


- 1 Department of Anesthesiology, Medical College of Wisconsin, Milwaukee, WI, USA
- 2 Cardiovascular Research Center, Medical College of Wisconsin, Milwaukee, WI, USA
- 3 Department of Pharmacology and Toxicology, Medical College of Wisconsin, Milwaukee, WI, USA
- 4 Department of Physiology, Medical College of Wisconsin, Milwaukee, WI, USA
- 5 Research Service, Veterans Affairs Medical Center, Milwaukee, WI, USA
- 6 Department of Biomedical Engineering, Marquette University, Milwaukee, WI, USA
The mitochondrion is a vital component in cellular energy metabolism and intracellular signaling processes. Mitochondria are involved in a myriad of complex signaling cascades regulating cell death vs. survival. Importantly, mitochondrial dysfunction and the resulting oxidative and nitrosative stress are central in the pathogenesis of numerous human maladies including cardiovascular diseases, neurodegenerative diseases, diabetes, and retinal diseases, many of which are related. This review will examine the emerging understanding of the role of mitochondria in the etiology and progression of cardiovascular diseases and will explore potential therapeutic benefits of targeting the organelle in attenuating the disease process. Indeed, recent advances in mitochondrial biology have led to selective targeting of drugs designed to modulate or manipulate mitochondrial function, to the use of light therapy directed to the mitochondrial function, and to modification of the mitochondrial genome for potential therapeutic benefit. The approach to rationally treat mitochondrial dysfunction could lead to more effective interventions in cardiovascular diseases that to date have remained elusive. The central premise of this review is that if mitochondrial abnormalities contribute to the etiology of cardiovascular diseases (e.g., ischemic heart disease), alleviating the mitochondrial dysfunction will contribute to mitigating the severity or progression of the disease. To this end, this review will provide an overview of our current understanding of mitochondria function in cardiovascular diseases as well as the potential role for targeting mitochondria with potential drugs or other interventions that lead to protection against cell injury.
Introduction
Mitochondria are abundant and dynamic organelles that not only produce ATP for cellular function, but also participate in a number of intracellular processes such as cell division, the initiation of mitochondrial signaling pathways, modulation of cytosolic metabolic pathways, and modulation of cytosolic Ca2+ signals and concentration, and ultimately determination of cell life or death. In addition, mitochondria are a continuous source of superoxide anions ( ) and their reactive oxygen species (ROS) products (Stowe and Camara, 2009; Camara et al., 2010b; Koopman et al., 2010), particularly during cell injury. The rekindled interest in the organelle is ascribed in part to its role as a “friend and a foe.” This “new” role of mitochondria is crucial in understanding their utility as potential targets for therapies in mitigating diseases process.
) and their reactive oxygen species (ROS) products (Stowe and Camara, 2009; Camara et al., 2010b; Koopman et al., 2010), particularly during cell injury. The rekindled interest in the organelle is ascribed in part to its role as a “friend and a foe.” This “new” role of mitochondria is crucial in understanding their utility as potential targets for therapies in mitigating diseases process.
Overall cellular function is dependent on O2 consumption by functioning mitochondria to produce energy with minimal electron leak to generate  Under physiological conditions, mitochondria exist in a metabolic and cellular ion homeostatic state that is crucial for normal myocardial function. Deviation from this physiological set-point can result for example, in abnormally high mitochondrial [Ca2+] (mCa2+) and increased oxidative stress. Oxidative damage to mitochondrial membranes, enzymes, and electron transport chain (ETC) components culminate in impaired mitochondrial ATP production, which facilitates mitochondrial permeability transition pore (mPTP) opening (Waldmeier et al., 2003) that leads to cellular apoptosis and necrosis. Therefore, increases in mCa2+ and ROS generation are two important underlying factors in the etiology of many disease states, notably cardiovascular diseases including heart failure, ischemia-reperfusion (I/R) injury, and other vascular disease processes. A salient observation of mitochondria-related diseases is that even though the fundamental pathological mechanisms remain unclear, there is much evidence that mitochondrial dysfunction is a contributing factor at some level of every pathogenic process (Duchen, 2004).
Under physiological conditions, mitochondria exist in a metabolic and cellular ion homeostatic state that is crucial for normal myocardial function. Deviation from this physiological set-point can result for example, in abnormally high mitochondrial [Ca2+] (mCa2+) and increased oxidative stress. Oxidative damage to mitochondrial membranes, enzymes, and electron transport chain (ETC) components culminate in impaired mitochondrial ATP production, which facilitates mitochondrial permeability transition pore (mPTP) opening (Waldmeier et al., 2003) that leads to cellular apoptosis and necrosis. Therefore, increases in mCa2+ and ROS generation are two important underlying factors in the etiology of many disease states, notably cardiovascular diseases including heart failure, ischemia-reperfusion (I/R) injury, and other vascular disease processes. A salient observation of mitochondria-related diseases is that even though the fundamental pathological mechanisms remain unclear, there is much evidence that mitochondrial dysfunction is a contributing factor at some level of every pathogenic process (Duchen, 2004).
It is evident then that knowledge of mitochondrial function in normal and pathological states is crucial not only for our understanding of the basic cause of cardiovascular disorders, but also for developing therapeutic strategies for attenuating a given disease process. For example, attenuating electron transport, preventing mPTP opening (Wenzel et al., 2008), or activating the putative mitochondrial K+ channels (mitoKATP and mitoKCa), are potential ways to attenuate cardiac and vascular dysfunction by ameliorating the underlying mitochondriopathic disturbances (Doughan et al., 2008; Wenzel et al., 2008). The central premise of this review is simply that if mitochondrial abnormalities contribute to a pathological state (i.e., directly or indirectly), then alleviating the mitochondrial dysfunction should attenuate the severity or progression of the disease (Camara et al., 2010b). Hence, the main objective here is to present a concept that the mitochondria in all cells of the cardiovascular system can be potentially targeted for therapeutic intervention in treating cardiovascular diseases. This attention is warranted because cardiovascular diseases are a leading cause of death in the world. This review will focus in Part 1 on basic mitochondrial biology and its integration with the cytosol, metabolic regulation, structure and function, and the emerging yet essential role of mitochondria in cardiovascular disease in tandem with potential mitochondrial approaches for intervention. Separate sections on selected mitochondria therapeutic approaches in specific cardiovascular maladies will be presented in Part 2, followed by limitations and conclusions.
Part 1
Myocardial Substrate Metabolism and Regulation – Integrating Cell and Mitochondrial Function
Although this review emphasizes the important role of the mitochondrion in cardiovascular diseases and the emerging pursuit of mitochondrial-directed therapies, it is reasonable to first describe what is currently known about myocardial cell metabolism. Also, we would be remiss not to summarize the pioneering work of several research groups (Garland et al., 1963; Randle et al., 1963, 1970; Bing, 1965; Opie, 1965; Williamson, 1965, 1966; England and Randle, 1967; Bremer, 1969; Bremer and Wojtczak, 1972; Neely et al., 1972; Oram et al., 1973; Neely and Morgan, 1974) who sought to understand the mechanical/metabolic integration of the cytosolic processes (energy use) with mitochondrial function (energy supply) during the basal cardiac state and during increases in cardiac workload, hypoxia and ischemia.
It is well known that phasic contractility and relaxation are dependent on the transient influx and efflux of Na+, K+ and Ca2+ to generate the action potential and trigger Ca2+ release from the sarcoplasmic reticulum (SR). The phasic increase and decrease in [Ca2+] with each heartbeat precedes each contraction and relaxation phase, which causes spikes in energy demand. How is this demand met? We now typically divide cardiac energy metabolism into three components (Neubauer, 2007) (1) substrate utilization [cell substrate uptake and fatty acid (FA) breakdown by β-oxidation and pyruvate by glycolysis yielding acetyl carbons on coenzyme A (CoA)], which enters into the TCA cycle to produce NADH, FADH2, and CO2), (2) oxidative phosphorylation (OxPhos, transfer of electrons from NADH to O2 by respiratory complexes I–IV to create a proton motive force required for ATP production at complex V, and (3) energy transfer and utilization [translocation of matrix ATP via the adenine nucleotide transporter (ANT) of the IMM and its conversion to phosphocreatine (PCr) for transfer to myofibrils where it is reconverted to ATP by the creatine kinase (CK) energy shuttle system].
CK is a fast ATP-generating enzyme and a low-threshold sensor for ADP (Wallimann et al., 1992). Because mycoplasmic PCr is highly abundant, it serves as a temporal and spatial energy buffering transport system, which allows rapid ADP delivery for OxPhos with an increase in workload and a rapid absorption of ATP when workload decreases. Indeed the rate of ATP consumption is so exquisitely linked to the rate of oxidative phosphorylation that ATP, ADP, and PCr content remain nearly constant even when cardiac workload increases during intense exercise or catecholamine release (Balaban et al., 1986). This suggested that consumption of ATP did not directly control ATP synthesis.
The hypothesis that ATP, ADP, CrP, and Pi are not the primary feedback regulators of OxPhos, e.g., during an increase in workload and O2 consumption, is based on numerous data that the concentrations of these compounds do not change significantly (Neely et al., 1972; Raymond and Potreau, 1977; Katz et al., 1989; Balaban, 1990), i.e., the metabolic stability hypothesis. So alternatively it was proposed, and some data was provided, that (a) increases in mCa2+ stimulate various rate – limiting TCA cycle enzymes (Wan et al., 1989; McCormack et al., 1990), i.e., the Ca2+ stimulating hypothesis, (b) the PCr shuttle acts as temporal and spatial buffers for the ATP hydrolysis potential, i.e., the PCr buffering hypothesis (Saks et al., 2000), and (c) that the activities of enzymes and transporters involved in ATP synthesis change in synchrony with, and proportional to, changes in the rate of ATP utilization so that perturbations can not be observed, i.e., the parallel activation theory (Korzeniewski et al., 2005). The alternative hypothesis to the metabolic stability hypothesis is of course that despite the negative experimental data generated, organic phosphates and Pi indeed regulate cardiac metabolism. Wu et al. (2007, 2008) developed a computer model of cardiac energy metabolism that was able to adequately simulate the data of Katz et al. (1989) but they arrived at a different interpretation, i.e., that ATP synthesis is indeed feedback controlled by an increase in the products of ATP hydrolysis (Beard, 2006; Beard and Kushmerick, 2009). The simulations also agree with more recent experimental data (Ochiai et al., 2001; Zhang et al., 2005). The point here is that all hypotheses are subject to be disproven and alternate hypotheses should always be considered and tested even if the null hypothesis appears dominant.
Acetyl-CoA is the 2-carbon donor derived from FA or from glucose oxidation. Complete β-oxidation of a 12-carbon FA produces 96 mol ATP compared to 72 or 76 mol ATP by glycolytic oxidation of a 12-carbon disaccharide. The different moles of ATP produced by glycolysis depends on the use of the glycerol phosphate shuttle vs. the malate shuttle, respectively, which transfers reducing equivalents from the cytosol to the matrix where they are oxidized to produce energy. This transfer is slow and rate limiting in glycolysis so mitochondrial FA oxidation is much faster (Williamson et al., 1976; Saks et al., 2006b). The major task of the TCA cycle is to carry out the oxidation of the acetyl group of acetyl-CoA to CO2 and H2O with the subsequent production of reducing equivalents to maintain the proton motive force and energy transfer from electrons to drive ATP synthesis.
Indeed, from earlier work (Williamson, 1965, 1966) it is now well understood (Saks et al., 2006b) that during an increase in workload, ATP production and respiration are increased due to feedback signaling via the CK shuttle that leads to a decrease in the mitochondrial content of acetyl-CoA. This decrease reflects increases in the rates of respiration and NADH oxidation relative to acyl-carnitine transport into mitochondria and β-oxidation. It is now known that acetyl-CoA is transferred into the cytoplasm during high workloads via the carnitine-acetyl carrier where it is converted by acetyl-CoA carboxylase (ACC) to malonyl-CoA, an important inhibitor of carnitine-acetyl-CoA (palmitoyl) transferase (CPT), and then converted back to acetyl-CoA by malonyl-CoA decarboxylase (MCD) during reduced workloads. Therefore, an increase in workload increases the rate of acetyl-CoA consumption with a concomitant increase in malonyl-CoA content. This mechanism prevents excess FA β-oxidation intermediates when the workload decreases. This implies that malonyl-CoA is a feedback regulator of FA oxidation by allowing acyl-CoA entry into the matrix only when it is needed (Opie, 1998). Thus the decrease in malonyl-CoA (and increase in acetyl-CoA) is not the reason, but rather the consequence of an increase in workload and FA oxidation (Saks et al., 2006b).
In broad terms this process of multiple feedback steps in regulating substrate utilization, OxPhos, and energy transfer and utilization at varying workloads, is called metabolic homeostasis, and its study, metabolic control analysis. Identifying the specific rate controlling reactions is cumbersome and typically carried out by measuring depletion of substrates and accumulation of products and their associated flux rates. Stanley et al. (2005) and Neubauer (2007) have reviewed recently how substrates are metabolized by mitochondria in the normal and failing heart. Metabolic Control Analysis is a methodology that allows one to quantitate the control of flux and metabolites through a metabolic pathway (Kacser and Burns, 1973; Heinrich and Rapoport, 1974; Fell, 1992). This approach has been applied to a great variety of systems – from bacteria to mammalian and plant cells – functioning at steady state (Fell, 1996; Cortassa et al., 2002) but recently also to heart cells and trabeculae under working conditions (Cortassa et al., 2009).
Cardiac Metabolic Control – Historical Perspective
Randle et al. (1963) long ago first recognized that cells utilize either FA or glucose depending on substrate availability and that the presence of FA markedly inhibits glycolysis; this is known as the glucose–fatty acid cycle or the “Randle cycle.” It has been long known that FAs contain more energy per mass unit than glucose (Newsholm and Start, 1973). Neely et al. (1972) were among the first to recognize that increased cardiac work load was accompanied by moderately increased glycolysis, marked fatty acid oxidation, and enhanced TCA cycle turnover, but not by changes in PCr, ATP, ADP, or 5′-AMP (later unequivocally verified by Balaban et al., 1986 using NMR). Although perfusion of hearts only with glucose exhibited marked pyruvate oxidation, those perfused with glucose+acetate showed inhibited glycolysis and inhibited pyruvate oxidation. They concluded that TCA cycle turnover was not solely dependent on the availability of a given substrate but that the normally preferred substrate was FA.
In a subsequent review, Neely and Morgan (1974) further described the regulation of substrate utilization and energy metabolism in the heart. They pointed out that the availability of higher energy FA in aerobic perfused hearts suppressed lower energy glucose utilization at several steps in the glycolytic pathway, i.e., blocked glucose transport, inhibited hexokinase by its product glucose-6-P, phosphofructokinase (PFK) by citrate (Garland et al., 1963; England and Randle, 1967), and pyruvate dehydrogenase (PDH) by acetyl-CoA and NADH (Bremer, 1969; Randle et al., 1970). They described how FA uptake and oxidation are controlled by FA availability, the rate of acetyl-CoA oxidation (low energy rates), and the rate of acyl translocation across the IMM (high energy rates), which in turn is dependent on the activity of CPT. They discovered that at higher workloads acetyl-CoA levels were progressively decreased and concluded that FA oxidation becomes limited (Oram et al., 1973); they suggested that acyl translocation and transfer to matrix CoASH or CPT activity, rather than β-oxidation, was the limiting step to FA utilization. They discussed how the NAD-linked dehydrogenase reactions of the TCA cycle ultimately determine the rates of oxidation of both glucose and FA and adjust the rate of utilization of each substrate to the availability of the other.
Although the preferred substrate for the heart was found to be FA (80–100%), Neely and Morgan (1974) discussed that under hypoxic or anoxic conditions the heart switches primarily from FA to glucose; but under ischemic conditions the switch to glucose is limited due to shortage of substrate. They also indicated that a hypoxia/ischemia-induced rise in NADH inhibits glyceraldehyde-3-P dehydrogenase thus restricting glycolysis. During I/R, diminished O2 supply for respiration and OxPhos leads to rapid decline of ATP and PCr, and the concomitant increase in Pi, as a consequence of PCr hydrolysis, stimulate anaerobic ATP generation with an increase in glycolysis and lactate production (Oram et al., 1973; Wallimann et al., 1992). Whitmer et al. (1978) later reported that decreased O2 delivery to the heart caused a fall in tissue content of acetyl-CoA, acetyl-carnitine, CoASH, and carnitine, indicating that β-oxidation becomes limited under these conditions. Thus the heart has the ability to switch from FA to glucose oxidation during anaerobic metabolism, which yields substrate (cytosolic) level phosphorylation (2 mol ATP/mole glucose when pyruvate is formed from phosphoenolpyruvate) on its pathway to form acetyl-CoA.
In a 1979 review Williamson (1979) discussed how feedback regulation from the TCA cycle and the respiratory chain to PDH is likely determined primarily by alterations of the intra-mitochondrial NADH/NAD+ and acetyl-CoA/CoASH ratios acting separately or in concert, and not by the intra-mitochondrial ATP/ADP ratio because this did not appear to change appreciably with changes in cardiac activity, as noted by others. He noted that the malate shuttle is slow and limits maximal glucose oxidation. He additionally described how the activities of NAD-linked TCA enzymes pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, and isocitrate dehydrogenase are regulated by the NADH/NAD+ ratio. As this ratio decreases with increased workload, and the cytosolic ATP/ADP ratio tends to fall and Pi to increase at the expense of PCr, respiration increases, thus increasing the flux of TCA cycle intermediates (just enough acetyl-CoA enters the matrix (Saks et al., 2006b) with increases in FA and/or glucose oxidation, which is highly regulated by feedback mechanisms. There are other excellent older and newer reviews (Bing, 1965; Williamson, 1979; Opie, 1998; Randle, 1998; Nicholls and Fergunson, 2002) and original articles (Garland et al., 1963; Williamson, 1965, 1966, 1979; England and Randle, 1967; Bremer, 1969; Bremer and Wojtczak, 1972) that summarize and update this seminal early work on cardiac metabolic homeostasis.
Mitochondrial DNA and Protein Transport as Sites for Therapeutic Intervention
Mitochondrial biogenesis and regulation of mitochondrial function result from a complex process that involves the coordinated expression of both mitochondrial and nuclear genes (Camara et al., 2010b). Much of the mitochondrial DNA (mtDNA) is used to code for proteins that are key components of the energy-production system. Indeed, mitochondrial DNA contributes 13 subunits to ETC complexes. Cells depleted of mtDNA, called ρ0-cells, lack some of the critical subunits of the ETC complexes, which result in a defective respiratory system as evidenced by an insufficient mitochondrial membrane potential (ΔΨm; Camara et al., 2010b). The nucleus regulates numerous mitochondrial functions because the nuclear DNA (nDNA) encodes a majority of mitochondrial proteins. For example, complexes I and III are coded by the nDNA and the mtDNA (Papa et al., 2008) whereas complex II is coded only by nDNA. Several complex IV subunits, depending on the tissue type, are also mitochondria-derived (Lenka et al., 1998; Vijayasarathy et al., 1998). The mitochondrial proteins coded by the nuclear genome promote increased synthesis of mitochondrial encoded proteins and replication of mtDNA (Duchen, 2004). For example, the nuclear respiratory factor-1 activates the transcription of a number of the nuclear-encoded components of the ETC and also regulates mitochondrial transcription factor A, which is responsible for the transcription of mtDNA-encoded genes (Csiszar et al., 2009b).
Nuclear-encoded proteins targeted for mitochondria are imported by a complex system of import proteins (TOM and TIM, outer and inner membrane translocases, respectively). Import of nuclear-encoded proteins or precursor peptides relies on the proper function of the mitochondrial protein uptake machinery. A coordinated protein import system is important for mitochondrial maintenance and biogenesis and any disruption can lead to cell dysfunction and demise, including cells in the cardiovascular system (Duchen, 2004). The protein import pathway has been used to direct proteins into mitochondria based on specific targeting sequences (Schapira, 2006). A defect in a specific protein or damage to the import system can contribute to other defects in mitochondrial function and underlie at least some cardiovascular disorders. Therefore, knowledge of mitochondrial protein import is likely key to understanding the molecular mechanisms of many disease states, and it also opens the possibility of therapeutic intervention by targeting the imported proteins or their translocases.
Outer Mitochondrial Membrane as a Target for Potential Therapeutic Agents
The elaborate structure of a mitochondrion is important for its normal functioning, so its structural elements are potential therapeutic targets. Each mitochondrion consists of two specialized membranes dividing the organelle into a narrow intermembrane space (IMS) bordered by the outer mitochondrial membrane (OMM) and the inner mitochondrial membrane (IMM) that encloses the matrix (Figure 1). The OMM is a relatively simple phospholipid structure permeable to small molecules and ions; it contains many proteins including monoamine oxidase (MAO) and the voltage dependent anion channel (VDAC).
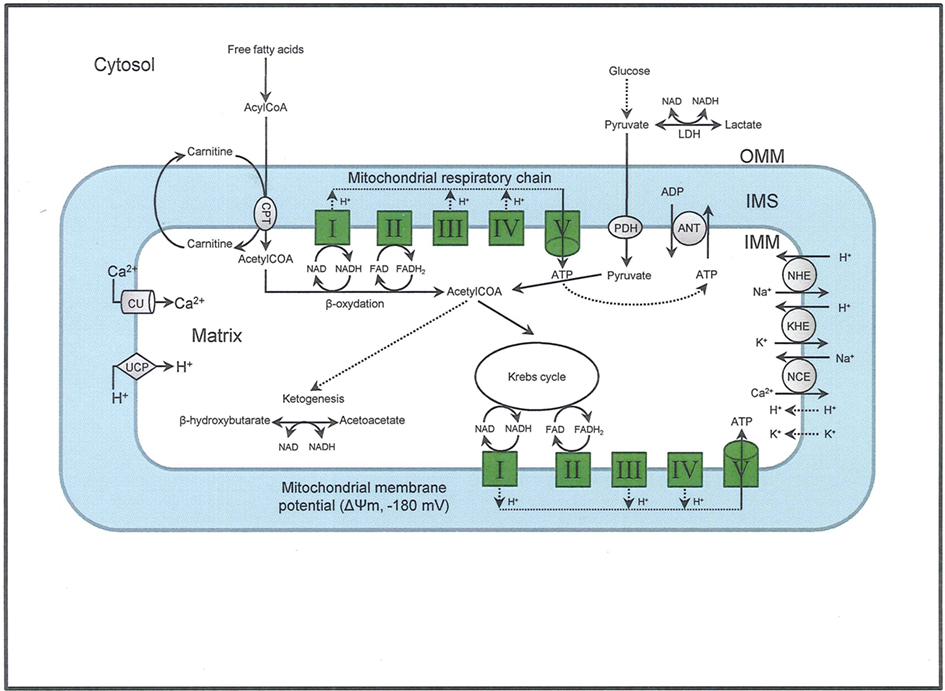
Figure 1. The figure shows the basic structural components of the ETC complexes (I, II, III, IV, and V) the sites of proton pumping after electron transfer from complex I and complex II during mitochondria substrate oxidation, that leads to the generation of ATP. Fatty acid oxidation and Krebs cycle generate NADH and FADH2 needed to energize mitochondria and establish the mitochondrial membrane potential (ΔΨm; −180 to −200 mV). The ΔΨm can be modulated be uncoupling proteins (UCP). Phosphate is imported and exported through the adenine nucleotide translocase (ANT) and this is one way to regulate matrix phosphate levels. Substrate uptake is mediated through mitochondrial inner membrane (IMM) proteins [e.g., carnitine palmitoyl transferase (CPT) and pyruvate dehydrogenase (PDH)]. Ca2+, which may modulate mitochondrial respiration is taken up via the calcium uniporter (CaU). Other components of the IMM include the putative transporters/exchangers. These include the Na+/H+ exchanger (NHE), the K+/H+ exchanger (KHE) and the Na+/Ca2+ exchanger (NCE), some of which have been characterized. The putative Ca2+/H+ exchanger (the so-called Na independent Ca exchanger or NICE; not shown) is considered a likely regulator of ion flux dynamics.
The OMM is also a major source of ROS and MAO (mostly the A isoform), with a large presence in the OMM, contributes to the ROS generated by mitochondria under pathological conditions. The MAOs are mitochondrial flavoenzymes that catalyze the oxidative deamination of catecholamines and biogenic amines, like dopamine, and serotonin (5-HT). In the process, ROS (H2O2 from  ) is generated (Kaludercic et al., 2010a,b), which exert hypertrophic effects or aggravate I/R injury (Kaludercic et al., 2010a; Nediani et al., 2011). During I/R or heart failure, the heart is exposed to chronic neuro-hormonal or peripheral hemodynamic distress and this leads to elevated circulating catecholamines, resulting in MAO-induced ROS production. In the heart, it has been reported that 5-HT accumulates during I/R and contributes to the progression of myocardial damage and injury independent of 5-HT receptor mediated activity (Bianchi et al., 2005), and in the kidney MAO-induced H2O2 production during dopamine degradation was reported to mediate cell apoptosis (Bianchi et al., 2005). A possible mechanism for MAO-mediated cell death is that MAO-dependent ROS generation induces an increase in the Bax/Bcl-2 ratio and mitochondrial cytochrome c release, leading to cardiac cell apoptosis (Bianchi et al., 2005). In a recent study, Kaludercic et al. (2010b) reported that in addition to 5HT, NE catabolism by MAO plays a prominent role in cardiac hypertrophy. In their study, Kaludercic et al. (2010b) reported that MAO enhanced NE metabolism in transverse aortic constriction in mice, which was also associated with exacerbated oxidative stress, chamber dilation, and reduced systolic function. These observations provide evidence for the pivotal role that mitochondrial MAO has in these disease processes and provides a therapeutic potential for their inhibition. The targeting of mitochondrial MAO for its potential therapeutic effect derives in part from the finding that MAO inhibition can counteract oxidative stress and mitochondrial-induced cell death while reducing myocardial damage due to I/R injury (Bianchi et al., 2005; Morin et al., 2009).
) is generated (Kaludercic et al., 2010a,b), which exert hypertrophic effects or aggravate I/R injury (Kaludercic et al., 2010a; Nediani et al., 2011). During I/R or heart failure, the heart is exposed to chronic neuro-hormonal or peripheral hemodynamic distress and this leads to elevated circulating catecholamines, resulting in MAO-induced ROS production. In the heart, it has been reported that 5-HT accumulates during I/R and contributes to the progression of myocardial damage and injury independent of 5-HT receptor mediated activity (Bianchi et al., 2005), and in the kidney MAO-induced H2O2 production during dopamine degradation was reported to mediate cell apoptosis (Bianchi et al., 2005). A possible mechanism for MAO-mediated cell death is that MAO-dependent ROS generation induces an increase in the Bax/Bcl-2 ratio and mitochondrial cytochrome c release, leading to cardiac cell apoptosis (Bianchi et al., 2005). In a recent study, Kaludercic et al. (2010b) reported that in addition to 5HT, NE catabolism by MAO plays a prominent role in cardiac hypertrophy. In their study, Kaludercic et al. (2010b) reported that MAO enhanced NE metabolism in transverse aortic constriction in mice, which was also associated with exacerbated oxidative stress, chamber dilation, and reduced systolic function. These observations provide evidence for the pivotal role that mitochondrial MAO has in these disease processes and provides a therapeutic potential for their inhibition. The targeting of mitochondrial MAO for its potential therapeutic effect derives in part from the finding that MAO inhibition can counteract oxidative stress and mitochondrial-induced cell death while reducing myocardial damage due to I/R injury (Bianchi et al., 2005; Morin et al., 2009).
The VDAC is a principal site for the exchange of metabolites between the IMS and the cytosol. The VDAC is a regulated conduit that provides an access route into the IMS for ions and a variety of cellular metabolites such as ATP and ADP (Okada et al., 2004). Decreased phosphorylation of VDAC, which is induced by inhibition of glycogen synthase kinase, can reduce ATP entry into mitochondria (Das et al., 2008). As a major gateway in and out of the mitochondrion, VDAC mediates an intimate dichotomy between metabolism and cell death (Ujwal et al., 2008). The mitochondrial-mediated cell death pathway involves an array of OMM and cytosolic proteins that include the hexokinases (I and II), the 4-chlorodiazepine (CDZ) sensitive peripheral benzodiazepine receptor (PBR), and the Bcl-2 protein family that alternatively promote or prevent cell injury (Bryson et al., 2002; Pastorino et al., 2005; Pastorino and Hoek, 2008; Ujwal et al., 2008). Specifically, PBR interacts with mPTP through VDAC and regulates apoptosis as it blocks the anti-apoptotic effect of Bcl-2 family proteins (Kroemer et al., 2007). CDZ reduces ischemia-induced arrhythmias and cytochrome c release independent of mPTP opening by inhibiting PBR (Camara et al., 2010b). Perfusion of rat hearts with a cell-permeable peptide corresponding to the BH4 domain of Bcl-XL, which is reported to close VDAC and to prevent the VDAC-mediated release of cytochrome c, was shown to inhibit CK release and to reduce myocardial cell death (Morin et al., 2009). Thus the OMM supports protein-to-protein interactions between the anti-apoptotic and pro-apoptotic proteins.
The hexokinase (HK)-VDAC is another OMM protein-to-protein interaction that offers an intriguing target to selectively provide cytoprotection against oxidative damage. Accumulating evidence has shown that HK translocation to mitochondria plays a crucial role in promoting cell survival, when needed, and to instigate its removal if cell death is required, as in cancer cells (Camara et al., 2010b). Indeed, in malignant tumor cells, unlike normal cells, elevated oncogenic kinase signaling favors the binding of HK to VDAC by Akt-dependent phosphorylation of HK (Pastorino et al., 2005). Enhanced association between HK and VDAC provides protection against permeabilization of the OMM and resistance to apoptosis (Galluzzi et al., 2008; Pastorino and Hoek, 2008). However, it is noted that the binding to the VDAC is not a prerequisite for HK binding to OMM because the enzyme binds to OMM in yeast strains that are deficient in VDAC (Shoshan-Barmatz et al., 2008).
Nonetheless, a possible therapeutic strategy that targets mitochondrial metabolism involves enhancing HK–VDAC interactions. For example, it was reported that closure of VDAC by HK prevents mPTP opening and blocks apoptosis (Azoulay-Zohar et al., 2004). Other studies show that increased HKII binding to VDAC prevents the pro-apoptotic Bcl-2 family proteins from binding to the OMM, and so helps to promote, for example, cancer cell survival (Costantini et al., 2000; Gogvadze et al., 2009; Mathupala et al., 2009). Indeed, HK is frequently overexpressed in some human tumors and it binds to VDAC more tightly in cancer cells than in their normal cell counterparts (Pastorino et al., 2005). This protection may be prevented, as a therapeutic approach, by adding 3-bromopyruvate, an inhibitor of HK activity and HK binding to mitochondria, the latter most likely by preventing the HK-VDAC interaction (Pedersen et al., 2002; Chen et al., 2009). In non-malignant conditions, partial knockout of HKII has been associated with increased cell death in a mouse model of cardiac I/R injury (Smeele et al., 2010). For the heart it is suggested that the mitochondria-HK interaction is indeed an integral part of cardioprotection because many cardioprotective interventions, such as ischemic preconditioning (IPC) and anesthetic preconditioning, increase mitochondria-HK binding (Zuurbier et al., 2009). The translocation and increased binding of HKII to cardiac mitochondria can be invoked in the mechanism of cardioprotection by IPC and opioids (Smeele et al., 2010). HKII binding to mitochondria to decrease pro-apoptotic Bcl-2 family protein binding to OMM, as well as decreasing ROS production may contribute together to improving mitochondrial and cell function during reperfusion after ischemia.
Taken together, these studies suggest that the mitochondrial HK interaction plays a key role in promoting cellular survival; thus targeting its binding to the mitochondrion may offer a novel strategy to potentiate the efficacy of other modes of cardiovascular intervention. A mitochondria-targeted therapy designed for the OMM as a potential therapeutic maneuver is highly relevant and is the subject of intense research. An understanding of the interaction between the cytosolic proteins, glycolytic or apoptotic, and the OMM should help to optimize therapies for treating degenerative diseases, including ischemic heart disease and heart failure. These mitochondrial related diseases and the potential targeting of the organelle as a mitigating factor will be discussed in detail in the coming sections.
Inner Mitochondrial Membrane and Matrix as Sites for Potential Therapeutic Agents
The IMM is convoluted into a large number of infoldings called cristae. As a consequence, the IMM has a large surface area that increases the efficiency of the chemical reactions that take place at its inner surface (Koopman et al., 2010). The IMM is relatively impermeable and allows only certain small molecules (ions mostly) to pass through. The major phospholipid component of the IMM, cardiolipin, is intimately involved in several mitochondrial bioenergetic functions as well as in the regulation of cytochrome c content and function. IMM proteases, by targeting mitochondrial fusion proteins (discussed below), also regulate the release of cytochrome c release from mitochondria (Frezza et al., 2006). Apoptosis inducing factor (AIF) is also released from the IMM, but by proteolytic cleavage catalyzed by mitochondrial calpain and preceded by mitochondrial ROS emission (Norberg et al., 2010). I/R mediated cell death can be prevented by calpain inhibitors and by MnSOD (Norberg et al., 2010). Damage to cardiolipin has been implicated in numerous pathological conditions, including cardiovascular abnormalities. Paradies et al. (2009) reported that cardiolipin content observed in mitochondria from I/R hearts paralleled the fall in complex I activity.
