 Jörg Striessnig
Jörg Striessnig- Department of Pharmacology and Toxicology, Institute of Pharmacy, Center for Molecular Biosciences Innsbruck, University of Innsbruck, Innsbruck, Austria
This review summarizes our current knowledge of human disease-relevant genetic variants within the family of voltage gated Ca2+ channels. Ca2+ channelopathies cover a wide spectrum of diseases including epilepsies, autism spectrum disorders, intellectual disabilities, developmental delay, cerebellar ataxias and degeneration, severe cardiac arrhythmias, sudden cardiac death, eye disease and endocrine disorders such as congential hyperinsulinism and hyperaldosteronism. A special focus will be on the rapidly increasing number of de novo missense mutations identified in the pore-forming α1-subunits with next generation sequencing studies of well-defined patient cohorts. In contrast to likely gene disrupting mutations these can not only cause a channel loss-of-function but can also induce typical functional changes permitting enhanced channel activity and Ca2+ signaling. Such gain-of-function mutations could represent therapeutic targets for mutation-specific therapy of Ca2+-channelopathies with existing or novel Ca2+-channel inhibitors. Moreover, many pathogenic mutations affect positive charges in the voltage sensors with the potential to form gating-pore currents through voltage sensors. If confirmed in functional studies, specific blockers of gating-pore currents could also be of therapeutic interest.
Introduction
Decreasing cost for next-generation sequencing (NGS) has allowed to diagnose pathogenic mutations in an increasing number of patients affected by genetic diseases. Genome-wide association studies mainly identify disease-associated common genetic variants, which explain only a very small proportion of overall phenotypic variance. In contrast, trio-based whole-exome or whole-genome sequencing genetic studies in well-phenotyped patient cohorts can detect rare, recurrent damaging variants of large effect size that can account for most phenotypic variance and can be considered causative (Sullivan and Geschwind, 2019). Such variants explain a wide variety of symptoms, especially in early-onset sporadic pediatric genetic disorders (Consortium et al., 2013; Allen et al., 2017; Satterstrom et al., 2020). This informs not only diagnosis and genetic counseling, but can also refine a patients’ prognosis, initiate surveillance for other symptoms in multisystem disorders and guide therapeutic decision-making (Helbig et al., 2016; Weber et al., 2017; Noebels, 2019). In addition, the identification of multiple risk genes within defined signaling networks provides valuable novel insight into dysregulated molecular pathways underlying a pathology.
Disorders that have strongly benefited from NGS – based genetic diagnostics are neuropsychiatric and neurodevelopmental disorders, including epilepsies, in particular childhood epilepsies, autism spectrum disorders (ASD), and intellectual disability (ID) with a large collective contribution of rare, large-effect pathogenic variants (Allen et al., 2017; Howard and Baraban, 2017; Weber et al., 2017; Myers et al., 2020; Satterstrom et al., 2020). Rare large-effect variants in the same gene may not be specific for one of these disorders (e.g., ASD, Myers et al., 2020) and other sources of genetic variation, such as polygenic risk from common variants or genetic mosaicism, as well as environmental factors can affect phenotypic expression. Whereas the significance of a novel de novo mutation identified only in a single individual is difficult to interpret, the identification of a recurrent pathogenic variant strongly supports a large genetic contribution to the phenotype in an individual within this disease spectrum. In some cases, this can even guide therapies either with new drugs or by repurposing drugs already licensed for other indications. Although often experimental in nature and mainly symptomatic, such individualized therapies can enhance the quality of life for affected individuals and their caregivers. For example, everolimus has been repurposed for the treatment of Tuberosclerosis TSC1 and TSC2 (Krueger et al., 2016; Samueli et al., 2016), and 5-hydroxytryptophan is used to treat patients with DOPA-responsive dystonia (DRD, OMIM #128230) caused by Sepiapterin Reductase Deficiency (Bainbridge et al., 2011). Among patients with epilepsy referred to diagnostic whole exome sequencing the diagnostic yield of characterized disease genes is > 30% (Helbig et al., 2016; Symonds and McTague, 2020) and genetic diagnosis has significant impact on patient management, such as choosing or avoiding certain antiepileptic medications (Symonds and McTague, 2020).
Some rare disease phenotypes are associated with only a single or a few defective genes (like TSC or DRD). More common, pathophysiologically more complex, and clinically often overlapping phenotypes such as ASD, childhood epilepsies, ID or cardiac arrhythmias are associated with a larger number of risk genes (e.g., over 100 in ASD and more than 650 in ID; Kochinke et al., 2016; Satterstrom et al., 2020). However, most of them account only for a very small fraction of affected individuals. This increases the probability that a new sporadic variant of unknown pathogenicity is found in one of these genes. In the case of de novo mutations (DNMs, newly formed during gamete development or very early in embryonic development, not inherited from the parents), bioinformatics pipelines can help to separate likely disease - causing from other variants. This involves eliminating variants exceeding a defined allele frequency in likely unaffected control populations as, for example, available in the gnomAD database (Karczewski et al., 2020). Validated DNMs can then be classified according to the predicted impact of the genetic variation on gene and protein function. A protein loss of function (pLOF) can reliably be predicted from de novo deleterious, likely gene-disrupting mutations, such as nonsense, stop gain, splice site or frameshift variants. In contrast, missense DNMs change the amino acid composition of a protein but usually do not prevent its complete translation. Therefore, their functional consequences are more difficult to predict: they can be functionally silent, induce a decrease (loss-of-function, LOF) or an increase (gain-of-function, GOF) of protein function. On average each offspring harbors at least one de novo protein – coding missense variant (Gratten et al., 2013; Krumm et al., 2015). Therefore, in diseases with many risk genes (see above) the probability of observing a new missense DNM in one of them by chance is high but its pathogenicity remains often unclear, even if present in a known high risk gene. Therefore, standards and guidelines for the interpretation of sequence variants have been developed by the American College of Medical Genetics (ACMG, Richards et al., 2015) to classify variants according to their predicted pathogenicity (“pathogenic”, “likely pathogenic”, “uncertain significance”, “likely benign”, and “benign”). Ideally, functional assays are available to test for GOF or LOF changes, which can support classification as is the case for voltage-gated Ca2+-channel (Cavs, see section “Functional Analysis of Protein-Coding Missense Variants”).
This review summarizes our current knowledge of disease-relevant genetic variants within the family of voltage gated Ca2+-channels. A special focus will be on missense DNMs in their pore-forming α1-subunits, which are increasingly reported in NGS studies and can help to explain sporadic cases of Ca2+-channel-associated pathologies. These include epilepsies, ASD, ID, developmental delay, cerebellar ataxias, severe cardiac arrhythmias, sudden cardiac death and endocrine disorders such as congential hyperinsulinism and hyperaldosteronism. Since potential personalized therapeutic approaches depend on whether a missense DNM induces a loss- or gain of channel function, we will discuss the complex question of how altered gating of these ion channels can translate into such functional changes.
First, this review provides a brief overview about the ten members of the Cav-channel family and highlights the most common macroscopic gating changes induced by single amino acid substitutions. Finally, the spectrum of variants known to cause Ca2+-channelopathies is described. Since the protein structures of the pore-forming Ca2+-channel α1-subunits are highly conserved (Wu et al., 2016; Zhao et al., 2019a, b; Catterall et al., 2020), we will also discuss if the location and functional effects in one of the Cav family members can help to predict pathogenicity of a novel variant in another Cav. This could refine predictions based on the ACMG guidelines and thus help in genetic counseling.
The Voltage-Gated Ca2+-Channel Family
Excellent reviews have recently been published on this topic (Zamponi et al., 2015; Catterall et al., 2020; Dolphin and Lee, 2020; Ablinger et al., 2020) and valuable detailed information on the physiology, tissue distribution and pharmacology of these channels can be found in the NC-IUPHAR Guide to Pharmacology1 (Alexander et al., 2019).
Cavs are found in all electrically excitable cells. Voltage-dependent Ca2+ influx through Cavs serves a dual function in cell signaling. It generates intracellular Ca2+ signals controlling Ca2+-dependent cellular processes. In addition, Ca2+ ion inward current also drives membrane depolarization and thus directly contributes to a cell’s electrical activity (Zamponi et al., 2015). Consequently, Cavs are essential for key physiological functions, such as learning and memory, neurotransmitter and hormone release, muscle contraction, cardiac pacemaking and sensory functions, including hearing and visual function (Zamponi et al., 2015; Nanou and Catterall, 2018; Pangrsic et al., 2018). Ten types of Cavs are formed by different pore-forming α1-subunits (Figure 1), each encoded by a separate gene (Zamponi et al., 2015; Alexander et al., 2019) (Table 1). Differences in their biophysical properties, tissue expression, subcellular targeting, association with other interacting proteins in signalosomes and in their modulation by other signaling pathways generates the functional diversity required for the many physiological functions they support (Zamponi et al., 2015; Nanou and Catterall, 2018; Liu et al., 2020). Extensive alternative splicing and association with various modulatory accessory subunits (β1-β4, α2δ1-α2δ4), posttranslational modifications and RNA-editing (Huang et al., 2012; Zamponi et al., 2015; Li et al., 2017; Loh et al., 2020) further fine-tunes their biophysical and pharmacological properties (Buraei and Yang, 2010; Zamponi et al., 2015; Ablinger et al., 2020). This requirement for tight functional control explains why even minor changes in the activity of these channels induced by genetic variants can cause human disease.
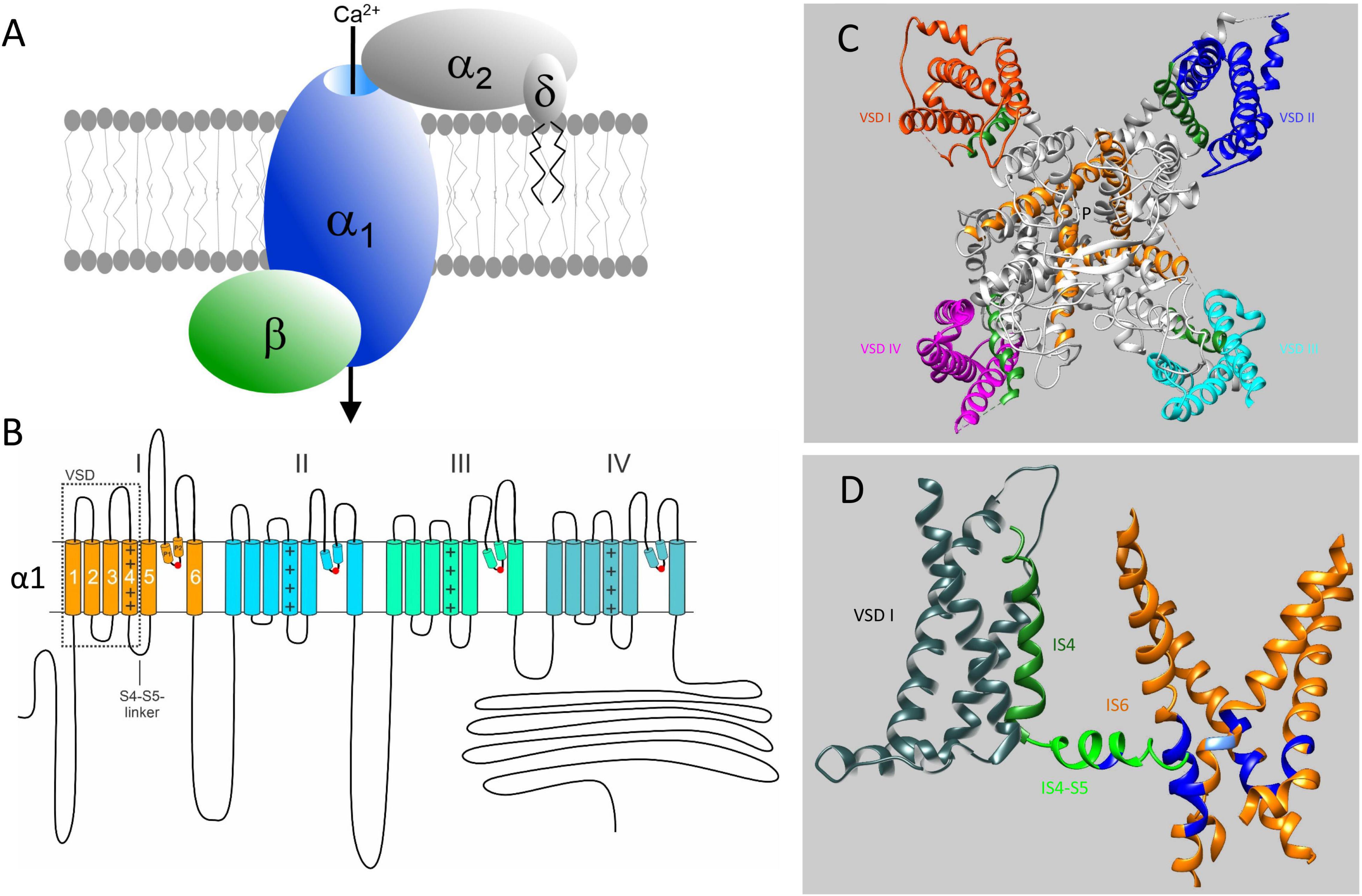
Figure 1. Subunit structure of voltage-gated Ca2+ channels and their mutation-sensitive regions. (A) The pore-forming α1-subunit determines most of the biophysical properties of voltage-gated Ca2+ channels and also carries the binding domains for subtype-selective drugs and toxins. β-and α2δ-subunits associate with α1-subunits of Cav1 and Cav2 channels but not with Cav3 T-type channels. They support channel trafficking to the membrane, fine-tune gating properties but appear to have also channel-independent functions. Here we only discuss pathogenic mutations in α1-subunit genes. (B) Transmembrane folding topology of α1-subunits. Four homologous domains (I-IV) each form a voltage sensing domain (VSD, S1-S4; dotted box). The S6 helices together with the connecting S5-S6 linkers of each domain contribute to the formation of a single central Ca2+-selective pore. The S4-S5 linkers in each domain transmit the voltage-dependent conformational changes of the S4-helix movements to the cytoplasmic side of the pore by interactions with adjacent S4-S5 – linkers (Hofer et al., 2020) and S6 helices (Wu et al., 2016; Catterall et al., 2020). Mutations neutralizing the voltage-sensing positive charges in each voltage sensor can open an additional ion conducting pathway (termed ω-pore) through the voltage –sensing domain, which can conduct pathogenic gating pore currents (see Figure 5 and text for details). P1 and P2 are helices contributing to the formation of the external part of the pore. They coordinate the formation of the Ca2+-selectivity filter formed by four negatively charged amino acid residues indicated by the red circles. (C) Cryo-electron microscopy structure of the Cav1.1 calcium channel complex purified from rabbit skeletal muscle (PDB 5GJV). Only a top view of the pore-forming α1-subunit is shown to illustrate the position of the four voltage-sensing domains (VSD I – IV, highlighted in different colors). The voltage-sensing positively charged S4 helices of the VSD domains are shown in green (positive charges are not indicated). The central pore-forming region including the intracellular activation gate are formed by the S6 helices indicated in orange. P indicates the ion conducting pathway. The structure is in a presumably inactivated state with S4 voltage-sensors “up” and the activation gate closed (Wu et al., 2016). (D) Most of the missense mutations (in particular GOF mutations) causing Ca2+-channelopathies occur in regions important for voltage-dependent channel gating. These functional modules consists of S4 (green) and the cytoplasmic S4-S5 linkers (light green), which are tightly coupled through multiple interactions (not illustrated) to the activation gate formed by the four S6 helices (orange). For clarity the VSD (gray), S4 and S4-S5 linker are only shown for domain I together with all 4 S6 helices (orange). Positions where pathogenic mutations occur in all Cavs (Figures 3, 4) are indicated in blue. In IS4-S5 this represents the position of the FHM1 mutation S218L (CACNA1A) and the CSNB2 mutation S229P (CACNA1F), which both cause GOF (type 2 gating; see text for details). The position of the Timothy Syndrome mutation G402R/S (CACNA1C) is indicated in light blue. Note that pathogenic GOF mutations at the same position also occur in Cav1.3, Cav1.4, and Cav2.3 (Figure 3). The schemes in (C,D) were generated using UCSF Chimera 1.13.1 (Pettersen et al., 2004). The position of mutations in the Cav1.1 α1-subunit is shown based on the sequence alignments in Figures 3, 4 and does not account for potential differences in the folding structure of the different α1-subunits.

Table 1. Voltage-gated Ca2+ channels: classification and human genetic diseases.
Based on sequence homology between their pore-forming α1-subunits (Figure 1) as well as their functional and pharmacological properties, three Cav families are distinguished: Cav1, Cav2 and Cav3 (Table 1; Alexander et al., 2019).
Cav1.1-Cav1.4 α1- subunits form the family of L-type Ca2+-channels (Cav1). They have unique sensitivity to low nanomolar concentrations of dihydropyridine (DHP) Ca2+-channel blockers (Alexander et al., 2019; Table 1). Cav1.2 and Cav1.3 are expressed, often together, in most electrically excitable cells (Zamponi et al., 2015). In contrast, Cav1.1 and Cav1.4 expression and function is largely restricted to skeletal muscle (Cav1.1) and the retina (Cav1.4). Cav1.2 controls cardiac inotropy and arterial vascular tone (Zamponi et al., 2015). It is the main therapeutic target for Ca2+-channel blockers (such as amlodipine, felodipine), which are approved since decades for the treatment of angina and hypertension (Striessnig and Ortner, 2020). Cav1.3 activates at more negative voltages than Cav1.2 (Koschak et al., 2001; Zamponi et al., 2015). Therefore it serves as a pacemaker channel in the sinoatrial and AV-node (Marger et al., 2014) and is essential for cochlear inner hair cell function and hearing (Zamponi et al., 2015). Cav1.2 and Cav1.3 are both present in central neurons, predominantly postsynaptically at dendritic spines. Both channels control the short- and long-term regulation of neuronal activity in several brain circuits, and contribute to different types of learning, memory and emotional behaviors (Zamponi et al., 2015; Kabir et al., 2017; Nanou and Catterall, 2018).
Members of the Cav2 family (Cav2.1-Cav2.3, giving rise to P/Q-, N- and R-type voltage-gated Ca2+ currents) are also located at pre-synaptic active zones and support fast neurotransmitter release in neurons. Together with L-type channels they also trigger hormone release in endocrine cells (Zamponi et al., 2015).
Low-voltage activated Cav3 channels (Cav3.1-Cav3.3) comprise the family of T-type Ca2+-channels. They activate and inactivate at more negative membrane potentials than Cav1 (including Cav1.3) and Cav2 (Perez-Reyes and Lory, 2006; Zamponi et al., 2015). This negative operation range allows them to be active at subthreshold voltages, and provides them with a prominent role for integration and control of neuronal firing patterns (Kim et al., 2001; Zamponi et al., 2015).
Recently, the cryo-EM structures of the pore-forming α1-subunits of two members of the Cav family have been solved in complex with selective channel blockers: Cav1.1 (Wu et al., 2016; Zhao et al., 2019b; Gao and Yan, 2020) and the evolutionary more distantly related Cav3.1 (Zhao et al., 2019a) channel. Together with previous work on related channels (Catterall et al., 2020), these structures provide exciting new information about the molecular details involved in voltage-sensing, channel gating (Figures 1C,D, based on the cryo-EM structure of Cav1.1; Wu et al., 2016) and ion permeation as well as isoform-specific functional and pharmacological differences. As will be discussed below, these common structural features can be instrumental for predicting functional consequences of disease-causing mutations across different α1-subunits.
Gain- and Loss- of Ca2+-Channel Function Induced by Missense Mutations
For proper function and subcellular targeting Cav1 and Cav2 channels require association with one of four different β- and one of four different α2δ-subunits (Figure 1A; Buraei and Yang, 2010; Zamponi et al., 2015; Ablinger et al., 2020). Therefore, genetic variants in any of these accessory subunits can indirectly affect channel function. Signaling changes resulting from genetic variation in these subunits are mechanistically more complex to interpret, because any of the four different β- and of the four different α2δ isoforms may associate with the different Cav1 and Cav2 α1- subunits in a given cell (such as a neuron). Moreover, these subunits also appear to serve functions independent of Cavs (Hofmann et al., 2015; Dolphin, 2018; Striessnig, 2018). This review therefore exclusively focuses on channelopathies resulting from genetic variants in the pore forming α1-subunit genes. These can be directly related to the known physiological functions of a particular channel type and thus provide more specific hints for molecular disease mechanisms.
