been linked to several processes essential in brain homeostasis such as neurotransmitter release, synaptic function, and plasticity. However, despite the efforts made in this direction, the main function of α-syn is still unknown. Moreover, α-syn became a protein of interest for neurologists and neuroscientists when mutations in its gene were found associated with Parkinson’s disease (PD) and even more when α-syn protein deposits were observed in the brain of PD, dementia with Lewy bodies (DLB), and multiple system atrophy (MSA) patients. At present, the abnormal accumulation of α-syn constitutes one of the pathological hallmarks of these disorders, also referred to as α-synucleinopathies, and it is used for post-mortem diagnostic criteria. Whether α-syn aggregation is cause or consequence of the pathogenic events underlying α-synucleinopathies remains unclear and under discussion. Recently, different in vitro and in vivo studies have shown the ability of pathogenic α-syn to spread between cells, not only within the CNS but also from peripheral locations such as the gut, salivary glands, and through the olfactory network into the CNS, inducing abnormal misfolding of endogenous α-syn and leading to neurodegeneration and motor and cognitive impairment in animal models. Thus, it has been suggested that α-syn should be considered a prion protein. Here we present an update of what we know about α-syn function, aggregation and spreading, and its role in neurodegeneration. We also discuss the rationale and findings supporting the hypothetical prion nature of α-syn, its weaknesses, and future perspectives for research and the development of disease-modifying therapies.
Introduction
α-synuclein (α-syn) is one of the most abundant proteins in the nervous system, encoded by the SNCA gene. With a molecular mass of approximately 15 kDa, α-syn is composed of 140 amino acid residues (Lee and Trojanowski, 2006; Bendor et al., 2013). α-syn presents three domains: the C-terminal domain, the central domain, or “non-amyloid component” (NAC), which permits its oligomerization due to its hydrophobic composition, and the N-terminal domain, that forms an alpha helix allowing α-syn-lipid interaction (Lashuel et al., 2013). However, despite the efforts made since its discovery, its precise native structure is still unclear and under discussion. It has been proposed that, in physiological conditions, α-syn is mainly found in its monomeric form, either as a soluble intrinsically disordered protein in the cytoplasm of neurons (Binolfi et al., 2012; Fauvet et al., 2012; Theillet et al., 2016), or in an alpha-helical conformation (Bartels et al., 2011; Wang et al., 2011), or a combination of the two (Burré et al., 2013). Moreover, α-syn can undergo a variety of post-translational modifications (PTM; i.e., phosphorylation, glycation, glycosylation, acetylation) that may affect protein structure and function (Giasson et al., 2000; Theillet et al., 2016; Burré et al., 2018; Meade et al., 2019).
At the cellular level, α-syn can be found in different cellular organelles, including synaptic terminals and the nucleus of neurons (Maroteaux et al., 1988), mitochondria (Li et al., 2007; Devi et al., 2008; Parihar et al., 2008), endoplasmic reticulum (Hoozemans et al., 2007; Guardia-Laguarta et al., 2014), Golgi apparatus (Gosavi et al., 2002; Mori et al., 2002), and in the endolysosomal system (Lee et al., 2004). In this regard, since its discovery, α-syn has been associated or involved in different cellular processes, such as neurotransmission, calcium homeostasis, vesicle transport, mitochondrial function, and gene regulation (Figure 1). However, the physiological function of α-syn in those subcellular compartments is not fully understood. It has been shown that α-syn is involved in the regulation of the membrane lipid content and curvature, therefore playing a role in vesicle budding and trafficking (Chandra et al., 2003; Varkey et al., 2010). Moreover, α-syn plays a role in SNARE complex assembly at presynaptic terminals (Burré et al., 2010). Studies based on in situ proximity ligation assay (PLA) in neurons have demonstrated proximity between α-syn and SNARE proteins in cell bodies and neurites, including VAMP-2, SNAP-25, and syntaxin-1 (Almandoz-Gil et al., 2018). α-syn is also involved in clathrin-mediated endocytosis (Ben Gedalya et al., 2009) and in dopamine content and metabolism (Sidhu et al., 2004; Yu et al., 2005; Al-Wandi et al., 2010). In association with its nuclear localization, α-syn functions as a regulator of gene expression through its direct interaction with DNA (Pinho et al., 2019), chromatin (Vasquez et al., 2017), and transcription factors involved in development, mitochondrial homeostasis and metabolism of energy (Zheng et al., 2010; Decressac et al., 2012; Eschbach et al., 2015; Davidi et al., 2020), as well as through epigenetic mechanism including DNA methylation or histone acetylation (Surguchev and Surguchov, 2017). Furthermore, α-syn directly controls mitochondrial activity and function by modulating membrane potential, calcium homeostasis, cytochrome release, ATP production, and mitochondrial fusion and fission (Vicario et al., 2018). Interestingly, α-syn is also present in blood cells including red blood cells, peripheral blood mononuclear cells, platelets, and in the plasma. However, the role of α-syn in those cells is unclear (Barbour et al., 2008).
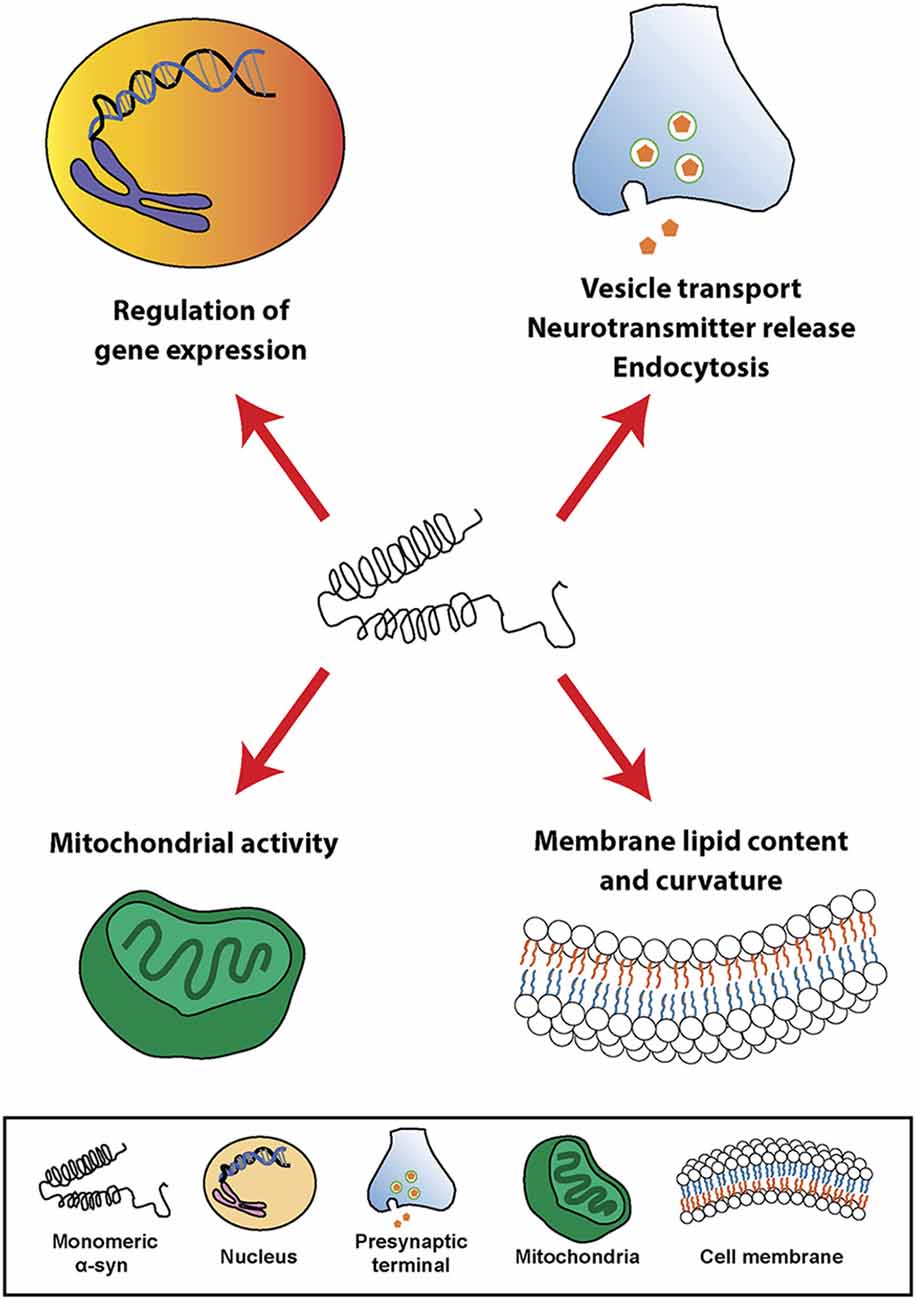
Figure 1. Physiological roles of α-synuclein (α-syn). Schematic overview including some of the cellular processes linked to α-syn in physiological conditions.
The functions and the structure of α-syn have being intensively investigated during the last decades, leading to a better knowledge of the protein and its association with crucial pathways on different cell types in the central nervous system (CNS), however α-syn research remains a priority not only for neuroscientist, but also for clinicians and pharmaceutical companies. The fact that α-syn aggregation and accumulation constitute the main pathological hallmark of a group of neurodegenerative disorders, referred to as α-synucleinopathies, and that mutations of its gene (SNCA) are associated with familiar forms of these disorders, has led α-syn to become one of the “most wanted” proteins in the biomedical field. Pre-clinical studies in vitro and in vivo have shown than α-syn oligomeric species and aggregates induce the impairment of different processes essential for cellular homeostasis, not only in neurons, but also in other cell types. Moreover, inoculation of α-syn oligomers, fibrils or aggregates in animal models, demonstrated the capability of pathogenic forms of α-syn to spread in a prion-like manner, inducing in some cases changes that replicated human pathology, including the clinical presentation. As a result of those observations, some authors have proposed that α-syn should be considered a prion protein. In this review, we discuss the role of α-syn in neurodegeneration, the different findings supporting the prion nature of α-syn, and its consequences for future therapeutic strategies.
α-Syn and Neurodegeneration: α-Synucleionopathies
The abnormal aggregation and accumulation of α-syn constitute the main pathological hallmark of Parkinson’s disease (PD), multiple system atrophy (MSA), and dementia with Lewy bodies (DLB; Spillantini et al., 1997, 1998; Goedert and Spillantini, 1998). These three disorders are also known as α-synucleinopathies. Recently, pure autonomic failure (PAF) and REM sleep behavior disorder (RBD) have been suggested to represent prodromal α-synucleinopathies commonly preceding PD or MSA and showing α-syn aggregation in the peripheral and autonomic nervous system (Arai et al., 2000; Kaufmann et al., 2001, 2004; Iranzo et al., 2013; Barber et al., 2017). Polymeropoulos et al. (1996, 1997) first described the association between α-syn and PD, when they found that individuals with familiar forms of this disorder presented mutations in the SNCA gene (Polymeropoulos et al., 1996, 1997). Soon afterwards Spillantini et al. (1997, 1998) showed that the abnormal protein aggregates observed in α-synucleinopathies were strongly immunoreactive for α-syn (Spillantini et al., 1997, 1998; Goedert and Spillantini, 1998). Moreover, some SNCA gene allelic variants (i.e., SNCA locus triplication) were sufficient to develop a severe early onset PD and DLB (Singleton et al., 2003; Orme et al., 2018; Zafar et al., 2018). Since then, multiple in vitro and in vivo studies have tried to decipher the mechanisms underlying α-syn misfolding and aggregation, the following cellular and physiological consequences, and their influence in the human pathology.
α-synucleinopathies present a preference for affecting motor, cognitive and autonomic areas, leading to motor dysfunction, cognitive impairment and/or autonomic failure (Marti et al., 2003). Phenotypic diversity, symptom severity and disease duration varies among these disorders based on the affectation of different neuroanatomical regions, selective vulnerability of different cell types and the extent of the pathology (Marti et al., 2003). Pathologically, PD and DLB are characterized by the abnormal accumulation of α-syn in the cytoplasm of neurons, in the so-called Lewy bodies (LBs; Spillantini et al., 1998). Associated with the presence of LBs in cortical and subcortical areas, post-mortem evaluation of PD and DLB brains show a preferential loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc), neuronal loss in the locus coeruleus (LC) and cortical atrophy (Dickson et al., 2009; Cheng et al., 2010; Jellinger, 2018). In addition to that, other neuronal populations in the brainstem accumulate LBs and degenerate, probably leading to many of the secondary clinical features of PD and DLB, such as autonomic dysfunction or sleep disturbances (Coon et al., 2018; Outeiro et al., 2019). In PAF, typical LBs are present in sympathetic ganglia, distal sympathetic nerves, substantia nigra, and locus ceruleus. In correlation with the presence of LBs, PAF patients present a significant unmyelinated nerve fiber density compared to healthy controls and neuronal degeneration in preganglionic sympathetic intermediolateral column and paravertebral ganglia (Kaufmann et al., 2001; Thaisetthawatkul, 2016). In MSA, however, α-syn mainly (but not exclusively) accumulates throughout the neocortex, hippocampus, brainstem, spinal cord, and dorsal root ganglia in the cytoplasm of oligodendrocytes, in the so-called glial cytoplasmic inclusions (GCIs), associating with striatonigral degeneration (SND), olivopontocerebellar atrophy and cell loss in LC and brainstem nuclei, leading to parkinsonism, cerebellar symptoms and autonomic dysfunction (Fanciulli and Wenning, 2015).
Despite the different clinical features of α-synucleinopathies, these disorders share characteristics such as neurodegeneration, neuroinflammation and demyelination, among others. Based on in vitro and in vivo studies, α-syn seems to play a central role in the pathological changes through a direct interaction with components of cellular pathways essential for cell homeostasis (Figure 2). Thus, pathological α-syn is able to bind and inhibit lysosomal function (Mazzulli et al., 2016; Flavin et al., 2017), proteasomal activity (Stefanis et al., 2001; Tanaka et al., 2001; Petrucelli et al., 2002; Snyder et al., 2003; Lindersson et al., 2004; Emmanouilidou et al., 2010), to impair axonal transport (Volpicelli-Daley, 2017), and to induce Ca2+ dyshomeostasis (Danzer et al., 2007; Chen Y. et al., 2015; Angelova et al., 2016) and mitochondrial dysfunction (Di Maio et al., 2016; Ganjam et al., 2019; Wang et al., 2019; Park et al., 2020). Latter promotes the generation of reactive oxygen species (ROS), and therefore oxidative stress, which may lead to mitochondrial damage, the release of cytochrome c to the cytoplasm, and cell death (Hsu et al., 2000; Smith et al., 2005; Parihar et al., 2008; Reeve et al., 2015; Tapias et al., 2017; Ludtmann et al., 2018). Associated with its nuclear localization or its capacity to promote oxidative stress, recent studies suggested an involvement of α-syn in DNA damage (Paiva et al., 2017; Vasquez et al., 2017; Milanese et al., 2018; Schaser et al., 2019). Pathological α-syn also promotes pathogenic redistribution of membrane proteins (Shrivastava et al., 2015, 2020) and interferes with the normal function of cell membranes, facilitating diffusion of molecules and ions, specially Ca2+, and inducing cell damage (Tosatto et al., 2012; Tsigelny et al., 2012; Angelova et al., 2016; Fusco et al., 2017; Dong et al., 2018). The impairment of all these cellular processes may be especially significant in dopaminergic neurons, since dopamine promotes the formation of pathological α-syn species (Cappai et al., 2005; Lee et al., 2011; Mor et al., 2017). Thus, a multi-hit model has been proposed, in which the synergistic interaction between α-syn, dopamine and Ca2+ in neurons may underlie neuronal loss in SNpc and LC (Post et al., 2018). Neurodegeneration in the LC may induce a direct loss of norepinephrine (NE), however, α-syn is also able to translocate to the nucleus of NE-producing cells and interfere with transcription of dopamine ß-hydroxylase, the final enzyme in NE biosynthesis, disrupting NE production and leading to a reduction of neurotrophic factor signaling, indirectly altering innate and adaptive immune response and worsening central and peripheral inflammation (Butkovich et al., 2018).
