 Ian N. Sabir1,2*
Ian N. Sabir1,2* Nan Ma3
Nan Ma3 Victoria J. Jones4
Victoria J. Jones4 Catharine A. Goddard5
Catharine A. Goddard5 Yanmin Zhang1,5
Yanmin Zhang1,5 Asli Kalin3
Asli Kalin3 Andrew A. Grace1,5
Andrew A. Grace1,5 Christopher L.-H. Huang1,5
Christopher L.-H. Huang1,5
- 1Physiological Laboratory, Department of Physiology, Development and Neuroscience, University of Cambridge, Cambridge, UK
- 2Hammersmith Hospital, London, UK
- 3John Radcliffe Hospital, Headington, Oxford, UK
- 4Papworth Hospital, Papworth Everard, Cambridge, UK
- 5Department of Biochemistry, University of Cambridge, Cambridge, UK
The relationship between alternans and arrhythmogenicity was studied in genetically modified murine hearts modeling catecholaminergic polymorphic ventricular tachycardia (CPVT) during Langendorff perfusion, before and after treatment with catecholamines and a β-adrenergic antagonist. Heterozygous (RyR2p/s) and homozygous (RyR2s/s) RyR2-P2328S hearts, and wild-type (WT) controls, were studied before and after treatment with epinephrine (100 nM and 1 μM) and propranolol (100 nM). Monophasic action potential recordings demonstrated significantly greater incidences of arrhythmia in RyR2s/p and RyR2s/s hearts as compared to WTs. Arrhythmogenicity in RyR2s/s hearts was associated with alternans, particularly at short baseline cycle lengths. Both phenomena were significantly accentuated by treatment with epinephrine and significantly diminished by treatment with propranolol, in full agreement with clinical expectations. These changes took place, however, despite an absence of changes in mean action potential durations, ventricular effective refractory periods or restitution curve characteristics. Furthermore pooled data from all hearts in which arrhythmia occurred demonstrated significantly greater alternans magnitudes, but similar restitution curve slopes, to hearts that did not demonstrate arrhythmia. These findings thus further validate the RyR2-P2328S murine heart as a model for human CPVT, confirming an alternans phenotype in common with murine genetic models of the Brugada syndrome and the congenital long-QT syndrome type 3. In contrast to these latter similarities, however, this report demonstrates the dissociation of alternans from changes in the properties of restitution curves for the first time in a murine model of a human arrhythmic syndrome.
Introduction
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a hereditary arrhythmic disorder characterized by ventricular tachyarrhythmias precipitated by exercise, stress or catecholamine infusion (Wehrens et al., 2003). Such arrhythmias may potentially lead to ventricular fibrillation, resulting in syncope and sudden cardiac death (Priori et al., 2001). CPVT is a highly lethal condition, with Kaplan Meyer analyses confirming mortality rates of 30–33% by age 35 (Priori et al., 2001). Current therapies are unsatisfactory but include regular administration of β-adrenoceptor antagonists (Leenhardt et al., 1995) and insertion of automatic implantable cardioverter defibrillators (Priori et al., 2001).
CPVT is associated with mutations in the cardiac ryanodine receptor (RyR2) (Marks et al., 2002). Six distinct RyR2 mutations, occurring in the absence of structural abnormalities, echocardiographic evidence of cardiac failure or QT prolongation, have been reported to result in CPVT (Laitinen et al., 2001; Priori et al., 2001). Of these, a hereditary P2328S mutation has been shown to alter FKBP12.6 binding following PKA phosphorylation in HEK293 cells (Lehnart et al., 2004). Mice genetically modified to lack FKBP12.6 and proposed as a model for CPVT demonstrate arrhythmia in association with alternans in action potential duration (APD) (Lehnart et al., 2006); both these phenomena could be prevented by pharmacological maneuvers expected to increase the binding affinity of FKBP12.6 for RyR2. Such associations between alternans and ventricular arrhythmia are well established in both historical and contemporary literature (Lewis, 1911; Gold et al., 2000; Weiss et al., 2006), with electrocardiographic alternans having been reported in carriers of mutations associated with CPVT (Aizawa et al., 2006).
The present study extends correlations between alternans and arrhythmia (Weiss et al., 2006) to genetically modified hearts carrying the P2328S mutation and thus representing a true genetic model of CPVT (Goddard et al., 2008). It thus: (1) confirms previous reports of elevated incidences of arrhythmia in RyR2-P2328S hearts as compared to wild-type (WT) controls (Goddard et al., 2008); (2) correlates such arrhythmogenicity with a tendency to exhibit alternans, in agreement with observations both in humans with CPVT (Aizawa et al., 2006) and in murine genetic models of other hereditary arrhythmic syndromes and (3) demonstrates that treatment with adrenergic agonists and antagonists, known to be pro- and anti-arrhythmic respectively in humans with CPVT (Leenhardt et al., 1995), suppresses and accentuates alternans as might be expected. It then proceeds to demonstrate that such alternans occurs in the absence of changes in either (4) mean action potential properties, (5) refractory periods or (6) restitution curve characteristics, in sharp contrast to findings in those other murine models (Sabir et al., 2008a).
Materials and Methods
Genetically modified mice heterozygous (RyR2p/s) and homozygous (RyR2s/s) for the P2328S mutation in the central region of the RyR2 gene (Goddard et al., 2008), along with WT controls, were housed in an animal facility at 21 ± 1°C with 12-h light/dark cycles. All mice were fed sterile chow (RM3 Maintenance Diet, SDS, Witham, Essex, UK) and given free access to water. All procedures were carried out in accordance with the UK Animals (Scientific Procedures) Act 1986. Bicarbonate-buffered Krebs–Henseleit solutions (119 mM NaCl, 25 mM NaHCO3, 4 mM KCl, 1.2 mM KH2PO4, 1 mM MgCl2, 1.8 mM CaCl2, 10 mM glucose and 2 mM Na-pyruvate) were used. These were bubbled with 95% O2/5% CO2 (British Oxygen Company, Manchester, UK) to achieve a physiological pH of 7.4 and prevent the precipitation of calcium carbonate. Drug-containing solutions were prepared by adding epinephrine (Sigma-Aldrich, Poole, UK) to final concentrations of either 100 nM or 1 μM as stated and propranolol (Sigma-Aldrich, Poole, UK) to a final concentration of 100 nM, as used on previous occasions (Tosaka et al., 2003; Thomas et al., 2007a).