Functional Analysis of Protein-Coding Missense Variants
As mentioned above, the functional consequences of protein-coding missense DNMs (including in-frame deletions/insertions) are more difficult to predict and different amino acid substitutions at the same position may even cause opposite functional effects (Hofer et al., 2020). While missense DNMs may also induce a LOF by generating functionally silent channels (e.g., by an in-frame glycine insertion in Cav1.3 α1; Baig et al., 2011), they can have more complex effects by affecting channel gating or ion conductance. In this case, it is difficult to predict if a given gating change enhances or reduces channel activity in the context of a cell’s characteristic firing pattern. It is very important that we keep this in mind when referring to a “GOF” phenotype. It only implies that functional changes induced by a mutation can permit enhanced channel activity and Ca2+ influx under certain circumstances, such as during a given electrical firing pattern of a cell. As outlined below, it does not necessarily exclude that the same “GOF” variant can also reduce channel activity in a different functional context.
Analysis of mutation-induced functional changes in cells expressing Cavs is complicated by the fact that tissues cannot be routinely obtained from patients (skeletal muscle biopsies are an exception). Moreover, individual Cavs are difficult to study in native tissues, because often currents of many different Cav types contribute to the total Ca2+ current in a cell. This requires isolation of the current component of interest, which is possible for some (e.g., Cav2.1-mediated P/Q-type currents in cerebellar Purkinje cells, Cav1.2-mediated L-type currents in cardiomyocytes; see below) but very difficult for others (e.g., Cav1.3-mediated L-type currents, Cav2.3-mediated R-type currents; Forti and Pietrobon, 1993; Sinnegger-Brauns et al., 2004). Moreover, currents recorded in native cells expressing heterozygous variants are “contaminated” by wildtype currents, making subtle changes of mutant current components more difficult to detect than in a heterologous system (Miki et al., 2008).
These restrictions require expression of missense variants in mammalian heterologous systems, such as HEK-293 or baby hamster kidney cells, and quantification of functional changes in patch-clamp experiments. In general, it appears that macroscopic gating changes observed in heterologous expression systems reproduce the changes observed in native cells reasonably well. This is evident from animal models harboring specific human mutations (Drum et al., 2014; Rose et al., 2014; Calorio et al., 2019) or from human cells differentiated from patient-derived induced pluripotent stem cells (iPSCs; e.g., neuron-like or cardiomyocyte-like cells; Yazawa et al., 2011; Krey et al., 2013; Song et al., 2017; Estes et al., 2019; Chavali et al., 2019). In contrast, it is less clear how well changes of current density and of channel protein expression observed in heterologous overexpression systems reflect changes of current density in native tissues (Chavali et al., 2019). Obviously, many other regulatory factors absent in heterologous expression systems, in particular cell-type specific protein-protein interactions, affect channel protein stability and its plasma membrane-targeting in their native environment.
Special attention should also be paid to missense mutations located in alternatively spliced exons, which may restrict the mutation only to channels expressing this exon. Conversely, a single missense mutation may even promote the expression of the exon in which it is located. This has recently been shown in iPSC-derived neural progenitor cells and neurons (Panagiotakos et al., 2019) generated from patients with Timothy syndrome (see below). The GOF mutation G406R in exon 8a of CACNA1C (Cav1.2α1; see section on CACNA1C below) inhibits the developmental splicing switch to exon 8 – containing Cav1.2 channels, which do not harbor the mutation. This favors extended use of the mutant, exon 8a-containing channel permitting enhanced mutant channel activity during brain development. This is likely due to the position of the mutation in the splice acceptor site of exon 8a (Panagiotakos et al., 2019).
Channel Loss of Function Mutations
Like for other proteins (see above), genetic variants can cause disease (or increase disease risk) by disrupting channel function. This can be predicted for likely gene-disrupting mutations expected to prevent the synthesis of functional α1-subunit proteins (pLOF). If no protein is made from the defective allele, heterozygous LOF mutations may cause disease resulting from haploinsufficiency. However, if a defective (e.g., truncated) protein is still synthesized it may even exert a dominant negative effect on the wildtype channel produced from the unaffected allele. This requires a mechanism enabling functional coupling between (wildtype and mutant) α1-subunits. In addition, the mutant RNA must escape nonsense-mediated RNA decay (Holbrook et al., 2004). In the case of Cavs such dominant negative effects appear likely: substantial expression of mRNA transcripts from mutated alleles of both frameshift and nonsense mutations (in the case of Cav2.1) has been reported in humans (Sintas et al., 2017; Balck et al., 2018). Moreover, binding of misfolded mutants to wildtype α1-subunits occurs in vitro thereby promoting channel degradation (Mezghrani et al., 2008; Page et al., 2010). It is therefore possible that some heterozygous LOF mutations will reduce channel activity even below 50%, a consideration especially important for understanding the pathology of Cav2.1 channelopathies (see below). However, it should be kept in mind that the in vitro studies were carried out with cDNA constructs in heterologous overexpression systems with yet unclear relevance for the in vivo situation, and the detection of transcripts of the mutant alleles is still very indirect evidence.
Common Macroscopic Gating Changes Induced by Protein-Coding Missense Mutations
Missense-mutations can affect the macroscopic gating properties of Cavs in a complex manner, evident as changes of the voltage-dependence of current activation and inactivation. These alterations are very similar across different Cavs. For further discussion Figure 2 illustrates the most frequently observed gating changes in a simplified manner as type 1 – 4, although overlaps exist between them (Pinggera et al., 2018; Ortner and Striessnig, 2020). A gating change unambiguously interpretable as a phenotype permitting enhanced channel function (GOF) is illustrated in panel A (“type 1”). Its hallmark is an almost complete failure of a large fraction of current to inactivate during depolarization. This is often quantified in whole-cell patch-clamp experiments during square pulse depolarizations (ΔV) from a negative holding potential (hp) to the maximum of the voltage of maximal inward current (Vmax, Figure 3A, right panel). This failure to inactivate can even prevent meaningful measurements of steady-state inactivation. At the same time, the half-maximal activation voltage (V0,5,act) can be either unchanged or shifted to more negative voltages (blue curves, Figure 2A, left panel). Note that the time course of inactivation is controlled by Ca2+-dependent as well as voltage-dependent inactivation. The latter occurs in the absence of Ca2+ and with Ba2+ as charge carrier (for reviews see Ben-Johny et al., 2015; Hardie and Lee, 2016). For L-type channels the differential effects on Ca2+- and voltage-dependent inactivation have been quantified for some mutations, showing that this pronounced slowing of inactivation is mainly due to reduced voltage-dependent inactivation (Splawski et al., 2004; Pinggera et al., 2018). Why can we be confident that this type of macroscopic gating change in HEK-293 cells can also translate to enhanced cellular Ca2+ signaling in vivo? As mentioned below, a type 1 gating change has been reported for Cav1.2 channels in LQT8 and explains the prolongation of the cardiac action potential driven by the failure of the channel to inactivate (Yazawa et al., 2011; Drum et al., 2014; Estes et al., 2019). Moreover, in Cav1.3 channels type 1 gating is associated with ASD (G407R, Pinggera et al., 2015) and a more severe neurodevelopmental syndrome (G403D, Scholl et al., 2013; Figure 3A) which cannot be explained by a Cav1.3 channel LOF (Ortner and Striessnig, 2020). In addition, the GOF phenotype is supported by somatic mutations in Cav1.3 (G403R,G403D) in aldosterone-producing adenomas (APAs), in which enhanced Ca2+ entry is known to drive excess Ca2+-dependent aldosterone production (Boulkroun et al., 2020). When expressed in HEK-293 cells, some type 1 mutations result in smaller current amplitudes but, due to slow inactivation, mutant currents exceed the amplitude of wildtype currents upon prolonged depolarization (Figure 2A, right panel, dashed line; Pinggera et al., 2018). Accordingly, expression of such a type 1 Cav1.3 channel mutant (G407R) in cultured GLT muscle cells enhanced intracellular transients (Pinggera et al., 2015).
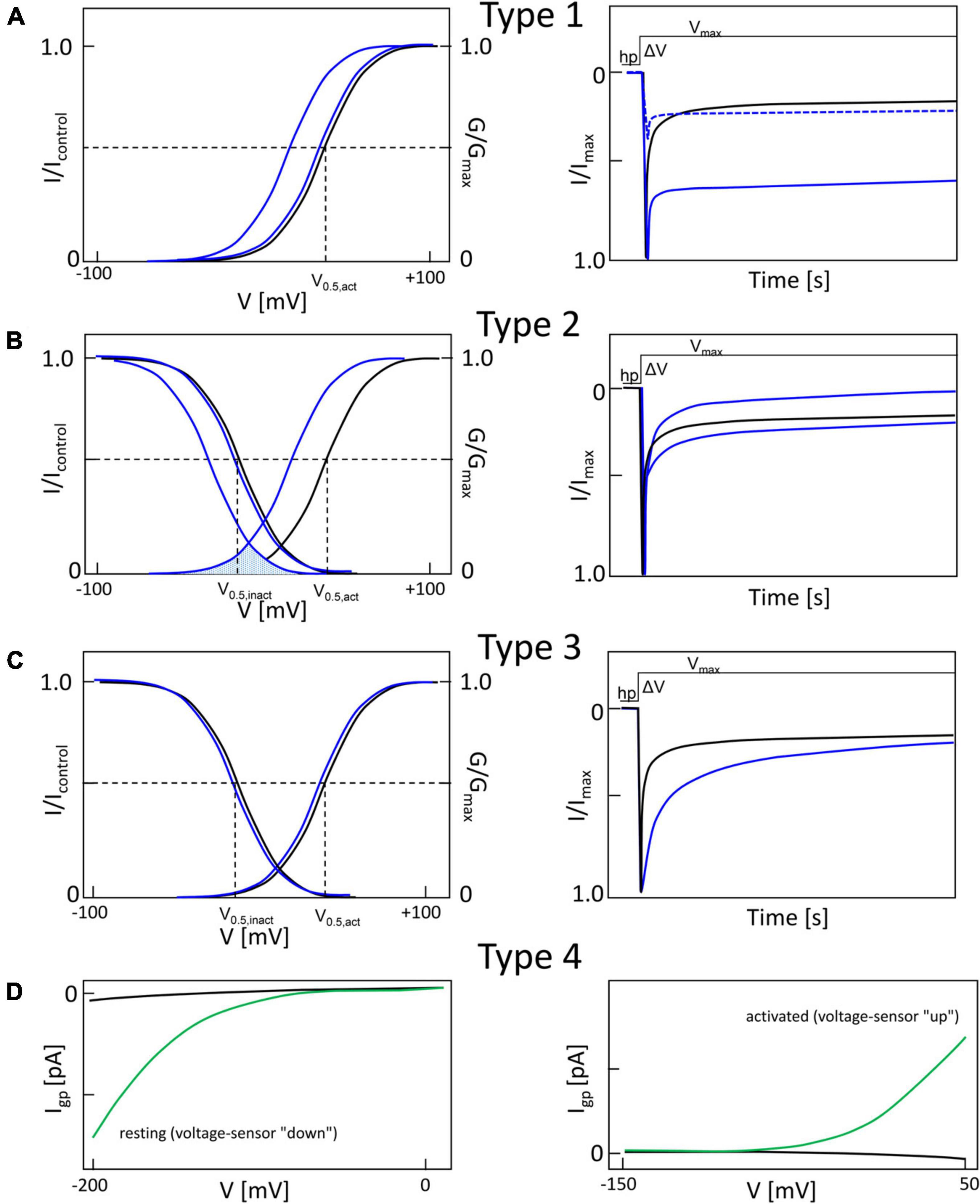
Figure 2. Typical gain-of-function macroscopic gating changes described for inherited or de novo missense mutations in voltage-gated Ca2+ channel α1-subunits. To facilitate discussion the most frequently observed gating changes are classified into types 1-4. For details see text. Mutant current properties are indicated in blue (A–C) or green curves (D), wildtype current in black. (A) Type 1 is characterized by the appearance of a large fraction of a non-inactivating current component (as e.g., during long depolarizations from a negative holding potential to the potential of maximal inward current; right panel). Changes in the voltage-dependence of activation gating (left) may or may not be present (blue curves, left). Note that even if maximal inward current is reduced by a mutation, current amplitude may exceed wildtype current later during depolarization due to the slowly inactivating current component (right, dotted curve). (B) The main feature of type 2 changes is a strong shift of the voltage-dependence of activation to more negative voltages independent of smaller effects on current inactivation during depolarizations. The voltage-dependence of inactivation may or may not be shifted to more negative voltages (left). The voltage range in which steady-state inward current (“window current”) may be observed (i.e., the overlap of voltage-dependence of activation and inactivation curves) is indicated for mutant current (shaded) (C). Some mutations do not affect the voltage-dependence of gating (left) but cause a slowing of inactivation (weaker than in type 1). (D) As described in Figure 5, gating pore currents are enabled by mutations of an S4 gating charge (green lines). The position of the mutation relative to the hydrophobic constriction site (HCS, Figure 5) determines if the pore is open during the hyperpolarized “down”-state of the S4 helix at negative voltages (left) or during the depolarized “up”-state at positive voltages (right). In the “down” state, at potentials near the K+-equilibrium potential, inward gating-pore currents would be primarily carried by Na+. In the “up” state, at potentials positive to the activation threshold of the channel, potassium outward gating-pore current predominates. However, upon fast repolarization of the action potential to negative potentials Na+ inward gating-pore current may predominate until the voltage sensor moves back to the “down” position” (especially relevant in “slowly inactivated” Na+-channels,” for details see ref. Sokolov et al., 2008). G/Gmax, normalized conductance (steady-state activation); I/Imax, normalized inward current (steady-state inactivation). V0.5,act, V0.5,inact, half maximal voltages of activation and inactivation; hp, holding potential; ΔV, depolarization from a negative holding potential to the voltage of maximal inward current (Vmax).
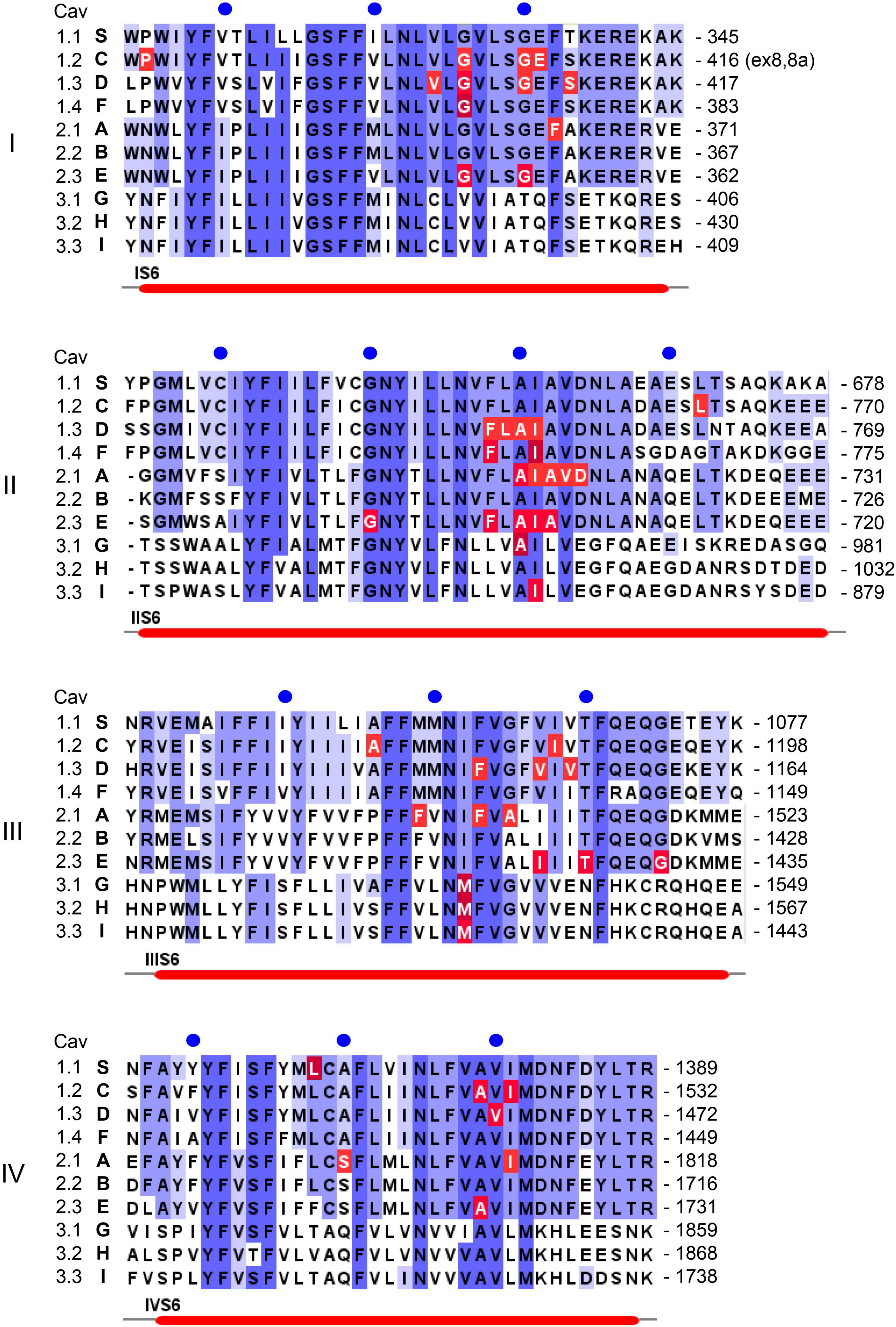
Figure 3. Position of pathogenic mutations within S6 helices of the ten different Ca2+ channel α1-subunits. Sequence alignment and labeling is as described in legend to Figure 4.
Figure 2B illustrates another typical pathological pattern, termed “type 2” for further discussion. Its characteristic feature is a shift of the activation voltage range to more negative voltages. The steady-state inactivation voltage range is either unchanged (e.g., Cav1.3-V401L, Cav2.1-S218L; Tottene et al., 2005; Pinggera et al., 2017) or is shifted to more negative potentials (e.g., Cav1.3-S652L, Cav3.1-A961T, Cav3.2-M1549V; Scholl et al., 2015; Chemin et al., 2018; Hofer et al., 2020) (Figures 3, 4). As a consequence, the voltage-range at which channels can conduct a steady-state Ca2+ inward current (the so-called “window current”, i.e., the voltage range at which the steady-state activation and inactivation curves overlap) is also shifted to more negative voltages (see legend to Figure 3B). The time course of Ca2+-current inactivation may be accelerated (e.g., Cav1.3-S652L, Cav2.1-V714A; Kraus et al., 1998; Hofer et al., 2020) or slowed (Cav1.3 –V401L, Cav2.1-S218L, Cav3.1-A961T, Cav3.2-M1549V; Tottene et al., 2005; Scholl et al., 2015; Pinggera et al., 2017; Chemin et al., 2018). A slowing of current deactivation upon repolarization can also occur but is not illustrated here (Cav1.3-S652L, Cav1.3-A749G, Limpitikul et al., 2016; Hofer et al., 2020) (Figures 3, 4). In L-type channels, this type of gating change appears to be associated with reduced Ca2+-dependent inactivation (as shown for Cav1.3 mutations V401L, S652L, A749G; Limpitikul et al., 2016; Pinggera et al., 2017; Hofer et al., 2020). While the negative shift of activation voltage is compatible with a GOF, more negative steady-state inactivation and the faster current inactivation time course can can lead to reduced channel activity. This LOF will be mainly relevant in cells, which, on average, rest at a membrane potential positive enough to allow more channels to inactivate along the more negative steady-state inactivation voltage-dependence. In addition, the faster inactivation time course would mainly be relevant if the cell’s firing pattern involves prolonged depolarized states. Therefore, it appears that more negative activation and window current are sufficient to induce GOF disease phenotypes, like in Cav2.1 for Familial hemiplegic Migraine Type I (FHM1; e.g., V714L, T501M, Hans et al., 1999; Carreño et al., 2013), in Cav1.3 for severe neurodevelopmental disorders (e.g., S652L, A749G, Ortner and Striessnig, 2020), in Cav1.2 for Timothy Syndrome (e.g., I1166T, Boczek et al., 2015) and in other Cavs (see below).