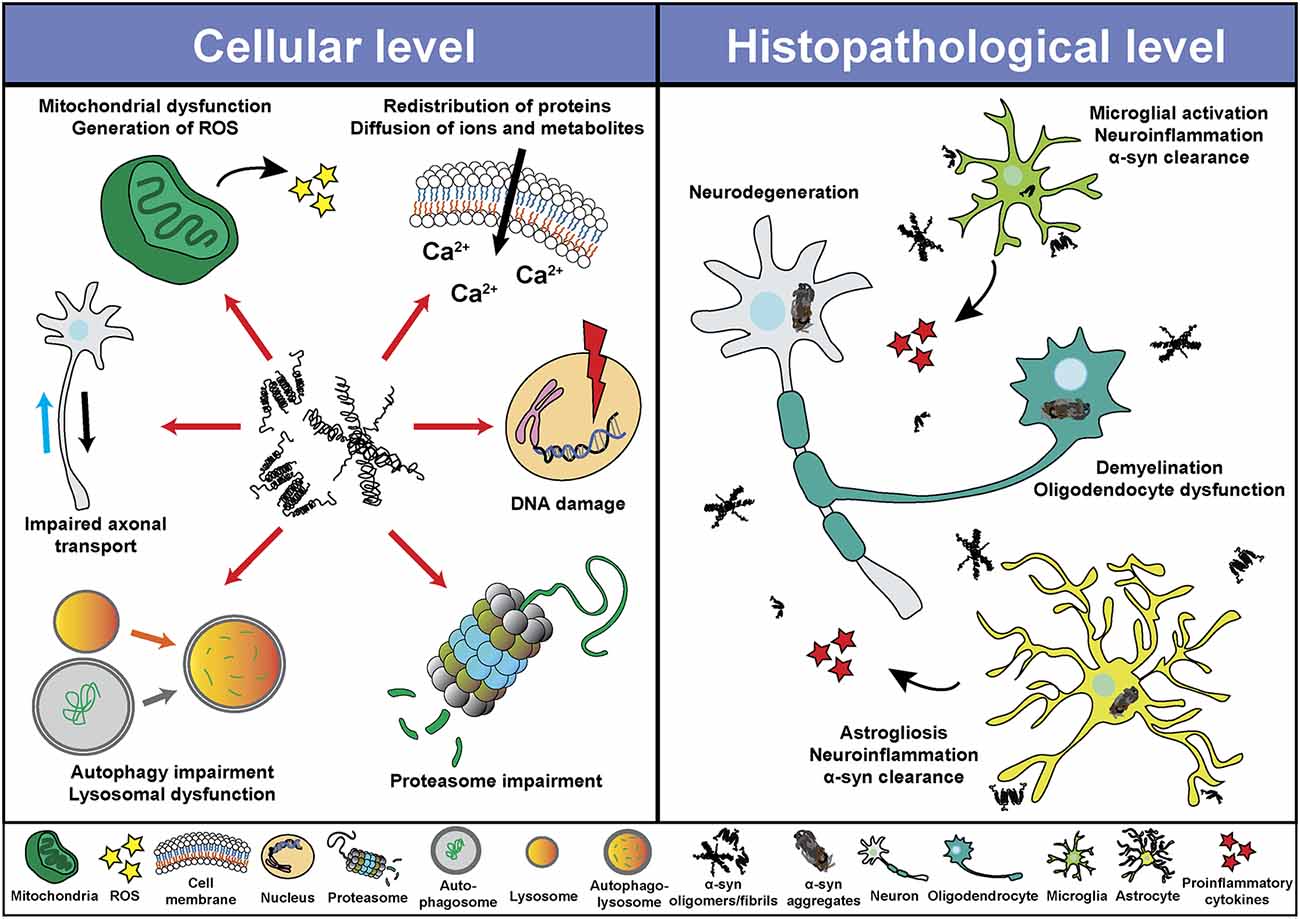
Figure 2. Pathological roles of α-syn. Schematic overview including some of the cellular processes (left) and histopathological events (right) associated with the presence of pathogenic α-syn species.
Different in vivo and in vitro studies have also shown the ability of pathological α-syn to induce synaptic dysfunction, both at the presynaptic and postsynaptic terminal (Ghiglieri et al., 2018). At presynaptic level, pathological α-syn seems to affect synaptic vesicle pool, trafficking and endocytosis by interfering with the exo-endocytotic cycling machinery, thus suggesting the possible association between α-syn pathology and impaired synaptic function even in the absence of cell death (Bridi and Hirth, 2018; Froula et al., 2018; Wu et al., 2019). At a postsynaptic level, in vivo studies have shown that pathological α-syn reduces striatal dopamine, NMDA receptor-mediated synaptic currents, impairs early memory and motor learning, and prevents long-term potentiation of cholinergic interneurons and spiny projection neurons in the striatum even at early stages of the disease (Tozzi et al., 2016; Phan et al., 2017; Giordano et al., 2018; Durante et al., 2019).
Pathological α-syn can also modulate neuroinflammation (Figure 2) directly through different mechanism including the activation of microglial cells (Austin et al., 2006; Su et al., 2008; Theodore et al., 2008; Fellner et al., 2013; Choi et al., 2020), which induces a robust pro-inflammatory response (Kim et al., 2013; Sarkar et al., 2020) leading to increased α-syn aggregation and potentiating neurodegeneration (Zhang et al., 2005; Stefanova et al., 2007). In this regard, microgliosis has been shown in post-mortem studies and in PET imaging analysis of patients diagnosed with α-synucleinopathies (McGeer et al., 1988a; Gerhard et al., 2003, 2006; Doorn et al., 2014; Stokholm et al., 2017; Surendranathan et al., 2018). In addition, microgliosis paralleled system degeneration in patients and was associated with α-syn inclusions (Imamura et al., 2003; Ishizawa et al., 2004, 2008; Croisier et al., 2005; Kubler et al., 2019). A recent study with PD patients engrafted with foetal embryonic dopaminergic neurons showed microglia activation in all grafts before α-syn aggregates were detected (Olanow et al., 2019). The authors speculated that microglial activation could have been initiated by the presence of endogenous pathological α-syn in patients, by an immune reaction due to the implantation of foreign tissue or by the surgical procedure itself (Olanow et al., 2019). Moreover, we recently showed that targeting α-syn oligomerization in an animal model for MSA led to a decrease of α-syn aggregates which significantly correlated with neuroprotection and a reduction in microglial activation (Heras-Garvin et al., 2019).
Similar to microglial cells, pathological α-syn also induces activation of astrocytes (Figure 2), aggravating neuroinflammation (Lee et al., 2010; Fellner et al., 2013; Radford et al., 2015). Astrogliosis has also been described in α-synucleinopathies paralleling the neurodegenerative process (Ozawa et al., 2004; Miklossy et al., 2006; Radford et al., 2015). Moreover, α-syn inclusions are often found in the cytoplasm of astrocytes and may lead to astrocyte dysfunction (Song et al., 2009; Halliday and Stevens, 2011; Sorrentino et al., 2019). Even a role of the adaptive immune system in PD has been proposed. Post-mortem evaluation showed higher numbers of T cells in the ventral midbrain of PD brains and mouse models than in healthy controls, suggesting a targeted extravasation, and supporting a role for T cells in disease pathogenesis (McGeer et al., 1988b; Theodore et al., 2008; Brochard et al., 2009; Sommer et al., 2018). Recent studies showed the presence of α-syn-specific T cells in PD patients, and therefore an autoimmune response to α-syn (Sulzer et al., 2017; Garretti et al., 2019). However, at present, α-syn-specific T cells have only been detected in the periphery of PD patients. Whether these cells are able to infiltrate into the CNS and cause dopaminergic neuron death remains to be determined.
Although most research on the pathogenic role of α-syn have been focused in neurons, microglia and astrocytes, there have also been studies that evaluated the deleterious effect of pathogenic α-syn in oligodendrocytes and oligodendroglial precursor cells (OPCs), which are essential to understand the cellular and molecular consequences of α-syn accumulation in MSA (Figure 2). Over-expression of α-syn or the addition of exogenous pathological α-syn severely impaired myelin formation in vitro and in vivo (Ettle et al., 2016; Mandel et al., 2017; Mavroeidi et al., 2019), corroborating the demyelination observed in MSA brains (Song et al., 2007; Don et al., 2014; Ettle et al., 2016). This effect seems to be mediated by the interference of α-syn with the expression of proteins associated with cholesterol and membrane biogenesis (Ettle et al., 2016; Kaji et al., 2018), and by the partial sequestration of the myelin basic protein (MBP) into GCIs in a process orchestrated by endogenous α-syn and the the oligodendroglial-specific phosphoprotein p25α (Mavroeidi et al., 2019). Moreover, α-syn impairs OPC maturation in vitro (Ettle et al., 2014) and in animal models of MSA, where an age-dependent increase of dividing OPCs within the striatum was observed (May et al., 2014). Latter was confirmed after postmortem analysis in MSA patients, revealing α-syn within OPCs and an increased number of striatal OPCs compared to healthy controls (May et al., 2014). α-syn also induces a distinct gene expression profile in oligodendrocytes, including upregulation of cytokines important for myeloid cell attraction and proliferation (Schafferer et al., 2016; Hoffmann et al., 2019), which constitutes an early crosstalk between neuroinflammation and α-syn-mediated oligodendrocyte dysfunction.
Overall, multiple cell populations and pathways can be affected by the abnormal misfolding, aggregation and accumulation of α-syn. Determining which events are primary or secondary in the pathogenesis of α-synucleinopathies will be essential in order to define potential strategies to prevent cell damage and, finally, neurodegeneration.
Aggregation and Spreading of α-Syn
As stated before, the normal conformation of α-syn is either a disordered monomer, alpha-helical, or a combination of the two. But, under pathological conditions, monomers can convert to β-sheets by recruiting additional monomers, aggregating and giving rise to oligomers that, eventually, form protofilaments and amyloid fibrils (Figure 3), which can adopt different morphologies such as spherical, chain-like, annular (pore-like structure), and tubular (Cremades and Dobson, 2018). To define the process through which α-syn oligomers give rise to pathological inclusions is challenging due to their transient nature and their ability to rapidly recruit monomeric α-syn to form fibrils. Nevertheless, the recent description that α-syn oligomers can be kinetically trapped made possible to compare the contribution of oligomers and fibrils to different α-synucleinopathy phenotypes (Chen S. W. et al., 2015; Iljina et al., 2016; De Oliveira and Silva, 2019; Froula et al., 2019). It is believed that the formation of α-syn fibrils results from interactions between monomers and oligomers that are thermodynamically favorable and stabilizing (Alam et al., 2019). Despite several studies have associated the presence of α-syn oligomers and fibrillar species with cytotoxicity and neurodegeneration, thus supporting the idea that these α-syn forms are the responsible for the neurodegenerative process observed in α-synucleinoptahies (Alam et al., 2019), a recent publication by Mahul-Mellier et al. (2020) proposed that the process of LB formation, rather than simply fibril formation, is one of the major drivers of neurodegeneration through disruption of cellular functions, inducing mitochondria damage and deficits, and synaptic dysfunction. Therefore, which α-syn species or processes associated with α-syn aggregation and accumulation are the most toxic is still a matter of debate.
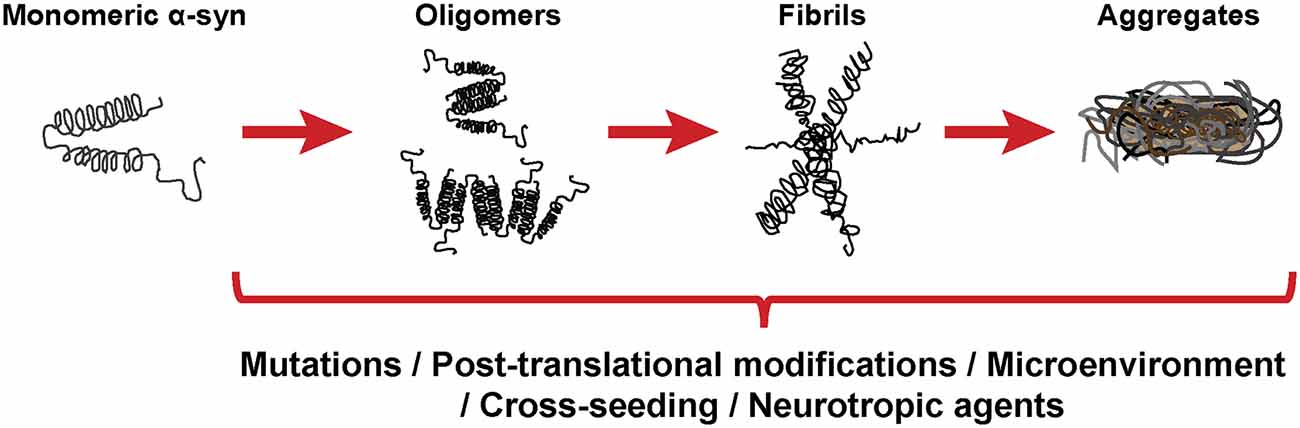
Figure 3. α-synuclein aggregation process. Schematic overview of the hypothetical aggregation process of monomeric α-syn and the different events that can induce its misfolding and promote aggregation.
Mutations in the SNCA gene associated with familiar forms of α-synucleinopathies, such as A30P or A53T, are more prone to aggregate and form fibrillar α-syn oligomers (Lashuel et al., 2002; Bengoa-Vergniory et al., 2017). Different events promote α-syn aggregation (Figure 3), including low pH (Buell et al., 2014). Indeed, neutral pH inhibits the growth of α-syn fibrils, while acidic pH increases it (Grey et al., 2011). Other micro-environmental factors affecting α-syn aggregation include metal ions (Uversky et al., 2001), polyamines (Krasnoslobodtsev et al., 2012; Holmes et al., 2013), proteoglycans (Lashuel et al., 2013), nucleic acids (Hegde and Rao, 2007), poly-ADP ribose (Kam et al., 2018) and fatty acids (Fecchio et al., 2018). α-syn PTM play also a role in its aggregation process. Thus, glycation of α-syn potentiate toxicity and aggregation (Vicente Miranda et al., 2017). Phosphorylation at various sites contributes to α-syn aggregation (Zhang et al., 2019). In particular, phosphorylation at residue Ser129 constitutes one of the most studied α-syn PTM, however, there is contradictory data about its role in pathology, as it was shown to be both protective and toxic (Arawaka et al., 2017; Zhang et al., 2019). In physiological conditions, α-syn is constitutively acetylated in its N-terminal, which seems to make α-syn more resistant to oligomerization and aggregation (Gonzalez et al., 2019). C-terminal truncation of α-syn has been associated with an increase in self-assembly properties and constitutes 10–25% of the LBs (Li et al., 2005; Wang et al., 2016). As a result of oxidative stress, the generation of nitric oxide induces nitration of tyrosine residues of α-syn, promoting aggregation (Giasson et al., 2000). SUMOylation inhibits ubiquitin-mediated degradation of α-syn, which leads to an increase of α-syn concentration and aggregation (Rott et al., 2017). Lastly, the aggregation process of α-syn seems to be affected by the presence of other proteins. Although homotypic seeding is the preferred form of aggregation, α-syn can bind other natively unfolded proteins such as tau, PrPC or Aβ, in a process termed heterotypic cross-seeding, which accelerate aggregation and neuropathology (Clinton et al., 2010; Ono et al., 2012; Guo et al., 2013; Katorcha et al., 2017; Bassil et al., 2020). However, despite our knowledge about the different processes that may be involved in its abnormal aggregation, the initial event that triggers α-syn oligomer formation and accumulation remains unknown.
In 2003, after post-mortem evaluation of PD brains, Braak et al. (2003a) staged the pathology of this disorder. In that study, they defined the earliest phases of the disease, characterized by the presence of α-syn pathology in the anterior olfactory nucleus and in the dorsal motor nucleus of the vagal in the absence of motor symptoms. Based on those findings, Braak et al. (2003b) developed a theory in which PD might be initiated when a neurotropic agent enters the body by nasal route, through the olfactory epithelium to the olfactory bulb, or by gastric route, infiltrating the gut to the submucous plexus and traveling along preganglionic parasympathetic fibers to the dorsal motor nucleus of the vagus nerve. The presence of LBs in the enteric and peripheral nervous systems supported their claim (Braak et al., 2006, 2007; Shannon et al., 2012). The LB pathology would then travel through the CNS. The authors proposed that PD could be divided into six different stages, each one defined by abnormal pathology in particular neurological structures and specific symptom severity. Thus, early stages would be characterized by non-motor symptoms, including hyposmia and autonomic dysfunction, mid-stages would be characterized by the presence of motor symptoms and later stages by the presence of cognitive impairment (Braak et al., 2004). In their theory, the authors proposed as the possible neurotropic agent a virus or a pathogen composed of fragments of misfolded α-syn that could possess unconventional prion-like properties (Braak et al., 2003b). Over the last decade, different studies have shown that α-syn is in fact not only able to aggregate, but also to spread and propagate the synucleinopathy in a prion-like fashion. This new hypothesis and line of research was initially inspired by the observation that grafts of embryonic dopamine neurons into the brains of human PD patients developed α-syn pathology (Kordower et al., 2008; Li et al., 2008). The authors hypothesized that pathological α-syn from the host was transferred into the young grafted neurons in a prion-like manner (Brundin et al., 2008). Since then, different groups have proven that α-syn is able to be taken up by cells, including dopaminergic neurons (Luk et al., 2009; Volpicelli-Daley et al., 2011), be transmitted from neuron to neuron (Desplats et al., 2009; Freundt et al., 2012), from neurons to oligodendrocytes (Reyes et al., 2014), and to induce the formation of aggregates that share some of the LB features, i.e., a high degree of phosphorylation and polyubiquitination (Volpicelli-Daley et al., 2011). An early work demonstrated the presence of α-syn in plasma and cerebrospinal fluid of PD patients, thus supporting the hypothesis that α-syn may enter cells from the extracellular space (El-Agnaf et al., 2003). Moreover, the inoculation of recombinant α-syn preformed-fibrils (PFFs) or patient-derived pathological α-syn in wildtype and transgenic animals resulted in the development of widespread synucleinopathy throughout synaptically connected networks, neurodegeneration and behavioral deficits, following in some cases a similar pattern to the one define in Braak’s hypothesis (Luk et al., 2012a,b; Masuda-Suzukake et al., 2013; Ulusoy et al., 2013; Holmqvist et al., 2014; Recasens et al., 2014; Paumier et al., 2015; Abdelmotilib et al., 2017; Manfredsson et al., 2018; Rey et al., 2018; Chu et al., 2019; Kim S. et al., 2019; Challis et al., 2020). Additional studies demonstrated that the spreading of α-syn pathology depended on the misfolding and recruitment of endogenous α-syn (Volpicelli-Daley et al., 2011; Kaji et al., 2018; Kim S. et al., 2019; Wu et al., 2019). Furthermore, a recent publication suggested that the inoculation site may determine the synucleinopathy, since autonomic ganglionic injection of α-syn fibrils resulted in a model of PAF (Wang et al., 2020). However, the mechanism involved in α-syn internalization remains unclear. In the last years, several membrane protein receptors have been proposed to play an important role in α-syn uptake, including lymphocyte-activation gene 3 (Mao et al., 2016), Rab proteins (Masaracchia et al., 2018) and flotillin-1 and dopamine transporter (DAT; Kobayashi et al., 2019).