A Langendorff-perfusion protocol previously validated for use with murine hearts (Balasubramaniam et al., 2003) was used. In brief, mice were killed by cervical dislocation (Schedule 1: UK Animals [Scientific Procedures] Act 1986). Hearts were then quickly excised and placed in ice-cold bicarbonate-buffered Krebs–Henseleit solution. Short sections of aorta were then rapidly cannulated under the surface of the ice-cold buffer solution and attached using an aneurysm clip (Harvard Apparatus, Edenbridge, Kent, UK) to a custom-made 21-gauge cannula. Fresh Krebs–Henseleit solution was passed through 200 and 5 μm filters (Millipore, Watford, UK) and warmed to 37ºC using a water jacket and circulator (Techne model C-85A, Cambridge, UK). A peristaltic pump (Watson-Marlow Bredel model 505S, Falmouth, Cornwall, UK) in constant flow mode was then used to perfuse hearts with this solution at 2–2.5 ml/min in a retrograde fashion. Hearts were regarded as suitable for experimentation if they regained a healthy pink color and began to contract spontaneously on re-warming.
An epicardial monophasic action potential (MAP) electrode (Hugo Sachs, Harvard Apparatus, UK) was placed against the basal left ventricular epicardium. Signals were amplified, band-pass filtered (0.5 Hz to 1 kHz: Neurolog, Digitimer, Welwyn Garden City, Hertfordshire, UK) and digitized at a sampling frequency of 5 kHz (micro1401, Cambridge Electronic Design, Cambridge, UK). Data analysis was performed using Spike II (Cambridge Electronic Design, Cambridge, UK).
A bipolar platinum stimulating electrode (1 mm inter-pole spacing) was placed against the basal surface of the right ventricular epicardium. Square-wave stimuli (Grass S48 stimulator, Grass-Telefactor, Slough, UK) of 2-ms duration and amplitudes of twice the diastolic excitation threshold were initially applied to hearts at a constant baseline cycle length (BCL) of 125 ms. Recordings met quality assurance criteria defined previously, with one action potential occurring in response to each stimulus, each action potential demonstrating a rapid upstroke phase that reached a consistent amplitude and a smooth re-polarization phase and the baseline remaining stable between action potentials (Knollmann et al., 2001; Fabritz et al., 2003). APD was quantified at 90% re-polarization (APD90), APD0 being defined as the peak upstroke potential difference and APD100 as the diastolic potential difference. Notably, inclusion or exclusion of the upstroke phase did not make a significant (P > 0.05) difference to results under any set of experimental conditions studied. Before subsequent recordings were made hearts were then exposed to test solutions for 10 min while stimulation was continued.
MAPs were recorded during regular stimulation at a BCL of 85, 95, 105, 115, and 125 ms and the data obtained used to assess the magnitude of alternans present. Such alternans was defined as a sequence of ten or more action potentials the duration of which were shorter then longer (or vice versa) than in the previous beat. A detection margin of 0.2 ms was used, reflecting the sampling sensitivity of the Spike II data acquisition software utilized. An extrasystolic stimulation protocol (Saumarez and Grace, 2000) previously adapted for use in murine hearts (Head et al., 2005) was then applied. This comprised regular stimuli delivered at a BCL of 125 ms interrupted by extrasystolic stimuli imposed after every eighth regular stimulus at delays progressively decremented with each successive stimulus cycle in 1 ms steps from the BCL. Such cycles were repeated until the extrasystolic stimuli either initiated arrhythmia or failed to initiate a MAP, the ventricular effective refractory period (VERP) having been reached. Hearts were then subjected to an adapted dynamic pacing protocol (Koller et al., 1998; Sabir et al., 2008b). This comprised cycles each of 100 stimuli, initially delivered at a BCL of 175 ms. BCL was decremented in 5 ms steps with each successive stimulus cycle until a reproducible sequence of consistently shaped MAP waveforms was no longer obtained. This dynamic pacing protocol was also used to induce arrhythmia, with arrhythmia being defined as a > 5 s period of morphologically irregular waveforms continuing in the absence of stimulation.
Recordings were made in the absence of pharmacological agents, following addition of epinephrine, following addition of propranolol, and following addition of both epinephrine and propranolol. Hearts were exposed to reagent-containing solutions for 10 min during stimulation at a constant BCL of 125 ms before data were collected.
All data are presented as means ± standard errors of the means and include the numbers of hearts studied. Comparisons between data sets used analysis of variance (ANOVA) and the Chi-squared test to make multiple comparisons between continuous and discrete data respectively (significance thresholds set at P ≤ 0.05). A Levenberg–Marquardt algorithm (OriginPro 7.5, OriginLab, Northampton, MA, USA) was used for curve fitting.
Results
The experiments first confirmed the presence or absence of arrhythmogenic phenomena in intact Langendorff-perfused WT murine hearts, as well as in hearts heterozygous (RyR2p/s) and homozygous (RyR2s/s) for a mutation known to result in CPVT in humans. This confirmed and extended previous reports of an increase in incidence of arrhythmia with gene dosage in RyR2-P2328S mice (Goddard et al., 2008) using a dynamic pacing protocol.
Figure 1 gives incidences of arrhythmia in WT, RyR2p/s and RyR2s/s hearts under the various pharmacological and stimulation conditions employed. Figure 2 illustrates the recordings obtained, summarizing these results by showing arrhythmic waveforms in situations where these were observed at a significantly (P < 0.05) higher incidence than in WT controls. Incidences of arrhythmia increased both with gene dosage, with effects noticeable in the RyR2s/s rather than the RyR2p/s hearts, and following treatment with epinephrine (see Figure 1, top panel). Treatment with propranolol significantly decreased arrhythmia incidence in epinephrine-treated hearts but had no effect in controls. There were no significant differences in incidence of arrhythmia between groups treated with propranolol. We thus confirm previous reports of increases in the incidence of arrhythmia with mutant gene dosage in RyR2-P2328S mice (Goddard et al., 2008), as well as the pro-arrhythmic and anti-arrhythmic effects of epinephrine and propranolol respectively.
