


- Department of Pharmacology and Toxicology, Michigan State University, East Lansing, MI, USA
Norepinephrine (NE) is thought to mediate its effects through G-protein coupled receptors. However, previous studies have shown that NE and another primary amine, serotonin, also have the ability to exert effects in a receptor-independent manner. We hypothesized that the enzyme transglutaminase II (TG II) has the ability to modify proteins with NE and that this modification is physiologically relevant. As our model we used rat aortic and vena cava tissues, two tissues that depend on NE to modulate vascular tone. Immunohistochemical and immunocytochemical staining showed that NE and TG II are present in smooth muscle cells of these tissues. Western analysis shows aorta and vena cava homogenate proteins are recognized by an antibody raised against NE conjugated to bovine serum albumin (NE-BSA). NE and α-actin colocalize in cultured aorta and vena cava smooth muscle cells. Freshly dissociated smooth muscle cells from these vessels were able to take up NE-biotin. In isolated tissue baths, inhibition of TG II with cystamine (0.5 mM) completely abolished NE-induced contraction in the aorta but only attenuated the receptor-independent contractant KCl (max contraction to 100 mM KCl in cystamine treated = 88.8 ± 7.0% of vehicle treated, p < 0.05). In the vena cava, contraction to NE was abolished with 0.1 mM cystamine and KCl contraction was attenuated (max contraction to 100 mM KCl in cystamine treated = 54.8 ± 7.0% of vehicle treated, p < 0.05). Taken together, these results show that vascular smooth muscle cells take up and utilize NE for the modification of proteins, and that this modification may play an important role in vascular contraction.
Introduction
Norepinephrine (NE), a member of the class of primary amines known as catecholamines, has a number of different roles throughout the body. One of these functions is as a mediator of vascular tone through its actions as a neurotransmitter of the sympathetic nervous system. The physiological responses of NE have been attributed to its actions on adrenergic receptors, a class of G-protein coupled receptors that are the primary targets for the two catecholamines NE and epinephrine. Adrenergic receptors were first divided into two main groups based on their excitatory (α-adrenergic receptors) or inhibitory (β-adrenergic receptors) effects in the vasculature (Ahlquist, 1948). The vasoconstrictive effects of the catecholamines NE and epinephrine are mediated through their interactions with α-adrenergic receptors, while their vasodilatory effects are mediated through their interactions with β-adrenergic receptors (Ahlquist, 1948).
While many of the effects of primary amines are mediated through their receptors, there have been reports where their effects cannot be explained by receptor activation. This includes the inability of adrenergic and serotonergic antagonists to block NE- and serotonin-induced reduction of endothelial cell barrier function (Bottaro et al., 1986). In addition to this, serotonin receptor antagonists do not affect serotonin-induced proliferation (Guilluy et al., 2009) and hypertrophy (Lee et al., 1994; Eddahibi et al., 2001) of bovine pulmonary smooth muscle cells.
Recently, a novel, receptor-independent mode for signal transduction of amines has been described. Serotonylation is the covalent linkage of serotonin to a protein through a glutamyl-amide bond (Figure 1). This reaction is catalyzed by the enzyme transglutaminase II (TG II), an enzyme that also covalently links proteins through a ε-(γ-glutamyl)lysine bond. This modification was first shown to occur within the platelet, where small GTPases become serotonylated, constitutively activating them and leading to the internalization of the serotonin reuptake transporter (Walther et al., 2003). Subsequently, serotonylation of proteins has been shown to occur on the small GTPases Rab3a and Rab27a in the pancreatic beta cell, rendering them constitutively active and facilitating insulin secretion (Paulmann et al., 2009). Serotonylation also occurs to proteins of vascular smooth muscle cells, where it has been shown to accelerate RhoA degradation (Guilluy et al., 2007) and modify aortic contraction (Watts et al., 2009).
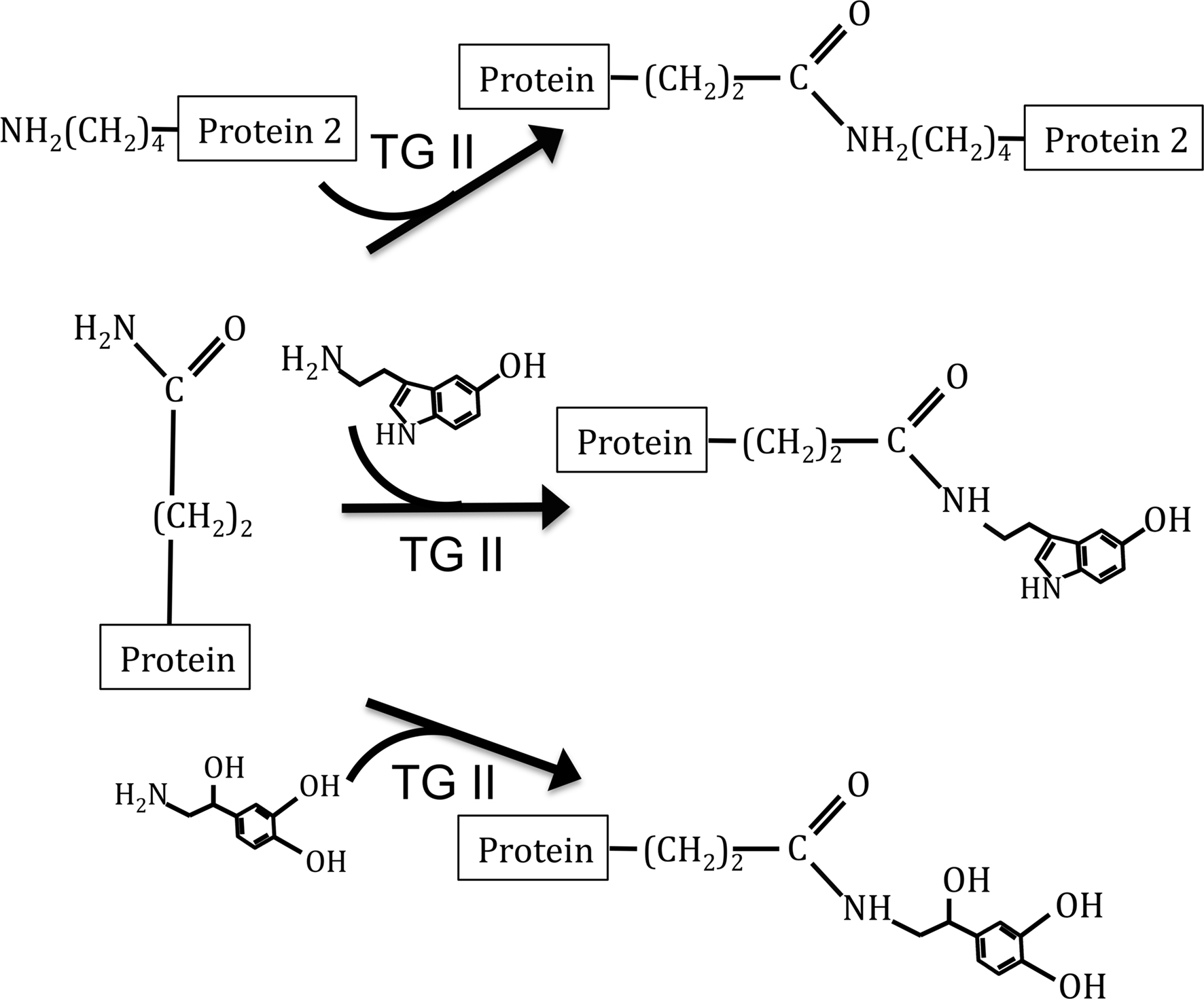
Figure 1. Depiction of TG II catalyzed reactions. Top reaction depicts protein cross-linking reaction of TG II. The middle reaction depicts the addition of serotonin to proteins, a modification known as serotonylation. Bottom reaction depicts our hypothesized modification, the covalent linkage of NE to proteins.