In addition, recent publications have demonstrated the existence of different strains of pathological α-syn which cause distinct synucleinopathies, targeting distinct brain regions and cell types (Bousset et al., 2013; Peelaerts et al., 2015; Rey et al., 2019; Lau et al., 2020). In particular, MSA and PD strains seem to have different structural and seed properties that may be used as differential diagnostic criteria and that could explain the more aggressive and rapid progression of MSA compared to other synucleinopathies (Peng et al., 2018b; Candelise et al., 2019; Yamasaki et al., 2019; Schweighauser et al., 2020; Shahnawaz et al., 2020; Van Der Perren et al., 2020). It is not yet clear how the different α-syn strains are generated, but the cellular micro-environment may play an important role (Candelise et al., 2020). Thus, a recent publication by the group of Virginia Lee showed that the α-syn extracted from GCIs presents a different proteolytic profile and more potent biological activity than the α-syn from LBs and that the specific cellular milieu of oligodendrocytes is responsible for the transformation of misfolded α-syn into the MSA strain (Peng et al., 2018a).
The identification of the mechanisms associated or responsible for α-syn uptake, processing, aggregation and release, and the contribution of different α-syn strains to inclusion formation and to the impairment of cellular pathways will be essential to completely understand the pathogenic process underlying α-synucleinopathies, to identify novel targets for disease modification, the development of new therapeutic strategies and to generate possible biomarkers which would allow the early diagnosis of these disorders.
α-Syn, a Prion Protein?
The term prion was first described by Stanley Prusiner as a “small proteinaceous infectious particle which is resistant to inactivation by most procedures that modify nucleic acids” and resulted from the blend of “protein” and “infection” (Prusiner, 1982). Therefore, prions should constitute the underlying cause of an infectious disease that can be transmitted among individuals from the same or different species, and not a secondary consequence of the disease (Prusiner, 1998). In this regard, based on in vivo and in vitro studies, α-syn seems to exhibit some of the properties of prion proteins (Figure 4), such as the ability to undergo misfolding and to self-propagate, inducing aggregation of endogenous α-syn (Hijaz and Volpicelli-Daley, 2020), to exist as distinct strains (Bousset et al., 2013; Peelaerts et al., 2015; Candelise et al., 2019; Yamasaki et al., 2019; Lau et al., 2020; Schweighauser et al., 2020; Shahnawaz et al., 2020) and to be resistant to inactivation by formaldehyde (Schweighauser et al., 2015; Woerman et al., 2018). As discussed before, the fact that α-syn aggregates were found in human brain regions affected by the disease and that some studies, like the one conducted by Braak et al. (2004) showed the presence of inclusions in peripheral organs and described an association between disease stage and Lewy pathology, led the authors to propose that α-synucleinopathies could initiate in the gut or the olfactory bulb and slowly spread to the CNS (Beach et al., 2009). This evidence and the data demonstrating the ability of α-syn to propagate from the gut and other peripheral organs in animal models (Kim S. et al., 2019; Lohmann et al., 2019) as well as the finding of LBs in grafted dopaminergic neurons in PD patients (Kordower et al., 2008; Li et al., 2008) supported the prion nature of α-syn.
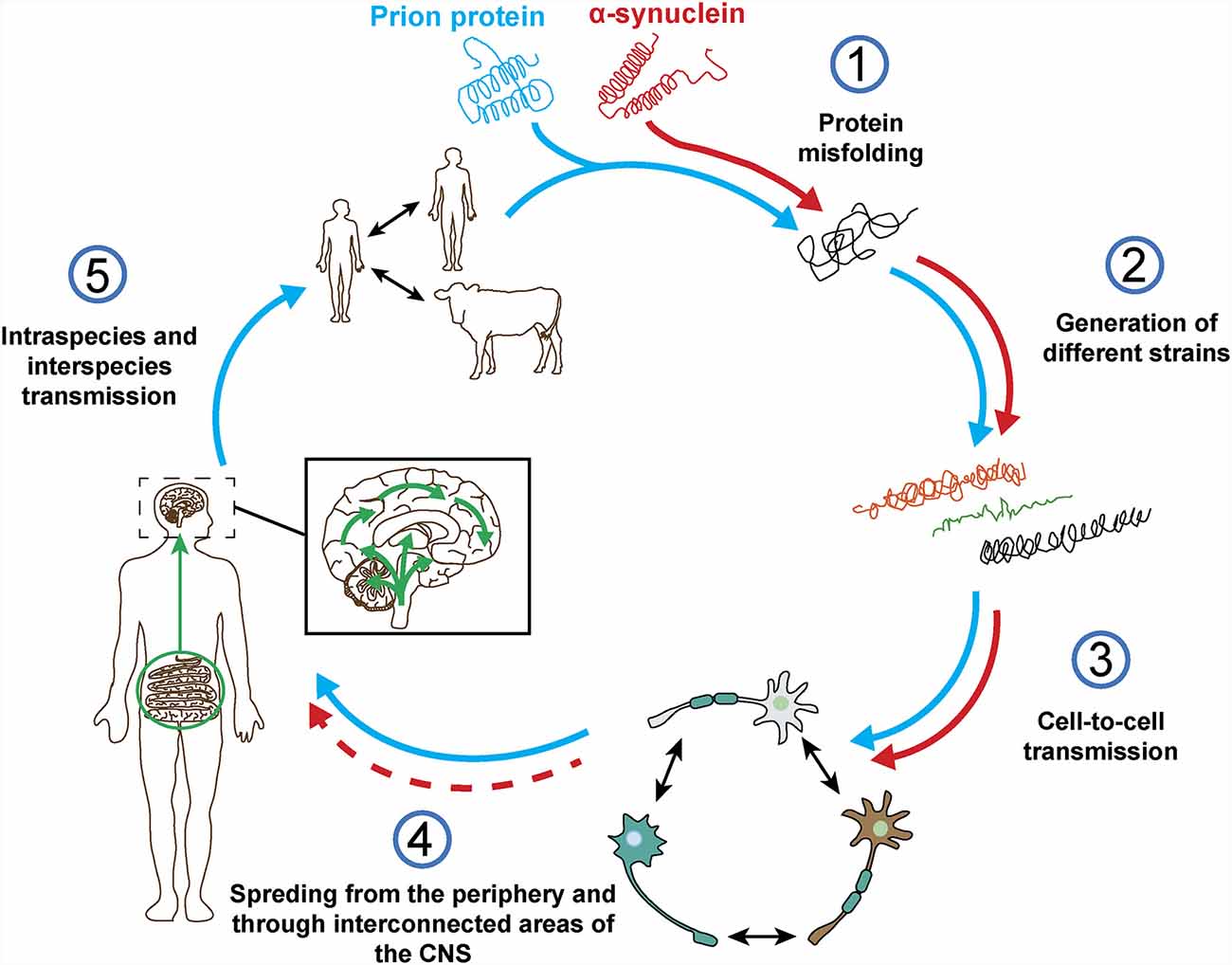
Figure 4. Prion protein vs. α-synuclein: current knowledge. Both proteins can undergo misfolding (1) which leads to the formation of different pathogenic strains (2). The pathogenic forms of both proteins can be transferred cell-to-cell (3) and spread from the periphery into the CNS in vivo (4). However, based on post-mortem data from a-synucleinopathy patients and in contrast to the prion protein, it is not clear how and where does α-synuclein pathology start and if the spreading observed in in vivo studies really happens in humans (4). Finally, unlike prion protein, no evidence of intraspecies or interpecies transmission of α-syn has ever been reported (5).
However, there is evidence that challenges some of these observations and the prion hypothesis for α-syn (Figure 4). On one hand, subsequent post-mortem studies showed that only half of PD cases present a pattern of Lewy pathology consistent with Braak’s staging and that an important number do not even follow it (Kalaitzakis et al., 2008; Jellinger, 2009; Halliday et al., 2012). In many cases, the distribution of α-syn inclusions is sparse and discrete, even at late stages of the disease (Beach et al., 2009; Dijkstra et al., 2014), and some patients do not show aggregates at all (Berg et al., 2014). Moreover, no correlation has been observed between Braak’s staging of Lewy pathology and severity of clinical progression (Jellinger, 2009). In addition, cells within the same region are not equally affected by Lewy pathology, which is only observed in a small percentage of neurons, mainly affecting catecholaminergic and cholinergic but not GABAergic neurons. Furthermore, the strength of connectivity between brain areas does not correlate with Lewy pathology patterns (Surmeier et al., 2017) and, in MSA, the trans-synaptic propagation of α-syn pathology proposed for PD cannot explain the spatial distribution of GCIs (Armstrong et al., 2004; Dhillon et al., 2019; Kaji et al., 2020).
The ability of α-syn to propagate and generate aggregates in humans is also a matter of debate since additional transplantation studies did not show α-syn accumulation in the engrafted dopaminergic cells (Mendez et al., 2008; Hallett et al., 2014). Interestingly, the main difference between these studies and the ones showing Lewy pathology was the use of dissociated neurons instead of cellular aggregates or tissue pieces. Moreover, independently of the absence or presence of α-syn aggregates, transplanted neurons generally survive and remain healthy and functional for decades (Li et al., 2016). The appearance of pathology in the engrafted cells has been, however, associated with an extensive microglial activation when cellular aggregates or tissue pieces are used (Kordower et al., 2008; Li et al., 2008). This effect was not observed when dissociated neurons were transplanted (Mendez et al., 2008). Nevertheless, it is still unclear from where was the pathology observed in transplanted cells coming from, and why the number of neurons affected did not change with time (Kordower et al., 2008; Li et al., 2008, 2016). It has been proposed that the graft’s micro-environment together with the already stressed environment from the host may trigger the pathology in those cells (Surmeier et al., 2017). In fact, post-mortem studies have shown that brain regions presenting α-syn inclusions do not necessarily develop neuronal loss, in contrast, neurodegeneration sometimes occurs in areas with no α-syn pathology or before it appears (Leak et al., 2019). Furthermore, in vitro studies have shown that mutations in α-syn, such as A53E and G51D, which are associated with familiar forms of PD and MSA, attenuate aggregation and delay the pathology (Fares et al., 2014; Ghosh et al., 2014). Interestingly, different neuropathological reports have shown an absence of Lewy pathology in most patients with familiar forms of parkin-related PD and in a proportion of PD patients with mutations in the LRRK2 gene, which are respectively the most common cause of recessive and dominant forms of PD, and a small proportion of sporadic PD cases (Kalia and Lang, 2015; Tolosa et al., 2020).
Bonafide prions can spread from extraneural sites into the CNS and the molecular processes underlying their spreading have been evaluated in detail (Kara et al., 2018). Regarding α-syn, recent studies have demonstrated the presence of inclusions in peripheral sites, including cells of the enteric nervous system, nerve cells and fibers in the skin and submandibular salivary glands of α-synucleinopathies patients (Beach et al., 2010, 2016; Shannon et al., 2012; Kim J. Y. et al., 2019). Therefore, these data support the hypothesis that α-syn pathology may start in the periphery and ascend towards the CNS in a similar way to prions. In addition, it was reported that vagotomy reduces the risk of developing PD (Svensson et al., 2015), that may be explained by a disruption of α-syn spreading from the periphery to the brain. However, these observations were not replicated in a different study with the same cohort but extended follow-up (Tysnes et al., 2015). Therefore, the debate on the periphery as the possible initiation site of α-syn pathology remains unsolved, especially considering that: (i) α-syn is mainly expressed in the brain (Lashuel et al., 2013); (ii) some peripheral regions with α-syn pathology do not present a clear exposure route, such as the cardiac sympathetic nerve (Orimo et al., 2008); (iii) no cases have been found in post-mortem whole body studies with α-syn inclusions in the periphery without the brain being affected (Lionnet et al., 2018); and (iv) peripheral α-syn aggregates from PD patients show lack of pathogenic potential (Recasens et al., 2018; Figure 4).
Other exogenous and endogenous factors may trigger the pathological changes underlying α-synucleinopathies and α-syn aggregation (Figure 3). Thus, gut dysbiosis and microbiota have been associated with PD (Fitzgerald et al., 2019). Moreover, recent studies demonstrated that amyloid proteins produced by gut bacteria can induce α-syn aggregation and seeding (Sampson et al., 2016, 2020), and that viral infection disrupts cellular proteostasis and causes α-syn aggregation (Marreiros et al., 2020). Several authors have therefore suggested that neurodegeneration and the neurological deficits observed in α-synucleinopathies may be triggered by factors other than α-syn, like inflammation or local micro environmental conditions (Engelender and Isacson, 2017) and that selective vulnerability of neuronal subpopulations may better explain the generation of α-syn inclusions throughout disease progression, rather than prion-like transmission (Surmeier et al., 2017). In this regard, a protective role of α-syn aggregates has been suggested, where they may be a consequence of the disease process rather than the cause (Olanow et al., 2004; Sian-Hulsmann et al., 2015). Based on these observations, and the fact that severity of clinical symptoms correlates with neurodegeneration rather than with α-syn inclusions in humans (Beach et al., 2009), the role of α-syn in these pathologies remains unclear.
It has to be mention that, to date, despite longitudinal studies with α-synucleinopathies patients have demonstrated clinical and biological changes throughout disease progression, they have not been able to show meaningful concordance between them, making difficult to properly define disease stages and, therefore, to evaluate the association between α-syn pathology and disease progression (Espay et al., 2020). In addition to that, the absence of significant correlation between α-syn pathology and cell loss in brain areas affected in these disorders could be due to a lack of sensitivity of current immunohistochemical methods. Further studies evaluating the presence of α-syn oligomers or early aggregation stages by novel techniques such as AS-PLA, RT-QuIC or PMCA may lead to a better understanding of the role of α-syn in α-synucleinopathies and clarify the possible pathological routes of α-syn propagation (Bengoa-Vergniory et al., 2017; Shahnawaz et al., 2020).
Finally, no evidences of α-syn transmission between humans has ever been reported, including iatrogenic (Irwin et al., 2013; Beekes et al., 2014), and there is no epidemiological data supporting this possibility for any α-synucleinopathy (Rajput et al., 2016; Figure 4). No evidences of transmission from animal to human have been found either (Wenning et al., 2018). Historically, prion infections have been a rarity whereas α-synucleinopathies present an important incidence that remains relatively stable over time in various ethnical populations (Pringsheim et al., 2014; Leak et al., 2019), however, in vivo studies show that the level of infectivity of α-syn aggregates per amount of protein might be orders of magnitude lower than that of prions (Watts, 2019).
Overall, despite α-syn shares several characteristics with prions, the absence of infectivity, the lack of human studies supporting the hypothesis and the existence of contradictory data must be taken in consideration. Thus, the term “prionoid” has been proposed to designate proteins that share propagative mechanisms with prions but may not be transmissible between individuals (Kara et al., 2018). At present, the most probable explanation regarding the start and progression of α-synucleionopathies would be that they are the consequence of a multifactorial process. This process would include the occurrence of exogenous and cellular micro-environmental changes in the affected regions that may trigger and modulate the pathology, and the presence of different α-syn strains that are able to spread through the organism in a cell type/susceptibility-dependent manner leading to different neuroanatomical spreading patterns.
Futures Perspectives in α-Syn Research and Therapy
Although our knowledge about α-syn, its physiological functions, structure, properties and its pathological role in α-synucleinopathies have been extensively studied, there are still gaps that must be filled to completely understand the nature and the importance of this protein.