Cardiolipin and cytochrome c can form into a complex called cytochrome c-cardiolipin peroxidase, which under oxidative stress and oxidation of cardiolipin can lead to the release of cytochrome c and other pro-apoptotic factors from the IMS to the cytosol (Chen et al., 2010). Therefore, targeting this complex, as demonstrated with nitric oxide (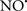 ), has proven somewhat valuable in protecting against oxidation of cardiolipin, and as a result helps to preserve cytochrome c (Vlasova et al., 2006). Indeed, in a comprehensive study, Vlasova et al. (2006) reported that
), has proven somewhat valuable in protecting against oxidation of cardiolipin, and as a result helps to preserve cytochrome c (Vlasova et al., 2006). Indeed, in a comprehensive study, Vlasova et al. (2006) reported that 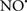 acts as an effective inhibitor of peroxidase activity on the cytochrome c-cardiolipin complex, and in this way preserves cardiolipin integrity. The authors observed that
acts as an effective inhibitor of peroxidase activity on the cytochrome c-cardiolipin complex, and in this way preserves cardiolipin integrity. The authors observed that 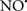 acted as an effective inhibitor of the peroxidase activity of cytochrome c without significantly altering the cardiolipin molecule itself. Similarly, the dominant antioxidant effect of
acted as an effective inhibitor of the peroxidase activity of cytochrome c without significantly altering the cardiolipin molecule itself. Similarly, the dominant antioxidant effect of 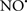 on the peroxidation reaction catalyzed by myoglobin and hemoglobin is reduction of oxo-ferryl reactive species (Gorbunov et al., 1995). This observation along with those by Shidoji et al. (1999) showed that cardiolipin peroxidation products resulted in a lack of effective binding of cytochrome c to cardiolipin. This suggests that reducing the catalytic peroxidase activity of the complex by
on the peroxidation reaction catalyzed by myoglobin and hemoglobin is reduction of oxo-ferryl reactive species (Gorbunov et al., 1995). This observation along with those by Shidoji et al. (1999) showed that cardiolipin peroxidation products resulted in a lack of effective binding of cytochrome c to cardiolipin. This suggests that reducing the catalytic peroxidase activity of the complex by 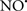 reduces the hydroperoxide products that enhance peroxidation of the lipid. It is therefore likely that
reduces the hydroperoxide products that enhance peroxidation of the lipid. It is therefore likely that 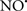 prevents the oxidation of cardiolipin in part by inhibiting the damaging effects of the peroxidase activities of the cytochrome c-cardiolipin complex that occurs during ischemia and other oxidative stress. Thus, inhibiting the peroxidase activity of the complex may be a pathway by which
prevents the oxidation of cardiolipin in part by inhibiting the damaging effects of the peroxidase activities of the cytochrome c-cardiolipin complex that occurs during ischemia and other oxidative stress. Thus, inhibiting the peroxidase activity of the complex may be a pathway by which 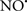 attenuates cardiolipin oxidation (Shidoji et al., 1999); it is also reported that
attenuates cardiolipin oxidation (Shidoji et al., 1999); it is also reported that 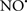 could be an effective scavenger of peroxyl and other radicals (Chamulitrat, 2001). These observations indicate that the cytochrome c-cardiolipin peroxidase complex is an important factor in oxidative-mediated damage and represents a viable option to be considered in targeting mitochondria therapeutically.
could be an effective scavenger of peroxyl and other radicals (Chamulitrat, 2001). These observations indicate that the cytochrome c-cardiolipin peroxidase complex is an important factor in oxidative-mediated damage and represents a viable option to be considered in targeting mitochondria therapeutically.
The adaptor protein p66Shc is an IMS protein located in the ETC segment between complex III and IV. The regulation of p66Shc is not fully understood. At physiological states, the protein is silent but is activated in response to pro-apoptotic stimuli. Recent studies have shown that p66Shc is involved in e− transfer with reduced cytochrome c, and so it can modulate ROS production and the cardiovascular oxidative stress response (Shoshan-Barmatz et al., 2008; Camara et al., 2010b). Mitochondrial p66Shc can be induced, for example, by hypertrophic stimuli and it is a signaling link between cellular stress and mitochondrial apoptotic activity (Guo et al., 2009). Activation of the protein occurs via H2O2 dependent phosphorylation of the Ser36 residue, with its ultimate translocation to mitochondria. Once in mitochondria, p66Shc also alters mCa2+ handling, and ultimately can induce apoptosis (Pinton et al., 2007). In addition to stimulating release of ROS, p66Shc can decrease MnSOD expression levels by activating the Akt-dependent phosphorylation of Forkhead transcription factor (FOXO3a) pathway, a regulator of MnSOD expression. Consistent with these mechanisms, mice deficient in mitochondrial p66Shc genes display increased levels of FOXO3a, and decreased ROS production and decreased cardiac I/R injury (Carpi et al., 2009; Di Lisa et al., 2009).
It was proposed recently that the therapeutic targeting of ETC complex IV as a strategy to mitigate I/R injury may worsen outcome in part because of the increased levels of reduced cytochrome c (Chen et al., 2010), a substrate for p66Shc mediated ROS generation. It is possible that pharmacological therapies that inhibit mitochondrial p66Shc expression or action may act as panacea for clinical disorders characterized by increased oxidative stress, e.g., atherosclerosis, I/R injury and cardiac heart failure. However, Di Lisa et al. (2009) noted in a recent review that at present the translation of experimental findings into the clinical realm is limited by the lack of drugs that might be effective against a direct p66Shc mediated ROS production.
The IMM also contains the adenine nucleotide translocator (ANT), which along with VDAC, is believed to play a role in the formation, or more likely, the regulation of the mPTP. The ANT has been shown to be a prominent target for oxidative and nitrosative stress and so appears to be an important component in mitochondria-related cell death and diseases. ANT interacts with both the anti-apoptotic and pro-apoptotic BCl-2 protein families, which then act as allosteric modulators of the ATP/ADP anti-porter (Marzo et al., 1998; Brenner et al., 2000; Belzacq et al., 2003). Another potential binding partner of ANT is mitochondrial creatine kinase (mCK), which together with other enzymes is part of a phosphotransfer network that is crucial for the coupling of OxPhos with ATP-consuming processes (Saks et al., 2006a). Enhancing phosphotransfer from ATP to creatine in close proximity to ANT may be beneficial in preventing mPTP opening and reducing ROS production under stress conditions (Dolder et al., 2003; Meyer et al., 2006). The underlying mechanism may be the result of conformational changes in ANT and a more state 3 like respiration due to increased ADP production. A therapeutic approach could involve the design of creatine analogs that would increase the turnover of ATP to ADP in proximity to ANT. Disruptions of ANT activity, on the other hand, can result in inhibition of ATP/ADP anti-porter activity, excess ROS production, cytosolic ATP depletion, and mitochondria depolarization and apoptosis (Don et al., 2003). Clinically, dysfunction of ANT has been proposed to be involved in dilated cardiomyopathy (Dorner et al., 1997). Thus, preserving ANT functional integrity may provide a unique opportunity for pharmacological or molecular genetic maneuvers that target this protein to preserve mitochondrial function during cell injury.
Under physiological conditions, mitochondria exhibit a high IMM potential (ΔΨm) that is initiated by proton pumping from the matrix with simultaneous e− transfer along the ETC; ΔΨm is also maintained by various cation channels and exchangers that regulate cation fluxes. The trans-membrane cation movement through specialized cation transporters and exchangers are therefore essential for creating the resulting mitochondrial bioenergetic state (Figure 1). Mitochondrial cation anti-porters/exchangers (H+-linked) regulate the osmotic differential across the IMM. This is necessary in part because of a low cationic permeability and a strong H+ electrochemical gradient that is established by e− transfer and cation exchangers to prevent osmotic overload; these conditions are necessary for efficient OxPhos (Camara et al., 2010b). The trans-membrane cation movement through specialized cation transport and exchangers is therefore essential for creating the resulting mitochondrial bioenergetic state (Figure 1).
Alterations in the dynamic regulation of ion fluxes underlie numerous abnormalities of mitochondrial related function. For instance, mCa2+ uptake through the Ca2+ uniporter (CaU) is largely dependent on ΔΨm. This mCa2+ uptake may have profound consequences for mitochondrial function, such as altering cellular respiration and mediating cell death by apoptosis or necrosis (Sack, 2006a; Camara et al., 2010b). A small increase in mCa2+ during increased workload is thought to be necessary for activating TCA cycle enzymes to furnish the reducing equivalents necessary to match energy demand with supply (Camara et al., 2010b). However, high m[Ca2+], as observed during cardiac I/R, can impair ATP synthesis and lead to a loss of ion homeostasis, stimulation of ROS generation, mPTP opening, matrix swelling, and OMM rupture (Jones et al., 2003; Teshima et al., 2003b). Cumulatively, all these factors result in collapse of ΔΨm, release of signaling molecules (cytochrome c, AIF, and Smac/Diablo), and exacerbation of ROS production, resulting in a catastrophic vicious cycle of mitochondrial functional decline, and ultimately cell death (Brookes et al., 2004; Muravchick and Levy, 2006). This subject matter along with the role of other mitochondrial proteins intimately involved in regulating mitochondrial free ion concentrations and matrix pH, have been discussed extensively in our recent comprehensive review article (Camara et al., 2010b).
It is important to note that the ΔΨm is about two times larger than the sarcolemmal membrane potential. This provides a unique chemical opportunity for selectively targeting the mitochondrion for therapeutic purpose. In fact, this unique attribute of mitochondria has been employed to direct the so-called “mitochondriotropic” drugs to target the matrix (Brookes et al., 2004). For example, Smith et al. (1999) reported that complexing vitamin E with the triphenyl-phosphonium (TPP+) cation augmented mitochondrial uptake of the complex. The cationic metalloporphyrin superoxide dismutase mimetic Mn (III) meso-tetrakis (N-ethylpyridium-2-yl) porphyrin (MnIII TE-2-Pyp5+) has been shown to accumulate in mitochondria derived from mice cardiac myocytes following systemic injection (Spasojevic et al., 2007). We have shown that mitochondrial superoxide ( ) production and heart damage resulting from prolonged cold I/R was attenuated when the permeant MnSOD mimetic manganese (III) tetrakis (4-benzoic acid)porphyrin (MnTBAP) was perfused into the coronary arteries along with catalase and glutathione (GSH) before ischemia (Camara et al., 2010b). However, the effectiveness of these drugs to mitigate cardiovascular abnormalities in the clinical setting has yet to be determined.
) production and heart damage resulting from prolonged cold I/R was attenuated when the permeant MnSOD mimetic manganese (III) tetrakis (4-benzoic acid)porphyrin (MnTBAP) was perfused into the coronary arteries along with catalase and glutathione (GSH) before ischemia (Camara et al., 2010b). However, the effectiveness of these drugs to mitigate cardiovascular abnormalities in the clinical setting has yet to be determined.
Mitochondrial Permeability Transition Pore Opening as a Potential Therapeutic Target
A key aspect of mitochondrial involvement in cell demise is control of the opening of the mPTP. Crompton et al. (1987) were the first to report that mPTP opening plays a crucial role in myocardial I/R injury. The mPTP is a highly dynamic, non-selective pore for which the precise structural identity is not well defined. Suffice it to state, however, that the pore is thought to span the OMM and the IMM and that it mediates the lethal permeability changes of the IMM and OMM that leads to cell apoptosis. The mPTP may allow limited and transient passage of electrolytes, small molecules, and metabolites. But irreversible opening of the pore leads to release of apoptotic protein factors (Kim et al., 2003; Martinez-Ruiz et al., 2008; Sun et al., 2008), and a loss of NAD+ results in impaired capacity to restore redox signaling and  scavenging (Di Lisa et al., 2001). A detailed description on the putative constituents and functionality of the pore, physiological or pathophysiological, has been discussed in our recent comprehensive review article (Camara et al., 2010b) and so will not be discussed further here. Indeed, mPTP opening is especially sensitive to oxidative stress since it is favored by decrease in NADPH/NADH and GSH/GSSG ratio (Di Lisa et al., 2009).
scavenging (Di Lisa et al., 2001). A detailed description on the putative constituents and functionality of the pore, physiological or pathophysiological, has been discussed in our recent comprehensive review article (Camara et al., 2010b) and so will not be discussed further here. Indeed, mPTP opening is especially sensitive to oxidative stress since it is favored by decrease in NADPH/NADH and GSH/GSSG ratio (Di Lisa et al., 2009).
As noted above, recent evidence suggests that at low conductance states the pore might contribute to the exchange of small metabolites between the cytosol and matrix, a process that is mainly controlled by mitochondrial solute carriers (Fulda et al., 2010). In this case the opening of the pore is considered a “physiological valve,” which alleviates mCa2+ overload, for example, as a consequence of a brief surge of Ca2+ within a localized microdomain of the mitochondria–endoplasmic reticulum (Smaili and Russell, 1999) (Figure 2). Mitochondria play an essential role in buffering physiological loads of Ca2+ upon insult, whereas the mPTP provides a fast Ca2+ extrusion mechanism in response to excessive levels of Ca2+ (Sareen et al., 2007). In addition, the brief opening and closing of the pore (flickering) are implicated in providing protection against cellular injury (Kindler et al., 2003). This dual role of the mPTP in the survival and death of the cell will therefore be critical in selective targeting of the pore for therapeutic interventions.
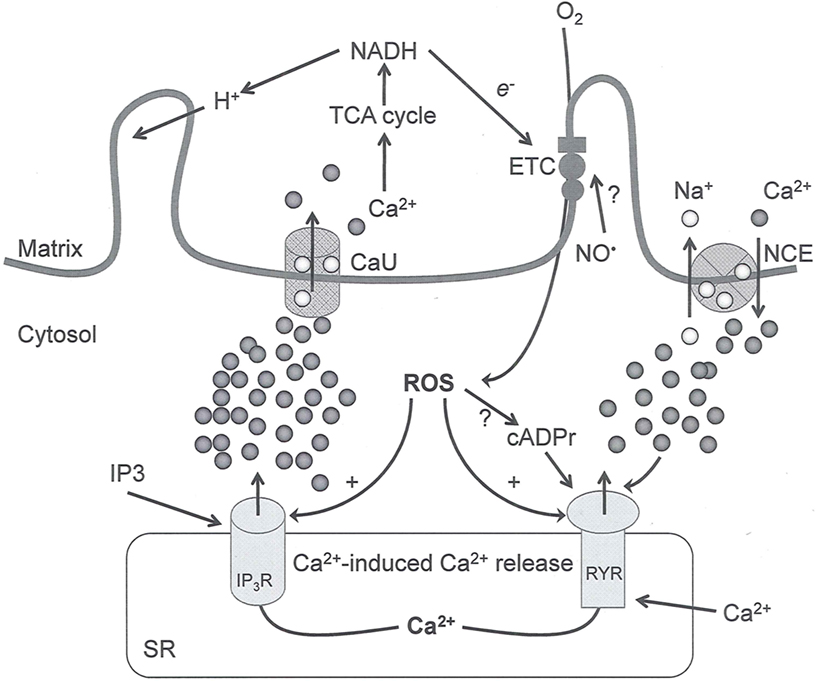
Figure 2. Interaction between mitochondria and SR Ca2+-release channels. This structural arrangement allows mitochondria and SR to be connected by tether proteins (not shown), which allows regulation of matrix Ca2+ that may modulate mitochondrial activity. Mitochondria take up Ca2+ ions entering the cytosol from internal stores (SR) of the local “microdomain” via signaling molecules that include Ca2+ and ROS. After cell stimulation, inositol 1,4,5-trisphosphate (IP3) and cADP ribose (cADPr) release calcium from SR through their respective receptors [IP3R and ryanodine receptors (RyR)]. Local increases in cytosolic Ca2+ concentrations are buffered by mitochondrial uptake through the calcium uniporter (CaU) and possible alternative routes, such as the putative Ca2+/H+ exchanger [the so-called Na+ independent Ca2+ exchanger (NICE, or CHE); not shown]. Intra-mitochondrial Ca2+ may activate synthesis of reduced substrates (NADH) by metabolic pathways and accelerate the electron transport chain (ETC); a resulting increase in ROS production may in turn facilitate Ca2+ release by sensitization of IP3R and RyR. The increase in respiration is associated with enhanced proton pumping and subsequently increase state 3 respiration with perhaps increase CHE activity so that more Ca2+ is taken up from the localized “microdomain.” Other putative mechanisms that increase ROS generation are modifications of ETC complexes by mCa2+ and inhibition of the ETC by nitric oxide ( ). The
). The 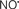 can be increased by Ca2+-induced
can be increased by Ca2+-induced  synthase. Mitochondrial can also support Ca2+ release by efflux via Na+/Ca2+ exchange (NCE). The delicate balance in the regulation of Ca2+ in the “microdomain” is crucial in the pathology of mitochondria-related cardiovascular complications. Reproduced and modified by permission from Camello-Almaraz et al. (2006).
synthase. Mitochondrial can also support Ca2+ release by efflux via Na+/Ca2+ exchange (NCE). The delicate balance in the regulation of Ca2+ in the “microdomain” is crucial in the pathology of mitochondria-related cardiovascular complications. Reproduced and modified by permission from Camello-Almaraz et al. (2006).
Pathologically, mPTP opening is initiated by excess mCa2+, enhanced ROS production, and increased Pi. Adenine nucleotides, Mg2+, and matrix H+ restrict the pore from opening (Brookes et al., 2004; Duchen, 2004; Halestrap et al., 2004, 2007; O’Rourke, 2007). It is proposed that I/R promotes mPTP opening in two phases: (i) mPTP priming during ischemia occurs as a progressive IMM leak and is accompanied by depressed ETC function in the setting of FA accumulation and loss of cytochrome c and ROS scavengers; and (ii) the triggering of mPTP at reperfusion as determined by the interplay of ΔΨm with rising mCa2+, with ROS and matrix H+ (Honda et al., 2005). During ischemia pore opening is minimized, but on reperfusion conditions such as increased matrix Ca2+, Pi, and ROS favor long-lasting pore opening. Sustained and prolonged opening of the mPTP can lead to excessive H2O entry into the matrix, matrix swelling, and increased OMM permeability (via oligomerization of Bid or Bax) or rupture of the OMM and IMM. Figure 3 summarizes the deleterious changes in mitochondrial function during and after cardiac I/R associated with mPTP opening, permeabilization of the OMM, and changes in bioenergetic state associated with individual components of mitochondria that are potential targets for therapeutic intervention.
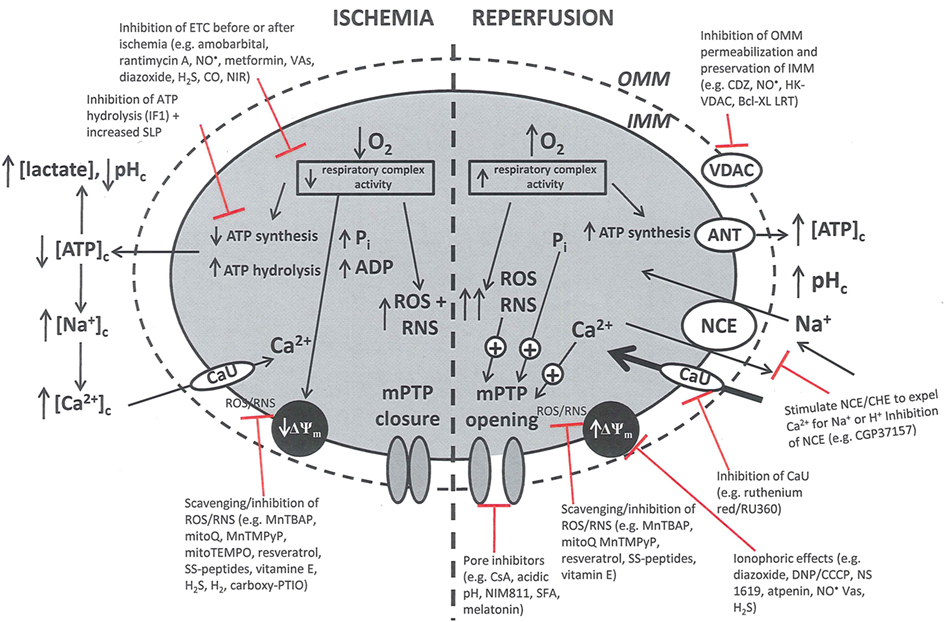
Figure 3. Simplified schema of changes in cytosolic and mitochondrial function during cardiac I/R injury and site of mitochondrial-targeted therapies with potential to reduce mitochondrial and cell damage. Targets are (a) the outer mitochondrial membrane (OMN), (e.g., putative mPTP components VDAC, ANT, etc.); (b) the inner mitochondrial membrane (IMM), [e.g., ANT, Ca2+ uniporter (CaU), Na+/Ca2+ exchanger (NCE), Ca2+/H+ exchanger (CHE)]; (c) the membrane potential (ΔΨm); (d) electron transport (complexes I–V); and (e) ATP synthesis/hydrolysis (complex V). Abbreviations: ETC, electron transport chain; SLP, substrate level phosphorylation; ROS, reactive oxygen species; RNS, reactive nitrogen species; mPTP, membrane permeability transition pore; ANT, adenine nucleotide translocator; VDAC, voltage dependent anion channel. Compounds: VAs, volatile anesthetics; CDZ, chlorodiazepine; HK, hexokinase; CGP, CGP37175: DNP, dinitrophenol; MnTBAP, Mn(II)tetrakis(4-benzoate) porphyrin chlorine; CsA, cyclosporin A; NIM811, N-methyl-4-isoleucine-cyclosporin; SS, Sezto–Schiller tetrapeptides; SFA, sanglifehrin; LRT, lipid replacement therapy; CCCP, carbonyl cyanide p-(trifluoromethoxy) phenylhydrazine, MnTMPyP, Mn(III)tetrakis[(1-methyl-4-pyridyl)-porphyrin]; IF1, inhibitory factor 1; CO, carbon monoxide; NIRL, near infrared light.
As noted the exact molecular composition of the mPTP remains elusive and mysterious. The previous model structure of the mPTP put forward by Halestrap (2010) and Di Lisa and Bernardi (2006) involves the combination of the ANT in the IMM, the VDAC in the OMM, and the regulatory protein cyclophilin D (CypD) in the matrix (Camara et al., 2010b). In addition, a benzodiazepine receptor, HK, and mitochondrial mCK are thought to act as regulatory components of the pore (Honda et al., 2005). Associated with the outer leaflet of the IMM, in the IMS, is the CK. As mentioned earlier, mCK, under physiological conditions, is crucial in catalyzing the transphosphorylation of creatine by ATP to phosphocreatine and ADP. In apoptotic-induced cell death, mCK may facilitate contact between VDAC and ANT to form the pore at the contact site between the IMM and the OMM (Muravchick and Levy, 2006).
Interestingly, recent studies have provided evidence that exclude the ANT and VDAC as components of the mPTP. For example, genetic knockout studies showed that mPTP opening could occur in the absence of VDAC and ANT; nevertheless, they could both play a regulatory role. VDAC plays a critical role in the maintenance of mitochondrial function, and ultimately preserves cellular function, while ANT appears to function as a Pi and Ca2+ sensitive mPTP regulator. IPC protection could be mediated by the phosphorylation and inhibition of GSK-3β, a downstream target of Akt, thereby, inhibiting mPTP opening in part by binding to ANT (Nishihara et al., 2007). CypD was first identified as ANT-binding protein that mediated the inhibitory effect of cyclosporin A (CsA) on mPTP. The role of CypD in the regulation of cell survival or death is evidenced by the finding that cells lacking the Ppif gene that encodes the protein are protected from necrotic caspase-independent cell death, but not from caspase-dependent apoptosis (Kim et al., 2003; Baines et al., 2005; Basso et al., 2005).
Prolonged opening or a large conductance of the pore has been associated with numerous pathological conditions, e.g., stroke accompanied by brain ischemia (Matsukawa et al., 2009). These actions of the mPTP apply in most instances of cell damage and suggest that pharmacological agents, or any other maneuvers that minimize pore opening, might minimize the extent of permanent damage that arises. For example, Pravdic et al. (2010) reported recently that in vivo isoflurane-induced post-conditioning prevented mPTP opening in part by mild inhibition of mitochondrial respiration, depolarization of mitochondria, and acidification of matrix pH. These events may lead to better preservation of mitochondrial bioenergetics and minimize Ca2+ overload and cell death (Pravdic et al., 2010). Another example is that melatonin, a well-known antioxidant, has also been shown to protect against reperfusion injury by inhibiting mPTP opening (Petrosillo et al., 2009) (Figure 3). In this case, mitochondria isolated from rat hearts exposed to melatonin before I/R were less sensitive than mitochondria isolated from control I/R hearts to mPTP opening as demonstrated by their resistance to Ca2+-induced pore opening. CsA, the well-known inhibitor of mPTP opening, had effects similar to melatonin (Petrosillo et al., 2009).
In a recent limited human clinical trial it was suggested that CsA can reduce infarct size and improve recovery of contractile function on reperfusion (Gomez et al., 2009). The antidepressant drug nortriptyline was also reported to exert its neuroprotective effects against cerebral I/R injury in part by delaying mPTP opening secondary to resisting Ca2+ overload (Zhang et al., 2008). Similarly, the derivative of CsA, NIM811, and the non-immunosuppressive sanglifehrin (SFA) inhibited mPTP opening (Theruvath et al., 2008; Figure 3) and prevented cytochrome c release. In this way it is thought that normal e− transfer to complex IV (see below) and the scavenging potential of cytochrome c (discussed below) is also preserved. A clinical trial reported that targeting mPTP with post-conditioning or CsA at the moment of angioplasty improved contractile function and reduced cell death in patients with acute myocardial infarction (Ruiz-Meana et al., 2010). These studies do provide some encouragement for a clinical approach to delay or prevent irreversible mPTP opening. It is obvious that our understanding of the structural components and activity of the pore are crucial in our attempts to pharmacologically target the pore as a potential site for therapy (Figure 3). It is thus noteworthy that achieving control of the mPTP during the disease process is an important goal from the perspective of minimizing the early loss of functional and structural integrity, and for maximizing salvage following the initial injury.
Therapeutic Potential of Interfering with Electron Transport Chain and Oxidative Phosphorylation
The IMM contains enzyme complexes and transport systems that are important in the transduction of energy from substrates that provide for the normal functioning of the cell. Under aerobic conditions, the ETC complexes allow proton translocation, and in the process, function to convey e− along and down a redox potential (Stowe and Camara, 2009; Lenaz and Genova, 2010; Lenaz et al., 2010). Electrons are transferred from NADH and FADH2 from complex I and II, respectively, to downstream redox centers (Figure 1). The complexes are thought to be dispersed randomly in large groupings called respirasomes or supercomplexes, which are held in place by cardiolipin and which are functionally connected to coenzyme Q and cytochrome c (Stowe and Camara, 2009; Lenaz and Genova, 2010; Lenaz et al., 2010). The supercomplex is thought to improve the efficiency of OxPhos by eliminating the diffusion of substrates and products between individual complexes (Paradies et al., 2009). The ubiquinone moiety is redox active, accepting two e− and two H+ in its reduction to ubiquinol (James et al., 2005). Concomitant to the e− transport, H+ ions are pumped from the matrix into the IMS (Figure 1). The energy released during the process is efficiently utilized to phosphorylate ADP to ATP by translocation of H+ from the IMS to the matrix through complex V (F1/F0 ATP synthase; Stowe and Camara, 2009; Camara et al., 2010b). Hence OxPhos results in transient dissipation of ΔΨm.
Cardiomyocytes, like neurons, depend almost entirely on OxPhos for their function. Therefore, structural preservation of the ETC complexes/supercomplexes is paramount in maintaining a high bioenergetic status to provide efficient cellular homeostasis with minimal ROS production (Camara et al., 2010b). In fact, mitochondrial defects encompassing complex I–IV of the ETC likely characterize a large number of human diseases (Cohen and Kesler, 1999a,b), at least in part. In a recent preliminary study, we reported that cardiac I/R disrupted the supercomplex structures and that ranolazine, a late Na+ channel blocker and an anti-anginal drug, preserved the integrity of the complexes following I/R (Gadicherla et al., 2010). This preservation of the mitochondrial supercomplexes by ranolazine, after I/R, was associated with improved functional recovery and more normalized mitochondrial bioenergetics in the isolated beating heart (Aldakkak et al., 2009).
Indeed, the targeting of mitochondrial ETC complexes to achieve cardioprotection has become an attractive approach compared to other remedies for protection against cardiac injury. For example, amobarbital and volatile anesthetics attenuate complex I activity, diazoxide reduces complex II activity, and hydrogen sulfide attenuates complex IV activity (discussed below and shown in Figure 3). Because of the abundance of mitochondria in cardiomyocytes, these mitochondria represent a major source of 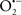 (discussed below); on the other hand, mitochondria are also major targets of ROS. Therefore the successful targeting of mitochondrial complexes for a therapeutic purpose is in part ascribed to the vulnerability of the complexes and cardiolipin to oxidative stress (Paradies et al., 1997; Lesnefsky et al., 2001).
(discussed below); on the other hand, mitochondria are also major targets of ROS. Therefore the successful targeting of mitochondrial complexes for a therapeutic purpose is in part ascribed to the vulnerability of the complexes and cardiolipin to oxidative stress (Paradies et al., 1997; Lesnefsky et al., 2001).
A defect in several of the respiratory complexes can be considered a potential source or inducer of 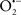 during I/R. Paradies et al. (2004, 2009) reported that a decrease in complex I activity, as observed in I/R hearts, was associated with a decrease in state 3 respiration. Others have coined the name “complex I syndrome” in which complex I inactivation is associated with protein nitration and oxidative damage to mitochondrial proteins and phospholipids (Navarro and Boveris, 2009). On the other hand, a limitation of e− transfer during ischemia to complex III, a major source of
during I/R. Paradies et al. (2004, 2009) reported that a decrease in complex I activity, as observed in I/R hearts, was associated with a decrease in state 3 respiration. Others have coined the name “complex I syndrome” in which complex I inactivation is associated with protein nitration and oxidative damage to mitochondrial proteins and phospholipids (Navarro and Boveris, 2009). On the other hand, a limitation of e− transfer during ischemia to complex III, a major source of 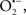 represents a new concept to limit mitochondrial damage specifically during I/R (Lesnefsky et al., 2001; Chen et al., 2006, 2008; Aldakkak et al., 2008a). In fact, we observed that attenuating complex I activity (e− flux) with the reversible inhibitor amobarbital protected the heart, and mitochondria in particular, from I/R injury when the drug was present only during ischemia (Aldakkak et al., 2008a). A recent study by Chen et al. (2010) showed that amobarbital given before ischemia preserved mitochondrial respiration in isolated mitochondria with glutamate and TMPD-ascorbate, whereas sodium azide inhibition was not protective. The authors inferred that a specific region in the ETC, most likely between complex III and IV, leads to mitochondrial damage during ischemia (Chen et al., 2010). Although the Qo site of complex III is implicated in most of the ROS during I/R, Chen et al. (2010) postulated that the protection by antimycin A against I/R injury indicates that the specific site that elicits mitochondrial damage during ischemia is likely located in the segment between complex III and complex IV.
represents a new concept to limit mitochondrial damage specifically during I/R (Lesnefsky et al., 2001; Chen et al., 2006, 2008; Aldakkak et al., 2008a). In fact, we observed that attenuating complex I activity (e− flux) with the reversible inhibitor amobarbital protected the heart, and mitochondria in particular, from I/R injury when the drug was present only during ischemia (Aldakkak et al., 2008a). A recent study by Chen et al. (2010) showed that amobarbital given before ischemia preserved mitochondrial respiration in isolated mitochondria with glutamate and TMPD-ascorbate, whereas sodium azide inhibition was not protective. The authors inferred that a specific region in the ETC, most likely between complex III and IV, leads to mitochondrial damage during ischemia (Chen et al., 2010). Although the Qo site of complex III is implicated in most of the ROS during I/R, Chen et al. (2010) postulated that the protection by antimycin A against I/R injury indicates that the specific site that elicits mitochondrial damage during ischemia is likely located in the segment between complex III and complex IV.
Other strategies for targeting the mitochondrial complexes have potential to be worthwhile approaches. For example, Hanley et al. (Hanley et al., 2002, 2003; Hanley and Daut, 2005) reported that the putative mitoKATP channel opener diazoxide may protect against I/R injury in part by inhibiting complex II, which could lead to less e− transfer to complex III and decrease reverse e− flow to complex I, and in the process minimize 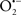 generation. In contrast, Wojtovich and Brookes (2009) reported recently that atpenin, believed to be a specific complex II inhibitor, increased post-ischemic recovery and decreased infarct size in a mitoKATP (blocked by 5-hydroxydecanoate) channel sensitive manner. The authors observed that although atpenin interacts with complex II, it had no direct effect to decrease complex II activity and that it is indeed a potent mitoKATP channel activator (Wojtovich and Brookes, 2009). Thus, the benefit of targeting the mitochondrial ETC or membrane channels is that it provides an alternative approach to cardioprotection against I/R injury (Chen et al., 2007; Nadtochiy et al., 2007).
generation. In contrast, Wojtovich and Brookes (2009) reported recently that atpenin, believed to be a specific complex II inhibitor, increased post-ischemic recovery and decreased infarct size in a mitoKATP (blocked by 5-hydroxydecanoate) channel sensitive manner. The authors observed that although atpenin interacts with complex II, it had no direct effect to decrease complex II activity and that it is indeed a potent mitoKATP channel activator (Wojtovich and Brookes, 2009). Thus, the benefit of targeting the mitochondrial ETC or membrane channels is that it provides an alternative approach to cardioprotection against I/R injury (Chen et al., 2007; Nadtochiy et al., 2007).
The structural integrity of the IMM is equally important in maintaining mitochondrial function to achieve normal and efficient OxPhos. As noted above, cardiolipin, a lipid high in unsaturated fatty acids content, is highly susceptible to oxidative and nitrosative stress (Armstrong, 2008). The loss of ETC complex activities has been ascribed to degradation of cardiolipin. For example, cardiolipin dysfunction can lead to impaired complex V activity, which would lead to impaired ATP levels, to a derangement of cellular ion homeostasis, and eventually to cell death. In a brain anoxia and reoxygenation model, mitochondrial lipid peroxidation, cardiolipin damage and cytochrome c release, and collapse of ΔΨm, were observed in isolated mitochondria, independent of mPTP opening (Morin et al., 2003b). Thus therapeutic targeting of cardiolipin is a reasonable alternative approach. Indeed, lipid replacement therapy (LRT) has gained significant traction recently as a therapeutic approach to restore mitochondrial membrane integrity and function as a means to mitigate oxidative damage (Figure 3). LRT involves actual replacement of damaged lipids of cellular and mitochondrial membranes. Fusion of mitochondria from I/R hearts, which contain lower levels of cardiolipin, with liposomes containing cardiolipin restored cardiolipin levels and preserved complex activities (Paradies et al., 2009). The restoration or preservation of the integrity of the IMM, specifically its cardiolipin content, is likely to be beneficial in restoring cardiac function. Overall, these studies highlight an emerging paradigm that restoration of mitochondrial structural integrity may improve ETC efficiency and hinder activation of apoptotic mechanisms.