We hypothesized that TG II utilizes the primary amine NE as a substrate for protein modification (Figure 1) and that this modification has physiological significance in the vasculature. As our model, we chose aortic and vena cava tissues from the rat, two tissues in which depend on NE via the sympathetic nervous system to control vascular tone. In order to follow the addition of NE to proteins, we utilized a biotin-conjugated NE (NE-biotin) in several of our experiments.
Materials and Methods
Animal Use
Male Sprague–Dawley rats (Charles River Laboratories, Inc., Portage, MI, USA) were used. Rats were anesthetized with pentobarbital (60 mg kg−1, i.p.) prior to removal of tissues. Procedures were performed in accordance of Michigan State University with the approval of the Institutional Animal Use and Care Committee.
Immunohistochemistry
Paraffin-embedded tissue sections were dewaxed, unmasked and treated as previously described (Ni et al., 2008). Primary antibodies used were: anti-TG II (1:1000; mouse monoclonal TG100, LabVision, Fremont CA, USA) and anti-NE-bovine serum albumin (NE-BSA) (1:500; rabbit polyclonal, ab8887, Abcam, Cambridge, MA, USA). Primary antibodies were left out of some experiments and tissues were developed in the presence of only secondary antibody.
Dissociation of Vascular Smooth Muscle Cells
Thoracic aorta and vena cava tissues were isolated, dissected and cleaned as previously described (Thakali et al., 2008). Cells were resuspended in Dulbecco’s phosphate buffered saline (DPBS; 2.57 mM KCl, 1.47 mM KH2PO4, 137.93 mM NaCl, 8.06 mM Na2HPO4-7H2O, pH 7.2) plus sodium nitroprusside (872 nM). Cells were made to adhere to poly-lysine (50 μ g/mL) coated coverslips using the Shandon Cytospin 4 Centrifuge (Thermo Scientific, Waltham, MA, USA).
Cell Culture
Aortic and vena cava smooth muscle cells were derived from explants of the thoracic aorta or vena cava. Cells were fed with DMEM supplemented with 10% fetal bovine serum and 1% penicillin/streptomyocin. Cells were plated to cover slips for immunocytochemical experiments. Aortic cells used were between passages 2 and 6; vena cava cells used were from passages 2 or 3. All explants stained positive for α-actin, smooth muscle cell specific (1:1000; mouse monoclonal, FITC conjugate, F3777, Sigma, St. Louis, MO, USA).
Immunocytochemistry
For experiments using NE-biotin, cells were equilibrated in physiological salt solution (PSS: 103 mM NaCl; 4.7 mM KCl; 1.13 mM KH2PO4; 1.17 mM MgSO4-7H20; 1.6 mM CaCl2-2H2O; 14.9 mM NaHCO3; 5.5 mM dextrose and 0.03 mM CaNa2 EDTA) plus the monoamine oxidase inhibitor pargyline (10 μ M) for 30 min and then incubated with either NE-biotin (12.7 μ M, IBL, Hamburg, Germany) or vehicle (N,N-dimethylformamide) for 1 h. When analyzed by NMR, NE-biotin was found to be a mixture of species, with the biotin attached to the primary amine and hydroxyl groups. The ratio of these species was unable to be determined. Thus, the concentration of NE-biotin used is the maximum possible. This is reasonable, as NE-biotin was used to track the location of NE qualitatively, and not quantitatively. Freshly dissociated smooth muscle cells were then rinsed in PBS and fixed in Zamboni’s fixative for 20 min, followed by permeabilization by incubating in 0.1% Trition X-100 for 20 min. Cultured smooth muscle cells were rinsed and then fixed with acetone for 1 min. Cells not used in NE-biotin experiments were immediately fixed. Primary antibodies used were anti-NE-BSA (1:500; rabbit polyclonal, ab8887, Abcam, Cambridge, MA, USA), anti-TG II (1:1000; rabbit polyclonal, ab421, Abcam, Cambridge, MA, USA), and anti-α-actin (1:1000; mouse monoclonal, FITC conjugate, F3777, Sigma, St. Louis, MO, USA) in PBS. Secondary antibodies used were Cy3-conjugated Affini Pure goat anti-rabbit (1:1000; IgG, Jackson ImmunoResearch Laboratories Inc., West Grove, PA, USA) and Rhodamine Red-X-conjugated Streptavidin (1:1000; Jackson ImmunoResearch Laboratories Inc., West Grove, PA, USA). After incubation in secondary, cover slips were blotted dry and mounted on slides using Prolong Gold medium with DAPI (Invitrogen, Carlsbad, CA, USA). Slides were viewed and photographed on a Nikon Eclipse Ti-S microscope using NIS Elements BR 3.00 software. The light source was a Nikon intensilight C-HGFI, and the camera a Nikon Digital Sight DS-Qi1. Slides were viewed under a 60× Nikon Plan Apo oil-immersion objective using non-drying immersion oil, type A. Three Nikon filters were used: UV-2EC 96310 (lot number: C109990), HYQ TRITC 96321 (lot number: C112237) and HYQ FITC 96320 (lot number: C111294). Excitation ranges were: UV-2EC 340-380 nm, HYQ TRITC 528-552 nm, and HYQ FITC 450-490 nm. Emission ranges were: UV-2EC 435–485, HYQ TRITC 580–632, and HYQ FITC 515–555. Images were unaltered when combined into the overlay image. Analysis of the images was done in ImageJ (Rasband, 1997–2009), using the JACoP plugin (Bolte and Cordelieres, 2006) to find Van Steensel’s cross-correlation functions (CCF) with a pixel shift of δ = ± 20. Van Steensel’s CCFs are formed by shifting the image in one channel in the x-direction pixel by pixel relative to the second channel and calculating a Pearson’s coefficient for each shift. The Pearson’s coefficients are then plotted as the function of pixel shifts (δx). Colocalization events are indicated by a peak at δ = 0, while partial colocalization will shift the peak slightly to the left or right. Noise and differing intensities between the two channels will reduce the maximum peak of the CCF from one and also increase the width of the peak at half maximum.