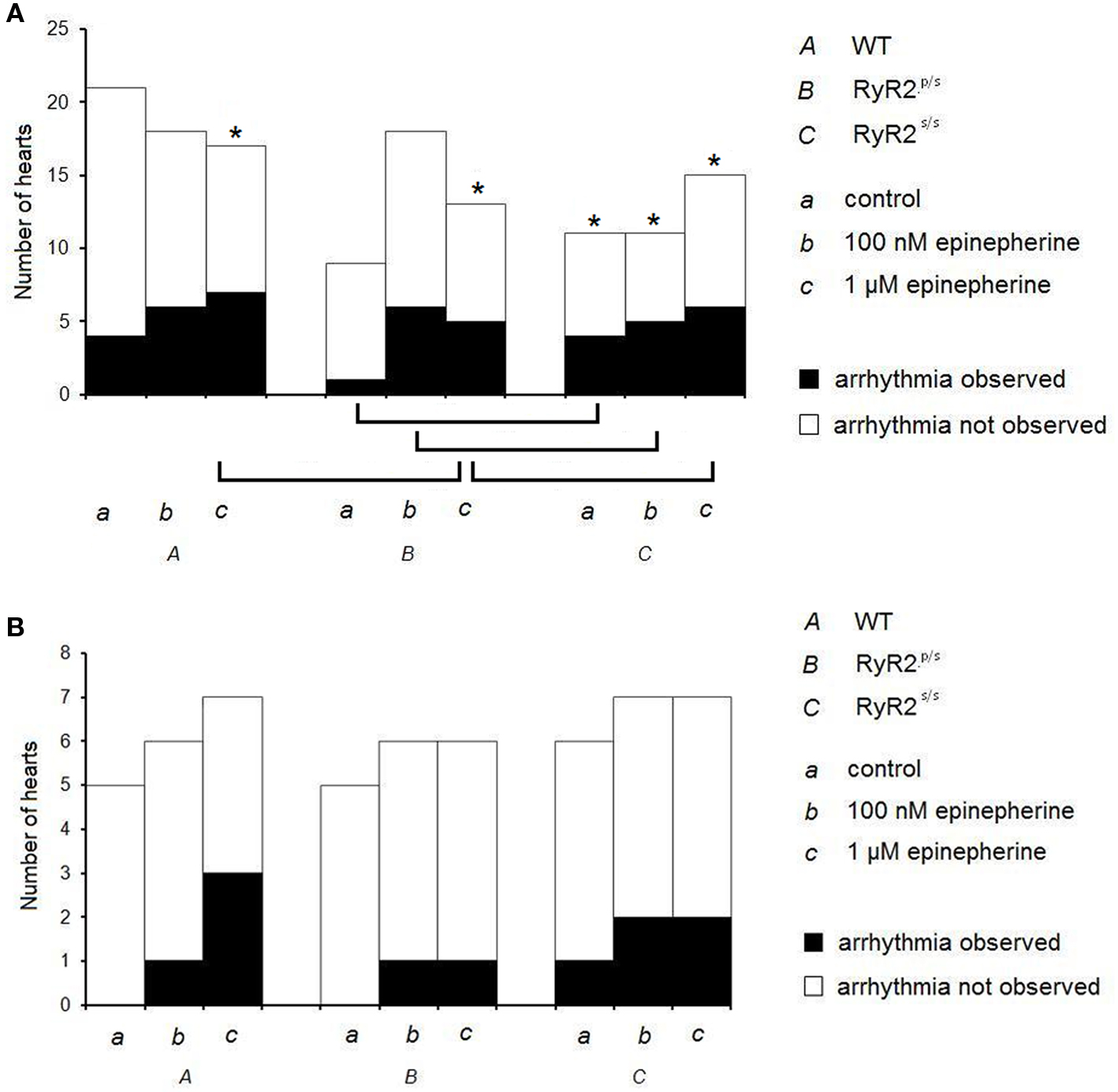
Figure 1. Incidences of arrhythmia in WT, RyR2p/s and RyR2p/s hearts. Incidences of ventricular arrhythmia occurring in WT (A), RyR2p/s (B) and RyR2p/s (C) hearts treated with control solution (a) and with solutions containing 100 nM epinephrine (b) and 1 μM epinephrine (c). The top panel shows traces obtained in the absence of propranolol; the bottom panel shows data obtained in the presence of 100 nM propranolol. Asterisks in the top panel indicate situations in which incidence of arrhythmia was significantly (P < 0.05) higher than in WT controls; brackets indicate significant (P < 0.05) differences in incidence of arrhythmia between other sets of conditions.
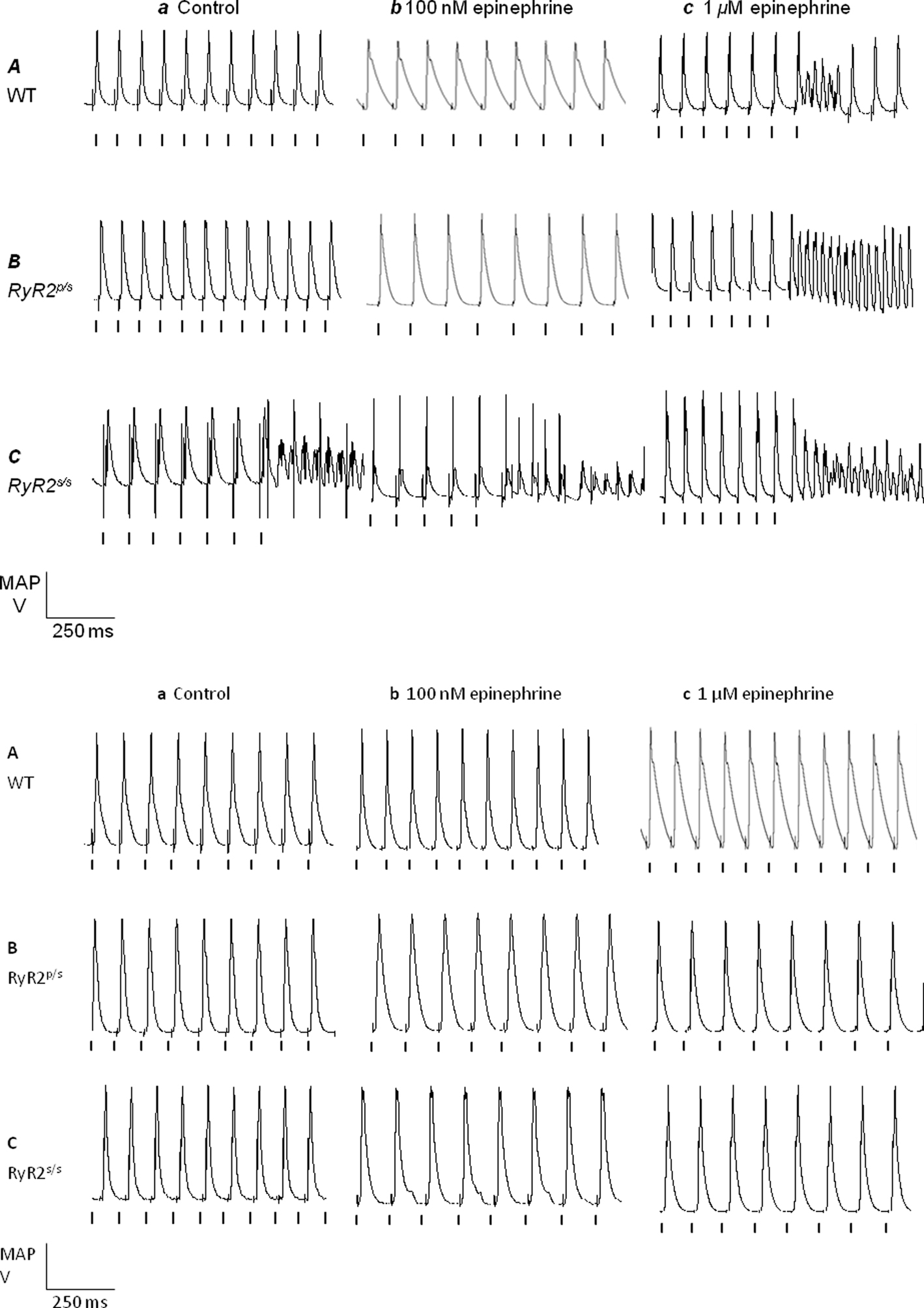
Figure 2. Arrhythmia in WT, RyR2p/s and RyR2p/s hearts. Monophasic action potential recordings obtained from WT (A), RyR2p/s (B) and RyR2p/s(C) hearts treated with control solution (a) and with solutions containing 100 nM epinephrine (b) and 1 μM epinephrine (c). The top panel shows traces obtained in the absence of propranolol; the bottom panel shows traces obtained in the presence of 100 nM propranolol. Traces show arrhythmia where this was observed significantly (P < 0.05) more frequently than in WT controls (see Figure 1). Vertical lines indicate the timing of stimuli.