As alluded to before, mitochondrial electrophysiology is crucial in determining whether there is mitochondrial function or dysfunction. An increase in K+ flux via the putative mitoKATP channel may lead to an increase in matrix volume and may result in more efficient ATP synthesis (Garlid et al., 2003) and better cellular preservation. In a recent study (Stowe et al., 2006) we provided novel evidence for a regulatory role of the putative mitoKCa on mitochondrial function and its cardioprotective value against I/R injury. Mitochondrial ROS scavengers abolished the cardioprotection implying a key role of ROS in activation of the putative mitoKCa channel-mediated cardioprotection. Putative activation of mitoKATP channels, for example with diazoxide, which as noted above has other effects on mitochondria, also has cardioprotective effects; preventing mCa2+ overload and subsequently preserving ΔΨm are considered as possible mechanisms (Teshima et al., 2003a). The implications for the involvement of mitochondrial channels and/or exchangers in cellular damage portend a potential therapeutic utility in the management of ischemic disease states.
Therapeutic Potential of Uncoupling Proteins
Uncoupling proteins (UCPs), which are integral membrane proteins located in the IMM (Figure 1), are important physiological regulators of mitochondrial function and redox potential; thus UCPs are also regulators of 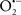 production. Uncoupled OxPhos results from e− transfer and H+ influx into the matrix independent of ADP phosphorylation (Sack, 2006b). UCPs stimulate mitochondrial respiration by partially dissipating the H+ gradient across the IMM, which tends to destabilize ΔΨm. Excessive ROS accumulation commonly occurs in state 4 respiration due to the high electrochemical gradient and limited e− transfer. One of the effects of UCPs is to prevent buildup of ROS by partially dissipating the ΔΨm (Teshima et al., 2003a; Sack, 2006b). The physiologic role of uncoupled respiration is best exemplified by the adaptive thermogenesis induced by UCPs in brown fat. Because UCPs are capable of reducing mitochondrial ROS formation by modulating ΔΨm (Haines et al., 2010), they have an impact to reduce the damaging effects of ROS during cardiac I/R or hypoxia injury (Fiskum et al., 2004; Barreiro et al., 2009).
production. Uncoupled OxPhos results from e− transfer and H+ influx into the matrix independent of ADP phosphorylation (Sack, 2006b). UCPs stimulate mitochondrial respiration by partially dissipating the H+ gradient across the IMM, which tends to destabilize ΔΨm. Excessive ROS accumulation commonly occurs in state 4 respiration due to the high electrochemical gradient and limited e− transfer. One of the effects of UCPs is to prevent buildup of ROS by partially dissipating the ΔΨm (Teshima et al., 2003a; Sack, 2006b). The physiologic role of uncoupled respiration is best exemplified by the adaptive thermogenesis induced by UCPs in brown fat. Because UCPs are capable of reducing mitochondrial ROS formation by modulating ΔΨm (Haines et al., 2010), they have an impact to reduce the damaging effects of ROS during cardiac I/R or hypoxia injury (Fiskum et al., 2004; Barreiro et al., 2009).
There are four UCPs variants (1–4) believed to induce inward H+ leak in energized mitochondria (Jezek et al., 1998; Stuart et al., 2001; Brookes, 2005; Stowe and Camara, 2009) (Figure 1) and thereby to uncouple OxPhos. This uncoupling is well established for UCP1 with its central role in thermogenesis whereas the functions of the other isoforms are still being debated. UCP2 and UCP3 are present in the heart and provide some cardioprotection (Sack, 2006b). Knocking out UCP2 increased brain damage after middle cerebral artery occlusion (Haines et al., 2010). Other studies show that UCP2 overexpression is inversely correlated with caspase-3 activation in neurons (Deierborg et al., 2008). In rat neonatal cardiomyocytes, UCP2 overexpression confers tolerance to oxidative stress via diminished mCa2+ uptake and reduced ROS production (Sack, 2006b). Some lipid peroxidation products, such as 4-hydroxy-trans-2-noneal (HNE), may induce partial uncoupling of mitochondria through UCPs and are thereby also reported to initiate protective mechanisms (Boss et al., 1997; Echtay et al., 2003).
It is worth repeating that during myocardial I/R injury or heart failure an increase in catecholamine concentration activates FA utilization in cardiomyocytes by β-oxidation. With increased β-oxidation there is a concomitant increase in mitochondrial ROS, but gene expression of UCPs is also activated so that tends to reduce ΔΨm and ROS production. So it is interesting that chronic treatment with trimetazidine, which partially blocks β-oxidation in the matrix, has been shown to mitigate ischemic or heart failure disease (Ashrafian et al., 2007), but this may be counterintuitive as β-oxidation furnishes more energy per carbon than glycolysis.
In a recent study we (Heinen et al., 2007) suggested that a small H+ leak brought on by the putative mitoKCa channel opening NS1619 in cardiomyocytes led to ROS production without substantially changing ΔΨm. It is possible that this “mild” uncoupling may contribute to the protection of the heart against I/R injury as shown by NS1619 (Stowe et al., 2006). We showed that the NS1619-induced protection in isolated hearts was mediated by preservation of mitochondrial redox state, reduced mCa2+ loading, decreased deleterious ROS production, and better functional recovery; this protection was abolished when hearts were treated either with MnTBAP, an antioxidant, or paxilline, a KCa channel antagonist (Camara et al., 2010b). It has also been suggested that mitoKATP channel openers (Murata et al., 2001) and volatile anesthetics (Sedlic et al., 2010a,b) are cardioprotective by virtue of inducing a “small amount” of ROS production and mild uncoupling of mitochondria (Figure 3).
In addition to stimulating UCPs to ameliorate pathological ROS production and protect against oxidative injury, there is the potential therapeutic use of low doses of artificial uncouplers to target the mitochondrion. One such drug is the protonophore dinitrophenol (DNP), which has been utilized to treat obesity (Camara et al., 2010b). However DNP, if used in excess can markedly reduce ΔΨm, so its low therapeutic index limits its utility as a viable therapeutic option (Blaikie et al., 2006). Similarly, low concentrations of carbonyl cyanide p-(trifluoromethoxy) phenylhydrazine have been reported to protect by preventing mPTP opening (Teshima et al., 2003b) (Figure 3). Taken together, these findings suggest that a better knowledge of the ionic mechanism of H+ leak by UCPs and synthetic uncouplers could provide the basis for better design of mitochondriotropic drugs to reduce cellular injury following oxidative stress. Indeed, animal studies of heart failure and transgenic mouse models have suggested that UCPs may be important in the endogenous response of heart muscles to oxidant stress and that UCPs may be a potential target for heart disease therapy (Laskowski and Russell, 2008).
Targeting Mitochondrial ROS and RNS Generation as Potential Therapies
In this and the subsequent section, ROS (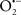 , H2O2 and
, H2O2 and 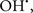 etc.) and RNS (
etc.) and RNS (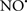 and ONOO−, etc.) generation and scavenging will be discussed. Under the physiologic state, “small amounts” of
and ONOO−, etc.) generation and scavenging will be discussed. Under the physiologic state, “small amounts” of 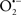 are formed during mitochondrial respiration, but which are maintained at acceptable levels by endogenous matrix antioxidant defenses (Tsutsui et al., 2006) (Figure 4). A “small amount” of ROS is important in normal cellular function. In excitable tissue, especially cardiac and neuronal, mitochondria represent a major source of
are formed during mitochondrial respiration, but which are maintained at acceptable levels by endogenous matrix antioxidant defenses (Tsutsui et al., 2006) (Figure 4). A “small amount” of ROS is important in normal cellular function. In excitable tissue, especially cardiac and neuronal, mitochondria represent a major source of 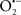 as a consequence of large mitochondrial numbers and a high level of OxPhos (Cadenas et al., 1977; Cadenas and Davies, 2000; Andreyev et al., 2005). H2O2, a relatively inactive compound, is generated from dismutation of
as a consequence of large mitochondrial numbers and a high level of OxPhos (Cadenas et al., 1977; Cadenas and Davies, 2000; Andreyev et al., 2005). H2O2, a relatively inactive compound, is generated from dismutation of 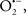 and in a Fe-dependent Fenton reaction, it generates the toxic radical group Fe=Oo (Stowe and Camara, 2009). When administered on reperfusion the Fe chelator desferrioxamine was found to improve function during cardiac I/R (Lesnefsky et al., 1990; Dobsak et al., 2002; Liachenko et al., 2003).
and in a Fe-dependent Fenton reaction, it generates the toxic radical group Fe=Oo (Stowe and Camara, 2009). When administered on reperfusion the Fe chelator desferrioxamine was found to improve function during cardiac I/R (Lesnefsky et al., 1990; Dobsak et al., 2002; Liachenko et al., 2003).
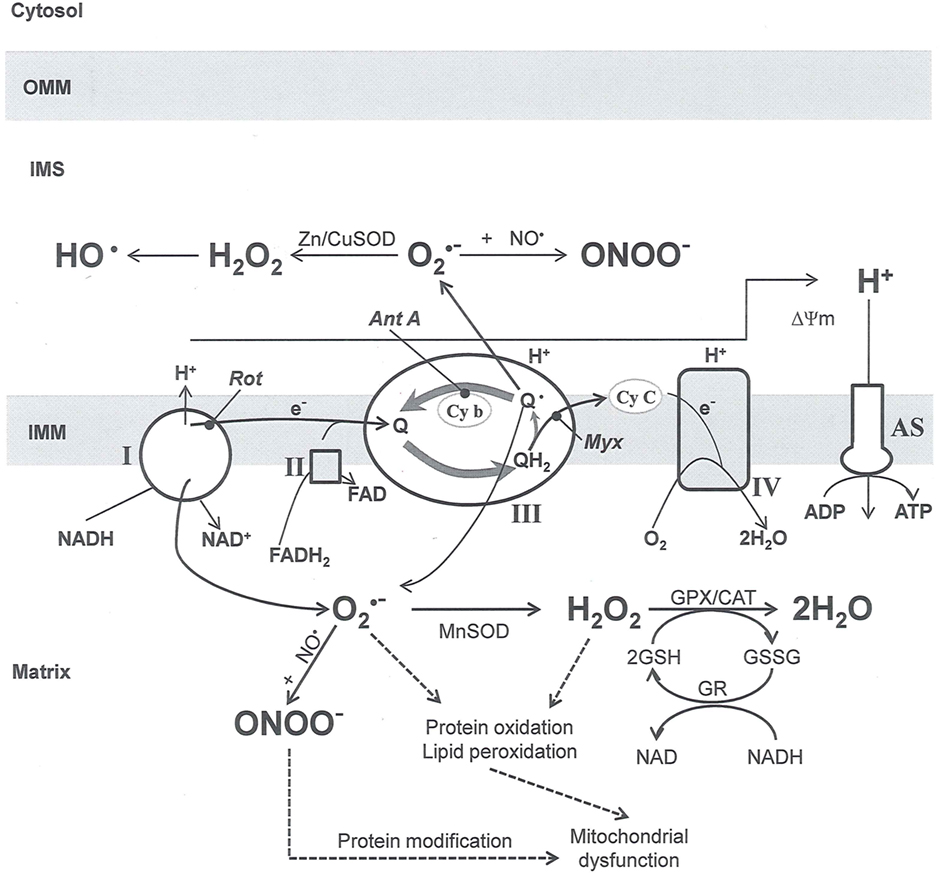
Figure 4. Principal mechanisms for mitochondrial reactive oxygen species (ROS) generation. Reduced substrates synthesized in metabolic pathways supply e− to complex I and complex II of the ETC. The major centers for superoxide (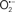 ) formation are complex I and complex III, although small amounts may also be formed at complex II and complex IV (omitted for clarity). The electron carrier of complex III ubiquinone (Q) is reduced to ubiquinol (QH2), which transfers an electron to cytochrome c (Cy C) through a Fe protein (not shown) inhibited by myxothiazol (Myx). The resulting semiubiquinone (Qo) is oxidized back to ubiquinone by cytochrome b (Cy b), and can also transfer e− to O2 to form
) formation are complex I and complex III, although small amounts may also be formed at complex II and complex IV (omitted for clarity). The electron carrier of complex III ubiquinone (Q) is reduced to ubiquinol (QH2), which transfers an electron to cytochrome c (Cy C) through a Fe protein (not shown) inhibited by myxothiazol (Myx). The resulting semiubiquinone (Qo) is oxidized back to ubiquinone by cytochrome b (Cy b), and can also transfer e− to O2 to form  Myx reduces
Myx reduces  production because it blocks Qo formation, whereas antimycin A (Ant A) enhances it by increasing Qo levels. The drug rotenone (Rot) inhibits electron flow distal to O2 generation, enhancing its production. The
production because it blocks Qo formation, whereas antimycin A (Ant A) enhances it by increasing Qo levels. The drug rotenone (Rot) inhibits electron flow distal to O2 generation, enhancing its production. The  generated by Myx and Ant A are topographically oriented, whereas the
generated by Myx and Ant A are topographically oriented, whereas the  generated by Myx are directed to the matrix where they are scavenged;
generated by Myx are directed to the matrix where they are scavenged; 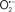 generated by Ant A are released into the IMS and so escape the powerful matrix scavengers. The figure also depicts the dismutation of ROS and the reaction
generated by Ant A are released into the IMS and so escape the powerful matrix scavengers. The figure also depicts the dismutation of ROS and the reaction 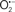 and
and 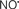 to generate ONOO−. A better understanding of the topographical sites of
to generate ONOO−. A better understanding of the topographical sites of 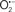 generation and the role it would have in a disease process could be important in targeted scavenging of ROS to mitigate or prevent the initiation or progression of a particular-mitochondria-related dysfunction and cardiovascular abnormalities. Modified reproduction with permission (Camello-Almaraz et al., 2006). SOD, superoxide dismutase; H2O2, hydrogen peroxide; ONOO−, peroxynitrite;
generation and the role it would have in a disease process could be important in targeted scavenging of ROS to mitigate or prevent the initiation or progression of a particular-mitochondria-related dysfunction and cardiovascular abnormalities. Modified reproduction with permission (Camello-Almaraz et al., 2006). SOD, superoxide dismutase; H2O2, hydrogen peroxide; ONOO−, peroxynitrite; 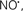 nitric oxide; ΔΨm: mitochondrial potential; AS, ATP synthase; OMM, outer mitochondrial membrane; IMM, inner mitochondrial membrane; GSH and GSSG reduced and oxidized glutathione, respectively; GPX, GR and CAT glutathione peroxidase, glutathione reductase and catalase, respectively.
nitric oxide; ΔΨm: mitochondrial potential; AS, ATP synthase; OMM, outer mitochondrial membrane; IMM, inner mitochondrial membrane; GSH and GSSG reduced and oxidized glutathione, respectively; GPX, GR and CAT glutathione peroxidase, glutathione reductase and catalase, respectively.
Many more recent observations indicate that the targeting of mitochondrial 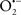 as a therapeutic goal in cellular dysfunction requires a careful balancing of the “good” (physiological) and “bad” (pathological)
as a therapeutic goal in cellular dysfunction requires a careful balancing of the “good” (physiological) and “bad” (pathological) 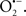 Small amounts of ROS have been implicated in pre- and post-conditioning cytoprotection as evidenced by the loss of protection in the presence of scavengers (Stowe and Camara, 2009; Camara et al., 2010b). When ROS production exceeds the capacity of detoxification and repair pathways, oxidative damage to proteins, DNA (mtDNA and nDNA), and membrane lipids such as cardiolipin occurs. In post-mitotic cells the continuous accumulation of oxidative damage triggers the accumulation of non-functional mitochondria, a process that could lead to mitochondrial autophagy. These conditions can culminate in impaired ATP generation and cell death (James et al., 2005). Hence, mitochondrial ROS generation and oxidative damage are potential targets (Figure 3) to apply pharmacological and molecular approaches for therapeutic interventions (James et al., 2005) as discussed in detail below.
Small amounts of ROS have been implicated in pre- and post-conditioning cytoprotection as evidenced by the loss of protection in the presence of scavengers (Stowe and Camara, 2009; Camara et al., 2010b). When ROS production exceeds the capacity of detoxification and repair pathways, oxidative damage to proteins, DNA (mtDNA and nDNA), and membrane lipids such as cardiolipin occurs. In post-mitotic cells the continuous accumulation of oxidative damage triggers the accumulation of non-functional mitochondria, a process that could lead to mitochondrial autophagy. These conditions can culminate in impaired ATP generation and cell death (James et al., 2005). Hence, mitochondrial ROS generation and oxidative damage are potential targets (Figure 3) to apply pharmacological and molecular approaches for therapeutic interventions (James et al., 2005) as discussed in detail below.
Although 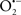 is generated from several sources in the mitochondrion, the main source of mitochondrial
is generated from several sources in the mitochondrion, the main source of mitochondrial 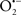 appears to be within the IMM of the ETC, in particular at complexes I and III (Turrens and Boveris, 1980; Cadenas and Davies, 2000; Chen et al., 2003, 2007). However, the specific sites and mechanisms of
appears to be within the IMM of the ETC, in particular at complexes I and III (Turrens and Boveris, 1980; Cadenas and Davies, 2000; Chen et al., 2003, 2007). However, the specific sites and mechanisms of 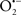 generation remain unsettled (Turrens et al., 1985; Ambrosio et al., 1993; Babcock and Varotsis, 1993; Chen et al., 2003). In intact mitochondria from normal hearts, ROS are formed from complex I when e− flux is backward from complex II, or at the ubisemiquinone site of complex III (Camara et al., 2010b), where a radical intermediate is formed through the Q-cycle of the complex, particularly at the Qo site (Figure 4). Indeed experiments have shown that complex III generates
generation remain unsettled (Turrens et al., 1985; Ambrosio et al., 1993; Babcock and Varotsis, 1993; Chen et al., 2003). In intact mitochondria from normal hearts, ROS are formed from complex I when e− flux is backward from complex II, or at the ubisemiquinone site of complex III (Camara et al., 2010b), where a radical intermediate is formed through the Q-cycle of the complex, particularly at the Qo site (Figure 4). Indeed experiments have shown that complex III generates 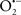 through the oxidation of ubisemiquinone, which faces the IMS, while complex I mediated
through the oxidation of ubisemiquinone, which faces the IMS, while complex I mediated 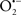 generation is directed mostly toward the strongly scavenging matrix side (Chen et al., 2003, 2007, 2008).
generation is directed mostly toward the strongly scavenging matrix side (Chen et al., 2003, 2007, 2008).
In cardiac I/R injury, impaired complex I can enhance 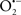 generation as a result of increased e− leak as e− transfer is impeded (Camara et al., 2010b). When FADH2 related substrates are used and an e− enters the ETC at complex II,
generation as a result of increased e− leak as e− transfer is impeded (Camara et al., 2010b). When FADH2 related substrates are used and an e− enters the ETC at complex II, 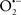 may be generated by reverse e− transfer to the FMN site of complex I (Chen et al., 2007; Zoccarato et al., 2007; Stowe and Camara, 2009). Although complex I is a site for
may be generated by reverse e− transfer to the FMN site of complex I (Chen et al., 2007; Zoccarato et al., 2007; Stowe and Camara, 2009). Although complex I is a site for 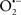 generation (Chen et al., 2008), complex III may become a major site for
generation (Chen et al., 2008), complex III may become a major site for 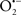 generation (Chen et al., 2008) in cardiac cells under ischemic conditions. Ischemia damages complex III by functionally altering the Fe–S protein subunits (Chen et al., 2007). As will be discussed, an increase in
generation (Chen et al., 2008) in cardiac cells under ischemic conditions. Ischemia damages complex III by functionally altering the Fe–S protein subunits (Chen et al., 2007). As will be discussed, an increase in 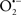 emission (
emission (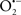 generation –
generation – 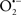 -scavenging) under pathological conditions can also occur as a consequence of depletion or a defect in the mitochondrial antioxidant system (Camara et al., 2010b). The increase in ROS production can contribute to a self-regenerating ROS production facilitated by ROS-induced ROS release (Zorov et al., 2000; Brady et al., 2006b; Stowe and Camara, 2009). This increase in oxidative stress will result in further damage to mitochondria. A point is reached where the scavenging system almost completely collapses and emission of ROS is further perpetuated in a vicious cycle.
-scavenging) under pathological conditions can also occur as a consequence of depletion or a defect in the mitochondrial antioxidant system (Camara et al., 2010b). The increase in ROS production can contribute to a self-regenerating ROS production facilitated by ROS-induced ROS release (Zorov et al., 2000; Brady et al., 2006b; Stowe and Camara, 2009). This increase in oxidative stress will result in further damage to mitochondria. A point is reached where the scavenging system almost completely collapses and emission of ROS is further perpetuated in a vicious cycle.
The free radical 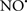 that is released from the vascular endothelium probably exerts an important role in regulating mitochondrial respiration.
that is released from the vascular endothelium probably exerts an important role in regulating mitochondrial respiration. 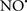 and its derivatives have emerged as significant players in the control of mitochondrial function and dysfunction (Camara et al., 2010b). Some of the effects of RNS include: (1) competitive inhibition of cytochrome oxidase (complex IV), (2) partial inactivation of complex I, (3) activation of mPTP, and (4) stimulation of mitochondrial biogenesis (Camara et al., 2010a).
and its derivatives have emerged as significant players in the control of mitochondrial function and dysfunction (Camara et al., 2010b). Some of the effects of RNS include: (1) competitive inhibition of cytochrome oxidase (complex IV), (2) partial inactivation of complex I, (3) activation of mPTP, and (4) stimulation of mitochondrial biogenesis (Camara et al., 2010a).
The inhibition of complex I by S-nitrosation has surfaced recently as an important mechanism in cardioprotection by IPC due to the direct action of 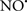 on mitochondria independent of its effects on the soluble guanylate cyclase signaling pathway (Burwell et al., 2009). In a way, the partial inhibitory effect of
on mitochondria independent of its effects on the soluble guanylate cyclase signaling pathway (Burwell et al., 2009). In a way, the partial inhibitory effect of 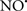 on complex I is analogous to the protective effect afforded by amobarbital on complex I (Aldakkak et al., 2008a). However, the effect of
on complex I is analogous to the protective effect afforded by amobarbital on complex I (Aldakkak et al., 2008a). However, the effect of 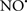 to bind to complex I can also result in increased
to bind to complex I can also result in increased 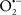 and H2O2 (Camara et al., 2010a). Furthermore,
and H2O2 (Camara et al., 2010a). Furthermore, 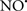 reacts with
reacts with 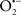 to generate the potent RNS product peroxynitrite (ONOO−; Figure 4).
to generate the potent RNS product peroxynitrite (ONOO−; Figure 4).
The main source of 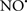 in cardiac mitochondria likely arises from the vascular endothelium, but there are other potential cytosolic sources, such as nerve terminals or mitochondria.
in cardiac mitochondria likely arises from the vascular endothelium, but there are other potential cytosolic sources, such as nerve terminals or mitochondria. 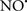 has multiple targets in mitochondria including metalloproteins containing Fe–S clusters that are known to be highly sensitive to a direct oxidative modification by
has multiple targets in mitochondria including metalloproteins containing Fe–S clusters that are known to be highly sensitive to a direct oxidative modification by 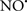 ; other important targets are the lipid thiols. Like
; other important targets are the lipid thiols. Like 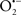 the role of
the role of 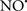 and its products in the cell is akin to a “double-edged” sword.
and its products in the cell is akin to a “double-edged” sword. 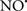 can act both as a vasodilatory and cardioprotective molecule and as a facilitator of cellular injury.
can act both as a vasodilatory and cardioprotective molecule and as a facilitator of cellular injury. 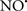 can act directly on mitochondria, independent of guanylate cyclase, to protect tissue/organs, or it can provide protection by way of a
can act directly on mitochondria, independent of guanylate cyclase, to protect tissue/organs, or it can provide protection by way of a 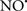 -mediated signaling cascade. But, the most important feature
-mediated signaling cascade. But, the most important feature 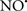 of in mitochondria is likely the competitive, reversible inhibition of O2 binding at complex IV. The
of in mitochondria is likely the competitive, reversible inhibition of O2 binding at complex IV. The 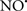 inhibition of O2 binding is reversible as this depends on the concentration of the two gases in the mitochondrion (Camara et al., 2010b).
inhibition of O2 binding is reversible as this depends on the concentration of the two gases in the mitochondrion (Camara et al., 2010b).
Although there is considerable support for the concept that 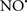 per se is cardioprotective, the exact mechanisms for this effect remains to be unraveled. Under physiological conditions, when the O2 concentration is low,
per se is cardioprotective, the exact mechanisms for this effect remains to be unraveled. Under physiological conditions, when the O2 concentration is low, 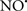 may effectively act as a regulator of the mitochondrial ETC to induce reversible inhibition of e− transfer. In this situation,
may effectively act as a regulator of the mitochondrial ETC to induce reversible inhibition of e− transfer. In this situation, 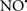 generation during hypoxia may lower mitochondrial respiration as an adaptation to lower O2 availability (Harrois et al., 2009). In addition, inhibition of complex IV by
generation during hypoxia may lower mitochondrial respiration as an adaptation to lower O2 availability (Harrois et al., 2009). In addition, inhibition of complex IV by 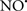 is thought to elicit cardioprotection by preserving the limited supply of O2.
is thought to elicit cardioprotection by preserving the limited supply of O2. 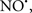 in limited amounts, may also provide protection against I/R injury in part by preserving cardiolipin (discussed above under IMM), which is essential for complex IV function, and to minimize cytochrome c release and other apoptotic factors (Gorbunov et al., 1997; Osipov et al., 2006; Vlasova et al., 2006). Of course, nitration of amino acid residues in the ETC complexes and other proteins (VDAC, ANT), likely leads to altered enzyme function. Nitrosation of complex I on the other hand, has been shown to protect against I/R injury (Nadtochiy et al., 2007). In a recent study, Cheng et al. (2011) reported that
in limited amounts, may also provide protection against I/R injury in part by preserving cardiolipin (discussed above under IMM), which is essential for complex IV function, and to minimize cytochrome c release and other apoptotic factors (Gorbunov et al., 1997; Osipov et al., 2006; Vlasova et al., 2006). Of course, nitration of amino acid residues in the ETC complexes and other proteins (VDAC, ANT), likely leads to altered enzyme function. Nitrosation of complex I on the other hand, has been shown to protect against I/R injury (Nadtochiy et al., 2007). In a recent study, Cheng et al. (2011) reported that 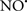 caused a concentration-dependent biphasic inhibitory effect of the cardiac VDAC that was incorporated into a planer lipid bilayer. The implications of these novel findings is that post-translational modification of VDAC by
caused a concentration-dependent biphasic inhibitory effect of the cardiac VDAC that was incorporated into a planer lipid bilayer. The implications of these novel findings is that post-translational modification of VDAC by 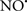 and other nitrogen containing molecules may be important in regulating the function of the channel, thereby preserving mitochondrial function and cell survival. Lastly,
and other nitrogen containing molecules may be important in regulating the function of the channel, thereby preserving mitochondrial function and cell survival. Lastly, 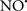 is reported to decrease mCa2+ overload by binding to complex I and this appears to be associated with a decrease in ΔΨm (Solaini and Harris, 2005; Burwell and Brookes, 2008).
is reported to decrease mCa2+ overload by binding to complex I and this appears to be associated with a decrease in ΔΨm (Solaini and Harris, 2005; Burwell and Brookes, 2008). 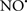 is a mediator of preconditioning through several putative pathways (Camara et al., 2010b). It is plausible that some of the protective effects of
is a mediator of preconditioning through several putative pathways (Camara et al., 2010b). It is plausible that some of the protective effects of 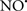 can also be attributed to a coordinated series of responses by modest levels of
can also be attributed to a coordinated series of responses by modest levels of 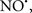 which indirectly enhances mitochondrial function by increasing local blood supply to enhance O2 and substrate delivery to mitochondria (Camara et al., 2010b).
which indirectly enhances mitochondrial function by increasing local blood supply to enhance O2 and substrate delivery to mitochondria (Camara et al., 2010b).
At higher concentrations, 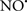 appears to shift the balance from cytoprotection toward damage and apoptosis. Increased mitochondrial
appears to shift the balance from cytoprotection toward damage and apoptosis. Increased mitochondrial 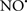 may induce cell death by necrosis and apoptosis via mPTP opening (Solaini and Harris, 2005). Indeed, as noted elsewhere, ONOO− is thought to be an important mediator of tissue injury in myocardial ischemia-reperfusion (Camara et al., 2002). The most damaging effects of
may induce cell death by necrosis and apoptosis via mPTP opening (Solaini and Harris, 2005). Indeed, as noted elsewhere, ONOO− is thought to be an important mediator of tissue injury in myocardial ischemia-reperfusion (Camara et al., 2002). The most damaging effects of 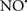 activity on mitochondria are attributed to an increased susceptibility of cardiolipin to ONOO− (Chen et al., 2007). It is important to note that while
activity on mitochondria are attributed to an increased susceptibility of cardiolipin to ONOO− (Chen et al., 2007). It is important to note that while 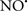 mainly reacts reversibly at complex IV, ONOO− is known to affect complexes I, II, and V by irreversible nitration of the tyrosine residues (Duchen, 2004). Thus conditions leading to excess ONOO− will promote enhanced mitochondrial lipid/protein damage, Ca2+ uptake and swelling, rupture or increased permeabilization of OMM and cytochrome c release. ONOO− also can directly oxidize glutathione (GSH; (Harrois et al., 2009).
mainly reacts reversibly at complex IV, ONOO− is known to affect complexes I, II, and V by irreversible nitration of the tyrosine residues (Duchen, 2004). Thus conditions leading to excess ONOO− will promote enhanced mitochondrial lipid/protein damage, Ca2+ uptake and swelling, rupture or increased permeabilization of OMM and cytochrome c release. ONOO− also can directly oxidize glutathione (GSH; (Harrois et al., 2009).
The balance between physiologically relevant levels and pathological concentrations of 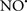 is critical in the role of this gas in mitochondrial function (Kim et al., 1999) and points out the difficulty in maintaining a delicate balance between
is critical in the role of this gas in mitochondrial function (Kim et al., 1999) and points out the difficulty in maintaining a delicate balance between 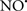 generation and ONOO− production during development of potential therapeutic approaches to target mitochondria. Given that ONOO− is an intermediate formed from
generation and ONOO− production during development of potential therapeutic approaches to target mitochondria. Given that ONOO− is an intermediate formed from 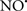 and
and 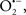 inhibition of either
inhibition of either 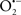 or
or 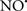 generation will attenuate the levels of ONOO− and minimize mitochondrial damage (Camara et al., 2010b). For example, overexpression of MnSOD has been shown to protect neuronal-like cells against
generation will attenuate the levels of ONOO− and minimize mitochondrial damage (Camara et al., 2010b). For example, overexpression of MnSOD has been shown to protect neuronal-like cells against 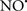 -dependent cell injury, tyrosine nitration, and lipid peroxidation. In addition, overexpression of nNOS initiates
-dependent cell injury, tyrosine nitration, and lipid peroxidation. In addition, overexpression of nNOS initiates 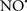 -dependent neuronal cell death, which is alleviated in cells enriched in MnSOD (Radi et al., 2001). However, the chemical reaction between
-dependent neuronal cell death, which is alleviated in cells enriched in MnSOD (Radi et al., 2001). However, the chemical reaction between 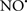 and
and 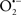 is faster than the reaction with MnSOD (Ferdinandy and Schulz, 2003) and ONOO− mediated inactivation of the enzyme amplifies
is faster than the reaction with MnSOD (Ferdinandy and Schulz, 2003) and ONOO− mediated inactivation of the enzyme amplifies 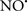 and ONOO−-dependent mitochondrial oxidative stress (Radi et al., 2001). It is reported that ONOO− can inhibit mitochondrial aconitase (Hausladen and Fridovich, 1994) and stimulate the release of AIF and other apoptogenic factors from mitochondria, which in turn leads to DNA fragmentation or increased cytochrome c peroxidase activity that can worsen the oxidative damage of mitochondrial proteins and membranes (Szabo et al., 2007). Overall, a targeted approach against excess ONOO− and
and ONOO−-dependent mitochondrial oxidative stress (Radi et al., 2001). It is reported that ONOO− can inhibit mitochondrial aconitase (Hausladen and Fridovich, 1994) and stimulate the release of AIF and other apoptogenic factors from mitochondria, which in turn leads to DNA fragmentation or increased cytochrome c peroxidase activity that can worsen the oxidative damage of mitochondrial proteins and membranes (Szabo et al., 2007). Overall, a targeted approach against excess ONOO− and 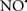 with allowance for “small” concentrations of
with allowance for “small” concentrations of 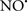 as a signaling mediator for cytoprotection may be an important initial strategy to protect against cardiac cell injury from oxidative/nitrosative stress (Camara et al., 2010b). Indeed at low concentrations even ONOO− has also been reported to be cardioprotective (Nossuli et al., 1998).
as a signaling mediator for cytoprotection may be an important initial strategy to protect against cardiac cell injury from oxidative/nitrosative stress (Camara et al., 2010b). Indeed at low concentrations even ONOO− has also been reported to be cardioprotective (Nossuli et al., 1998).
Low levels of ONOO−, however, may actually provide protection (Camara et al., 2010b) through its decomposition products, NO2 and NO3 (Szabo et al., 2007), by reacting with glutathione to form S-nitrosoglutathione (Nossuli et al., 1998), and/or by acting as an intermediate in preconditioning (Ferdinandy and Schulz, 2003). The earliest evidence in favor of a protective effect of ONOO− is its decomposition to other nitrogen compounds by the Fe(III) porphyrin compounds in the carrageenan-induced paw edema model (Szabo et al., 2007). Since then, other studies have reported ONOO− furnishes protection against diabetes-associated impairment of erectile dysfunction and I/R injury. In I/R injury the cardioprotective effect were demonstrated by reduced infarct size and improved contractile function (Ronson et al., 1999; Szabo et al., 2007).
