 Nadine J. Ortner
Nadine J. Ortner- Department of Pharmacology and Toxicology, Institute of Pharmacy, University of Innsbruck, Innsbruck, Austria
The loss of dopamine (DA)-producing neurons in the substantia nigra pars compacta (SN) underlies the core motor symptoms of the progressive movement disorder Parkinson's disease (PD). To date, no treatment to prevent or slow SN DA neurodegeneration exists; thus, the identification of the underlying factors contributing to the high vulnerability of these neurons represents the basis for the development of novel therapies. Disrupted Ca2+ homeostasis and mitochondrial dysfunction seem to be key players in the pathophysiology of PD. The autonomous pacemaker activity of SN DA neurons, in combination with low cytosolic Ca2+ buffering, leads to large somatodendritic fluctuations of intracellular Ca2+ levels that are linked to elevated mitochondrial oxidant stress. L-type voltage-gated Ca2+ channels (LTCCs) contribute to these Ca2+ oscillations in dendrites, and LTCC inhibition was beneficial in cellular and in vivo animal models of PD. However, in a recently completed phase 3 clinical trial, the dihydropyridine (DHP) LTCC inhibitor isradipine failed to slow disease progression in early PD patients, questioning the feasibility of DHPs for PD therapy. Novel evidence also suggests that R- and T-type Ca2+ channels (RTCCs and TTCCs, respectively) represent potential PD drug targets. This short review aims to (re)evaluate the therapeutic potential of LTCC, RTCC, and TTCC inhibition in light of novel preclinical and clinical data and the feasibility of available Ca2+ channel blockers to modify PD disease progression. I also summarize their cell-specific roles for SN DA neuron function and describe how their gating properties allow activity (and thus their contribution to stressful Ca2+ oscillations) during pacemaking.
Introduction
The primary motor symptoms of the neurodegenerative disorder Parkinson's disease (PD) are caused by a progressive loss of dopamine (DA)-producing neurons in the substantia nigra pars compacta (SN) and associated striatal DA depletion (Obeso et al., 2017). Although PD was first described in 1817 (Parkinson, 2002), to date only symptomatic treatments, but still no cure or disease-modifying therapy, exist (Schulz et al., 2016; Obeso et al., 2017). To develop an effective treatment, it is essential to understand the contributing factors and the disease-underlying cellular mechanisms. Next to globally acting factors (e.g., toxins, aging, and genetic mutations), cell-autonomous ones have also been proposed and widely studied (Poewe et al., 2017; Surmeier et al., 2017b). Neighboring DA neurons in the ventral tegmental area (VTA) share many of the intrinsic properties but are spared in PD (Dauer and Przedborski, 2003). Both are autonomous pacemakers, but in vulnerable SN DA neurons, large oscillations of intracellular Ca2+ levels accompany pacemaking (Wilson and Callaway, 2000; Chan et al., 2007; Guzman et al., 2009, 2018; Hage and Khaliq, 2015). In contrast, no or much smaller Ca2+ transients were detected in VTA neurons (Guzman et al., 2010, 2018; Benkert et al., 2019) that rely on a different pacemaking mechanism with less contribution of Ca2+ currents (Khaliq and Bean, 2010; Philippart et al., 2016). Ca2+ influx is important to modulate neuronal excitability and to activate Ca2+-dependent physiological processes, but the rhythmic Ca2+ load in SN DA neurons also triggers mitochondrial oxidative stress (Guzman et al., 2010, 2018; Surmeier et al., 2017a). Disrupted Ca2+ homeostasis and mitochondrial dysfunction are considered key players in PD pathophysiology (Schapira, 2008; Zaichick et al., 2017; Zampese and Surmeier, 2020), and many mutations causing inherited forms of PD affect proteins associated with mitochondrial homeostasis and stress responses (Park et al., 2018). Thus, reducing the activity-related Ca2+ load and associated mitochondrial stress in SN DA neurons represents a feasible strategy to increase their resistance to degenerative stressors. L-, R-, and T-type voltage-gated Ca2+ channels (LTCCs, RTCCs, and TTCCs, respectively) contribute to the stress-inducing cytosolic Ca2+ oscillations, and different approaches to decrease their activity produced promising protective effects in preclinical models of PD. However, a phase 3 clinical trial evaluating the efficacy of LTCC inhibition in early PD patients recently failed. This short review aims to give an overview of the (patho)physiological roles of Ca2+ channel activity in SN DA neurons and to (re)evaluate the therapeutic potential of Ca2+ channel inhibition (in light of novel preclinical and clinical evidence) and the availability of clinically applicable selective drugs.
Voltage-Gated Ca2+ Channels in SN DA Neurons
Plasmalemmal voltage-gated Ca2+ channels mediate controlled Ca2+ influx in response to membrane depolarization and contribute to important functions within the sensory, cardiac, endocrine, and nervous systems (Zamponi et al., 2015). Three main families (Cav1/Cav2/Cav3) and 10 individual isoforms are distinguished based on the biophysical and pharmacological properties of the pore-forming α1 subunit: Cav1.1–Cav1.4 (LTCCs), Cav2.1 (P/Q-type), Cav2.2 (N-type), Cav2.3 (RTCCs), and Cav3.1–3.3 (TTCCs) (Catterall, 2011; Zamponi et al., 2015). They are further classified into high-voltage (HVA; Cav1 and Cav2) and low-voltage activated (LVA; Cav3), and only HVA channels require the association with auxiliary β- and α2δ subunits for proper function (Dolphin, 2016; Figure 1A). This functional complexity (further fine-tuned by alternative splicing), their distinct tissue distribution, and subcellular localization enable them to differentially contribute to cellular processes (Dolphin, 2012, 2016; Zamponi et al., 2015).
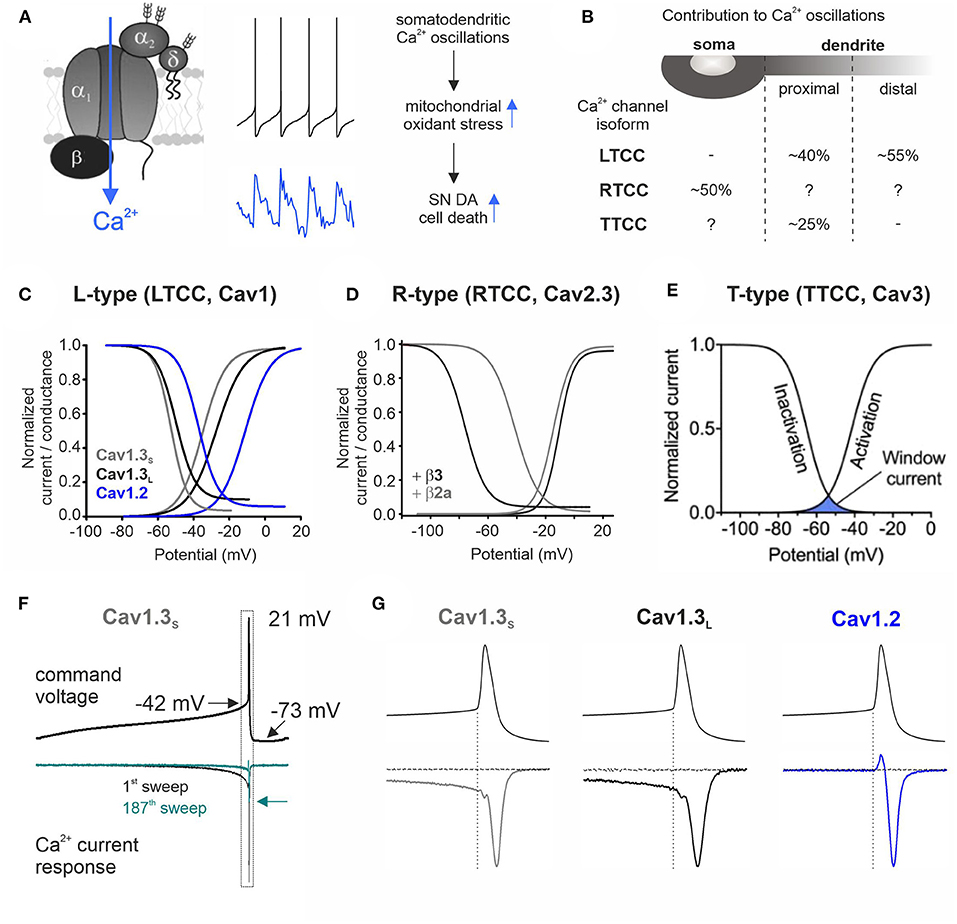