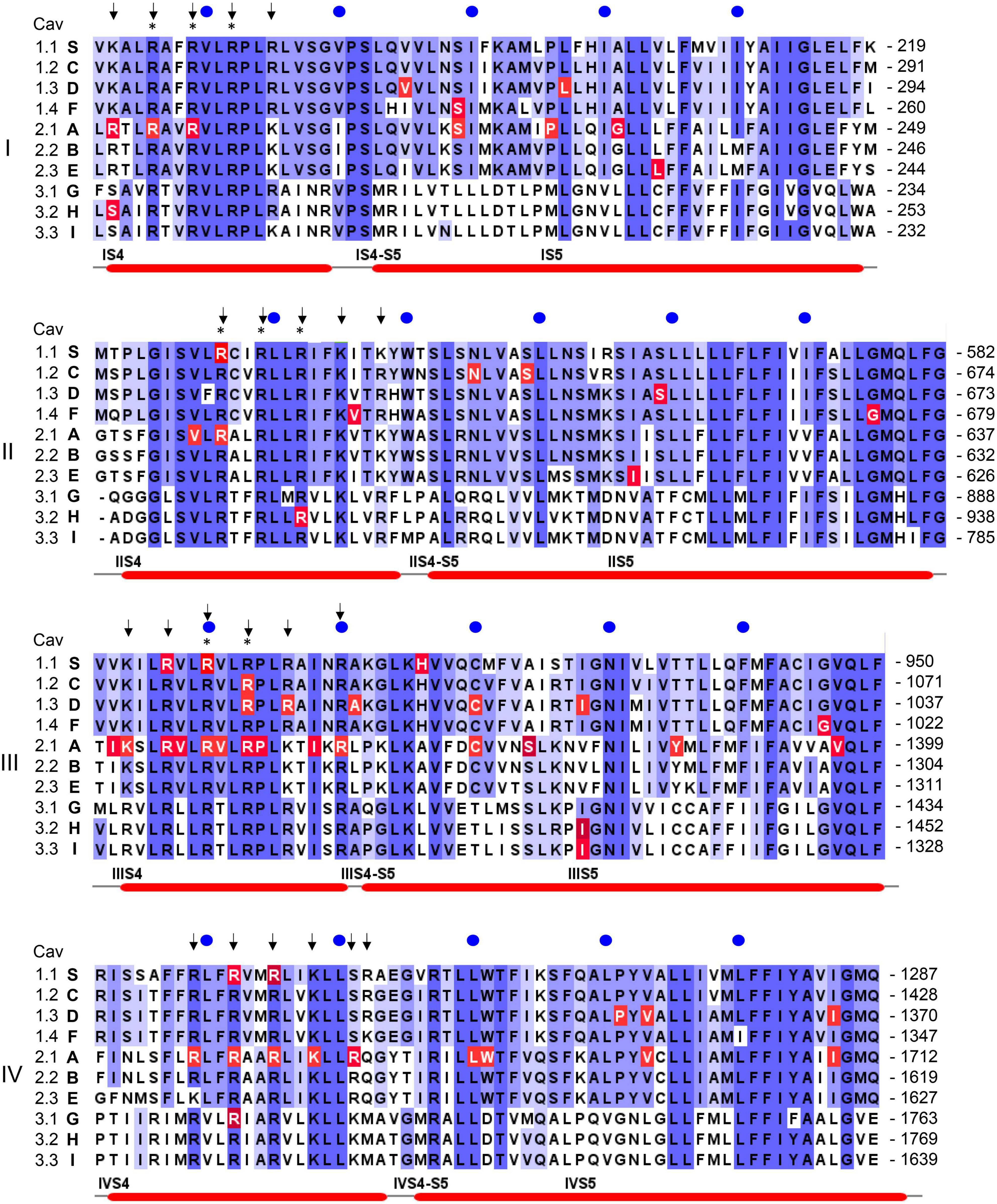
Figure 4. Position of pathogenic mutations within S4, the S4-S5 linkers and S5 helices of the ten different Ca2+ channel α1-subunits. The sequence alignment for the human α1-subunits was generated using Clustal omega and Jalview software (www.jalview.org; Waterhouse et al., 2009). Conserved residues are highlighted in blue. The accession numbers are identical to the alignment in Wu et al. (2016): CACNA1S (S, Cav1.1): Q13698; CACNA1C [C, Cav1.2; contains exon 8 (Splawski et al., 2005)]: Q13936; CACNA1D (D, Cav1.3): Q01668; CACNA1F (F, hCav1.4): O60840; CACNA1A (A, Cav2.1): O00555; CACNA1B (B, Cav2.2): Q00975; CACNA1E (E, hCav2.3): Q15878; CACNA1G (G, hCav3.1): O43497; CACNA1H (H, hCav3.2): O95180; CACNA1I (I, hCav3.3: Q9P0 × 4. All amino acid positions in the text are numbered according to the above Uniprot accession numbers and may differ in the original publications or other databases. Arrows indicate the position of positively charged residues (gating charges) in S4 helices, asterisks indicate the presence of pathogenic gating pore mutations in human Nav1.4 (SCNA4) or Nav1.5 (SCNA5) Na+-channels. Numbers on the right indicate the amino acid position of the last residue in each line, blue circles are 10 residues apart. Red lines on the bottom denote secondary structural elements of the rabbit Cav1.1 channel (Wu et al., 2016) with labels placed at the start of a feature. Residues affected by a mutation are highlighted in red. Note that not all highlighted mutations are discussed in the text. For information on mutations in all Ca2+ channels refer to the Uniprot-database and to reviews cited in the text.
Type 3 changes refer to slowing of the inactivation time course alone as a GOF feature without detectable changes in the voltage-dependence of gating (Figure 2C) and has also been found for some pathogenic mutations (e.g., Cav1.3-P1336R, Azizan et al., 2013).
In addition to macroscopic gating changes, protein-coding missense variants can also alter ion permeation through the channels by affecting unitary conductance or ion selectivity. Especially mutations close to the channels’ selectivity filter can alter single channel conductance, open probability and the voltage-dependence of single channel gating. The selectivity filter is formed by four conserved negatively charged residues (red dots in Figure 1B) held in place by two supporting pore helices (P1, P2 in Figure 1B, Wu et al., 2016; Catterall et al., 2020) in the S5-S6 linker of each domain. This has been studied for the Cav2.1 FHM1 variant T666M (2 residues N-terminal of the selectivity filter glutamate; Hans et al., 1999; Tottene et al., 2002). Mutations in the activation gate formed by the cytoplasmic ends of the S6 helices also affect ion selectivity and single channel conductance (e.g., Cav2.1-V714L, Cav1.4-I756T in IIS6; Figure 3; Hans et al., 1999; Tottene et al., 2002; Williams et al., 2020). A dramatic effect on ion selectivity has recently been reported for CACNA1F mutation I756T (Figure 3), which depends on C-terminal splicing and the associated β-subunit isoform (Williams et al., 2020).
Pathogenic variants directly hitting the negative charges forming the Ca2+ selectivity filter are rare but have been reported for Cav1.2 (E1135K) associated with LQT8 and Cav2.1 associated with a Spinocerebellar Ataxia Type 6 (SCA6) phenotype (not illustrated; see below). This can create a channel population permeable for monovalent cations with Na+-mediated inward currents and K+-mediated outward currents. It can explain the prolongation of the cardiac action potential in cardiomyocyte-like cells derived from human induced pluripotent stem cells expressing the E1135K LQT8 variant (Ye et al., 2019).
Pathogenic Gating Pore Currents
In addition to altering the ion flow through the canonical central pore, missense mutations can even generate a new ion-conducting pathway theoretically within each of the four VSDs (Figure 1C; Cannon, 2017; Groome et al., 2017; Jiang et al., 2018; Männikkö et al., 2018; Jiang D. et al., 2019). Whereas the pore-lining S6 helices of all four homologous α1-subunit domains together form the single, central Ca2+-selective pore (orange helices in Figures 1C,D), the voltage-sensing apparatus consists of four separate but cooperatively gating VSDs (Pantazis et al., 2014; Catterall et al., 2020). Each of the VSDs exists as a four-helix bundle (S1-S4 helices; Catterall et al., 2020). The S4 helix contains at least 4 positively charged arginine or lysine residues (gating charges, Figure 5; arrows in Figure 4) spaced at three (or four) residue intervals. These charges sense changes in transmembrane voltage and depolarization drives them outward across the transmembrane electric field (Figure 5; Catterall et al., 2020). The intracellularly connected S4-S5 linkers (illustrated for domain I in Figure 1D) open the pore by transmitting the S4 movements to the so-called “activation gate”, the narrow part of the pore formed by the S6 helices on their cytoplasmic ends (Figures 1C,D; Wu et al., 2016; Catterall et al., 2020). While moving through the membrane, the S4 positive charges are stabilized by ion pairs formed with negative countercharges from neighboring helices (NC in Figure 5) in- and outside of a hydrophobic constriction site (HCS in Figure 5) whereby ion pair partners are exchanged dynamically during this “sliding helix movement” (Figure 5; Wu et al., 2016; Catterall et al., 2020). The HCS helps to shield the structure from the extracellular hydrophilic milieu and prevents transmembrane flux of water and ions through the VSD. Positive charges are stabilized around the HCS and thereby also prevent ion flux (Figure 5). However, if one of these positive charges is mutated and replaced by a smaller or hydrophilic amino acid this seal can open, allowing water molecules to enter. The resulting pore can be large enough to conduct either measurable Na+ and K+ currents or proton currents if a positive gating charge is replaced by a protonable histidine (Figure 5). When open, this artificial pore (“gating-pore” or “ω-pore”) can conduct outward or inward current depending on the driving forces imposed by transmembrane voltage. Depending on the position of a mutation within the S4 helix a “gating pore current” can develop in the resting state when the outermost gating charges are affected. As illustrated in Figure 5, these residues are close to the HCS when the S4 helix is “down” and, when mutated, allow formation of a water wire through which ions can pass. In contrast, inner gating charges are close to the HCS when the S4 helix is “up.” Their neutralization can support gating pore current when S4 is “up,” which is the case during the open and inactivated state until repolarization forces S4 helices again into their “down” state (Sokolov et al., 2008; Cannon, 2017; Groome et al., 2017; Jiang et al., 2018; Männikkö et al., 2018; Jiang D. et al., 2019). Notably, also variants outside the S4 helix can induce gating pore currents by inducing conformational rearrangements within the VSD (Fuster et al., 2017; Monteleone et al., 2017; Flucher, 2020; see section on CACNA1S below).
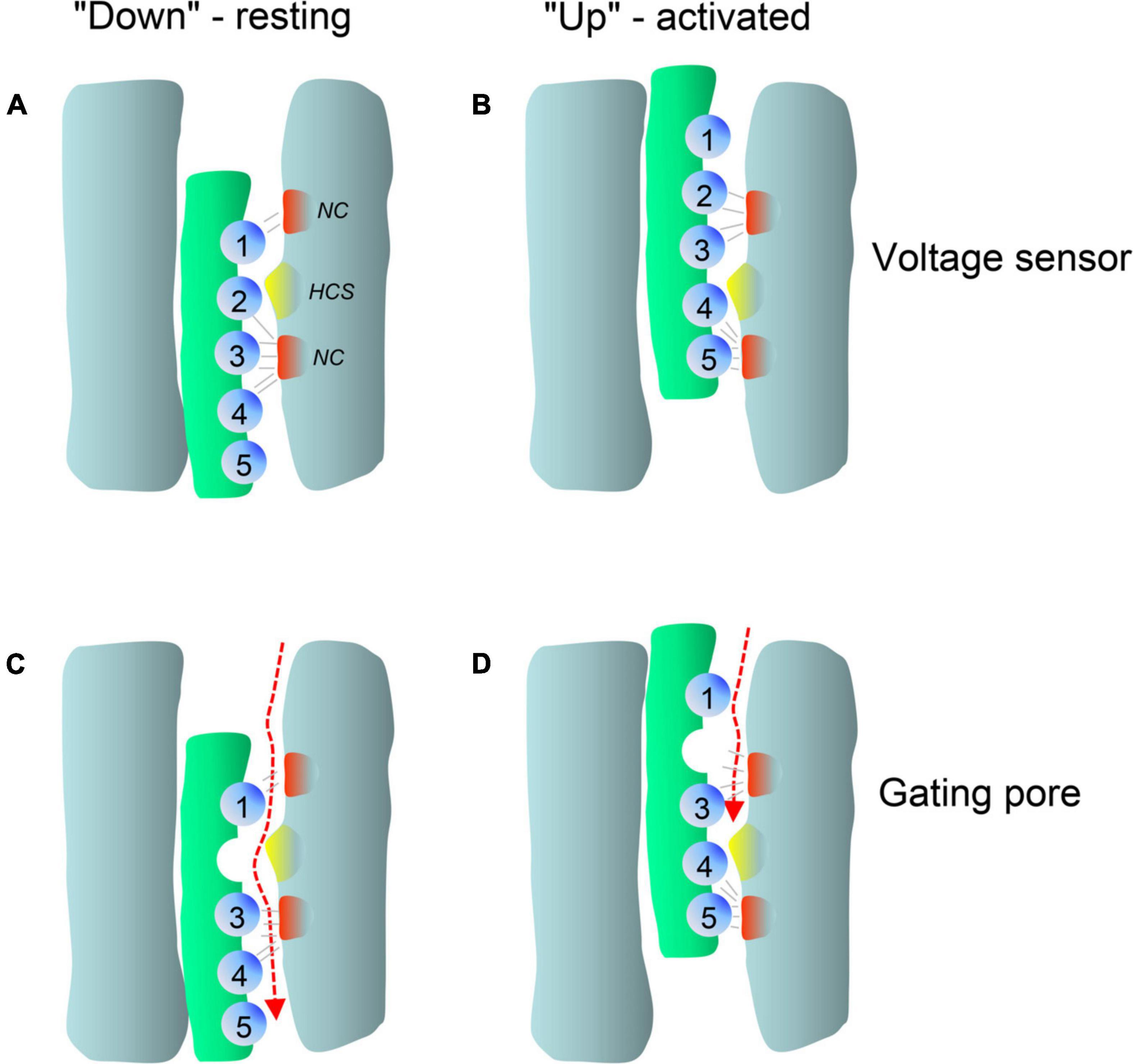
Figure 5. Membrane potential dependent conformations of the voltage-sensor allow pathogenic gating pore current. Simplified scheme illustrating the membrane potential dependent conformations of a single voltage-sensor and the effects of a mutation in gating charge R2. Membrane-spanning S4 helices are shown in green, positively charged residues (mostly arginines spaced at three residue intervals) as blue spheres. Clusters of extracellular and intracellular negative counter-charges (NC) forming ion pair interactions with the gating charges are shown in red. The hydrophobic constriction site is indicated in yellow (HCS). (A) In the resting state, the positively charged S4 helix is pulled inside by the negative resting potential (A, S4 “down”). (B) Upon depolarization, the S4 segment moves outward according to a sliding-helix model (Catterall et al., 2020; B, S4 “up”), and transports positive gating charges through the HCS. Inside and outside the HCS the arginine side chains are stabilized by forming ion pairs with negative charges within the VSD (gray lines). Upon S4 movement the ion pair partners are exchanged and the large arginine side chains seal the VSD and prevent formation of a water filled space through which ions can flow (Catterall et al., 2020). Replacement of a positive gating charge by a smaller or hydrophilic residue can disrupt this seal as exemplified here for a neutralizing mutation in R2 (C). This position of the mutation permits an inward gating-pore current during the hyperpolarized “down”-state of the S4 helix at negative voltages but not during the depolarized “up”-state at positive voltages (D). In contrast, mutations of one of the inner gating charges would permit gating pore current to flow in the activated but not the resting position of S4 (for X-ray structures of mutant voltage-gated Na+-channels see Jiang et al., 2018; Jiang D. et al., 2019).
In addition to pathogenic gating pore current, S4 mutations can also impair voltage-sensor movement thus affecting channel gating and Ca2+ inward current through the canonical central pore (Jurkat-Rott et al., 2012). However, in the case of Periodic Paralysis mutations in Cav1.1 Ca2+- (CACNA1S) and Nav1.4 Na+- channels (SCN4A) only the gating-pore currents are disease-relevant (see section on CACNA1S below). The function and structure of VSDs is highly conserved among voltage-gated ion channels, including Cavs, Na+- (Navs) and K+-channels. Therefore, S4 mutations in Cav1.1 or in Nav1.4 VSDs can both cause Periodic Paralysis in humans (Cannon, 2017; Groome et al., 2017; Jiang et al., 2018). Notably, this has important implications also for the discussion of the functional consequences of variants found in other Ca2+-channelopathies, since VSD mutations in the same conserved position of different Cavs may cause similar gating-pore currents and perhaps even similar phenotypes. Indeed, when discussing a number of gating charge mutations for the individual channels below, we need to consider the possibility that gating pore currents may explain the pathogenic phenotype better than alterations of macroscopic Ca2+ currents (e.g., Cav1.3 – R990H, Cav3.1 – R1715H, Cav3.2 R890H; Figure 4). As a take home message, for gating charge missense variants the possibility of a gating pore current leading to abnormal channel function should always also be considered as a pathogenic mechanism independent of the mutation’s effect on the macroscopic Ca2+ current.
Other Mechanisms
Especially for variants outside the classical regions responsible for ion permeation, gating and voltage-sensing other molecular pathogenic mechanisms have to be considered. This includes sites for protein interactions with one of the channel’s accessory subunits or with other proteins. Several examples exist: the (expected) LOF phenotype of the Episodic Ataxia CACNA1A variant L437F may be explained by reduced affinity for β-subunits (which are essential for channel function, Buraei and Yang, 2010), because this mutation occurs within the α1-subunit’s cytoplasmic I-II linker where β-subunits bind (Van Petegem et al., 2004). In LQT8 families Cav1.2 channel variants were found to cluster within four-residues (857-860, a PRPR-motif, not illustrated) in the cytosolic II–III loop region and enhance channel activity through a so far unknown mechanism. Recently, crystal structures and homology modeling provided compelling evidence that this region binds SH3-domain-containing proteins (e.g., STAC) and LQT8 variants are expected to weaken this interaction (Mellor et al., 2019). Another example is the slow inactivation of the Cav1.2-G406R type 1 mutation, which appears to be mediated by cyclin-dependent kinase 5 (CDK-5; Song et al., 2017).
These examples highlight another weakness of heterologous expression systems in which mutations affecting protein-protein interactions are less likely to be detected.
Ca2+-Channelopathies
How do these GOF or LOF mechanisms translate into different clinical syndromes? As mentioned above, we only focus on disorders in which rare single missense variants explain high disease risk. We will not discuss variants weakly contributing to polygenic disease risk.
Cav1.1 (CACNA1S)
Cav1.1 is the only relevant Cav species expressed in adult skeletal muscle. Due to the slow activation kinetics of the adult splice variant and its positive activation voltage-range (Tuluc and Flucher, 2011) Ca2+ entry through the channel does not directly contribute to fast excitation-contraction coupling in skeletal muscle (Zamponi et al., 2015). Instead, fast conformational changes of its VSDs open the ryanodine receptor and induce fast SR Ca2+ release through the cytoplasmic linker connecting domains II and III (Figure 1) and the associated essential EC-coupling protein STAC3 (for review see Flucher and Campiglio, 2019).
Heterozygous LOF mutations in the Cav1.1 α1-subunit appear clinically silent, unless variants on the second allele reduce function further. This is the case in recessively inherited CACNA1S-associated Congenital Myopathy (Schartner et al., 2017; Flucher, 2020), a disorder characterized by marked neonatal hypotonia, generalized weakness with pronounced axial involvement, variable symptoms such as respiratory and swallowing issues, delayed motor development and peculiar histological features in muscle biopsies (Schartner et al., 2017). Interestingly, heterozygous, dominantly inherited protein-coding missense mutations were also identified in 3 unrelated families located in functionally sensitive regions of the channel (with P742Q and P742S in the II-III linker; not illustrated) expected to weaken channel coupling to the ryanodine receptor (Schartner et al., 2017). Like with recessive mutations, Western blots of patient muscle biopsies showed that the dominant missense variants also induced a strong decrease in overall Cav1.1 protein expression below a level expected from haploinsuffiency. This suggests a dominant-negative effect on wildtype protein expression (Schartner et al., 2017). It is therefore tempting to speculate that, as described for CACNA1A LOF variants (see section on CACNA1A below; Mezghrani et al., 2008), this may result from the interaction of wildtype with mutant Cav1.1 α1-subunits and the formation of complexes susceptible to early protein degradation.
As mentioned above, CACNA1S variants causing Hypokalemic (HypoPP1, familial autosomal dominant inheritance or sporadic de novo) and Normokalemic Periodic Paralysis (NormoPP, Table 1) are the most relevant also for our understanding of the potential functional effects of gating charge mutations in other Cav isoforms. HypoPP1 mutations affect arginines of S4 voltage sensors in domains II–IV and produce an anomalous gating pore leak current (the positions of the mutations are highlighted in red in Figure 4). Like gating pore currents in Nav1.4, these currents, and not changes in the function of the central pore, cause attacks of weakness in HypoPP1 (Cannon, 2017; Flucher, 2020). Seven of the 9 known variants affect arginines. Exceptions are V876E in IIIS3, which also produces gating pore currents (Fuster et al., 2017; Monteleone et al., 2017) and H916Q in the IIIS4-S5 linker (Flucher, 2020; Figure 4). In addition to CACNA1S arginine mutations (Flucher, 2020), gating pore currents have so far only been investigated and confirmed experimentally for Cav1.3-R990H, which therefore is expected to induce a GOF phenotype (see section on CACNA1D below) despite a LOF gating change (Monteleone et al., 2017). It is therefore possible that S4 gating charge variants in other Cav isoforms also generate disease-relevant gating pore currents. Figure 4 illustrates that outside of Cav1.1 α1, disease-associated mutations were reported in a total of at least 17 S4 gating charges in 5 different Cav isoforms (Cav1.2, Cav1.3, Cav2.1, Cav3.1, Cav3.2). Many of them are located in the same homologous positions as Cav1.1 HypoPP1 mutations or pathogenic Nav1.4 (HypoPP2) or Nav1.5 (arrhythmias and dilatated cardiomyopathy) mutations (indicated by asterisks in Figure 4; Groome et al., 2017; Jiang D. et al., 2019; Kubota et al., 2020).