In cardiomyocytes, ONOO− protection may be mediated in part by nitration of PKCε, which in turn activates mitoKATP channels resulting in inhibition of mPTP opening. ONOO− conversion to 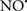 in the presence of hemoglobin, or decomposition of nitrite in the presence of myoglobin nitrite reductase to form
in the presence of hemoglobin, or decomposition of nitrite in the presence of myoglobin nitrite reductase to form 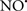 , may all lead to protection by inhibiting complex IV or by activation of mitoKATP, to block mPTP opening (Gladwin and Kim-Shapiro, 2008; Nediani et al., 2011). As noted above,
, may all lead to protection by inhibiting complex IV or by activation of mitoKATP, to block mPTP opening (Gladwin and Kim-Shapiro, 2008; Nediani et al., 2011). As noted above, 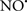 can also inhibit the ETC thus tending to preserve O2 gradients during ischemia and to favor a more controlled resumption of respiration during reperfusion to minimize ROS production (Nediani et al., 2011). Clearly, the mechanism of protection by nitrogen containing molecules, such as nitrites, await further characterization, and the reproducibility of this effect in multiple animal models of I/R suggests nitrite as a novel potential therapy for ischemic heart diseases (Nediani et al., 2011).
can also inhibit the ETC thus tending to preserve O2 gradients during ischemia and to favor a more controlled resumption of respiration during reperfusion to minimize ROS production (Nediani et al., 2011). Clearly, the mechanism of protection by nitrogen containing molecules, such as nitrites, await further characterization, and the reproducibility of this effect in multiple animal models of I/R suggests nitrite as a novel potential therapy for ischemic heart diseases (Nediani et al., 2011).
Potential Therapeutic Value of Mitochondrial ROS Scavenging
Under physiologic conditions, scavengers or antioxidants can attenuate the toxic effects of ROS and RNS. This is because mitochondria are endowed with an elaborate and well-defined multi-leveled antioxidant defense system of enzymes and non-enzymes to quench mitochondrial 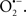 The scavenging system includes the matrix MnSOD and glutathione (GSH; Figure 4) and thioredoxin (TRXSH2) systems, cytochrome c, peroxidase, catalase, and others. A detailed review of ROS scavengers, which is beyond the scope of this review, has been discussed in recent comprehensive review articles by two of the authors (Stowe and Camara, 2009; Camara et al., 2010b).
The scavenging system includes the matrix MnSOD and glutathione (GSH; Figure 4) and thioredoxin (TRXSH2) systems, cytochrome c, peroxidase, catalase, and others. A detailed review of ROS scavengers, which is beyond the scope of this review, has been discussed in recent comprehensive review articles by two of the authors (Stowe and Camara, 2009; Camara et al., 2010b).
The association of increased ROS generation in various pathological conditions has made development of the ideal antioxidant therapy to target the mitochondrion an important goal (Camara et al., 2010b). Recent evidence suggests that targeting 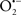 emission during I/R requires adoption of alternative approaches, i.e., focusing on the
emission during I/R requires adoption of alternative approaches, i.e., focusing on the 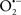 generation that occurs during ischemia as well as
generation that occurs during ischemia as well as 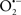 the emission during reperfusion (Camara et al., 2010b)
the emission during reperfusion (Camara et al., 2010b) 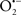 . generation during ischemia sets the stage for an increase
. generation during ischemia sets the stage for an increase 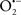 in emission during reperfusion as a mechanism of cellular injury (Chen et al., 2007, 2008; Aldakkak et al., 2008a). As noted before, blockade of complex I and III with amobarbital and antimycin A, respectively, can protect mitochondria against ischemic damage (Chen et al., 2010). Clinically, this is a relevant area of research because patients with active myocardial ischemia could theoretically receive pharmacologic therapies that target the spatial and temporal aspects of ROS production. Pharmaceutical agents that provide ROS scavenging systems are most effective if they address the problem at its source, in this case within the IMM (Hoye et al., 2008). Overall, a better understanding of the sources and direction of
in emission during reperfusion as a mechanism of cellular injury (Chen et al., 2007, 2008; Aldakkak et al., 2008a). As noted before, blockade of complex I and III with amobarbital and antimycin A, respectively, can protect mitochondria against ischemic damage (Chen et al., 2010). Clinically, this is a relevant area of research because patients with active myocardial ischemia could theoretically receive pharmacologic therapies that target the spatial and temporal aspects of ROS production. Pharmaceutical agents that provide ROS scavenging systems are most effective if they address the problem at its source, in this case within the IMM (Hoye et al., 2008). Overall, a better understanding of the sources and direction of 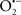 generation from the mitochondrion is crucial to an understanding of the potential of particular antioxidants used to mitigate oxidative stress and cellular damage. However, effective delivery of these antioxidants into the cytosol or matrix as a therapy remains elusive and problematic. Attempts to address these physical limitations are being pursued by reformulations of different therapeutic antioxidants based on the much larger ΔΨm than cell membrane potential.
generation from the mitochondrion is crucial to an understanding of the potential of particular antioxidants used to mitigate oxidative stress and cellular damage. However, effective delivery of these antioxidants into the cytosol or matrix as a therapy remains elusive and problematic. Attempts to address these physical limitations are being pursued by reformulations of different therapeutic antioxidants based on the much larger ΔΨm than cell membrane potential.
Because MnSOD is located primarily in the matrix (Figure 4), it plays an essential role in protecting against oxidative stress, and is therefore a critical component of normal development and survival. Its only known function is to detoxify 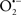 to H2O2. MnSOD is a nuclear-encoded protein that is transported to the mitochondria via a targeting sequence through the import machinery. A number of studies have shown better cell survival under oxidative stress, e.g., lipopolysaccharide treatment, by increasing the level of MnSOD (Czaja et al., 1994; Rogers et al., 2001). In SOD2+/− mice, brain infarct size is greater than wild type after focal cerebral ischemia. Accordingly, overexpression of MnSOD results in neuroprotection (Duckles et al., 2006).
to H2O2. MnSOD is a nuclear-encoded protein that is transported to the mitochondria via a targeting sequence through the import machinery. A number of studies have shown better cell survival under oxidative stress, e.g., lipopolysaccharide treatment, by increasing the level of MnSOD (Czaja et al., 1994; Rogers et al., 2001). In SOD2+/− mice, brain infarct size is greater than wild type after focal cerebral ischemia. Accordingly, overexpression of MnSOD results in neuroprotection (Duckles et al., 2006).
Mitochondrial MnSOD mimetics (non-protein) have been developed to allow uptake into the mitochondrion to scavenge ROS. The mitochondrial superoxide dismutase mimetics MnTBAP and MnTE-2-Py5+ are permeable to the IMM and are capable of scavenging 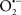 and ONOO− (Saba et al., 2007). MnTBAP was shown to improve survival in MnSOD knock out mice (Benov et al., 1998) and to decrease mitochondrial ROS production (Camara et al., 2004). Another cell-permeable MnSOD mimetic, Mn(III)tetrakis[(1-methyl-4-pyridyl)-porphyrin] (MnTMPyP), has both MnSOD and catalase effects (Wang et al., 2004). Indeed, we observed that when MnTPyP was given before prolonged cold ischemia, it protected the isolated heart from ROS damage during I/R (Camara et al., 2007). The variant form of the porphyrin MnTE-2-Py5+, MnTnHex-2-Py5+, given 24 h before ischemia, preserved endogenous MnSOD and F0/F1 ATP synthase activities, with a concomitant increase in renal functional recovery. This action was unlike the classic MnSOD mimetics (Saba et al., 2007). However, it is unfortunate to note that attempts to incorporate these antioxidants in human cardiovascular preservation techniques remain either untested or are largely disappointing (O’Connor and Gutterman, 2010). One would hope in time, and with rigorous testing and reformulations, the potential beneficial role of synthetic scavengers could lead to novel and useful mitochondriotropic antioxidants (Camara et al., 2010b).
and ONOO− (Saba et al., 2007). MnTBAP was shown to improve survival in MnSOD knock out mice (Benov et al., 1998) and to decrease mitochondrial ROS production (Camara et al., 2004). Another cell-permeable MnSOD mimetic, Mn(III)tetrakis[(1-methyl-4-pyridyl)-porphyrin] (MnTMPyP), has both MnSOD and catalase effects (Wang et al., 2004). Indeed, we observed that when MnTPyP was given before prolonged cold ischemia, it protected the isolated heart from ROS damage during I/R (Camara et al., 2007). The variant form of the porphyrin MnTE-2-Py5+, MnTnHex-2-Py5+, given 24 h before ischemia, preserved endogenous MnSOD and F0/F1 ATP synthase activities, with a concomitant increase in renal functional recovery. This action was unlike the classic MnSOD mimetics (Saba et al., 2007). However, it is unfortunate to note that attempts to incorporate these antioxidants in human cardiovascular preservation techniques remain either untested or are largely disappointing (O’Connor and Gutterman, 2010). One would hope in time, and with rigorous testing and reformulations, the potential beneficial role of synthetic scavengers could lead to novel and useful mitochondriotropic antioxidants (Camara et al., 2010b).
In general, cellular antioxidant defenses depend on the reduction potential of the electron carriers and the reducing capacity of linked redox couples in the matrix [NADH/NAD+ and FADH2/FAD] and cytoplasm (Stowe and Camara, 2009; Camara et al., 2010b). The balance between oxidized and reduced species, i.e., GSH/GSSG and pyridine nucleotides (NADH/NAD, NAD(P)H/NADP), determine the cellular redox status. Oxidative stress and redox status are widely considered as major facets of mitochondria-related diseases/abnormalities. In fact, one defense against I/R-induced ROS accumulation and damage may involve preservation of the NAD(P)H pool (Camara et al., 2010b). An adequate NAD(P)H/NAD ratio, maintained through an NADH kinase and transhydrogenase (Hanukoglu and Rapoport, 1995; Outten and Culotta, 2003)-dependent mechanism, preserves the mitochondrial NAD(P)H pool required to maintain the redox status necessary for effective scavenging. Thus the redox homeostasis in mitochondria is regulated by the coordinated activity of various antioxidant mechanisms including the GSH, thioredoxin (TRX; Camara et al., 2010b), and other redox systems. Therefore, these thiol-reducing agents are critically involved in defense against large increases in 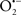 emission (Sen, 1998; Ueda et al., 1998; Lin and Beal, 2006; Stowe and Camara, 2009).
emission (Sen, 1998; Ueda et al., 1998; Lin and Beal, 2006; Stowe and Camara, 2009).
GSH is a small molecule with a thiol (-SH) residue of cysteine as its active site. Under conditions of oxidative stress, large amounts of GSH are rapidly oxidized to GSSG. The enzyme glutathione reductase (GR) uses NAD(P)H/NADH as its source of e− to regenerate GSH. Hence an increased concentration of NAD(P)H/NADH relative to NADP+/NAD+ promotes the production of GSH via GR (Figure 4). GSH protects mitochondria from extra-mitochondrial ROS and detoxifies 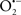 in a non-enzymatic fashion. Mitochondria appear to be most susceptible to damage during a GSH-depleted state (Stowe and Camara, 2009; Camara et al., 2010b). Inhibition of GSH transport results in depletion of mitochondrial GSH and increased oxidative damage (Aon et al., 2007) and subsequent cellular injury. In addition to protection against ROS, GSH is an ideal scavenger of HNE and other lipid peroxides (Calabrese et al., 2005).
in a non-enzymatic fashion. Mitochondria appear to be most susceptible to damage during a GSH-depleted state (Stowe and Camara, 2009; Camara et al., 2010b). Inhibition of GSH transport results in depletion of mitochondrial GSH and increased oxidative damage (Aon et al., 2007) and subsequent cellular injury. In addition to protection against ROS, GSH is an ideal scavenger of HNE and other lipid peroxides (Calabrese et al., 2005).
Mitochondrial thioredoxin reductase-2 (TrxR-2) is present in large concentrations in the brain, heart, and liver (Rybnikova et al., 2000). TRX is reduced by TrxR-2, which utilizes the mitochondrial NADH/NAD redox state similar to GR as a source of reducing equivalents. TRX protected against H2O2-induced cytotoxicity, improved post-ischemic ventricular recovery, and reduced infarct size when compared to hearts from wild type mice (Ebrahimian and Touyz, 2008; Ebrahimian et al., 2008). The TRX system has also been shown to have a potential therapeutic value against cardiac hypertrophy and cardiac failure (Stowe and Camara, 2009; Camara et al., 2010b).
The contribution to scavenging potential of mitochondria by these redox proteins is therefore dependent on normal mitochondrial bioenergetic function and the availability of reducing equivalents. Thus, maintaining a large pool of reductants like GSH and TRX requires mitochondria to regenerate the reduced state via the NADH/NAD+ redox pair after detoxifying the ROS. Hence, supplying exogenous GSH or TRX-2 may be protective, but only if there are also sufficient reducing equivalents.
Catalase is present in the cardiac cell mitochondrial matrix in extremely small amounts (Andreyev et al., 2005). Catalase protects mitochondria against intra- and extra-mitochondrial generated H2O2 (Andreyev et al., 2005). GPx, ubiquitously expressed in mammalian tissues and present in the mitochondrial matrix, seems to be the dominant H2O2 detoxifying agent in the heart. Catalase or GPx coupled to GR converts H2O2 to H2O (Fruehauf and Meyskens, 2007) to mitigate oxidative stress. Overexpression of GPx improves left ventricular pressure in mice after myocardial infarction (Laskowski and Russell, 2008). Recent studies have also shown that targeting catalase to the mitochondria can reduce cardiac pathology (e.g., cardiomyopathy with age) consistent with the effect of accumulated endogenous ROS that may decrease heart functional capacity with aging (Treuting et al., 2008).
Lastly, cytochrome c, which is one of the mediators of apoptosis, can accept or donate an e− depending on the redox state of its heme (Fe). In this way it is also a scavenger of 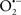 (Camara et al., 2010b). The reduced cytochrome c is reinstituted by donating its e− to complex IV. In I/R injury, loss of cytochrome c through mPTP or OMM permeabilization, as a result of oxidative damage to cardiolipin and membrane proteins, inhibits or slows down respiration, which leads to increased e− leak (Zhao et al., 2003; Li et al., 2006); the outcome is a vicious cycle of more
(Camara et al., 2010b). The reduced cytochrome c is reinstituted by donating its e− to complex IV. In I/R injury, loss of cytochrome c through mPTP or OMM permeabilization, as a result of oxidative damage to cardiolipin and membrane proteins, inhibits or slows down respiration, which leads to increased e− leak (Zhao et al., 2003; Li et al., 2006); the outcome is a vicious cycle of more 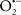 production and more cell damage. In the cytochrome c deficient Keilin–Hartree heart muscle model, Zhao et al. (2003) showed that e− transfer was impaired and
production and more cell damage. In the cytochrome c deficient Keilin–Hartree heart muscle model, Zhao et al. (2003) showed that e− transfer was impaired and 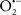 generation was significantly greater in the mutant than in the wild type mouse. Reconstituting cytochrome c in the cytochrome c deficient hearts resulted in less
generation was significantly greater in the mutant than in the wild type mouse. Reconstituting cytochrome c in the cytochrome c deficient hearts resulted in less 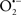 accumulation. Therefore, an adequate concentration of cytochrome c in the ETC process is necessary to maintain ROS at physiological levels (Zhao et al., 2003). Hence, maintaining the structural and functional integrity of the IMM and cytochrome c levels could represent a potential strategy for mitigating mitochondria dysfunction (Camara et al., 2010b) (Figure 3).
accumulation. Therefore, an adequate concentration of cytochrome c in the ETC process is necessary to maintain ROS at physiological levels (Zhao et al., 2003). Hence, maintaining the structural and functional integrity of the IMM and cytochrome c levels could represent a potential strategy for mitigating mitochondria dysfunction (Camara et al., 2010b) (Figure 3).
Thus it can be summarized here that under pathological conditions a good understanding of the balance between the 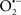 producing side and its scavenging side is crucial for an effective therapy (Camara et al., 2010b). In ischemia the excess
producing side and its scavenging side is crucial for an effective therapy (Camara et al., 2010b). In ischemia the excess  appear to be confined to the production side and less to the detoxifying side. This is evident by the apparent intactness of the scavenging capability of GPx and MnSOD in isolated mitochondria during the ischemic insult (Starke et al., 1997; Jin et al., 2005). In the failing heart, on the other hand, increase ROS production is attributed in part to decreased expression of MnSOD and catalase (Laskowski and Russell, 2008). With these findings, the importance of decreasing oxidant stress in the ischemic and failing heart prior to reaching a critical mass that leads to irreparable damage is evident. Hence targeting the ETC to attenuate e− transfer and consequently to decrease the e− leak may represent a better strategy than attempting to change the matrix antioxidant capacity during ischemia (Camara et al., 2010b).
appear to be confined to the production side and less to the detoxifying side. This is evident by the apparent intactness of the scavenging capability of GPx and MnSOD in isolated mitochondria during the ischemic insult (Starke et al., 1997; Jin et al., 2005). In the failing heart, on the other hand, increase ROS production is attributed in part to decreased expression of MnSOD and catalase (Laskowski and Russell, 2008). With these findings, the importance of decreasing oxidant stress in the ischemic and failing heart prior to reaching a critical mass that leads to irreparable damage is evident. Hence targeting the ETC to attenuate e− transfer and consequently to decrease the e− leak may represent a better strategy than attempting to change the matrix antioxidant capacity during ischemia (Camara et al., 2010b).
Maintaining Intra-Mitochondrial and Intra-Organellar Interactions as a Potential Therapeutic Strategy
It is believed that in certain cell types, mitochondria are organized in a reticular system of interconnected organelles, whereas in cardiac cells they are thought to exist as individual units capable of undergoing dynamic fusion and fission (Dorn and Scorrano, 2010). These are processes that are regulated by fusion and fission proteins (Archer et al., 2008; Camara et al., 2010a), and they play a major role in regulating mitochondrial function and cellular metabolism. However, the occurrence of mitochondrial fusion and fission in the healthy adult heart is presently unclear, as these myocytes do not normally divide. Furthermore, Collins et al. (2002), Aon et al. (2003, 2006b), and a several other investigators (Zorov et al., 2000, 2004; Vendelin et al., 2005), have reported that fusion and fission are neither required for normal mitochondrial function in cardiomyocytes, nor in all cell types. Consistent with these observations, Beraud et al. (2009) in an elaborate study, examined quantitatively the structural organization of mitochondrial dynamics and movement in two different types of heart cells, i.e., adult cardiomyocytes and non-beating HL-1 (NB HL-1) cells, and reported that NB HL-1 cells, but not the adult cardiomyocytes, showed fusion and fission. The dynamic fission and fusion in the NB HL-1 cells were observed only in dividing cells, which undergo rapid fusion–fission with formation of reticular networks. The observed differences between adult cardiomyocytes and NB HL-1 cells, according to the authors, are related to the specific structural organization of the cells, e.g., a variance in the mitochondria-cytoskeletal architecture (Beraud et al., 2009). From their findings, the authors (Beraud et al., 2009) contended that fusion and fission are demonstrated only in cultured cells of higher organisms or in yeast when these cells are actively dividing. They also argued that if mitochondria were fused in the cell, isolation of mitochondria should disrupt all mitochondrial membranes and release TCA cycle intermediates and render the mitochondrion unresponsive to TCA cycle linked susbtrates, e.g., pyruvate and glutamate (Beraud et al., 2009).
Nevertheless, fission and fusion have been implicated in a variety of biological processes, physiological, e.g., embryonic and neonatal development and metabolism, and pathological, e.g., I/R injury, heart failure, “diabetic” heart, vascular dysfunction, and apoptosis (Ong and Hausenloy, 2010; Williamson et al., 2010). Therefore, fusion and fission may be important in the etiology and/or propagation of mitochondrial dysfunction and ultimately cellular disorder, in some vascular cell types, such as endothelial cells. For example, fission might reduce propagation of mitochondrial ROS leading to less lipid peroxidation and loss of ΔΨm, whereas fusion might facilitate spread of ROS and lead to mitochondrial destruction. Fission can also prevent cell death induced by Ca2+-dependent apoptotic stimuli that require mitochondria to amplify the lethal signals of this second messenger (Dorn and Scorrano, 2010). However, abnormal fission results in fragmented and bioenergetically impaired mitochondria. Fission also leads to destabilization of mitochondria while fusion may promote stable mitochondrial architecture and function. Yu et al. (2008) reported, for the first time, that maintenance of the tubular mitochondrial network by inhibiting mitochondrial fission in a sustained hyperglycemic condition normalized cellular ROS levels and thwarted mPTP opening and subsequent cell death. Therefore, an understanding of this complex dynamic interaction between mitochondria, and between mitochondria and other organelles, is central to understanding the underlying causes of cellular dysfunction. It may also provide a potential for discovering novel therapeutic strategies for treating cardiovascular diseases (Ong and Hausenloy, 2010) using alternative protective mechanisms against oxidative stress.
Mitochondria can fuse with each other with the support of the cytoskeleton. Changes in the cytoskeletal architecture have recently been documented in patients with end-stage dilated and ischemic cardiomyopathies and, as such, may represent a common path for contractile dysfunction in the failing heart (Camara et al., 2010a). Mitochondria are also capable of fragmenting into more individual mitochondria and this may be beneficial or harmful to the cell. For instance, mitochondria fission or failure of fusion appears to engage apoptotic mechanisms leading to cell degeneration (Duchen, 2004). Oxidative stress is one of the major triggers of impaired mitochondrial dynamics. Substantial evidence indicates that mitochondrial fission occurs as a result of oxidative stress, which leads to loss of ΔΨm and consequently to impaired mitochondrial function and increased cell vulnerability. For example, acute exposure of myocytes to H2O2 leads to p53-dependent mitochondrial network fragmentation and the extent of the mitochondrial fragmentation (Li et al., 2010) correlates with a decrease in ΔΨm (Camara et al., 2010b).
Indeed, in normal cells ΔΨm is required for fusion. Specific fission and fusion proteins regulate this dynamic relationship by maintaining the IMM and by controlling cytochrome c release (Camara et al., 2010b). In cells derived from disease states, the compromise in ΔΨm is a consequence of defects in ETC complexes (McBride et al., 2006; Taguchi et al., 2007). This dynamic disintegration is observed in numerous mitochondrial associated cytopathies, including neurodegenerative diseases (Kwong et al., 2007), pulmonary artery hypertension (Archer et al., 2008), and I/R injury (Aon et al., 2006a; Brady et al., 2006a). In pulmonary artery hypertension a decrease in mitofusin proteins contributes to disruption of mitochondrial fusion (Archer et al., 2008). Therefore, maintaining mitochondrial fusion by expression of fusion proteins, while minimizing the levels of fission proteins (Wang et al., 2009), could represent a novel therapeutic approach to improving cell survival during oxidative and nitrosative stress. In many diseases, in which a less polarized ΔΨm may contribute to mitochondrial instability, a strategy to maintain integrity of the ETC with normal e− flux is a therapeutic option (Camara et al., 2010b).
Mitochondria also have anatomical and functional connectivity with other organelles such as the nucleus and the SR. These dynamic functional and structural interconnections are important for the normal coordination of cellular function. It is therefore anticipated that derangement of this dynamic link could be involved in pathologic states associated with cellular dysfunction. In some cell types, e.g., myocytes, mitochondria lie in very close apposition to the SR, and so are exposed to microdomains of high cytosolic [Ca2+] following local Ca2+ release from the SR. Evidence has emerged that there is a preferential coupling of Ca2+ transport from SR directly to mitochondria (Sharma et al., 2000), thereby providing a means for coupling cardiac muscle excitation to oxidative energy production, possibly through activation of Ca2+ sensitive mitochondrial dehydrogenases (Csordas et al., 2001; Hajnoczky and Csordas, 2010). Mitochondria–SR communication is therefore a crucial component in the normal function of mitochondria within the cell. The anatomical proximity between mitochondria and ER conversely creates a local mitochondrial [ATP] within the microdomain required for SR Ca2+ regulation (Duchen, 2004; Szabadkai and Duchen, 2008; Figure 2). This dynamic relationship highlights the importance of Ca2+ signaling between the two compartments. For example, the low conductance form of the Ca2+-induced mPTP opening leads to Ca2+ extrusion from mitochondria; this may serve as an amplification loop for Ca2+-induced Ca2+-release from the ER or SR stores (Pereira et al., 2009).
In the heart the inter-fibrillar mitochondria are closely intertwined with the SR to provide functional communication between the local ATP and the site for contraction (Dalen, 1989). Therefore, this proximity of mitochondria to Ca2+ release sites has functional consequences for intracellular Ca2+ signaling, capacitative Ca2+ influx, and cellular function (Duchen, 2004) and dysfunction (Camara et al., 2010b). Mitochondrial Ca2+ dynamics as well as emission of ROS are thus two key important events during the course of cellular injury and cell death. It has been proposed that these interactions are modulated in part by locally generated mitochondrial ROS, which is believed to act as the trigger to regulate the Ca2+ flux between the organelles (Waypa et al., 2002; Walsh et al., 2009) (Figure 2). A strategy of combined protection of mitochondria-SR dynamic anatomic and functional relationship may represent an innovative therapeutic approach to enhance organ viability (Du et al., 2008).
Examples of Potential Therapeutic Compounds to Protect Mitochondrial Function
Given the marked importance of mitochondrial events during the initial critical phases of apoptosis, or in the propagation of many disease processes, the appropriate design of mitochondriotropic drugs is crucial in developing promising, novel strategies to mitigate cell injury and to prevent cell death. Because of the importance of this organelle in disease processes, either secondary or primary, a new field has emerged in the clinical realm that recognizes “Mitochondrial Medicine.” This is a new and rapidly developing medical research field. In recent years, the scope of the Society of Mitochondrial Medicine has expanded to pay increased attention to promote research and education for the diagnosis, treatment, and cure of mitochondrial disorders.
Mitochondria are ideal targets for therapeutic modification because they are key producers of ATP and ROS production, whereby an imbalance of the two factors can lead to apoptosis. As a first step in designing a mitochondria-specific therapy, it is foremost important to develop a drug that can access the matrix. One general solution is to utilize the large ΔΨm and to attach a molecule, for example one with antioxidant properties, onto a lipoid vehicle that can penetrate membranes (Hoye et al., 2008). With a ΔΨm of −180 mV under state 4 conditions, mitochondria-targeted drugs are preferentially taken up into the mitochondrion, with little or no accumulation in the cytosol (Camara et al., 2010b).
To achieve this goal, numerous approaches have been attempted. For example, the lipophilic agent TPP+, mentioned earlier, has been used to increase the incorporation of antioxidants and other therapeutic impermeants into mitochondria. The appended positively charged phosphonium moiety induces about a 1000-fold uptake of these compounds into the matrix over the cytoplasm. Indeed antioxidants that accumulate within the matrix provide better protection from oxidative injury than untargeted antioxidants (Milagros Rocha and Victor, 2007). Examples include mitochondria-targeted ubiquinone (MitoQ), mitoTEMPO, and idebenone, currently under study for their possible use as therapies to decrease oxidative damage in human pathological conditions (Maroz et al., 2009). These drugs accumulate in the mitochondria and could enhance ETC function, scavenge ROS, and prevent oxidation of cardiolipin, all of which may result in better functioning of mitochondria (Schapira, 2006; Pope et al., 2008). CoQ10 is a natural antioxidant and a highly mobile e− carrier between complexes I and II to III. It is redox active and has been effective in reducing oxidative stress. It may provide some protection for patients with mitochondrial cardiomyopathies (Sokol and Treem, 1999).
Another mitochondrial-targeted antioxidant, the cationic SOD2 mimetic MnTBAP, can accumulate in cardiac mitochondria after systemic injection (Spasojevic et al., 2007). We showed that MnTBAP reversed the protection provided by the putative mitoKCa channel agonist NS 1619 (Stowe et al., 2006) and by IPC (Kevin et al., 2003). This demonstrates the fine line between scavenging harmful ROS and the ROS that are essential for signaling purposes. The mitochondrial-targeted scavenger mitoTEMPO decreased  and attenuated AII-induced hypertension, whereas when a similar dose of the non-targeted TEMPOL was given it was ineffective. The effect of mitoTEMPO was mimicked by overexpression of MnSOD (Dikalova et al., 2010). In an unrelated study, it was reported that SOD1 and mitoTEMPO prevented translocation of Bax to mitochondria and preserved mitochondrial integrity during simulated oxidative stress in tubular epithelial cells (Liang et al., 2010).
and attenuated AII-induced hypertension, whereas when a similar dose of the non-targeted TEMPOL was given it was ineffective. The effect of mitoTEMPO was mimicked by overexpression of MnSOD (Dikalova et al., 2010). In an unrelated study, it was reported that SOD1 and mitoTEMPO prevented translocation of Bax to mitochondria and preserved mitochondrial integrity during simulated oxidative stress in tubular epithelial cells (Liang et al., 2010).
Metformin, an anti-diabetic drug also accumulates in the matrix of energized mitochondria due to its positive charge (Owen et al., 2000). Metformin has a broad spectrum of effects that include fat breakdown and an increase in glucose transport and utilization (Zmijewski et al., 2008). Metformin has also been shown to protect against I/R injury (Calvert et al., 2008), possibly through the direct effects of the drug on complex I (Brunmair et al., 2004; Zmijewski et al., 2008), another action analogous to the action of amobarbital on mitochondria (Aldakkak et al., 2008a). Recent evidence suggests that metformin also can protect against oxidative stress. Metformin, via AMP-activated protein kinase, was reported to upregulate the expression/activity of thioredoxin, which, e.g., can attenuate the ROS generated by an enhanced FA load (Nediani et al., 2011). In the murine model of heart failure caused by I/R injury, metformin was effective in decreasing left ventricular dysfunction. This could make metformin a suitable mitochondria-directed therapy for protection against I/R injury and other cardiac vascular complications, particularly in the non-diabetic patient with heart failure (Nediani et al., 2011).
Another drug that also directly targets the mitochondrion is resveratrol (Sareen et al., 2006). Mitochondria represent a powerful target for resveratrol in the nervous system (Raval et al., 2008). Resveratrol is a polyphenolic compound derived from some grapes and roots; it is a natural antioxidant. It has been shown to reduce oxidative stress developed during reperfusion of the ischemic heart; it may also be in part effective against hypertension, cardiomyopathy, and heart failure (Das et al., 2010). Resveratrol has been shown to induce  synthesis in the setting of ischemic reperfused heart, brain, and kidney, where it may lower the oxidative damage (Das et al., 2010). It has also been demonstrated that resveratrol can improve mitochondrial function and provide cardioprotection by stimulating the sirtuin 1 (SIRT1)-dependent deacetylation of the transcriptional co-activator peroxisome proliferator-activated receptor-γ co-activator 1α (PGC-1α) in a NAD+-dependent manner. Because sirtuin depends on cellular NAD+ availability, mitochondrial regulation of NAD+ levels may contribute to an increase in sirtuin mediated PGC-1α deacetylation. Activation of PGC-1α co-activates nuclear transcription factors involved in OxPhos, increases mitochondrial biogenesis and enhances
synthesis in the setting of ischemic reperfused heart, brain, and kidney, where it may lower the oxidative damage (Das et al., 2010). It has also been demonstrated that resveratrol can improve mitochondrial function and provide cardioprotection by stimulating the sirtuin 1 (SIRT1)-dependent deacetylation of the transcriptional co-activator peroxisome proliferator-activated receptor-γ co-activator 1α (PGC-1α) in a NAD+-dependent manner. Because sirtuin depends on cellular NAD+ availability, mitochondrial regulation of NAD+ levels may contribute to an increase in sirtuin mediated PGC-1α deacetylation. Activation of PGC-1α co-activates nuclear transcription factors involved in OxPhos, increases mitochondrial biogenesis and enhances  detoxification enzymes MnSOD and catalase (Das et al., 2010; Fulda et al., 2010), all of which contribute to protection. At high concentrations resveratrol has been shown to act as a pro-oxidant (Raval et al., 2008); it could also lead to inhibition of mitochondrial ATP synthesis by binding to F1-ATPase, or by triggering mPTP opening, which would eventually contribute to cell injury. This suggests that the therapeutic role of resveratrol, as with other drugs, may be dependent on the concentration of the drug (Szende et al., 2000; Morin et al., 2003a).
detoxification enzymes MnSOD and catalase (Das et al., 2010; Fulda et al., 2010), all of which contribute to protection. At high concentrations resveratrol has been shown to act as a pro-oxidant (Raval et al., 2008); it could also lead to inhibition of mitochondrial ATP synthesis by binding to F1-ATPase, or by triggering mPTP opening, which would eventually contribute to cell injury. This suggests that the therapeutic role of resveratrol, as with other drugs, may be dependent on the concentration of the drug (Szende et al., 2000; Morin et al., 2003a).
There has recently emerged a novel class of cell-permeable antioxidant peptides that are selectively partitioned into the mitochondrion independent of the ΔΨm. These series of peptides, known as Szeto–Schiller (SS)-peptides can selectively target the IMM. For example, SS-31, which scavenges matrix H2O2 and ONOO−, reduced cytochrome c release and minimized mitochondrial swelling; its efficacy has been demonstrated in vivo by its effect to reduce cardiac and brain I/R injury in animal models (Cho et al., 2007; Thomas et al., 2007; Szeto, 2008a,b).
Other novel pharmacological agents may effect organ protection by optimizing substrate metabolism (discussed later) or by maintaining or improving oxidative capacity. However, it is clear that the subject of mitochondrial pharmacology is vast and the discussion presented here represents only a sampling of what has been uncovered to date. Nonetheless, the notion to use mitochondrial-targeted therapeutics as an alternative approach is becoming an increasingly attractive strategy for treating cardiovascular diseases. One can predict that there will be whole classes of these so-called “mitochondriotropic drugs” under development in the near future.
Potential Therapeutic Implications Directed Toward Mitochondrial Ca2+ Handling
Mitochondrial Ca2+ handling plays a key physiological and pathophysiological role in the control of cellular function and dysfunction. Mitochondrial Ca2+ overload is a deleterious consequence of cardiac I/R injury and so mitochondrial Ca2+ handling is an important target for potential therapy (Camara et al., 2010b). As noted previously, the primary source of mCa2+ is external to the cell or from Ca2+ stored in the SR, following an increase in cytosolic Ca2+ uptake. Although modest increases in m[Ca2+] enhance enzymatic activity and subsequently OxPhos, massive mCa2+ loading can disable ATP synthesis. Under simulated or true I/R conditions the excess mCa2+ may contribute to mPTP opening and destabilization of cellular ionic homeostasis, and eventually to cell death by necrosis or apoptosis. However, the targeting of mCa2+ dynamics (uptake and release) for therapeutic purpose creates a difficult challenge that could impede the way forward in this area. These impediments include the structural identification of the CaU, NCE, NHE and a sodium-independent Ca2+ exchanger (NICE), and the myriad of other purported molecular structures that regulate the dynamic Ca2+ flux between the SR and mitochondria (see above).