The characterization of α-syn interactions in the different subcellular compartments, not only in the CNS, but also in peripheral organs may be essential to fully understand its functions, the cellular and systemic changes that underlie α-synucleinopathies, and the possible deleterious consequences of therapeutic strategies targeting α-syn. The combination of novel super-resolution imaging, proteomics and computational techniques constitute a powerful approach to define α-syn interactome (Brás et al., 2020). A complete and detailed identification of all α-syn PTM, the different pathways involved in its clearance and the role of glial cells in this process represent other important targets for further studies (Lopes Da Fonseca et al., 2015; Gonzalez et al., 2019; Tremblay et al., 2019). Regarding the pathological role of α-syn, the absence of specific biomarkers and PET tracers constitute, at present, one of the most important disadvantages to identify its spatial and temporal contribution to disease progression in α-synucleinopathy patients. In addition, the biochemical composition and the molecular architecture of α-syn strains and inclusions and the cellular and environmental factors modulating their formation in the different disorders have not yet been completely revealed, mainly due to the lack of proper and highly sensitive protocols and tools. Elucidating the native conformation of α-syn and the different pathogenic strains by cryo-electron microscopy (Chen S. W. et al., 2015; Li et al., 2018; Guerrero-Ferreira et al., 2019), nuclear magnetic resonance (Galvagnion et al., 2019) or super-resolution imaging techniques (Nugent et al., 2017; Shahmoradian et al., 2019) will be essential to develop alternative drug-based disease modifying therapies. The establishment of brain banks of high-quality post-mortem brain tissue, CSF, plasma and fibroblast from clinically and pathologically well-defined patients and healthy controls will improve the potential of future studies. Moreover, the incorporation of induced-pluripotent stem cells (iPSCs) to those banks, to develop human patient-specific in vitro models of the disease, should be considered. Finally, further genetic and epigenetic studies will also provide important insights into the mechanism underlying α-synucleinopathies, the differences between them, as well as novel therapeutic targets.
Though the exact role of α-syn in the pathology has not yet been fully elucidated, an important number of preclinical studies and clinical trials have been focused on targeting directly α-syn and α-syn aggregation, or indirectly through the modulation of processes associated with its accumulation. The use of small molecules to target α-syn aggregation have shown positive results in preclinical models of α-synucleionopathies by reducing aggregation and accumulation of α-syn inclusions, neurodegeneration and motor symptoms (Finkelstein et al., 2017; Price et al., 2018; Heras-Garvin et al., 2019; Wegrzynowicz et al., 2019). Antibiotics such as rifampicin also reduces α-syn aggregation and fibrillization (Li et al., 2004) and showed positive results in animal models (Ubhi et al., 2008). Different preclinical studies have demonstrated that α-syn immunotherapy enhances α-syn clearance, prevents spreading and neuronal loss and demyelination and improves motor function in animal models (Masliah et al., 2011; Mandler et al., 2014, 2015). Enhancing α-syn degradation by modulating cellular pathways and the activation of glial cell types associated with α-syn clearance constitute another promising strategy (Malagelada et al., 2010; Gao et al., 2019; Perrino et al., 2019). Stimulation of microglial-dependent clearance of α-syn reduces the number of inclusions, ameliorates motor symptoms and shows a neuroprotective effect in in vivo models of MSA and PD (Park et al., 2016; Venezia et al., 2017; Choi et al., 2020). Intriguingly, microglial activation and neuroinflammation can also promote α-syn aggregation and induce neurodegeneration, accelerating disease progression (Lema Tome et al., 2013), therefore, reduction of microglial activation have also been used for disease modification (Liu et al., 2019). Recently, the use of antisense nucleotides (ASO) to selectively inhibit proteins associated with α-syn pathology in specific brain regions or cell types has been proposed as an alternative approach to reduce α-syn accumulation in PD models (Zhao et al., 2017; Alarcón-Arís et al., 2018). This is particularly important since altering or removing α-syn gene expression in previous studies has shown profound and deleterious intracellular and developmental effects that in some cases could induce neuronal loss (Perez and Hastings, 2004; Benskey et al., 2016).
Most of these therapeutic strategies are currently under evaluation in human clinical trials (Heras-Garvin and Stefanova, 2020). Unfortunately, some of them have not shown ability to slow or halt disease progression (Heras-Garvin and Stefanova, 2020). The distinct efficacies observed between preclinical studies, based on in vitro and in vivo models, and the data from human studies could be explained by the neurobiological differences among species, but may be a consequence of current limitations to recruit patients at early stages of the disease, when significant neurodegeneration and motor symptoms are not yet present, to misdiagnosis and to the absence of specific biomarkers for α-syn.
Conclusions
It has been a long journey since α-syn was discovered 30 years ago. Many in vitro and in vivo studies have contributed to a better understanding of this versatile and multifaceted protein, its functions and its potential role in neurodegeneration. Moreover, the analysis of human material has indeed supported the idea that α-syn may contribute in some extent to the pathogenic events underlying the α-synucleionopathies. However, whether α-syn is the cause or main force driving these disorders or a mere secondary event is still a matter of debate. The use of novel highly sensitive tools and techniques in future studies, combined with the recruitment and collection of human material from well-characterized and documented cohorts, will definitely help us to finally define the pathogenic role of α-syn and to improve the development of proper therapeutic strategies.
Author Contributions
AH-G: conception and design, drafting and revising the manuscript, and final approval of the submission. NS: conception and design, revising the manuscript, and final approval of the submission. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a grant of the Austrian Science Funds (FWF) F4414 and the grant “Fit for Science” of the Tyrol County.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abdelmotilib, H., Maltbie, T., Delic, V., Liu, Z., Hu, X., Fraser, K. B., et al. (2017). α-Synuclein fibril-induced inclusion spread in rats and mice correlates with dopaminergic Neurodegeneration. Neurobiol. Dis. 105, 84–98. doi: 10.1016/j.nbd.2017.05.014
Alam, P., Bousset, L., Melki, R., and Otzen, D. E. (2019). α-synuclein oligomers and fibrils: a spectrum of species, a spectrum of toxicities. J. Neurochem. 150, 522–534. doi: 10.1111/jnc.14808
Alarcón-Arís, D., Recasens, A., Galofre, M., Carballo-Carbajal, I., Zacchi, N., Ruiz-Bronchal, E., et al. (2018). Selective α-synuclein knockdown in monoamine neurons by intranasal oligonucleotide delivery: potential therapy for Parkinson’s disease. Mol. Ther. 26, 550–567. doi: 10.1016/j.ymthe.2017.11.015
Almandoz-Gil, L., Persson, E., Lindstrom, V., Ingelsson, M., Erlandsson, A., and Bergstrom, J. (2018). In situ proximity ligation assay reveals co-localization of α-synuclein and SNARE proteins in murine primary neurons. Front. Neurol. 9:180. doi: 10.3389/fneur.2018.00180
Al-Wandi, A., Ninkina, N., Millership, S., Williamson, S. J., Jones, P. A., and Buchman, V. L. (2010). Absence of α-synuclein affects dopamine metabolism and synaptic markers in the striatum of aging mice. Neurobiol. Aging 31, 796–804. doi: 10.1016/j.neurobiolaging.2008.11.001
Angelova, P. R., Ludtmann, M. H., Horrocks, M. H., Negoda, A., Cremades, N., Klenerman, D., et al. (2016). Ca2+ is a key factor in α-synuclein-induced neurotoxicity. J. Cell Sci. 129, 1792–1801. doi: 10.1242/jcs.180737
Arai, K., Kato, N., Kashiwado, K., and Hattori, T. (2000). Pure autonomic failure in association with human α-synucleinopathy. Neurosci. Lett. 296, 171–173. doi: 10.1016/s0304-3940(00)01623-2
Arawaka, S., Sato, H., Sasaki, A., Koyama, S., and Kato, T. (2017). Mechanisms underlying extensive Ser129-phosphorylation in α-synuclein aggregates. Acta Neuropathol. Commun. 5:48. doi: 10.1186/s40478-017-0452-6
Armstrong, R. A., Lantos, P. L., and Cairns, N. J. (2004). Spatial patterns of α-synuclein positive glial cytoplasmic inclusions in multiple system atrophy. Mov. Disord. 19, 109–112. doi: 10.1002/mds.10637
Austin, S. A., Floden, A. M., Murphy, E. J., and Combs, C. K. (2006). α-synuclein expression modulates microglial activation phenotype. J. Neurosci. 26, 10558–10563. doi: 10.1523/JNEUROSCI.1799-06.2006
Barber, T. R., Lawton, M., Rolinski, M., Evetts, S., Baig, F., Ruffmann, C., et al. (2017). Prodromal Parkinsonism and neurodegenerative risk stratification in REM sleep behavior disorder. Sleep 40:zsx071. doi: 10.1093/sleep/zsx071
Barbour, R., Kling, K., Anderson, J. P., Banducci, K., Cole, T., Diep, L., et al. (2008). Red blood cells are the major source of α-synuclein in blood. Neurodegener. Dis. 5, 55–59. doi: 10.1159/000112832
Bartels, T., Choi, J. G., and Selkoe, D. J. (2011). α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477, 107–110. doi: 10.1038/nature10324
Bassil, F., Brown, H. J., Pattabhiraman, S., Iwasyk, J. E., Maghames, C. M., Meymand, E. S., et al. (2020). Amyloid-β (Aβ) plaques promote seeding and spreading of α-synuclein and tau in a mouse model of Lewy body disorders with Aβ pathology. Neuron 105, 260.e6–275.e6. doi: 10.1016/j.neuron.2019.10.010
Beach, T. G., Adler, C. H., Lue, L., Sue, L. I., Bachalakuri, J., Henry-Watson, J., et al. (2009). Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol. 117, 613–634. doi: 10.1007/s00401-009-0538-8
Beach, T. G., Adler, C. H., Serrano, G., Sue, L. I., Walker, D. G., Dugger, B. N., et al. (2016). Prevalence of submandibular gland synucleinopathy in Parkinson’s disease, dementia with Lewy bodies and other Lewy body disorders. J. Parkinsons Dis. 6, 153–163. doi: 10.3233/JPD-150680
Beach, T. G., Adler, C. H., Sue, L. I., Vedders, L., Lue, L., White Iii, C. L., et al. (2010). Multi-organ distribution of phosphorylated α-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 119, 689–702. doi: 10.1007/s00401-010-0664-3
Beekes, M., Thomzig, A., Schulz-Schaeffer, W. J., and Burger, R. (2014). Is there a risk of prion-like disease transmission by Alzheimer- or Parkinson-associated protein particles? Acta Neuropathol. 128, 463–476. doi: 10.1007/s00401-014-1324-9
Bendor, J. T., Logan, T. P., and Edwards, R. H. (2013). The function of α-synuclein. Neuron 79, 1044–1066. doi: 10.1016/j.neuron.2013.09.004
Ben Gedalya, T., Loeb, V., Israeli, E., Altschuler, Y., Selkoe, D. J., and Sharon, R. (2009). α-synuclein and polyunsaturated fatty acids promote clathrin-mediated endocytosis and synaptic vesicle recycling. Traffic 10, 218–234. doi: 10.1111/j.1600-0854.2008.00853.x
Bengoa-Vergniory, N., Roberts, R. F., Wade-Martins, R., and Alegre-Abarrategui, J. (2017). α-synuclein oligomers: a new hope. Acta Neuropathol. 134, 819–838. doi: 10.1007/s00401-017-1755-1
Benskey, M. J., Perez, R. G., and Manfredsson, F. P. (2016). The contribution of α-synuclein to neuronal survival and function—implications for Parkinson’s disease. J. Neurochem. 137, 331–359. doi: 10.1111/jnc.13570
Berg, D., Postuma, R. B., Bloem, B., Chan, P., Dubois, B., Gasser, T., et al. (2014). Time to redefine PD? Introductory statement of the MDS Task Force on the definition of Parkinson’s disease. Mov. Disord. 29, 454–462. doi: 10.1002/mds.25844
Binolfi, A., Theillet, F. X., and Selenko, P. (2012). Bacterial in-cell NMR of human α-synuclein: a disordered monomer by nature? Biochem. Soc. Trans. 40, 950–954. doi: 10.1042/bst20120096
Bousset, L., Pieri, L., Ruiz-Arlandis, G., Gath, J., Jensen, P. H., Habenstein, B., et al. (2013). Structural and functional characterization of two α-synuclein strains. Nat. Commun. 4:2575. doi: 10.3990/1.9789036529310
Braak, H., Del Tredici, K., Rub, U., De Vos, R. A., Jansen Steur, E. N., and Braak, E. (2003a). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211. doi: 10.1016/s0197-4580(02)00065-9
Braak, H., Rüb, U., Gai, W. P., and Del Tredici, K. (2003b). Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Transm. 110, 517–536. doi: 10.1007/s00702-002-0808-2
Braak, H., De Vos, R. A., Bohl, J., and Del Tredici, K. (2006). Gastric α-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci. Lett. 396, 67–72. doi: 10.1016/j.neulet.2005.11.012
Braak, H., Ghebremedhin, E., Rub, U., Bratzke, H., and Del Tredici, K. (2004). Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 318, 121–134. doi: 10.1007/s00441-004-0956-9
Braak, H., Sastre, M., Bohl, J. R., De Vos, R. A., and Del Tredici, K. (2007). Parkinson’s disease: lesions in dorsal horn layer I, involvement of parasympathetic and sympathetic pre- and postganglionic neurons. Acta Neuropathol. 113, 421–429. doi: 10.1007/s00401-007-0193-x
Brás, I. C., Dominguez-Meijide, A., Gerhardt, E., Koss, D., Lazaro, D. F., Santos, P. I., et al. (2020). Synucleinopathies: where we are and where we need to go. J. Neurochem. 153, 433–454. doi: 10.1111/jnc.14965
Bridi, J. C., and Hirth, F. (2018). Mechanisms of α-synuclein induced synaptopathy in Parkinson’s disease. Front. Neurosci. 12:80. doi: 10.3389/fnins.2018.00080
Brochard, V., Combadière, B., Prigent, A., Laouar, Y., Perrin, A., Beray-Berthat, V., et al. (2009). Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J. Clin. Invest. 119, 182–192. doi: 10.1172/JCI36470
Brundin, P., Li, J. Y., Holton, J. L., Lindvall, O., and Revesz, T. (2008). Research in motion: the enigma of Parkinson’s disease pathology spread. Nat. Rev. Neurosci. 9, 741–745. doi: 10.1038/nrn2477
Buell, A. K., Galvagnion, C., Gaspar, R., Sparr, E., Vendruscolo, M., Knowles, T. P., et al. (2014). Solution conditions determine the relative importance of nucleation and growth processes in α-synuclein aggregation. Proc. Natl. Acad. Sci. U S A 111, 7671–7676. doi: 10.1073/pnas.1315346111
Burré, J., Sharma, M., and Sudhof, T. C. (2018). Cell biology and pathophysiology of α-synuclein. Cold Spring Harb. Perspect. Med. 8:a024091. doi: 10.1101/cshperspect.a024091
Burré, J., Sharma, M., Tsetsenis, T., Buchman, V., Etherton, M. R., and Sudhof, T. C. (2010). α-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667. doi: 10.1126/science.1195227