Figure 1. Contribution to stressful somatodendritic Ca2+ oscillations, gating properties, and Ca2+ current responses of different Ca2+ channel isoforms during SN DA neuronal pacemaking. (A) Schematic of a plasmalemmal Ca2+ channel complex consisting of the pore-forming α1 subunit and auxiliary β and α2δ subunits [only HVA α1 subunits require auxiliary subunits for proper function (Dolphin, 2016)]. Membrane depolarization triggers channel opening and subsequent Ca2+ influx. The middle panel shows mouse SN DA neuron pacemaking (top) with the associated intracellular Ca2+ transients (bottom; blue) recorded at the cell soma (taken from Ortner et al., 2017). Somatodendritic Ca2+ oscillations have been linked to increased mitochondrial oxidative stress, implicated in the high vulnerability of SN DA neurons in PD (Guzman et al., 2010). (B) Contribution of different Ca2+ channel isoforms to intracellular Ca2+ oscillations in distinct cellular compartments (soma and proximal and distal dendrites) (Ortner et al., 2017; Guzman et al., 2018; Benkert et al., 2019). With a higher isradipine concentration [5 μM (Guzman et al., 2018) compared to 1 μM, 10 nM, or chronic in vivo isradipine application resulting in plasma levels of ~5 nM isradipine in the abovementioned refs], a complete inhibition of distal dendritic Ca2+ transients was observed (Guzman et al., 2009, 2010). Hyphen indicates no contribution found and question mark indicates not determined. (C–E) The voltage-conductance (activation) curve describes at which potential a certain Ca2+ channel isoform opens (activation threshold) and is in a Ca2+ conductive state. The voltage dependence of inactivation gives the proportion of inactivated (non-available) channels at a certain membrane potential. The overlap of these two curves defines the window current (see E) that represents the voltage range at which the respective Ca2+ channel is steadily active and can thus create a constant background influx of Ca2+. (C) Cav1.2 or Cav1.3 α1 with β3 and α2δ1 (2 mM Ca2+; modified from Ortner et al., 2017). Alternative splicing of the Cav1.3 C-terminus results in functionally distinct long (Cav1.3L, black) and short splice variants (Cav1.3s, gray; Singh et al., 2008; Bock et al., 2011; Tan et al., 2011). (D) Cav2.3e α1 with β3 (black) or β2a (gray) and α2δ1 (2 mM Ca2+). Association with membrane-bound β2a shifts the voltage dependence of inactivation of Cav2.3 channels ~35 mV toward more positive potentials (Olcese et al., 1994; Jones et al., 1998; Yasuda et al., 2004; Miranda-Laferte et al., 2012). (E) Voltage dependence of activation and inactivation of TTCCs with indicated window current (blue area; taken from Weiss and Zamponi, 2019a). (F) Representative Ca2+ current traces (ICa, lower panel) through Cav1.3S in response to a murine SN DA neuron action potential command voltage shown above (2 mM Ca2+, tsA201 cells; modified from Ortner et al., 2017). Simulated pacemaking (2.5 Hz) resulted in a decrease of ICa to ~20% in steady state (cyan trace, arrow indicates decreased peak ICa) through all three investigated Cav1 subtypes (Cav1.2, Cav1.3S, and Cav1.3L). (G) Enlargement of the action potential spike region indicated in (F) (dotted rectangle). During the interspike interval (before the spike threshold indicated by the dotted vertical line), only Cav1.3 variants conducted Ca2+ while all LTCCs showed Ca2+ influx in response to the action potential spike. The horizontal current trace shows full LTCC inhibition at the end of the recording (3 μM isradipine).
Rodent SN DA neurons express all voltage-gated Ca2+ channel isoforms, except Cav1.1 and Cav1.4 (restricted to skeletal muscle and retina, respectively) (Cardozo and Bean, 1995; Chan et al., 2007; Sinnegger-Brauns et al., 2009; Dufour et al., 2014; Brichta et al., 2015; Shin, 2015; Philippart et al., 2016; Ortner et al., 2017; Guzman et al., 2018; Benkert et al., 2019; Verma and Ravindranath, 2019). The recording of individual current components in intact SN DA neurons is complicated, but in somatic nucleated outside-out patches from juvenile rat SN DA neurons, Ca2+ currents of all expressed isoforms were found, with a large LTCC contribution that was higher compared to VTA (Philippart et al., 2016). On the transcript level, RTCC Cav2.3 channels are most abundant in mouse SN DA neurons and levels increase with age (Benkert et al., 2019), while LTCCs (Cav1.2/Cav1.3) get downregulated in an age-dependent manner (Branch et al., 2014; Ortner et al., 2017; Benkert et al., 2019). Of the LVA TTCCs, Cav3.1, and Cav3.2 predominate in mouse SN DA neurons (Poetschke et al., 2015; Guzman et al., 2018; Benkert et al., 2019) and immunohistochemical stainings suggest a rise of somatodendritic TTCCs during development (Dufour et al., 2014) [also shown for Cav1.3, but antibody specificity was not demonstrated in brain (SN) tissue lacking Cav1.3]. SN DA neurons are constantly active and fire action potentials in a tonic single spike or transient high-frequency burst mode in vivo, resulting in axonal and somatodendritic DA release (Grace and Bunney, 1984a,b; Chiodo, 1988; Paladini and Roeper, 2014). Cav2 N- and P/Q-type channels drive fast presynaptic neurotransmission, but LTCCs, RTCCs, and TTCCs seem to also contribute to DA release from axonal and/or somatodendritic locations in rodent SN DA neurons (Bergquist and Nissbrandt, 2003; Chen et al., 2006; Brimblecombe et al., 2015; Yee et al., 2019). In vitro, even in complete synaptic isolation, SN DA neurons maintain an intrinsically generated regular pacemaker activity (0.5–4 Hz). Inhibition of Na+ channels by tetrodotoxin abolished spike generation and revealed slow oscillatory membrane depolarizations (“SOPs,” slow oscillatory potentials; Fujimura and Matsuda, 1989; Yee et al., 2019) that were absent in neighboring VTA neurons and abolished upon LTCC inhibition (Chan et al., 2007). However, LTCCs are not required for pacemaker generation but rather stabilize precision and robustness of pacemaking (Guzman et al., 2009, 2010; Poetschke et al., 2015; Ortner et al., 2017). Similarly, pharmacological inhibition of TTCCs decreased pacemaker precision (juvenile) and frequency (adult mice) (Wolfart and Roeper, 2002; Poetschke et al., 2015), while Cav2.3 knockout or its partial pharmacological inhibition reduced spike amplitude and afterhyperpolarization (AHP) (Benkert et al., 2019). Ca2+ influx through voltage-gated Ca2+ channels can drive depolarization, but its coupling to K+ conductances [e.g., Ca2+-sensitive small conductance K+ (SK) or A-type K+ channels] can also trigger the opposite—a functional coupling important for rhythmic activity (Wolfart and Roeper, 2002; Ji and Shepard, 2006; Duda et al., 2016). During in vivo high DA states, Cav1.3 can sensitize the DA-D2 autoreceptor response resulting in activation of G protein-coupled GIRK2 K+ channels and inhibition of spiking (Dragicevic et al., 2014). Moreover, voltage-gated Ca2+ channels can activate various cellular Ca2+-dependent signaling processes, including gene expression as shown for LTCCs (Catterall, 2011; Ma et al., 2012).