The experiments then demonstrated alternans in RyR2-P2328S hearts for the first time, a finding in full agreement with clinical reports (Aizawa et al., 2006). Figure 3 plots alternans magnitude, the mean difference in APD (quantified at 90% re-polarization, APD90) between successive odd- and even-numbered action potentials, against BCL. Hearts were studied before (diamonds) and after addition of 100 nM (squares) or 1 μM (triangles) epinephrine both before (Figure 3, top panel) and after (Figure 3, bottom panel) introduction of propranolol. Alternans magnitude tended to increase with decreasing BCL in agreement with previous findings (Walker et al., 2003), yielding significantly larger values at the shortest (85 ms) as compared to the longest (125 ms, P < 0.05) BCLs studied under all conditions. All subsequent comparisons between alternans magnitudes were made using data obtained at a BCL of 85 ms.
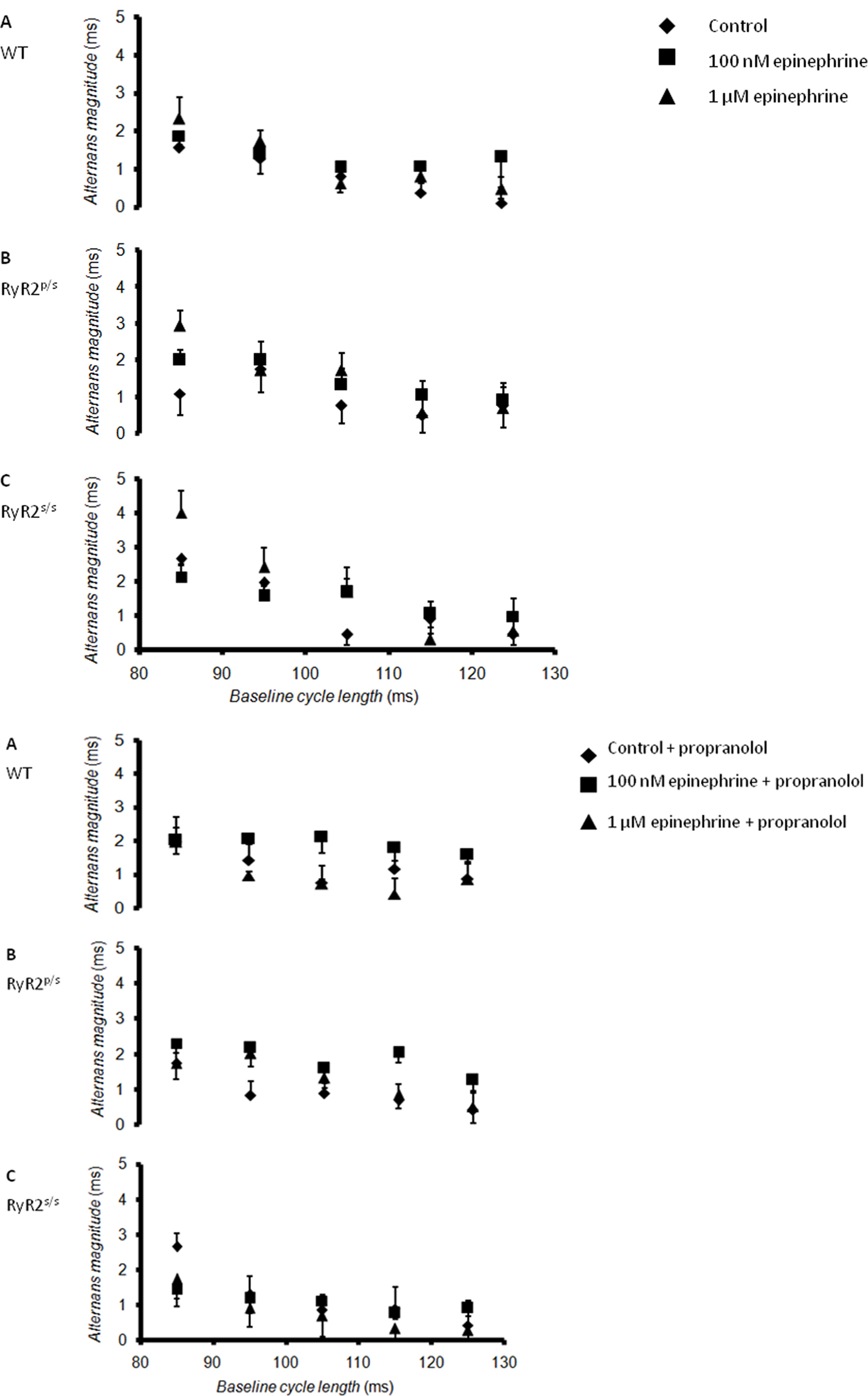
Figure 3. Relationships between alternans magnitude and baseline cycle length. Relationships between alternans magnitude, defined as the difference between APD (at 90% re-polarization, APD90) for successive odd- and even-numbered action potentials, and BCL in WT (A), RyR2p/s (B) and RyR2s/s (C) hearts treated with control solution (diamonds) and with solutions containing 100 nM epinephrine (squares) and 1 μM epinephrine (triangles). The top panel shows traces obtained in the absence of propranolol; the bottom panel shows traces obtained in the presence of 100 nM propranolol.
WT hearts demonstrated alternans only at the shortest BCL studied, as in previous reports (Sabir et al., 2008b). Exposure to epinephrine, whether at concentration of 100 nM or 1 μM, significantly (P < 0.05) increased alternans magnitude. Before pharmacological manipulation, RyR2p/s hearts demonstrated alternans magnitudes statistically indistinguishable from values obtained in WTs. Following treatment with epinephrine, however, alternans magnitude was significantly (P < 0.05) greater than seen in equivalent groups of WT hearts. RyR2s/s hearts showed significantly (P < 0.05) greater alternans magnitudes than either WT or RyR2p/s hearts, irrespective of the pharmacological conditions studied. Treatment with epinephrine again increased alternans magnitude, with values being significantly (P < 0.05) larger than found in WT or RyR2p/s hearts. While treatment with propranolol tended to increase alternans magnitude when applied to WT hearts in the absence of epinephrine, it significantly (P < 0.05) decreased alternans magnitude in all other cases.