For missense variants causing other skeletal muscle disorders (Malignant Hyperthermia Susceptibility) the disease-inducing mechanism is less well understood. Malignant Hyperthermia Susceptibility mutations sensitize RyR1 Ca2+ release to activation by caffeine and volatile anesthetics but their location in different regions of the channel currently provides no clue about the molecular mechanism (see reference Flucher, 2020 for details).
Cav1.2 (CACNA1C)
Cav1.2 GOF mutations can cause both a syndromic (Timothy syndrome, TS) and non-syndromic form of long-QT syndrome (LQT8). In contrast, LOF mutations shorten the cardiac action potential and are associated with Brugada syndrome and short-QT syndrome (LQTS; Antzelevitch and Yan, 2015; Priori et al., 2015; Antzelevitch et al., 2017). Excellent articles have reviewed this topic recently (Antzelevitch et al., 2017; Marcantoni et al., 2020). Like in other Cavs, GOF mutations are located preferentially in the functionally most sensitive regions of the gating apparatus, the S4 helices, the coupling machinery of the S4-S5 linker and the activation gate formed by the S6 helices (Figure 1D). Examples for regions affected by mutations in different Cavs are indicated in blue in Figure 1D.
Timothy syndrome is a rare systemic disorder originally described as the heart-hand syndrome (Reichenbach et al., 1992). This refers to the typical diagnostic findings of LQTS, often leading to fatal arrhythmias at young age, and unilateral or bilateral cutaneous syndactyly of fingers or toes. In surviving individuals other characteristic clinical features were described compatible with a multi-organ disease. In most cases facial anomalies (including depressed nasal bridge, low-set ears, thin vermilion of the upper lip), congenital heart defects and developmental delay (language, motor, and generalized cognitive impairment) are present. Autism, seizures, and intellectual disability as well as hypoglycemia were also reported (Splawski et al., 2004). These symptoms are in accordance with the known role of Cav1.2 for cardiac function, neuronal development and plasticity (Krey et al., 2013; Zamponi et al., 2015), craniofacial development (Ramachandran et al., 2013), and insulin secretion (Sinnegger-Brauns et al., 2004; Zamponi et al., 2015).
Classical TS is caused by GOF mutations in Cav1.2. Originally, only mutations in alternatively spliced exons 8a (G406R, TS1) and 8 (G406R, G402S, TS2) had been described (Splawski et al., 2004; Splawski et al., 2005) (Figure 3; exon 8a sequence). All of them cause GOF by a pronounced reduction of (voltage-dependent) channel inactivation (type 1, Figure 2A). This can explain a sustained depolarizing Ca2+ influx with broadening of the action potential and QT-prolongation. A more severe LQT phenotype (QTc > 620 ms) has been reported in TS2. This was explained by the higher abundance of exon 8 in heart and brain. Interestingly, the first two TS2 patients had no syndactyly, one of the diagnostic features of the syndrome, but were affected by facial dysmorphism and developmental delay. The phenotype in the patient with the G402S mutation was milder. It was attributed to somatic mosaicism with the mutation being much less abundant in oral mucosa than in blood DNA (Splawski et al., 2005). This was an important observation because it shows that the severity of the phenotype is also determined by mutation load. Mosaicism is also evident from severely affected TS1 and TS2 cases who inherited the mutation from a parent with germline mosaicism. The parent had either no symptoms (Splawski et al., 2004), or, upon full medical work-up, revealed diagnostic features, such as syndactyly with and without LQT8 (Etheridge et al., 2011; Dufendach et al., 2013; Fröhler et al., 2014; Baurand et al., 2017). Therefore mosaicism has to be considered in the interpretation of phenotype-genotype relationships (Lim et al., 2017; Myers et al., 2018; Cao et al., 2019) not only of CACNA1C-associated disorders but also of other Ca2+-channelopathies (e.g., CACNA1A, Cao et al., 2019; see below).
In addition to the classical TS mutations, four mutations outside exon 8 and 8a were reported. A GOF phenotype has either been proven in functional experiments or can be assumed based on their locations in S6 or the S4-S5 linkers. The affected individuals presented as atypical TS with LQT8 but some other clinical features of TS missing, such as syndactyly (S643F, type 1; Figure 4; Ozawa et al., 2018), or facial dysmorphisms (E407A, adjacent to G406R, Figure 3; Colson et al., 2019). In other cases, an even broader phenotypic spectrum was reported, including osteopenia, cerebral and cerebellar atrophy, intractable irritability (I1186T, type 2; Figure 3; Boczek et al., 2015) or intractable seizures and stroke (A1521G, type 2, Figure 3; Gillis et al., 2012).
Two additional variants deserve attention because their clinical phenotypes are even more distinct, R1024G and E1135K: R1024G (Figure 4) was reported in a 5-year-old girl with profound developmental and speech delay, TS-like craniofacial dysmorphism and syndactyly but without LQTS upon repeated ECG measurements (Kosaki et al., 2018). Pulmonary hypertension was also diagnosed. Although no functional studies have been performed, the position of this variant is intriguing. It neutralizes one of the positive gating charges in voltage sensor IIIS4. It corresponds to the R990G and R990H mutations in Cav1.3 (Figure 4; somatic mutations in APAs, see section on CACNA1D below). In Cav1.3 R990H destabilizes the VSD and can conduct a depolarizing gating pore current at negative membrane potentials (Monteleone et al., 2017). VSD mutations R1135H and R1135C in Nav1.4, which cause HypoPP2 (see above, position indicated by asterisk in Figure 4C) also occur in the equivalent positions. One possible scenario therefore is that R1024G also induces a gating pore current in Cav1.2. Assuming that it destabilizes the VSD in a similar manner as R990H in Cav1.3 it could contribute depolarizing current at more negative potentials. In arterial smooth muscle cells increased arterial muscular tone could explain pulmonary hypertension, whereas in neurons enhanced subthreshold neuronal depolarization could drive abnormal neuronal firing underlying the severe neurodevelopmental phenotype. However, upon activation of the Cav1.2 voltage-sensors (S4 “up” in open and inactivated channels) during the plateau of the cardiac AP, this gating pore current is expected to turn off and may thus not affect AP duration and QT-interval.
E1135K (not illustrated) was found in a 14-year-old male proband with idiopathic QT prolongation, autism spectrum disorder and unexplained hyperglycemia (Ye et al., 2019). This mutation reverses one of the four essential negative charges of the channel’s selectivity filter. Functional expression in tsA-201 cells confirmed the expected conversion of a Ca2+-selective channel into a non-selective cation channel with marked increase in both peak and persistent inward Na+- and outward K+-currents (Ye et al., 2019). Its expression in human iPSC-derived cardiomyocytes prolonged the cardiac AP. By favoring membrane depolarization, it can therefore be considered a GOF variant in the heart and likely also in neurons, which could affect neuronal excitability and underlie the autism phenotype. The mutation may also account for the “unexplained” hyperglycemia reported in this patient due to reduced insulin secretion. An increased membrane depolarization of the pancreatic β-cell could result in an increased basal intracellular Ca2+ level and reduced glucose-induced insulin secretion as observed upon genetic deletion of the KATP K+-channel subunits (Miki et al., 1998). Alternatively, while mediating a non-selective pore current, the mutated channels are not expected to conduct Ca2+. This could impair Ca2+-dependent processes depending on Cav1.2, including insulin secretion (Zamponi et al., 2015; Mastrolia et al., 2017).
In addition to TS, GOF Cav1.2 mutations can also cause a non-syndromic form of LQT8. LQT8 can be regarded as part of a CACNA1C disease spectrum because with some variants the clinical phenotype can be either LQT8 alone (G402S, Mellor et al., 2019), LQT with syndactyly but without other TS-typical features (G402S, I1186T; Fröhler et al., 2014; Wemhöner et al., 2015) or Timothy syndrome. Somatic mosaicism and/or genetic background can explain these variable phenotypes. Many of the LQT8-associated variants were confirmed to induce GOF (Hennessey et al., 2014; Fukuyama et al., 2014b; Boczek et al., 2015; Wemhöner et al., 2015; Landstrom et al., 2016; Chavali et al., 2019; Estes et al., 2019) with slowing of the inactivation time course and increased late currents. Introduction of LQT8 mutations into human iPSC-derived cardiomyocytes confirmed the broadening of the action potential in the heterozygous state (Estes et al., 2019; Chavali et al., 2019). It is possible that mutations shifting the voltage-dependence of activation and inactivation to more negative voltages favor more severe extracardiac manifestations (in particular the nervous system; Marcantoni et al., 2020). However, some variants may predominantly affect the function of cardiac channels. This has recently been shown for a cluster of variants (affecting residues P857, R858, R860; not illustrated) located within the long cytoplasmic II-III – linker without obvious role in channel gating and conductance. However, this proline-rich region serves as a binding site for SH3-domain containing proteins, such as STAC proteins, known to affect the expression and function of L-type channels (Mellor et al., 2019; Flucher and Campiglio, 2019). For a complete list of LQT8 mutations see recent reviews (Giudicessi and Ackerman, 2016; Zhang et al., 2018; Marcantoni et al., 2020).
LOF CACNA1C variants are associated with so-called J-wave syndromes (named after the typical electrocardiographic “J-wave”), consisting of the Brugada syndrome and short-QT syndromes (Priori et al., 2015; Antzelevitch and Yan, 2015; Antzelevitch et al., 2017). Variants leading to altered Cav1.2 activity in the heart were also reported in β- (CACNB2 gene) and α2δ1 (CACNA2D1) subunits (Antzelevitch et al., 2007; Burashnikov et al., 2010). LOF CACNA1C variants are protein-coding variants predominantly located in the cytoplasmic regions of the channel and it is less clear how they induce the observed reduction in Ca2+ current when heterologously expressed. Synonymous variants in splice sites inducing nonsense-mediated RNA-decay have also been described (Fukuyama et al., 2014a). Interestingly, unlike with pathogenic CACNA1F and CACNA1A LOF mutations (see below), no protein-truncating variants have been reported so far. This could indicate that these phenotypes are not simply due to haploinsufficiency.
Cav1.3 (CACNA1D)
Current evidence from knockout mouse models and human genetics indicate that heterozygous Cav1.3 LOF is unlikely to cause a disease phenotype. About 20 heterozygous LOF genotypes2 are listed for mostly healthy control individuals in the gnomAD database. Only homozygous Cav1.3-deficiency causes sinoatrial node dysfunction and deafness in mice (Platzer et al., 2000; Zamponi et al., 2015). The same phenotype (Sinoatrial Node Dysfunction and Deafness, Table 1) is observed in humans with a homozygous LOF mutations in exon 8 (also referred to as exon 8b; Baig et al., 2011), which is the predominant splice variant in brain, sinoatrial node and the cochlea (Baig et al., 2011; Zamponi et al., 2015; Liaqat et al., 2019).
In contrast, heterozygous de novo GOF missense mutations cause human disease by affecting multiple Cav1.3-dependent physiological functions. Since we have recently reviewed the disease spectrum associated with CACNA1D GOF variants (Ortner and Striessnig, 2020), this is only briefly summarized here.
In most affected individuals germline missense DNMs lead to a severe neurodevelopmental disorder with ID, developmental delay, seizures, auto-aggressive behaviors, delayed or no speech development, muscle hypotonia and autism spectrum disorder (V401L, S652L, A749T; Figure 3). In even more severe cases congenital hyperinsulinemic hypoglycemia and/or congenital aldosteronism (Ortner and Striessnig, 2020) can be present at birth (V259A, G403D, I750M, L271H; Figures 3, 4). A syndrome, Primary Aldosteronism with Seizures and Neurological Abnormalities (PASNA) is part of this disease spectrum. Less severe cases were diagnosed with ASD with (A749G) and without (G407R) mild intellectual impairment (Figure 3). So far, for these Cav1.3 – linked disorders type 1 and 2 GOF gating changes were reported for nine mutations in 12 individuals (Ortner and Striessnig, 2020) but unpublished cases with pathogenic mutations exist and are currently being characterized. For a detailed list of mutations and associated phenotypes see Ortner and Striessnig (2020). Like most other GOF mutations, they are located in the activation gate (cytoplasmic region of S6 helices) as well as in the S5-S6 linkers, see above and Figures 1D, 3, 4). Interestingly, the positions of G403R and G403D (exon 8) and G407R (exon 8a) are identical to the Timothy syndrome mutations G402 and G406 (see above) in CACNA1C and also cause very similar gating defects (type 1; Scholl et al., 2013; Pinggera et al., 2018). When present as somatic mutations in APAs, these variants induce excessive Ca2+-dependent aldosterone production resulting in drug-resistant hypertension (Azizan et al., 2013; Scholl et al., 2013). Some of these mutations affect the same amino acid or are even identical to the ones identified germline. Importantly, some of the germline mutations exhibit increased sensitivity to DHP LTCC blockers (Hofer et al., 2020). Future clinical research should therefore test the potential of these drugs to improve symptoms in severely affected individuals. Preliminary evidence suggests that muscle hypotonia may respond to treatment with DHPs (De Mingo Alemany et al., 2020).
Cav1.4 (CACNA1F)
Cav1.4 L-type calcium channels are essential for tonic glutamate release from retinal photoreceptors (as is Cav1.3 in cochlear inner hair cells; Zamponi et al., 2015; Pangrsic et al., 2018). Their α1-subunit gene (CACNA1F) was originally discovered as the target of LOF mutations causing X-linked Congenital Stationary Night Blindness Type 2 (CSNB2), a non-progressive vision disorder with characteristic clinical findings (Zamponi et al., 2015). Since then a large number of CACNA1F LOF mutations, most of them protein-truncating variants, have been reported to be associated with CSNB2 as well as other eye disorders, including Åland Island Eye Disease, Cone-Rod Dystrophy, X-linked retinal disorder, and Night Blindness-Associated Transient Tonic Downgaze, which share a variety of clinical symptoms (Hope et al., 2005; Stockner and Koschak, 2013) (Table 1). As expected for an X-linked disorder females are rarely affected. The absence of the channel protein or the absence of functioning channels leads to abnormal cellular organization of synapses within the retina and defective neurotransmission between photoreceptors and second-order neurons (Zamponi et al., 2015).
Interestingly, a number of missense variants have also been identified. While some of them decrease channel function or expression (Hoda et al., 2006; Stockner and Koschak, 2013), others show the typical gating changes compatible with a gain of channel function. For example, G369D in domain I and F753C and I756T in the activation gate of domain II (Figure 3) cause a dramatic type 2 gating change and slow inactivation as expected from their locations (Hemara-Wahanui et al., 2005; Hoda et al., 2005; Peloquin et al., 2007). One plausible explanation of how these gating changes can actually decrease Cav1.4 channel activity is that the pronounced negative shift of voltage-dependent activation gating moves the channel out of its optimal operating range within the voltage window during light exposure (about − 55 mV) and darkness (up to −36 mV). During illumination a larger fraction of mutated Cav1.4 channels may already be activated thus limiting the maximal gain during depolarization in the dark (Stockner and Koschak, 2013). This is another example demonstrating that a given “GOF” gating change can either increase or decrease channel signaling depending on the context of a cell’s electrical activity pattern (see above).
Cav2.1 (CACNA1A)
Cav2.1 channels are widely expressed in the central (including thalamocortical, cortical, cerebellar neurons) and peripheral nervous system. Together with Cav2.2 channels they are tightly coupled to fast neurotransmitter release at presynaptic terminals, including motoneurons (Zamponi et al., 2015). Homozygous knockout mice develop rapidly progressive neurological symptoms with ataxia and dystonia and die a few weeks after birth (Jun et al., 1999; Fletcher et al., 2001). Therefore, homozygous LOF of Cav2.1 is also expected to be incompatible with life in humans. Accordingly, no homozygous CACNA1A pLOF variants are reported in the gnomAD database.
CACNA1A mutations cause several dominantly inherited or sporadic neurologic disorders with a wide spectrum of neurological symptoms (Spacey, 2015): hemiplegic migraine with aura (including severe forms with cerebral edema and coma), cerebellar signs (including episodic, permanent, early or late onset progressive ataxia, cerebellar atrophy, nystagmus, vertigo, dysarthria), seizures, and developmental delay. Although symptoms often overlap, the three main neurological CACNA1A disorders reveal some genotype-phenotype correlation. Episodic ataxia type 2 (EA-2) typically starts in childhood or early adolescence and is characterized by paroxysmal attacks of ataxia and other cerebellar signs and can be associated with other symptoms including hemiplegia and headache (Spacey, 2015). Attacks may last from minutes to days. The majority of EA-2-associated mutations are pLOF mutations or missense mutations, which typically reduce current amplitudes and/or lead to LOF gating changes. When expressed as trunctated proteins in vitro some of them also exert a dominant negative effect on wildtype channels (see above). Excellent reviews summarizing the position of pLOF and missense LOF variants associated with EA-2 have been published recently (Eising and van den Maagdenberg, 2017; Tyagi et al., 2020).
In contrast, GOF mutations cause Familial (FHM-1) or Sporadic (SHM-1) Hemiplegic Migraine Type 1, a form of migraine with aura, which, in addition to other aura symptoms, is characterized by the presence of motor weakness, i.e. hemiparesis (Jen, 2015).
FHM-1-associated mutations mostly exhibit GOF gating changes with enhanced channel activity at more negative voltages (Tottene et al., 2002; Pietrobon and Striessnig, 2003). They are mainly located at the positions within voltage-sensors (e.g., R192Q, R195K, V581L/M, R583Q, K1343Q, R1352Q, R1667W, K1670R), the S4-S5 linkers (e.g., S2181L, C1369Y, L1682P, W1683R) and the cytoplasmic S6 activation gates (e.g., F363S, V714A, D715E, F1506S, I1810L; Figures 3, 4) where other GOF channelopathy mutations are typically located (Eising and van den Maagdenberg, 2017; Tyagi et al., 2020). Moreover, some variants associated with FHM-1 also neutralize S4 gating charges, including positions in which also Cav1.1 and Nav1.4 gating-pores were reported (e.g., R195K, R583Q, K1343Q, R1352Q; asterisks in Figure 4; note that R195K in Cav2.1 and R219K in Nav1.4 are both charge-retaining substitutions; Ducros et al., 2001; Kubota et al., 2020) and may therefore affect neuronal excitability also through gating pore currents (Jurkat-Rott et al., 2012). This hypothesis should be tested experimentally and by molecular modeling like for other Cavs (Monteleone et al., 2017; Flucher, 2020).
As a monogenic form of migraine, FHM-1 offered important insight into the pathophysiology of migraine with aura (Pietrobon and Striessnig, 2003; Goadsby et al., 2017; Brennan and Pietrobon, 2018). Human CACNA1A GOF mutations (R192Q, S218L; Figure 4), when introduced in mice, promote cortical spreading depression, a self-propagating wave of neuronal and glial depolarization that spreads slowly across the cerebral cortex and underlies the aura symptoms in humans (Pietrobon and Striessnig, 2003; Brennan and Pietrobon, 2018). Moreover, enhanced excitatory, but not inhibitory synaptic transmission and alterations of cortical excitatory-inhibitory balance can explain the altered sensory and pain processing in FHM1 (Tottene et al., 2019) and likely also in more common forms of migraine.
Unlike EA-2, Spinocerebellar Ataxia Type 6 (SCA-6) is characterized by adult onset, slowly progressive rather than episodic cerebellar ataxia, dysarthria, and nystagmus with a mean age of onset between 43 and 52 years (Du and Gomez, 2018; Casey and Gomez, 2019). Genetic diagnosis of SCA-6 requires documentation of an expanded polyglutamine repeat within the C-terminus of the Cav2.1 α1-subunit. There is strong experimental evidence that, unlike the other Ca2+-channelopathies described here, SCA6 is not caused by a dysfunction of the Cav2.1 channel itself, but instead by a separate protein, the transcription factor α1ACT. This protein is also encoded by the CACNA1A gene due to the presence of an internal ribosomal entry site (IRES) within its mRNA (for a detailed review see Du and Gomez, 2018). α1ACT is important for normal perinatal cerebellar developmental and motor function (Du and Gomez, 2018). Wildtype α1ACT, but not α1ACT-containing SCA6 poly-glutamine expansions (α1ACTSCA6), can rescue neurological deficits when transgenically expressed in Cav2.1 knockout mice (Du and Gomez, 2018). α1ACTSCA6 is also toxic to cultured cells and when expressed transgenically in Purkinje cells (Du and Gomez, 2018).