Voltage-Gated Ca2+ Channels Contribute to Stressful Somatodendritic Ca2+ Oscillations
The slow SN DA neuron pacemaker activity with its broad action potentials triggers large fluctuations of intracellular Ca2+ levels throughout the somatodendritic compartment (Chan et al., 2007; Figure 1A, middle panel). Dendritic Ca2+ transients occur despite spike inhibition or failure (Chan et al., 2007; Guzman et al., 2018), and their rising phase starts before the action potential threshold (Guzman et al., 2018). This points to a contribution of a Ca2+ conductance that is active at subthreshold potentials. Cav1.3 and TTCCs activate well before the spike threshold (as shown for Cav1.3 in tsA201 cells; Ortner et al., 2017; Figure 1G), and their specific gating properties create a “window current” permitting a constant background Ca2+ influx through a small fraction of non-inactivated channels (Figures 1C,E) in the average membrane potential range of SN DA neurons (Figure 1F). Pharmacological experiments in mouse brain slices found that LTCCs mediate Ca2+ transients in the dendrites (with a higher contribution in distal compartments) (Guzman et al., 2009, 2010, 2018) but not in the soma (Ortner et al., 2017), while TTCCs account for ~25% of proximal dendritic transients (soma not tested) (Guzman et al., 2018; Figure 1B). LTCC dihydropyridine (DHP) inhibitors act on both LTCC isoforms (with higher potency for Cav1.2) (Koschak et al., 2001; Xu and Lipscombe, 2001) and thus cannot be used to discriminate Cav1.2- and Cav1.3-mediated effects. However, using a Cav1.3-specific shRNA-mediated knockdown approach (Guzman et al., 2018), Cav1.3 was identified as the major LTCC isoform underlying the Ca2+ oscillations. Similar effects were observed by knockdown and inhibition with DHPs, particularly before spike onset (Guzman et al., 2018) where Cav1.2 is not yet active (Ortner et al., 2017; Figures 1C,G). In the soma, HVA Cav2.3 channels mediate a large proportion of the Ca2+ oscillations [~50% in Cav2.3−/− mice; ~25% upon partial pharmacological inhibition by 100 nM SNX-482 (Benkert et al., 2019); dendrites not investigated; Figure 1B]. Cav1.2 and Cav2.3 start to activate at approximately−40 mV (Figures 1C,D) and conduct Ca2+ only in response to the strong depolarization of the action potential spike (shown for Cav1.2 in tsA201 cells; Ortner et al., 2017; Figure 1G). In contrast to Cav1.2, Cav2.3 channels typically inactivate at comparatively negative membrane potentials (black trace, steady-state inactivation curve in Figure 1D), which predicts almost complete channel inactivation during SN DA neuron pacemaking. Interaction with membrane-bound β2 splice variants (β2a and β2e) (Buraei and Yang, 2010) was shown to shift Cav2.3 voltage dependence of inactivation toward more positive potentials (Olcese et al., 1994; Jones et al., 1998; Yasuda et al., 2004; Miranda-Laferte et al., 2012, 2014) (gray trace, Figure 1D) and could thereby facilitate Cav2.3 steady-state availability during SN DA neuron activity. β2 accounts for ~30% of β subunits in mouse SN DA neurons (Brichta et al., 2015; Shin, 2015); however, if and to which extent these β2 splice variants are expressed in SN DA neurons and regulate Cav2.3 RTCCs is not known.
Experiments in transgenic mice (mito-GFP mouse) that allow to monitor the oxidation state of mitochondria established a link between Ca2+ oscillations and elevated mitochondrial oxidative stress (Guzman et al., 2010, 2018; Figure 1A). Dendritic Ca2+ transients and associated mitochondrial oxidation increased with age (Guzman et al., 2018) and were almost absent in neighboring resistant VTA DA neurons (Guzman et al., 2010, 2018; Benkert et al., 2019). In addition, the already high basal oxidant stress level in SN DA neurons was further exacerbated in a genetic PD mouse model (DJ-1 knockout) (Guzman et al., 2010). Interestingly, factors contributing to vulnerability described in SN DA neurons, i.e., slow pacemaking, cytosolic Ca2+ oscillations, low intracellular Ca2+ buffering (Foehring et al., 2009), and elevated levels of mitochondrial oxidant stress, are also found in other vulnerable non-DA neurons (Surmeier et al., 2017b; Zampese and Surmeier, 2020).
Besides the metabolic challenging pacemaker activity, burst firing is also associated with high Ca2+ influx and intracellular Ca2+ levels (Hage and Khaliq, 2015; Philippart et al., 2016; Ortner et al., 2017), and membrane hyperpolarizations (e.g., during post-burst pauses) allow channels to recover from inactivation, in particular TTCCs. This mechanism underlies the large TTCC-mediated Ca2+ currents and the resulting afterdepolarizations that facilitate rebound spiking (Evans et al., 2017; Tracy et al., 2018). Interestingly, in PD, cells in the ventrolateral part of the SN are particularly prone to cell death and lateral SN DA neurons also show high in vivo bursting (Schiemann et al., 2012; Farassat et al., 2019). In line, knockout of KATP K+ channels reduced in vivo burst firing and conferred protection in two PD mouse models (Liss et al., 2005; Schiemann et al., 2012).
Neuroprotection by LTCC Inhibition: Preclinical and Clinical Evidence
Over the last years, LTCCs (and particularly Cav1.3) were considered the main voltage-gated Ca2+ channel subtype underlying stressful Ca2+ oscillations and thus a major driver of SN DA neuronal cell death (Surmeier et al., 2017a; Guzman et al., 2018; Liss and Striessnig, 2019). Epidemiological studies found that the intake of brain-permeable DHP LTCC inhibitors (antihypertensives) reduced the risk to develop PD (Gudala et al., 2015; Lang et al., 2015; Mullapudi et al., 2016). DHPs have been extensively studied in preclinical PD models and showed promising protective effects in most (but not all) studies (Table 1; reviewed in Leandrou et al., 2019; Liss and Striessnig, 2019). The diverging outcomes of DHP treatment in toxin-based PD animal models have been recently discussed in great detail (Liss and Striessnig, 2019), but no definite explanation has been found. Briefly, in eight out of 13 reports, DHPs significantly reduced mitochondrial-targeting toxin-induced SN DA cell death in mice, rats, and primates (Table 1), but the experimental design of all studies varied, complicating an overall conclusion. Differences included the used PD model (6-OHDA and MPTP), animals (species, strain, age, and sex), treatment regimen (DHP, treatment onset, route of administration, and dosing interval), readout (approach and methodology), and plasma concentrations (if even reported) (Liss and Striessnig, 2019). Thus, a standardized approach in future studies would help to better interpret and compare obtained results. In addition, one recent study (not included in Liss and Striessnig, 2019) tested the DHP felodipine in a genetic PD mouse model (expressing the A53T mutant α-synuclein) and found protective effects on SN DA neuron survival and motor deficits and autophagy-induced clearance of disease-associated proteins from brain (Siddiqi et al., 2019). Noteworthy, in contrast to other DHPs like isradipine (Uchida et al., 1997), felodipine accumulated in the brain with ~2 to 5-fold higher brain levels compared to plasma (Siddiqi et al., 2019). Epidemiological and preclinical evidence, and the availability of safe and clinically approved LTCC DHP inhibitors, prompted the phase 3 STEADY-PD III clinical trial with the DHP isradipine as treatment in early PD patients (336 patients, randomized, double blind, and placebo controlled; Biglan et al., 2017), which however did not reach its primary endpoint (Parkinson Study Group STEADY-PD III Investigators, 2020). Several aspects may have contributed.
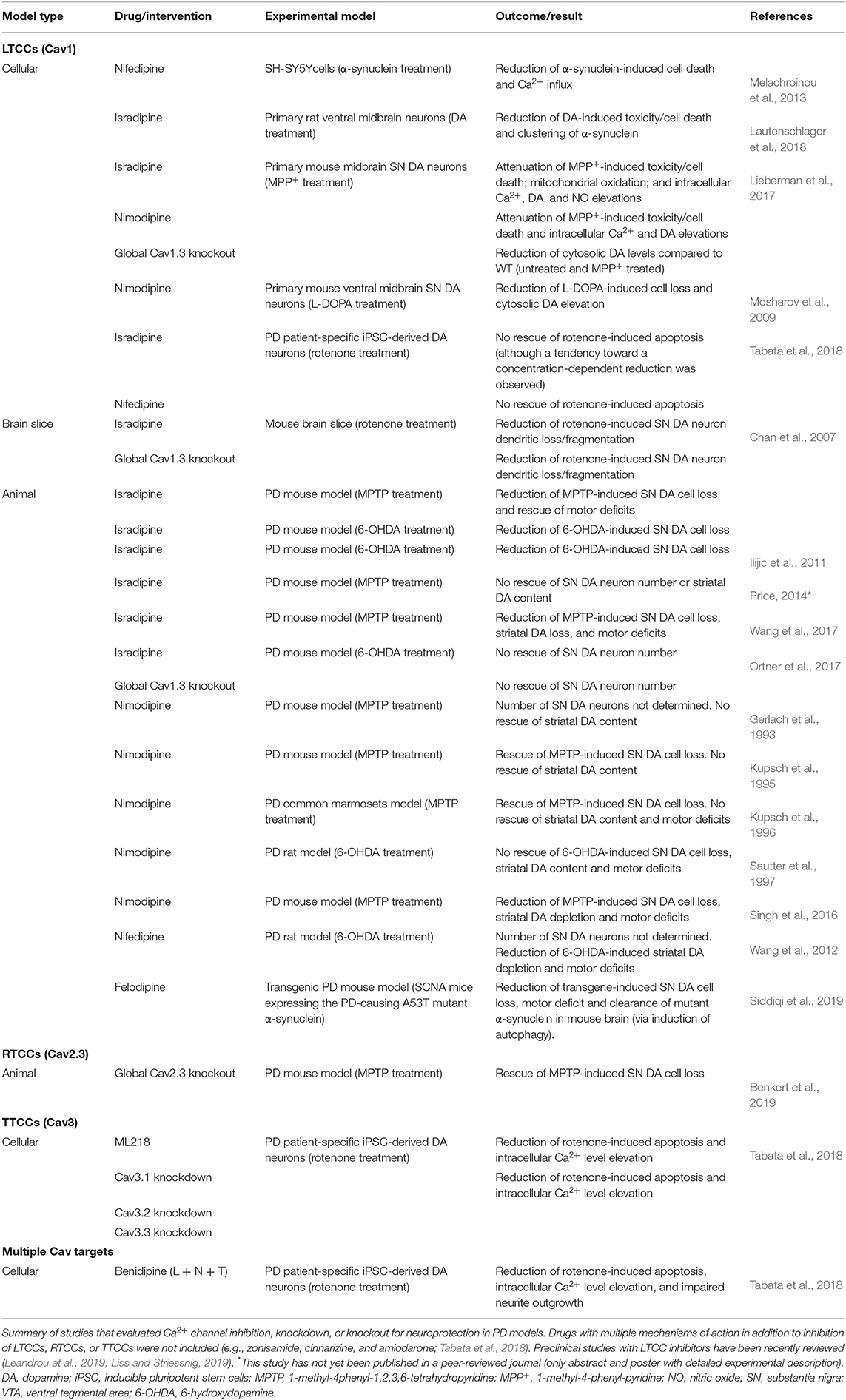
Table 1. Studies that directly tested neuroprotection by Ca2+ channel inhibition, knockdown, or knockout in cellular and animal PD models.