Western Analysis
Protein isolation and western blotting procedures were performed as previously described using standard SDS-PAGE conditions and transferring proteins to either nitrocellulose (for TG II) or PVDF-FL (for NE) membranes (Ni et al., 2008). Primary antibodies used were anti-NE-BSA (1:500; rabbit polyclonal, ab8887, Abcam, Cambridge, MA, USA), anti-TG II (1:1000; mouse monoclonal TG100, LabVision, Fremont, CA, USA) and anti-β-actin (1:1000; mouse monoclonal, A2228, Sigma, St. Louis, MO, USA). Secondaries used were IR dye 680 donkey anti-mouse (1:1000; Li-Cor Biosciences, Lincoln, NE, USA) and IR dye 800 goat anti-Rabbit (1:1000; Li-Cor Biosciences, Lincoln, NE, USA) for the blot probed with anti-NE-BSA antibody, and sheep peroxidase-linked anti-mouse (1:2000; GE Healthcare, Piscataway, NJ, USA) for the TG II blot. The blot probed with the anti-NE-BSA antibody was blocked using Odyssey blocking buffer (Li-Cor Biosciences, Lincoln, NE, USA) and was analyzed using the Odyssey infrared imaging system (Li-Cor Biosciences, Lincoln, NE, USA). The blot probed with the anti-TG II antibody was blocked in 4% w/v chick egg ovalbumin and placed in ECL (Amersham Life Sciences, Arlington Heights, IL, USA) prior to exposure to film. The film was scanned and placed within the figure without modification using Adobe Photoshop. Densitometric analysis was done using ImageJ.
Isometric Contraction
Endothelial cell-intact rings of aorta and vena cava tissues were hung in tissue baths for isometric tension recordings using Grass transducers and PowerLab data Acquisitions (Colorado Springs, CO, USA). Rings were pulled to optimum resting tension (4000 mg aorta, 1000 mg vena cava), and equilibrated for 1 h, with washing, prior to exposure to compounds. Tissue baths contained 37°C, aerated (95% O2/CO2) PSS. To test viability, aortic rings were treated with an initial concentration of 10 μ M phenylephrine (PE), an α1-adrenergic receptor agonist. Since the vena cava does not contract to PE, vena cava rings were treated with an initial concentration of 10 μ M NE. All tissues had an intact endothelial layer, evidenced by a robust (>50%) relaxation to acetylcholine (1 μ M) in tissues contracted to half-maximum with PE (aorta) or contracted to 10 μ M PGF2α (vena cava). Tissues were incubated with vehicle (water) or the TG II inhibitor cystamine (0.01–0.1 mM) for 30–60 min prior to cumulative addition of NE (10−9−10−5 M). Tissues were then washed, incubated in the same concentration of cystamine or vehicle for 30–60 min prior to the cumulative addition of the non-receptor mediated contractant potassium chloride (KCl, 6–100 mM). Data are reported as a percentage of the initial contraction to PE (aorta) or NE (vena cava).
Results
TG II and NE are Present in Smooth Muscle Cells from Aorta and Vena Cava Tissues
Immunohistochemical staining of NE in rat aortic and vena cava tissues shows positive staining in all layers of these two tissues (Figure 2A). TG II staining, in contrast, appears to be localized to the smooth muscle layers of the media in both aortic and vena cava tissues (Figure 2B). Immunocytochemical staining of freshly dissociated smooth muscle cells, cells that mediate vascular contraction, confirmed TG II was present in this cell type for both the aorta (Figure 2C, left) and the vena cava (Figure 2C, right), as was NE (Figure 2D, aorta left, vena cava right). In the vena cava, cell types besides smooth muscle cells also stained for NE (Figure 2D, right). For both aortic and venous smooth muscle cells, the image overlays show colocalization of α-actin with TG II (Figure 2C) and NE (Figure 2D).