Suffice it to say, however, that attempts have been being undertaken to target mCa2+ in order to mitigate oxidative damage or I/R injury. Therapeutic approaches to reduce mCa2+ uptake leading to protection may involve direct targeting of the CaU or the mitochondrial exchangers that regulate m[Ca2+], or may involve indirect approaches that reduce Ca2+ release in the mitochondria-SR microdomain. For example, attenuation of mCa2+ overload and the subsequent reduction in the sensitivity of the mPTP opening has been attributed in part to inhibition of NHE or the CaU. Blocking NHE preserved ATP levels during cardiac I/R by improving state 3 respiration (Javadov et al., 2008) and preventing mitochondrial Ca2+ overload (Camara et al., 2010b). In a recent study we also reported that activation or inhibition of NHE might act through mitochondria as shown by changes in redox state, ROS generation, and mCa2+ levels. We concluded from this study that the protection afforded by NHE inhibition may be due at least in part to inhibited mNHE, as well as sarcolemmal NHE. This notion is consistent with the observation that cariporide blocked matrix acidification and ATP depletion during simulated ischemia in cardiac myocytes (Ruiz-Meana et al., 2003). Under these conditions, one would expect a decrease in matrix pH with concomitant mitochondrial depolarization leading to diminished Ca2+ uptake.
The mNCE is another putative mitochondrial Ca2+ transporter, which is likely functionally similar to the sarcolemmal NCE, and it is sensitive to cations and a variety of pharmacological agents such as diltiazem and more specifically CGP37157 (Rizzuto et al., 2009). Like the sarcolemmal NCE, the mNCE can operate in both directions depending on the trans-membrane cation concentrations. During metabolic inhibition an increase in mCa2+ levels was observed despite a reduced ΔΨm, which would limit uptake through the CaU (Hoppe, 2010). Indeed, it was reported that the rise in mCa2+ observed during metabolic inhibition was minimized by CGP37157. This mode of action may be particularly significant in diseased states with elevation of cytosolic Ca2+ and Na2+ levels during heart failure (Hoppe, 2010). In addition, strategies to enhance extrusion of mCa2+ through the exchanger could lead to diminished mCa2+ load and reduce mitochondrial damage. This would make the targeting of mNCE a potential therapeutic target for mitigating Ca2+ overload during ischemia. However, direct targeting of these drugs to mitochondria with greater efficacy than on sarcolemmal exchangers remains elusive in so far as the molecular identity of the exchanger/transporters remains obscure.
The CaU, due to its close anatomical and physiological proximity to the SR RyR, is implicated in both normal cellular function and injury. Indeed, targeting the CaU in the local domain has been shown to mediate protection against I/R injury in various models. It has been shown to mediate IPC and to be directly involved in protection. Studies have shown that blocking the CaU with ruthenium red (RR) or RU360 prior to ischemia decreased the incidence of ventricular fibrillation (Brown and O’Rourke, 2010). We reported in preliminary studies that RR improved function and reduced mCa2+ load following I/R. Interestingly, we also observed in preliminary studies that RR significantly reduced the worsening of mCa2+ overload that was instituted by the perfusion of hearts with pH 8 buffer (Aldakkak et al., 2008c), a condition that would maximally stimulate NHE and lead to increased cytosolic Ca2+ secondary to activation of NCE. However, as noted by Brown and O’Rourke (2010) in a recent review, it is difficult to draw conclusions on the effects of blocking the CaU due to the confounding effects of the ruthenium compounds on cellular Ca2+ fluxes, for example, blocking SR Ca2+ release. Even the more specific RU360 is trapped in controversy related to its membrane permeability. Until then, it is opined that future research with novel compounds that lack these pleiotropic/permeability issues will provide better insights into the role of CaU in cardioprotection.
SR stress results from a perturbation of SR homeostasis and has been implicated in cardiac contractile dysfunction associated with I/R and heart failure as a result of SR-induced intracellular [Ca2+] changes. Evidence suggests that IP3R-mediated release of Ca2+ from the SR appears to be a key-sensitizing step in various apoptotic processes (Rizzuto et al., 2009). In early apoptosis, Ca2+ released from the SR activates mPTP; with the subsequent release of cytochrome c, this leads to an unrestrained increase in the local cytosolic [Ca2+] (Romagnoli et al., 2007). This might provide a direct mechanism for more matrix Ca2+ uptake. Ca2+ released via the IP3R is imported across the OMM through the VDAC (Rizzuto et al., 2009). Overexpression of VADC enhances mCa2+ uptake (Rizzuto et al., 2009) and this could offset the balance of mCa2+. All together, it is conceivable that interrupting the interaction between mitochondria and SR could mitigate (or promote) mitochondrial-mediated cellular damage. Thus targeting the dynamic Ca2+ flux within the microdomain, pharmacologically or by genetic manipulations, could provide potential therapeutic alternatives for reducing mCa2+ overload. Indeed, inhibition of SR Ca2+ release kinetics was proposed to reduce mCa2+ uptake and prevent collapse of ΔΨm or the release of cytochrome c (Rizzuto et al., 2009). Akt phosphorylation of the receptor inhibits Ca2+ flux from the SR to mitochondria (Szado et al., 2008) and thereby inhibits apoptosis. Mao et al. (2008) in an elaborate study reported that the compound darbepoetin alfa, via activation of Akt, improved cardiac function and reduced cardiomyocyte apoptosis in a rabbit model of autoimmune cardiomyopathy. Therefore intrinsic activation of the Akt could be a potential therapeutic strategy in attenuating the transfer of Ca2+ during SR-mitochondrial stress and changes in Ca2+ levels within the microdomain. It has been suggested recently that mitochondria-SR crosstalk may play a greater role than previously suspected in cardiac hypertrophy and the progression to heart failure. It is possible that in heart failure therapeutic manipulations involving an increase sarcoplasmic reticulum Ca2+ ATPase (SERCA), or impaired function of its endogenous inhibitor phospholamban, could reduce the Ca2+ transient (Dorn and Scorrano, 2010) and reduce m[Ca2+] by which to improve OxPhos with adequate ATP production.
The mitochondrial K channels (ATP-sensitive or Ca2+ activated) have been shown to have potential cardioprotective roles. The opening of mitoKATP or mitoKCa channels has been shown to be beneficial for cell survival under certain stress conditions, including I/R and heart failure (Ruiz-Meana et al., 2010). Opening of mitoKCa channels appear to protect hearts against infarction (Xu et al., 2002; Stowe et al., 2006) and similarly, activation of the mitoKATP has provided protection against I/R injury (Camara et al., 2010a). The precise mechanism for this protection has yet to be fully elucidated. However, it is postulated that the opening of these channels produces both an increase in mitochondrial volume and a certain degree of uncoupling (mild depolarization) that reduces mCa2+ overload (Ruiz-Meana et al., 2010) and protects against apoptosis/necrosis. We reported that activation of the mitoKCa channel with NS 1619 resulted in cardioprotection from I/R injury and that this was associated with reduced mCa2+ loading, better preservation of the redox state, and improved functional return (Stowe et al., 2006). The activation of mitoKATP channels has also been proposed to mitigate mCa2+ loading as a mechanism of protection against I/R injury (Brown and O’Rourke, 2010).
Part 2
Mitochondrial Dysfunction in Endothelial Cells, Heart Failure, and I/R Injury
Given the fundamental function of mitochondria in cellular bioenergetics and oxidative stress, we have stressed that dysfunction within this organelle underlies the pathologic mechanisms of many diseases. In this section, a brief discussion of the role of mitochondria in vascular diseases, heart failure, and I/R injury will be presented. Potential therapeutic approaches that are targeted to mitochondria by which to mitigate cardiovascular damage also will be discussed. When mitochondrial ATP production is impaired tissues that rely heavily on high levels of ATP, such as the heart, skeletal muscle, and brain, are most affected. Depletion of ATP can lead, for example, to dissipation of intracellular ionic homeostasis, cell swelling, and subsequent cell death by necrosis. In heart failure, defects have been purported to occur in ETC complexes or in components of the OxPhos machinery. These alterations are manifested frequently as cardiomyopathy (Rosca et al., 2008). Concomitant with impaired ETC function and OxPhos, increased 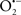 generation and mCa2+ overload occur and lead to a “vicious perpetual cycle” of mitochondrial damage and dysfunction (Loeb et al., 2005); ultimately mitochondrial functional decline will lead to cell damage and organ failure.
generation and mCa2+ overload occur and lead to a “vicious perpetual cycle” of mitochondrial damage and dysfunction (Loeb et al., 2005); ultimately mitochondrial functional decline will lead to cell damage and organ failure.
Mitochondrial DNA is most susceptible to mutations because of its lack of histones and a limited repair mechanism. Increased oxidative stress leads to genetic mutations. Indeed, diseases related to mtDNA defects appear to be more prominent in brain and in skeletal and cardiac muscles where defects accumulate more extensively than in rapidly dividing cells (Muravchick and Levy, 2006). It has been reported that in heart failure mtDNA damage leads to dysfunction and loss of mitochondrial mass (Murray et al., 2007). Mitochondrial related diseases have been discussed extensively in the literature (Duchen, 2004) and in our recent comprehensive review (Camara et al., 2010b).
Treatments Targeted to Endothelial and Vascular Cell Mitochondrial Dysfunction
Endothelial cells play a major role in vascular diseases and so their mitochondria are crucial in both the etiology and in the amelioration of the disease process. A primary role of the vasculature is in facilitating the delivery of O2 and energetic substrates for powering the high metabolic demands of the cardiomyocytes; endothelial cells are also critical in the development and manifestation of several types of cardiovascular disease that develop when the vasculature becomes damaged (Pipinos et al., 2006). Damage to endothelial cells can lead to atherosclerosis and to vascular complications associated with diabetes and other peripheral artery diseases. The manifestations of peripheral artery diseases are presumed to be due to reduced blood flow and vessel compliance. However, an increasing number of reports have implicated suboptimal bioenergetic function from impaired endothelial cell mitochondria as an additional etiologic factor (Pipinos et al., 2006). The defective mitochondrial function in these vascular disorders may be analogous to mitochondrial associated myopathies with increased deleterious ROS production (Pipinos et al., 2006).
Under normal conditions, endothelial-derived 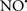 may be able to modulate cardiac contractility, heart rate, coronary flow, and myocardial energetics. In pathological situations this role may be extended to regulation of cardiomyocyte metabolism and determination of cell survival. Unlike the cardiomyocyte, the endothelial cell is dependent more on glycolytic ATP production (cytosolic substrate level phosphorylation) and less on mitochondrial OxPhos. Endothelial cells have far less mitochondrial density than cardiomyocytes (5 vs. 40% cell volume, respectively). Consequently, mitochondrial ROS generation is likely much less than in myocytes. However, studies have indicated that vascular and endothelial cell mitochondria can represent a significant source of ROS, which in addition to cytosolic NAD(P)H oxidase (Nox) and xanthine oxidase-dependent ROS sources, contribute to the vicious cycle of ROS production and subsequent endothelial dysfunction (Dikalova et al., 2010). Increased vessel wall Nox-derived
may be able to modulate cardiac contractility, heart rate, coronary flow, and myocardial energetics. In pathological situations this role may be extended to regulation of cardiomyocyte metabolism and determination of cell survival. Unlike the cardiomyocyte, the endothelial cell is dependent more on glycolytic ATP production (cytosolic substrate level phosphorylation) and less on mitochondrial OxPhos. Endothelial cells have far less mitochondrial density than cardiomyocytes (5 vs. 40% cell volume, respectively). Consequently, mitochondrial ROS generation is likely much less than in myocytes. However, studies have indicated that vascular and endothelial cell mitochondria can represent a significant source of ROS, which in addition to cytosolic NAD(P)H oxidase (Nox) and xanthine oxidase-dependent ROS sources, contribute to the vicious cycle of ROS production and subsequent endothelial dysfunction (Dikalova et al., 2010). Increased vessel wall Nox-derived 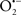 is indeed an important determinant of endothelial dysfunction in experimental angiotensin II (AII)-induced hypertension and genetic hypertension (Cave et al., 2006). Moreover, in a recent study, Dikalova et al. (2010) reported that endothelial cell mitochondria are the initial sources of ROS in response to AII-stimulated vascular dysfunction, whereas Nox is secondarily activated. This Nox-dependent ROS production could lead to a pathological process of mitochondria-Nox axis in a feed-forward loop of deleterious ROS production (O’Connor and Gutterman, 2010).
is indeed an important determinant of endothelial dysfunction in experimental angiotensin II (AII)-induced hypertension and genetic hypertension (Cave et al., 2006). Moreover, in a recent study, Dikalova et al. (2010) reported that endothelial cell mitochondria are the initial sources of ROS in response to AII-stimulated vascular dysfunction, whereas Nox is secondarily activated. This Nox-dependent ROS production could lead to a pathological process of mitochondria-Nox axis in a feed-forward loop of deleterious ROS production (O’Connor and Gutterman, 2010).
It is evident that a loss of endothelial-derived 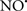 bioavailability is a critical component in the etiology of vascular dysfunction and is an independent risk marker for future cardiovascular complications.
bioavailability is a critical component in the etiology of vascular dysfunction and is an independent risk marker for future cardiovascular complications. 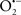 inactivation of
inactivation of 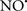 by the uncoupling of NOS, contributes to vascular endothelial dysfunction. In addition to its indirect actions through inactivation of
by the uncoupling of NOS, contributes to vascular endothelial dysfunction. In addition to its indirect actions through inactivation of 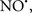
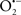 also likely exerts a direct action on vascular endothelium (Cave et al., 2006). Increased endothelial
also likely exerts a direct action on vascular endothelium (Cave et al., 2006). Increased endothelial 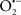 and the concomitant production of ONOO− are likely to result in nitration of mitochondrial proteins that cause further damage to the organelle. It has been proposed that one of the major culprits in vessel damage is H2O2-mediated damage of the ETC (O’Connor and Gutterman, 2010), in which case reduction of H2O2 to H2O by antioxidants would help to ameliorate the damage. Damage to endothelial cell structure or function is considered to be among the primary causes of atherosclerosis (Davidson and Duchen, 2007) and other peripheral artery diseases (Pipinos et al., 2006). mPTP opening is prominent in endothelial cell-induced death leading to atherosclerotic lesions (Davidson and Duchen, 2007), so the mPTP is a potential target for minimizing the progression of atherosclerosis. In addition, endothelial cell mCa2+ overload, diminished levels of MnSOD activity, and decreases in ETC complex activities, have all been implicated in the events that lead to endothelial and vascular cell injury and death (Pipinos et al., 2006; Davidson and Duchen, 2007).
and the concomitant production of ONOO− are likely to result in nitration of mitochondrial proteins that cause further damage to the organelle. It has been proposed that one of the major culprits in vessel damage is H2O2-mediated damage of the ETC (O’Connor and Gutterman, 2010), in which case reduction of H2O2 to H2O by antioxidants would help to ameliorate the damage. Damage to endothelial cell structure or function is considered to be among the primary causes of atherosclerosis (Davidson and Duchen, 2007) and other peripheral artery diseases (Pipinos et al., 2006). mPTP opening is prominent in endothelial cell-induced death leading to atherosclerotic lesions (Davidson and Duchen, 2007), so the mPTP is a potential target for minimizing the progression of atherosclerosis. In addition, endothelial cell mCa2+ overload, diminished levels of MnSOD activity, and decreases in ETC complex activities, have all been implicated in the events that lead to endothelial and vascular cell injury and death (Pipinos et al., 2006; Davidson and Duchen, 2007).
A thorough understanding of the etiology of vascular dysfunction, specifically as it pertains to the role of mitochondria, may enable development of improved therapies for vascular diseases. Although treatment with antioxidants or with SOD mimetics has been effective in reducing oxidative stress and improving endothelial function, the results from human studies have been singularly disappointing (Cave et al., 2006; O’Connor and Gutterman, 2010). For example, instead of using antioxidants that are not targeted to sites of ROS (e.g., vitamins C and E), mitochondria-targeted ROS scavengers or other “mitochondriotropic” agents (discussed above) might be more effective (Cave et al., 2006; Dikalova et al., 2010; O’Connor and Gutterman, 2010). Recent treatment strategies that target mitochondrial sources of ROS over cytosolic sources of ROS (O’Connor and Gutterman, 2010) have proven more effective in dampening endothelial dysfunction and minimizing vascular impairment (Dikalova et al., 2010). It is thought that the ability of targeted delivery in this case would also minimize the potential for untoward effects of other therapies that are less specific (Dikalova et al., 2010).
Other potential strategies include pathways that regulate mitochondrial biogenesis. As noted above, resveratrol is one such agent. Impaired mitochondrial endothelial cell function in a disease process, e.g., diabetes, may impair bioenergetics, which could underlie a defect in endothelial paracrine factors essential for maintaining vascular tone (Csiszar et al., 2009a). Resveratrol has been shown to induce mitochondrial biogenesis in the aortas of diabetic mice and to reduce mitochondrial ROS; this suggests the potential for targeting endothelial mitochondria in metabolic diseases (Csiszar et al., 2009a,b). Interestingly, recent studies have demonstrated the presence of estrogen receptors on mitochondria and that estrogen stimulates increase ATP production while reducing ROS generation in cerebral arteries (Duckles et al., 2006). Estrogen also increases transcription of mitochondrial genes that encode subunits of complex IV (Duckles et al., 2006). These effects could be significant in certain age-related cardiovascular diseases such as stroke (Duckles et al., 2006). Resveratrol and statin drugs also induce endothelial NOS to restore endothelial 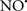 levels appropriate for vasodilatation. This vascular response is central to the achievement of integrated increases or redistribution of blood flow among specific vascular beds (Cave et al., 2006; Davidson and Duchen, 2007).
levels appropriate for vasodilatation. This vascular response is central to the achievement of integrated increases or redistribution of blood flow among specific vascular beds (Cave et al., 2006; Davidson and Duchen, 2007).
Potential for Mitochondria-Targeted Strategies for the Failing Heart
Heart failure progressively limits the pumping capacity of the heart and endothelial and vascular dysfunction are important components in this process (Davidson and Duchen, 2007) because of decreases in vascular diameter and compliance (coronary reserve) which reduces tolerance to exercise (Cave et al., 2006). Chronic heart failure occurs frequently as a consequence of longstanding hypertension or ischemic heart disease (Cave et al., 2006) and is a leading cause of morbidity and mortality in Western industrialized countries. Despite extensive studies, the fundamental mechanisms responsible for the development and progression of heart failure have yet to be fully elucidated (Tsutsui et al., 2006). A growing body of evidence supports a role for oxidative stress and redox signaling in the pathophysiology of heart failure (Cave et al., 2006). Indeed a number of recent studies have indicated that mitochondria are central to the pathophysiologic mechanism of the failing heart. Mitochondrial released ROS play a major role in mitochondrial bioenergetic dysfunction and contribute to myocardial apoptosis, which with cell necrosis and fibrotic cell invasion, underlie the anatomical progression of heart failure (Marin-Garcia and Goldenthal, 2008; Chen and Knowlton, 2010). Increased levels of mitochondrial network fragmentation are also observed in damaged heart muscle, thus suggesting the significance of the mitochondrial reticular network in the etiology and progression of the disease (Shen et al., 2007).
The inexorable loss of terminally differentiated cardiomyocytes and the subsequent invasion of fibrotic tissue are pivotal to progression of heart diseases, especially ischemic and dilated cardiomyopathies and congestive heart failure (Shen et al., 2007). During failure the heart decompensates because of an imbalance between energy production and use that leads to a state of energy deficit (Camara et al., 2010a). Abnormalities in energy transfer may occur in failing cardiac muscle and correlate with clinical symptoms and mortality (Murray et al., 2007). In general, the progression of heart failure can be characterized by diminished energy metabolism, poor Ca2+ handling, ROS generation, and apoptotic cell death. Alterations in mitochondrial function with dysregulation of mitochondrial biogenesis, and defects in ETC complexes I and III (Rosca et al., 2008), have been long suspected in heart failure. Indeed, therapies aimed at protecting mitochondrial function have shown some promise in heart failure patients and animal models of heart failure (Murray et al., 2007). For example, in mouse hearts exposed to chronic systolic pressure overload induced by transverse aortic constriction, the PGC-1α−/− knockout caused moderate but significant decreases in MnSOD and TRX2, with subsequent increases in oxidative stress markers, e.g., 4-HNE. Furthermore, treatment with the MnSOD mimetic MnTMPyP significantly reduced the transverse aortic constriction-induced left ventricular hypertrophy and dysfunction in PGC-1α−/− deficient mice (Lu et al., 2010).
The mitochondrial content and hence oxidative capacity of cardiac muscle cells are diminished in heart failure (Garnier et al., 2003). The result is limited work capacity in the failing heart due to limited free mitochondrial energy (Ochiai et al., 2001; Gong et al., 2003). Indeed, in mitochondria isolated from patients exhibiting severe cardiomyopathies there is a significant decrease in state 3 respiration and a lower respiratory control index (Sharov et al., 2000). Thus the altered metabolic pattern observed in heart failure has a clear impact on the energetic state of the heart. The potential consequences of a diminished energetic state include an impaired ability of the heart to work and respond to acute and chronic stresses (Camara et al., 2010b).
Although the idea of treating metabolic cardiac dysfunction is not new, the targeting of mitochondria has garnered significant interest recently. Cardiac metabolic homeostasis was summarized near the beginning of this review. The failing heart is characterized by enhanced glycolysis, impaired pyruvate oxidation, and reduced FA oxidation. Glycolysis generates pyruvate and if this pyruvate cannot be utilized by PDH to fuel OxPhos only 2 ATP molecules/glucose are realized. As discussed above, PDH is regulated by multiple factors including PDH kinase (PDK) and PDH phosphatase (Beadle and Frenneaux, 2010); PDK, whose activity is low depending on NADH and acetyl-CoA coming from FA β-oxidation (Nediani et al., 2011), is thus a key regulator of glucose oxidation. Activated PDK phosphorylates and inhibits PDH and thereby blocks pyruvate uptake into mitochondria and prevents its oxidation. During acute ischemia, there is also a block at the level of PDH. It was reported that activation of PDK can lead to right ventricular dysfunction and that reduction of enzyme function not only restores right ventricular function, but also enhances glucose oxidation (Piao et al., 2010). Selected and precise control of these mitochondrial metabolic enzymes and glucose transporters (GLUT) represent possible therapeutic targets to modulate substrate utilization (Beadle and Frenneaux, 2010). Indeed, a variety of metabolically targeted therapies have already been applied clinically in an attempt to improve cardiac metabolic function in human heart failure. These include, but are not limited to, an approach to reverse abnormal metabolism by stimulating PDH by inhibiting PDK directly with dichloroacetate (DCA) and leelamine HCl, or indirectly with ranolazine and trimetazidine (Cadoudal et al., 2008). DCA can restore ΔΨm by inhibiting PDK and thereby activating PDH with a concomitant increase in intra-mitochondrial acetyl-CoA (Archer et al., 2008) (Figure 1). DCA has been safely used in children with inherited mitochondrial disorders (Bersin and Stacpoole, 1997) and lactic acidosis (Archer et al., 2008).
As discussed earlier, the adult healthy heart derives most of its energy from oxidation of FA, and FA oxidation strongly inhibits glucose oxidation; this is because acetyl-CoA derived from FA β-oxidation inhibits PDH activity so glycolysis is halted (Beadle and Frenneaux, 2010). Thus, by maintaining acetyl-CoA removal by FA β-oxidation, upregulation of PDK facilitates the continuous uptake of long chain fatty acyl-CoA (transformed from malonyl-CoA) into the mitochondrial matrix for oxidation, preventing their accumulation in the cytoplasm, where they would exert toxic effects on cardiac function (Nediani et al., 2011). However, despite its potential benefits, attempting to control β-oxidation of FA has numerous drawbacks and limitations. For example, although β-oxidation of FA produces more energy per carbon than glycolysis, it requires a higher consumption of O2 (Nediani et al., 2011).
In heart failure there appears to be a downregulation of genes controlling mitochondria biogenesis and of enzymes involved in FA oxidation (Nediani et al., 2011). The mitochondrial content and hence oxidative capacity of both skeletal and cardiac muscle cells are diminished in heart failure (Camara et al., 2010a), i.e., “starvation in the land of plenty.” However, the concept that FAs are bad for contractile function in the failing or diseased heart is challenged by other studies. For example, recent reports show that decreased FA oxidation during heart failure could be maladaptive (Cheng et al., 2004), and that increasing FA availability aids the failing heart (Duda et al., 2008; Muoio and Newgard, 2008). In this case, patients with dilated cardiomyopathy challenged with acute FA depletion demonstrated enhanced glucose oxidation but decreased cardiac work and efficiency (Tuunanen et al., 2006a,b). A crucial distinction between physiological and pathological cardiac remodeling (the later being a process ultimately progressing to heart failure) is the ability of the heart to match workload with an increased ATP production (see earlier discussion).
In an attempt to increase the efficiency of energy production in the failing heart, several drugs that inhibit FA oxidation and thereby allow glycolysis and SLP have been subject to testing. For example, etomoxir, originally developed as an anti-diabetic drug to switch energy metabolism from FA to glucose by inhibiting the FA transporter CPT1, has been clinically tested for treatment of chronic heart failure (Wolf, 1992; Kerner et al., 1994). However, long-term use of this drug is associated with toxicity and ironically, heart failure (Stanley et al., 1997). The CPT1 inhibitors oxfenicine and perhexiline have been shown to delay the development of terminal heart failure and attenuate the adverse hemodynamic changes (Abozguia et al., 2006). These drugs are known to activate expression of genes encoding key mitochondrial proteins involved in cardiac energy metabolism. For example, oxfenicine in heart failure has been shown to down regulate the transcription of CPT1 and to prevent a decrease of TCA cycle enzyme activity (Abozguia et al., 2006). Other drugs such as trimetazidine, which blocks the key enzyme in FA oxidation, as well as ranolazine, are reported to directly attenuate β-oxidation enzyme activity and potentially other metabolic enzymes (Stanley et al., 1997; Bristow, 2000). Hence, the process of metabolic remodeling could serve as a target to restore metabolic flexibility and thereby facilitate improved myocardial contractility (Taegtmeyer et al., 2005; Kolwicz and Tian, 2009). This approach of optimizing energy substrate preference in the heart is a new and potentially promising approach to treat cardiac dysfunction associated with angina pectoris (Lam and Lopaschuk, 2007) and other cardiac abnormalities.
As noted above, mitochondria isolated from failing hearts display increased ROS production in association with signs of oxidative stress-related damage due to accumulation of lipid peroxidation by-products and mutations or deletions of mtDNA (Ide et al., 2001; Burelle et al., 2009). Other studies show decreases in the expression and activity of catalase and MnSOD in heart failure (Laskowski and Russell, 2008). Evidence also shows increased mitochondrial membrane permeability and a consequent increase in release of cytochrome c and down stream pro-caspases; all of these factors indicate that the mitochondria-induced apoptotic pathway is a requirement to cardiomyocyte death by apoptosis (Burelle et al., 2009). From these observations the importance of decreasing oxidative and nitrosative stress by decreases in ROS and RNS generation and the successful use of antioxidant therapy in heart failure can not be overstated (Tsutsui et al., 2006). Indeed, ROS and RNS scavengers have been shown to attenuate the structural and functional abnormalities associated with heart failure in a rat model (Li et al., 2009). In that study Li et al. (2009) reported that the antioxidant α-lipoic acid attenuated mitochondria-dependent apoptosis in diabetic cardiomyopathy. In addition, UCP levels are often decreased in animal models of heart failure and in human clinical samples (Laskowski and Russell, 2008). Targeting the UCPs therefore may represent a promising approach to managing heart failure.
Potential for Treating Mitochondria in Cardiac I/R Injury
Some aspects of this subject were discussed in other sections and also were discussed extensively in two recent review articles (Chen et al., 2007; Camara et al., 2010b) so this topic will be only briefly addressed. I/R has been shown to result in mitochondrial dysfunction, so the importance of mitochondria as both targets and mediators of I/R injury is well recognized (Chen et al., 2007). During ischemia, ETC complex activities and the functions of ANT and CPT1 are depressed as a result of damage to cardiolipin and increased H+ leak, thereby compromising ΔΨm and ATP supply (Paradies et al., 2009). During ischemia, anaerobic glycolysis partially generates the needed ATP with a concomitant increase in intracellular acidosis, but eventually impaired ATP-dependent ion pumps that are required to maintain ion homeostasis fail. Intracellular acidosis drives the Na+/H+ exchanger and this leads to increased intracellular [Na+] and a subsequent increase in intracellular [Ca2+] after activation of the Na+/Ca+ exchanger in reverse mode. These alterations in cytosolic [Na+] and [Ca2+] also induce dysregulation of ion homeostasis in mitochondria resulting in mCa2+ overload (discussed above), which in turn results in mitochondrial depolarization that helps to limit further Ca2+ uptake via the CaU. Upon reperfusion however, repolarization of ΔΨm coupled with increased cytosolic Ca2+ leads to a further increase in mCa2+ load via the CaU. With recovery of matrix pH (less acidic), low ATP levels, and a high Pi, on reperfusion, opening of the mPTP can occur. Thus, CsA and other mPTP inhibitors may lead to cardioprotection, but only if administered on reperfusion (Duchen, 2004). Five potential strategic approaches to protect mitochondria against cardiac I/R injury are given in the following five sections (not discussed in our previous comprehensive review article (Camara et al., 2010a).
Attenuation of Mitochondrial Bioenergetics as a Potential Strategy to Protect Against I/R Injury
Many studies now show that increases in mitochondrial ROS and m[Ca2+] occur nearly simultaneously during ischemia (Vanden Hoek et al., 1997; Kevin et al., 2003; Riess et al., 2004; Stowe et al., 2006; Camara et al., 2007; Aldakkak et al., 2008a,b) and that their reduction on reperfusion are indicative of protection. A pioneering study by Ganote et al. (1976) showed that inhibiting mitochondrial respiration could decrease contraction band formation and attenuate enzyme release during reoxygenation; they suggested that resumption of mitochondrial metabolism during reoxygenation could initially lead to deleterious consequences (Ganote et al., 1976). Either restriction of oxidative metabolism during the early reperfusion period, use of a hypoxic reperfusate (Kasamaki et al., 1997; Serviddio et al., 2005), or inhibition of ETC activity (Ambrosio et al., 1993), attenuated mitochondrial and cardiac damage as evidenced by decreased cardiac function and accumulation of oxidatively damaged lipids. In a recent insightful review, Burwell and Brookes (2008) proposed the concept of “a more gradual wake-up of mitochondrial function” to protect the myocardium from a burst of ROS and mCa2+ overload following I/R. This concept of gradual awakening on reperfusion is based on the premise that reversible inhibition of metabolism by blocking ETC complexes and glycolysis, and the slow reintroduction of electrons to the ETC, will reduce damage (Burwell and Brookes, 2008).
Many findings support the physiologic significance of excess mitochondrial ROS production in cardiac injury during reperfusion (Chen et al., 2007). With knowledge of some of the endpoints of cardiac injury there are now a variety of strategies to target mitochondria that are postulated to interrupt the link between mitochondrial damage during ischemia and mitochondria-mediated damage during reperfusion. Interestingly, we have observed preliminarily that resveratrol, a ROS scavenger (Bastianetto et al., 2000), is more effective in protecting against I/R injury when administered briefly on reperfusion, whereas ranolazine, the anti-anginal agent believed to reversibly inhibit complex I (Wyatt et al., 1995), was more effective against I/R injury when given briefly before ischemia (Aldakkak et al., 2009). Consistent with our observation with resveratrol in the isolated beating heart, Morin et al. (2003a) reported that resveratrol was more effective when administered during reoxygenation in isolated brain mitochondria. These data suggest drugs that scavenge ROS may be more protective when administered on reperfusion, whereas blockers of ETC are more effective when given just before ischemia. Thus a potentially effective combination may be the selective administration of the differently acting drugs either before or after ischemia.
Other viable strategies that may work by modulating mitochondrial bioenergetics include the use of putative K+ channel agonists like NS1619 (discussed above) and uncouplers like DNP (Aldakkak et al., 2010; Sedlic et al., 2010b). Sedlic et al. (2010b) reported recently that isoflurane, like DNP, may protect cardiomyocytes in part via a mild decrease in ΔΨm, which attenuated ROS production under stress and led to a delay in mPTP opening. Overexpression of UCPs in mice as well as chemical uncoupling of mitochondria in rats markedly reduced infarct volume following focal cerebral I/R (Duckles et al., 2006). However, clinical use of chemical uncoupling (e.g., DNP) has been limited by its toxicity (Camara et al., 2010b).
Modulation of TCA Cycle Function as a Potential Strategy to Protect Mitochondria during I/R Injury
It has been suggested that glycolytically derived ATP is preferentially used to activate Ca2+ uptake into the ER by Ca2+ ATPase and to activate the Na+/K+ ATPase that prevents intracellular Na+ accumulation during ischemia (Beadle and Frenneaux, 2010). As discussed earlier, limited O2 availability during ischemia or hypoxia leads to a shift to glucose metabolism from mitochondrial OxPhos to cytosolic substrate level phosphorylation (SLP; anaerobic glycolysis); but prolonged ischemia leads to accumulation of lactic acid and depletion of NAD+. To sustain glycolysis under prolonged anaerobic conditions, lactate is excreted from the cell as a waste product. In this scenario, when mitochondrial function is compromised and ΔΨm is reduced as in I/R, F0/F1 ATP synthase acts in reverse thereby hydrolyzing ATP in an attempt to reestablish ΔΨm. In this case cytosolic ATP is consumed to pump H+ out of the matrix to attempt to restore ΔΨm; however, this is counterproductive for ATP-dependent cellular processes, e.g., contraction/relaxation of myofibrils and for establishing cellular ionic homeostasis. In addition, restoration of ΔΨm may cause further matrix Ca2+ accumulation, which could lead to the perpetual and vicious cycle of cellular injury.