Alternans magnitude in pooled data from arrhythmic hearts was significantly greater than in hearts that did not demonstrate arrhythmia (4.2 ± 0.5 ms, n = 55 vs. 2.8 ± 0.3 ms, n = 126; P < 0.05), confirming an association between alternans and arrhythmia under the experimental conditions studied.
Alternans and arrhythmia in murine models of a number of hereditary (Thomas et al., 2007a,b) and acquired (Sabir et al., 2007) arrhythmic disorders have been associated with changes in APD, VERP and restitution curve slopes. The present experiments thus proceeded to study these parameters, the latter two for the first time in RyR2-P2328S mutant hearts.
First, APD90 was recorded through a wide range of BCLs before and after exposure to epinephrine and to propranolol, separately and in combination. APD90 increased with increasing BCL, as previously established (Sabir et al., 2007) but did not differ significantly between WT, RyR2p/s and RyR2s/s hearts at any BCL. Furthermore, epinephrine treatment left APD90 unchanged apart from in two cases in WT (P < 0.05), but not RyR2p/s or RyR2s/s, hearts. Finally, propranolol did not significantly affect APD90 at any BCL. These observations fully agree with previous observations of electrocardiographic QT interval in patients with CPVT (Priori et al., 2001).
Secondly, VERP values were obtained under the full range of experimental conditions studied using an extrasystolic stimulation protocol. Neither these genetic nor pharmacological manipulations resulted in significant changes in VERP in any case.
Thirdly, recent studies had associated alternans and arrhythmia in murine hearts with increases in the slopes of restitution curves relating APD90 to the preceding diastolic interval (DI), given by the difference between the preceding BCL and APD90 (Sabir et al., 2008a,b) (illustrated in Figure 4).
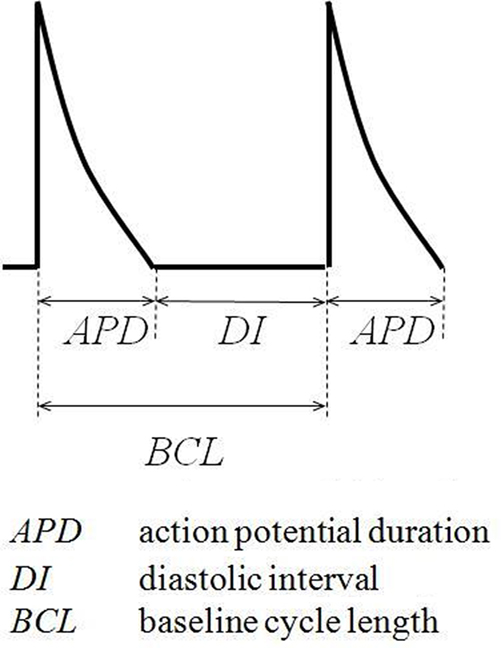
Figure 4. Cartoon representation of parameters used to construct restitution curves.
Such curves were constructed using APD90 and DI data obtained during the dynamic pacing protocol. They could not be derived from parameters recorded during treatment with 1 μM epinephrine as the high incidence of arrhythmia did not permit acquisition of the appropriate data. Their slopes were assessed from fits to a simple, mono-exponential growth function of the form:

y representing APD90, x representing DI and y0 and A and τ being constants obtained by least-squares fitting to the experimental values of APD90 and DI. Fits were made using a least-squares method.
Figure 5 shows an example of such a curve obtained from WT hearts under control conditions.
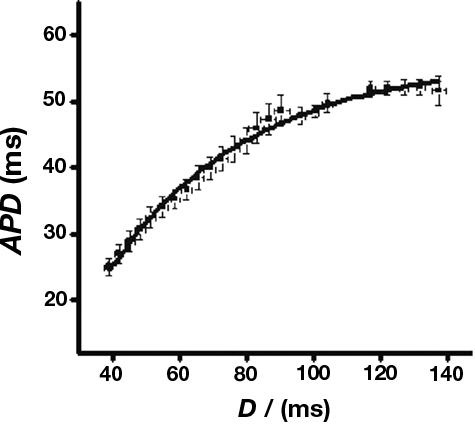
Figure 5. Sample restitution curve. Sample curve obtained from WT hearts in the absence of pharmacological manipulations. Data were fitted with a curve of the form y = y0 + A(1−e−x/τ) where y0 = −11.9 ± 3.6, A = 60.0 ± 2.6, τ = 57.8 ± 5.4, χ2 = 0.98 (n = 7).
The corresponding slopes are then given by:

Figure 6 summarizes the maximum values of slopes obtained at the shortest BCLs studied.
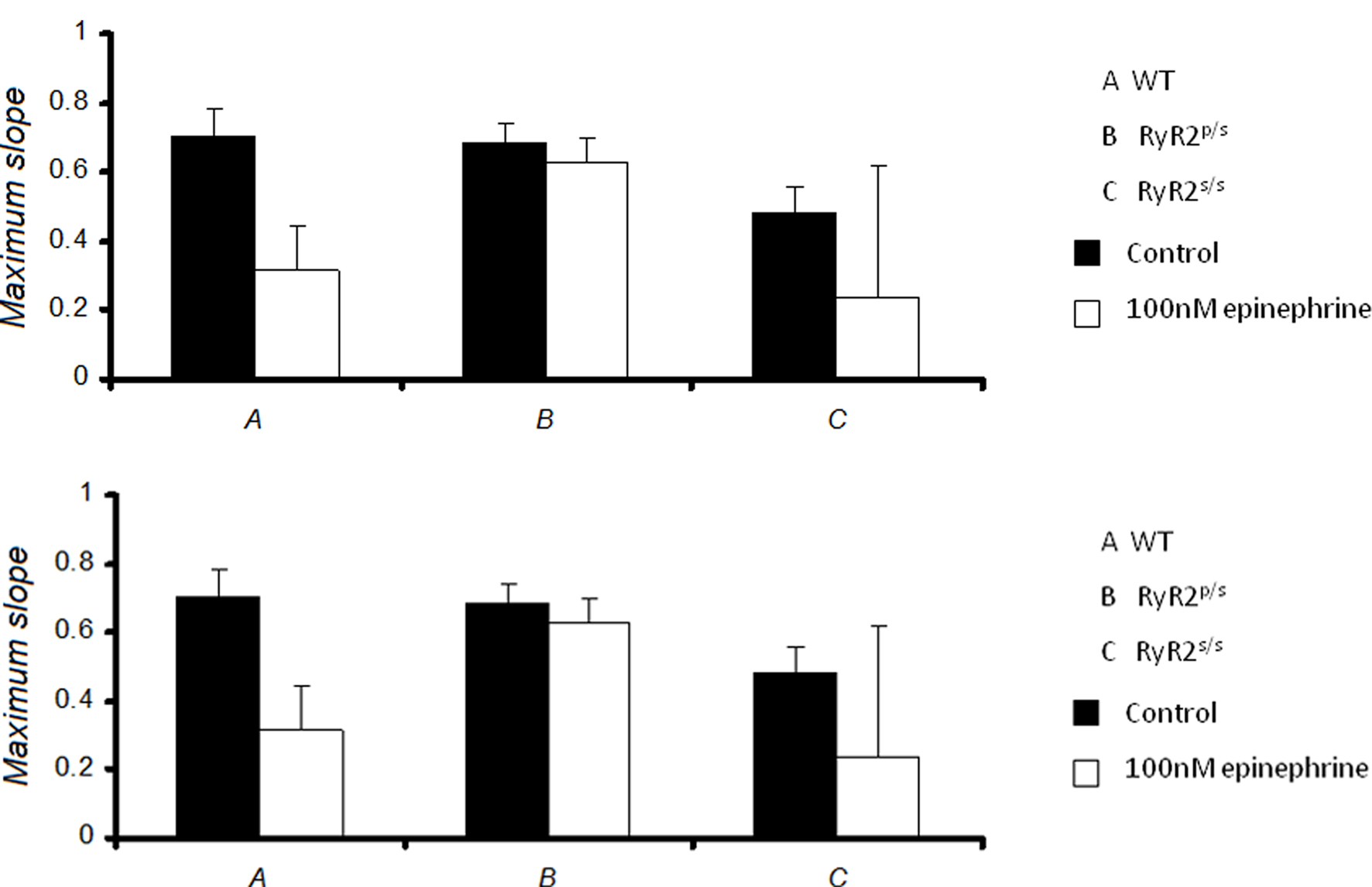
Figure 6. Maximum slopes of restitution curves. Maximum slopes obtained from restitution curves in WT (A), RyR2p/s (B) and RyR2s/s (C) hearts treated with control solution (■) and with solution containing 100 nM epinephrine (□), prior to (top panel) and after (bottom panel) addition of 100 nM propranolol. Maximum slopes did not differ significantly between experimental groups.