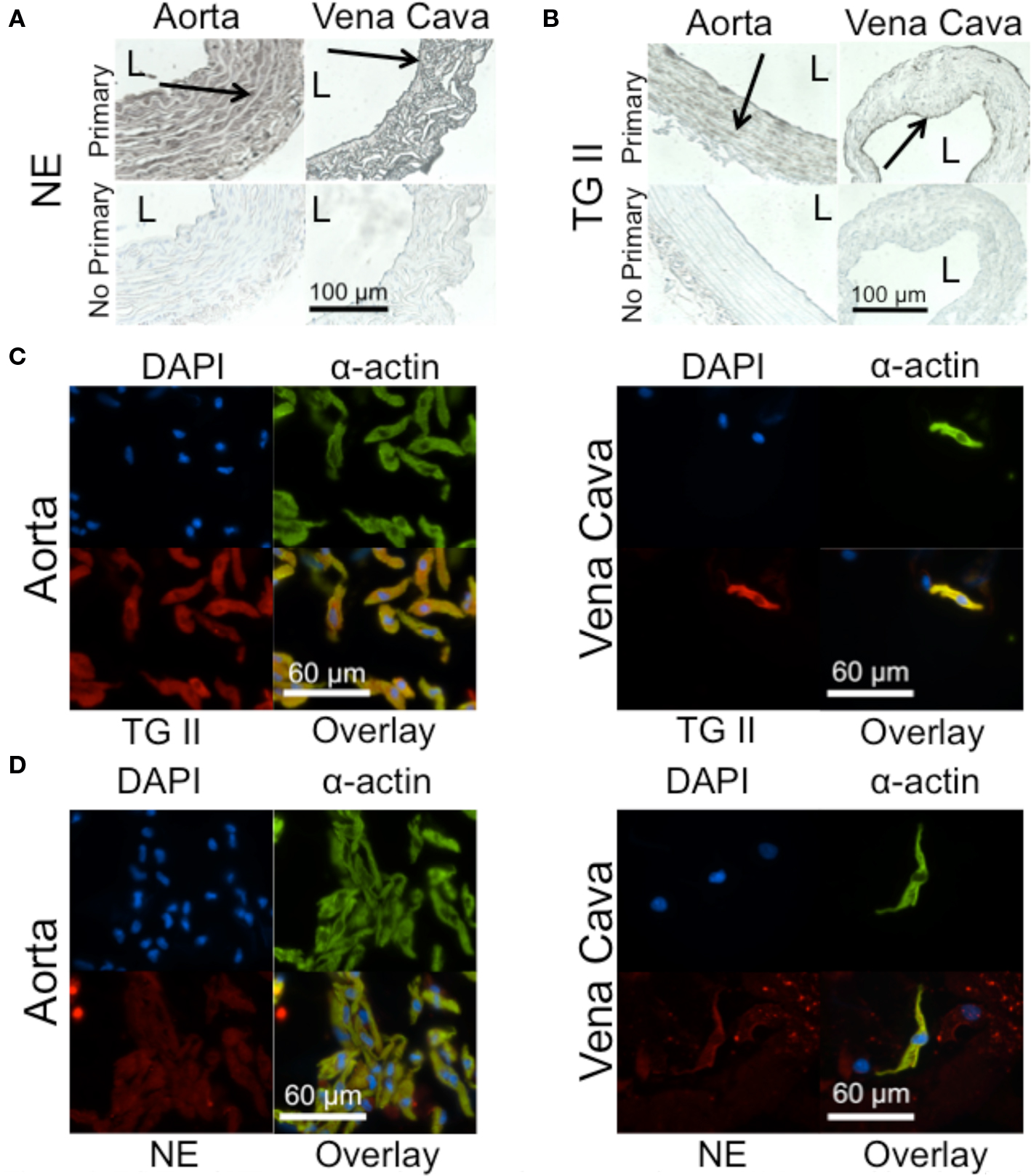
Figure 2. TG II and NE are present in aorta and vena cava tissues. (A) Immunohistochemical staining of NE in a rat aorta (left) and vena cava (right). L = lumen. Arrows point to areas of staining. (B) Immunohistochemical staining of TG II in the rat aorta (left) and vena cava (right). (C) Immunocytochemical staining of TG II (red) in freshly dissociated smooth muscle cells of the aorta (left) and vena cava (right). Smooth muscle cells were identified with a smooth muscle cell-specific α-actin antibody (green). Nuclei (blue) were identified using 4’,6-diamidino-2-phenylindole (DAPI). (D) Immunocytochemical staining of NE (red) in freshly dissociated smooth muscle cells of the aorta (left) and vena cava (right). Images are representative of tissues taken from four to five different rats.
NE is Attached to Proteins of the Vasculature
Western analysis of aorta and vena cava homogenates showed protein bands that were recognized by an antibody raised against NE-BSA (Figure 3A). Because this antibody was made against NE that had been conjugated to a protein, it will preferentially bind to proteins that have been covalently modified by NE. Because these proteins were from tissues not exposed to exogenous NE after being removed from the animal, they reflect a physiological state of protein amidation, supporting our hypothesis that proteins are naturally amidated by NE. β-actin was probed for to ensure equal loading of protein. Using ImageJ, densitometric analysis of the blot shows the differences between aorta and vena cava band intensities (Figure 3B). Values for band intensities were normalized to β-actin.
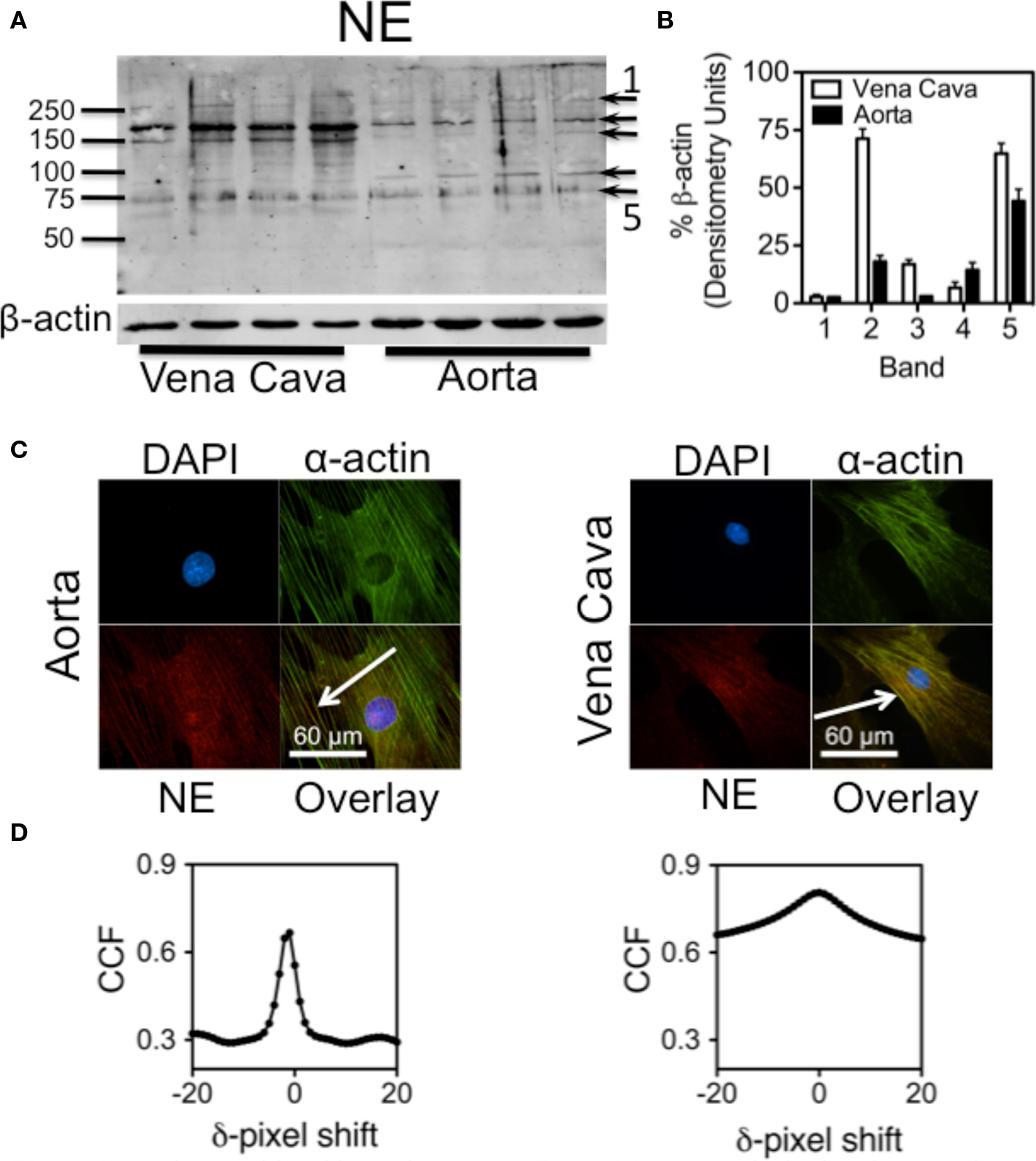
Figure 3. Proteins amidated by endogenous NE. (A) Western analysis showing proteins recognized by an anti-NE-BSA antibody in rat aorta and vena cava homogenates not exposed to exogenous NE. (B) Densitometry of the five darkest bands, (arrows in A) from the Western analysis. Each band was normalized to β-actin. (C) Images of cultured smooth muscle cells from rat aorta (left) and vena cava (right). Cells were stained for nuclei (DAPI, blue, top left), α-actin (green, top right) and NE (red. bottom left). Overlay of the three images is bottom right. Colocalization is indicated by the yellow color. Arrows point to areas of colocalization. Images are representative of cells cultured from four different rats. (D) Van Steensel’s cross-correlation functions of α-actin and NE overlays. Peaks at or near δ = 0 indicate a colocalization event. Graphs were made using the images above them, and are representative of graphs made of images from the cells of four different rats.