Burré, J., Vivona, S., Diao, J., Sharma, M., Brunger, A. T., and Sudhof, T. C. (2013). Properties of native brain α-synuclein. Nature 498, E4–6; discussion E6–E7. doi: 10.1038/nature12125
Butkovich, L. M., Houser, M. C., and Tansey, M. G. (2018). α-synuclein and noradrenergic modulation of immune cells in Parkinson’s disease pathogenesis. Front. Neurosci. 12:626. doi: 10.3389/fnins.2018.00626
Candelise, N., Schmitz, M., Llorens, F., Villar-Pique, A., Cramm, M., Thom, T., et al. (2019). Seeding variability of different α-synuclein strains in synucleinopathies. Ann. Neurol. 85, 691–703. doi: 10.1002/ana.25446
Candelise, N., Schmitz, M., Thune, K., Cramm, M., Rabano, A., Zafar, S., et al. (2020). Effect of the micro-environment on α-synuclein conversion and implication in seeded conversion assays. Transl. Neurodegener. 9:5. doi: 10.1186/s40035-019-0181-9
Cappai, R., Leck, S. L., Tew, D. J., Williamson, N. A., Smith, D. P., Galatis, D., et al. (2005). Dopamine promotes α-synuclein aggregation into SDS-resistant soluble oligomers via a distinct folding pathway. FASEB J. 19, 1377–1379. doi: 10.1096/fj.04-3437fje
Challis, C., Hori, A., Sampson, T. R., Yoo, B. B., Challis, R. C., Hamilton, A. M., et al. (2020). Gut-seeded α-synuclein fibrils promote gut dysfunction and brain pathology specifically in aged mice. Nat. Neurosci. 23, 327–336. doi: 10.1038/s41593-020-0589-7
Chandra, S., Chen, X., Rizo, J., Jahn, R., and Sudhof, T. C. (2003). A broken α-helix in folded α-synuclein. J. Biol. Chem. 278, 15313–15318. doi: 10.1074/jbc.M213128200
Chen, S. W., Drakulic, S., Deas, E., Ouberai, M., Aprile, F. A., Arranz, R., et al. (2015). Structural characterization of toxic oligomers that are kinetically trapped during α-synuclein fibril formation. Proc. Natl. Acad. Sci. U S A 112, E1994–E2003. doi: 10.1073/pnas.1421204112
Chen, Y., Yang, W., Li, X., Li, X., Yang, H., Xu, Z., et al. (2015). α-Synuclein-induced internalization of NMDA receptors in hippocampal neurons is associated with reduced inward current and Ca(2+) influx upon NMDA stimulation. Neuroscience 300, 297–306. doi: 10.1016/j.neuroscience.2015.05.035
Cheng, H. C., Ulane, C. M., and Burke, R. E. (2010). Clinical progression in Parkinson disease and the neurobiology of axons. Ann. Neurol. 67, 715–725. doi: 10.1002/ana.21995
Choi, I., Zhang, Y., Seegobin, S. P., Pruvost, M., Wang, Q., Purtell, K., et al. (2020). Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat. Commun. 11:1386. doi: 10.1038/s41467-020-15119-w
Chu, Y., Muller, S., Tavares, A., Barret, O., Alagille, D., Seibyl, J., et al. (2019). Intrastriatal α-synuclein fibrils in monkeys: spreading, imaging and neuropathological changes. Brain 142, 3565–3579. doi: 10.1093/brain/awz296
Clinton, L. K., Blurton-Jones, M., Myczek, K., Trojanowski, J. Q., and Laferla, F. M. (2010). Synergistic interactions between Aβ, τ, and α-synuclein: acceleration of neuropathology and cognitive decline. J. Neurosci. 30, 7281–7289. doi: 10.1523/jneurosci.0490-10.2010
Coon, E. A., Cutsforth-Gregory, J. K., and Benarroch, E. E. (2018). Neuropathology of autonomic dysfunction in synucleinopathies. Mov. Disord. 33, 349–358. doi: 10.1002/mds.27186
Cremades, N., and Dobson, C. M. (2018). The contribution of biophysical and structural studies of protein self-assembly to the design of therapeutic strategies for amyloid diseases. Neurobiol. Dis. 109, 178–190. doi: 10.1016/j.nbd.2017.07.009
Croisier, E., Moran, L. B., Dexter, D. T., Pearce, R. K., and Graeber, M. B. (2005). Microglial inflammation in the parkinsonian substantia nigra: relationship to α-synuclein deposition. J Neuroinflammation 2:14. doi: 10.1186/1742-2094-2-14
Danzer, K. M., Haasen, D., Karow, A. R., Moussaud, S., Habeck, M., Giese, A., et al. (2007). Different species of α-synuclein oligomers induce calcium influx and seeding. J. Neurosci. 27, 9220–9232. doi: 10.1523/jneurosci.2617-07.2007
Davidi, D., Schechter, M., Elhadi, S. A., Matatov, A., Nathanson, L., and Sharon, R. (2020). α-synuclein translocates to the nucleus to activate retinoic-acid-dependent gene transcription. iScience 23:100910. doi: 10.2139/ssrn.3480698
De Oliveira, G. A. P., and Silva, J. L. (2019). α-synuclein stepwise aggregation reveals features of an early onset mutation in Parkinson’s disease. Commun. Biol. 2:374. doi: 10.1038/s42003-019-0598-9
Decressac, M., Kadkhodaei, B., Mattsson, B., Laguna, A., Perlmann, T., and Bjorklund, A. (2012). α-Synuclein-induced down-regulation of Nurr1 disrupts GDNF signaling in nigral dopamine neurons. Sci. Transl. Med. 4:163ra156. doi: 10.1126/scitranslmed.3004676
Desplats, P., Lee, H. J., Bae, E. J., Patrick, C., Rockenstein, E., Crews, L., et al. (2009). Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc. Natl. Acad. Sci. U S A 106, 13010–13015. doi: 10.1073/pnas.0903691106
Devi, L., Raghavendran, V., Prabhu, B. M., Avadhani, N. G., and Anandatheerthavarada, H. K. (2008). Mitochondrial import and accumulation of α-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 283, 9089–9100. doi: 10.1074/jbc.m710012200
Dhillon, J. S., Trejo-Lopez, J. A., Riffe, C., Mcfarland, N. R., Hiser, W. M., Giasson, B. I., et al. (2019). Dissecting α-synuclein inclusion pathology diversity in multiple system atrophy: implications for the prion-like transmission hypothesis. Lab. Invest. 99, 982–992. doi: 10.1038/s41374-019-0198-9
Dickson, D. W., Braak, H., Duda, J. E., Duyckaerts, C., Gasser, T., Halliday, G. M., et al. (2009). Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. Lancet Neurol. 8, 1150–1157. doi: 10.1016/S1474-4422(09)70238-8
Dijkstra, A. A., Voorn, P., Berendse, H. W., Groenewegen, H. J., Netherlands Brain, B., Rozemuller, A. J., et al. (2014). Stage-dependent nigral neuronal loss in incidental Lewy body and Parkinson’s disease. Mov. Disord. 29, 1244–1251. doi: 10.1002/mds.25952
Di Maio, R., Barrett, P. J., Hoffman, E. K., Barrett, C. W., Zharikov, A., Borah, A., et al. (2016). α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 8:342ra378. doi: 10.1126/scitranslmed.aaf3634
Don, A. S., Hsiao, J. H., Bleasel, J. M., Couttas, T. A., Halliday, G. M., and Kim, W. S. (2014). Altered lipid levels provide evidence for myelin dysfunction in multiple system atrophy. Acta Neuropathol. Commun. 2:150. doi: 10.1186/s40478-014-0150-6
Dong, C., Hoffmann, M., Li, X., Wang, M., Garen, C. R., Petersen, N. O., et al. (2018). Structural characteristics and membrane interactions of tandem α-synuclein oligomers. Sci. Rep. 8:6755. doi: 10.1038/s41598-018-25133-0
Doorn, K. J., Moors, T., Drukarch, B., Van De Berg, W., Lucassen, P. J., and Van Dam, A. M. (2014). Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson’s disease patients. Acta Neuropathol. Commun. 2:90. doi: 10.1186/s40478-014-0090-1
Durante, V., De Iure, A., Loffredo, V., Vaikath, N., De Risi, M., Paciotti, S., et al. (2019). α-synuclein targets GluN2A NMDA receptor subunit causing striatal synaptic dysfunction and visuospatial memory alteration. Brain 142, 1365–1385. doi: 10.1093/brain/awz065
El-Agnaf, O. M., Salem, S. A., Paleologou, K. E., Cooper, L. J., Fullwood, N. J., Gibson, M. J., et al. (2003). α-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J. 17, 1945–1947. doi: 10.1096/fj.03-0098fje
Emmanouilidou, E., Stefanis, L., and Vekrellis, K. (2010). Cell-produced α-synuclein oligomers are targeted to and impair, the 26S proteasome. Neurobiol. Aging 31, 953–968. doi: 10.1016/j.neurobiolaging.2008.07.008
Engelender, S., and Isacson, O. (2017). The threshold theory for Parkinson’s disease. Trends Neurosci. 40, 4–14. doi: 10.1016/j.tins.2016.10.008
Eschbach, J., Von Einem, B., Müller, K., Bayer, H., Scheffold, A., Morrison, B. E., et al. (2015). Mutual exacerbation of peroxisome proliferator-activated receptor γ coactivator 1α deregulation and α-synuclein oligomerization. Ann. Neurol. 77, 15–32. doi: 10.1002/ana.24294
Espay, A. J., Kalia, L. V., Gan-Or, Z., Williams-Gray, C. H., Bedard, P. L., Rowe, S. M., et al. (2020). Disease modification and biomarker development in Parkinson disease: revision or reconstruction? Neurology 94, 481–494. doi: 10.1212/WNL.0000000000009107
Ettle, B., Kerman, B. E., Valera, E., Gillmann, C., Schlachetzki, J. C., Reiprich, S., et al. (2016). α-Synuclein-induced myelination deficit defines a novel interventional target for multiple system atrophy. Acta Neuropathol. 132, 59–75. doi: 10.1007/s00401-016-1572-y
Ettle, B., Reiprich, S., Deusser, J., Schlachetzki, J. C., Xiang, W., Prots, I., et al. (2014). Intracellular α-synuclein affects early maturation of primary oligodendrocyte progenitor cells. Mol. Cell. Neurosci. 62, 68–78. doi: 10.1016/j.mcn.2014.06.012
Fanciulli, A., and Wenning, G. K. (2015). Multiple-system atrophy. N. Engl. J. Med. 372, 249–263. doi: 10.1056/NEJMra1311488
Fares, M. B., Ait-Bouziad, N., Dikiy, I., Mbefo, M. K., Jovicic, A., Kiely, A., et al. (2014). The novel Parkinson’s disease linked mutation G51D attenuates in vitro aggregation and membrane binding of α-synuclein and enhances its secretion and nuclear localization in cells. Hum. Mol. Genet. 23, 4491–4509. doi: 10.1093/hmg/ddu165
Fauvet, B., Mbefo, M. K., Fares, M. B., Desobry, C., Michael, S., Ardah, M. T., et al. (2012). α-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J. Biol. Chem. 287, 15345–15364. doi: 10.1074/jbc.M111.318949
Fecchio, C., Palazzi, L., and De Laureto, P. P. (2018). α-synuclein and polyunsaturated fatty acids: molecular basis of the interaction and implication in neurodegeneration. Molecules 23:1531. doi: 10.3390/molecules23071531
Fellner, L., Irschick, R., Schanda, K., Reindl, M., Klimaschewski, L., Poewe, W., et al. (2013). Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia 61, 349–360. doi: 10.1002/glia.22437
Finkelstein, D. I., Billings, J. L., Adlard, P. A., Ayton, S., Sedjahtera, A., Masters, C. L., et al. (2017). The novel compound PBT434 prevents iron mediated neurodegeneration and α-synuclein toxicity in multiple models of Parkinson’s disease. Acta Neuropathol. Commun. 5:53. doi: 10.1186/s40478-017-0456-2
Fitzgerald, E., Murphy, S., and Martinson, H. A. (2019). α-synuclein pathology and the role of the microbiota in Parkinson’s disease. Front. Neurosci. 13:369. doi: 10.3389/fnins.2019.00369
Flavin, W. P., Bousset, L., Green, Z. C., Chu, Y., Skarpathiotis, S., Chaney, M. J., et al. (2017). Endocytic vesicle rupture is a conserved mechanism of cellular invasion by amyloid proteins. Acta Neuropathol. 134, 629–653. doi: 10.1007/s00401-017-1722-x
Freundt, E. C., Maynard, N., Clancy, E. K., Roy, S., Bousset, L., Sourigues, Y., et al. (2012). Neuron-to-neuron transmission of α-synuclein fibrils through axonal transport. Ann. Neurol. 72, 517–524. doi: 10.1002/ana.23747
Froula, J. M., Castellana-Cruz, M., Anabtawi, N. M., Camino, J. D., Chen, S. W., Thrasher, D. R., et al. (2019). Defining α-synuclein species responsible for Parkinson’s disease phenotypes in mice. J. Biol. Chem. 294, 10392–10406. doi: 10.1074/jbc.RA119.007743
Froula, J. M., Henderson, B. W., Gonzalez, J. C., Vaden, J. H., Mclean, J. W., Wu, Y., et al. (2018). α-Synuclein fibril-induced paradoxical structural and functional defects in hippocampal neurons. Acta Neuropathol. Commun. 6:35. doi: 10.1186/s40478-018-0537-x
Fusco, G., Chen, S. W., Williamson, P. T. F., Cascella, R., Perni, M., Jarvis, J. A., et al. (2017). Structural basis of membrane disruption and cellular toxicity by α-synuclein oligomers. Science 358, 1440–1443. doi: 10.1126/science.aan6160
Galvagnion, C., Topgaard, D., Makasewicz, K., Buell, A. K., Linse, S., Sparr, E., et al. (2019). Lipid dynamics and phase transition within α-synuclein amyloid fibrils. J. Phys. Chem. Lett. 10, 7872–7877. doi: 10.1021/acs.jpclett.9b03005
Ganjam, G. K., Bolte, K., Matschke, L. A., Neitemeier, S., Dolga, A. M., Hollerhage, M., et al. (2019). Mitochondrial damage by α-synuclein causes cell death in human dopaminergic neurons. Cell Death Dis. 10:865. doi: 10.1038/s41419-019-2091-2
Gao, J., Perera, G., Bhadbhade, M., Halliday, G. M., and Dzamko, N. (2019). Autophagy activation promotes clearance of α-synuclein inclusions in fibril-seeded human neural cells. J. Biol. Chem. 294, 14241–14256. doi: 10.1074/jbc.ra119.008733
Garretti, F., Agalliu, D., Lindestam Arlehamn, C. S., Sette, A., and Sulzer, D. (2019). Autoimmunity in Parkinson’s disease: the role of α-synuclein-specific T cells. Front. Immunol. 10:303. doi: 10.3389/fimmu.2019.00303
Gerhard, A., Banati, R. B., Goerres, G. B., Cagnin, A., Myers, R., Gunn, R. N., et al. (2003). [11C](R)-PK11195 PET imaging of microglial activation in multiple system atrophy. Neurology 61, 686–689. doi: 10.1212/01.wnl.0000078192.95645.e6
Gerhard, A., Pavese, N., Hotton, G., Turkheimer, F., Es, M., Hammers, A., et al. (2006). In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 21, 404–412. doi: 10.1016/j.nbd.2005.08.002
Ghiglieri, V., Calabrese, V., and Calabresi, P. (2018). α-synuclein: from early synaptic dysfunction to neurodegeneration. Front. Neurol. 9:295. doi: 10.3389/fneur.2018.00295
Ghosh, D., Sahay, S., Ranjan, P., Salot, S., Mohite, G. M., Singh, P. K., et al. (2014). The newly discovered Parkinson’s disease associated Finnish mutation (A53E) attenuates α-synuclein aggregation and membrane binding. Biochemistry 53, 6419–6421. doi: 10.1021/bi5010365
Giasson, B. I., Duda, J. E., Murray, I. V., Chen, Q., Souza, J. M., Hurtig, H. I., et al. (2000). Oxidative damage linked to neurodegeneration by selective α-synuclein nitration in synucleinopathy lesions. Science 290, 985–989. doi: 10.1126/science.290.5493.985
Giordano, N., Iemolo, A., Mancini, M., Cacace, F., De Risi, M., Latagliata, E. C., et al. (2018). Motor learning and metaplasticity in striatal neurons: relevance for Parkinson’s disease. Brain 141, 505–520. doi: 10.1093/brain/awx351
Goedert, M., and Spillantini, M. G. (1998). Lewy body diseases and multiple system atrophy as alpha-synucleinopathies. Mol. Psychiatry 3, 462–465. doi: 10.1038/sj.mp.4000458
Gonzalez, N., Arcos-Lopez, T., Konig, A., Quintanar, L., Menacho Marquez, M., Outeiro, T. F., et al. (2019). Effects of α-synuclein post-translational modifications on metal binding. J. Neurochem. 150, 507–521. doi: 10.1111/jnc.14721
Gosavi, N., Lee, H. J., Lee, J. S., Patel, S., and Lee, S. J. (2002). Golgi fragmentation occurs in the cells with prefibrillar α-synuclein aggregates and precedes the formation of fibrillar inclusion. J. Biol. Chem. 277, 48984–48992. doi: 10.1074/jbc.m208194200
Grey, M., Linse, S., Nilsson, H., Brundin, P., and Sparr, E. (2011). Membrane interaction of α-synuclein in different aggregation states. J. Parkinsons Dis. 1, 359–371. doi: 10.3233/JPD-2011-11067
Guardia-Laguarta, C., Area-Gomez, E., Rub, C., Liu, Y., Magrane, J., Becker, D., et al. (2014). α-Synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 34, 249–259. doi: 10.1523/JNEUROSCI.2507-13.2014