A novel concept in mitochondria-mediated cytoprotection is that enhanced SLP within the matrix (in contrast to cytosolic SLP from pyruvate) represents an endogenous rescue mechanism that supports the reversal of the F0F1 ATP synthase to maintain ΔΨm while preserving cytosolic ATP. The matrix contains several enzyme sites capable of SLP; these include α-ketoglutarate dehydrogenase (Schwimmer et al., 2005) and succinyl-CoA synthase (SULCA). SUCLA is an enzyme that catalyzes the reversible conversion of succinyl-CoA and ADP or GDP to ATP or GTP (Kibbey et al., 2007; Phillips et al., 2009). Hence, this could represent a source of ATP during ischemia, primarily by SLP (Camara et al., 2010a). Therefore, activation of these pathways could be crucial in periods of energy deprivation when complex V reverts to ATP consumption to maintain ΔΨm as observed during I/R. In this way, cytosolic ATP levels from glycolysis might be preserved for maintaining the operation of ATP-dependent ion exchangers, e.g., Na+/K+ ATPase and Ca2+ ATPase, needed to maintain cellular cytosolic ion gradients, while reverse ATP synthase at complex V supports, at least in part, the ΔΨm. Therefore, cells may be salvaged as long as matrix SLP is able to provide ATP for use by complex V and by limiting the consumption of cytosolic ATP. Increasing the generation of matrix SLP may be a worthwhile, but temporary, strategy to mitigate mitochondrial dysfunction at times of impaired OxPhos. For example, overexpression or stimulation of SUCLA at a time of severe energy deficit may provide the needed ATP for mitochondrial preservation while sparing cytosolic ATP. Indeed, the putative mitoKATP channel agonist diazoxide may in part mediate protection by facilitating the binding of IF1, an endogenous regulator of F0F1 ATP synthase activity. Upon binding to complex V, IF1 blocks ATP synthase-mediated ATP hydrolysis. In this way diazoxide could contribute to cardioprotection by preserving ATP during ischemia (Szewczyk et al., 2010).
Modulation of Mitochondrial ROS Generation as a Potential Protective Strategy during I/R Injury
Some aspects of this topic have been discussed earlier. Nonetheless, the association of 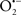 generation with I/R injury has made the development of antioxidants for therapeutic uses a plausible goal (Camara et al., 2010b). Therapeutic strategies to limit the rate of mitochondrial ROS generation or to increase the rate of ROS scavenging may be useful adjuvants to conventional therapies designed to protect the heart from I/R damage. We reported that administering a cocktail of mitochondrial scavengers (MnTBAP + glutathione + catalase) prior to cold cardiac ischemia and warm reperfusion provided protection against mCa2+ overload and ROS production and that this helped to preserve cardiac function (Camara et al., 2007). Artificial antioxidants such as SOD mimetics may be more potent than their natural counterparts, but their usefulness in clinical trials has not been determined (Muravchick and Levy, 2006; Schapira, 2006). Needless to say, mitochondria-targeted ROS scavengers, such as ubiquinone and vitamin E, have shown some efficacy against cell damage associated with I/R injury. However, clinical trials using α-tocopherol (vitamin E), ascorbate (vitamin C), CoQ, and α-lipoic acid, have so far yielded ambiguous results (Camara et al., 2010b).
generation with I/R injury has made the development of antioxidants for therapeutic uses a plausible goal (Camara et al., 2010b). Therapeutic strategies to limit the rate of mitochondrial ROS generation or to increase the rate of ROS scavenging may be useful adjuvants to conventional therapies designed to protect the heart from I/R damage. We reported that administering a cocktail of mitochondrial scavengers (MnTBAP + glutathione + catalase) prior to cold cardiac ischemia and warm reperfusion provided protection against mCa2+ overload and ROS production and that this helped to preserve cardiac function (Camara et al., 2007). Artificial antioxidants such as SOD mimetics may be more potent than their natural counterparts, but their usefulness in clinical trials has not been determined (Muravchick and Levy, 2006; Schapira, 2006). Needless to say, mitochondria-targeted ROS scavengers, such as ubiquinone and vitamin E, have shown some efficacy against cell damage associated with I/R injury. However, clinical trials using α-tocopherol (vitamin E), ascorbate (vitamin C), CoQ, and α-lipoic acid, have so far yielded ambiguous results (Camara et al., 2010b).
Modulation of Gaseous Signaling Molecules as a Potential Protective Strategy during I/R Injury
In addition to the endogenous enzymes synthesizing or releasing 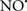 and
and 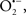 carbon monoxide (CO) and hydrogen sulfide (H2S) gases are also present in small amounts; they share many of the “friends-and-foe” characteristics of ROS and RNS (Baumgart et al., 2009). The role of
carbon monoxide (CO) and hydrogen sulfide (H2S) gases are also present in small amounts; they share many of the “friends-and-foe” characteristics of ROS and RNS (Baumgart et al., 2009). The role of 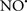 has been discussed before in several sections and a detailed discussion of its role in cardioprotection can be found in our recent comprehensive articles (Stowe and Camara, 2009; Camara et al., 2010b), and will not be discussed further here. As gaseous second messengers, they also share some common biological targets, for example heme-proteins and KATP channels (Burwell et al., 2009). At physiologic concentrations CO and H2S may help to protect against many pathologic conditions including cardiovascular diseases. At higher concentrations, however, these gases are toxic with deleterious effects on the cell (Lefer, 2007). A recent review article discusses the interplay between the gases at the level of mitochondrial complexes (Cooper and Brown, 2008; Calvert et al., 2010).
has been discussed before in several sections and a detailed discussion of its role in cardioprotection can be found in our recent comprehensive articles (Stowe and Camara, 2009; Camara et al., 2010b), and will not be discussed further here. As gaseous second messengers, they also share some common biological targets, for example heme-proteins and KATP channels (Burwell et al., 2009). At physiologic concentrations CO and H2S may help to protect against many pathologic conditions including cardiovascular diseases. At higher concentrations, however, these gases are toxic with deleterious effects on the cell (Lefer, 2007). A recent review article discusses the interplay between the gases at the level of mitochondrial complexes (Cooper and Brown, 2008; Calvert et al., 2010).
H2S may be endogenously synthesized in small amounts in the heart, and like 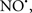 it competitively inhibits O2 binding to complex IV, an effect that could promote myocardial cell survival during I/R (Bian et al., 2006; Lefer, 2007). Considered a poisonous gas at high concentrations, H2S is being recognized as an inorganic mediator with a potentially broad therapeutic potential (Baumgart et al., 2009). It could develop as a useful mediator of cardiovascular homeostasis and cytoprotection. For example, H2S has been implicated in IPC cardioprotection via activation of both mitoKATP and sarcolemmal KATP channels; the exogenous H2S donor, NaHS, was shown to increase cell viability and to improve cardiac function during I/R (Bian et al., 2006); 5-HD abolished the protective effects of H2S (Baumgart et al., 2009). Another possible mechanism of protection by H2S is scavenging of ROS, by which it is believed to reduce arrhythmias during I/R (Bian et al., 2006). Similar cytoprotective effects against oxidative stress have been observed in the brain where H2S protected neurons from oxidative stress by increasing the production of the antioxidant GSH (Kimura and Kimura, 2004). As a reducing agent, H2S may react with H2O2 and in this way scavenges ROS (Kimura and Kimura, 2004). However, the preferred route of therapeutic administration of H2S remains unclear.
it competitively inhibits O2 binding to complex IV, an effect that could promote myocardial cell survival during I/R (Bian et al., 2006; Lefer, 2007). Considered a poisonous gas at high concentrations, H2S is being recognized as an inorganic mediator with a potentially broad therapeutic potential (Baumgart et al., 2009). It could develop as a useful mediator of cardiovascular homeostasis and cytoprotection. For example, H2S has been implicated in IPC cardioprotection via activation of both mitoKATP and sarcolemmal KATP channels; the exogenous H2S donor, NaHS, was shown to increase cell viability and to improve cardiac function during I/R (Bian et al., 2006); 5-HD abolished the protective effects of H2S (Baumgart et al., 2009). Another possible mechanism of protection by H2S is scavenging of ROS, by which it is believed to reduce arrhythmias during I/R (Bian et al., 2006). Similar cytoprotective effects against oxidative stress have been observed in the brain where H2S protected neurons from oxidative stress by increasing the production of the antioxidant GSH (Kimura and Kimura, 2004). As a reducing agent, H2S may react with H2O2 and in this way scavenges ROS (Kimura and Kimura, 2004). However, the preferred route of therapeutic administration of H2S remains unclear.
CO, like 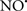 and H2S, reversibly binds to the reduced complex IV where it competes with O2; CO may block O2 binding at complex IV in vivo (Zuckerbraun et al., 2007) and lead to cardioprotection against I/R injury, whereas Na azide, a cyanide containing compound which also inhibits complex IV, was found to be ineffective in protecting the heart (Chen et al., 2010). Taken together, Chen et al. (2010) posits that there are inconsistencies regarding potential cardioprotection provided by complex IV inhibition. It may be that CO has other cytoprotective effects other than binding to complex IV. Indeed, in I/R, CO has been reported to increase ATP and GTP levels to maintain efficient energy utilization within the heart during the ischemic period (Lavitrano et al., 2004), which correlates to a greater ease by which the heart can generate force on reperfusion and minimize the incidence of fibrillation. Although the cause–effect relationship of CO with nucleotide concentrations is unclear, this metabolic modulation to protect the heart may be analogous to SLP-induced protection discussed previously. CO could also provide protection by preserving mitochondrial NADH, i.e., decrease oxidation of the reduced redox state, via its action on the ETC. The increase in NADH may reflect the possibility of decreased O2 consumption, resulting in an overall lower metabolic state and a reduction in appearance in apoptotic markers (Lavitrano et al., 2004). Endogenous CO generated by overexpression of heme oxygenase (HO)-1 was shown to stimulate MnSOD and generate H2O2, which in turn activated signaling molecules that lead to mitochondrial biogenesis (Piantadosi et al., 2008) and cardioprotection.
and H2S, reversibly binds to the reduced complex IV where it competes with O2; CO may block O2 binding at complex IV in vivo (Zuckerbraun et al., 2007) and lead to cardioprotection against I/R injury, whereas Na azide, a cyanide containing compound which also inhibits complex IV, was found to be ineffective in protecting the heart (Chen et al., 2010). Taken together, Chen et al. (2010) posits that there are inconsistencies regarding potential cardioprotection provided by complex IV inhibition. It may be that CO has other cytoprotective effects other than binding to complex IV. Indeed, in I/R, CO has been reported to increase ATP and GTP levels to maintain efficient energy utilization within the heart during the ischemic period (Lavitrano et al., 2004), which correlates to a greater ease by which the heart can generate force on reperfusion and minimize the incidence of fibrillation. Although the cause–effect relationship of CO with nucleotide concentrations is unclear, this metabolic modulation to protect the heart may be analogous to SLP-induced protection discussed previously. CO could also provide protection by preserving mitochondrial NADH, i.e., decrease oxidation of the reduced redox state, via its action on the ETC. The increase in NADH may reflect the possibility of decreased O2 consumption, resulting in an overall lower metabolic state and a reduction in appearance in apoptotic markers (Lavitrano et al., 2004). Endogenous CO generated by overexpression of heme oxygenase (HO)-1 was shown to stimulate MnSOD and generate H2O2, which in turn activated signaling molecules that lead to mitochondrial biogenesis (Piantadosi et al., 2008) and cardioprotection.
Interestingly, hydrogen gas (H2) has been proposed as a potential therapeutic agent with both anti-apoptotic and antioxidant features (Wood and Gladwin, 2007). Inhaled H2 gas can reach its target rapidly and provide protection against I/R (Hayashida et al., 2008). Thus H2 gas may be a promising strategy to mitigate I/R injury (Hayashida et al., 2008); but its use is limited by its highly explosive nature (Baumgart et al., 2009). All together, these studies provide a rationale for targeting mitochondria with low concentrations of gases that compete with O2 at complex IV, although as noted, this may also be detrimental if administered at the inappropriate time and concentration. The defined physiological role of these gases makes them potential pharmacological candidates to target mitochondria in the treatment of cardiovascular diseases. Specific overexpression of those enzymes that generate some of these gases could be a feasible approach to mitigating some disease states, particularly in high energy consuming organs.
Protecting Cardiac Mitochondria with Near Infrared Light during Cardiac I/R Injury
Mitochondria are sensitive targets for therapy with red light. Low energy photon irradiation by light in the far red to near infrared spectral range (630–1000 nm; NIR) emitted using low energy lasers or light emitting diode arrays (LED) has been found to provide good cardioprotection. In experimental models of myocardial infarction low energy lasers (803 nm) reduced infarct size 50–70% 4–6 weeks after permanent occlusion of the left descending coronary artery in rats and dogs (Oron et al., 2001). Low-level laser application up-regulated inducible NOS and vascular endothelial growth factor and these changes were associated with cardioprotection and enhanced angiogenesis (Tuby et al., 2006).
We demonstrated recently that acute application of NIR (670 nm) at the point of reperfusion or reoxygenation resulted in a significant decrease in infarct size, or protection of cardiomyocytes, respectively (Lohr et al., 2009; Zhang et al., 2009). Indices of apoptosis, including caspase 3 activity, annexin binding, and the release of cytochrome c from mitochondria into the cytosol, were decreased after NIR treatment. NIR increased 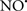 levels in cardiomyocytes, and the protective effect of NIR was completely reversed by the
levels in cardiomyocytes, and the protective effect of NIR was completely reversed by the 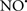 scavenger carboxy-PTIO and by oxyhemoglobin, but only partially blocked by the NOS inhibitor L-NMMA. Mitochondrial metabolism, measured by ATP synthase activity, was increased by NIR, and
scavenger carboxy-PTIO and by oxyhemoglobin, but only partially blocked by the NOS inhibitor L-NMMA. Mitochondrial metabolism, measured by ATP synthase activity, was increased by NIR, and 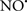 -induced inhibition of O2 consumption with substrates for complex I or complex IV was reversed by exposure to NIR (Zhang et al., 2009).
-induced inhibition of O2 consumption with substrates for complex I or complex IV was reversed by exposure to NIR (Zhang et al., 2009).
Mitochondrial cytochromes have been proposed as photoacceptors for NIR light energy and mitochondrial redox changes have been proposed as potential mediators of the biological effects of NIR (Hallen et al., 1993; Hallen and Brzezinski, 1994; Wong-Riley et al., 2005). The action spectrum of NIR light for stimulation of cytochrome c oxidase activity parallels the NIR absorption spectrum of the oxidized form of complex IV (Karu et al., 2004). NIR increased mitochondrial metabolism, including cytochrome c oxidase activity and elevated cellular ATP content (Karu et al., 1995). Interestingly, NIR reversed the toxic effects of KCN, an irreversible inhibitor of complex IV, in primary neurons (Wong-Riley et al., 2005) and improved retinal function in an animal model of mitochondrial dysfunction induced by methanol-derived formate, a reversible cytochrome c oxidase inhibitor (Eells et al., 2004).
The mechanism behind this metabolic enhancement of challenged mitochondria through NIR remains controversial. Changes in the redox properties and acceleration of e− transfer at the center of complex IV have been suggested as well as enhanced 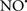 release (Karu, 2008). The direct actions of light reversal of
release (Karu, 2008). The direct actions of light reversal of 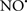 mediated complex IV inhibition, with its resulting increase in respiration and ATP production, could be effective in ameliorating reperfusion injury. In contrast, we demonstrated recently, both in purified systems and in myocardium, that NIR can release
mediated complex IV inhibition, with its resulting increase in respiration and ATP production, could be effective in ameliorating reperfusion injury. In contrast, we demonstrated recently, both in purified systems and in myocardium, that NIR can release 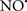 from nitrosyl hemoglobin or myoglobin (Lohr et al., 2009). NIR-induced release of
from nitrosyl hemoglobin or myoglobin (Lohr et al., 2009). NIR-induced release of 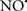 might contribute to protection from reperfusion injury through actions on mitochondria and
might contribute to protection from reperfusion injury through actions on mitochondria and 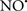 -dependent signaling pathways. As noted above, an inhibitory effect of
-dependent signaling pathways. As noted above, an inhibitory effect of 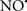 on ETC, particularly complexes I and IV, has been suggested to increase cellular viability in myocardial I/R injury by decreasing ROS generation during reperfusion (Hendgen-Cotta et al., 2008). Although NOS produces the bulk of
on ETC, particularly complexes I and IV, has been suggested to increase cellular viability in myocardial I/R injury by decreasing ROS generation during reperfusion (Hendgen-Cotta et al., 2008). Although NOS produces the bulk of 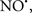 there is increasing interest in NOS independent generation of
there is increasing interest in NOS independent generation of 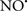 in vivo, particularly during hypoxia or anoxia, where low O2 tensions limit NOS activity and when the nitrite reductase activity of myoglobin and hemoglobin are highest (Gladwin and Kim-Shapiro, 2008). Interventions such as NIR that can increase
in vivo, particularly during hypoxia or anoxia, where low O2 tensions limit NOS activity and when the nitrite reductase activity of myoglobin and hemoglobin are highest (Gladwin and Kim-Shapiro, 2008). Interventions such as NIR that can increase 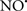 bioavailability in a localized manner have significant therapeutic potential against heart disease without systemic side effects. Thus, the effect of NIR on the heart, and specifically on mitochondria, may depend on a delicate balance between stimulation and attenuation of mitochondrial metabolism.
bioavailability in a localized manner have significant therapeutic potential against heart disease without systemic side effects. Thus, the effect of NIR on the heart, and specifically on mitochondria, may depend on a delicate balance between stimulation and attenuation of mitochondrial metabolism.
Limitations and Conclusion
As mitochondria are the most prominent source of intracellular ROS and RNS, they have been implicated in both defensive and offensive aspects of the cell; but selective targeting of mitochondria with mitochondrial-targeted agents is likely to pose a quandary. Targeting mitochondria for therapeutic purposes poses a dilemma of how to protect, while at the same time, preserve the normal aspects of cellular functions that maintain viability. This quandary has been observed by Muravchick and Levy (2006) in a recent review article noting that, “therapeutic strategies that suppress or block the effects of putative pro-apoptotic agents may produce unintended interruptions of other cell functions and actually compromise viability.”
The aim of this review was to provide and summarize a growing body of evidence that mitochondria are intimately involved in cell survival and cell death and indeed in the etiology of cardiovascular diseases. This role renders mitochondria as unique targets for pharmacological and non-pharmacological interventions. Currently, most efforts along these lines focus on preventing the mitochondrial oxidative damage that arises from ischemic or hypoxic conditions. Examples of mitochondria-targeted maneuvers include: decreasing mPTP opening; decreasing ΔΨm to reduce 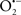 production; moderate inhibition of complexes upstream of complex IV to reduce ROS production from complex III; limiting cytosolic ATP consumption during cell distress with increased matrix SLP; and increasing signaling gas molecules with, for instance, photobiomodulation therapy. In addition, mitochondria are targets for gene therapy in case of diseases caused by mutations of mtDNA or nuclear DNA (Wojtczak and Zablocki, 2008). The timing, conditions of drug deliver, and drug concentration, will be crucial in assessing whether the treatment is beneficial or deleterious to mitochondrial function during and after cell injury.
production; moderate inhibition of complexes upstream of complex IV to reduce ROS production from complex III; limiting cytosolic ATP consumption during cell distress with increased matrix SLP; and increasing signaling gas molecules with, for instance, photobiomodulation therapy. In addition, mitochondria are targets for gene therapy in case of diseases caused by mutations of mtDNA or nuclear DNA (Wojtczak and Zablocki, 2008). The timing, conditions of drug deliver, and drug concentration, will be crucial in assessing whether the treatment is beneficial or deleterious to mitochondrial function during and after cell injury.
It is evident that much attention has been focused on developing mitochondria-targeted drugs that may accumulate in mitochondria without affecting other parts of the cell. Therefore, novel approaches with more tissue specific mitochondria-targeted approaches or interventions that will furnish a greater efficacy and selectivity with fewer undesirable effects are being pursued. Use of mitochondriotropic drugs seems to be the most promising approach to prevent or treat mitochondrial diseases associated with cardiovascular dysfunction. However, due to lingering concerns in drug delivery to appropriate organs and possible untoward effects, these techniques will need to be improved and refined over time. Lastly, effective genetic and molecular approaches to provide better-targeted delivery of desired therapeutic agent, although promising, remains at best a rudimentary notion to date (Camara et al., 2010b).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank Mohammed Aldakkak MD for his help with the figures shown in this review. This work was supported in part by grants from the National Institutes of Health (HL095122, HL089514, HL098490, P01GM066730); the American Heart Association (0855940G); and the Veterans Administration (Merit Review 8204-05P).
Abbreviations
5-HD, 5-hydroxydecanoic acid; ACC, acetyl-CoA carboxylase; Acetyl-CoA, acetyl coenzyme A; ANT, adenine nucleotide translocase; CaU, calcium uniporter; CK, creatine kinase; CPT, carnitine-acetyl-CoA (palmitoyl) transferase; CsA, cyclosporin A; ETC, electron transport chain; FA, Fatty acids; FADH2, flavin adenine dinucleotide (reduced); GPx, glutathione peroxidase; GR, glutathione reductase; GSH, glutathione (reduced); H2O2, hydrogen peroxide; HKI and HKII, hexokinase I and II; HNE, 4-hydroxy-trans-2-noneal; IMM, inner mitochondrial membrane; IMS, intermembrane space; IPC, ischemic preconditioning; KATP, ATP-sensitive K+ channel; KCa, Ca2+ sensitive K+ channel; LRT, lipid replacement therapy; MCD, malonyl-CoA decarboxylase; MnIII TE-2-Pyp5+, Mn (III) meso-tetrakis (N-ethylpyridium-2-yl) porphyrin; MnSOD, manganese superoxide dismutase; MnTBAP, Mn(II)tetrakis(4-benzoate) porphyrin chlorine; MnTPyP, Mn(III)tetrakis [(1-methyl-4-pyridyl)-porphyrin]; mPTP, mitochondrial permeability transition pore; mtDNA, mitochondrial DNA; NADH, nicotinamide adenine dinucleotide (reduced); NICE, sodium-independent calcium exchanger; NIM811, N-methyl-4-isoleucine-cyclosporin; nitric oxide radical; superoxide anion radical; OMM, outer mitochondrial membrane; ONOO–, peroxynitrite; OxPhos, oxidative phosphorylation; PBR, peripheral benzodiazepine receptor; PCr, phosphocreatine; PDH, pyruvate dehydrogenase; PDK, pyruvate dehydrogenase kinase; PFK, phosphofructokinase; PGC-1α, peroxisome proliferator-activated receptor-gamma co-activator-1α; Q, coenzyme Q10, ubiquinone, quinone; RNS, reactive nitrogen species; ROS, reactive oxygen species; RR, ruthenium red; RyR, ryanodine receptor; SLP, substrate level phosphorylation; SNO-MPG, S-nitroso-2-mercaptopropionyl glycine; SOD, superoxide dismutase; SR, sarcoplasmic reticulum; TOM and TIM, outer and inner membrane translocases, respectively; TPP+, triphenyl-phosphonium; TRX, thioredoxin; TRXSH2, thioredoxin (reduced); UCP, uncoupling proteins; VDAC, voltage dependent anion channel; ΔΨm, mitochondrial trans-membrane potential.
References
Abozguia, K., Clarke, K., Lee, L., and Frenneaux, M. (2006). Modification of myocardial substrate use as a therapy for heart failure. Nat. Clin. Pract. Cardiovasc. Med. 3, 490–498.
Aldakkak, M., Heisner, J. S., Camara, A. K. S., Haumann, J., and Stowe, D. F. (2009). Ranolazine, a late sodium current inhibitor, reduces ischemia-induced superoxide emission and improves functional recovery in guinea pig hearts. FASEB J. Abstract 23, :93.17.
Aldakkak, M., Stowe, D. F., Chen, Q., Lesnefsky, E. J., and Camara, A. K. (2008a). Inhibited mitochondrial respiration by amobarbital during cardiac ischaemia improves redox state and reduces matrix Ca2+ overload and ROS release. Cardiovasc. Res. 77, 406–415.
Aldakkak, M., Stowe, D. F., Heisner, J. S., Spence, M., and Camara, A. K. (2008b). Enhanced Na+/H+ exchange during ischemia and reperfusion impairs mitochondrial bioenergetics and myocardial function. J. Cardiovasc. Pharmacol. 52, 236–244.
Aldakkak, M., Stowe, D. F., Heisner, J. S., Haumann, J., and Camara, A. K. S. (2008c). Blocking mitochondrial Ca2+ uniporter activity during activated Na+/H+ exchange reduces Ca2+ loading but does little to better protect function on reperfusion. FASEB J. Abstract 22, :730.24.
Aldakkak, M., Stowe, D. F., Cheng, Q., Kwok, W. M., and Camara, A. K. (2010). Mitochondrial matrix K+ flux independent of large-conductance Ca2+-activated K+ channel opening. Am. J. Physiol. Cell Physiol. 298, C530–C541.
Ambrosio, G., Zweier, J. L., Duilio, C., Kuppusamy, P., Santoro, G., Elia, P. P., Tritto, I., Cirillo, P., Condorelli, M., and Chiariello, M. (1993). Evidence that mitochondrial respiration is a source of potentially toxic oxygen free radicals in intact rabbit hearts subjected to ischemia and reflow. J. Biol. Chem. 268, 18532–18541.
Andreyev, A. Y., Kushnareva, Y. E., and Starkov, A. A. (2005). Mitochondrial metabolism of reactive oxygen species. Biochemistry Mosc. 70, 200–214. [Review].
Aon, M. A., Cortassa, S., Akar, F. G., and O’Rourke, B. (2006a). Mitochondrial criticality: a new concept at the turning point of life or death. Biochim. Biophys. Acta 1762, 232–240.
Aon, M. A., Cortassa, S., and O’Rourke, B. (2006b). The fundamental organization of cardiac mitochondria as a network of coupled oscillators. Biophys. J. 91, 4317–4327.
Aon, M. A., Cortassa, S., Maack, C., and O’Rourke, B. (2007). Sequential opening of mitochondrial ion channels as a function of glutathione redox thiol status. J. Biol. Chem. 282, 21889–21900.
Aon, M. A., Cortassa, S., Marban, E., and O’Rourke, B. (2003). Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J. Biol. Chem. 278, 44735–44744.
Archer, S. L., Gomberg-Maitland, M., Maitland, M. L., Rich, S., Garcia, J. G., and Weir, E. K. (2008). Mitochondrial metabolism, redox signaling, and fusion: a mitochondria-ROS-HIF-1alpha-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am. J. Physiol. Heart Circ. Physiol. 294, H570–H578.
Ashrafian, H., Frenneaux, M. P., and Opie, L. H. (2007). Metabolic mechanisms in heart failure. Circulation 116, 434–448.
Azoulay-Zohar, H., Israelson, A., Abu-Hamad, S., and Shoshan-Barmatz, V. (2004). In self-defence: hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. Biochem. J. 377(Pt 2), 347–355.
Babcock, G. T., and Varotsis, C. (1993). Discrete steps in dioxygen activation – the cytochrome oxidase/O2 reaction. J. Bioenerg. Biomembr. 25, 71–80.
Baines, C. P., Kaiser, R. A., Purcell, N. H., Blair, N. S., Osinska, H., Hambleton, M. A., Brunskill, E. W., Sayen, M. R., Gottlieb, R. A., Dorn, G. W., Robbins, J., and Molkentin, J. D. (2005). Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434, 658–662.
Balaban, R. S. (1990). Regulation of oxidative phosphorylation in the mammalian cell. Am. J. Physiol. 258(Pt 1), C377–C389.
Balaban, R. S., Kantor, H. L., Katz, L. A., and Briggs, R. W. (1986). Relation between work and phosphate metabolite in the in vivo paced mammalian heart. Science 232, 1121–1123.
Barreiro, E., Garcia-Martinez, C., Mas, S., Ametller, E., Gea, J., Argiles, J. M., Busquets, S., and Lopez-Soriano, F. J. (2009). UCP3 overexpression neutralizes oxidative stress rather than nitrosative stress in mouse myotubes. FEBS Lett. 583, 350–356.
Basso, E., Fante, L., Fowlkes, J., Petronilli, V., Forte, M. A., and Bernardi, P. (2005). Properties of the permeability transition pore in mitochondria devoid of cyclophilin D. J. Biol. Chem. 280, 18558–18561.
Bastianetto, S., Zheng, W. H., and Quirion, R. (2000). Neuroprotective abilities of resveratrol and other red wine constituents against nitric oxide-related toxicity in cultured hippocampal neurons. Br. J. Pharmacol. 131, 711–720.
Baumgart, K., Georgieff, M., Radermacher, P., and Calzia, E. (2009). Cardioprotection by hydrogen sulfide: suspended animation, inflammation, and apoptosis. Shock 31, 218–219.
Beadle, R. M., and Frenneaux, M. (2010). Modification of myocardial substrate utilisation: a new therapeutic paradigm in cardiovascular disease. Heart 96, 824–830.
Beard, D. A. (2006). Modeling of oxygen transport and cellular energetics explains observations on in vivo cardiac energy metabolism. PLoS Comput. Biol. 2, e107. doi: 10.1371/journal.pcbi.0020107
Beard, D. A., and Kushmerick, M. J. (2009). Strong inference for systems biology. PLoS Comput. Biol. 5, e1000459. doi: 10.1371/journal.pcbi.1000459
Belzacq, A. S., Vieira, H. L., Verrier, F., Vandecasteele, G., Cohen, I., Prevost, M. C., Larquet, E., Pariselli, F., Petit, P. X., Kahn, A., Rizzuto, R., Brenner, C., and Kroemer, G. (2003). Bcl-2 and Bax modulate adenine nucleotide translocase activity. Cancer Res. 63, 541–546.
Benov, L., Sztejnberg, L., and Fridovich, I. (1998). Critical evaluation of the use of hydroethidine as a measure of superoxide anion radical. Free Radic. Biol. Med. 25, 826–831.
Beraud, N., Pelloux, S., Usson, Y., Kuznetsov, A. V., Ronot, X., Tourneur, Y., and Saks, V. (2009). Mitochondrial dynamics in heart cells: very low amplitude high frequency fluctuations in adult cardiomyocytes and flow motion in non beating Hl-1 cells. J. Bioenerg. Biomembr. 41, 195–214.
Bersin, R. M., and Stacpoole, P. W. (1997). Dichloroacetate as metabolic therapy for myocardial ischemia and failure. Am. Heart J. 134(Pt 1), 841–855.
Bian, J. S., Yong, Q. C., Pan, T. T., Feng, Z. N., Ali, M. Y., Zhou, S., and Moore, P. K. (2006). Role of hydrogen sulfide in the cardioprotection caused by ischemic preconditioning in the rat heart and cardiac myocytes. J. Pharmacol. Exp. Ther. 316, 670–678.
Bianchi, P., Kunduzova, O., Masini, E., Cambon, C., Bani, D., Raimondi, L., Seguelas, M. H., Nistri, S., Colucci, W., Leducq, N., and Parini, A. (2005). Oxidative stress by monoamine oxidase mediates receptor-independent cardiomyocyte apoptosis by serotonin and postischemic myocardial injury. Circulation 112, 3297–3305.
Blaikie, F. H., Brown, S. E., Samuelsson, L. M., Brand, M. D., Smith, R. A., and Murphy, M. P. (2006). Targeting dinitrophenol to mitochondria: limitations to the development of a self-limiting mitochondrial protonophore. Biosci. Rep. 26, 231–243.
Boss, O., Samec, S., Paoloni-Giacobino, A., Rossier, C., Dulloo, A., Seydoux, J., Muzzin, P., and Giacobino, J. P. (1997). Uncoupling protein-3: a new member of the mitochondrial carrier family with tissue-specific expression. FEBS Lett. 408, 39–42.
Brady, N. R., Hamacher-Brady, A., and Gottlieb, R. A. (2006a). Proapoptotic BCL-2 family members and mitochondrial dysfunction during ischemia/reperfusion injury, a study employing cardiac HL-1 cells and GFP biosensors. Biochim. Biophys. Acta 1757, 667–678.
Brady, N. R., Hamacher-Brady, A., Westerhoff, H. V., and Gottlieb, R. A. (2006b). A wave of reactive oxygen species (ROS)-induced ROS release in a sea of excitable mitochondria. Antioxid. Redox Signal. 8, 1651–1665.
Bremer, J. (1969). Pyruvate dehydrogenase, substrate specificity and product inhibition. Eur. J. Biochem. 8, 535–540.
Bremer, J., and Wojtczak, A. B. (1972). Factors controlling the rate of fatty acid-oxidation in rat liver mitochondria. Biochim. Biophys. Acta 280, 515–530.
Brenner, C., Cadiou, H., Vieira, H. L., Zamzami, N., Marzo, I., Xie, Z., Leber, B., Andrews, D., Duclohier, H., Reed, J. C., and Kroemer, G. (2000). Bcl-2 and Bax regulate the channel activity of the mitochondrial adenine nucleotide translocator. Oncogene 19, 329–336.
Bristow, M. (2000). Etomoxir: a new approach to treatment of chronic heart failure. Lancet 356, 1621–1622.
Brookes, P. S. (2005). Mitochondrial H+ leak and ROS generation: an odd couple. Free Radic. Biol. Med. 38, 12–23. [Review].
Brookes, P. S., Yoon, Y., Robotham, J. L., Anders, M. W., and Sheu, S. S. (2004). Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 287, C817–C833. [Review].
Brown, D. A., and O’Rourke, B. (2010). Cardiac mitochondria and arrhythmias. Cardiovasc. Res. 88, 241–249.
Brunmair, B., Staniek, K., Gras, F., Scharf, N., Althaym, A., Clara, R., Roden, M., Gnaiger, E., Nohl, H., Waldhausl, W., and Furnsinn, C. (2004). Thiazolidinediones, like metformin, inhibit respiratory complex I: a common mechanism contributing to their antidiabetic actions? Diabetes 53, 1052–1059.
Bryson, J. M., Coy, P. E., Gottlob, K., Hay, N., and Robey, R. B. (2002). Increased hexokinase activity, of either ectopic or endogenous origin, protects renal epithelial cells against acute oxidant-induced cell death. J. Biol. Chem. 277, 11392–11400.
Burelle, Y., Khairallah, M., Ascah, A., Allen, B. G., Deschepper, C. F., Petrof, B. J., and Des Rosiers, C. (2009). Alterations in mitochondrial function as a harbinger of cardiomyopathy: lessons from the dystrophic heart. J. Mol. Cell. Cardiol. 48, 310–321.