Substantial phenotypic overlap is observed for these three CACNA1A disorders, complicating the interpretation of this genotype-phenotype relationship. For example, a large fraction of FHM-1 families with GOF variants report cerebellar signs ranging from nystagmus to progressive, usually late-onset mild ataxia (Ducros et al., 2001; Jen, 1993). EA-2 individuals (i.e., with LOF variants) may also eventually develop interictal symptoms and cerebellar signs such as truncal ataxia and nystagmus. Such late-onset progressive ataxia makes the clinical phenotype similar to SCA-6 (Di Cristofori et al., 2012). In some families progressive ataxia due to missense variants and without expanded polyglutamine repeat was the major clinical feature, with some family members also diagnosed with EA-2 (Yue et al., 1997; Jen et al., 2004; Romaniello et al., 2010).
Congential ataxia, with presentation before the age of 2 years, is mostly associated with GOF variants, many of them also found in FHM-1 patients. Examples are S218L, D715E, I1810L, V1396M (Figures 3, 4) and T666M (close to selectivity filter in domain II, not illustrated). For an excellent detailed recent review on congential ataxia see reference Izquierdo-Serra et al. (2020).
Interestingly, a slowly progressing non-episodic spinocerebellar ataxia without migraine symptoms differing from SCA-6 and EA-2 has been associated with the mutation E668K. This mutation reverses the negative charge of the selectivity filter glutamate in domain II, similar to CACNA1C mutation E1135K (domain III, not illustrated) mentioned above. Non-selective ion currents and loss of Ca2+-conductance through this mutant may therefore contribute to the observed atypical phenotype.
CACNA1A mutations have also been associated with severe neurodevelopmental syndromes including early-onset epilepsy (Early Infantile Epileptic Encephalopathy 42) and other neurocognitive (intellectual impairment, learning disabilities) and neurological symptoms, such as ataxia, nystagmus, hypotonia (Damaj et al., 2015; Epi4K Consortium, 2016; Epperson et al., 2018; Jiang X. et al., 2019). It appears that both CACNA1A haploinsufficiency (like in EA-2; Damaj et al., 2015; Jiang X. et al., 2019) and FHM1-like GOF gating changes (A711T, A713T, Ser218L, Figure 3; Epi4K Consortium, 2016; Epperson et al., 2018; Jiang X. et al., 2019) can cause this phenotype. Recent studies indicate that neuropsychiatric and developmental symptoms are common in patients with episodic CACNA1A disorders and should be considered in the differential diagnosis of otherwise unexplained developmental delay (Indelicato et al., 2019; Humbertclaude et al., 2020).
Taken together, in vitro gating changes indicating increased activity of Cav2.1 at more negative voltages is mostly associated with FHM1/SHM1 and congenital ataxias, whereas channel LOF causes EA-2. However, both GOF and LOF mutations are associated with progressive ataxia and epilepsy. At present, there is no clear explanation for this observation. One hint could come from the finding that Purkinje cell-specific Cacna1a knockout is sufficient to cause cerebellar ataxia in mice (Mark et al., 2011; Todorov et al., 2012). This strongly suggests that reduced Cav2.1 activity is responsible for this phenotype. It is therefore possible that, as outlined above, certain gating changes can cause both, GOF (e.g., by shifting activation voltage to more negative potentials) and LOF (e.g., by shifting inactivation voltage also to more negative potentials and/or by reducing maximal channel open probability at more positive voltages) (Tottene et al., 2002). This could lead to a scenario where GOF changes predominate at e.g., cortical synaptic terminals and LOF changes at Purkinje cell bodies. At presynaptic active zones the higher open probability at threshold voltages due to type 2 gating changes could facilitate nanodomain Ca2+ influx and glutamate release and underlie the enhanced neuronal excitability predisposing to migraine attacks (Tottene et al., 2002). At the same time, global cellular Cav2.1 current density during action potentials at cell bodies and dendrites of cerebellar neurons may be reduced (LOF), the latter predisposing to ataxia and cerebellar neurodegeneration.
Another open question is how missense mutations inducing only a partial LOF cause EA-2. In a knock-in mouse model the EA-2-associated missense mutation F1403C (F1406C in Rose et al., 2014) reduced Cav2.1 current density in Purkinje cells of homozygous knock-in mice by about 70%, as predicted from expression in HEK-293 cells. However, this mutation failed to induce a motor phenotype as observed in other recessive Cacna1a mouse mutant lines with mutations resulting in similar biophysical alterations (Rose et al., 2014).
Cav2.2 (CACNA1B)
Like Cav2.1-mediated P/Q-type currents, Cav2.2-mediated N-type Ca2+ currents also play an essential role for presynaptic neurotransmitter release. However, in contrast to Cav2.1, homozygous knockout mice show only minor abnormalities. This includes reduced pain hypersensitivity in models of inflammatory and neuropathic pain, hyperactivity, increased aggression and altered blood pressure control (Kim et al., 2009; Zamponi et al., 2015). This suggests that homozygous LOF may also not cause severe disease in humans. However, this is in contrast to recent reports, describing bi-allelic LOF variants in an autosomal recessive, complex and progressive neurologic disorder with severe neurodevelopmental delay and developmental regression, drug-resistant epileptic encephalopathy, postnatal microcephaly, hypotonia, and a non-epileptic hyperkinetic movement disorder (Neurodevelopmental disorder with seizures and non-epileptic hyperkinetic movements, NEDNEH; Gorman et al., 2019). This was confirmed in three unrelated families with different protein-truncating mutations. A possible association between a form of autosomal dominant myoclonic dystonia and the CACNA1B variant R1389H has also been postulated (Groen et al., 2014). However, its pathogenic potential is questionable, because it is a common variant (1:500) observed at comparable frequencies in affected and in neurologically healthy individuals (Mencacci et al., 2015). Similarly, the significance of two variants associated with adult-onset isolated focal dystonia is unclear (Cocos et al., 2020).
Taken together, current evidence suggests that heterozygous Cav2.2 LOF does not confer high risk for disease but that homozygous LOF due to protein truncating mutations can cause a severe neurodevelopmental disorder in humans.
Interestingly, unlike for other Cavs, so far no GOF variants have been reported for Cav2.2. However, rare genetic variants located within the gating-sensitive regions of Cav2.2 (Figure 1D) are reported in apparently healthy control individuals in the gnomAD database at high allele numbers. Some of these variants are even located in the identical homologous positions causing typical and disease-associated GOF gating changes in other Cavs. Variant V351E (2 residues upstream of the Timothy syndrome mutations in CACNA1C) is located in exactly the same position as mutation V401L (Figure 3), which causes a pronounced type 2 gating change in Cav1.3 and is associated with a severe neurodevelopmental phenotype (Ortner and Striessnig, 2020). Variants S215A and S215L affect the same serine residue in the IS4-S5 linker causing GOF changes in Cav2.1 (S218L) and Cav1.4 (S229P; highlighted in blue in Figure 1D) (Tottene et al., 2005; Hoda et al., 2005). Also, variants V1408I and N1409S in IIIS6 are surrounded by GOF variants in Cav1.2, Cav1.3, Cav2.1, Cav3.1 and Cav3.2 (Figure 3). Possible explanations are that these variants are either functionally silent or induce a LOF phenotype, which, as mentioned above, is not expected to cause disease in the heterozygous state. Alternatively, these variants may induce robust GOF changes that are not compatible with life and cause intrauterine death. Functional analysis of these variants in heterologous expression systems could answer this interesting question.
Cav2.3 (CACNA1E)
Helbig and colleagues recently described de novo CACNA1E missense variants causing Cav2.3 GOF in 30 individuals with Developmental and Epileptic Encephalopathies (Helbig et al., 2018). Most affected individuals presented with pharmacoresistant seizures beginning in the first year of life, profound developmental impairment, hyperkinetic movement disorders, severe axial hypotonia and some had spastic quadriplegia. Most were non-verbal and non-ambulatory. Developmental regression occurred in 9/30 individuals often in association with seizure onset. Two recurrent variants G352R and A702T accounted for half of the cohort and with a severe phenotype. Out of the 14 variants four were functionally tested and showed type 2 gating behavior with slight slowing of inactivation. They are located within the typical regions involved in channel gating (Figures 1D, 3).
The same study also reported 3 individuals with variants introducing premature termination codons predicted to result in haploinsufficiency. These individuals had a milder phenotype, achieved independent ambulation at 15–18 months and acquired single words. Two of them had epilepsy (Helbig et al., 2018). One of them inherited the mutation from the apparently healthy father. Due to the small number of cases and the fact that a few heterozygous pLOF variants are also reported in gnomAD (“non-neuro” subset), the disease-causing potential of these variants is at present less clear.
T-Type Ca2+ Channels
Based on the expression pattern of T-type channels and their special functional properties (low-voltage activated and inactivated, window current at negative voltages, recruitment upon hyperpolarization enabling low-threshold spike/rebound bursting observed in seen in many types of neurons) together with the known anti-epileptic properties of T-type channel blockers makes these channels major suspects for genetic causes of epilepsy. Cav3.1 and Cav3.2 knockout protects mice from epilepsies (Weiss and Zamponi, 2019; Lory et al., 2020). However, so far the association of most genetic variants in CACNA1G and CACNA1H and their functional mechanism of action in terms of GOF or LOF for idiopathic genetic epilepsies is less well understood (Weiss and Zamponi, 2019; Lory et al., 2020) and mutations in Cav3.1 or Cav3.2 causing high risk for epilepsy have not been described. However, changes in their activity (as shown for Cav3.1 in mice, Calhoun et al., 2016) may serve as genetic modifiers of other genetic risk factors for idiopathic genetic epilepsies. These are generally considered to have complex inheritance patterns with combinations of common with rare genetic risk variants required to cause disease. Some CACNA1G variants may therefore also interact with other genes, rather than being pathogenic per se (Weiss and Zamponi, 2019).
Excellent reviews summarizing all Cav3 channelopathies (except for CACNA1I, see below) have recently been published (Weiss and Zamponi, 2019; Lory et al., 2020).
Cav3.1 (CACNA1G)
De novo mutations A961T and M1531T were recently found in Childhood-Onset Cerebellar Atrophy (ChCA; Spinocerebellar Ataxia Type 42 (SCA42) early-onset with neurodevelopmental deficits) patients in unrelated pedigrees (Chemin et al., 2018; Barresi et al., 2020). ChCA is an early-onset neurodevelopmental disorder, with cerebellar atrophy, severe motor and cognitive impairments, and variable features including epilepsy and facial dysmorphisms. The mutations are located in “classical sites” of the S6 activation gates in domains II and III (Figure 2) and cause typical type 2 gating changes (Chemin et al., 2018).
In contrast, autosomal-dominant SCA42 is characterized by a slowly progressive ataxia with an onset mainly in young adulthood. Another CACNA1G mutation, R1715H, has been identified in SCA42 families by different groups (Coutelier et al., 2015; Morino et al., 2015; Hashiguchi et al., 2019). When expressed in HEK-293 cells (Coutelier et al., 2015; Morino et al., 2015) it caused a positive shift of the activation and steady-state inactivation voltage with a shift of the window current to more positive voltages. A mouse animal model of SCA42 harboring this mutation confirmed these gating changes in Purkinje cells and recapitulated the phenotype (adult onset, gait instability) and pathology (Purkinje cells degeneration) of the disease (Hashiguchi et al., 2019). In contrast to R1715H knockin in mice, heterozygous and homozygous knockout of Cav3.1 in mice does not cause progressive ataxia and cerebellar atrophy indicating that the observed gating change is not a simple LOF. Again, an alternative explanation could be that this mutation affects a gating charge in the VSD of domain IV (Figure 4). While this can explain the reduced voltage-sensing of the mutant channels, it could also generate an additional gating pore current resulting in a depolarizing drive. This is also supported by the finding that R1664Q located in the homologous position in CACNA1A also causes an early-onset and progressive form of ataxia not characterized by the typical episodic features of EA-2 (Tonelli et al., 2006; Luo et al., 2017). Moreover, this residue corresponds to R1239 in CACNA1S in which R1239H- and R1239G- induced gating pore currents cause HypoPP1 (see above, 91). If the CACNA1G R1715H variant induces gating pore-currents it should be classified as a “GOF” variant.
Another variant, M1574K, was found in three SCA42 patients from a Chinese family but it has not been functionally characterized yet. It is located within the III-IV-linker (not illustrated) of the channel, outside the classical regions causing gating defects (Li et al., 2018). It is located within a short alternatively spliced region (exon 25 splice variant, Latour et al., 2004) and may therefore not affect all Cav3.1 channel species in the cerebellum. However, alternative splicing in this region has been found to affect Cav3.1 recovery from inactivation (Latour et al., 2004). In the structurally highly related Cav3.2 channels alternative splicing in this position (exon 25) is required for permitting the functional effects of the adjacent R1584P mutation found in the GAERS (Genetic Absence Epilepsy Rats from Strasbourg) rat model of idiopathic generalized epilepsies (Powell et al., 2009). This GOF mutation in combination with exon 25 promotes recovery from channel inactivation and enhances channel expression by disrupting the interaction of the III-IV-linker with calnexin (Proft et al., 2017). Since calnexin also reduces the expression of Cav3.1 channels (Proft et al., 2017), it is possible that the introduction of a positive charge in the M1574K variant also affects channel function and/or expression. However, this hypothesis needs to be tested in functional and biochemical studies.
Cav3.2 (CACNA1H)
Like Cav1.3, Cav3.2 is important for mediating depolarization-induced aldosterone production. As a consequence, inherited (Familial Hyperaldosteronism Type 4) and de novo germline GOF missense mutations S196L, M1549I, M1549V (Figures 3, 4), P2083L (not illustrated) (Scholl et al., 2015; Daniil et al., 2016) and R890H (Figure 4, Wulczyn et al., 2019) cause hyperaldosteronism. Neurodevelopmental symptoms (such as mild ID, social skills alterations and learning) were also reported in carriers of the M1549 variants (Daniil et al., 2016). Two somatic variants were found in APAs (I1430T, V1951E; Daniil et al., 2016; Nanba et al., 2020). From the five mutations reported so far, three show typical type 2 (M1549I/V) or type-3 (S196L) gating changes. I1430T corresponds to the position of the CACNA1D APA mutations I1015S and I1015V (IIIS5) (Ortner and Striessnig, 2020).
Unlike other mutations, R890H caused a pronounced LOF phenotype when expressed in HEK-293 cells (Wulczyn et al., 2019), a puzzling finding considering the Ca2+-dependence of aldosterone secretion and the other disease-causing GOF mutations in CACNA1D and CACNA1H. However, a GOF in this mutation may again be due to the induction of gating-pore currents. This is strongly supported by the observation that pathogenic gating charge mutations in the homologous positions of Nav1.4 (e.g., R675Q; Sokolov et al., 2007) and of Nav1.5 channels (R814W, Moreau et al., 2015; asterisks in Figure 4) also conduct gating pore currents.
CACNA1H variants V1951E and P2093L are located in the cytoplasmic C-terminus and it is less clear how they affect channel function.
Cav3.3 (CACNA1I)
Although CACNA1I variants seem to contribute to the polygenic risk of neuropsychiatric disorders (including autism and schizophrenia, Lory et al., 2020; Ghoshal et al., 2020), rare missense mutations causing a monogenic disorder have only been published very recently (Ghaleb et al., 2021). The de novo missense variant (I860N, IIS6, Figure 3) was found in a girl with a severe neurodevelopmental disorder with epilepsy, muscle hypotonia, hyperexcitability, severe global developmental delay, no cognitive development and progressive brain atrophy. It induces typical type 2 GOF gating properties with significantly left-shifted voltage-dependence of activation and inactivation, increased window currents, slowed activation, inactivation, and deactivation of Cav3.3 currents. Another mutation in the same position, I860M, induced less severe biophysical changes and was present in a mother and her two children with intellectual disability but no epilepsy. Two other missense mutations, I1306T (de novo, IIIS5, Figure 4) in a 21-month-old female and M1425I (likely de novo, IIIS6, Figure 2) in an 8-year old male patient were also associated with seizures, severe developmental delay, hypotonia, severe speech impairment, and cortical visual impairment. Again, all these variants affect residues in homologous positions for which cause GOF channelopathies also in other Cavs (Figures 3, 4).
Inspection of the activation gate formed by IIS6 nicely demonstrates the accumulation of pathogenic GOF mutations within this structurally and functionally highly conserved region. Together with similar aggregations of mutations in other conserved regions of the gating apparatus (Figure 1D), functionally validated pathogenic mutations in one Cav should - in combination with other ACMG critera - help to predict the level of pathogenicity of newly diagnosed variants in another Cav.
Implications for Therapy
The appraisal of functional changes introduced by missense mutations in terms of gain- or loss-of Ca2+-channel function may guide the development of potential new therapies for Ca2+-channelopathies.
In particular, in case of pathogenic GOF mutations, specific inhibitors of the channel isoform affected by the mutation are obvious therapeutic candidates. An intriguing example was the discovery of heterozygous activating mutations in the genes encoding ATP-sensitive K+-channel subunits (KCNJ11, ABCC8; De Franco et al., 2020). By keeping pancreatic β-cells hyperpolarized they prevent insulin release and cause permanent neonatal diabetes and in about 20% of patients also neurodevelopmental delay (Gloyn et al., 2004; De Franco et al., 2020). In most patients switching from insulin injections to high oral doses of sulfonylureas, known well-tolerated blockers of these channels, increases quality of life, glycemic control and ameliorates some of the neurological disabilities (Ashcroft et al., 2017).
Unfortunately, not all channelopathies respond to such gene-specific pharmacotherapy. Long QT-syndromes, including LQT3 (Nav1.5 sodium channels, Arbelo et al., 2016) and LQT8 (see above), are such examples. Although LQT8 is caused by GOF mutations in Cav1.2 LTCCs, case reports of patients treated with the LTCC blockers verapamil or diltiazem for prevention of ventricular fibrillation episodes (Jacobs et al., 2006; Shah et al., 2012; Liu et al., 2016) showed mixed effects. Verapamil may not be the best choice for these patients because it is also a potent blocker of HERG K+-channels, favoring QT-prolongation (this explains why verapamil usually does not affect the QT-interval because it also blocks L-type channels; Zhang et al., 1999). DHP L-type channel blockers would be another option but due to their state-dependent action (with preferential inhibition of arterial smooth muscle channels and blood pressure lowering) they are only weak blockers of cardiac L-type channels (Zamponi et al., 2015). Moreover, pathogenic mutations may decrease the apparent sensitivity for DHP, as shown for nisoldipine-inhibition of the TS1 mutation G406R (3-fold increase in IC50; Splawski et al., 2004). Therefore, as suggested also for GOF Na+-channel mutations causing LQT3-syndrome (Arbelo et al., 2016), mutation-specific management may be necessary. This requires extensive characterization of the biophysical and biochemical properties of mutations (ideally in human iPSC-derived cells) to identify patient populations most likely to respond to a given channel blocker in prospective clinical trials. In contrast to the type-1 TS1 mutation G406R, the type-2 Cav1.3 mutation S652L increases the sensitivity to the DHP isradipine about 3-fold (Hofer et al., 2020). Therefore, carriers of this mutation should be the most likely candidates for clinical (off-label) testing. The recent observation that nifedipine may improve some neurological symptoms in a patient with a pathogenic Cav1.3 GOF mutation provides some hope for a therapeutic benefit of this strategy (De Mingo Alemany et al., 2020). Evidence for a mutation-specific therapeutic benefit also comes from the observation that some patients with an epileptic encephalopathy due to CACNA1E GOF mutations, who were resistant to multiple anti-epileptic drugs, reported significant improvement in seizure control with topiramate. This antiepileptic can inhibit Cav2.3 currents at therapeutically relevant concentrations (Kuzmiski et al., 2005).
Mutation-specific strategies would require new drugs that are capable of reversing some or all of the gating changes of Ca2+-channel GOF mutations. As a proof-of-concept it has already been shown that the tert-butyl dihydroquinone (BHQ) can modulate Cav2.1 channels in a manner that oppose altered channel gating of the FHM-1 mutation S218L (type 2, Figures 1D, 4; Inagaki et al., 2014). BHQ inhibited voltage-dependent activation thereby also reducing the enhanced current amplitudes in the mutant channel at negative voltages. At the Drosophila neuromuscular junctions expressing the S218L channel, BHQ largely restored the abnormally elevated evoked postsynaptic potentials and the impaired synaptic plasticity to the wildtype phenotypes.