Because freshly dissociated cells tended to be compacted (Figure 2D), cultured aortic and vena cava smooth muscle cells were used next to better determine subcellular localization of NE. Immunocytochemical staining with the anti-NE-BSA antibody showed that endogenous NE colocalized with α-actin fibers in cultured smooth muscle cells from aorta (Figure 3C, left) and vena cava (Figure 3C, right) tissues. Analysis of these images using Van Steensel’s CCF points toward colocalization of NE with α-actin (Figure 3D; peaks of Van Steensel cross-correlation function peaks of Figure 3B: aorta = 0.666 at δ = -1, vena cava = 0.805 at δ = 0). The CCF peak at zero in the vena cava explant indicates complete colocalization, while the CCF peak slightly aside from zero (at δ = −1) in the aortic explants is indicative of partial colocalization. The reduction of peaks from a CCF = 1 may indicate either noise in the channels, different intensities between the two channels, or both. However, it is still possible to identify a colocalization event.
NE-Biotin is Taken Up by Vascular Cells
To differentiate between NE that was already present, taken up or made inside the cell and to confirm that exogenous NE could be taken up, cells were treated with NE-biotin (12.7 μ M) for 30 min. Immunocytochemical staining with Rhodamine-Red-X-conjugated streptavidin showed NE-biotin was taken up by smooth muscle cells of the aorta (Figure 4, left) and, in the vena cava, by smooth muscle cells as well as by other non-identified vascular cells (Figure 4, right). This is consistent with endogenous NE staining in both vessel types (Figure 2D). Once taken up, NE-biotin was largely cytosolic. The overlays show colocalization of α-actin with NE-biotin, which suggests that NE-biotin is being covalently attached to α-actin.
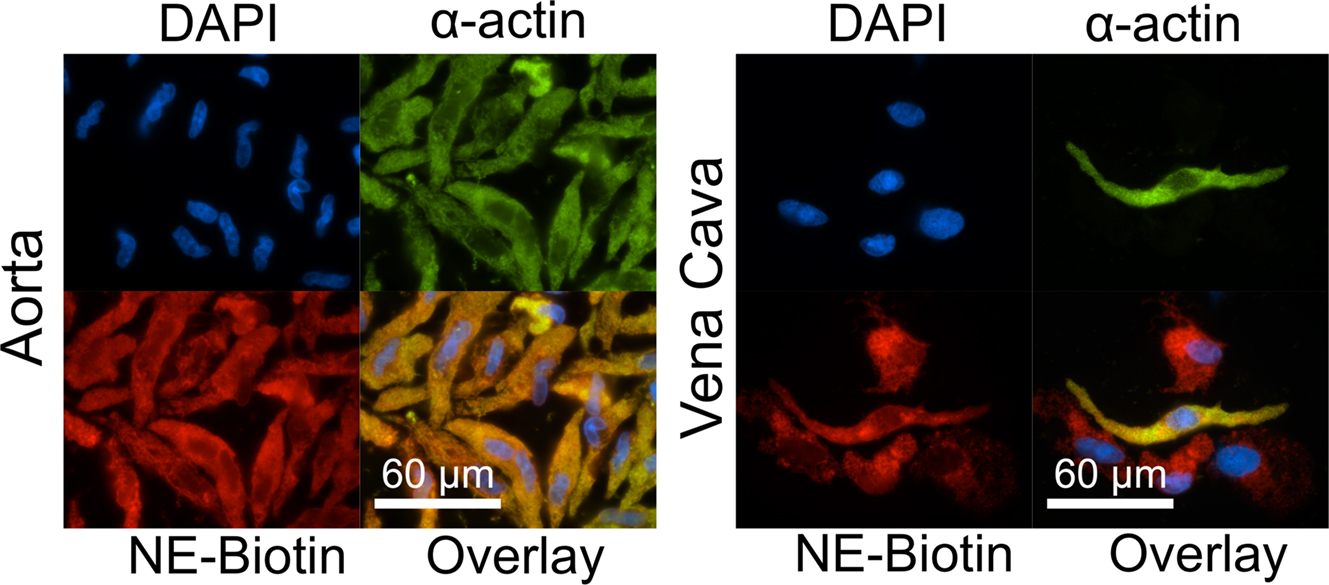
Figure 4. Aorta and vena cava tissues can take up and attach NE-biotin to proteins. Freshly dissociated smooth muscle cells of the aorta (left) and vena cava (right) were incubated with either 12.7 μ M NE-biotin or vehicle (not shown) for 30 min. Staining with a Rhodamine-Red-X conjugated streptavidin (red, bottom left) showed that NE-biotin entered the smooth muscle cells (indicated by α-actin staining, green, top right), and, in the vena cava, other vascular cells (red staining, bottom right of the vena cava). Nuclei are indicated by DAPI staining. Images are representative of tissues from four different rats.