Guerrero-Ferreira, R., Taylor, N. M., Arteni, A. A., Kumari, P., Mona, D., Ringler, P., et al. (2019). Two new polymorphic structures of human full-length α-synuclein fibrils solved by cryo-electron microscopy. eLife 8:e48907. doi: 10.7554/eLife.48907
Guo, J. L., Covell, D. J., Daniels, J. P., Iba, M., Stieber, A., Zhang, B., et al. (2013). Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell 154, 103–117. doi: 10.1016/j.cell.2013.05.057
Hallett, P. J., Cooper, O., Sadi, D., Robertson, H., Mendez, I., and Isacson, O. (2014). Long-term health of dopaminergic neuron transplants in Parkinson’s disease patients. Cell Rep. 7, 1755–1761. doi: 10.1016/j.celrep.2014.05.027
Halliday, G., Mccann, H., and Shepherd, C. (2012). Evaluation of the Braak hypothesis: how far can it explain the pathogenesis of Parkinson’s disease? Expert Rev. Neurother. 12, 673–686. doi: 10.1586/ern.12.47
Halliday, G. M., and Stevens, C. H. (2011). Glia: initiators and progressors of pathology in Parkinson’s disease. Mov. Disord. 26, 6–17. doi: 10.1002/mds.23455
Hegde, M. L., and Rao, K. S. (2007). DNA induces folding in α-synuclein: understanding the mechanism using chaperone property of osmolytes. Arch. Biochem. Biophys. 464, 57–69. doi: 10.1016/j.abb.2007.03.042
Heras-Garvin, A., and Stefanova, N. (2020). MSA: from basic mechanisms to experimental therapeutics. Parkinsonism Relat. Disord. 73, 94–104. doi: 10.1016/j.parkreldis.2020.01.010
Heras-Garvin, A., Weckbecker, D., Ryazanov, S., Leonov, A., Griesinger, C., Giese, A., et al. (2019). Anle138b modulates α-synuclein oligomerization and prevents motor decline and neurodegeneration in a mouse model of multiple system atrophy. Mov. Disord. 34, 255–263. doi: 10.1002/mds.27562
Hijaz, B. A., and Volpicelli-Daley, L. A. (2020). Initiation and propagation of α-synuclein aggregation in the nervous system. Mol. Neurodegener. 15:19. doi: 10.1186/s13024-020-00368-6
Hoffmann, A., Ettle, B., Battis, K., Reiprich, S., Schlachetzki, J. C. M., Masliah, E., et al. (2019). Oligodendroglial α-synucleinopathy-driven neuroinflammation in multiple system atrophy. Brain Pathol. 29, 380–396. doi: 10.1111/bpa.12678
Holmes, B. B., Devos, S. L., Kfoury, N., Li, M., Jacks, R., Yanamandra, K., et al. (2013). Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. U S A 110, E3138–E3147. doi: 10.1073/pnas.1301440110
Holmqvist, S., Chutna, O., Bousset, L., Aldrin-Kirk, P., Li, W., Bjorklund, T., et al. (2014). Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 128, 805–820. doi: 10.1007/s00401-014-1343-6
Hoozemans, J. J., Van Haastert, E. S., Eikelenboom, P., De Vos, R. A., Rozemuller, J. M., and Scheper, W. (2007). Activation of the unfolded protein response in Parkinson’s disease. Biochem. Biophys. Res. Commun. 354, 707–711. doi: 10.1016/j.bbrc.2007.01.043
Hsu, L. J., Sagara, Y., Arroyo, A., Rockenstein, E., Sisk, A., Mallory, M., et al. (2000). α-synuclein promotes mitochondrial deficit and oxidative stress. Am. J. Pathol. 157, 401–410. doi: 10.1016/s0002-9440(10)64553-1
Iljina, M., Garcia, G. A., Horrocks, M. H., Tosatto, L., Choi, M. L., Ganzinger, K. A., et al. (2016). Kinetic model of the aggregation of α-synuclein provides insights into prion-like spreading. Proc. Natl. Acad. Sci. U S A 113, E1206–E1215. doi: 10.1073/pnas.1524128113
Imamura, K., Hishikawa, N., Sawada, M., Nagatsu, T., Yoshida, M., and Hashizume, Y. (2003). Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol. 106, 518–526. doi: 10.1007/s00401-003-0766-2
Iranzo, A., Tolosa, E., Gelpi, E., Molinuevo, J. L., Valldeoriola, F., Serradell, M., et al. (2013). Neurodegenerative disease status and post-mortem pathology in idiopathic rapid-eye-movement sleep behaviour disorder: an observational cohort study. Lancet Neurol. 12, 443–453. doi: 10.1016/S1474-4422(13)70056-5
Irwin, D. J., Abrams, J. Y., Schonberger, L. B., Leschek, E. W., Mills, J. L., Lee, V. M., et al. (2013). Evaluation of potential infectivity of Alzheimer and Parkinson disease proteins in recipients of cadaver-derived human growth hormone. JAMA Neurol. 70, 462–468. doi: 10.1001/jamaneurol.2013.1933
Ishizawa, K., Komori, T., Arai, N., Mizutani, T., and Hirose, T. (2008). Glial cytoplasmic inclusions and tissue injury in multiple system atrophy: A quantitative study in white matter (olivopontocerebellar system) and gray matter (nigrostriatal system). Neuropathology 28, 249–257. doi: 10.1111/j.1440-1789.2007.00855.x
Ishizawa, K., Komori, T., Sasaki, S., Arai, N., Mizutani, T., and Hirose, T. (2004). Microglial activation parallels system degeneration in multiple system atrophy. J. Neuropathol. Exp. Neurol. 63, 43–52. doi: 10.1093/jnen/63.1.43
Jellinger, K. A. (2009). A critical evaluation of current staging of α-synuclein pathology in Lewy body disorders. Biochim. Biophys. Acta 1792, 730–740. doi: 10.1016/j.bbadis.2008.07.006
Jellinger, K. A. (2018). Dementia with Lewy bodies and Parkinson’s disease-dementia: current concepts and controversies. J. Neural Transm. 125, 615–650. doi: 10.1007/s00702-017-1821-9
Kaji, S., Maki, T., Ishimoto, T., Yamakado, H., and Takahashi, R. (2020). Insights into the pathogenesis of multiple system atrophy: focus on glial cytoplasmic inclusions. Transl. Neurodegener. 9:7. doi: 10.1186/s40035-020-0185-5
Kaji, S., Maki, T., Kinoshita, H., Uemura, N., Ayaki, T., Kawamoto, Y., et al. (2018). Pathological endogenous α-synuclein accumulation in oligodendrocyte precursor cells potentially induces inclusions in multiple system atrophy. Stem Cell Reports 10, 356–365. doi: 10.1016/j.stemcr.2017.12.001
Kalaitzakis, M. E., Graeber, M. B., Gentleman, S. M., and Pearce, R. K. (2008). The dorsal motor nucleus of the vagus is not an obligatory trigger site of Parkinson’s disease: a critical analysis of α-synuclein staging. Neuropathol. Appl. Neurobiol. 34, 284–295. doi: 10.1111/j.1365-2990.2007.00923.x
Kalia, L. V., and Lang, A. E. (2015). Parkinson’s disease. Lancet 386, 896–912. doi: 10.1016/S0140-6736(14)61393-3
Kam, T. I., Mao, X., Park, H., Chou, S. C., Karuppagounder, S. S., Umanah, G. E., et al. (2018). Poly(ADP-ribose) drives pathologic α-synuclein neurodegeneration in Parkinson’s disease. Science 362:eaat8407. doi: 10.1126/science.aat8407
Kara, E., Marks, J. D., and Aguzzi, A. (2018). Toxic protein spread in neurodegeneration: reality versus fantasy. Trends Mol. Med. 24, 1007–1020. doi: 10.1016/j.molmed.2018.09.004
Katorcha, E., Makarava, N., Lee, Y. J., Lindberg, I., Monteiro, M. J., Kovacs, G. G., et al. (2017). Cross-seeding of prions by aggregated α-synuclein leads to transmissible spongiform encephalopathy. PLoS Pathog. 13:e1006563. doi: 10.1371/journal.ppat.1006563
Kaufmann, H., Hague, K., and Perl, D. (2001). Accumulation of α-synuclein in autonomic nerves in pure autonomic failure. Neurology 56, 980–981. doi: 10.1212/wnl.56.7.980
Kaufmann, H., Nahm, K., Purohit, D., and Wolfe, D. (2004). Autonomic failure as the initial presentation of Parkinson disease and dementia with Lewy bodies. Neurology 63, 1093–1095. doi: 10.1212/01.wnl.0000138500.73671.dc
Kim, C., Ho, D. H., Suk, J. E., You, S., Michael, S., Kang, J., et al. (2013). Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 4:1562. doi: 10.1038/ncomms2534
Kim, J. Y., Illigens, B. M., Mccormick, M. P., Wang, N., and Gibbons, C. H. (2019). α-synuclein in skin nerve fibers as a biomarker for α-synucleinopathies. J. Clin. Neurol. 15, 135–142. doi: 10.3988/jcn.2019.15.2.135
Kim, S., Kwon, S. H., Kam, T. I., Panicker, N., Karuppagounder, S. S., Lee, S., et al. (2019). Transneuronal propagation of pathologic α-synuclein from the gut to the brain models Parkinson’s disease. Neuron 103, 627.e7–641.e7. doi: 10.1016/j.neuron.2019.05.035
Kobayashi, J., Hasegawa, T., Sugeno, N., Yoshida, S., Akiyama, T., Fujimori, K., et al. (2019). Extracellular α-synuclein enters dopaminergic cells by modulating flotillin-1-assisted dopamine transporter endocytosis. FASEB J. 33, 10240–10256. doi: 10.1096/fj.201802051r
Kordower, J. H., Chu, Y., Hauser, R. A., Freeman, T. B., and Olanow, C. W. (2008). Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med. 14, 504–506. doi: 10.1096/fj.201802051R
Krasnoslobodtsev, A. V., Peng, J., Asiago, J. M., Hindupur, J., Rochet, J. C., and Lyubchenko, Y. L. (2012). Effect of spermidine on misfolding and interactions of α-synuclein. PLoS One 7:e38099. doi: 10.1371/journal.pone.0038099
Kubler, D., Wachter, T., Cabanel, N., Su, Z., Turkheimer, F. E., Dodel, R., et al. (2019). Widespread microglial activation in multiple system atrophy. Mov. Disord. 34, 564–568. doi: 10.1002/mds.27620
Lashuel, H. A., Overk, C. R., Oueslati, A., and Masliah, E. (2013). The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 14, 38–48. doi: 10.1038/nrn3406
Lashuel, H. A., Petre, B. M., Wall, J., Simon, M., Nowak, R. J., Walz, T., et al. (2002). α-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J. Mol. Biol. 322, 1089–1102. doi: 10.1016/s0022-2836(02)00735-0
Lau, A., So, R. W. L., Lau, H. H. C., Sang, J. C., Ruiz-Riquelme, A., Fleck, S. C., et al. (2020). α-Synuclein strains target distinct brain regions and cell types. Nat. Neurosci. 23, 21–31. doi: 10.1038/s41593-019-0541-x
Leak, R. K., Frosch, M. P., Beach, T. G., and Halliday, G. M. (2019). α-synuclein: prion or prion-like? Acta Neuropathol. 138, 509–514. doi: 10.1007/s00401-019-02057-1
Lee, H. J., Baek, S. M., Ho, D. H., Suk, J. E., Cho, E. D., and Lee, S. J. (2011). Dopamine promotes formation and secretion of non-fibrillar α-synuclein oligomers. Exp. Mol. Med. 43, 216–222. doi: 10.3858/emm.2011.43.4.026
Lee, H. J., Khoshaghideh, F., Patel, S., and Lee, S. J. (2004). Clearance of α-synuclein oligomeric intermediates via the lysosomal degradation pathway. J. Neurosci. 24, 1888–1896. doi: 10.1523/jneurosci.3809-03.2004
Lee, H. J., Suk, J. E., Patrick, C., Bae, E. J., Cho, J. H., Rho, S., et al. (2010). Direct transfer of α-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem. 285, 9262–9272. doi: 10.1074/jbc.m109.081125
Lee, V. M., and Trojanowski, J. Q. (2006). Mechanisms of Parkinson’s disease linked to pathological α-synuclein: new targets for drug discovery. Neuron 52, 33–38. doi: 10.1016/j.neuron.2006.09.026
Lema Tome, C. M., Tyson, T., Rey, N. L., Grathwohl, S., Britschgi, M., and Brundin, P. (2013). Inflammation and α-synuclein’s prion-like behavior in Parkinson’s disease–is there a link? Mol. Neurobiol. 47, 561–574. doi: 10.1007/s12035-012-8267-8
Li, B., Ge, P., Murray, K. A., Sheth, P., Zhang, M., Nair, G., et al. (2018). Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel. Nat. Commun. 9:3609. doi: 10.1038/s41467-018-05971-2
Li, J., Zhu, M., Rajamani, S., Uversky, V. N., and Fink, A. L. (2004). Rifampicin inhibits α-synuclein fibrillation and disaggregates fibrils. Chem. Biol. 11, 1513–1521. doi: 10.1016/j.chembiol.2004.08.025
Li, J. Y., Englund, E., Holton, J. L., Soulet, D., Hagell, P., Lees, A. J., et al. (2008). Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 14, 501–503. doi: 10.1038/nm1746
Li, W., Englund, E., Widner, H., Mattsson, B., Van Westen, D., Latt, J., et al. (2016). Extensive graft-derived dopaminergic innervation is maintained 24 years after transplantation in the degenerating parkinsonian brain. Proc. Natl. Acad. Sci. U S A 113, 6544–6549. doi: 10.1073/pnas.1605245113
Li, W., West, N., Colla, E., Pletnikova, O., Troncoso, J. C., Marsh, L., et al. (2005). Aggregation promoting C-terminal truncation of α-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s disease-linked mutations. Proc. Natl. Acad. Sci. U S A 102, 2162–2167. doi: 10.1073/pnas.0406976102
Li, W. W., Yang, R., Guo, J. C., Ren, H. M., Zha, X. L., Cheng, J. S., et al. (2007). Localization of α-synuclein to mitochondria within midbrain of mice. Neuroreport 18, 1543–1546. doi: 10.1097/WNR.0b013e3282f03db4
Lindersson, E., Beedholm, R., Hojrup, P., Moos, T., Gai, W., Hendil, K. B., et al. (2004). Proteasomal inhibition by α-synuclein filaments and oligomers. J. Biol. Chem. 279, 12924–12934. doi: 10.1074/jbc.M306390200
Lionnet, A., Leclair-Visonneau, L., Neunlist, M., Murayama, S., Takao, M., Adler, C. H., et al. (2018). Does Parkinson’s disease start in the gut? Acta Neuropathol. 135, 1–12. doi: 10.1007/s00401-017-1777-8
Liu, C. Y., Wang, X., Liu, C., and Zhang, H. L. (2019). Pharmacological targeting of microglial activation: new therapeutic approach. Front. Cell. Neurosci. 13:514. doi: 10.3389/fncel.2019.00514
Lohmann, S., Bernis, M. E., Tachu, B. J., Ziemski, A., Grigoletto, J., and Tamguney, G. (2019). Oral and intravenous transmission of α-synuclein fibrils to mice. Acta Neuropathol. 138, 515–533. doi: 10.1007/s00401-019-02037-5
Lopes Da Fonseca, T., Villar-Pique, A., and Outeiro, T. F. (2015). The interplay between α-synuclein clearance and spreading. Biomolecules 5, 435–471. doi: 10.3390/biom5020435
Ludtmann, M. H. R., Angelova, P. R., Horrocks, M. H., Choi, M. L., Rodrigues, M., Baev, A. Y., et al. (2018). α-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat. Commun. 9:2293. doi: 10.1038/s41467-018-04422-2
Luk, K. C., Kehm, V., Carroll, J., Zhang, B., O’brien, P., Trojanowski, J. Q., et al. (2012a). Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338, 949–953. doi: 10.1126/science.1227157
Luk, K. C., Kehm, V. M., Zhang, B., O’brien, P., Trojanowski, J. Q., and Lee, V. M. (2012b). Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J. Exp. Med. 209, 975–986. doi: 10.1084/jem.20112457
Luk, K. C., Song, C., O’Brien, P., Stieber, A., Branch, J. R., Brunden, K. R., et al. (2009). Exogenous α-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. U S A 106, 20051–20056. doi: 10.1073/pnas.0908005106
Mahul-Mellier, A. L., Burtscher, J., Maharjan, N., Weerens, L., Croisier, M., Kuttler, F., et al. (2020). The process of Lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc. Natl. Acad. Sci. U S A 117, 4971–4982. doi: 10.1073/pnas.1913904117
Malagelada, C., Jin, Z. H., Jackson-Lewis, V., Przedborski, S., and Greene, L. A. (2010). Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J. Neurosci. 30, 1166–1175. doi: 10.1523/jneurosci.3944-09.2010
Mandel, R. J., Marmion, D. J., Kirik, D., Chu, Y., Heindel, C., Mccown, T., et al. (2017). Novel oligodendroglial α-synuclein viral vector models of multiple system atrophy: studies in rodents and nonhuman primates. Acta Neuropathol. Commun. 5:47. doi: 10.1186/s40478-017-0451-7
Mandler, M., Valera, E., Rockenstein, E., Mante, M., Weninger, H., Patrick, C., et al. (2015). Active immunization against α-synuclein ameliorates the degenerative pathology and prevents demyelination in a model of multiple system atrophy. Mol. Neurodegener. 10:10. doi: 10.1186/s13024-015-0008-9