Maximum slopes obtained from WT hearts under control conditions closely agreed with previous data (Sabir et al., 2008a,b). There was no significant difference in restitution curve slope between any experimental groups, however. Furthermore, pooled data from arrhythmic hearts gave a maximum slope that was not significantly different from that obtained from non-arrhythmic hearts (0.4 ± 0.2, n = 55 vs. 0.5 ± 0.2, n = 126). Thus alternans and arrhythmia were not associated with significant changes in the slopes of restitution curves. Thus, in contrast to the results of a previous study carried out in models of other hereditary arrhythmic syndromes (Sabir et al., 2008a), alternans and arrhythmia in this setting cannot be explained by alterations in the slopes of restitution curves.
Discussion
Catecholaminergic polymorphic ventricular tachycardia is a hereditary arrhythmic disorder attributed to mutations that affect the cardiac ryanodine receptor (RyR2) (Priori et al., 2001). Ventricular arrhythmia in CPVT patients has been associated with alternations in APD, alternans (Aizawa et al., 2006), a link already well established in other situations (Lewis, 1911; Gold et al., 2000; Weiss et al., 2006). The present study sought to determine if genetically modified mice incorporating a P2328S mutation in the RyR2 gene (Goddard et al., 2008), previously established to result in CPVT in some patients (Laitinen et al., 2001; Priori et al., 2001), exhibit alternans. It then proceeded to determine if such alternans and arrhythmia were associated with changes in action potential or refractory properties, or in the slopes of restitution curves (Nolasco and Dahlen, 1968), as is the case in murine models of other human arrhythmic syndromes (Thomas et al., 2007a; Sabir et al., 2008a,b). MAP recordings were obtained from the epicardia of Langendorff-perfused hearts both heterozygous (RyR2p/s) and homozygous (RyR2s/s) for the mutation, as well as from WT controls.
Previous studies conducted on genetically modified murine hearts modeling CPVT have used a range of maneuvers to demonstrate the presence of an arrhythmogenic phenotype. For example, Cerrone et al. (2005) used a combination of isoprenaline and caffeine. Goddard et al. (2008) later reported the qualitative presence of an arrhythmogenic phenotype in catecholamine-treated hearts incorporating the RyR2-P2328S mutation during the application of both regular and extrasystolic stimulation protocols. In the present study arrhythmogenicty was assessed during periods of rapid pacing as part of a dynamic pacing protocol. Results confirmed the arrhythmogenic effect of this mutation, as well as demonstrating a pro-arrhythmic effect from epinephrine and an anti-arrhythmic effect from propranolol, all in full agreement with expectations from clinical studies (Priori et al., 2002; Postma et al., 2005).
The experiments then sought to study alternans in these mice. Under all genetic and pharmacological conditions studied alternans magnitude, defined as the difference in APD between successive odd- and even-numbered action potentials, increased as BCL was decreased. This is in full agreement with previous work (Walker et al., 2003). In general, treatment with epinephrine increased, while treatment with propranolol decreased, alternans magnitude. Furthermore, RyR2s/s hearts demonstrated significantly larger alternans magnitudes than RyR2p/s hearts or WT controls, in fitting with the increased incidences of arrhythmia observed. This represents both the first experimental demonstration of alternans occurring in the murine heart as a result of exposure to catecholamines and the first demonstration of alternans in any experimental model of CPVT. Notably alternans magnitude in pooled data from arrhythmic hearts was significantly greater than that from hearts that did not demonstrate arrhythmia.
Subsequent experiments examined a possible basis for alternans in this setting. A large number of previous studies (Karagueuzian et al., 1993; Lee et al., 1996; Koller et al., 1998, 2000; Taggart et al., 2003; Pak et al., 2004) have associated alternans with steeply sloping restitution curves relating APD to the preceding DI (Nolasco and Dahlen, 1968). Furthermore, such steeply sloping restitution curves have been associated with arrhythmogenicity in humans with the Brugada syndrome (Narayan et al., 2007), another hereditary arrhythmic syndrome (Antzelevitch et al., 2003). Furthermore, similar associations have been made in murine genetic models of both the Brugada syndrome and the congenital long-QT syndrome type 3, as well as in hypokalemic murine hearts (Sabir et al., 2008a,b). In this study, however, neither the effect of RyR2-P2328S mutation, epinephrine nor propranolol were associated with changes in the slopes of restitution curves. Furthermore, the maximum slopes of restitution curves did not exceed unity, an established criterion for arrhythmogenesis (Nolasco and Dahlen, 1968), under any set of experimental conditions studied. Notably alternans has previously been reported in the absence of steeply sloping restitution curves (Pruvot et al., 2004; Goldhaber et al., 2005). In these cases alternations in [Ca2+]i has been suggested to affect both the inward current carried by the Na+/Ca2+ exchanger and L-type Ca2+ channel inactivation, thereby resulting in alternation in APD (Weiss et al., 2006). Such a mechanism has been suggested to underlie alternans in chronic heart failure, where hyperphosphorylation of RyR2 results in increased Ca2+ leak (Wehrens et al., 2005). Given that mutations underlying CPVT ultimately result in increased Ca2+ leak from the RyR2 (Lehnart et al., 2004), a similar mechanism may explain the alternans reported here.