Inhibition of Transglutaminase II Activity Abolishes NE-Induced Contraction
The physiological importance of NE-amidation of proteins is illustrated by the abolishment of NE-induced contraction in the presence of cystamine, a TG II inhibitor. Cystamine abolished NE-induced contraction in a concentration-dependent manner in both the aorta (Figure 5A) and vena cava (Figure 5B). Vena cava tissues proved to be more sensitive to cystamine inhibition, as concentrations of cystamine (0.1 mM) that merely attenuated NE-induced contraction in the aorta (max contraction to NE = 67.6 ± 3.9 % of vehicle) completely abolished contraction in the vena cava (Figure 5E). This difference cannot be explained by the amount of TG II present in each of the tissues, as the TG II protein content of aorta and vena cava homogenates did not differ significantly (Figure 5F). In contrast to NE-induced contraction, KCl-induced contraction, a receptor-independent contraction, was only modestly reduced by concentrations of cystamine that abolished NE-induced contraction in both the aorta (Figure 5C; max contraction to 100 mM KCl in 0.5 mM cystamine treated = 88.8 ± 7.5% of vehicle treated) and the vena cava (Figure 5D; max contraction to 100 mM KCl in 0.1 mM cystamine treated = 54.8 ± 21.2% of vehicle treated).
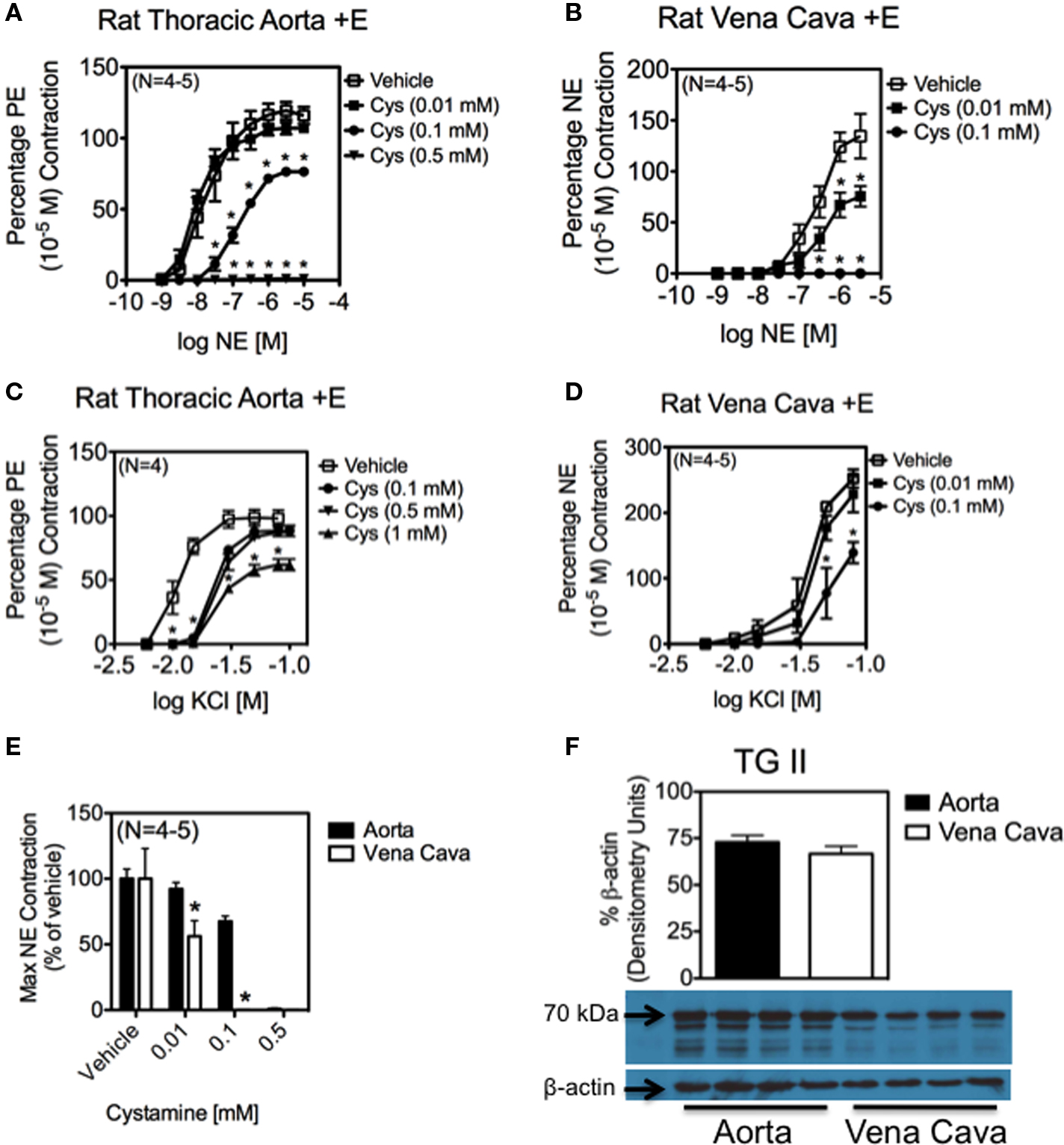
Figure 5. Inhibition of TG II abolishes contraction to NE in aortic and vena cava tissues. (A–D) Effect of vehicle (open symbol) and cystamine (0.01–1 mM) on contraction in the vena cava and aorta. NE-induced contraction was eventually abolished in the aorta (A) and vena cava (B). Contraction to KCI was attenuated, but not abolished in both the aorta (C) and vena cava (D). Cys = Cystamine * indicates statistical difference (p <0.05) from vehicle-treated values. Points and vertical lines represent means ± SEM for the number of animals in parentheses. (E) NE-induced vena cava contraction was more sensitive than aortic contraction to TG II inhibition. (F) Densitometry of a Western analysis demonstrating the presence of TG II (70 kDa) in rat aorta and vena cava homogenates. TG II was normalized to β-actin.