Mandler, M., Valera, E., Rockenstein, E., Weninger, H., Patrick, C., Adame, A., et al. (2014). Next-generation active immunization approach for synucleinopathies: implications for Parkinson’s disease clinical trials. Acta Neuropathol. 127, 861–879. doi: 10.1007/s00401-014-1256-4
Manfredsson, F. P., Luk, K. C., Benskey, M. J., Gezer, A., Garcia, J., Kuhn, N. C., et al. (2018). Induction of α-synuclein pathology in the enteric nervous system of the rat and non-human primate results in gastrointestinal dysmotility and transient CNS pathology. Neurobiol. Dis. 112, 106–118. doi: 10.1016/j.nbd.2018.01.008
Mao, X., Ou, M. T., Karuppagounder, S. S., Kam, T. I., Yin, X., Xiong, Y., et al. (2016). Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 353:aah3374. doi: 10.1126/science.aah3374
Maroteaux, L., Campanelli, J. T., and Scheller, R. H. (1988). Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 8, 2804–2815. doi: 10.1523/jneurosci.08-08-02804.1988
Marreiros, R., Muller-Schiffmann, A., Trossbach, S. V., Prikulis, I., Hansch, S., Weidtkamp-Peters, S., et al. (2020). Disruption of cellular proteostasis by H1N1 influenza A virus causes α-synuclein aggregation. Proc. Natl. Acad. Sci. U S A 117, 6741–6751. doi: 10.1073/pnas.1906466117
Marti, M. J., Tolosa, E., and Campdelacreu, J. (2003). Clinical overview of the synucleinopathies. Mov. Disord. 18, S21–S27. doi: 10.1002/mds.10559
Masaracchia, C., Hnida, M., Gerhardt, E., Lopes Da Fonseca, T., Villar-Pique, A., Branco, T., et al. (2018). Membrane binding, internalization and sorting of α-synuclein in the cell. Acta Neuropathol. Commun. 6:79. doi: 10.1186/s40478-018-0578-1
Masliah, E., Rockenstein, E., Mante, M., Crews, L., Spencer, B., Adame, A., et al. (2011). Passive immunization reduces behavioral and neuropathological deficits in an α-synuclein transgenic model of Lewy body disease. PLoS One 6:e19338. doi: 10.1371/journal.pone.0019338
Masuda-Suzukake, M., Nonaka, T., Hosokawa, M., Oikawa, T., Arai, T., Akiyama, H., et al. (2013). Prion-like spreading of pathological α-synuclein in brain. Brain 136, 1128–1138. doi: 10.1093/brain/awt037
Mavroeidi, P., Arvanitaki, F., Karakitsou, A. K., Vetsi, M., Kloukina, I., Zweckstetter, M., et al. (2019). Endogenous oligodendroglial α-synuclein and TPPP/p25α orchestrate α-synuclein pathology in experimental multiple system atrophy models. Acta Neuropathol. 138, 415–441. doi: 10.1007/s00401-019-02068-y
May, V. E., Ettle, B., Poehler, A. M., Nuber, S., Ubhi, K., Rockenstein, E., et al. (2014). α-Synuclein impairs oligodendrocyte progenitor maturation in multiple system atrophy. Neurobiol. Aging 35, 2357–2368. doi: 10.1016/j.neurobiolaging.2014.02.028
Mazzulli, J. R., Zunke, F., Isacson, O., Studer, L., and Krainc, D. (2016). α-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc. Natl. Acad. Sci. U S A 113, 1931–1936. doi: 10.1073/pnas.1520335113
McGeer, P. L., Itagaki, S., Boyes, B. E., and McGeer, E. G. (1988a). Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38, 1285–1291. doi: 10.1212/wnl.38.8.1285
McGeer, P. L., Itagaki, S., and McGeer, E. G. (1988b). Expression of the histocompatibility glycoprotein HLA-DR in neurological disease. Acta Neuropathol. 76, 550–557. doi: 10.1007/bf00689592
Meade, R. M., Fairlie, D. P., and Mason, J. M. (2019). α-synuclein structure and Parkinson’s disease—lessons and emerging principles. Mol. Neurodegener. 14:29. doi: 10.1186/s13024-019-0329-1
Mendez, I., Vinuela, A., Astradsson, A., Mukhida, K., Hallett, P., Robertson, H., et al. (2008). Dopamine neurons implanted into people with Parkinson’s disease survive without pathology for 14 years. Nat. Med. 14, 507–509. doi: 10.1038/nm1752
Miklossy, J., Doudet, D. D., Schwab, C., Yu, S., Mcgeer, E. G., and Mcgeer, P. L. (2006). Role of ICAM-1 in persisting inflammation in Parkinson disease and MPTP monkeys. Exp. Neurol. 197, 275–283. doi: 10.1016/j.expneurol.2005.10.034
Milanese, C., Cerri, S., Ulusoy, A., Gornati, S. V., Plat, A., Gabriels, S., et al. (2018). Activation of the DNA damage response in vivo in synucleinopathy models of Parkinson’s disease. Cell Death Dis. 9:818. doi: 10.1038/s41419-018-0848-7
Mor, D. E., Tsika, E., Mazzulli, J. R., Gould, N. S., Kim, H., Daniels, M. J., et al. (2017). Dopamine induces soluble α-synuclein oligomers and nigrostriatal degeneration. Nat. Neurosci. 20, 1560–1568. doi: 10.1038/nn.4641
Mori, F., Inenaga, C., Yoshimoto, M., Umezu, H., Tanaka, R., Takahashi, H., et al. (2002). α-synuclein immunoreactivity in normal and neoplastic Schwann cells. Acta Neuropathol. 103, 145–151. doi: 10.1007/s004010100443
Nugent, E., Kaminski, C. F., and Kaminski Schierle, G. S. (2017). Super-resolution imaging of α-synuclein polymorphisms and their potential role in neurodegeneration. Integr. Biol. 9, 206–210. doi: 10.1039/c6ib00206d
Olanow, C. W., Perl, D. P., Demartino, G. N., and Mcnaught, K. S. (2004). Lewy-body formation is an aggresome-related process: a hypothesis. Lancet Neurol. 3, 496–503. doi: 10.1016/s1474-4422(04)00827-0
Olanow, C. W., Savolainen, M., Chu, Y., Halliday, G. M., and Kordower, J. H. (2019). Temporal evolution of microglia and α-synuclein accumulation following foetal grafting in Parkinson’s disease. Brain 142, 1690–1700. doi: 10.1093/brain/awz104
Ono, K., Takahashi, R., Ikeda, T., and Yamada, M. (2012). Cross-seeding effects of amyloid β-protein and α-synuclein. J. Neurochem. 122, 883–890. doi: 10.1111/j.1471-4159.2012.07847.x
Orimo, S., Uchihara, T., Nakamura, A., Mori, F., Kakita, A., Wakabayashi, K., et al. (2008). Axonal α-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 131, 642–650. doi: 10.1093/brain/awm302
Orme, T., Guerreiro, R., and Bras, J. (2018). The genetics of dementia with Lewy bodies: current understanding and future directions. Curr. Neurol. Neurosci. Rep. 18:67. doi: 10.1007/s11910-018-0874-y
Outeiro, T. F., Koss, D. J., Erskine, D., Walker, L., Kurzawa-Akanbi, M., Burn, D., et al. (2019). Dementia with Lewy bodies: an update and outlook. Mol. Neurodegener. 14:5. doi: 10.1186/s13024-019-0306-8
Ozawa, T., Paviour, D., Quinn, N. P., Josephs, K. A., Sangha, H., Kilford, L., et al. (2004). The spectrum of pathological involvement of the striatonigral and olivopontocerebellar systems in multiple system atrophy: clinicopathological correlations. Brain 127, 2657–2671. doi: 10.1093/brain/awh303
Paiva, I., Pinho, R., Pavlou, M. A., Hennion, M., Wales, P., Schutz, A. L., et al. (2017). Sodium butyrate rescues dopaminergic cells from α-synuclein-induced transcriptional deregulation and DNA damage. Hum. Mol. Genet. 26, 2231–2246. doi: 10.1093/hmg/ddx114
Parihar, M. S., Parihar, A., Fujita, M., Hashimoto, M., and Ghafourifar, P. (2008). Mitochondrial association of α-synuclein causes oxidative stress. Cell Mol. Life Sci. 65, 1272–1284. doi: 10.1007/s00018-008-7589-1
Park, H. J., Oh, S. H., Kim, H. N., Jung, Y. J., and Lee, P. H. (2016). Mesenchymal stem cells enhance α-synuclein clearance via M2 microglia polarization in experimental and human parkinsonian disorder. Acta Neuropathol. 132, 685–701. doi: 10.1007/s00401-016-1605-6
Park, J. H., Burgess, J. D., Faroqi, A. H., Demeo, N. N., Fiesel, F. C., Springer, W., et al. (2020). α-synuclein-induced mitochondrial dysfunction is mediated via a sirtuin 3-dependent pathway. Mol. Neurodegener. 15:5. doi: 10.1101/357624
Paumier, K. L., Luk, K. C., Manfredsson, F. P., Kanaan, N. M., Lipton, J. W., Collier, T. J., et al. (2015). Intrastriatal injection of pre-formed mouse α-synuclein fibrils into rats triggers α-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol. Dis. 82, 185–199. doi: 10.1016/j.nbd.2015.06.003
Peelaerts, W., Bousset, L., Van Der Perren, A., Moskalyuk, A., Pulizzi, R., Giugliano, M., et al. (2015). α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522, 340–344. doi: 10.1038/nature14547
Peng, C., Gathagan, R. J., Covell, D. J., Medellin, C., Stieber, A., Robinson, J. L., et al. (2018a). Cellular milieu imparts distinct pathological α-synuclein strains in alpha-synucleinopathies. Nature 557, 558–563. doi: 10.1038/s41586-018-0104-4
Peng, C., Gathagan, R. J., and Lee, V. M. (2018b). Distinct α-Synuclein strains and implications for heterogeneity among alpha-Synucleinopathies. Neurobiol. Dis. 109, 209–218. doi: 10.1016/j.nbd.2017.07.018
Perez, R. G., and Hastings, T. G. (2004). Could a loss of α-synuclein function put dopaminergic neurons at risk? J. Neurochem. 89, 1318–1324. doi: 10.1111/j.1471-4159.2004.02423.x
Perrino, G., Wilson, C., Santorelli, M., and Di Bernardo, D. (2019). Quantitative characterization of α-synuclein aggregation in living cells through automated microfluidics feedback control. Cell Rep. 27, 916.e5–927.e5. doi: 10.1016/j.celrep.2019.03.081
Petrucelli, L., O’Farrell, C., Lockhart, P. J., Baptista, M., Kehoe, K., Vink, L., et al. (2002). Parkin protects against the toxicity associated with mutant α-synuclein: proteasome dysfunction selectively affects catecholaminergic neurons. Neuron 36, 1007–1019. doi: 10.1016/s0896-6273(02)01125-x
Phan, J. A., Stokholm, K., Zareba-Paslawska, J., Jakobsen, S., Vang, K., Gjedde, A., et al. (2017). Early synaptic dysfunction induced by α-synuclein in a rat model of Parkinson’s disease. Sci. Rep. 7:6363. doi: 10.1038/s41598-017-06724-9
Pinho, R., Paiva, I., Jercic, K. G., Fonseca-Ornelas, L., Gerhardt, E., Fahlbusch, C., et al. (2019). Nuclear localization and phosphorylation modulate pathological effects of α-synuclein. Hum. Mol. Genet. 28, 31–50. doi: 10.1093/hmg/ddy326
Polymeropoulos, M. H., Higgins, J. J., Golbe, L. I., Johnson, W. G., Ide, S. E., Di Iorio, G., et al. (1996). Mapping of a gene for Parkinson’s disease to chromosome 4q21–q23. Science 274, 1197–1199. doi: 10.1126/science.274.5290.1197
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., et al. (1997). Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. doi: 10.1126/science.276.5321.2045
Post, M. R., Lieberman, O. J., and Mosharov, E. V. (2018). Can interactions between α-synuclein, dopamine and calcium explain selective neurodegeneration in Parkinson’s disease? Front. Neurosci. 12:161. doi: 10.3389/fnins.2018.00161
Price, D. L., Koike, M. A., Khan, A., Wrasidlo, W., Rockenstein, E., Masliah, E., et al. (2018). The small molecule α-synuclein misfolding inhibitor, NPT200–11, produces multiple benefits in an animal model of Parkinson’s disease. Sci. Rep. 8:16165. doi: 10.1038/s41598-018-34490-9
Pringsheim, T., Jette, N., Frolkis, A., and Steeves, T. D. (2014). The prevalence of Parkinson’s disease: a systematic review and meta-analysis. Mov. Disord. 29, 1583–1590. doi: 10.1002/mds.25945
Prusiner, S. B. (1982). Novel proteinaceous infectious particles cause scrapie. Science 216, 136–144. doi: 10.1126/science.6801762
Prusiner, S. B. (1998). Prions. Proc. Natl. Acad. Sci. U S A 95, 13363–13383. doi: 10.1073/pnas.95.23.13363
Radford, R., Rcom-H’cheo-Gauthier, A., Wong, M. B., Eaton, E. D., Quilty, M., Blizzard, C., et al. (2015). The degree of astrocyte activation in multiple system atrophy is inversely proportional to the distance to α-synuclein inclusions. Mol. Cell. Neurosci. 65, 68–81. doi: 10.1016/j.mcn.2015.02.015
Rajput, A. H., Ferguson, L. W., Robinson, C. A., Guella, I., Farrer, M. J., and Rajput, A. (2016). Conjugal parkinsonism—clinical, pathology and genetic study. No evidence of person-to-person transmission. Parkinsonism Relat. Disord. 31, 87–90. doi: 10.1016/j.parkreldis.2016.07.011
Recasens, A., Carballo-Carbajal, I., Parent, A., Bove, J., Gelpi, E., Tolosa, E., et al. (2018). Lack of pathogenic potential of peripheral α-synuclein aggregates from Parkinson’s disease patients. Acta Neuropathol. Commun. 6:8. doi: 10.1186/s40478-018-0509-1
Recasens, A., Dehay, B., Bove, J., Carballo-Carbajal, I., Dovero, S., Perez-Villalba, A., et al. (2014). Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys. Ann. Neurol. 75, 351–362. doi: 10.1002/ana.24066
Reeve, A. K., Ludtmann, M. H., Angelova, P. R., Simcox, E. M., Horrocks, M. H., Klenerman, D., et al. (2015). Aggregated α-synuclein and complex I deficiency: exploration of their relationship in differentiated neurons. Cell Death Dis. 6:e1820. doi: 10.1038/cddis.2015.166
Rey, N. L., Bousset, L., George, S., Madaj, Z., Meyerdirk, L., Schulz, E., et al. (2019). α-Synuclein conformational strains spread, seed and target neuronal cells differentially after injection into the olfactory bulb. Acta Neuropathol. Commun. 7:221. doi: 10.1186/s40478-019-0859-3
Rey, N. L., George, S., Steiner, J. A., Madaj, Z., Luk, K. C., Trojanowski, J. Q., et al. (2018). Spread of aggregates after olfactory bulb injection of α-synuclein fibrils is associated with early neuronal loss and is reduced long term. Acta Neuropathol. 135, 65–83. doi: 10.1007/s00401-017-1792-9
Reyes, J. F., Rey, N. L., Bousset, L., Melki, R., Brundin, P., and Angot, E. (2014). α-synuclein transfers from neurons to oligodendrocytes. Glia 62, 387–398. doi: 10.1002/glia.22611
Rott, R., Szargel, R., Shani, V., Hamza, H., Savyon, M., Abd Elghani, F., et al. (2017). SUMOylation and ubiquitination reciprocally regulate α-synuclein degradation and pathological aggregation. Proc. Natl. Acad. Sci. U S A 114, 13176–13181. doi: 10.1073/pnas.1704351114
Sampson, T. R., Challis, C., Jain, N., Moiseyenko, A., Ladinsky, M. S., Shastri, G. G., et al. (2020). A gut bacterial amyloid promotes α-synuclein aggregation and motor impairment in mice. eLife 9:e53111. doi: 10.7554/eLife.53111
Sampson, T. R., Debelius, J. W., Thron, T., Janssen, S., Shastri, G. G., Ilhan, Z. E., et al. (2016). Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 167, 1469.e12–1480.e12. doi: 10.1016/j.cell.2016.11.018
Sarkar, S., Dammer, E. B., Malovic, E., Olsen, A. L., Raza, S. A., Gao, T., et al. (2020). Molecular signatures of neuroinflammation induced by α-synuclein aggregates in microglial cells. Front. Immunol. 11:33. doi: 10.3389/fimmu.2020.00033
Schafferer, S., Khurana, R., Refolo, V., Venezia, S., Sturm, E., Piatti, P., et al. (2016). Changes in the miRNA-mRNA regulatory network precede motor symptoms in a mouse model of multiple system atrophy: clinical implications. PLoS One 11:e0150705. doi: 10.1371/journal.pone.0150705
Schaser, A. J., Osterberg, V. R., Dent, S. E., Stackhouse, T. L., Wakeham, C. M., Boutros, S. W., et al. (2019). α-synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Sci. Rep. 9:10919. doi: 10.1038/s41598-019-47227-z
Schweighauser, M., Bacioglu, M., Fritschi, S. K., Shimshek, D. R., Kahle, P. J., Eisele, Y. S., et al. (2015). Formaldehyde-fixed brain tissue from spontaneously ill α-synuclein transgenic mice induces fatal alpha-synucleinopathy in transgenic hosts. Acta Neuropathol. 129, 157–159. doi: 10.1007/s00401-014-1360-5
Schweighauser, M., Shi, Y., Tarutani, A., Kametani, F., Murzin, A. G., Ghetti, B., et al. (2020). Structures of α-synuclein filaments from multiple system atrophy. Nature doi: 10.1038/s41586-020-2317-6 [Epub ahead of print].