Another relevant aspect to be considered for GOF mutations is the fact that increased channel activity may not only affect the electrical properties directly by enhancing depolarizing current but may also affect downstream Ca2+-dependent signals that could be accessible to therapeutic intervention. Enhanced Ca2+-channel signaling may activate Ca2+-dependent K+-channels, which are closely coupled to different types of Ca2+-channels in different cells (Berkefeld et al., 2006; Marcantoni et al., 2010). Another example exists for the TS1 mutation G406R. Roscovitine, a CDK kinase inhibitor, enhances the inactivation of LTCCs and partially reverses the abnormal, slow inactivation of G406R channels (Paşca et al., 2011; Yazawa et al., 2011 for references). It rescues the phenotypes in iPSC-derived cardiomyocytes (prolonged action potential duration) and neurons (aberrant activity-dependent gene-expression) from TS patients (Yazawa et al., 2011; Paşca et al., 2011; Song et al., 2017). In iPSC-derived cardiomyocytes this was explained by enhanced Ca2+-dependent activation of the ERK/EGR1 pathway by G406R, which drives overexpression of CDK5. CDK5 inhibition by roscovitine, dominant-negative CDK5 constructs or shRNAs alleviated the TS1 phenotype (Song et al., 2017), and may therefore guide a novel therapeutic approach.
Mutation-specific approaches could also target gating-pore currents. If, as speculated in this review, gating-pore currents contribute to pathology not only in Cav1.1 channelopathies (HypoPP, NormoPP) but also in other Cav channelopathies, then gating-pore blockers would be another attractive approach. Both toxins (e.g., spider toxin Hm-3 from Heriaeus melloteei, Männikkö et al., 2018) as well as guanidinium-derivatives (Sokolov et al., 2010) have already been shown to block gating-pore currents through Nav1.4 channel mutants causing periodic paralysis. Together with high-resolution crystal structures of channel mutants (Jiang et al., 2018), molecular dynamics simulations and pharmacophore modeling these studies provided valuable molecular templates for the further development of hit compounds specifically targeting gating-pores.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
The author’s work is supported by the Austrian Science Fund (FWF P27809, CavX/Doc-30-B30), the European Community (DP-ARDRE/H2020 Marie Skłodowska-Curie Actions COFUND 847681), and the University of Innsbruck.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author thanks Nadine J. Ortner, Petronel Tuluc and Bernhard E. Flucher for stimulating discussions and continuous support and Sylvia Boesch for neurological counseling. Figures 1C,D were generated with UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311. This work is devoted to Wilhelm Strießnig (1917–2020).
Footnotes
- ^ www.guidetopharmacology.org
- ^ Predicted from truncating mutations within the transmembrane portion of the channel protein (gnomAD Ver2.1.1).
References
Ablinger, C., Geisler, S. M., Stanika, R. I., Klein, C. T., and Obermair, G. J. (2020). Neuronal alpha2delta proteins and brain disorders. Pflugers. Arch. 472, 845–863. doi: 10.1007/s00424-020-02420-2
Alexander, S. P. H., Mathie, A., Peters, J. A., Veale, E. L., Striessnig, J., Kelly, E., et al. (2019). The concise guide to pharmacology 2019/20: ion channels. Br. J. Pharmacol. 176(Suppl. 1), S142–S228. doi: 10.1111/bph.14749
Allen, A. S., Bellows, S. T., Berkovic, S. F., Bridgers, J., Burgess, R., Cavalleri, G., et al. (2017). Ultra-rare genetic variation in common epilepsies: a case-control sequencing study. Lancet Neurol. 16, 135–143. doi: 10.1016/S1474-4422(16)30359-3
Antzelevitch, C., Pollevick, G. D., Cordeiro, J. M., Casis, O., Sanguinetti, M. C., Aizawa, Y., et al. (2007). Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 115, 442–449. doi: 10.1161/CIRCULATIONAHA.106.668392
Antzelevitch, C., and Yan, G.-X. (2015). J-wave syndromes: brugada and early repolarization syndromes. Heart Rhythm 12, 1852–1866. doi: 10.1016/j.hrthm.2015.04.014
Antzelevitch, C., Yan, G.-X., Ackerman, M. J., Borggrefe, M., Corrado, D., Guo, J., et al. (2017). J-Wave syndromes expert consensus conference report: emerging concepts and gaps in knowledge. Europace 19, 665–694. doi: 10.1093/europace/euw235
Arbelo, E., Sarquella-Brugada, G., and Brugada, J. (2016). Gene-specific therapy for congenital long QT syndrome. J. Am. Coll. Cardiol. 67, 1059–1061. doi: 10.1016/j.jacc.2015.12.030
Ashcroft, F. M., Puljung, M. C., and Vedovato, N. (2017). Neonatal diabetes and the KATP channel: from mutation to therapy. Trends Endocrinol. Metab. 28, 377–387. doi: 10.1016/j.tem.2017.02.003
Azizan, E. A. B., Poulsen, H., Tuluc, P., Zhou, J., Clausen, M. V., Lieb, A., et al. (2013). Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat. Genet. 45, 1055–1060. doi: 10.1038/ng.2716
Baig, S. M., Koschak, A., Lieb, A., Gebhart, M., Dafinger, C., et al. (2011). Loss of Ca(v)1.3 (CACNA1D) function in a human channelopathy with bradycardia and congenital deafness. Nat. Neurosci. 14, 77–84. doi: 10.1038/nn.2694
Bainbridge, M. N., Wiszniewski, W., Murdock, D. R., Friedman, J., Gonzaga-Jauregui, C., Newsham, I., et al. (2011). Whole-genome sequencing for optimized patient management. Sci. Transl. Med. 3:87re3. doi: 10.1126/scitranslmed.3002243
Balck, A., Tunc, S., Schmitz, J., Hollstein, R., Kaiser, F. J., and Brüggemann, N. (2018). A novel frameshift CACNA1A mutation causing episodic ataxia type 2. Cerebellum 17, 504–506. doi: 10.1007/s12311-018-0931-8
Barresi, S., Dentici, M. L., Manzoni, F., Bellacchio, E., Agolini, E., Pizzi, S., et al. (2020). Infantile-onset syndromic cerebellar ataxia and CACNA1G mutations. Pediatr. Neurol. 104, 40–45. doi: 10.1016/j.pediatrneurol.2019.09.005
Baurand, A., Falcon-Eicher, S., Laurent, G., Villain, E., Bonnet, C., Thauvin-Robinet, C., et al. (2017). Incomplete Timothy syndrome secondary to a mosaic mutation of the CACNA1C gene diagnosed using next-generation sequencing. Am. J. Med. Genet. A 173, 531–536. doi: 10.1002/ajmg.a.38045
Ben-Johny, M., Dick, I. E., Sang, L., Limpitikul, W. B., Kang, P. W., Niu, J., et al. (2015). Towards a unified theory of calmodulin regulation (Calmodulation) of voltage-gated calcium and sodium channels. Curr. Mol. Pharmacol. 8, 188–205. doi: 10.2174/1874467208666150507110359
Berkefeld, H., Sailer, C. A., Bildl, W., Rohde, V., Thumfart, J.-O., Eble, S., et al. (2006). BKCa-Cav channel complexes mediate rapid and localized Ca2+-activated K+ signaling. Science 314, 615–620. doi: 10.1126/science.1132915
Boczek, N. J., Miller, E. M., Ye, D., Nesterenko, V. V., Tester, D. J., Antzelevitch, C., et al. (2015). Novel timothy syndrome mutation leading to increase in CACNA1C window current. Heart Rhythm 12, 211–219. doi: 10.1016/j.hrthm.2014.09.051
Boulkroun, S., Fernandes-Rosa, F. L., and Zennaro, M. C. (2020). Old and new genes in primary aldosteronism. Best Pract. Res. Clin. Endocrinol. Metab. 34:101375. doi: 10.1016/j.beem.2020.101375
Brennan, K. C., and Pietrobon, D. (2018). A systems neuroscience approach to migraine. Neuron 97, 1004–1021. doi: 10.1016/j.neuron.2018.01.029
Buraei, Z., and Yang, J. (2010). The ß subunit of voltage-gated Ca2+ channels. Physiol. Rev. 90, 1461–1506. doi: 10.1152/physrev.00057.2009
Burashnikov, E., Pfeiffer, R., Barajas-Martinez, H., Delpón, E., Hu, D., Desai, M., et al. (2010). Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Heart Rhythm 7, 1872–1882. doi: 10.1016/j.hrthm.2010.08.026
Calhoun, J. D., Hawkins, N. A., Zachwieja, N. J., and Kearney, J. A. (2016). Cacna1g is a genetic modifier of epilepsy caused by mutation of voltage-gated sodium channel Scn2a. Epilepsia 57, e103–e107. doi: 10.1111/epi.13390
Calorio, C., Gavello, D., Guarina, L., Salio, C., Sassoè-Pognetto, M., Riganti, C., et al. (2019). Impaired chromaffin cell excitability and exocytosis in autistic Timothy syndrome TS2-neo mouse rescued by L-type calcium channel blockers. J. Physiol. 597, 1705–1733. doi: 10.1113/JP277487
Cannon, S. C. (2017). An atypical CaV1.1 mutation reveals a common mechanism for hypokalemic periodic paralysis. J. Gen. Physiol. 149, 1061–1064. doi: 10.1085/jgp.201711923
Cao, Y., Tokita, M. J., Chen, E. S., Ghosh, R., Chen, T., Feng, Y., et al. (2019). A clinical survey of mosaic single nucleotide variants in disease-causing genes detected by exome sequencing. Genome Med. 11:48. doi: 10.1186/s13073-019-0658-2
Carreño, O., Corominas, R., Serra, S. A., Sintas, C., Fernández-Castillo, N., Vila-Pueyo, M., et al. (2013). Screening of CACNA1A and ATP1A2 genes in hemiplegic migraine: clinical, genetic, and functional studies. Mol. Genet. Genomic Med. 1, 206–222. doi: 10.1002/mgg3.24
Casey, H. L., and Gomez, C. M. (2019). “Spinocerebellar Ataxia Type 6,” in GeneReviews® [Internet], eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. Bean, and K. Stephens (Seattle, WA: University of Washington).
Catterall, W. A., Lenaeus, M. J., and Gamal El-Din, T. M. (2020). Structure and pharmacology of voltage-gated sodium and calcium channels. Annu. Rev. Pharmacol. Toxicol. 60, 133–154. doi: 10.1146/annurev-pharmtox-010818-021757
Chavali, N. V., Kryshtal, D. O., Parikh, S. S., Wang, L., Glazer, A. M., Blackwell, D. J., et al. (2019). Patient-independent human induced pluripotent stem cell model: a new tool for rapid determination of genetic variant pathogenicity in long QT syndrome. Heart Rhythm 16, 1686–1695. doi: 10.1016/j.hrthm.2019.04.031
Chemin, J., Siquier-Pernet, K., Nicouleau, M., Barcia, G., Ahmad, A., Medina-Cano, D., et al. (2018). De novo mutation screening in childhood-onset cerebellar atrophy identifies gain-of-function mutations in the CACNA1G calcium channel gene. Brain 141, 1998–2013. doi: 10.1093/brain/awy145
Cocos, R., Raicu, F., Bajenaru, O. L., Olaru, I., Dumitrescu, L., and Popescu, B. O. (2020). CACNA1B gene variants in adult-onset isolated focal dystonia. Neurol. Sci. doi: 10.1007/s10072-020-04778-8 [Epub ahead of print].
Colson, C., Mittre, H., Busson, A., Leenhardt, A., Denjoy, I., Fressard, V., et al. (2019). Unusual clinical description of adult with Timothy syndrome, carrier of a new heterozygote mutation of CACNA1C. Eur. J. Med. Genet. 62:103648. doi: 10.1016/j.ejmg.2019.04.005
Consortium, E. K., Project, E. P. G., Allen, A. S., Berkovic, S. F., Cossette, P., Delanty, N., et al. (2013). De novo mutations in epileptic encephalopathies. Nature 501, 217–221. doi: 10.1038/nature12439
Coutelier, M., Blesneac, I., Monteil, A., Monin, M.-L., Ando, K., Mundwiller, E., et al. (2015). A recurrent mutation in CACNA1G Alters Cav3.1 T-type calcium-channel conduction and causes autosomal-dominant cerebellar Ataxia. Am. J. Hum. Genet. 97, 726–737. doi: 10.1016/j.ajhg.2015.09.007
Damaj, L., Lupien-Meilleur, A., Lortie, A., Riou, É, Ospina, L. H., Gagnon, L., et al. (2015). CACNA1A haploinsufficiency causes cognitive impairment, autism and epileptic encephalopathy with mild cerebellar symptoms. Eur. J. Hum. Genet. 23, 1505–1512. doi: 10.1038/ejhg.2015.21
Daniil, G., Fernandes-Rosa, F. L., Chemin, J., Blesneac, I., Beltrand, J., Polak, M., et al. (2016). CACNA1H mutations are associated with different forms of primary aldosteronism. EBioMedicine 13, 225–236. doi: 10.1016/j.ebiom.2016.10.002
De Franco, E., Saint-Martin, C., Brusgaard, K., Knight Johnson, A. E., Aguilar-Bryan, L., Bowman, P., et al. (2020). Update of variants identified in the pancreatic β-cell KATP channel genes KCNJ11 and ABCC8 in individuals with congenital hyperinsulinism and diabetes. Hum. Mutat. 41, 884–905. doi: 10.1002/humu.23995
De Mingo Alemany, M. C., Mifsud Grau, L., Moreno Macián, F., Ferrer Lorente, B., and León Cariñena, S. (2020). A de novo CACNA1D missense mutation in a patient with congenital hyperinsulinism, primary hyperaldosteronism and hypotonia. Channels 14, 175–180. doi: 10.1080/19336950.2020.1761171
Di Cristofori, A., Fusi, L., Gomitoni, A., Grampa, G., and Bersano, A. (2012). R583Q CACNA1A variant in SHM1 and ataxia: case report and literature update. J. Headache Pain 13, 419–423. doi: 10.1007/s10194-012-0444-7
Dolphin, A. C. (2018). Voltage-gated calcium channel alpha 2delta subunits: an assessment of proposed novel roles. F1000Res. 7:F1000 Faculty Rev-1830. doi: 10.12688/f1000research.16104.1
Dolphin, A. C., and Lee, A. (2020). Presynaptic calcium channels: specialized control of synaptic neurotransmitter release. Nat. Rev. Neurosci. 21, 213–229. doi: 10.1038/s41583-020-0278-2
Drum, B. M. L., Dixon, R. E., Yuan, C., Cheng, E. P., and Santana, L. F. (2014). Cellular mechanisms of ventricular arrhythmias in a mouse model of Timothy syndrome (long QT syndrome 8). J. Mol. Cell Cardiol. 66, 63–71. doi: 10.1016/j.yjmcc.2013.10.021
Du, X., and Gomez, C. M. (2018). “Spinocerebellar ataxia type 6: molecular mechanisms and calcium channel genetics,” in Polyglutamine Disorders, eds C. Nóbrega and L. Pereira de Almeida (Cham: Springer).
Ducros, A., Denier, C., Joutel, A., Cecillon, M., Lescoat, C., Vahedi, K., et al. (2001). The clinical spectrum of familial hemiplegic migraine associated with mutations in a neuronal calcium channel. N. Engl. J. Med. 345, 17–24. doi: 10.1056/nejm200107053450103
Dufendach, K. A., Giudicessi, J. R., Boczek, N. J., and Ackerman, M. J. (2013). Maternal mosaicism confounds the neonatal diagnosis of type 1 Timothy syndrome. Pediatrics 131, e1991–e1995. doi: 10.1542/peds.2012-2941
Eising, E., and van den Maagdenberg, A. M. J. M. (2017). “Hemiplegic migraine and channel disorders,” in Reference Module in Neuroscience and Biobehavioral Psychology, (Amsterdam: Elsevier). doi: 10.1016/B978-0-12-809324-5.01969-6
Epi4K Consortium. (2016). De novo mutations in SLC1A2 and CACNA1A are important causes of epileptic encephalopathies. Am. J. Hum. Genet. 99, 287–298. doi: 10.1016/j.ajhg.2016.06.003
Epperson, M. V., Haws, M. E., Standridge, S. M., and Gilbert, D. L. (2018). An atypical Rett syndrome phenotype due to a novel missense mutation in CACNA1A. J. Child Neurol. 33, 286–289. doi: 10.1177/0883073818754987
Estes, S. I., Ye, D., Zhou, W., Dotzler, S. M., Tester, D. J., Bos, J. M., et al. (2019). Characterization of the CACNA1C-R518C missense mutation in the pathobiology of long-QT syndrome using human induced pluripotent stem cell cardiomyocytes shows action potential prolongation and L-type calcium channel perturbation. Circ. Genom. Precis. Med. 12:e002534. doi: 10.1161/CIRCGEN.119.002534
Etheridge, S. P., Bowles, N. E., Arrington, C. B., Pilcher, T., Rope, A., Wilde, A. A. M., et al. (2011). Somatic mosaicism contributes to phenotypic variation in Timothy syndrome. Am. J. Med. Genet. A 155A, 2578–2583. doi: 10.1002/ajmg.a.34223
Fletcher, C. F., Tottene, A., Lennon, V. A., Wilson, S. M., Dubel, S. J., Paylor, R., et al. (2001). Dystonia and cerebellar atrophy in Cacna1a null mice lacking P/Q calcium channel activity. FASEB J. 15, 1288–1290. doi: 10.1096/fj.00-0562fje
Flucher, B. E. (2020). Skeletal muscle CaV1.1 channelopathies. Pflügers Arch. 472, 739–754. doi: 10.1007/s00424-020-02368-3
Flucher, B. E., and Campiglio, M. (2019). STAC proteins: the missing link in skeletal muscle EC coupling and new regulators of calcium channel function. Biochim. Biophys. Acta Mol. Cell Res. 1866, 1101–1110. doi: 10.1016/j.bbamcr.2018.12.004
Forti, L., and Pietrobon, D. (1993). Functional diversity of L-type calcium channels in rat cerebellar neurons. Neuron 10, 437–450. doi: 10.1016/0896-6273(93)90332-l
Fröhler, S., Kieslich, M., Langnick, C., Feldkamp, M., Opgen-Rhein, B., Berger, F., et al. (2014). Exome sequencing helped the fine diagnosis of two siblings afflicted with atypical Timothy syndrome (TS2). BMC Med. Genet. 15:48. doi: 10.1186/1471-2350-15-48
Fukuyama, M., Ohno, S., Wang, Q., Shirayama, T., Itoh, H., and Horie, M. (2014a). Nonsense-mediated mRNA decay due to a CACNA1C splicing mutation in a patient with Brugada syndrome. Heart Rhythm 11, 629–634. doi: 10.1016/j.hrthm.2013.12.011
Fukuyama, M., Wang, Q., Kato, K., Ohno, S., Ding, W.-G., Toyoda, F., et al. (2014b). Long QT syndrome type 8: novel CACNA1C mutations causing QT prolongation and variant phenotypes. Europace 16, 1828–1837. doi: 10.1093/europace/euu063
Fuster, C., Perrot, J., Berthier, C., Jacquemond, V., Charnet, P., and Allard, B. (2017). Na leak with gating pore properties in hypokalemic periodic paralysis V876E mutant muscle Ca channel. J. Gen. Physiol. 149, 1139–1148. doi: 10.1085/jgp.201711834
Gao, S., and Yan, N. (2020). Structural basis of the modulation of the voltage-gated calcium ion channel Cav 1.1 by dihydropyridine compounds. Angew. Chem. Int. Ed. Engl. doi: 10.1002/anie.202011793 [Epub ahead of print].
Ghaleb, Y. E., Schneeberger, P. E., Fernández-Quintero, M. L., Geisler, S. M., Pelizzari, S., Polstra, A. M., et al. (2021). CACNA1I gain-of-function mutations differentially affect channel gating and cause neurodevelopmental disorders. Brain. (in press).