Discussion
The importance of NE in vascular physiology is well established. Both arteries and veins are innervated by the sympathetic nervous system, and NE is critical for maintaining vascular tone. The current model of NE action is dependent on its interaction with an adrenergic receptor. However, other primary amines, such as serotonin, elicit biological effects that are independent of receptor binding (Eddahibi et al., 2001; Guilluy et al., 2009). Since TG II can recognize and utilize serotonin as a substrate, other primary amines such as NE can also be candidates for TG II-mediated amidation events. The present work suggests that this is indeed the case, and presents a novel mechanism through which NE elicits a biological response in the vasculature. The importance of this work is exemplified in the alteration of sympathetic nervous system activity in several models of hypertension (Luo et al., 2003; King et al., 2008), and in that sympathetic nervous system activity is commonly increased in human essential hypertension (Guyenet, 2006).
TG II and Smooth Muscle Cells
Other studies have shown that TG II is present in the aortic smooth muscle cells (Lai et al., 2007) and the present work confirms this. Our study shows that TGII is also present in venous smooth muscle cells (Figure 2C), which is a novel finding. Immunohistochemical staining showed that NE was present in virtually all layers of both the aorta and vena cava (Figure 2A). TG II, on the other hand, was primarily localized to the smooth muscle layers of these tissues (Figure 2B). Immunocytochemical staining confirmed that NE and TG II both exist in smooth muscle cells of the vasculature, and that NE was present in additional, unidentified cell types (Figures 2C,D), that probably correspond to the non-smooth muscle cell layers of aorta and vena cava that stained positive for NE in Figure 2A. This puts the tools for protein amidation to NE together in vascular smooth cells, making these cells likely candidates in which this process can occur. The possibility of smooth muscle cells to take up NE was demonstrated in their ability to incorporate NE-biotin into the cell when incubated with it (Figure 4).
Although staining of NE was observed in freshly dissociated smooth muscle cells, its exact cellular localization was difficult to visualize due to the compacted nature of these cells. To overcome this, cultured smooth muscle cells, which are more diffuse, were used. Immunocytochemical staining showed that while NE existed throughout the cell, it had a high correlation with α-actin fibers (Figure 3C). Colocalization was assessed using the JACoP plugin in ImageJ. Van Steensel’s cross-correlation function showed a peak near or at zero, suggesting a colocalization event had occurred. This was similar to what we had reported earlier with serotonin (Watts et al., 2009). NE’s colocalization with α-actin fibers is significant because isometric contraction of vascular smooth muscle cells is an α-actin dependent process (Ogden et al., 2006).
Physiological Relevance
To show that protein amidation to NE is physiologically relevant, it is necessary for protein amidation to occur naturally and modify biological activity. Protein bands of rat aorta and vena cava homogenates not exposed to either the monoamine oxidase inhibitor pargyline or exogenous NE were recognized by an antibody raised against NE, suggesting NE amidation to proteins naturally occurs (Figure 3A). Biological function of this process was assayed by measuring isometric contraction to NE in the presence and absence of cystamine, which inhibits TG II by acting as a competitive substrate (Figures 5A,B). NE-induced contraction was abolished by cystamine while contraction to the receptor-independent contractile agent KCl was only slightly reduced (Figures 5C,D). KCl-induced contraction was important in showing that the tissues were viable at the concentration of cystamine used. KCl-treatment stimulates contraction by depolarizing the cell, resulting in Ca2+ influx through l-type calcium channels. Our data suggest that TG II function is critical for vascular contraction to NE; this may be through its ability to covalently attach NE to proteins. While it is possible that TG II is playing a more global role in influencing vascular contraction given that cystamine reduced KCl-induced contraction at higher concentrations, it should be noted that part of the KCl-induced contraction may be due to the NE that is present in sympathetic nerve terminals that are intrinsically a part of the isolated vessels. When KCl is added, these cells would also depolarize, resulting in the release of NE which then can be taken up and used as a substrate for TG II by the smooth muscle cells.
Another way that TG II is capable of influencing contraction is through its ability to act as a G protein, denoted as Gh (Griffin et al., 2002). This is especially likely with NE, as Gh couples to both the α1B and α1D -adrenergic receptors (Chen et al., 1996), two receptors that play a significant role in vascular contraction (David et al., 2010). This could also partially explain why NE-induced contraction in the aorta was abolished at a lower concentration than that used to abolish serotonin-induced contraction (Watts et al., 2009).
It is worth noting that vena cava tissues were more sensitive to TG II inhibition by cystamine than aortic tissues (Figure 5E). There are several ways that TG II could be acting in these two tissues to explain this difference. It may be that protein amidation is more important to contraction in the vena cava than in the aorta. This is supported by Western analysis showing that a blot probed with an anti-NE-BSA antibody (Figure 3A) showed much darker staining of vena cava protein bands of the same molecular weight than aorta, even when corrected for β-actin (Figure 3B). It is also possible that a larger percentage of the adrenergic receptors couple to Gh in the vena cava than in the aorta, and this in turn results in the greater sensitivity observed. Another explanation is that the difference in banding patterns seen in aorta and vena cava homogenates probed with an antibody against NE could mean that TG II is regulating different proteins in the two tissues. Finally, though Western analysis showed that aorta and vena cava homogenates had the same amount of TG II present when equal amounts of protein from these two tissues were analyzed (Figure 5F), this does not take into account that TG II was found primarily in the smooth muscle cell layer of these tissues. Because aorta tissues contain many layers smooth muscle cells while the vena cava only contains a single smooth muscle cell layer, homogenates from the aorta would contain more protein from smooth muscle cells than from other vascular cell types, while vena cava homogenates would contain more protein from other vascular cell types than from smooth muscle cells. This could indicate that vena cava vascular smooth muscle cells contain more TG II than aorta smooth muscle cells.
Limitations
There are a number of limitations that should be recognized in this study. First, the aorta and vena cava are large conduit vessels. As such, it is not possible to directly relate how this process may influence total peripheral resistance, which is largely controlled by smaller resistance arteries. A second limitation is in the use of cultured cells in this study, although this was mostly avoided by carrying out all experiments done in cultured cells in freshly dissociated smooth muscle cells as well. The study also focused on α-actin as a candidate for amidation. This was despite only a limited detection of protein bands at a molecular weight (around 42 kDa) of actins in our blot probed with an antibody raised against NE (Figure 3A). Identification of the proteins that are amidated by NE will lead to candidates that may be more likely substrates than actin for this event. These proteins may physically interact with α-actin, which would explain why we saw α-actin and NE colocalize (Figure 3C). Another issue is the apparent irreversibility of protein amidation carried out by TG II. This leads to the question of how protein amidation to NE is regulated. It may be that this modification is a terminal event for a protein.