Shahmoradian, S. H., Lewis, A. J., Genoud, C., Hench, J., Moors, T. E., Navarro, P. P., et al. (2019). Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 22, 1099–1109. doi: 10.1038/s41593-019-0423-2
Shahnawaz, M., Mukherjee, A., Pritzkow, S., Mendez, N., Rabadia, P., Liu, X., et al. (2020). Discriminating α-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 578, 273–277. doi: 10.1038/s41586-020-1984-7
Shannon, K. M., Keshavarzian, A., Mutlu, E., Dodiya, H. B., Daian, D., Jaglin, J. A., et al. (2012). α-synuclein in colonic submucosa in early untreated Parkinson’s disease. Mov. Disord. 27, 709–715. doi: 10.3410/f.717698045.793456802
Shrivastava, A. N., Bousset, L., Renner, M., Redeker, V., Savistchenko, J., Triller, A., et al. (2020). Differential membrane binding and seeding of distinct α-synuclein fibrillar polymorphs. Biophys. J. 118, 1301–1320. doi: 10.1016/j.bpj.2020.01.022
Shrivastava, A. N., Redeker, V., Fritz, N., Pieri, L., Almeida, L. G., Spolidoro, M., et al. (2015). α-synuclein assemblies sequester neuronal α3-Na+/K+-ATPase and impair Na+ gradient. EMBO J. 34, 2408–2423. doi: 10.15252/embj.201591397
Sian-Hulsmann, J., Monoranu, C., Strobel, S., and Riederer, P. (2015). Lewy bodies: a spectator or salient killer? CNS Neurol. Disord. Drug Targets 14, 947–955. doi: 10.2174/1871527314666150317225659
Sidhu, A., Wersinger, C., and Vernier, P. (2004). Does α-synuclein modulate dopaminergic synaptic content and tone at the synapse? FASEB J. 18, 637–647. doi: 10.1096/fj.03-1112rev
Singleton, A. B., Farrer, M., Johnson, J., Singleton, A., Hague, S., Kachergus, J., et al. (2003). α-Synuclein locus triplication causes Parkinson’s disease. Science 302:841. doi: 10.1126/science.1090278
Smith, W. W., Jiang, H., Pei, Z., Tanaka, Y., Morita, H., Sawa, A., et al. (2005). Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant α-synuclein-induced toxicity. Hum. Mol. Genet. 14, 3801–3811. doi: 10.1093/hmg/ddi396
Snyder, H., Mensah, K., Theisler, C., Lee, J., Matouschek, A., and Wolozin, B. (2003). Aggregated and monomeric α-synuclein bind to the S6’ proteasomal protein and inhibit proteasomal function. J. Biol. Chem. 278, 11753–11759. doi: 10.1074/jbc.m208641200
Sommer, A., Marxreiter, F., Krach, F., Fadler, T., Grosch, J., Maroni, M., et al. (2018). Th17 lymphocytes induce neuronal cell death in a human iPSC-based model of Parkinson’s disease. Cell Stem Cell 23, 123.e6–131.e6. doi: 10.1016/j.stem.2018.06.015
Song, Y. J., Halliday, G. M., Holton, J. L., Lashley, T., O’sullivan, S. S., Mccann, H., et al. (2009). Degeneration in different parkinsonian syndromes relates to astrocyte type and astrocyte protein expression. J. Neuropathol. Exp. Neurol. 68, 1073–1083. doi: 10.1097/nen.0b013e3181b66f1b
Song, Y. J., Lundvig, D. M., Huang, Y., Gai, W. P., Blumbergs, P. C., Hojrup, P., et al. (2007). p25α relocalizes in oligodendroglia from myelin to cytoplasmic inclusions in multiple system atrophy. Am. J. Pathol. 171, 1291–1303. doi: 10.1016/s1353-8020(08)70483-8
Sorrentino, Z. A., Giasson, B. I., and Chakrabarty, P. (2019). α-Synuclein and astrocytes: tracing the pathways from homeostasis to neurodegeneration in Lewy body disease. Acta Neuropathol. 138, 1–21. doi: 10.1007/s00401-019-01977-2
Spillantini, M. G., Crowther, R. A., Jakes, R., Hasegawa, M., and Goedert, M. (1998). α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. U S A 95, 6469–6473. doi: 10.1073/pnas.95.11.6469
Spillantini, M. G., Schmidt, M. L., Lee, V. M., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). α-synuclein in Lewy bodies. Nature 388, 839–840. doi: 10.1038/42166
Stefanis, L., Larsen, K. E., Rideout, H. J., Sulzer, D., and Greene, L. A. (2001). Expression of A53T mutant but not wild-type α-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release and autophagic cell death. J. Neurosci. 21, 9549–9560. doi: 10.1523/jneurosci.21-24-09549.2001
Stefanova, N., Reindl, M., Neumann, M., Kahle, P. J., Poewe, W., and Wenning, G. K. (2007). Microglial activation mediates neurodegeneration related to oligodendroglial alpha-synucleinopathy: implications for multiple system atrophy. Mov. Disord. 22, 2196–2203. doi: 10.1002/mds.21671
Stokholm, M. G., Iranzo, A., Ostergaard, K., Serradell, M., Otto, M., Svendsen, K. B., et al. (2017). Assessment of neuroinflammation in patients with idiopathic rapid-eye-movement sleep behaviour disorder: a case-control study. Lancet Neurol. 16, 789–796. doi: 10.1016/s1474-4422(17)30173-4
Su, X., Maguire-Zeiss, K. A., Giuliano, R., Prifti, L., Venkatesh, K., and Federoff, H. J. (2008). Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol. Aging 29, 1690–1701. doi: 10.1016/j.neurobiolaging.2007.04.006
Sulzer, D., Alcalay, R. N., Garretti, F., Cote, L., Kanter, E., Agin-Liebes, J., et al. (2017). T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature 546, 656–661. doi: 10.1038/nature22815
Surendranathan, A., Su, L., Mak, E., Passamonti, L., Hong, Y. T., Arnold, R., et al. (2018). Early microglial activation and peripheral inflammation in dementia with Lewy bodies. Brain 141, 3415–3427. doi: 10.1093/brain/awy265
Surguchev, A. A., and Surguchov, A. (2017). Synucleins and gene expression: ramblers in a crowd or cops regulating traffic? Front. Mol. Neurosci. 10:224. doi: 10.3389/fnmol.2017.00224
Surmeier, D. J., Obeso, J. A., and Halliday, G. M. (2017). Parkinson’s disease is not simply a prion disorder. J. Neurosci. 37, 9799–9807. doi: 10.1523/jneurosci.1787-16.2017
Svensson, E., Horváth-Puhó, E., Thomsen, R. W., Djurhuus, J. C., Pedersen, L., Borghammer, P., et al. (2015). Vagotomy and subsequent risk of Parkinson’s disease. Ann. Neurol. 78, 522–529. doi: 10.1002/ana.24448
Tanaka, Y., Engelender, S., Igarashi, S., Rao, R. K., Wanner, T., Tanzi, R. E., et al. (2001). Inducible expression of mutant α-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum. Mol. Genet. 10, 919–926. doi: 10.1093/hmg/10.9.919
Tapias, V., Hu, X., Luk, K. C., Sanders, L. H., Lee, V. M., and Greenamyre, J. T. (2017). Synthetic α-synuclein fibrils cause mitochondrial impairment and selective dopamine neurodegeneration in part via iNOS-mediated nitric oxide production. Cell. Mol. Life Sci. 74, 2851–2874. doi: 10.1007/s00018-017-2541-x
Thaisetthawatkul, P. (2016). Pure autonomic failure. Curr. Neurol. Neurosci. Rep. 16:74. doi: 10.1007/s11910-016-0673-2
Theillet, F. X., Binolfi, A., Bekei, B., Martorana, A., Rose, H. M., Stuiver, M., et al. (2016). Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature 530, 45–50. doi: 10.1038/nature16531
Theodore, S., Cao, S., Mclean, P. J., and Standaert, D. G. (2008). Targeted overexpression of human α-synuclein triggers microglial activation and an adaptive immune response in a mouse model of Parkinson disease. J. Neuropathol. Exp. Neurol. 67, 1149–1158. doi: 10.1097/nen.0b013e31818e5e99
Tolosa, E., Vila, M., Klein, C., and Rascol, O. (2020). LRRK2 in Parkinson disease: challenges of clinical trials. Nat. Rev. Neurol. 16, 97–107. doi: 10.1038/s41582-019-0301-2
Tosatto, L., Andrighetti, A. O., Plotegher, N., Antonini, V., Tessari, I., Ricci, L., et al. (2012). α-synuclein pore forming activity upon membrane association. Biochim. Biophys. Acta 1818, 2876–2883. doi: 10.1016/j.bbamem.2012.07.007
Tozzi, A., De Iure, A., Bagetta, V., Tantucci, M., Durante, V., Quiroga-Varela, A., et al. (2016). α-synuclein produces early behavioral alterations via striatal cholinergic synaptic dysfunction by interacting with GluN2D N-methyl-D-aspartate receptor subunit. Biol. Psychiatry 79, 402–414. doi: 10.1016/j.biopsych.2015.08.013
Tremblay, M. E., Cookson, M. R., and Civiero, L. (2019). Glial phagocytic clearance in Parkinson’s disease. Mol. Neurodegener. 14:16. doi: 10.1186/s13024-019-0314-8
Tsigelny, I. F., Sharikov, Y., Wrasidlo, W., Gonzalez, T., Desplats, P. A., Crews, L., et al. (2012). Role of α-synuclein penetration into the membrane in the mechanisms of oligomer pore formation. FEBS J. 279, 1000–1013. doi: 10.1111/j.1742-4658.2012.08489.x
Tysnes, O. B., Kenborg, L., Herlofson, K., Steding-Jessen, M., Horn, A., Olsen, J. H., et al. (2015). Does vagotomy reduce the risk of Parkinson’s disease? Ann. Neurol. 78, 1011–1012. doi: 10.1097/01.nt.0000475926.46736.74
Ubhi, K., Rockenstein, E., Mante, M., Patrick, C., Adame, A., Thukral, M., et al. (2008). Rifampicin reduces α-synuclein in a transgenic mouse model of multiple system atrophy. Neuroreport 19, 1271–1276. doi: 10.1097/wnr.0b013e32830b3661
Ulusoy, A., Rusconi, R., Perez-Revuelta, B. I., Musgrove, R. E., Helwig, M., Winzen-Reichert, B., et al. (2013). Caudo-rostral brain spreading of α-synuclein through vagal connections. EMBO Mol. Med. 5, 1119–1127. doi: 10.1002/emmm.201302475
Uversky, V. N., Li, J., and Fink, A. L. (2001). Metal-triggered structural transformations, aggregation and fibrillation of human α-synuclein. A possible molecular NK between Parkinson’s disease and heavy metal exposure. J. Biol. Chem. 276, 44284–44296. doi: 10.1074/jbc.m105343200
Van Der Perren, A., Gelders, G., Fenyi, A., Bousset, L., Brito, F., Peelaerts, W., et al. (2020). The structural differences between patient-derived α-synuclein strains dictate characteristics of Parkinson’s disease, multiple system atrophy and dementia with Lewy bodies. Acta Neuropathol. 139, 977–1000. doi: 10.1007/s00401-020-02157-3
Varkey, J., Isas, J. M., Mizuno, N., Jensen, M. B., Bhatia, V. K., Jao, C. C., et al. (2010). Membrane curvature induction and tubulation are common features of synucleins and apolipoproteins. J. Biol. Chem. 285, 32486–32493. doi: 10.1074/jbc.m110.139576
Vasquez, V., Mitra, J., Hegde, P. M., Pandey, A., Sengupta, S., Mitra, S., et al. (2017). Chromatin-bound oxidized α-synuclein causes strand breaks in neuronal genomes in in vitro models of Parkinson’s disease. J. Alzheimers Dis. 60, S133–S150. doi: 10.3233/jad-170342
Venezia, S., Refolo, V., Polissidis, A., Stefanis, L., Wenning, G. K., and Stefanova, N. (2017). Toll-like receptor 4 stimulation with monophosphoryl lipid A ameliorates motor deficits and nigral neurodegeneration triggered by extraneuronal alpha-synucleinopathy. Mol. Neurodegener. 12:52. doi: 10.1186/s13024-017-0195-7
Vicario, M., Cieri, D., Brini, M., and Cali, T. (2018). The close encounter between α-synuclein and mitochondria. Front. Neurosci. 12:388. doi: 10.3389/fnins.2018.00388
Vicente Miranda, H., Cassio, R., Correia-Guedes, L., Gomes, M. A., Chegao, A., Miranda, E., et al. (2017). Posttranslational modifications of blood-derived α-synuclein as biochemical markers for Parkinson’s disease. Sci. Rep. 7:13713. doi: 10.1038/s41598-017-14175-5
Volpicelli-Daley, L. A. (2017). Effects of α-synuclein on axonal transport. Neurobiol. Dis. 105, 321–327. doi: 10.1016/j.nbd.2016.12.008
Volpicelli-Daley, L. A., Luk, K. C., Patel, T. P., Tanik, S. A., Riddle, D. M., Stieber, A., et al. (2011). Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72, 57–71. doi: 10.1016/j.neuron.2011.08.033
Wang, W., Nguyen, L. T., Burlak, C., Chegini, F., Guo, F., Chataway, T., et al. (2016). Caspase-1 causes truncation and aggregation of the Parkinson’s disease-associated protein α-synuclein. Proc. Natl. Acad. Sci. U S A 113, 9587–9592. doi: 10.1073/pnas.1610099113
Wang, W., Perovic, I., Chittuluru, J., Kaganovich, A., Nguyen, L. T., Liao, J., et al. (2011). A soluble α-synuclein construct forms a dynamic tetramer. Proc. Natl. Acad. Sci. U S A 108, 17797–17802. doi: 10.1073/pnas.1113260108
Wang, X., Becker, K., Levine, N., Zhang, M., Lieberman, A. P., Moore, D. J., et al. (2019). Pathogenic α-synuclein aggregates preferentially bind to mitochondria and affect cellular respiration. Acta Neuropathol. Commun. 7:41. doi: 10.1186/s40478-019-0696-4
Wang, X. J., Ma, M. M., Zhou, L. B., Jiang, X. Y., Hao, M. M., Teng, R. K. F., et al. (2020). Autonomic ganglionic injection of α-synuclein fibrils as a model of pure autonomic failure alpha-synucleinopathy. Nat. Commun. 11:934. doi: 10.1038/s41467-019-14189-9
Watts, J. C. (2019). Calling α-synuclein a prion is scientifically justifiable. Acta Neuropathol. 138, 505–508. doi: 10.1007/s00401-019-02058-0
Wegrzynowicz, M., Bar-On, D., Calo, L., Anichtchik, O., Iovino, M., Xia, J., et al. (2019). Depopulation of dense α-synuclein aggregates is associated with rescue of dopamine neuron dysfunction and death in a new Parkinson’s disease model. Acta Neuropathol. 138, 575–595. doi: 10.1007/s00401-019-02023-x
Wenning, G., Trojanowski, J. Q., Kaufmann, H., Wisniewski, T., Rocca, W. A., and Low, P. A. (2018). Is multiple system atrophy an infectious disease? Ann. Neurol. 83, 10–12. doi: 10.1002/ana.25132
Woerman, A. L., Kazmi, S. A., Patel, S., Freyman, Y., Oehler, A., Aoyagi, A., et al. (2018). MSA prions exhibit remarkable stability and resistance to inactivation. Acta Neuropathol. 135, 49–63. doi: 10.1007/s00401-017-1762-2
Wu, Q., Takano, H., Riddle, D. M., Trojanowski, J. Q., Coulter, D. A., and Lee, V. M. (2019). α-synuclein (αSyn) preformed fibrils induce endogenous alphasyn aggregation, compromise synaptic activity and enhance synapse loss in cultured excitatory hippocampal neurons. J. Neurosci. 39, 5080–5094. doi: 10.1523/jneurosci.0060-19.2019
Yamasaki, T. R., Holmes, B. B., Furman, J. L., Dhavale, D. D., Su, B. W., Song, E. S., et al. (2019). Parkinson’s disease and multiple system atrophy have distinct α-synuclein seed characteristics. J. Biol. Chem. 294, 1045–1058. doi: 10.1074/jbc.RA118.004471
Yu, S., Uéda, K., and Chan, P. (2005). α-synuclein and dopamine metabolism. Mol. Neurobiol. 31, 243–254. doi: 10.1385/MN:31:1-3:243
Zafar, F., Valappil, R. A., Kim, S., Johansen, K. K., Chang, A. L. S., Tetrud, J. W., et al. (2018). Genetic fine-mapping of the Iowan SNCA gene triplication in a patient with Parkinson’s disease. NPJ Parkinsons Dis. 4:18. doi: 10.1038/s41531-018-0054-4
Zhang, J., Li, X., and Li, J. D. (2019). The roles of post-translational modifications on α-synuclein in the pathogenesis of Parkinson’s diseases. Front. Neurosci. 13:381. doi: 10.3389/fnins.2019.00381
Zhang, W., Wang, T., Pei, Z., Miller, D. S., Wu, X., Block, M. L., et al. (2005). Aggregated α-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J. 19, 533–542. doi: 10.1096/fj.04-2751com
Zhao, H. T., John, N., Delic, V., Ikeda-Lee, K., Kim, A., Weihofen, A., et al. (2017). LRRK2 antisense oligonucleotides ameliorate α-synuclein inclusion formation in a Parkinson’s disease mouse model. Mol. Ther. Nucleic Acids 8, 508–519. doi: 10.1016/j.omtn.2017.08.002
Keywords: α-synuclein, α-synucleinopathies, Parkinson’s disease, neurodegeneration, prion
Citation: Heras-Garvin A and Stefanova N (2020) From Synaptic Protein to Prion: The Long and Controversial Journey of α-Synuclein. Front. Synaptic Neurosci. 12:584536. doi: 10.3389/fnsyn.2020.584536
Received: 17 July 2020; Accepted: 26 August 2020;
Published: 21 September 2020.
Edited by:
Taher Darreh-Shori, Karolinska Institutet (KI), SwedenReviewed by:
Paolo Calabresi, Catholic University of the Sacred Heart, ItalyQihui Wu, University of Pennsylvania, United States
Copyright © 2020 Heras-Garvin and Stefanova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonio Heras-Garvin, YW50b25pby5oZXJhc0BpLW1lZC5hYy5hdA==; Nadia Stefanova, bmFkaWEuc3RlZmFub3ZhQGktbWVkLmFjLmF0