Protective DHP effects might require an earlier treatment onset, since at the time when first PD symptoms occur, pathologic mechanisms and neurodegeneration already started—which however would require reliable PD biomarkers (Poewe et al., 2017). In addition, the age-dependent decrease of LTCCs (Branch et al., 2014; Ortner et al., 2017) could limit the therapeutic window of DHPs in PD (especially in elderly patients), although disease state seems to affect LTCC expression [SN neurons from MPTP-treated mice (Verma and Ravindranath, 2019) and post-mortem brains of early-stage PD patients (Hurley et al., 2015) showed robust Cav1.3 levels, despite profound neuron loss]. When considering Cav1.3 as the primary target, the used maximal tolerable isradipine dose [10 mg/day; limited by Cav1.2-mediated peripheral side effects (Parkinson Study Group, 2013)] might be too low to sufficiently engage Cav1.3 LTCCs in SN DA neurons due to their low apparent drug sensitivity (Ortner et al., 2017). This especially applies to C-terminally short splice variants (Huang et al., 2013; Ortner et al., 2017) that are associated with higher Ca2+ influx (Singh et al., 2008; Bock et al., 2011; Tan et al., 2011; Figure 1C) and account for ~50% of Cav1.3 transcript in mouse SN DA neurons (Ortner et al., 2017; Verma and Ravindranath, 2019) (higher compared to the cortex and striatum; Verma and Ravindranath, 2019). In this context, the use of an immediate-release isradipine formulation in the STEADY-PD III trial was unfavorable, since slow-onset continuous-release tablets [as used in the phase 2 trial (Parkinson Study Group, 2013), NCT00753636] result in higher average steady-state plasma levels (Liss and Striessnig, 2019). Thus, strategies to increase DHP brain concentrations (Yiu and Knaus, 1996; Ji et al., 2017) or DHPs that accumulate in the brain (e.g., nimodipine and felodipine; Kupsch et al., 1996; Siddiqi et al., 2019) could be an option. Noteworthy, microglia-specific Cav1.2 knockdown augmented MPTP-induced SN DA neurodegeneration and motor deficits in mice, associated with enhanced activation of “neuroinflammatory” M1 microglia (Wang et al., 2019). Thus, Cav1.2 inhibition might even be disadvantageous, which highlights the need for the development of reliable Cav1.3-selective inhibitory drugs. So far, only one putatively selective compound has been described (Cp8 in Kang et al., 2012 and cp-PYT in Cooper et al., 2020) but showed diverging results in follow-up studies from other laboratories (Huang et al., 2014; Ortner et al., 2014). Nevertheless, micromolar concentrations of cp-PYT could lower dendritic transients to a similar extent as isradipine and did not affect Ca2+ currents in Cav1.3 knockout mice (Cooper et al., 2020), suggesting Cav1.3-selective inhibition [lack of non-LTCC modulation was also shown in adrenal mouse chromaffin cells (Ortner et al., 2014)]. In the meantime, mice expressing DHP-insensitive Cav1.2 channels could be used to mimic selective pharmacological Cav1.3 inhibition in vivo (Sinnegger-Brauns et al., 2004). Lastly, another important aspect is a potential compensation by other Ca2+ channel isoforms during chronic isradipine treatment, as found in Cav1.3-deficient mice [upregulation of Cav3.1 TTCCs (Poetschke et al., 2015)]. However, this was not observed with systemic isradipine treatment for 7–10 days in mice [3 μg/g/day; ~5 nM plasma isradipine (Guzman et al., 2018)], but longer exposure has not been tested.
RTCCs and TTCCs Emerge as Novel PD Drug Targets
Global Cav2.3 knockout fully prevented SN DA neuron degeneration in the gold-standard chronic low-dose MPTP/probenecid PD mouse model (Table 1) and profoundly reduced somatic Ca2+ oscillations (~50%, dendrites not tested) (Benkert et al., 2019). Although a direct proof for knockout-induced lowering of high mitochondrial stress levels is missing, a similar inhibition of LTCC-mediated Ca2+ transients in proximal and distal dendrites (~35–60%) was sufficient to lower mitochondrial oxidation (Guzman et al., 2010, 2018). The high Cav2.3 levels in vulnerable SN DA neurons (compared to VTA) and their increase with aging further strengthen a possible involvement of RTCCs in PD pathology. Unlike what is observed for Cav1.3 (Poetschke et al., 2015), loss of Cav2.3 did not trigger compensatory upregulation of other Ca2+ channels in SN DA neurons (Cav1.2, Cav1.3, and Cav3.1 were tested) (Benkert et al., 2019). In light of these promising findings, it is unfortunate that to date (like for Cav1.3) no selective RTCC inhibitors exist (Schneider et al., 2013). Note that the peptide toxin SNX-482 is selective for Cav2.3 channels only at low concentrations and also inhibits other channels at elevated concentrations (“Cav2.3-prevalent”) (Newcomb et al., 1998; Bourinet et al., 2001; Schneider et al., 2013; Kimm and Bean, 2014). As mentioned above, association of Cav2.3 channels with membrane-bound β2 splice variants (β2a/β2e) may stabilize sustained Cav2.3 activity—a possible explanation for the large contribution of Cav2.3 channels to somatic Ca2+ oscillations (~50%, Benkert et al., 2019). Interrupting this putative interaction in SN DA neurons might represent an alternative approach to lower Cav2.3-mediated Ca2+ load; however, if these β2 splice variants are indeed expressed in SN DA neurons and modulate Cav2.3 in these cells has not been studied yet. Since β subunits regulate other HVA channels as well, this targeting approach should ideally be Cav2.3-selective [as recently shown for the Cav2.2/β interaction, successfully reducing heterologously expressed and native Cav2.2 currents (Khanna et al., 2019)].
Evidence for a role of TTCCs in PD pathology comes from a study employing DA neurons derived from PD patient-specific iPSCs as in vitro PD model (Tabata et al., 2018). Compared to controls, PD patient-specific DA neurons showed pathological signs including reduced neurite length, enhanced oxidative stress, and elevated resting intracellular Ca2+ levels and apoptosis. Interestingly, levels of all three TTCC isoforms were also increased. Treatment with the mitochondrial toxin rotenone (“PD trigger”) aggravated the observed effects on cell morphology and survival, which could be prevented by drugs targeting TTCCs as well as individual knockout of all TTCC isoforms. Two of the TTCC inhibitory compounds have complex mechanism of action besides inhibition of TTCCs, but one drug, ML218, specifically blocks TTCCs (Xiang et al., 2011). Interestingly, while LTCC inhibition (nifedipine and isradipine) had no effect, the DHP benidipine that acts on L-, N-, and T-type Ca2+ channels could prevent rotenone-induced apoptosis. Although promising, proof of SN DA neuroprotection in in vivo PD models is still missing. TTCC inhibition has been explored for the treatment of parkinsonism, but mainly based on their involvement in abnormal burst discharges within the thalamocortical circuitry and associated motor effects (Tai et al., 2011; Kopecky et al., 2014; Yang et al., 2014; Galvan et al., 2016). Zonisamide, an unselective TTCC inhibitor, ameliorated symptoms in PD patients (Murata, 2010), but due to its broad mechanism of action, it is difficult to assign observations to TTCC inhibition. Selective and clinically suitable TTCC inhibitors exist (analgesic/antiepileptic drugs) (Weiss and Zamponi, 2019a,b) and showed good tolerability and safety in phase 2 clinical trials (Richard et al., 2019, 2020), and one phase 2 trial plans to evaluate the selective blocker CX-8998 as treatment for PD-associated tremor (ClinicalTrials.gov #NCT03436953).