Ghoshal, A., Uygun, D. S., Yang, L., McNally, J. M., Lopez-Huerta, V. G., Arias-Garcia, M. A., et al. (2020). Effects of a patient-derived de novo coding alteration of CACNA1I in mice connect a schizophrenia risk gene with sleep spindle deficits. Transl. Psychiatry 10:29. doi: 10.1038/s41398-020-0685-1
Gillis, J., Burashnikov, E., Antzelevitch, C., Blaser, S., Gross, G., Turner, L., et al. (2012). Long QT, syndactyly, joint contractures, stroke and novel CACNA1C mutation: expanding the spectrum of Timothy syndrome. Am. J. Med. Genet. A 158A, 182–187. doi: 10.1002/ajmg.a.34355
Giudicessi, J. R., and Ackerman, M. J. (2016). Calcium revisited: new insights into the molecular basis of long-QT syndrome. Circ. Arrhythm. Electrophysiol. 9:e002480. doi: 10.1161/CIRCEP.116.002480
Gloyn, A. L., Pearson, E. R., Antcliff, J. F., Proks, P., Bruining, G. J., Slingerland, A. S., et al. (2004). Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N. Engl. J. Med. 350, 1838–1849. doi: 10.1056/NEJMoa032922
Goadsby, P. J., Holland, P. R., Martins-Oliveira, M., Hoffmann, J., Schankin, C., and Akerman, S. (2017). Pathophysiology of migraine: a disorder of sensory processing. Physiol. Rev. 97, 553–622. doi: 10.1152/physrev.00034.2015
Gorman, K. M., Meyer, E., Grozeva, D., Spinelli, E., McTague, A., et al. (2019). Bi-allelic Loss-of-function CACNA1B mutations in progressive epilepsy-dyskinesia. Am. J. Hum. Genet. 104, 948–956. doi: 10.1016/j.ajhg.2019.03.005
Gratten, J., Visscher, P. M., Mowry, B. J., and Wray, N. R. (2013). Interpreting the role of de novo protein-coding mutations in neuropsychiatric disease. Nat. Genet. 45, 234–238. doi: 10.1038/ng.2555
Groen, J. L., Andrade, A., Ritz, K., Jalalzadeh, H., Haagmans, M., Bradley, T. E. J., et al. (2014). CACNA1B mutation is linked to unique myoclonus-dystonia syndrome. Hum. Mol. Genet. 24, 987–993. doi: 10.1093/hmg/ddu513
Groome, J. R., Moreau, A., and Delemotte, L. (2017). “Gating pore currents in sodium channels,” in Voltage-gated Sodium Channels: Structure, Function and Channelopathies, ed. M. Chahine (Cham: Springer).
Hans, M., Luvisetto, S., Williams, M. E., Spagnolo, M., Urrutia, A., Tottene, A., et al. (1999). Functional consequences of mutations in the human alpha1A calcium channel subunit linked to familial hemiplegic migraine. J. Neurosci. 19, 1610–1619. doi: 10.1523/jneurosci.19-05-01610.1999
Hardie, J., and Lee, A. (2016). Decalmodulation of Cav1 channels by CaBPs. Channels 10, 33–37. doi: 10.1080/19336950.2015.1051273
Hashiguchi, S., Doi, H., Kunii, M., Nakamura, Y., Shimuta, M., Suzuki, E., et al. (2019). Ataxic phenotype with altered CaV3.1 channel property in a mouse model for spinocerebellar ataxia 42. Neurobiol. Dis. 130:104516. doi: 10.1016/j.nbd.2019.104516
Helbig, K. L., Farwell Hagman, K. D., Shinde, D. N., Mroske, C., Powis, Z., Li, S., et al. (2016). Diagnostic exome sequencing provides a molecular diagnosis for a significant proportion of patients with epilepsy. Genet. Med. 18, 898–905. doi: 10.1038/gim.2015.186
Helbig, K. L., Lauerer, R. J., Bahr, J. C., Souza, I. A., Myers, C. T., Uysal, B., et al. (2018). De novo pathogenic variants in CACNA1E cause developmental and epileptic encephalopathy with contractures, macrocephaly, and dyskinesias. Am. J. Hum. Genet. 103, 666–678. doi: 10.1016/j.ajhg.2018.09.006
Hemara-Wahanui, A., Berjukow, S., Hope, C. I., Dearden, P. K., Wu, S. B., Wilson-Wheeler, J., et al. (2005). A CACNA1F mutation identified in an X-linked retinal disorder shifts the voltage dependence of Cav1.4 channel activation. Proc. Natl. Acad. Sci. U.S.A. 102, 7553–7558. doi: 10.1073/pnas.0501907102
Hennessey, J. A., Boczek, N. J., Jiang, Y.-H., Miller, J. D., Patrick, W., Pfeiffer, R., et al. (2014). A CACNA1C variant associated with reduced voltage-dependent inactivation, increased CaV1.2 channel window current, and arrhythmogenesis. PLoS One 9:e106982. doi: 10.1371/journal.pone.0106982
Hoda, J. C., Zaghetto, F., Koschak, A., and Striessnig, J. (2005). Congenital stationary night blindness type 2 mutations S229P, G369D, L1068P, and W1440X alter channel gating or functional expression of Cav1.4 L-type calcium channels. J. Neurosci. 25, 252–259. doi: 10.1523/jneurosci.3054-04.2005
Hoda, J. C., Zaghetto, F., Singh, A., Koschak, A., and Striessnig, J. (2006). Effects of congenital stationary night blindness type 2 mutations R508Q and L1364H on Cav1.4 L-type calcium channel function and expression. J. Neurochem. 96, 1648–1658. doi: 10.1111/j.1471-4159.2006.03678.x
Hofer, N. T., Tuluc, P., Ortner, N. J., Nikonishyna, Y. V., Fernándes-Quintero, M. L., Liedl, K. R., et al. (2020). Biophysical classification of a CACNA1D de novo mutation as a high-risk mutation for a severe neurodevelopmental disorder. Mol. Autism 11:4. doi: 10.1186/s13229-019-0310-4
Hofmann, F., Belkacemi, A., and Flockerzi, V. (2015). Emerging alternative functions for the auxiliary subunits of the voltage-gated calcium channels. Curr. Mol. Pharmacol. 8, 162–168. doi: 10.2174/1874467208666150507110202
Holbrook, J. A., Neu-Yilik, G., Hentze, M. W., and Kulozik, A. E. (2004). Nonsense-mediated decay approaches the clinic. Nat. Genet. 36, 801–808. doi: 10.1038/ng1403
Hope, C. I., Sharp, D. M., Hemara-Wahanui, A., Sissingh, J. I., Lundon, P., Mitchell, E. A., et al. (2005). Clinical manifestations of a unique X-linked retinal disorder in a large New Zealand family with a novel mutation in CACNA1F, the gene responsible for CSNB2. Clin. Exp. Ophthalmol. 33, 129–136. doi: 10.1111/j.1442-9071.2005.00987.x
Howard, M. A., and Baraban, S. C. (2017). Catastrophic epilepsies of childhood. Annu. Rev. Neurosci. 40, 149–166. doi: 10.1146/annurev-neuro-072116-031250
Huang, H., Tan, B. Z., Shen, Y., Tao, J., Jiang, F., Sung, Y. Y., et al. (2012). RNA editing of the IQ domain in Cav1.3 channels modulates their Ca2+-dependent inactivation. Neuron 73, 304–316. doi: 10.1016/j.neuron.2011.11.022
Humbertclaude, V., Riant, F., Krams, B., Zimmermann, V., Nagot, N., Annequin, D., et al. (2020). Cognitive impairment in children with CACNA1A mutations. Dev. Med. Child Neurol. 62, 330–337. doi: 10.1111/dmcn.14261
Inagaki, A., Frank, C. A., Usachev, Y. M., Benveniste, M., and Lee, A. (2014). Pharmacological correction of gating defects in the voltage-gated Cav2.1 Ca2 channel due to a familial hemiplegic migraine mutation. Neuron 81, 91–102. doi: 10.1016/j.neuron.2013.10.056
Indelicato, E., Nachbauer, W., Karner, E., Eigentler, A., Wagner, M., Unterberger, I., et al. (2019). The neuropsychiatric phenotype in CACNA1A mutations: a retrospective single center study and review of the literature. Eur. J. Neurol. 26:66-e7. doi: 10.1111/ene.13765
Izquierdo-Serra, M., Fernández-Fernández, J. M., and Serrano, M. (2020). Rare CACNA1A mutations leading to congenital ataxia. Pflugers Arch. 472, 791–809. doi: 10.1007/s00424-020-02396-z
Jacobs, A., Knight, B. P., McDonald, K. T., and Burke, M. C. (2006). Verapamil decreases ventricular tachyarrhythmias in a patient with Timothy syndrome (LQT8). Heart Rhythm 3, 967–970. doi: 10.1016/j.hrthm.2006.04.024
Jen, J., Kim, G. W., and Baloh, R. W. (2004). Clinical spectrum of episodic ataxia type 2. Neurology 62, 17–22. doi: 10.1212/01.wnl.0000101675.61074.50
Jen, J. C. (1993). “Familial Hemiplegic Migraine,” in GeneReviews [Internet], eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. Bean, K. Stephens, et al. (Seattle, WA: University of Washington).
Jen, J. C. (2015). “Familial Hemiplegic Migraine,” in GeneReviews® [Internet], eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. Bean, and K. Stephens (Seattle, WA: University of Washington).
Jiang, D., Gamal El-Din, T. M., Ing, C., Lu, P., Pomes, R., Zheng, N., et al. (2018). Structural basis for gating pore current in periodic paralysis. Nature 557, 590–594. doi: 10.1038/s41586-018-0120-4
Jiang, D., Shi, H., Tonggu, L., Gamal El-Din, T. M., Lenaeus, M. J., Zhao, Y., et al. (2019). Structure of the cardiac sodium channel. Cell 180, 122–134.e10. doi: 10.1016/j.cell.2019.11.041
Jiang, X., Raju, P. K., D’Avanzo, N., Lachance, M., Pepin, J., Dubeau, F., et al. (2019). Both gain-of-function and loss-of-function de novo CACNA1A mutations cause severe developmental epileptic encephalopathies in the spectrum of Lennox-Gastaut syndrome. Epilepsia 60, 1881–1894. doi: 10.1111/epi.16316
Jun, K., Piedras-Renteria, E. S., Smith, S. M., Wheeler, D. B., Lee, S. B., Lee, T. G., et al. (1999). Ablation of P/Q-type Calcium channel currents, altered synaptic transmission, and progressive ataxia in mice lacking the alpha1A- subunit. Proc. Natl. Acad. Sci. U.S.A. 96, 15245–15250. doi: 10.1073/pnas.96.26.15245
Jurkat-Rott, K., Groome, J., and Lehmann-Horn, F. (2012). Pathophysiological role of omega pore current in channelopathies. Front. Pharmacol. 3:112. doi: 10.3389/fphar.2012.00112
Kabir, Z. D., Martinez-Rivera, A., and Rajadhyaksha, A. M. (2017). From gene to behavior: L-type calcium channel mechanisms underlying neuropsychiatric symptoms. Neurotherapeutics 14, 588–613. doi: 10.1007/s13311-017-0532-0
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. doi: 10.1038/s41586-020-2308-7
Kim, C., Jeon, D., Kim, Y.-H., Lee, C. J., Kim, H., and Shin, H.-S. (2009). Deletion of N-type Ca(2+) channel Ca(v)2.2 results in hyperaggressive behaviors in mice. J. Biol. Chem. 284, 2738–2745. doi: 10.1074/jbc.M807179200
Kim, D., Song, I., Keum, S., Lee, T., Jeong, M. J., Kim, S. S., et al. (2001). Lack of the burst firing of thalamocortical relay neurons and resistance to absence seizures in mice lacking alpha1G T-type Calcium channels. Neuron 31, 35–45. doi: 10.1016/s0896-6273(01)00343-9
Kochinke, K., Zweier, C., Nijhof, B., Fenckova, M., Cizek, P., Honti, F., et al. (2016). Systematic phenomics analysis deconvolutes genes mutated in intellectual disability into biologically coherent modules. Am. J. Hum. Genet. 98, 149–164. doi: 10.1016/j.ajhg.2015.11.024
Kosaki, R., Ono, H., Terashima, H., and Kosaki, K. (2018). Timothy syndrome-like condition with syndactyly but without prolongation of the QT interval. Am. J. Med. Genet. A 176, 1657–1661. doi: 10.1002/ajmg.a.38833
Koschak, A., Reimer, D., Huber, I., Grabner, M., Glossmann, H., Engel, J., et al. (2001). α1D (Cav1.3) subunits can form L-type calcium channels activating at negative voltages. J. Biol. Chem. 276, 22100–22106. doi: 10.1074/jbc.m101469200
Kraus, R. L., Sinnegger, M. J., Glossmann, H., Hering, S., and Striessnig, J. (1998). Familial hemiplegic migraine mutations change alpha1A Ca2+ channel kinetics. J. Biol. Chem. 273, 5586–5590. doi: 10.1074/jbc.273.10.5586
Krey, J. F., Pasca, S. P., Shcheglovitov, A., Yazawa, M., Schwemberger, R., Rasmusson, R., et al. (2013). Timothy syndrome is associated with activity-dependent dendritic retraction in rodent and human neurons. Nat. Neurosci. 16, 201–209. doi: 10.1038/nn.3307
Krueger, D. A., Wilfong, A. A., Mays, M., Talley, C. M., Agricola, K., Tudor, C., et al. (2016). Long-term treatment of epilepsy with everolimus in tuberous sclerosis. Neurology 87, 2408–2415. doi: 10.1212/WNL.0000000000003400
Krumm, N., Turner, T. N., Baker, C., Vives, L., Mohajeri, K., Witherspoon, K., et al. (2015). Excess of rare, inherited truncating mutations in autism. Nat. Genet. 47, 582–588. doi: 10.1038/ng.3303
Kubota, T., Wu, F., Vicart, S., Nakaza, M., Sternberg, D., Watanabe, D., et al. (2020). Hypokalaemic periodic paralysis with a charge-retaining substitution in the voltage sensor. Brain Commun. 2:fcaa103. doi: 10.1093/braincomms/fcaa103
Kuzmiski, J. B., Barr, W., Zamponi, G. W., and MacVicar, B. A. (2005). Topiramate inhibits the initiation of plateau potentials in CA1 neurons by depressing R-type calcium channels. Epilepsia 46, 481–489. doi: 10.1111/j.0013-9580.2005.35304.x
Landstrom, A. P., Boczek, N. J., Ye, D., Miyake, C. Y., De la Uz, C. M., Allen, H. D., et al. (2016). Novel long QT syndrome-associated missense mutation, L762F, in CACNA1C-encoded L-type calcium channel imparts a slower inactivation tau and increased sustained and window current. Int. J. Cardiol. 220, 290–298. doi: 10.1016/j.ijcard.2016.06.081
Latour, I., Louw, D. F., Beedle, A. M., Hamid, J., Sutherland, G. R., and Zamponi, G. W. (2004). Expression of T-type calcium channel splice variants in human glioma. Glia 48, 112–119. doi: 10.1002/glia.20063
Li, G., Wang, J., Liao, P., Bartels, P., Zhang, H., Yu, D., et al. (2017). Exclusion of alternative exon 33 of CaV1.2 calcium channels in heart is proarrhythmogenic. Proc. Natl. Acad. Sci. U.S.A. 114, E4288–E4295. doi: 10.1073/pnas.1617205114
Li, X., Zhou, C., Cui, L., Zhu, L., Du, H., Liu, J., et al. (2018). A case of a novel CACNA1G mutation from a Chinese family with SCA42: a case report and literature review. Medicine 97:e12148. doi: 10.1097/MD.0000000000012148
Liaqat, K., Schrauwen, I., Raza, S. I., Lee, K., Hussain, S., Chakchouk, I., et al. (2019). Identification of CACNA1D variants associated with sinoatrial node dysfunction and deafness in additional Pakistani families reveals a clinical significance. J. Hum. Genet. 64, 153–160. doi: 10.1038/s10038-018-0542-8
Lim, E. T., Uddin, M., De Rubeis, S., Chan, Y., Kamumbu, A. S., Zhang, X., et al. (2017). Rates, distribution and implications of postzygotic mosaic mutations in autism spectrum disorder. Nat. Neurosci. 20, 1217–1224. doi: 10.1038/nn.4598
Limpitikul, W. B., Dick, I. E., Ben-Johny, M., and Yue, D. T. (2016). An autism-associated mutation in CaV1.3 channels has opposing effects on voltage- and Ca(2+)-dependent regulation. Sci. Rep. 6:27235. doi: 10.1038/srep27235
Liu, G., Papa, A., Katchman, A. N., Zakharov, S. I., Roybal, D., Hennessey, J. A., et al. (2020). Mechanism of adrenergic CaV1.2 stimulation revealed by proximity proteomics. Nature 577, 695–700. doi: 10.1038/s41586-020-1947-z
Liu, Y., Xue, Y., Wu, S., Duan, J., Lin, L., Wang, L., et al. (2016). Effect of verapamil in the treatment of type 2 long QT syndrome is not a dose-dependent pattern: a study from bedside to bench, and back. Eur. Heart J. Suppl. 18(Suppl. A), A37–A46. doi: 10.1093/eurheartj/suw006
Loh, K. W. Z., Liang, M. C., Soong, T. W., and Hu, Z. (2020). Regulation of cardiovascular calcium channel activity by post-translational modifications or interacting proteins. Pflugers Arch. 472, 653–667. doi: 10.1007/s00424-020-02398-x
Lory, P., Nicole, S., and Monteil, A. (2020). Neuronal Cav3 channelopathies: recent progress and perspectives. Pflügers Arch. 472, 831–844. doi: 10.1007/s00424-020-02429-7
Luo, X., Rosenfeld, J. A., Yamamoto, S., Harel, T., Zuo, Z., Hall, M., et al. (2017). Clinically severe CACNA1A alleles affect synaptic function and neurodegeneration differentially. PLoS Genet. 13:e1006905. doi: 10.1371/journal.pgen.1006905
Männikkö, R., Shenkarev, Z. O., Thor, M. G., Berkut, A. A., Myshkin, M. Y., Paramonov, A. S., et al. (2018). Spider toxin inhibits gating pore currents underlying periodic paralysis. Proc. Natl. Acad. Sci. U.S.A. 115, 4495–4500. doi: 10.1073/pnas.1720185115
Marcantoni, A., Calorio, C., Hidisoglu, E., Chiantia, G., and Carbone, E. (2020). Cav1.2 channelopathies causing autism: new hallmarks on Timothy syndrome. Pflugers Arch. 472, 775–789. doi: 10.1007/s00424-020-02430-0
Marcantoni, A., Vandael, D. H., Mahapatra, S., Carabelli, V., Sinnegger-Brauns, M. J., Striessnig, J., et al. (2010). Loss of Cav1.3 channels reveals the critical role of L-type and BK channel coupling in pacemaking mouse adrenal chromaffin cells. J. Neurosci. 30, 491–504. doi: 10.1523/JNEUROSCI.4961-09.2010
Marger, L., Mesirca, P., Alig, J., Torrente, A., Dübel, S., Engeland, B., et al. (2014). Functional roles of Ca v 1.3, Ca v 3.1 and HCN channels in automaticity of mouse atrioventricular cells. Channels 5, 251–261. doi: 10.4161/chan.5.3.15266
Mark, M. D., Maejima, T., Kuckelsberg, D., Yoo, J. W., Hyde, R. A., Shah, V., et al. (2011). Delayed postnatal loss of P/Q-type calcium channels recapitulates the absence epilepsy, dyskinesia, and ataxia phenotypes of genomic Cacna1a mutations. J. Neurosci. 31, 4311–4326. doi: 10.1523/JNEUROSCI.5342-10.2011
Mastrolia, V., Flucher, S. M., Obermair, G. J., Drach, M., Hofer, H., Renstrom, E., et al. (2017). Loss of alpha2delta-1 calcium channel subunit function increases the susceptibility for diabetes. Diabetes 66, 897–907. doi: 10.2337/db16-0336
Mellor, G. J., Panwar, P., Lee, A. K., Steinberg, C., Hathaway, J. A., Bartels, K., et al. (2019). Type 8 long QT syndrome: pathogenic variants in CACNA1C-encoded Cav1.2 cluster in STAC protein binding site. Europace 21, 1725–1732. doi: 10.1093/europace/euz215
Mencacci, N. E., R’bibo, L., Bandres-Ciga, S., Carecchio, M., Zorzi, G., Nardocci, N., et al. (2015). The CACNA1B R1389H variant is not associated with myoclonus-dystonia in a large European multicentric cohort. Hum. Mol. Genet. 24, 5326–5329. doi: 10.1093/hmg/ddv255
Mezghrani, A., Monteil, A., Watschinger, K., Sinnegger-Brauns, M. J., Barrère, C., Bourinet, E., et al. (2008). A destructive interaction mechanism accounts for dominant-negative effects of misfolded mutants of voltage-gated calcium channels. J. Neurosci. 28, 4501–4511. doi: 10.1523/JNEUROSCI.2844-07.2008
Miki, T., Nagashima, K., Tashiro, F., Kotake, K., Yoshitomi, H., Tamamoto, A., et al. (1998). Defective insulin secretion and enhanced insulin action in KATP channel-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 95, 10402–10406. doi: 10.1073/pnas.95.18.10402