We thus establish that genetically modified murine hearts modeling CPVT exhibit alternans and that this phenomenon is exacerbated by epinephrine suppressible by propranolol, fully reflecting clinical findings (Leenhardt et al., 1995; Wehrens et al., 2003). The presence of such alternans is not, however, associated with changes in APD, VERP or the slopes of restitution curves. CPVT has not previously been associated with consistent electrocardiographic features that might predict arrhythmic risk (Sumitomo et al., 2003). It is possible that the presence of alternans may represent a useful marker in patients suspected of harboring this highly lethal condition.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the James Baird Fund, the Frank Elmore Fund, the Medical Research Council, the Wellcome Trust and the British Heart Foundation for their generous assistance. We are also grateful to both Downing College and Murray Edwards College, Cambridge for their support.
References
Aizawa, Y., Komura, S., Okada, S., Chinushi, M., Aizawa, Y., Morita, H., and Ohe, T. (2006). Distinct U wave changes in patients with catecholaminergic polymorphic ventricular tachycardia (CPVT). Int. Heart J. 47, 381–389.
Antzelevitch, C., Burashnikov, A., and Di Diego, J. M. (2003). “Cellular and ionic mechanisms underlying arrhythmogenesis,” in Cardiac Repolarization: Bridging Basic and Clinical Science, eds I. Gussak, C. Antzelevitch, S. C. Hammill, and W. K. Shen (Totowa, NJ: Humana Press), 201–252.
Balasubramaniam, R., Grace, A. A., Saumarez, R. C., Vandenberg, J. I., and Huang, C. L. (2003). Electrogram prolongation and nifedipine-suppressible ventricular arrhythmias in mice following targeted disruption of KCNE1. J. Physiol. (Lond.) 552, 535–546.
Cerrone, M., Colombi, B., Santoro, M., di Barletta, M. R., Scelsi, M., Villani, L., Napolitano, C., and Priori, S. G. (2005). Bidirectional ventricular tachycardia and fibrillation elicited in a knock-in mouse model carrier of a mutation in the cardiac ryanodine receptor. Circ. Res. 96, e77–e82.
Fabritz, L., Kirchhof, P., Franz, M. R., Eckardt, L., Monnig, G., Milberg, P., Breithardt, G., and Haverkamp, W. (2003). Prolonged action potential durations, increased dispersion of repolarization, and polymorphic ventricular tachycardia in a mouse model of proarrhythmia. Basic Res. Cardiol. 98, 25–32.
Goddard, C., Ghais, N. S., Zhang, Y., Williams, A. J., Colledge, W. H., Grace, A. A., and Huang, C. L. (2008). Physiological consequences of the P2328S mutation in the ryanodine receptor (RyR2) gene in genetically modified murine hearts. Acta Physiol. (Oxf) 194, 123–140.
Gold, M. R., Bloomfield, D. M., Anderson, K. P., El-Sherif, N. E., Wilber, D. J., Groh, W. J., Estes, N. A., 3rd, Kaufman, E. S., Greenberg, M. L., and Rosenbaum, D. S. (2000). A comparison of T-wave alternans, signal averaged electrocardiography and programmed ventricular stimulation for arrhythmia risk stratification. J. Am. Coll. Cardiol. 36, 2247–2253.
Goldhaber, J. I., Xie, L. H., Duong, T., Motter, C., Khuu, K., and Weiss, J. N. (2005). Action potential duration restitution and alternans in rabbit ventricular myocytes: the key role of intracellular calcium cycling. Circ. Res. 96, 459–466.
Head, C. E., Balasubramaniam, R., Thomas, G., Goddard, C. A., Lei, M., Colledge, W. H., Grace, A. A., and Huang, C. L. (2005). Paced electrogram fractionation analysis of arrhythmogenic tendency in DeltaKPQ Scn5a mice. J. Cardiovasc. Electrophysiol. 16, 1329–1340.
Karagueuzian, H. S., Khan, S. S., Hong, K., Kobayashi, Y., Denton, T., Mandel, W. J., and Diamond, G. A. (1993). Action potential alternans and irregular dynamics in quinidine-intoxicated ventricular muscle cells. Implications for ventricular proarrhythmia. Circulation 87, 1661–1672.
Knollmann, B. C., Katchman, A. N., and Franz, M. R. (2001). Monophasic action potential recordings from intact mouse heart: validation, regional heterogeneity, and relation to refractoriness. J. Cardiovasc. Electrophysiol. 12, 1286–1294.
Koller, M. L., Riccio, M. L., and Gilmour, R. F., Jr. (1998). Dynamic restitution of action potential duration during electrical alternans and ventricular fibrillation. Am. J. Physiol. 275, H1635–H1642.
Koller, M. L., Riccio, M. L., and Gilmour, R. F., Jr. (2000). Effects of [K( + )](o) on electrical restitution and activation dynamics during ventricular fibrillation. Am. J. Physiol. Heart Circ. Physiol. 279, H2665–H2672.
Laitinen, P. J., Brown, K. M., Piippo, K., Swan, H., Devaney, J. M., Brahmbhatt, B., Donarum, E. A., Marino, M., Tiso, N., Viitasalo, M., Toivonen, L., Stephan, D. A., and Kontula, K. (2001). Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation 103, 485–490.
Lee, J. J., Kamjoo, K., Hough, D., Hwang, C., Fan, W., Fishbein, M. C., Bonometti, C., Ikeda, T., Karagueuzian, H. S., and Chen, P. S. (1996). Reentrant wave fronts in Wiggers’ stage II ventricular fibrillation. Characteristics and mechanisms of termination and spontaneous regeneration. Circ. Res. 78, 660–675.
Leenhardt, A., Lucet, V., Denjoy, I., Grau, F., Ngoc, D. D., and Coumel, P. (1995). Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation 91, 1512–1519.
Lehnart, S. E., Terrenoire, C., Reiken, S., Wehrens, X. H., Song, L. S., Tillman, E. J., Mancarella, S., Coromilas, J., Lederer, W. J., Kass, R. S., and Marks, A. R. (2006). Stabilization of cardiac ryanodine receptor prevents intracellular calcium leak and arrhythmias. Proc. Natl. Acad. Sci. U.S.A. 103, 7906–7910.
Lehnart, S. E., Wehrens, X. H., Laitinen, P. J., Reiken, S. R., Deng, S. X., Cheng, Z., Landry, D. W., Kontula, K., Swan, H., and Marks, A. R. (2004). Sudden death in familial polymorphic ventricular tachycardia associated with calcium release channel (ryanodine receptor) leak. Circulation 109, 3208–3214.
Marks, A. R., Priori, S., Memmi, M., Kontula, K., and Laitinen, P. J. (2002). Involvement of the cardiac ryanodine receptor/calcium release channel in catecholaminergic polymorphic ventricular tachycardia. J. Cell. Physiol. 190, 1–6.