Summary and Conclusion
There is accumulating evidence that voltage-gated Ca2+ channels represent an attractive drug target for the therapy of PD. Their contribution to large somatodendritic Ca2+ oscillations, associated with increased mitochondrial oxidative stress, seems a likely mechanism by which their activity contributes to SN DA neuron degeneration. Pharmacological and genetic strategies to decrease the activity of LTCCs, RTCCs, and TTCCs showed promising neuroprotective effects in preclinical models of PD, but a phase 3 clinical trial found no slowing of disease progression in early PD patients upon treatment with the DHP LTCC inhibitor isradipine. Although DHPs are safe, brain permeable, and clinically available, their lack of selectivity for Cav1.3, the more likely LTCC target for neuroprotection, and adverse Cav1.2-mediated effects limit their potential for PD therapy. Selective TTCC blockers exist, show good tolerability and safety, and could be repurposed for the therapy of PD; however, evidence for neuroprotection in in vivo PD models is still missing. In contrast, no selective inhibitors for Cav1.3 and Cav2.3 exist. The convincing preclinical data described in this review and the discovery of activity-enhancing mutations in neurological diseases (Ortner et al., 2020; Schneider et al., 2020; Weiss and Zamponi, 2020) highlight the urgent need for isoform-selective blockers that are suitable for clinical application. Further studies with available TTCC or multiple Ca2+ channel targeting blockers and, if once available, Cav1.3- and Cav2.3-selective inhibitors will help to uncover the full therapeutic potential of Ca2+ channel inhibition for neuroprotection in PD.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
This work was supported by the Erika-Cremer habilitation fellowship of the University of Innsbruck and the Austrian Science Fund (P27809).
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author wants to thank all colleagues for helpful discussions.
References
Benkert, J., Hess, S., Roy, S., Beccano-Kelly, D., Wiederspohn, N., Duda, J., et al. (2019). Cav2.3 channels contribute to dopaminergic neuron loss in a model of Parkinson's disease. Nat. Commun. 10:5094. doi: 10.1038/s41467-019-12834-x
Bergquist, F., and Nissbrandt, H. (2003). Influence of R-type (Cav2.3) and t-type (Cav3.1-3.3) antagonists on nigral somatodendritic dopamine release measured by microdialysis. Neuroscience 120, 757–764. doi: 10.1016/S0306-4522(03)00385-3
Biglan, K. M., Oakes, D., Lang, A. E., Hauser, R. A., Hodgeman, K., Greco, B., et al. (2017). A novel design of a phase III trial of isradipine in early Parkinson disease (STEADY-PD III). Ann. Clin. Transl. Neurol. 4, 360–368. doi: 10.1002/acn3.412
Bock, G., Gebhart, M., Scharinger, A., Jangsangthong, W., Busquet, P., Poggiani, C., et al. (2011). Functional properties of a newly identified C-terminal splice variant of Cav1.3 L-type Ca2+ channels. J. Biol. Chem. 286, 42736–42748. doi: 10.1074/jbc.M111.269951
Bourinet, E., Stotz, S. C., Spaetgens, R. L., Dayanithi, G., Lemos, J., Nargeot, J., et al. (2001). Interaction of SNX482 with domains III and IV inhibits activation gating of alpha(1E) (Ca(V)2.3) calcium channels. Biophys. J. 81, 79–88. doi: 10.1016/S0006-3495(01)75681-0
Branch, S. Y., Sharma, R., and Beckstead, M. J. (2014). Aging decreases L-type calcium channel currents and pacemaker firing fidelity in substantia nigra dopamine neurons. J. Neurosci. 34, 9310–9318. doi: 10.1523/JNEUROSCI.4228-13.2014
Brichta, L., Shin, W., Jackson-Lewis, V., Blesa, J., Yap, E. L., Walker, Z., et al. (2015). Identification of neurodegenerative factors using translatome-regulatory network analysis. Nat. Neurosci. 18, 1325–1333. doi: 10.1038/nn.4070
Brimblecombe, K. R., Gracie, C. J., Platt, N. J., and Cragg, S. J. (2015). Gating of dopamine transmission by calcium and axonal N-, Q-, T- and L-type voltage-gated calcium channels differs between striatal domains. J. Physiol. 593, 929–946. doi: 10.1113/jphysiol.2014.285890
Buraei, Z., and Yang, J. (2010). The ss subunit of voltage-gated Ca2+ channels. Physiol. Rev. 90, 1461–1506. doi: 10.1152/physrev.00057.2009
Cardozo, D. L., and Bean, B. P. (1995). Voltage-dependent calcium channels in rat midbrain dopamine neurons: modulation by dopamine and GABAB receptors. J. Neurophysiol. 74, 1137–1148. doi: 10.1152/jn.1995.74.3.1137
Catterall, W. A. (2011). Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 3:a003947. doi: 10.1101/cshperspect.a003947
Chan, C. S., Guzman, J. N., Ilijic, E., Mercer, J. N., Rick, C., Tkatch, T., et al. (2007). 'Rejuvenation' protects neurons in mouse models of Parkinson's disease. Nature 447, 1081–1086. doi: 10.1038/nature05865
Chen, A. P., Ohno, M., Giese, K. P., Kuhn, R., Chen, R. L., and Silva, A. J. (2006). Forebrain-specific knockout of B-raf kinase leads to deficits in hippocampal long-term potentiation, learning, and memory. J. Neurosci. Res. 83, 28–38. doi: 10.1002/jnr.20703
Chiodo, L. A. (1988). Dopamine-containing neurons in the mammalian central nervous system: electrophysiology and pharmacology. Neurosci. Biobehav. Rev. 12, 49–91. doi: 10.1016/S0149-7634(88)80073-3
Cooper, G., Kang, S., Perez-Rosello, T., Guzman, J. N., Galtieri, D., Xie, Z., et al. (2020). A single amino acid determines the selectivity and efficacy of selective negative allosteric modulators of CaV1.3 L-type calcium channels. ACS Chem. Biol. 15, 2539–2550. doi: 10.1021/acschembio.0c00577
Dauer, W., and Przedborski, S. (2003). Parkinson's disease: mechanisms and models. Neuron 39, 889–909. doi: 10.1016/S0896-6273(03)00568-3
Dolphin, A. C. (2012). Calcium channel auxiliary alpha2delta and beta subunits: trafficking and one step beyond. Nat. Rev. Neurosci. 13, 542–55. doi: 10.1038/nrn3311
Dolphin, A. C. (2016). Voltage-gated calcium channels and their auxiliary subunits: physiology and pathophysiology and pharmacology. J. Physiol. 94, 5369–5390. doi: 10.1113/JP272262
Dragicevic, E., Poetschke, C., Duda, J., Schlaudraff, F., Lammel, S., Schiemann, J., et al. (2014). Cav1.3 channels control D2-autoreceptor responses via NCS-1 in substantia nigra dopamine neurons. Brain 137, 2287–2302. doi: 10.1093/brain/awu131
Duda, J., Potschke, C., and Liss, B. (2016). Converging roles of ion channels, calcium, metabolic stress, and activity-pattern of substantia nigra dopaminergic neurons in health and Parkinson's disease. J. Neurochem. 139, 156-178. doi: 10.1111/jnc.13572
Dufour, M. A., Woodhouse, A., and Goaillard, J. M. (2014). Somatodendritic ion channel expression in substantia nigra pars compacta dopaminergic neurons across postnatal development. J. Neurosci. Res. 92, 981–999. doi: 10.1002/jnr.23382
Evans, R. C., Zhu, M., and Khaliq, Z. M. (2017). Dopamine inhibition differentially controls excitability of substantia nigra dopamine neuron subpopulations through T-type calcium channels. J. Neurosci. 37, 3704–3720. doi: 10.1523/JNEUROSCI.0117-17.2017
Farassat, N., Costa, K. M., Stojanovic, S., Albert, S., Kovacheva, L., Shin, J., et al. (2019). In vivo functional diversity of midbrain dopamine neurons within identified axonal projections. Elife 8:e48408. doi: 10.7554/eLife.48408
Foehring, R. C., Zhang, X. F., Lee, J. C., and Callaway, J. C. (2009). Endogenous calcium buffering capacity of substantia nigral dopamine neurons. J. Neurophysiol. 102, 2326–2333. doi: 10.1152/jn.00038.2009
Fujimura, K., and Matsuda, Y. (1989). Autogenous oscillatory potentials in neurons of the guinea pig substantia nigra pars compacta in vitro. Neurosci. Lett. 104, 53–57. doi: 10.1016/0304-3940(89)90328-5
Galvan, A., Devergnas, A., Pittard, D., Masilamoni, G., Vuong, J., Daniels, J. S., et al. (2016). Lack of antiparkinsonian effects of systemic injections of the specific T-type calcium channel blocker ML218 in MPTP-treated monkeys. ACS Chem. Neurosci. 7, 1543–1551. doi: 10.1021/acschemneuro.6b00186
Gerlach, M., Russ, H., Winker, J., Witzmann, K., Traber, J., Stasch, J. P., et al. (1993). Effects of nimodipine on the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced depletions in the biogenic amine levels in mice. Arzneimittelforschung 43, 413–415.