Burwell, L. S., and Brookes, P. S. (2008). Mitochondria as a target for the cardioprotective effects of nitric oxide in ischemia-reperfusion injury. Antioxid. Redox Signal. 10, 579–599.
Burwell, L. S., Nadtochiy, S. M., and Brookes, P. S. (2009). Cardioprotection by metabolic shut-down and gradual wake-up. J. Mol. Cell. Cardiol. 46, 804–810.
Cadenas, E., Boveris, A., Ragan, C. I., and Stoppani, A. O. (1977). Production of superoxide radicals and hydrogen peroxide by NADH-ubiquinone reductase and ubiquinol-cytochrome c reductase from beef-heart mitochondria. Arch. Biochem. Biophys. 180, 248–257.
Cadenas, E., and Davies, K. J. (2000). Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 29, 222–230. [Review].
Cadoudal, T., Distel, E., Durant, S., Fouque, F., Blouin, J. M., Collinet, M., Bortoli, S., Forest, C., and Benelli, C. (2008). Pyruvate dehydrogenase kinase 4: regulation by thiazolidinediones and implication in glyceroneogenesis in adipose tissue. Diabetes 57, 2272–2279.
Calabrese, V., Lodi, R., Tonon, C., D’Agata, V., Sapienza, M., Scapagnini, G., Mangiameli, A., Pennisi, G., Stella, A. M., and Butterfield, D. A. (2005). Oxidative stress, mitochondrial dysfunction and cellular stress response in Friedreich’s ataxia. J. Neurol. Sci. 233, 145–162.
Calvert, J. W., Coetzee, W. A., and Lefer, D. J. (2010). Novel insights into hydrogen sulfide-mediated cytoprotection. Antioxid. Redox Signal. 12, 1203–1217.
Calvert, J. W., Gundewar, S., Jha, S., Greer, J. J., Bestermann, W. H., Tian, R., and Lefer, D. J. (2008). Acute metformin therapy confers cardioprotection against myocardial infarction via AMPK-eNOS-mediated signaling. Diabetes 57, 696–705.
Camara, A., Aldakkak, M., Heisner, J. S., Rhodes, S. S., Riess, M. L., An, J., Heinen, A., and Stowe, D. F. (2007). ROS scavenging before 27oC ischemia protects hearts and reduces mitochondrial ROS, Ca2+ overload, and changes in redox state. Am. J. Physiol. Cell Physiol. 292, C2021–C2031.
Camara, A., Aldakkak, M., and Stowe, D. (2010a). “Mitochondria as potential therapeutic targets in mitochondria-related diseases,” in Mitochondrial Structure, Function and Dysfunction, ed. O. L. Svensson (Milwaukee: Nova Science), 471–552.
Camara, A., Lesnefsky, E., and Stowe, D. (2010b). Potential therapeutic benefits of strategies directed to mitochondria. Antioxid. Redox Signal. 13, 279–347.
Camara, A., Kevin, L., Novalija, E., Varadarajan, S., and Stowe, D. (2002). Cold perfusion of isolated hearts generates reactive oxygen species but reduces generation and release during and after cold ischemia. Circulation, 106(Suppl. 2), SII–202.
Camara, A. K., Riess, M. L., Kevin, L. G., Novalija, E., and Stowe, D. F. (2004). Hypothermia augments reactive oxygen species detected in the guinea pig isolated perfused heart. Am. J. Physiol. Heart Circ. Physiol. 286, H1289–H1299.
Camello-Almaraz, C., Gomez-Pinilla, P. J., Pozo, M. J., and Camello, P. J. (2006). Mitochondrial reactive oxygen species and Ca2+ signaling. Am. J. Physiol. Cell Physiol. 291, C1082–C1088. [Review].
Carpi, A., Menabo, R., Kaludercic, N., Pelicci, P., Di Lisa, F., and Giorgio, M. (2009). The cardioprotective effects elicited by p66(Shc) ablation demonstrate the crucial role of mitochondrial ROS formation in ischemia/reperfusion injury. Biochim. Biophys. Acta 1787, 774–780.
Cave, A. C., Brewer, A. C., Narayanapanicker, A., Ray, R., Grieve, D. J., Walker, S., and Shah, A. M. (2006). NADPH oxidases in cardiovascular health and disease. Antioxid. Redox Signal. 8, 691–728.
Chamulitrat, W. (2001). EPR studies of nitric oxide interactions of alkoxyl and peroxyl radicals in in vitro and ex vivo model systems. Antioxid. Redox Signal. 3, 177–187.
Chen, L., and Knowlton, A. A. (2010). Mitochondria and heart failure: new insights into an energetic problem. Minerva Cardioangiol. 58, 213–229.
Chen, Q., Camara, A. K., Stowe, D. F., Hoppel, C. L., and Lesnefsky, E. J. (2007). Modulation of electron transport protects cardiac mitochondria and decreases myocardial injury during ischemia and reperfusion. Am. J. Physiol. Cell Physiol. 292, C137–C147. [Review].
Chen, Q., Moghaddas, S., Hoppel, C. L., and Lesnefsky, E. J. (2006). Reversible blockade of electron transport during ischemia protects mitochondria and decreases myocardial injury following reperfusion. J. Pharmacol. Exp. Ther. 319, 1405–1412.
Chen, Q., Moghaddas, S., Hoppel, C. L., and Lesnefsky, E. J. (2008). Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am. J. Physiol. Cell Physiol. 294, C460–C466.
Chen, Q., Vazquez, E. J., Moghaddas, S., Hoppel, C. L., and Lesnefsky, E. J. (2003). Production of reactive oxygen species by mitochondria: central role of complex III. J. Biol. Chem. 278, 36027–36031.
Chen, Q., Yin, G., Stewart, S., Hu, Y., and Lesnefsky, E. J. (2010). Isolating the segment of the mitochondrial electron transport chain responsible for mitochondrial damage during cardiac ischemia. Biochem. Biophys. Res. Commun. 397, 656–660.
Chen, Z., Zhang, H., Lu, W., and Huang, P. (2009). Role of mitochondria-associated hexokinase II in cancer cell death induced by 3-bromopyruvate. Biochim. Biophys. Acta 1787, 553–560.
Cheng, L., Ding, G., Qin, Q., Huang, Y., Lewis, W., He, N., Evans, R. M., Schneider, M. D., Brako, F. A., Xiao, Y., Chen, Y. E., and Yang, Q. (2004). Cardiomyocyte-restricted peroxisome proliferator-activated receptor-delta deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat. Med. 10, 1245–1250.
Cheng, Q., Sedlic, F., Pravdic, D., Bosnjak, Z. J., and Kwok, W. M. (2011). Biphasic effect of nitric oxide on the cardiac voltage-dependent anion channel. FEBS Lett. 585, 328–334.
Cho, S., Szeto, H. H., Kim, E., Kim, H., Tolhurst, A. T., and Pinto, J. T. (2007). A novel cell-permeable antioxidant peptide, SS31, attenuates ischemic brain injury by down-regulating CD36. J. Biol. Chem. 282, 4634–4642.
Cohen, G., and Kesler, N. (1999a). Monoamine oxidase and mitochondrial respiration. J. Neurochem. 73, 2310–2315.
Cohen, G., and Kesler, N. (1999b). Monoamine oxidase inhibits mitochondrial respiration. Ann. N. Y. Acad. Sci. 893, 273–278.
Collins, T. J., Berridge, M. J., Lipp, P., and Bootman, M. D. (2002). Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J. 21, 1616–1627.
Cooper, C. E., and Brown, G. C. (2008). The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: chemical mechanism and physiological significance. J. Bioenerg. Biomembr. 40, 533–539.
Cortassa, S., Aon, M., Iglesisa, A., and Llyod, D. (2002). An Introduction to Metabolic and Cellular Engineering. Singapore: World Scientific, 244.
Cortassa, S., O’Rourke, B., Winslow, R. L., and Aon, M. A. (2009). Control and regulation of mitochondrial energetics in an integrated model of cardiomyocyte function. Biophys. J. 96, 2466–2478.
Costantini, P., Jacotot, E., Decaudin, D., and Kroemer, G. (2000). Mitochondrion as a novel target of anticancer chemotherapy. J. Natl. Cancer Inst. 92, 1042–1053.
Crompton, M., Costi, A., and Hayat, L. (1987). Evidence for the presence of a reversible Ca2+-dependent pore activated by oxidative stress in heart mitochondria. Biochem. J. 245, 915–918.
Csiszar, A., Labinskyy, N., Olson, S., Pinto, J. T., Gupte, S., Wu, J. M., Hu, F., Ballabh, P., Podlutsky, A., Losonczy, G., de Cabo, R., Mathew, R., Wolin, M. S., and Ungvari, Z. (2009a). Resveratrol prevents monocrotaline-induced pulmonary hypertension in rats. Hypertension 54, 668–675.
Csiszar, A., Labinskyy, N., Pinto, J. T., Ballabh, P., Zhang, H., Losonczy, G., Pearson, K., de Cabo, R., Pacher, P., Zhang, C., and Ungvari, Z. (2009b). Resveratrol induces mitochondrial biogenesis in endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 297, H13–H20.
Csordas, G., Thomas, A. P., and Hajnoczky, G. (2001). Calcium signal transmission between ryanodine receptors and mitochondria in cardiac muscle. Trends Cardiovasc. Med. 11, 269–275.
Czaja, M. J., Xu, J., Ju, Y., Alt, E., and Schmiedeberg, P. (1994). Lipopolysaccharide-neutralizing antibody reduces hepatocyte injury from acute hepatotoxin administration. Hepatology 19, 1282–1289.
Dalen, H. (1989). An ultrastructural study of the hypertrophied human papillary muscle cell with special emphasis on specific staining patterns, mitochondrial projections and association between mitochondria and SR. Virchows Arch. A, Pathol. Anat. Histopathol. 414, 187–198.
Das, D. K., Mukherjee, S., and Ray, D. (2010). Resveratrol and red wine, healthy heart and longevity. Heart Fail. Rev. 15, 467–477.
Das, S., Wong, R., Rajapakse, N., Murphy, E., and Steenbergen, C. (2008). Glycogen synthase kinase 3 inhibition slows mitochondrial adenine nucleotide transport and regulates voltage-dependent anion channel phosphorylation. Circ. Res. 103, 983–991.
Davidson, S. M., and Duchen, M. R. (2007). Endothelial mitochondria: contributing to vascular function and disease. Circ. Res. 100, 1128–1141.
Deierborg, T., Wieloch, T., Diano, S., Warden, C. H., Horvath, T. L., and Mattiasson, G. (2008). Overexpression of UCP2 protects thalamic neurons following global ischemia in the mouse. J. Cereb. Blood Flow Metab. 28, 1186–1195.
Di Lisa, F., and Bernardi, P. (2006). Mitochondria and ischemia-reperfusion injury of the heart: fixing a hole. Cardiovasc. Res. 70, 191–199.
Di Lisa, F., Kaludercic, N., Carpi, A., Menabo, R., and Giorgio, M. (2009). Mitochondrial pathways for ROS formation and myocardial injury: the relevance of p66(Shc) and monoamine oxidase. Basic Res. Cardiol. 104, 131–139.
Di Lisa, F., Menabo, R., Canton, M., Barile, M., and Bernardi, P. (2001). Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J. Biol. Chem. 276, 2571–2575.
Dikalova, A. E., Bikineyeva, A. T., Budzyn, K., Nazarewicz, R. R., McCann, L., Lewis, W., Harrison, D. G., and Dikalov, S. I. (2010). Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 107, 106–116.
Dobsak, P., Siegelova, J., Wolf, J. E., Rochette, L., Eicher, J. C., Vasku, J., Kuchtickova, S., and Horky, M. (2002). Prevention of apoptosis by deferoxamine during 4 hours of cold cardioplegia and reperfusion: in vitro study of isolated working rat heart model. Pathophysiology 9, 27.
Dolder, M., Walzel, B., Speer, O., Schlattner, U., and Wallimann, T. (2003). Inhibition of the mitochondrial permeability transition by creatine kinase substrates. Requirement for microcompartmentation. J. Biol. Chem. 278, 17760–17766.
Don, A. S., Kisker, O., Dilda, P., Donoghue, N., Zhao, X., Decollogne, S., Creighton, B., Flynn, E., Folkman, J., and Hogg, P. J. (2003). A peptide trivalent arsenical inhibits tumor angiogenesis by perturbing mitochondrial function in angiogenic endothelial cells. Cancer Cell 3, 497–509.
Dorn, G. W. II, and Scorrano, L. (2010). Two close, too close: sarcoplasmic reticulum-mitochondrial crosstalk and cardiomyocyte fate. Circ. Res. 107, 689–699.
Dorner, A., Schulze, K., Rauch, U., and Schultheiss, H. P. (1997). Adenine nucleotide translocator in dilated cardiomyopathy: pathophysiological alterations in expression and function. Mol. Cell. Biochem. 174, 261–269.
Doughan, A. K., Harrison, D. G., and Dikalov, S. I. (2008). Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ. Res. 102, 488–496.
Du, W., Li, W., Chen, G., Cao, H., Tang, H., Tang, X., Jin, Q., Sun, Z., Zhao, H., Zhou, W., He, S., Lv, Y., Zhao, J., and Zhang, X. (2008). Detection of known base substitution mutations in human mitochondrial DNA of MERRF and MELAS by biochip technology. Biosens. Bioelectron. 24, 2371–2376.
Duchen, M. R. (2004). Mitochondria in health and disease: perspectives on a new mitochondrial biology. Mol. Aspects Med. 25, 365–451.
Duckles, S. P., Krause, D. N., Stirone, C., and Procaccio, V. (2006). Estrogen and mitochondria: a new paradigm for vascular protection? Mol. Interv. 6, 26–35.
Duda, M. K., O’Shea, K. M., Lei, B., Barrows, B. R., Azimzadeh, A. M., McElfresh, T. E., Hoit, B. D., Kop, W. J., and Stanley, W. C. (2008). Low-carbohydrate/high-fat diet attenuates pressure overload-induced ventricular remodeling and dysfunction. J. Card. Fail. 14, 327–335.
Ebrahimian, T., Sairam, M. R., Schiffrin, E. L., and Touyz, R. M. (2008). Cardiac hypertrophy is associated with altered thioredoxin and ASK-1 signaling in a mouse model of menopause. Am. J. Physiol. Heart Circ. Physiol. 295, H1481–H1488.
Ebrahimian, T., and Touyz, R. M. (2008). Thioredoxin in vascular biology: role in hypertension. Antioxid. Redox Signal. 10, 1127–1136.
Echtay, K. S., Esteves, T. C., Pakay, J. L., Jekabsons, M. B., Lambert, A. J., Portero-Otin, M., Pamplona, R., Vidal-Puig, A. J., Wang, S., Roebuck, S. J., and Brand, M. D. (2003). A signalling role for 4-hydroxy-2-nonenal in regulation of mitochondrial uncoupling. EMBO J. 22, 4103–4110.
Eells, J. T., Wong-Riley, M. T., VerHoeve, J., Henry, M., Buchman, E. V., Kane, M. P., Gould, L. J., Das, R., Jett, M., Hodgson, B. D., Margolis, D., and Whelan, H. T. (2004). Mitochondrial signal transduction in accelerated wound and retinal healing by near-infrared light therapy. Mitochondrion 4, 559–567.
England, P. J., and Randle, P. J. (1967). Effectors of rat-heart hexokinases and the control of rates of glucose phosphorylation in the perfused rat heart. Biochem. J. 105, 907–920.
Fell, D. A. (1992). Metabolic control analysis: a survey of its theoretical and experimental development. Biochem. J. 286(Pt 2), 313–330.
Ferdinandy, P., and Schulz, R. (2003). Nitric oxide, superoxide, and peroxynitrite in myocardial ischaemia-reperfusion injury and preconditioning. Br. J. Pharmacol. 138, 532–543.
Fiskum, G., Rosenthal, R. E., Vereczki, V., Martin, E., Hoffman, G. E., Chinopoulos, C., and Kowaltowski, A. (2004). Protection against ischemic brain injury by inhibition of mitochondrial oxidative stress. J. Bioenerg. Biomembr. 36, 347–352.
Frezza, C., Cipolat, S., Martins de Brito, O., Micaroni, M., Beznoussenko, G. V., Rudka, T., Bartoli, D., Polishuck, R. S., Danial, N. N., De Strooper, B., and Scorrano, L. (2006). OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126, 177–189.
Fruehauf, J. P., and Meyskens, F. L. Jr. (2007). Reactive oxygen species: a breath of life or death? Clin. Cancer Res. 13, 789–794. [Review].
Fulda, S., Galluzzi, L., and Kroemer, G. (2010). Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 9, 447–464.
Gadicherla, A., Yang, M., Camara, A., Aldakkak, M., Boelens, A., Wakim, B., and Stowe, D. (2010). Ranolazine preserves the integrity of mitochondrial supercomplexes. Biophys. J. 98, 56a. [Abstract].
Galluzzi, L., Kepp, O., Tajeddine, N., and Kroemer, G. (2008). Disruption of the hexokinase-VDAC complex for tumor therapy. Oncogene 27, 4633–4635.
Ganote, C. E., Worstell, J., and Kaltenbach, J. P. (1976). Oxygen-induced enzyme release after irreversible myocardial injury. Effects of cyanide in perfused rat hearts. Am. J. Pathol. 84, 327–350.
Garland, P. B., Randle, P. J., and Newsholme, E. A. (1963). Citrate as an intermediary in the inhibition of phosphofructokinase in rat heart muscle by fatty acids, ketone bodies, pyruvate, diabetes, and starvation. Nature 200, 169–170.
Garlid, K. D., Dos Santos, P., Xie, Z. J., Costa, A. D., and Paucek, P. (2003). Mitochondrial potassium transport: the role of the mitochondrial ATP-sensitive K+ channel in cardiac function and cardioprotection. Biochim. Biophys. Acta 1606, 1–21.
Garnier, A., Fortin, D., Delomenie, C., Momken, I., Veksler, V., and Ventura-Clapier, R. (2003). Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. J. Physiol. 551(Pt 2), 491–501.
Gladwin, M. T., and Kim-Shapiro, D. B. (2008). The functional nitrite reductase activity of the heme-globins. Blood 112, 2636–2647.
Gogvadze, V., Orrenius, S., and Zhivotovsky, B. (2009). Mitochondria as targets for cancer chemotherapy. Semin. Cancer Biol. 19, 57–66.
Gomez, L., Li, B., Mewton, N., Sanchez, I., Piot, C., Elbaz, M., and Ovize, M. (2009). Inhibition of mitochondrial permeability transition pore opening: translation to patients. Cardiovasc. Res. 83, 226–233.
Gong, G., Liu, J., Liang, P., Guo, T., Hu, Q., Ochiai, K., Hou, M., Ye, Y., Wu, X., Mansoor, A., From, A. H., Ugurbil, K., Bache, R. J., and Zhang, J. (2003). Oxidative capacity in failing hearts. Am. J. Physiol. Heart Circ. Physiol. 285, H541–H548.
Gorbunov, N. V., Osipov, A. N., Day, B. W., Zayas-Rivera, B., Kagan, V. E., and Elsayed, N. M. (1995). Reduction of ferrylmyoglobin and ferrylhemoglobin by nitric oxide: a protective mechanism against ferryl hemoprotein-induced oxidations. Biochemistry 34, 6689–6699.
Gorbunov, N. V., Yalowich, J. C., Gaddam, A., Thampatty, P., Ritov, V. B., Kisin, E. R., Elsayed, N. M., and Kagan, V. E. (1997). Nitric oxide prevents oxidative damage produced by tert-butyl hydroperoxide in erythroleukemia cells via nitrosylation of heme and non-heme iron. Electron paramagnetic resonance evidence. J. Biol. Chem. 272, 12328–12341.
Guo, J., Gertsberg, Z., Ozgen, N., and Steinberg, S. F. (2009). p66Shc links alpha1-adrenergic receptors to a reactive oxygen species-dependent AKT-FOXO3A phosphorylation pathway in cardiomyocytes. Circ. Res. 104, 660–669.
Haines, B. A., Mehta, S. L., Pratt, S. M., Warden, C. H., and Li, P. A. (2010). Deletion of mitochondrial uncoupling protein-2 increases ischemic brain damage after transient focal ischemia by altering gene expression patterns and enhancing inflammatory cytokines. J. Cereb. Blood Flow Metab. 30, 1825–1833.
Hajnoczky, G., and Csordas, G. (2010). Calcium signalling: fishing out molecules of mitochondrial calcium transport. Curr. Biol. 20, R888–R891.
Halestrap, A. P. (2010). A pore way to die: the role of mitochondria in reperfusion injury and cardioprotection. Biochem. Soc. Trans. 38, 841–860.
Halestrap, A. P., Clarke, S. J., and Javadov, S. A. (2004). Mitochondrial permeability transition pore opening during myocardial reperfusion – a target for cardioprotection. Cardiovasc. Res. 61, 372–385.
Halestrap, A. P., Clarke, S. J., and Khaliulin, I. (2007). The role of mitochondria in protection of the heart by preconditioning. Biochim. Biophys. Acta 1767, 1007–1031.
Hallen, S., and Brzezinski, P. (1994). Light-induced structural changes in cytochrome c oxidase: implication for the mechanism of electron and proton gating. Biochim. Biophys. Acta 1184, 207–218.
Hallen, S., Oliveberg, M., and Brzezinski, P. (1993). Light-induced structural changes in cytochrome c oxidase. Measurements of electrogenic events and absorbance changes. FEBS Lett. 318, 134–138.
Hanley, P. J., and Daut, J. (2005). KATP channels and preconditioning: a re-examination of the role of mitochondrial KATP channels and an overview of alternative mechanisms. J. Mol. Cell. Cardiol. 39, 17–50.
Hanley, P. J., Gopalan, K. V., Lareau, R. A., Srivastava, D. K., von Meltzer, M., and Daut, J. (2003). Beta-oxidation of 5-hydroxydecanoate, a putative blocker of mitochondrial ATP-sensitive potassium channels. J. Physiol. 547(Pt 2), 387–393.
Hanley, P. J., Mickel, M., Loffler, M., Brandt, U., and Daut, J. (2002). KATP channel-independent targets of diazoxide and 5-hydroxydecanoate in the heart. J. Physiol. 542(Pt 3), 735–741.
Hanukoglu, I., and Rapoport, R. (1995). Routes and regulation of NADPH production in steroidogenic mitochondria. Endocr. Res. 21, 231–241.
Harrois, A., Huet, O., and Duranteau, J. (2009). Alterations of mitochondrial function in sepsis and critical illness. Curr. Opin. Anaesthesiol. 22, 143–149.
Hausladen, A., and Fridovich, I. (1994). Superoxide and peroxynitrite inactivate aconitases, but nitric oxide does not. J. Biol. Chem. 269, 29405–29408.
Hayashida, K., Sano, M., Ohsawa, I., Shinmura, K., Tamaki, K., Kimura, K., Endo, J., Katayama, T., Kawamura, A., Kohsaka, S., Makino, S., Ohta, S., Ogawa, S., and Fukuda, K. (2008). Inhalation of hydrogen gas reduces infarct size in the rat model of myocardial ischemia-reperfusion injury. Biochem. Biophys. Res. Commun. 373, 30–35.
Heinen, A., Camara, A. K., Aldakkak, M., Rhodes, S. S., Riess, M. L., and Stowe, D. F. (2007). Mitochondrial Ca2+-induced K+ influx increases respiration and enhances ROS production while maintaining membrane potential. Am. J. Physiol. Cell Physiol. 292, C148–C156.
Heinrich, R., and Rapoport, T. A. (1974). A linear steady-state treatment of enzymatic chains. General properties, control and effector strength. Eur. J. Biochem. 42, 89–95.
Hendgen-Cotta, U. B., Merx, M. W., Shiva, S., Schmitz, J., Becher, S., Klare, J. P., Steinhoff, H. J., Goedecke, A., Schrader, J., Gladwin, M. T., Kelm, M., and Rassaf, T. (2008). Nitrite reductase activity of myoglobin regulates respiration and cellular viability in myocardial ischemia-reperfusion injury. Proc. Natl. Acad. Sci. U.S.A. 105, 10256–10261.
Honda, H. M., Korge, P., and Weiss, J. N. (2005). Mitochondria and ischemia/reperfusion injury. Ann. N. Y. Acad. Sci. 1047, 248–258.
Hoye, A. T., Davoren, J. E., Wipf, P., Fink, M. P., and Kagan, V. E. (2008). Targeting mitochondria. Acc. Chem. Res. 41, 87–97.
Ide, T., Tsutsui, H., Hayashidani, S., Kang, D., Suematsu, N., Nakamura, K., Utsumi, H., Hamasaki, N., and Takeshita, A. (2001). Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ. Res. 88, 529–535.
James, A. M., Cocheme, H. M., Smith, R. A., and Murphy, M. P. (2005). Interactions of mitochondria-targeted and untargeted ubiquinones with the mitochondrial respiratory chain and reactive oxygen species. Implications for the use of exogenous ubiquinones as therapies and experimental tools. J. Biol. Chem. 280, 21295–21312.
Javadov, S., Choi, A., Rajapurohitam, V., Zeidan, A., Basnakian, A. G., and Karmazyn, M. (2008). NHE-1 inhibition-induced cardioprotection against ischaemia/reperfusion is associated with attenuation of the mitochondrial permeability transition. Cardiovasc. Res. 77, 416–424.
Jezek, P., Engstova, H., Zackova, M., Vercesi, A. E., Costa, A. D., Arruda, P., and Garlid, K. D. (1998). Fatty acid cycling mechanism and mitochondrial uncoupling proteins. Biochim. Biophys. Acta 1365, 319–327.
Jin, Z. Q., Zhou, H. Z., Cecchini, G., Gray, M. O., and Karliner, J. S. (2005). MnSOD in mouse heart: acute responses to ischemic preconditioning and ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 288, H2986–H2994.
Jones, S. P., Teshima, Y., Akao, M., and Marban, E. (2003). Simvastatin attenuates oxidant-induced mitochondrial dysfunction in cardiac myocytes. Circ. Res. 93, 697–699.
Kaludercic, N., Carpi, A., Menabo, R., Di Lisa, F., and Paolocci, N. (2010a). Monoamine oxidases (MAO) in the pathogenesis of heart failure and ischemia/reperfusion injury. Biochim. Biophys. Acta. PMID 20975995.
Kaludercic, N., Takimoto, E., Nagayama, T., Feng, N., Lai, E. W., Bedja, D., Chen, K., Gabrielson, K. L., Blakely, R. D., Shih, J. C., Pacak, K., Kass, D. A., Di Lisa, F., and Paolocci, N. (2010b). Monoamine oxidase A-mediated enhanced catabolism of norepinephrine contributes to adverse remodeling and pump failure in hearts with pressure overload. Circ. Res. 106, 193–202.
Karu, T. I. (2008). Mitochondrial signaling in mammalian cells activated by red and near-IR radiation. Photochem. Photobiol. 84, 1091–1099.
Karu, T. I., Pyatibrat, L. V., and Afanasyeva, N. I. (2004). A novel mitochondrial signaling pathway activated by visible-to-near infrared radiation. Photochem. Photobiol. 80, 366–372.
Karu, T., Pyatibrat, L., and Kalendo, G. (1995). Irradiation with He-Ne laser increases ATP level in cells cultivated in vitro. J. Photochem. Photobiol. B Biol. 27, 219–223.
Kasamaki, Y., Guo, A. C., and McDonald, T. F. (1997). Protection by hypoxic preconditioning against hypoxia-reoxygenation injury in guinea-pig papillary muscles. Cardiovasc. Res. 34, 313–322.
Katz, L. A., Swain, J. A., Portman, M. A., and Balaban, R. S. (1989). Relation between phosphate metabolites and oxygen consumption of heart in vivo. Am. J. Physiol. 256(Pt 2), H265–H274.
Kerner, J., Zaluzec, E., Gage, D., and Bieber, L. L. (1994). Characterization of the malonyl-CoA-sensitive carnitine palmitoyltransferase (CPTo) of a rat heart mitochondrial particle. Evidence that the catalytic unit is CPTi. J. Biol. Chem. 269, 8209–8219.
Kevin, L. G., Camara, A. K., Riess, M. L., Novalija, E., and Stowe, D. F. (2003). Ischemic preconditioning alters real-time measure of O2 radicals in intact hearts with ischemia and reperfusion. Am. J. Physiol. Heart Circ. Physiol. 284, H566–H574.
Kibbey, R. G., Pongratz, R. L., Romanelli, A. J., Wollheim, C. B., Cline, G. W., and Shulman, G. I. (2007). Mitochondrial GTP regulates glucose-stimulated insulin secretion. Cell Metab. 5, 253–264.
Kim, G. T., Chun, Y. S., Park, J. W., and Kim, M. S. (2003). Role of apoptosis-inducing factor in myocardial cell death by ischemia-reperfusion. Biochem. Biophys. Res. Commun. 309, 619–624.
Kim, Y. M., Bombeck, C. A., and Billiar, T. R. (1999). Nitric oxide as a bifunctional regulator of apoptosis. Circ. Res. 84, 253–256.
Kimura, Y., and Kimura, H. (2004). Hydrogen sulfide protects neurons from oxidative stress. FASEB J. 18, 1165–1167.
Kindler, D. D., Thiffault, C., Solenski, N. J., Dennis, J., Kostecki, V., Jenkins, R., Keeney, P. M., and Bennett, J. P. Jr. (2003). Neurotoxic nitric oxide rapidly depolarizes and permeabilizes mitochondria by dynamically opening the mitochondrial transition pore. Mol. Cell. Neurosci. 23, 559–573.
Kolwicz, S. C. Jr. and Tian, R. (2009). Metabolic therapy at the crossroad: how to optimize myocardial substrate utilization? Trends Cardiovasc. Med. 19, 201–207.
Koopman, W. J., Nijtmans, L. G., Dieteren, C. E., Roestenberg, P., Valsecchi, F., Smeitink, J. A., and Willems, P. H. (2010). Mammalian mitochondrial complex I: biogenesis, regulation, and reactive oxygen species generation. Antioxid. Redox Signal. 12, 1431–1470.
Korzeniewski, B., Noma, A., and Matsuoka, S. (2005). Regulation of oxidative phosphorylation in intact mammalian heart in vivo. Biophys. Chem. 116, 145–157.
Kroemer, G., Galluzzi, L., and Brenner, C. (2007). Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 87, 99–163.
Kwong, J. Q., Henning, M. S., Starkov, A. A., and Manfredi, G. (2007). The mitochondrial respiratory chain is a modulator of apoptosis. J. Cell Biol. 179, 1163–1177.
Lam, A., and Lopaschuk, G. D. (2007). Anti-anginal effects of partial fatty acid oxidation inhibitors. Curr. Opin. Pharmacol. 7, 179–185.
Laskowski, K. R., and Russell, R. R. III. (2008). Uncoupling proteins in heart failure. Curr. Heart Fail Rep. 5, 75–79.
Lavitrano, M., Smolenski, R. T., Musumeci, A., Maccherini, M., Slominska, E., Di Florio, E., Bracco, A., Mancini, A., Stassi, G., Patti, M., Giovannoni, R., Froio, A., Simeone, F., Forni, M., Bacci, M. L., D’Alise, G., Cozzi, E., Otterbein, L. E., Yacoub, M. H., Bach, F. H., and Calise, F. (2004). Carbon monoxide improves cardiac energetics and safeguards the heart during reperfusion after cardiopulmonary bypass in pigs. FASEB J. 18, 1093–1095.
Lefer, D. J. (2007). A new gaseous signaling molecule emerges: cardioprotective role of hydrogen sulfide. Proc. Natl. Acad. Sci. U.S.A. 104, 17907–17908.
Lenaz, G., Baracca, A., Barbero, G., Bergamini, C., Dalmonte, M. E., Del Sole, M., Faccioli, M., Falasca, A., Fato, R., Genova, M. L., Sgarbi, G., and Solaini, G. (2010). Mitochondrial respiratory chain super-complex I-III in physiology and pathology. Biochim. Biophys. Acta 1797, 633–640.
Lenaz, G., and Genova, M. L. (2010). Structure and organization of mitochondrial respiratory complexes: a new understanding of an old subject. Antioxid. Redox Signal. 12, 961–1008.
Lenka, N., Vijayasarathy, C., Mullick, J., and Avadhani, N. G. (1998). Structural organization and transcription regulation of nuclear genes encoding the mammalian cytochrome c oxidase complex. Prog. Nucleic Acid Res. Mol. Biol. 61, 309–344.
Lesnefsky, E. J., Moghaddas, S., Tandler, B., Kerner, J., and Hoppel, C. L. (2001). Mitochondrial dysfunction in cardiac disease: ischemia – reperfusion, aging, and heart failure. J. Mol. Cell. Cardiol. 33, 1065–1089.
Lesnefsky, E. J., Repine, J. E., and Horwitz, L. D. (1990). Deferoxamine pretreatment reduces canine infarct size and oxidative injury. J. Pharmacol. Exp. Ther. 253, 1103–1109.
Li, C. J., Zhang, Q. M., Li, M. Z., Zhang, J. Y., Yu, P., and Yu, D. M. (2009). Attenuation of myocardial apoptosis by alpha-lipoic acid through suppression of mitochondrial oxidative stress to reduce diabetic cardiomyopathy. Chin. Med. J. 122, 2580–2586.
Li, J., Donath, S., Li, Y., Qin, D., Prabhakar, B. S., and Li, P. (2010). miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS Genet. 6, e1000795. doi: 10.1371/journal.pgen.1000795
Li, Q., Sato, E. F., Kira, Y., Nishikawa, M., Utsumi, K., and Inoue, M. (2006). A possible cooperation of SOD1 and cytochrome c in mitochondria-dependent apoptosis. Free Radic. Biol. Med. 40, 173–181.
Liachenko, S., Tang, P., and Xu, Y. (2003). Deferoxamine improves early postresuscitation reperfusion after prolonged cardiac arrest in rats. J. Cereb. Blood Flow Metab. 23, 574–581.
Liang, H. L., Sedlic, F., Bosnjak, Z., and Nilakantan, V. (2010). SOD1 and MitoTEMPO partially prevent MPTP, necrosis and mitochondrial apoptosis following ATP depletion-recovery. Free Radic. Biol. Med. 49, 1550–1560.
Lin, M. T., and Beal, M. F. (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795.