In summary, this work is the first to show NE being used as a substrate by TG II to modify proteins. These studies suggest that protein amidation by NE occurs naturally, and is a biologically relevant process that is important for vascular contraction on both sides of the vasculature.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Bolte, S., and Cordelieres, F. P. (2006). A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224, 213–232.
Bottaro, D., Shepro, D., Peterson, S., and Hechtman, H. B. (1986). Serotonin, norepinephrine, and histamine mediation of endothelial cell barrier function in vitro. J. Cell. Physiol. 128, 189–194.
Bylund, D. B., Bond, R. A., Eikenburg, D. C., Paul Hieble, J., Hills, R., Minneman, K. P., and Parra, S. (2010). Adrenoceptors. IUPHAR database (IUPHAR-DB). Accessed on August 18, 2010 from http://www.iuphar-db.org/DATABASE/FamilyMenuForward?familyId=4 (Last modified on July 21, 2010).
Chen, S., Lin, F., Iismaa, S., Lee, K. N., Birckbichler, P. J., and Graham, R. M. (1996). Alpha1-adrenergic receptor signaling via Gh is subtype specific and independent of its transglutaminase activity. J. Biol. Chem. 271, 32385–32391.
Eddahibi, S., Humbert, M., Fadel, E., Raffestin, B., Darmon, M., Capron, F., Simonneau, G., Dartevelle, P., Hamon, M., and Adnot, S. (2001). Serotonin transporter overexpression is responsible for pulmonary artery smooth muscle hyperplasia in primary pulmonary hypertension. J. Clin. Invest. 108, 1141–1150.
Griffin, M., Casadio, R., and Bergamini, C. M. (2002). Transglutaminases: nature’s biological glues. Biochem. J. 368, 377–396.
Guilluy, C., Eddahibi, S., Agard, C., Guignabert, C., Izikki, M., Tu, L., Savale, L., Humbert, M., Fadel, E., Adnot, S., Loirand, G., and Pacaud, P. (2009). RhoA and Rho kinase activation in human pulmonary hypertension: role of 5-HT signaling. Am. J. Respir. Crit. Care Med. 179, 1151–1158.
Guilluy, C., Rolli-Derkinderen, M., Tharaux, P. L., Melino, G., Pacaud, P., and Loirand, G. (2007). Transglutaminase-dependent RhoA activation and depletion by serotonin in vascular smooth muscle cells. J. Biol. Chem. 282, 2918–2928.
King, A. J., Novotny, M., Swain, G. M., and Fink, G. D. (2008). Whole body norepinephrine kinetics in ANG II-salt hypertension in the rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294, R1262–R1267.
Lai, T. S., Liu, Y., Li, W., and Greenberg, C. S. (2007). Identification of two GTP-independent alternatively spliced forms of tissue transglutaminase in human leukocytes, vascular smooth muscle, and endothelial cells. FASEB J. 21, 4131–4143.
Lee, S. L., Wang, W. W., Lanzillo, J. J., and Fanburg, B. L. (1994). Serotonin produces both hyperplasia and hypertrophy of bovine pulmonary artery smooth muscle cells in culture. Am. J. Physiol. 266, L46–L52.
Luo, M., Hess, M. C., Fink, G. D., Olson, L. K., Rogers, J., Kreulen, D. L., Dai, X., and Galligan, J. J. (2003). Differential alterations in sympathetic neurotransmission in mesenteric arteries and veins in DOCA-salt hypertensive rats. Auton Neurosci 104, 47–57.
Ni, W., Geddes, T. J., Priestley, J. R., Szasz, T., Kuhn, D. M., and Watts, S. W. (2008). The existence of a local 5-hydroxytryptaminergic system in peripheral arteries. Br. J. Pharmacol. 154, 663–674.
Ogden, K., Thompson, J. M., Hickner, Z., Huang, T., Tang, D. D., and Watts, S. W. (2006). A new signaling paradigm for serotonin: use of Crk-associated substrate in arterial contraction. Am. J. Physiol. Heart Circ. Physiol. 291, H2857–H2863.
Paulmann, N., Grohmann, M., Voigt, J. P., Bert, B., Vowinckel, J., Bader, M., Skelin, M., Jevsek, M., Fink, H., Rupnik, M., and Walther, D. J. (2009). Intracellular serotonin modulates insulin secretion from pancreatic beta-cells by protein serotonylation. PLoS Biol. 7, e1000229. doi: 10.1371/journal.pbio.1000229.
Rasband, W. S. (1997–2009). ImageJ. Bethesda, MD: U. S. National Institutes of Health. http://rsb.info.nih.gov/ij/ Pharmacological endothelin receptor interaction does not occur in veins from ET(B) receptor deficient rats. Vascul. Pharmacol. 49, 6–13.
Thakali, K., Galligan, J. J., Fink, G. D., Gariepy, C. E., and Watts, S. W. (2008). Pharmacological endothelin receptor interaction does not occur in veins from ET(B) receptor deficient rats. Vascul. Pharmacol. 49, 6–13.
Walther, D. J., Peter, J. U., Winter, S., Holtje, M., Paulmann, N., Grohmann, M., Vowinckel, J., Alamo-Bethencourt, V., Wilhelm, C. S., Ahnert-Hilger, G., and Bader, M. (2003). Serotonylation of small GTPases is a signal transduction pathway that triggers platelet alpha-granule release. Cell 115, 851–862.
Keywords: norepinephrine, transglutaminase, vascular smooth muscle cells, amidation
Citation: Johnson KB, Thompson JM and Watts SW (2010) Modification of proteins by norepinephrine is important for vascular contraction. Front. Physio. 1:131. doi: 10.3389/fphys.2010.00131
Received: 26 May 2010;
Paper pending published: 16 July 2010;
Accepted: 09 August 2010;
Published online: 04 October 2010
Edited by:
R. C. Webb, Medical College of Georgia, USAReviewed by:
R. C. Webb, Medical College of Georgia, USADavid Hardy, Medical College of Georgia, USA
Copyright: © 2010 Johnson, Thompson and Watts. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Stephanie W. Watts, Michigan State University, B445 Life Sciences, East Lansing, MI 48824, USA. e-mail:d2F0dHNzQG1zdS5lZHU=