Grace, A. A., and Bunney, B. S. (1984a). The control of firing pattern in nigral dopamine neurons: burst firing. J. Neurosci. 4, 2877–2890. doi: 10.1523/JNEUROSCI.04-11-02877.1984
Grace, A. A., and Bunney, B. S. (1984b). The control of firing pattern in nigral dopamine neurons: single spike firing. J. Neurosci. 4, 2866–2876. doi: 10.1523/JNEUROSCI.04-11-02866.1984
Gudala, K., Kanukula, R., and Bansal, D. (2015). Reduced risk of Parkinson's disease in users of calcium channel blockers: a meta-analysis. Int. J. Chronic Dis. 2015:697404. doi: 10.1155/2015/697404
Guzman, J. N., Ilijic, E., Yang, B., Sanchez-Padilla, J., Wokosin, D., Galtieri, D., et al. (2018). Systemic isradipine treatment diminishes calcium-dependent mitochondrial oxidant stress. J. Clin. Invest. 128, 2266–2280. doi: 10.1172/JCI95898
Guzman, J. N., Sanchez-Padilla, J., Chan, C. S., and Surmeier, D. J. (2009). Robust pacemaking in substantia nigra dopaminergic neurons. J. Neurosci. 29, 11011–11019. doi: 10.1523/JNEUROSCI.2519-09.2009
Guzman, J. N., Sanchez-Padilla, J., Wokosin, D., Kondapalli, J., Ilijic, E., Schumacker, P. T., et al. (2010). Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 468, 696–700. doi: 10.1038/nature09536
Hage, T. A., and Khaliq, Z. M. (2015). Tonic firing rate controls dendritic Ca2+ signaling and synaptic gain in substantia nigra dopamine neurons. J. Neurosci. 35, 5823–5836. doi: 10.1523/JNEUROSCI.3904-14.2015
Huang, H., Ng, C. Y., Yu, D., Zhai, J., Lam, Y., and Soong, T. W. (2014). Modest Cav1.342-selective inhibition by compound 8 is β-subunit dependent. Nat. Commun. 5:4481. doi: 10.1038/ncomms5481
Huang, H., Yu, D., and Soong, T. W. (2013). C-Terminal alternative splicing of CaV1.3 channels distinctively modulates their dihydropyridine sensitivity. Mol. Pharmacol. 84, 643–653. doi: 10.1124/mol.113.087155
Hurley, M. J., Gentleman, S. M., and Dexter, D. T. (2015). Calcium CaV1 channel subtype mRNA expression in Parkinson's disease examined by in situ hybridization. J. Mol. Neurosci. 55, 715–724. doi: 10.1007/s12031-014-0410-8
Ilijic, E., Guzman, J. N., and Surmeier, D. J. (2011). The L-type channel antagonist isradipine is neuroprotective in a mouse model of Parkinson's disease. Neurobiol. Dis. 43, 364–371. doi: 10.1016/j.nbd.2011.04.007
Ji, B., Wang, M., Gao, D., Xing, S., Li, L., Liu, L., et al. (2017). Combining nanoscale magnetic nimodipine liposomes with magnetic resonance image for Parkinson's disease targeting therapy. Nanomedicine 12, 237–253. doi: 10.2217/nnm-2016-0267
Ji, H., and Shepard, P. D. (2006). SK Ca2+-activated K+ channel ligands alter the firing pattern of dopamine-containing neurons in vivo. Neuroscience 140, 623–633. doi: 10.1016/j.neuroscience.2006.02.020
Jones, L. P., Wei, S. K., and Yue, D. T. (1998). Mechanism of auxiliary subunit modulation of neuronal alpha1E calcium channels. J. Gen. Physiol. 112, 125–143. doi: 10.1085/jgp.112.2.125
Kang, S., Cooper, G., Dunne, S. F., Dusel, B., Luan, C. H., Surmeier, D. J., et al. (2012). Cav1.3-selective L-type calcium channel antagonists as potential new therapeutics for Parkinson's disease. Nat. Commun. 3:1146. doi: 10.1038/ncomms2149
Khaliq, Z. M., and Bean, B. P. (2010). Pacemaking in dopaminergic ventral tegmental area neurons: depolarizing drive from background and voltage-dependent sodium conductances. J. Neurosci. 30, 7401–7413. doi: 10.1523/JNEUROSCI.0143-10.2010
Khanna, R., Yu, J., Yang, X., Moutal, A., Chefdeville, A., Gokhale, V., et al. (2019). Targeting the CaVα-CaVβ interaction yields an antagonist of the N-type CaV2.2 channel with broad antinociceptive efficacy. Pain 160, 1644–1661. doi: 10.1097/j.pain.0000000000001524
Kimm, T., and Bean, B. P. (2014). Inhibition of A-type potassium current by the peptide toxin SNX-482. J. Neurosci. 34, 9182–9189. doi: 10.1523/JNEUROSCI.0339-14.2014
Kopecky, B. J., Liang, R., and Bao, J. (2014). T-type calcium channel blockers as neuroprotective agents. Pflugers Arch. 466, 757–765. doi: 10.1007/s00424-014-1454-x
Koschak, A., Reimer, D., Huber, I., Grabner, M., Glossmann, H., Engel, J., et al. (2001). alpha 1D (Cav1.3) subunits can form l-type Ca2+ channels activating at negative voltages. J. Biol. Chem. 276, 22100–22106. doi: 10.1074/jbc.M101469200
Kupsch, A., Gerlach, M., Pupeter, S. C., Sautter, J., Dirr, A., Arnold, G., et al. (1995). Pretreatment with nimodipine prevents MPTP-induced neurotoxicity at the nigral, but not at the striatal level in mice. Neuroreport 6, 621–625. doi: 10.1097/00001756-199503000-00009
Kupsch, A., Sautter, J., Schwarz, J., Riederer, P., Gerlach, M., and Oertel, W. H. (1996). 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity in non-human primates is antagonized by pretreatment with nimodipine at the nigral, but not at the striatal level. Brain Res. 741, 185–196. doi: 10.1016/S0006-8993(96)00917-1
Lang, Y., Gong, D., and Fan, Y. (2015). Calcium channel blocker use and risk of Parkinson's disease: a meta-analysis. Pharmacoepidemiol. Drug Saf. 24, 559–566. doi: 10.1002/pds.3781
Lautenschlager, J., Stephens, A. D., Fusco, G., Strohl, F., Curry, N., Zacharopoulou, M., et al. (2018). C-terminal calcium binding of alpha-synuclein modulates synaptic vesicle interaction. Nat. Commun. 9:712. doi: 10.1038/s41467-018-03111-4
Leandrou, E., Emmanouilidou, E., and Vekrellis, K. (2019). Voltage-gated calcium channels and alpha-synuclein: implications in Parkinson's disease. Front. Mol. Neurosci. 12:237. doi: 10.3389/fnmol.2019.00237
Lieberman, O. J., Choi, S. J., Kanter, E., Saverchenko, A., Frier, M. D., Fiore, G. M., et al. (2017). α-Synuclein-dependent calcium entry underlies differential sensitivity of cultured SN and VTA dopaminergic neurons to a parkinsonian neurotoxin. eNeuro 4. doi: 10.1523/ENEURO.0167-17.2017
Liss, B., Haeckel, O., Wildmann, J., Miki, T., Seino, S., and Roeper, J. (2005). K-ATP channels promote the differential degeneration of dopaminergic midbrain neurons. Nat. Neurosci. 8, 1742–1751. doi: 10.1038/nn1570
Liss, B., and Striessnig, J. (2019). The potential of L-type calcium channels as a drug target for neuroprotective therapy in Parkinson's disease. Annu. Rev. Pharmacol. Toxicol. 59, 263–289. doi: 10.1146/annurev-pharmtox-010818-021214
Ma, H., Cohen, S., Li, B., and Tsien, R. W. (2012). Exploring the dominant role of Cav1 channels in signaling to the nucleus. Biosci. Rep. 33, 97–101. doi: 10.1042/BSR20120099
Melachroinou, K., Xilouri, M., Emmanouilidou, E., Masgrau, R., Papazafiri, P., Stefanis, L., et al. (2013). Deregulation of calcium homeostasis mediates secreted alpha-synuclein-induced neurotoxicity. Neurobiol. Aging 34, 2853–2865. doi: 10.1016/j.neurobiolaging.2013.06.006
Miranda-Laferte, E., Ewers, D., Guzman, R. E., Jordan, N., Schmidt, S., and Hidalgo, P. (2014). The N-terminal domain tethers the voltage-gated calcium channel beta2e-subunit to the plasma membrane via electrostatic and hydrophobic interactions. J. Biol. Chem. 289, 10387–10398. doi: 10.1074/jbc.M113.507244