Narayan, S. M., Kim, J., Tate, C., and Berman, B. J. (2007). Steep restitution of ventricular action potential duration and conduction slowing in human Brugada syndrome. Heart Rhythm 8, 1087–1089.
Nolasco, J. B., and Dahlen, R. W. (1968). A graphic method for the study of alternation in cardiac action potentials. J. Appl. Physiol. 25, 191–196.
Pak, H. N., Hong, S. J., Hwang, G. S., Lee, H. S., Park, S. W., Ahn, J. C., Moo Ro, Y., and Kim, Y. H. (2004). Spatial dispersion of action potential duration restitution kinetics is associated with induction of ventricular tachycardia/fibrillation in humans. J. Cardiovasc. Electrophysiol. 15, 1357–1363.
Postma, A. V., Denjoy, I., Kamblock, J., Alders, M., Lupoglazoff, J. M., Vaksmann, G., Dubosq-Bidot, L., Sebillon, P., Mannens, M. M., Guicheney, P., and Wilde, A. A. (2005). Catecholaminergic polymorphic ventricular tachycardia: RYR2 mutations, bradycardia, and follow up of the patients. J. Med. Genet. 42, 863–870.
Priori, S. G., Napolitano, C., Memmi, M., Colombi, B., Drago, F., Gasparini, M., DeSimone, L., Coltorti, F., Bloise, R., Keegan, R., Cruz Filho, F. E., Vignati, G., Benatar, A., and DeLogu, A. (2002). Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation 106, 69–74.
Priori, S. G., Napolitano, C., Tiso, N., Memmi, M., Vignati, G., Bloise, R., Sorrentino, V., and Danieli, G. A. (2001). Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 103, 196–200.
Pruvot, E. J., Katra, R. P., Rosenbaum, D. S., and Laurita, K. R. (2004). Role of calcium cycling versus restitution in the mechanism of repolarization alternans. Circ. Res. 94, 1083–1090.
Sabir, I. N., Fraser, J. A., Cass, T. R., Grace, A. A., and Huang, C. (2007). A quantitative analysis of the effect of cycle length on arrhythmogenicity in hypokalaemic Langendorff-perfused murine hearts. Pflugers Arch. 454, 925–936.
Sabir, I. N., Li, L. M., Jones, V. J., Goddard, C. A., Grace, A. A., and Huang, C. L. (2008a). Criteria for arrhythmogenicity in genetically-modified Langendorff-perfused murine hearts modelling the congenital long QT syndrome type 3 and the Brugada syndrome. Pflugers Arch. 455, 637–651.
Sabir, I. N., Li, L. M., Grace, A. A., and Huang, C. L. (2008b). Restitution analysis of alternans and its relationship to arrhythmogenicity in hypokalaemic Langendorff-perfused murine hearts. Pflugers Arch. 455, 653–666.
Saumarez, R. C., and Grace, A. A. (2000). Paced ventricular electrogram fractionation and sudden death in hypertrophic cardiomyopathy and other non-coronary heart diseases. Cardiovasc. Res. 47, 11–22.
Sumitomo, N., Harada, K., Nagashima, M., Yasuda, T., Nakamura, Y., Aragaki, Y., Saito, A., Kurosaki, K., Jouo, K., Koujiro, M., Konishi, S., Matsuoka, S., Oono, T., Hayakawa, S., Miura, M., Ushinohama, H., Shibata, T., and Niimura, I. (2003). Catecholaminergic polymorphic ventricular tachycardia: electrocardiographic characteristics and optimal therapeutic strategies to prevent sudden death. Heart 89, 66–70.
Taggart, P., Sutton, P., Chalabi, Z., Boyett, M. R., Simon, R., Elliott, D., and Gill, J. S. (2003). Effect of adrenergic stimulation on action potential duration restitution in humans. Circulation 107, 285–289.
Thomas, G., Killeen, M. J., Grace, A. A., and Huang, C. L. (2007a). Pharmacological separation of early afterdepolarizations from arrhythmogenic substrate in DeltaKPQ Scn5a murine hearts modelling human long QT 3 syndrome. Acta Physiol (Oxf). 192, 505–517.
Thomas, G., Killeen, M. J., Gurung, I. S., Hakim, P., Balasubramaniam, R. N., Goddard, C. A., Grace, A. A., and Huang, C. L. (2007b). Mechanisms of ventricular arrhythmogenesis in mice following targeted disruption of KCNE1 modelling long QT syndrome 5. J. Physiol. (Lond.) 578, 99–114.
Tosaka, T., Casimiro, M. C., Rong, Q., Tella, S., Oh, M., Katchman, A. N., Pezzullo, J. C., Pfeifer, K., and Ebert, S. N. (2003). Nicotine induces a long QT phenotype in Kcnq1-deficient mouse hearts. J. Pharmacol. Exp. Ther. 306, 980–987.
Walker, M. L., Wan, X., Kirsch, G. E., and Rosenbaum, D. S. (2003). Hysteresis effect implicates calcium cycling as a mechanism of repolarization alternans. Circulation 108, 2704–2709.
Wehrens, X. H., Lehnart, S. E., Huang, F., Vest, J. A., Reiken, S. R., Mohler, P. J., Sun, J., Guatimosim, S., Song, L. S., Rosemblit, N., D’Armiento, J. M., Napolitano, C., Memmi, M., Priori, S. G., Lederer, W. J., and Marks, A. R. (2003). FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell 113, 829–840.
Wehrens, X. H., Lehnart, S. E., and Marks, A. R. (2005). Intracellular calcium release and cardiac disease. Annu. Rev. Physiol. 67, 69–98.
Keywords: sudden cardiac death, Brugada syndrome, arrhythmia, monophasic action potential, alternans, catecholaminergic polymorphic ventricular tachycardia, restitution curve, ryanodine receptor
Citation: Sabir IN, Ma N, Jones VJ, Goddard CA, Zhang Y, Kalin A, Grace AA and Huang CL-H (2010) Alternans in genetically modified Langendorff-perfused murine hearts modeling catecholaminergic polymorphic ventricular tachycardia. Front. Physio. 1:126. doi: 10.3389/fphys.2010.00126
Received: 17 June 2010;
Paper pending published: 02 July 2010;
Accepted: 29 July 2010;
Published online: 28 October 2010.
Edited by:
Sander Verheule, Maastricht University, NetherlandsReviewed by:
Ruben Coronel, Academic Medical Center, Netherlands;Sander Verheule, Maastricht University, Netherlands
Copyright: © 2010 Sabir, Ma, Jones, Goddard, Zhang, Kalin, Grace and Huang. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Ian N. Sabir, Physiological Laboratory, Department of Physiology, Development and Neuroscience, University of Cambridge, Downing Street, Cambridge CB2 3EG, Cambridge, UK. e-mail:aW5zMjBAY2FtLmFjLnVr