Miki, T., Zwingman, T. A., Wakamori, M., Lutz, C. M., Cook, S. A., Hosford, D. A., et al. (2008). Two novel alleles of tottering with distinct Ca(v)2.1 calcium channel neuropathologies. Neuroscience 155, 31–44. doi: 10.1016/j.neuroscience.2008.05.028
Monteleone, S., Lieb, A., Pinggera, A., Negro, G., Fuchs, J. E., Hofer, F., et al. (2017). Mechanisms responsible for omega-pore currents in Cav calcium channel voltage-sensing domains. Biophys. J. 113, 1485–1495. doi: 10.1016/j.bpj.2017.08.010
Moreau, A., Gosselin-Badaroudine, P., Boutjdir, M., and Chahine, M. (2015). Mutations in the voltage sensors of domains I and II of Nav1.5 that are associated with arrhythmias and dilated cardiomyopathy generate gating pore currents. Front. Pharmacol. 6:301. doi: 10.3389/fphar.2015.00301
Morino, H., Matsuda, Y., Muguruma, K., Miyamoto, R., Ohsawa, R., Ohtake, T., et al. (2015). A mutation in the low voltage-gated calcium channel CACNA1G alters the physiological properties of the channel, causing spinocerebellar ataxia. Mol. Brain 8:89. doi: 10.1186/s13041-015-0180-4
Myers, C. T., Hollingsworth, G., Muir, A. M., Schneider, A. L., Thuesmunn, Z., Knupp, A., et al. (2018). Parental mosaicism in “De novo” epileptic encephalopathies. N. Engl. J. Med. 378, 1646–1648. doi: 10.1056/nejmc1714579
Myers, S. M., Challman, T. D., Bernier, R., Bourgeron, T., Chung, W. K., Constantino, J. N., et al. (2020). Insufficient evidence for “Autism-Specific” genes. Am. J. Hum. Genet. 106, 587–595. doi: 10.1016/j.ajhg.2020.04.004
Nanba, K., Blinder, A. R., Rege, J., Hattangady, N. G., Else, T., Liu, C.-J., et al. (2020). Somatic CACNA1H mutation as a cause of aldosterone-producing adenoma. Hypertension 75, 645–649. doi: 10.1161/HYPERTENSIONAHA.119.14349
Nanou, E., and Catterall, W. A. (2018). Calcium channels, synaptic plasticity, and neuropsychiatric disease. Neuron 98, 466–481. doi: 10.1016/j.neuron.2018.03.017
Noebels, J. L. (2019). Predicting the impact of sodium channel mutations in human brain disease. Epilepsia 60(Suppl. 3), S8–S16. doi: 10.1111/epi.14724
Ortner, N. J., and Striessnig, J. (2020). De novo CACNA1D Ca2+ channelopathies: clinical phenotypes and molecular mechanism. Pflügers Arch. 472, 755–773. doi: 10.1007/s00424-020-02418-w
Ozawa, J., Ohno, S., Saito, H., Saitoh, A., Matsuura, H., and Horie, M. (2018). A novel CACNA1C mutation identified in a patient with Timothy syndrome without syndactyly exerts both marked loss- and gain-of-function effects. HeartRhythm Case Rep. 4, 273–277. doi: 10.1016/j.hrcr.2018.03.003
Page, K. M., Heblich, F., Margas, W., Pratt, W. S., Nieto-Rostro, M., Chaggar, K., et al. (2010). N terminus is key to the dominant negative suppression of Ca(V)2 calcium channels: implications for episodic ataxia type 2. J. Biol. Chem. 285, 835–844. doi: 10.1074/jbc.M109.065045
Panagiotakos, G., Haveles, C., Arjun, A., Petrova, R., Rana, A., Portmann, T., et al. (2019). Aberrant calcium channel splicing drives defects in cortical differentiation in Timothy syndrome. eLife 8:e51037. doi: 10.7554/eLife.51037
Pangrsic, T., Singer, J. H., and Koschak, A. (2018). Voltage-gated calcium channels: key players in sensory coding in the retina and the inner ear. Physiol. Rev. 98, 2063–2096. doi: 10.1152/physrev.00030.2017
Pantazis, A., Savalli, N., Sigg, D., Neely, A., and Olcese, R. (2014). Functional heterogeneity of the four voltage sensors of a human L-type calcium channel. Proc. Natl. Acad. Sci. U.S.A. 111, 18381–18386. doi: 10.1073/pnas.1411127112
Paşca, S. P., Portmann, T., Voineagu, I., Yazawa, M., Shcheglovitov, A., Paşca, A. M., et al. (2011). Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat. Med. 17, 1657–1662. doi: 10.1038/nm.2576
Peloquin, J. B., Rehak, R., Doering, C. J., and McRory, J. E. (2007). Functional analysis of congenital stationary night blindness type-2 CACNA1F mutations F742C, G1007R, and R1049W. Neuroscience 150, 335–345. doi: 10.1016/j.neuroscience.2007.09.021
Perez-Reyes, E., and Lory, P. (2006). Molecular biology of T-type calcium channels. CNS Neurol. Disord. Drug Targets 5, 605–609. doi: 10.2174/187152706779025508
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., et al. (2004). UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. doi: 10.1002/jcc.20084
Pinggera, A., Lieb, A., Benedetti, B., Lampert, M., Monteleone, S., Liedl, K. R., et al. (2015). CACNA1D de novo mutations in autism spectrum disorders activate Cav1.3 L-type calcium channels. Biol. Psychiatry 77, 816–822. doi: 10.1016/j.biopsych.2014.11.020
Pinggera, A., Mackenroth, L., Rump, A., Schallner, J., Beleggia, F., Wollnik, B., et al. (2017). New gain-of-function mutation shows CACNA1D as recurrently mutated gene in autism spectrum disorders and epilepsy. Hum. Mol. Genet. 26, 2923–2932. doi: 10.1093/hmg/ddx175
Pinggera, A., Negro, G., Tuluc, P., Brown, M. J., Lieb, A., and Striessnig, J. (2018). Gating defects of disease-causing de novo mutations in Cav1.3 Ca(2+) channels. Channels 12, 388–402. doi: 10.1080/19336950.2018.1546518
Platzer, J., Engel, J., Schrott-Fischer, A., Stephan, K., Bova, S., Chen, H., et al. (2000). Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell 102, 89–97. doi: 10.1016/s0092-8674(00)00013-1
Powell, K. L., Cain, S. M., Ng, C., Sirdesai, S., David, L. S., Kyi, M., et al. (2009). A Cav3.2 T-type calcium channel point mutation has splice-variant-specific effects on function and segregates with seizure expression in a polygenic rat model of absence epilepsy. J. Neurosci. 29, 371–380. doi: 10.1523/JNEUROSCI.5295-08.2009
Priori, S. G., Blomström-Lundqvist, C., Mazzanti, A., Blom, N., Borggrefe, M., Camm, J., et al. (2015). 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC)Endorsed by: association for European Paediatric and Congenital Cardiology (AEPC). Eur. Heart J. 36, 2793–2867. doi: 10.1093/eurheartj/ehv316
Proft, J., Rzhepetskyy, Y., Lazniewska, J., Zhang, F.-X., Cain, S. M., Snutch, T. P., et al. (2017). The Cacna1h mutation in the GAERS model of absence epilepsy enhances T-type Ca2+ currents by altering calnexin-dependent trafficking of Cav3.2 channels. Sci. Rep. 7:11513. doi: 10.1038/s41598-017-11591-5
Ramachandran, K. V., Hennessey, J. A., Barnett, A. S., Yin, X., Stadt, H. A., Foster, E., et al. (2013). Calcium influx through L-type CaV1.2 Ca2+ channels regulates mandibular development. J. Clin. Invest. 123, 1638–1646. doi: 10.1172/jci66903
Reichenbach, H., Meister, E. M., and Theile, H. (1992). [The heart-hand syndrome. A new variant of disorders of heart conduction and syndactylia including osseous changes in hands and feet]. Kinderarztl. Prax. 60, 54–56.
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Romaniello, R., Zucca, C., Tonelli, A., Bonato, S., Baschirotto, C., Zanotta, N., et al. (2010). A wide spectrum of clinical, neurophysiological and neuroradiological abnormalities in a family with a novel CACNA1A mutation. J. Neurol. Neurosurg. Psychiatry 81, 840–843. doi: 10.1136/jnnp.2008.163402
Rose, S. J., Kriener, L. H., Heinzer, A. K., Fan, X., Raike, R. S., van den Maagdenberg, A. M. J. M., et al. (2014). The first knockin mouse model of episodic ataxia type 2. Exp. Neurol. 261, 553–562. doi: 10.1016/j.expneurol.2014.08.001
Samueli, S., Abraham, K., Dressler, A., Gröppel, G., Mühlebner-Fahrngruber, A., Scholl, T., et al. (2016). Efficacy and safety of Everolimus in children with TSC - associated epilepsy – Pilot data from an open single-center prospective study. Orphanet J. Rare Dis. 11:145. doi: 10.1186/s13023-016-0530-z
Satterstrom, F. K., Kosmicki, J. A., Wang, J., Breen, M. S., De Rubeis, S., An, J.-Y., et al. (2020). Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180, 568–584. doi: 10.1016/j.cell.2019.12.036
Schartner, V., Romero, N. B., Donkervoort, S., Treves, S., Munot, P., Pierson, T. M., et al. (2017). Dihydropyridine receptor (DHPR, CACNA1S) congenital myopathy. Acta Neuropathol. 133, 517–533. doi: 10.1007/s00401-016-1656-8
Scholl, U. I., Goh, G., Stolting, G., de Oliveira, R. C., Choi, M., Overton, J. D., et al. (2013). Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat. Genet. 45, 1050–1054. doi: 10.1038/ng.2695
Scholl, U. I., Stölting, G., Nelson-Williams, C., Vichot, A. A., Choi, M., Loring, E., et al. (2015). Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism. eLife 4:e06315. doi: 10.7554/eLife.06315
Shah, D. P., Baez-Escudero, J. L., Weisberg, I. L., Beshai, J. F., and Burke, M. C. (2012). Ranolazine safely decreases ventricular and atrial fibrillation in Timothy syndrome (LQT8). Pacing Clin. Electrophysiol. 35, e62–e64. doi: 10.1111/j.1540-8159.2010.02913.x
Sinnegger-Brauns, M. J., Hetzenauer, A., Huber, I. G., Renstrom, E., Wietzorrek, G., Berjukov, S., et al. (2004). Isoform-specific regulation of mood behavior and pancreatic beta cell and cardiovascular function by L-type Calcium channels. J. Clin. Invest. 113, 1430–1439. doi: 10.1172/jci20208
Sintas, C., Carreño, O., Fernàndez-Castillo, N., Corominas, R., Vila-Pueyo, M., Toma, C., et al. (2017). Mutation spectrum in the CACNA1A gene in 49 patients with episodic ataxia. Sci. Rep. 7:2514. doi: 10.1038/s41598-017-02554-x
Sokolov, S., Scheuer, T., and Catterall, W. A. (2007). Gating pore current in an inherited ion channelopathy. Nature 446, 76–78. doi: 10.1038/nature05598
Sokolov, S., Scheuer, T., and Catterall, W. A. (2008). Depolarization-activated gating pore current conducted by mutant sodium channels in potassium-sensitive normokalemic periodic paralysis. Proc. Natl. Acad. Sci. U.S.A. 105, 19980–19985. doi: 10.1073/pnas.0810562105
Sokolov, S., Scheuer, T., and Catterall, W. A. (2010). Ion permeation and block of the gating pore in the voltage sensor of NaV1.4 channels with hypokalemic periodic paralysis mutations. J. Gen. Physiol. 136, 225–236. doi: 10.1085/jgp.201010414
Song, L., Park, S.-H. E., Isseroff, Y., Morikawa, K., and Yazawa, M. (2017). Inhibition of CDK5 alleviates the cardiac phenotypes in timothy syndrome. Stem Cell Rep. 9, 50–57. doi: 10.1016/j.stemcr.2017.05.028
Spacey, S. (2015). “Episodic ataxia type 2,” in GeneReviews§[Internet], eds M. P. Adam, H. H. Ardinger, R. A. Pagon, et al. (Seattle, WA; University of Washington).
Splawski, I., Timothy, K. W., Decher, N., Kumar, P., Sachse, F. B., Beggs, A. H., et al. (2005). Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc. Natl. Acad. Sci. U.S.A. 102, 8089–8096; discussion 8086-8088. doi: 10.1073/pnas.0502506102
Splawski, I., Timothy, K. W., Sharpe, L. M., Decher, N., Kumar, P., Bloise, R., et al. (2004). Cav 1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119, 19–31. doi: 10.1016/j.cell.2004.09.011
Stockner, T., and Koschak, A. (2013). What can naturally occurring mutations tell us about Ca(v)1.x channel function? Biochim. Biophys. Acta 1828, 1598–1607. doi: 10.1016/j.bbamem.2012.11.026
Striessnig, J. (2018). Getting a handle on CaV2.2 (N-type) voltage-gated Ca(2+) channels. Proc. Natl. Acad. Sci. U.S.A. 115, 12848–12850. doi: 10.1073/pnas.1818608115
Striessnig, J., and Ortner, N. J. (2020). “Ca2+ channel blockers,” in Encyclopedia of Molecular Pharmacology, eds S. Offermanns and W. Rosenthal (Cham: Springer).
Sullivan, P. F., and Geschwind, D. H. (2019). Defining the genetic, genomic, cellular, and diagnostic architectures of psychiatric disorders. Cell 177, 162–183. doi: 10.1016/j.cell.2019.01.015
Symonds, J. D., and McTague, A. (2020). Epilepsy and developmental disorders: next generation sequencing in the clinic. Eur. J. Paediatr. Neurol. 24, 15–23. doi: 10.1016/j.ejpn.2019.12.008
Todorov, B., Kros, L., Shyti, R., Plak, P., Haasdijk, E. D., Raike, R. S., et al. (2012). Purkinje cell-specific ablation of Cav2.1 channels is sufficient to cause cerebellar ataxia in mice. Cerebellum 11, 246–258. doi: 10.1007/s12311-011-0302-1
Tonelli, A., D’Angelo, M. G., Salati, R., Villa, L., Germinasi, C., Frattini, T., et al. (2006). Early onset, non fluctuating spinocerebellar ataxia and a novel missense mutation in CACNA1A gene. J. Neurol. Sci. 241, 13–17. doi: 10.1016/j.jns.2005.10.007
Tottene, A., Favero, M., and Pietrobon, D. (2019). Enhanced Thalamocortical Synaptic Transmission and Dysregulation of the Excitatory-Inhibitory Balance at the Thalamocortical Feedforward Inhibitory Microcircuit in a Genetic Mouse Model of Migraine. J. Neurosci. 39, 9841–9851. doi: 10.1523/JNEUROSCI.1840-19.2019
Tottene, A., Fellin, T., Pagnutti, S., Luvisetto, S., Striessnig, J., Fletcher, C., et al. (2002). Familial hemiplegic migraine mutations increase calcium influx through single human Cav2.1 channels and decrease maximal Cav2.1 current density in neurons. Proc. Natl. Acad. Sci. U.S.A. 99, 13284–13289. doi: 10.1073/pnas.192242399
Tottene, A., Pivotto, F., Fellin, T., Cesetti, T., van den Maagdenberg, A. M., and Pietrobon, D. (2005). Specific kinetic alterations of human CaV2.1 calcium channels produced by mutation S218L causing familial hemiplegic migraine and delayed cerebral edema and coma after minor head trauma. J. Biol. Chem. 280, 17678–17686. doi: 10.1074/jbc.m501110200
Tuluc, P., and Flucher, B. E. (2011). Divergent biophysical properties, gating mechanisms, and possible functions of the two skeletal muscle Ca(V)1.1 calcium channel splice variants. J Muscle Res. Cell Motil. 32, 249–256. doi: 10.1007/s10974-011-9270-9
Tyagi, S., Ribera, A. B., and Bannister, R. A. (2020). Zebrafish as a Model System for the Study of Severe CaV2.1 (α1A) Channelopathies. Front. Mol. Neurosci. 12:329. doi: 10.3389/fnmol.2019.00329
Van Petegem, F., Clark, K. A., Chatelain, F. C., and Minor, D. L. Jr. (2004). Structure of a complex between a voltage-gated calcium channel beta-subunit and an alpha-subunit domain. Nature 429, 671–675.
Waterhouse, A. M., Procter, J. B., Martin, D. M., Clamp, M., and Barton, G. J. (2009). Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191. doi: 10.1093/bioinformatics/btp033
Weber, Y. G., Biskup, S., Helbig, K. L., Von Spiczak, S., and Lerche, H. (2017). The role of genetic testing in epilepsy diagnosis and management. Expert Rev. Mol. Diagn. 17, 739–750. doi: 10.1080/14737159.2017.1335598
Weiss, N., and Zamponi, G. W. (2019). Genetic T-type calcium channelopathies. J. Med. Genet. 57, 1–10. doi: 10.1136/jmedgenet-2019-106163
Wemhöner, K., Friedrich, C., Stallmeyer, B., Coffey, A. J., Grace, A., Zumhagen, S., et al. (2015). Gain-of-function mutations in the calcium channel CACNA1C (Cav1.2) cause non-syndromic long-QT but not Timothy syndrome. J. Mol. Cell. Cardiol. 80, 186–195. doi: 10.1016/j.yjmcc.2015.01.002
Williams, B., Lopez, J. A., Maddox, J. W., and Lee, A. (2020). Functional impact of a congenital stationary night blindness type 2 mutation depends on subunit composition of Cav1.4 Ca(2+) channels. J. Biol. Chem. 295, 17215–17226. doi: 10.1074/jbc.RA120.014138
Wu, J., Yan, Z., Li, Z., Qian, X., Lu, S., Dong, M., et al. (2016). Structure of the voltage-gated calcium channel Cav1.1 at 3.6Å resolution. Nature 537, 191–196. doi: 10.1038/nature19321
Wulczyn, K., Perez-Reyes, E., Nussbaum, R. L., and Park, M. (2019). Primary aldosteronism associated with a germline variant in CACNA1H. BMJ Case Rep. 12:e229031. doi: 10.1136/bcr-2018-229031
Yazawa, M., Hsueh, B., Jia, X., Pasca, A. M., Bernstein, J. A., Hallmayer, J., et al. (2011). Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature 471, 230–234. doi: 10.1038/nature09855
Ye, D., Tester, D. J., Zhou, W., Papagiannis, J., and Ackerman, M. J. (2019). A pore-localizing CACNA1C-E1115K missense mutation, identified in a patient with idiopathic QT prolongation, bradycardia, and autism spectrum disorder, converts the L-type calcium channel into a hybrid nonselective monovalent cation channel. Heart Rhythm 16, 270–278. doi: 10.1016/j.hrthm.2018.08.030
Yue, Q., Jen, J. C., Nelson, S. F., and Baloh, R. W. (1997). Progressive ataxia due to a missense mutation in a calcium-channel gene. Am. J. Hum. Genet. 61, 1078–1087. doi: 10.1086/301613
Zamponi, G. W., Striessnig, J., Koschak, A., and Dolphin, A. C. (2015). The physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol. Rev. 67, 821–870. doi: 10.1124/pr.114.009654
Zhang, Q., Chen, J., Qin, Y., Wang, J., and Zhou, L. (2018). Mutations in voltage-gated L-type calcium channel: implications in cardiac arrhythmia. Channels 12, 201–218. doi: 10.1080/19336950.2018.1499368
Zhang, S., Zhou, Z., Gong, Q., Makielski, J. C., and January, C. T. (1999). Mechanism of block and identification of the verapamil binding domain to HERG potassium channels. Circ. Res. 84, 989–998. doi: 10.1161/01.RES.84.9.989
Zhao, Y., Huang, G., Wu, Q., Wu, K., Li, R., Lei, J., et al. (2019a). Cryo-EM structures of apo and antagonist-bound human Cav3.1. Nature 576, 492–497. doi: 10.1038/s41586-019-1801-3
Keywords: calcium-channels, channelopathies, mutations, neurodevelopmental disorders, gating pore currents
Citation: Striessnig J (2021) Voltage-Gated Ca2+-Channel α1-Subunit de novo Missense Mutations: Gain or Loss of Function – Implications for Potential Therapies. Front. Synaptic Neurosci. 13:634760. doi: 10.3389/fnsyn.2021.634760
Received: 28 November 2020; Accepted: 02 February 2021;
Published: 03 March 2021.
Edited by:
Filippo Tempia, University of Turin, ItalyReviewed by:
Norbert Weiss, Charles University, CzechiaDimitri M. Kullmann, University College London, United Kingdom
Copyright © 2021 Striessnig. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jörg Striessnig, am9lcmcuc3RyaWVzc25pZ0B1aWJrLmFjLmF0