Miranda-Laferte, E., Schmidt, S., Jara, A. C., Neely, A., and Hidalgo, P. (2012). A short polybasic segment between the two conserved domains of the beta2a-subunit modulates the rate of inactivation of R-type calcium channel. J. Biol. Chem. 287, 32588–32597. doi: 10.1074/jbc.M112.362509
Mosharov, E. V., Larsen, K. E., Kanter, E., Phillips, K. A., Wilson, K., Schmitz, Y., et al. (2009). Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron 62, 218–229. doi: 10.1016/j.neuron.2009.01.033
Mullapudi, A., Gudala, K., Boya, C. S., and Bansal, D. (2016). Risk of Parkinson's disease in the users of antihypertensive agents: an evidence from the meta-analysis of observational studies. J. Neurodegener. Dis. 2016:5780809. doi: 10.1155/2016/5780809
Murata, M. (2010). Zonisamide: a new drug for Parkinson's disease. Drugs Today 46, 251–258. doi: 10.1358/dot.2010.46.4.1490077
Newcomb, R., Szoke, B., Palma, A., Wang, G., Chen, X., Hopkins, W., et al. (1998). Selective peptide antagonist of the class E calcium channel from the venom of the tarantula Hysterocrates gigas. Biochemistry 37, 15353–15362. doi: 10.1021/bi981255g
Obeso, J. A., Stamelou, M., Goetz, C. G., Poewe, W., Lang, A. E., Weintraub, D., et al. (2017). Past, present, and future of Parkinson's disease: a special essay on the 200th Anniversary of the Shaking Palsy. Mov. Disord. 32, 1264–1310. doi: 10.1002/mds.27115
Olcese, R., Qin, N., Schneider, T., Neely, A., Wei, X., Stefani, E., et al. (1994). The amino terminus of a calcium channel beta subunit sets rates of channel inactivation independently of the subunit's effect on activation. Neuron 13, 1433–1438. doi: 10.1016/0896-6273(94)90428-6
Ortner, N. J., Bock, G., Dougalis, A., Kharitonova, M., Duda, J., Hess, S., et al. (2017). Lower affinity of isradipine for L-type Ca2+ channels during substantia nigra dopamine neuron-like activity: implications for neuroprotection in Parkinson's disease. J. Neurosci. 37, 6761–6777. doi: 10.1523/JNEUROSCI.2946-16.2017
Ortner, N. J., Bock, G., Vandael, D. H., Mauersberger, R., Draheim, H. J., Gust, R., et al. (2014). Pyrimidine-2,4,6-triones are a new class of voltage-gated L-type Ca2+ channel activators. Nat. Commun. 5:3897. doi: 10.1038/ncomms4897
Ortner, N. J., Kaserer, T., Copeland, J. N., and Striessnig, J. (2020). De novo CACNA1D Ca2+ channelopathies: clinical phenotypes and molecular mechanism. Pflugers Arch. 472, 755–773. doi: 10.1007/s00424-020-02418-w
Paladini, C. A., and Roeper, J. (2014). Generating bursts (and pauses) in the dopamine midbrain neurons. Neuroscience 282C, 109–121. doi: 10.1016/j.neuroscience.2014.07.032
Park, J. S., Davis, R. L., and Sue, C. M. (2018). Mitochondrial dysfunction in Parkinson's disease: new mechanistic insights and therapeutic perspectives. Curr. Neurol. Neurosci. Rep. 18:21. doi: 10.1007/s11910-018-0829-3
Parkinson Study Group (2013). Phase II safety, tolerability, and dose selection study of isradipine as a potential disease-modifying intervention in early Parkinson's disease (STEADY-PD). Mov. Disord. 28, 1823–1831. doi: 10.1002/mds.25639
Parkinson Study Group STEADY-PD III Investigators (2020). Isradipine versus placebo in early Parkinson disease: a randomized trial. Ann. Intern. Med. 172, 591-598. doi: 10.7326/M19-2534
Parkinson, J. (2002). An essay on the shaking palsy. J. Neuropsychiatry Clin. Neurosci. 14, 223–236; discussion: 222. doi: 10.1176/jnp.14.2.223
Philippart, F., Destreel, G., Merino-Sepulveda, P., Henny, P., Engel, D., and Seutin, V. (2016). Differential somatic Ca2+ channel profile in midbrain dopaminergic neurons. J. Neurosci. 36, 7234–7245. doi: 10.1523/JNEUROSCI.0459-16.2016
Poetschke, C., Dragicevic, E., Duda, J., Benkert, J., Dougalis, A., DeZio, R., et al. (2015). Compensatory T-type Ca2+ channel activity alters D2-autoreceptor responses of substantia nigra dopamine neurons from Cav1.3 L-type Ca2+ channel KO mice. Sci. Rep. 5:13688. doi: 10.1038/srep13688
Poewe, W., Seppi, K., Tanner, C. M., Halliday, G. M., Brundin, P., Volkmann, J., et al. (2017). Parkinson disease. Nat. Rev. Dis. Primers 3:17013. doi: 10.1038/nrdp.2017.13
Price, C. J. (2014). Evaluation of Isradipine for Neuroprotection in the MPTP/p Mouse Model of Parkinson's Disease. Washington, DC: Society for Neuroscience.
Richard, M., Kaufmann, P., Kornberger, R., and Dingemanse, J. (2019). First-in-man study of ACT-709478, a novel selective triple T-type calcium channel blocker. Epilepsia 60, 968–978. doi: 10.1111/epi.14732
Richard, M., Kaufmann, P., Ort, M., Kornberger, R., and Dingemanse, J. (2020). Multiple-ascending dose study in healthy subjects to assess the pharmacokinetics, tolerability, and CYP3A4 interaction potential of the T-type calcium channel blocker ACT-709478, a potential new antiepileptic drug. CNS Drugs 34, 311–323. doi: 10.1007/s40263-019-00697-1
Sautter, J., Kupsch, A., Earl, C. D., and Oertel, W. H. (1997). Degeneration of pre-labelled nigral neurons induced by intrastriatal 6-hydroxydopamine in the rat: behavioural and biochemical changes and pretreatment with the calcium-entry blocker nimodipine. Exp. Brain Res. 117, 111–119. doi: 10.1007/s002210050204
Schapira, A. H. (2008). Mitochondria in the aetiology and pathogenesis of Parkinson's disease. Lancet Neurol. 7, 97–109. doi: 10.1016/S1474-4422(07)70327-7
Schiemann, J., Schlaudraff, F., Klose, V., Bingmer, M., Seino, S., Magill, P. J., et al. (2012). K-ATP channels in dopamine substantia nigra neurons control bursting and novelty-induced exploration. Nat. Neurosci. 15, 1272–1280. doi: 10.1038/nn.3185
Schneider, T., Dibue, M., and Hescheler, J. (2013). How “pharmacoresistant” is Cav2.3, the major component of voltage-gated R-type Ca2+ channels? Pharmaceuticals 6, 759–776. doi: 10.3390/ph6060759
Schneider, T., Neumaier, F., Hescheler, J., and Alpdogan, S. (2020). Cav2.3 R-type calcium channels: from its discovery to pathogenic de novo CACNA1E variants: a historical perspective. Pflugers Arch. 472, 811–816. doi: 10.1007/s00424-020-02395-0
Schulz, J. B., Hausmann, L., and Hardy, J. (2016). 199 years of Parkinson disease - what have we learned and what is the path to the future? J. Neurochem. 139(Suppl. 1), 3–7. doi: 10.1111/jnc.13733
Shin, W. (2015). RNASeq of DA neurons from SNpc and VTA. Available online at: https://figshare.com/articles/dataset/RNASeq_of_DA_neurons_from_SNpc_and_VTA/926519
Siddiqi, F. H., Menzies, F. M., Lopez, A., Stamatakou, E., Karabiyik, C., Ureshino, R., et al. (2019). Felodipine induces autophagy in mouse brains with pharmacokinetics amenable to repurposing. Nat. Commun. 10:1817. doi: 10.1038/s41467-019-09494-2
Singh, A., Gebhart, M., Fritsch, R., Sinnegger-Brauns, M. J., Poggiani, C., Hoda, J. C., et al. (2008). Modulation of voltage- and Ca2+-dependent gating of Cav1.3 L-type calcium channels by alternative splicing of a C-terminal regulatory domain. J. Biol. Chem. 283, 20733–20744. doi: 10.1074/jbc.M802254200
Singh, A., Verma, P., Balaji, G., Samantaray, S., and Mohanakumar, K. P. (2016). Nimodipine, an L-type calcium channel blocker attenuates mitochondrial dysfunctions to protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced parkinsonism in mice. Neurochem. Int. 99, 221–232. doi: 10.1016/j.neuint.2016.07.003
Sinnegger-Brauns, M. J., Hetzenauer, A., Huber, I. G., Renstrom, E., Wietzorrek, G., Berjukov, S., et al. (2004). Isoform-specific regulation of mood behavior and pancreatic beta cell and cardiovascular function by L-type Ca2+ channels. J. Clin. Invest. 113, 1430–1439. doi: 10.1172/JCI20208
Sinnegger-Brauns, M. J., Huber, I. G., Koschak, A., Wild, C., Obermair, G. J., Einzinger, U., et al. (2009). Expression and 1,4-dihydropyridine-binding properties of brain L-type calcium channel isoforms. Mol. Pharmacol. 75, 407–414. doi: 10.1124/mol.108.049981
Surmeier, D. J., Halliday, G. M., and Simuni, T. (2017a). Calcium, mitochondrial dysfunction and slowing the progression of Parkinson's disease. Exp. Neurol. 298(Pt B), 202–209.doi: 10.1016/j.expneurol.2017.08.001
Surmeier, D. J., Obeso, J. A., and Halliday, G. M. (2017b). Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 18, 101–113. doi: 10.1038/nrn.2016.178
Tabata, Y., Imaizumi, Y., Sugawara, M., Andoh-Noda, T., Banno, S., Chai, M., et al. (2018). T-type calcium channels determine the vulnerability of dopaminergic neurons to mitochondrial stress in familial Parkinson disease. Stem Cell Rep. 11, 1171–1184. doi: 10.1016/j.stemcr.2018.09.006
Tai, C. H., Yang, Y. C., Pan, M. K., Huang, C. S., and Kuo, C. C. (2011). Modulation of subthalamic T-type Ca2+ channels remedies locomotor deficits in a rat model of Parkinson disease. J. Clin. Invest. 121, 3289–3305. doi: 10.1172/JCI46482
Tan, B. Z., Jiang, F., Tan, M. Y., Yu, D., Huang, H., Shen, Y., et al. (2011). Functional characterization of alternative splicing in the C terminus of L-type CaV1.3 channels. J. Biol. Chem. 286, 42725–42735. doi: 10.1074/jbc.M111.265207
Tracy, M. E., Tesic, V., Stamenic, T. T., Joksimovic, S. M., Busquet, N., Jevtovic-Todorovic, V., et al. (2018). CaV3.1 isoform of T-type calcium channels supports excitability of rat and mouse ventral tegmental area neurons. Neuropharmacology 135, 343–354. doi: 10.1016/j.neuropharm.2018.03.028
Uchida, S., Yamada, S., Nagai, K., Deguchi, Y., and Kimura, R. (1997). Brain pharmacokinetics and in vivo receptor binding of 1,4-dihydropyridine calcium channel antagonists. Life Sci. 61, 2083–2090. doi: 10.1016/S0024-3205(97)00881-3
Verma, A., and Ravindranath, V. (2019). CaV1.3 L-type calcium channels increase the vulnerability of substantia nigra dopaminergic neurons in MPTP mouse model of Parkinson's disease. Front. Aging Neurosci. 11:382. doi: 10.3389/fnagi.2019.00382
Wang, Q. M., Xu, Y. Y., Liu, S., and Ma, Z. G. (2017). Isradipine attenuates MPTP-induced dopamine neuron degeneration by inhibiting up-regulation of L-type calcium channels and iron accumulation in the substantia nigra of mice. Oncotarget 8, 47284-47295. doi: 10.18632/oncotarget.17618
Wang, R., Ma, Z., Wang, J., and Xie, J. (2012). L-type Cav1.2 calcium channel is involved in 6-hydroxydopamine-induced neurotoxicity in rats. Neurotox. Res. 21, 266–270. doi: 10.1007/s12640-011-9271-x
Wang, X., Saegusa, H., Huntula, S., and Tanabe, T. (2019). Blockade of microglial Cav1.2 Ca2+ channel exacerbates the symptoms in a Parkinson's disease model. Sci. Rep. 9:9138. doi: 10.1038/s41598-019-45681-3
Weiss, N., and Zamponi, G. W. (2019a). T-type calcium channels: from molecule to therapeutic opportunities. Int. J. Biochem. Cell Biol. 108, 34–39. doi: 10.1016/j.biocel.2019.01.008
Weiss, N., and Zamponi, G. W. (2019b). T-type channel druggability at a crossroads. ACS Chem. Neurosci. 10, 1124–1126. doi: 10.1021/acschemneuro.9b00031
Weiss, N., and Zamponi, G. W. (2020). Genetic T-type calcium channelopathies. J. Med. Genet. 57, 1–10. doi: 10.1136/jmedgenet-2019-106163
Wilson, C. J., and Callaway, J. C. (2000). Coupled oscillator model of the dopaminergic neuron of the substantia nigra. J. Neurophysiol. 83, 3084–3100. doi: 10.1152/jn.2000.83.5.3084
Wolfart, J., and Roeper, J. (2002). Selective coupling of T-type calcium channels to SK potassium channels prevents intrinsic bursting in dopaminergic midbrain neurons. J. Neurosci. 22, 3404–3413. doi: 10.1523/JNEUROSCI.22-09-03404.2002
Xiang, Z., Thompson, A. D., Brogan, J. T., Schulte, M. L., Melancon, B. J., Mi, D., et al. (2011). The discovery and characterization of ML218: a novel, centrally active T-type calcium channel inhibitor with robust effects in STN neurons and in a rodent model of Parkinson's disease. ACS Chem. Neurosci. 2, 730–742. doi: 10.1021/cn200090z
Xu, W., and Lipscombe, D. (2001). Neuronal Cav1.3α1 L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J. Neurosci. 21, 5944–5951. doi: 10.1523/JNEUROSCI.21-16-05944.2001
Yang, Y. C., Tai, C. H., Pan, M. K., and Kuo, C. C. (2014). The T-type calcium channel as a new therapeutic target for Parkinson's disease. Pflugers Arch. 466, 747–755. doi: 10.1007/s00424-014-1466-6
Yasuda, T., Chen, L., Barr, W., McRory, J. E., Lewis, R. J., Adams, D. J., et al. (2004). Auxiliary subunit regulation of high-voltage activated calcium channels expressed in mammalian cells. Eur. J. Neurosci. 20, 1–13. doi: 10.1111/j.1460-9568.2004.03434.x
Yee, A. G., Forbes, B., Cheung, P. Y., Martini, A., Burrell, M. H., Freestone, P. S., et al. (2019). Action potential and calcium dependence of tonic somatodendritic dopamine release in the substantia nigra pars compacta. J. Neurochem. 148, 462–479. doi: 10.1111/jnc.14587
Yiu, S., and Knaus, E. E. (1996). Synthesis, biological evaluation, calcium channel antagonist activity, and anticonvulsant activity of felodipine coupled to a dihydropyridine-pyridinium salt redox chemical delivery system. J. Med. Chem. 39, 4576–4582. doi: 10.1021/jm960531r
Zaichick, S. V., McGrath, K. M., and Caraveo, G. (2017). The role of Ca2+ signaling in Parkinson's disease. Dis. Model. Mech. 10, 519–535. doi: 10.1242/dmm.028738
Zampese, E., and Surmeier, D. J. (2020). Calcium, bioenergetics, Parkinson's disease. Cells 9:2045. doi: 10.3390/cells9092045
Keywords: voltage-gated Ca2+ channels, Parkinson's disease, Ca2+ oscillations, Ca2+ channel blockers, L-type Ca2+ channels, R-type Ca2+ channels, T-type Ca2+ channels
Citation: Ortner NJ (2021) Voltage-Gated Ca2+ Channels in Dopaminergic Substantia Nigra Neurons: Therapeutic Targets for Neuroprotection in Parkinson's Disease? Front. Synaptic Neurosci. 13:636103. doi: 10.3389/fnsyn.2021.636103
Received: 30 November 2020; Accepted: 25 January 2021;
Published: 26 February 2021.
Edited by:
Sigismund Huck, Medical University of Vienna, AustriaReviewed by:
Norbert Weiss, Charles University, CzechiaPaolo Calabresi, Catholic University of the Sacred Heart, Italy
Copyright © 2021 Ortner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nadine J. Ortner, bmFkaW5lLm9ydG5lckB1aWJrLmFjLmF0