Loeb, L. A., Wallace, D. C., and Martin, G. M. (2005). The mitochondrial theory of aging and its relationship to reactive oxygen species damage and somatic mtDNA mutations. Proc. Natl. Acad. Sci. U.S.A. 102, 18769–18770.
Lohr, N. L., Keszler, A., Pratt, P., Bienengraber, M., Warltier, D. C., and Hogg, N. (2009). Enhancement of nitric oxide release from nitrosyl hemoglobin and nitrosyl myoglobin by red/near infrared radiation: potential role in cardioprotection. J. Mol. Cell. Cardiol. 47, 256–263.
Lu, Z., Xu, X., Hu, X., Fassett, J., Zhu, G., Tao, Y., Li, J., Huang, Y., Zhang, P., Zhao, B., and Chen, Y. (2010). PGC-1alpha regulates expression of myocardial mitochondrial antioxidants and myocardial oxidative stress after chronic systolic overload. Antioxid. Redox Signal. 13, 1011–1022.
Mao, W., Iwai, C., Liu, J., Sheu, S. S., Fu, M., and Liang, C. S. (2008). Darbepoetin alfa exerts a cardioprotective effect in autoimmune cardiomyopathy via reduction of ER stress and activation of the PI3K/Akt and STAT3 pathways. J. Mol. Cell. Cardiol. 45, 250–260.
Marin-Garcia, J., and Goldenthal, M. J. (2008). Mitochondrial centrality in heart failure. Heart Fail. Rev. 13, 137–150.
Maroz, A., Anderson, R. F., Smith, R. A., and Murphy, M. P. (2009). Reactivity of ubiquinone and ubiquinol with superoxide and the hydroperoxyl radical: implications for in vivo antioxidant activity. Free Radic. Biol. Med. 46, 105–109.
Martinez-Ruiz, G., Maldonado, V., Ceballos-Cancino, G., Grajeda, J. P., and Melendez-Zajgla, J. (2008). Role of Smac/DIABLO in cancer progression. J. Exp. Clin. Cancer Res. 27, 48.
Marzo, I., Brenner, C., Zamzami, N., Jurgensmeier, J. M., Susin, S. A., Vieira, H. L., Prevost, M. C., Xie, Z., Matsuyama, S., Reed, J. C., and Kroemer, G. (1998). Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science 281, 2027–2031.
Mathupala, S. P., Ko, Y. H., and Pedersen, P. L. (2009). Hexokinase-2 bound to mitochondria: cancer’s stygian link to the “Warburg Effect” and a pivotal target for effective therapy. Semin. Cancer Biol. 19, 17–24.
Matsukawa, N., Yasuhara, T., Hara, K., Xu, L., Maki, M., Yu, G., Kaneko, Y., Ojika, K., Hess, D. C., and Borlongan, C. V. (2009). Therapeutic targets and limits of minocycline neuroprotection in experimental ischemic stroke. BMC Neurosci. 10, 126. doi: 10.1186/1471-2202-10-126
McBride, H. M., Neuspiel, M., and Wasiak, S. (2006). Mitochondria: more than just a powerhouse. Curr. Biol. 16, R551–R560.
McCormack, J. G., Halestrap, A. P., and Denton, R. M. (1990). Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol. Rev. 70, 391–425.
Meyer, L. E., Machado, L. B., Santiago, A. P., da-Silva, W. S., De Felice, F. G., Holub, O., Oliveira, M. F., and Galina, A. (2006). Mitochondrial creatine kinase activity prevents reactive oxygen species generation: antioxidant role of mitochondrial kinase-dependent ADP re-cycling activity. J. Biol. Chem. 281, 37361–37371.
Milagros Rocha, M., and Victor, V. M. (2007). Targeting antioxidants to mitochondria and cardiovascular diseases: the effects of mitoquinone. Med. Sci. Monit. 13, RA132–RA145.
Morin, C., Zini, R., Albengres, E., Bertelli, A. A., Bertelli, A., and Tillement, J. P. (2003a). Evidence for resveratrol-induced preservation of brain mitochondria functions after hypoxia-reoxygenation. Drugs Exp. Clin. Res. 29, 227–233.
Morin, C., Zini, R., and Tillement, J. P. (2003b). Anoxia-reoxygenation-induced cytochrome c and cardiolipin release from rat brain mitochondria. Biochem. Biophys. Res. Commun. 307, 477–482.
Morin, D., Assaly, R., Paradis, S., and Berdeaux, A. (2009). Inhibition of mitochondrial membrane permeability as a putative pharmacological target for cardioprotection. Curr. Med. Chem. 16, 4382–4398.
Muoio, D. M., and Newgard, C. B. (2008). Fatty acid oxidation and insulin action: when less is more. Diabetes 57, 1455–1456.
Murata, M., Akao, M., O’Rourke, B., and Marban, E. (2001). Mitochondrial ATP-sensitive potassium channels attenuate matrix Ca2+ overload during simulated ischemia and reperfusion: possible mechanism of cardioprotection. Circ. Res. 89, 891–898.
Muravchick, S., and Levy, R. J. (2006). Clinical implications of mitochondrial dysfunction. Anesthesiology 105, 819–837.
Murray, A. J., Edwards, L. M., and Clarke, K. (2007). Mitochondria and heart failure. Curr. Opin. Clin. Nutr. Metab. Care 10, 704–711.
Nadtochiy, S. M., Burwell, L. S., and Brookes, P. S. (2007). Cardioprotection and mitochondrial S-nitrosation: effects of S-nitroso-2-mercaptopropionyl glycine (SNO-MPG) in cardiac ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 42, 812–825.
Navarro, A., and Boveris, A. (2009). Brain mitochondrial dysfunction and oxidative damage in Parkinson’s disease. J. Bioenerg. Biomembr. 41, 517–521.
Nediani, C., Raimondi, L., Borchi, E., and Cerbai, E. (2011). Nitric oxide/reactive oxygen species generation and nitroso/redox imbalance in heart failure: from molecular mechanisms to therapeutic implications. Antioxid. Redox Signal. 14, 289–331.
Neely, J. R., Denton, R. M., England, P. J., and Randle, P. J. (1972). The effects of increased heart work on the tricarboxylate cycle and its interactions with glycolysis in the perfused rat heart. Biochem. J. 128, 147–159.
Neely, J. R., and Morgan, H. E. (1974). Relationship between carbohydrate and lipid metabolism and the energy balance of heart muscle. Annu. Rev. Physiol. 36, 413–459.
Newsholm, E., and Start, C. (1973). Regulation in Metabolism, Chapter 5. London: John Wiley and Sons.
Nishihara, M., Miura, T., Miki, T., Tanno, M., Yano, T., Naitoh, K., Ohori, K., Hotta, H., Terashima, Y., and Shimamoto, K. (2007). Modulation of the mitochondrial permeability transition pore complex in GSK-3beta-mediated myocardial protection. J. Mol. Cell. Cardiol. 43, 564–570.
Norberg, E., Orrenius, S., and Zhivotovsky, B. (2010). Mitochondrial regulation of cell death: processing of apoptosis-inducing factor (AIF). Biochem. Biophys. Res. Commun. 396, 95–100.
Nossuli, T. O., Hayward, R., Jensen, D., Scalia, R., and Lefer, A. M. (1998). Mechanisms of cardioprotection by peroxynitrite in myocardial ischemia and reperfusion injury. Am. J. Physiol. 275(Pt 2), H509–H519.
Ochiai, K., Zhang, J., Gong, G., Zhang, Y., Liu, J., Ye, Y., Wu, X., Liu, H., Murakami, Y., Bache, R. J., Ugurbil, K., and From, A. H. (2001). Effects of augmented delivery of pyruvate on myocardial high-energy phosphate metabolism at high workstate. Am. J. Physiol. Heart Circ. Physiol. 281, H1823–H1832.
O’Connor, P. M., and Gutterman, D. D. (2010). Resurrecting hope for antioxidant treatment of cardiovascular disease: focus on mitochondria. Circ. Res. 107, 9–11.
Okada, S. F., O’Neal, W. K., Huang, P., Nicholas, R. A., Ostrowski, L. E., Craigen, W. J., Lazarowski, E. R., and Boucher, R. C. (2004). Voltage-dependent anion channel-1 (VDAC-1) contributes to ATP release and cell volume regulation in murine cells. J. Gen. Physiol. 124, 513–526.
Ong, S. B., and Hausenloy, D. J. (2010). Mitochondrial morphology and cardiovascular disease. Cardiovasc. Res. 88, 16–29.
Opie, L. (1998). Heart Physiology: From Cell to Circulation. Philadelphia: Lippincott-Raven Publishers, 44–63.
Opie, L. H. (1965). Coronary flow rate and perfusion pressure as determinants of mechanical function and oxidative metabolism of isolated perfused rat heart. J. Physiol. 180, 529–541.
Oram, J. F., Bennetch, S. L., and Neely, J. R. (1973). Regulation of fatty acid utilization in isolated perfused rat hearts. J. Biol. Chem. 248, 5299–5309.
Oron, U., Yaakobi, T., Oron, A., Mordechovitz, D., Shofti, R., Hayam, G., Dror, U., Gepstein, L., Wolf, T., Haudenschild, C., and Haim, S. B. (2001). Low-energy laser irradiation reduces formation of scar tissue after myocardial infarction in rats and dogs. Circulation 103, 296–301.
Osipov, A. N., Stepanov, G. O., Vladimirov, Y. A., Kozlov, A. V., and Kagan, V. E. (2006). Regulation of cytochrome C peroxidase activity by nitric oxide and laser irradiation. Biochemistry (Mosc). 71, 1128–1132.
Outten, C. E., and Culotta, V. C. (2003). A novel NADH kinase is the mitochondrial source of NADPH in Saccharomyces cerevisiae. EMBO J. 22, 2015–2024.
Owen, M. R., Doran, E., and Halestrap, A. P. (2000). Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 348(Pt 3), 607–614.
Papa, S., De Rasmo, D., Scacco, S., Signorile, A., Technikova-Dobrova, Z., Palmisano, G., Sardanelli, A. M., Papa, F., Panelli, D., Scaringi, R., and Santeramo, A. (2008). Mammalian complex I: a regulable and vulnerable pacemaker in mitochondrial respiratory function. Biochim. Biophys. Acta 1777, 719–728.
Paradies, G., Petrosillo, G., Paradies, V., and Ruggiero, F. M. (2009). Role of cardiolipin peroxidation and Ca2+ in mitochondrial dysfunction and disease. Cell Calcium 45, 643–650.
Paradies, G., Petrosillo, G., Pistolese, M., Di Venosa, N., Federici, A., and Ruggiero, F. M. (2004). Decrease in mitochondrial complex I activity in ischemic/reperfused rat heart: involvement of reactive oxygen species and cardiolipin. Circ. Res. 94, 53–59.
Paradies, G., Petrosillo, G., and Ruggiero, F. M. (1997). Cardiolipin-dependent decrease of cytochrome c oxidase activity in heart mitochondria from hypothyroid rats. Biochim. Biophys. Acta 1319, 5–8.
Pastorino, J. G., and Hoek, J. B. (2008). Regulation of hexokinase binding to VDAC. J. Bioenerg. Biomembr. 40, 171–182.
Pastorino, J. G., Hoek, J. B., and Shulga, N. (2005). Activation of glycogen synthase kinase 3beta disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 65, 10545–10554.
Pedersen, P. L., Mathupala, S., Rempel, A., Geschwind, J. F., and Ko, Y. H. (2002). Mitochondrial bound type II hexokinase: a key player in the growth and survival of many cancers and an ideal prospect for therapeutic intervention. Biochim. Biophys. Acta 1555, 14–20.
Pereira, C. V., Moreira, A. C., Pereira, S. P., Machado, N. G., Carvalho, F. S., Sardao, V. A., and Oliveira, P. J. (2009). Investigating drug-induced mitochondrial toxicity: a biosensor to increase drug safety? Curr. Drug Saf. 4, 34–54.
Petrosillo, G., Colantuono, G., Moro, N., Ruggiero, F. M., Tiravanti, E., Di Venosa, N., Fiore, T., and Paradies, G. (2009). Melatonin protects against heart ischemia-reperfusion injury by inhibiting mitochondrial permeability transition pore opening. Am. J. Physiol. Heart Circ. Physiol. 297, H1487–H1493.
Phillips, D., Aponte, A. M., French, S. A., Chess, D. J., and Balaban, R. S. (2009). Succinyl-CoA synthetase is a phosphate target for the activation of mitochondrial metabolism. Biochemistry 48, 7140–7149.
Piantadosi, C. A., Carraway, M. S., Babiker, A., and Suliman, H. B. (2008). Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ. Res. 103, 1232–1240.
Piao, L., Fang, Y. H., Cadete, V. J., Wietholt, C., Urboniene, D., Toth, P. T., Marsboom, G., Zhang, H. J., Haber, I., Rehman, J., Lopaschuk, G. D., and Archer, S. L. (2010). The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: resuscitating the hibernating right ventricle. J. Mol. Med. 88, 47–60.
Pinton, P., Rimessi, A., Marchi, S., Orsini, F., Migliaccio, E., Giorgio, M., Contursi, C., Minucci, S., Mantovani, F., Wieckowski, M. R., Del Sal, G., Pelicci, P. G., and Rizzuto, R. (2007). Protein kinase C beta and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science 315, 659–663.
Pipinos, I. I., Judge, A. R., Zhu, Z., Selsby, J. T., Swanson, S. A., Johanning, J. M., Baxter, B. T., Lynch, T. G., and Dodd, S. L. (2006). Mitochondrial defects and oxidative damage in patients with peripheral arterial disease. Free Radic. Biol. Med. 41, 262–269.
Pope, S., Land, J. M., and Heales, S. J. (2008). Oxidative stress and mitochondrial dysfunction in neurodegeneration; cardiolipin a critical target? Biochim. Biophys. Acta 1777, 794–799.
Pravdic, D., Mio, Y., Sedlic, F., Pratt, P. F., Warltier, D. C., Bosnjak, Z. J., and Bienengraeber, M. (2010). Isoflurane protects cardiomyocytes and mitochondria by immediate and cytosol-independent action at reperfusion. Br. J. Pharmacol. 160, 220–232.
Radi, R., Peluffo, G., Alvarez, M. N., Naviliat, M., and Cayota, A. (2001). Unraveling peroxynitrite formation in biological systems. Free Radic. Biol. Med. 30, 463–488.
Randle, P. J. (1998). Regulatory interactions between lipids and carbohydrates: the glucose fatty acid cycle after 35 years. Diabetes Metab. Rev. 14, 263–283.
Randle, P. J., England, P. J., and Denton, R. M. (1970). Control of the tricarboxylate cycle and its interactions with glycolysis during acetate utilization in rat heart. Biochem. J. 117, 677–695.
Randle, P. J., Garland, P. B., Hales, C. N., and Newsholme, E. A. (1963). The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1, 785–789.
Raval, A. P., Lin, H. W., Dave, K. R., Defazio, R. A., Della Morte, D., Kim, E. J., and Perez-Pinzon, M. A. (2008). Resveratrol and ischemic preconditioning in the brain. Curr. Med. Chem. 15, 1545–1551.
Raymond, G., and Potreau, D. (1977). Barium ions and excitation-contraction coupling of frog single muscle fibres under controlled current and voltage. J. Physiol. (Paris) 73, 617–631.
Riess, M. L., Camara, A. K., Kevin, L. G., An, J., and Stowe, D. F. (2004). Reduced reactive O2 species formation and preserved mitochondrial NADH and [Ca2+] levels during short-term 17°C ischemia in intact hearts. Cardiovasc. Res. 61, 580–590.
Rizzuto, R., Marchi, S., Bonora, M., Aguiari, P., Bononi, A., De Stefani, D., Giorgi, C., Leo, S., Rimessi, A., Siviero, R., Zecchini, E., and Pinton, P. (2009). Ca2+ transfer from the ER to mitochondria: when, how and why. Biochim. Biophys. Acta 1787, 1342–1351.
Rogers, R. J., Monnier, J. M., and Nick, H. S. (2001). Tumor necrosis factor-alpha selectively induces MnSOD expression via mitochondria-to-nucleus signaling, whereas interleukin-1beta utilizes an alternative pathway. J. Biol. Chem. 276, 20419–20427.
Romagnoli, A., Aguiari, P., De Stefani, D., Leo, S., Marchi, S., Rimessi, A., Zecchini, E., Pinton, P., and Rizzuto, R. (2007). Endoplasmic reticulum/mitochondria calcium cross-talk. Novartis Found. Symp. 287, 122–131; discussion 131–139.
Ronson, R. S., Nakamura, M., and Vinten-Johansen, J. (1999). The cardiovascular effects and implications of peroxynitrite. Cardiovasc. Res. 44, 47–59.
Rosca, M. G., Vazquez, E. J., Kerner, J., Parland, W., Chandler, M. P., Stanley, W., Sabbah, H. N., and Hoppel, C. L. (2008). Cardiac mitochondria in heart failure: decrease in respirasomes and oxidative phosphorylation. Cardiovasc. Res. 80, 30–39.
Ruiz-Meana, M., Fernandez-Sanz, C., and Garcia-Dorado, D. (2010). The SR-mitochondria interaction: a new player in cardiac pathophysiology. Cardiovasc. Res. 88, 30–39.
Ruiz-Meana, M., Garcia-Dorado, D., Pina, P., Inserte, J., Agullo, L., and Soler-Soler, J. (2003). Cariporide preserves mitochondrial proton gradient and delays ATP depletion in cardiomyocytes during ischemic conditions. Am. J. Physiol. Heart Circ. Physiol. 285, H999–H1006.
Rybnikova, E., Damdimopoulos, A. E., Gustafsson, J. A., Spyrou, G., and Pelto-Huikko, M. (2000). Expression of novel antioxidant thioredoxin-2 in the rat brain. Eur. J. Neurosci. 12, 1669–1678.
Saba, H., Batinic-Haberle, I., Munusamy, S., Mitchell, T., Lichti, C., Megyesi, J., and MacMillan-Crow, L. A. (2007). Manganese porphyrin reduces renal injury and mitochondrial damage during ischemia/reperfusion. Free Radic. Biol. Med. 42, 1571–1578.
Sack, M. N. (2006a). Exploring mitochondria in the intact ischemic heart: advancing technologies to image intracellular function. Circulation 114, 1452–1454.
Sack, M. N. (2006b). Mitochondrial depolarization and the role of uncoupling proteins in ischemia tolerance. Cardiovasc. Res. 72, 210–219.
Saks, V., Dzeja, P., Schlattner, U., Vendelin, M., Terzic, A., and Wallimann, T. (2006a). Cardiac system bioenergetics: metabolic basis of the Frank-Starling law. J. Physiol. 571(Pt 2), 253–273.
Saks, V., Favier, R., Guzun, R., Schlattner, U., and Wallimann, T. (2006b). Molecular system bioenergetics: regulation of substrate supply in response to heart energy demands. J. Physiol. 577(Pt 3), 769–777.
Saks, V. A., Kongas, O., Vendelin, M., and Kay, L. (2000). Role of the creatine/phosphocreatine system in the regulation of mitochondrial respiration. Acta Physiol. Scand. 168, 635–641.
Sareen, D., Darjatmoko, S. R., Albert, D. M., and Polans, A. S. (2007). Mitochondria, calcium, and calpain are key mediators of resveratrol-induced apoptosis in breast cancer. Mol. Pharmacol. 72, 1466–1475.
Sareen, D., van Ginkel, P. R., Takach, J. C., Mohiuddin, A., Darjatmoko, S. R., Albert, D. M., and Polans, A. S. (2006). Mitochondria as the primary target of resveratrol-induced apoptosis in human retinoblastoma cells. Invest. Ophthalmol. Vis. Sci. 47, 3708–3716.
Schwimmer, C., Lefebvre-Legendre, L., Rak, M., Devin, A., Slonimski, P. P., di Rago, J. P., and Rigoulet, M. (2005). Increasing mitochondrial substrate-level phosphorylation can rescue respiratory growth of an ATP synthase-deficient yeast. J. Biol. Chem. 280, 30751–30759.
Sedlic, F., Pravdic, D., Hirata, N., Mio, Y., Sepac, A., Camara, A. K., Wakatsuki, T., Bosnjak, Z. J., and Bienengraeber, M. (2010a). Monitoring mitochondrial electron fluxes using NAD(P)H-flavoprotein fluorometry reveals complex action of isoflurane on cardiomyocytes. Biochim. Biophys. Acta 1797, 1749–1758.
Sedlic, F., Sepac, A., Pravdic, D., Camara, A. K., Bienengraeber, M., Brzezinska, A. K., Wakatsuki, T., and Bosnjak, Z. J. (2010b). Mitochondrial depolarization underlies delay in permeability transition by preconditioning with isoflurane: roles of ROS and Ca2+. Am. J. Physiol., Cell Physiol. 299, C506–C515.
Sen, C. K. (1998). Redox signaling and the emerging therapeutic potential of thiol antioxidants. Biochem. Pharmacol. 55, 1747–1758.
Serviddio, G., Di Venosa, N., Federici, A., D’Agostino, D., Rollo, T., Prigigallo, F., Altomare, E., Fiore, T., and Vendemiale, G. (2005). Brief hypoxia before normoxic reperfusion (postconditioning) protects the heart against ischemia-reperfusion injury by preventing mitochondria peroxyde production and glutathione depletion. FASEB J. 19, 354–361.
Sharma, V. K., Ramesh, V., Franzini-Armstrong, C., and Sheu, S. S. (2000). Transport of Ca2+ from sarcoplasmic reticulum to mitochondria in rat ventricular myocytes. J. Bioenerg. Biomembr. 32, 97–104.
Sharov, V. G., Todor, A. V., Silverman, N., Goldstein, S., and Sabbah, H. N. (2000). Abnormal mitochondrial respiration in failed human myocardium. J. Mol. Cell. Cardiol. 32, 2361–2367.
Shen, T., Zheng, M., Cao, C., Chen, C., Tang, J., Zhang, W., Cheng, H., Chen, K. H., and Xiao, R. P. (2007). Mitofusin-2 is a major determinant of oxidative stress-mediated heart muscle cell apoptosis. J. Biol. Chem. 282, 23354–23361.
Shidoji, Y., Hayashi, K., Komura, S., Ohishi, N., and Yagi, K. (1999). Loss of molecular interaction between cytochrome c and cardiolipin due to lipid peroxidation. Biochem. Biophys. Res. Commun. 264, 343–347.
Shoshan-Barmatz, V., Keinan, N., and Zaid, H. (2008). Uncovering the role of VDAC in the regulation of cell life and death. J. Bioenerg. Biomembr. 40, 183–191.
Smaili, S. S., and Russell, J. T. (1999). Permeability transition pore regulates both mitochondrial membrane potential and agonist-evoked Ca2+ signals in oligodendrocyte progenitors. Cell Calcium 26, 121–130.
Smeele, K. M., Eerbeek, O., Koeman, A., Bezemer, R., Ince, C., Heikkinen, S., Laakso, M., de Haan, A., Schaart, G., Drost, M. R., Hollmann, M. W., and Zuurbier, C. J. (2010). Partial hexokinase II knockout results in acute ischemia-reperfusion damage in skeletal muscle of male, but not female, mice. Pflugers Arch. 459, 705–712.
Smith, R. A., Porteous, C. M., Coulter, C. V., and Murphy, M. P. (1999). Selective targeting of an antioxidant to mitochondria. Eur. J. Biochem. 263, 709–716.
Sokol, R. J., and Treem, W. R. (1999). Mitochondria and childhood liver diseases. J. Pediatr. Gastroenterol. Nutr. 28, 4–16.
Solaini, G., and Harris, D. A. (2005). Biochemical dysfunction in heart mitochondria exposed to ischaemia and reperfusion. Biochem. J. 390(Pt 2), 377–394.
Spasojevic, I., Chen, Y., Noel, T. J., Yu, Y., Cole, M. P., Zhang, L., Zhao, Y., St Clair, D. K., and Batinic-Haberle, I. (2007). Mn porphyrin-based superoxide dismutase (SOD) mimic, MnIIITE-2-PyP5+, targets mouse heart mitochondria. Free Radic. Biol. Med. 42, 1193–1200.
Stanley, W. C., Lopaschuk, G. D., and McCormack, J. G. (1997). Regulation of energy substrate metabolism in the diabetic heart. Cardiovasc. Res. 34, 25–33.
Stanley, W. C., Recchia, F. A., and Lopaschuk, G. D. (2005). Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 85, 1093–1129.
Starke, D. W., Chen, Y., Bapna, C. P., Lesnefsky, E. J., and Mieyal, J. J. (1997). Sensitivity of protein sulfhydryl repair enzymes to oxidative stress. Free Radic. Biol. Med. 23, 373–384.
Stowe, D. F., Aldakkak, M., Camara, A. K. S., Riess, M. L., Heinen, A., Varadarajan, S. G., and Jiang, M. T. (2006). Cardiac mitochondrial preconditioning by big Ca2+-sensitive K+ channel opening requires superoxide radical generation. Am. J. Physiol. Heart Circ. Physiol. 290, H434–H440.
Stowe, D. F., and Camara, A. K. S. (2009). Mitochondrial reactive oxygen species production in excitable cells: modulators of mitochondrial and cell function Antioxid. Redox Signal. 11, 1373–1396. [Comprehensive review].
Stuart, J. A., Cadenas, S., Jekabsons, M. B., Roussel, D., and Brand, M. D. (2001). Mitochondrial proton leak and the uncoupling protein 1 homologues. Biochim. Biophys. Acta 1504, 144–158.
Sun, H., Nikolovska-Coleska, Z., Yang, C. Y., Qian, D., Lu, J., Qiu, S., Bai, L., Peng, Y., Cai, Q., and Wang, S. (2008). Design of small-molecule peptidic and nonpeptidic Smac mimetics. Acc. Chem. Res. 41, 1264–1277.
Szabadkai, G., and Duchen, M. R. (2008). Mitochondria: the hub of cellular Ca2+ signaling. Physiology (Bethesda) 23, 84–94.
Szabo, C., Ischiropoulos, H., and Radi, R. (2007). Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat. Rev. Drug Discov. 6, 662–680.
Szado, T., Vanderheyden, V., Parys, J. B., De Smedt, H., Rietdorf, K., Kotelevets, L., Chastre, E., Khan, F., Landegren, U., Soderberg, O., Bootman, M. D., and Roderick, H. L. (2008). Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc. Natl. Acad. Sci. U.S.A. 105, 2427–2432.
Szende, B., Tyihak, E., and Kiraly-Veghely, Z. (2000). Dose-dependent effect of resveratrol on proliferation and apoptosis in endothelial and tumor cell cultures. Exp. Mol. Med. 32, 88–92.
Szeto, H. H. (2008a). Development of mitochondria-targeted aromatic-cationic peptides for neurodegenerative diseases. Ann. N. Y. Acad. Sci. 1147, 112–121.
Szeto, H. H. (2008b). Mitochondria-targeted cytoprotective peptides for ischemia-reperfusion injury. Antioxid. Redox Signal. 10, 601–619.
Szewczyk, A., Kajma, A., Malinska, D., Wrzosek, A., Bednarczyk, P., Zablocka, B., and Dolowy, K. (2010). Pharmacology of mitochondrial potassium channels: dark side of the field. FEBS Lett. 584, 2063–2069.
Taegtmeyer, H., Wilson, C. R., Razeghi, P., and Sharma, S. (2005). Metabolic energetics and genetics in the heart. Ann. N. Y. Acad. Sci. 1047, 208–218.
Taguchi, N., Ishihara, N., Jofuku, A., Oka, T., and Mihara, K. (2007). Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 282, 11521–11529.
Teshima, Y., Akao, M., Baumgartner, W. A., and Marban, E. (2003a). Nicorandil prevents oxidative stress-induced apoptosis in neurons by activating mitochondrial ATP-sensitive potassium channels. Brain Res. 990, 45–50.
Teshima, Y., Akao, M., Jones, S. P., and Marban, E. (2003b). Uncoupling protein-2 overexpression inhibits mitochondrial death pathway in cardiomyocytes. Circ. Res. 93, 192–200.
Theruvath, T. P., Zhong, Z., Pediaditakis, P., Ramshesh, V. K., Currin, R. T., Tikunov, A., Holmuhamedov, E., and Lemasters, J. J. (2008). Minocycline and N-methyl-4-isoleucine cyclosporin (NIM811) mitigate storage/reperfusion injury after rat liver transplantation through suppression of the mitochondrial permeability transition. Hepatology 47, 236–246.
Thomas, D. A., Stauffer, C., Zhao, K., Yang, H., Sharma, V. K., Szeto, H. H., and Suthanthiran, M. (2007). Mitochondrial targeting with antioxidant peptide SS-31 prevents mitochondrial depolarization, reduces islet cell apoptosis, increases islet cell yield, and improves posttransplantation function. J. Am. Soc. Nephrol. 18, 213–222.
Treuting, P. M., Linford, N. J., Knoblaugh, S. E., Emond, M. J., Morton, J. F., Martin, G. M., Rabinovitch, P. S., and Ladiges, W. C. (2008). Reduction of age-associated pathology in old mice by overexpression of catalase in mitochondria. J. Gerontol. A Biol. Sci. Med. Sci. 63, 813–822.
Tsutsui, H., Ide, T., and Kinugawa, S. (2006). Mitochondrial oxidative stress, DNA damage, and heart failure. Antioxid. Redox Signal. 8, 1737–1744.
Tuby, H., Maltz, L., and Oron, U. (2006). Modulations of VEGF and iNOS in the rat heart by low level laser therapy are associated with cardioprotection and enhanced angiogenesis. Lasers Surg. Med. 38, 682–688.
Turrens, J. F., Alexandre, A., and Lehninger, A. L. (1985). Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch. Biochem. Biophys. 237, 408–414.
Turrens, J. F., and Boveris, A. (1980). Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem. J. 191, 421–427.
Tuunanen, H., Engblom, E., Naum, A., Nagren, K., Hesse, B., Airaksinen, K. E., Nuutila, P., Iozzo, P., Ukkonen, H., Opie, L. H., and Knuuti, J. (2006a). Free fatty acid depletion acutely decreases cardiac work and efficiency in cardiomyopathic heart failure. Circulation 114, 2130–2137.
Tuunanen, H., Engblom, E., Naum, A., Scheinin, M., Nagren, K., Airaksinen, J., Nuutila, P., Iozzo, P., Ukkonen, H., and Knuuti, J. (2006b). Decreased myocardial free fatty acid uptake in patients with idiopathic dilated cardiomyopathy: evidence of relationship with insulin resistance and left ventricular dysfunction. J. Card. Fail. 12, 644–652.
Ueda, S., Nakamura, H., Masutani, H., Sasada, T., Yonehara, S., Takabayashi, A., Yamaoka, Y., and Yodoi, J. (1998). Redox regulation of caspase-3(-like) protease activity: regulatory roles of thioredoxin and cytochrome c. J. Immunol. 161, 6689–6695.
Ujwal, R., Cascio, D., Colletier, J. P., Faham, S., Zhang, J., Toro, L., Ping, P., and Abramson, J. (2008). The crystal structure of mouse VDAC1 at 2.3 A resolution reveals mechanistic insights into metabolite gating. Proc. Natl. Acad. Sci. U.S.A. 105, 17742–17747.
Vanden Hoek, T. L., Li, C., Shao, Z., Schumacker, P. T., and Becker, L. B. (1997). Significant levels of oxidants are generated by isolated cardiomyocytes during ischemia prior to reperfusion. J. Mol. Cell. Cardiol. 29, 2571–2583.
Vendelin, M., Beraud, N., Guerrero, K., Andrienko, T., Kuznetsov, A. V., Olivares, J., Kay, L., and Saks, V. A. (2005). Mitochondrial regular arrangement in muscle cells: a “crystal-like” pattern. Am. J. Physiol. Cell Physiol. 288, C757–C767.
Vijayasarathy, C., Biunno, I., Lenka, N., Yang, M., Basu, A., Hall, I. P., and Avadhani, N. G. (1998). Variations in the subunit content and catalytic activity of the cytochrome c oxidase complex from different tissues and different cardiac compartments. Biochim. Biophys. Acta 1371, 71–82.
Vlasova, I. I., Tyurin, V. A., Kapralov, A. A., Kurnikov, I. V., Osipov, A. N., Potapovich, M. V., Stoyanovsky, D. A., and Kagan, V. E. (2006). Nitric oxide inhibits peroxidase activity of cytochrome c.cardiolipin complex and blocks cardiolipin oxidation. J. Biol. Chem. 281, 14554–14562.
Waldmeier, P. C., Zimmermann, K., Qian, T., Tintelnot-Blomley, M., and Lemasters, J. J. (2003). Cyclophilin D as a drug target. Curr. Med. Chem. 10, 1485–1506.
Wallimann, T., Wyss, M., Brdiczka, D., Nicolay, K., and Eppenberger, H. M. (1992). Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: the ‘phosphocreatine circuit’ for cellular energy homeostasis. Biochem. J. 281(Pt 1), 21–40.
Walsh, C. B. S., Voronica, S., Chvanov, M., Petersen, O. H., and Tepikin, T. (2009). Modulation of calcium signalling by mitochondria. Biochim. Biophys. Acta, 1787, 1374–1382.
Wan, B., LaNoue, K. F., Cheung, J. Y., and Scaduto, R. C. Jr. (1989). Regulation of citric acid cycle by calcium. J. Biol. Chem. 264, 13430–13439.
Wang, H., Lim, P. J., Karbowski, M., and Monteiro, M. J. (2009). Effects of overexpression of huntingtin proteins on mitochondrial integrity. Hum. Mol. Genet. 18, 737–752.
Wang, T., Liu, B., Qin, L., Wilson, B., and Hong, J. S. (2004). Protective effect of the SOD/catalase mimetic MnTMPyP on inflammation-mediated dopaminergic neurodegeneration in mesencephalic neuronal-glial cultures. J. Neuroimmunol. 147, 68–72.
Waypa, G. B., Marks, J. D., Mack, M. M., Boriboun, C., Mungai, P. T., and Schumacker, P. T. (2002). Mitochondrial reactive oxygen species trigger calcium increases during hypoxia in pulmonary arterial myocytes. Circ. Res. 91, 719–726.
Wenzel, P., Mollnau, H., Oelze, M., Schulz, E., Wickramanayake, J. M., Muller, J., Schuhmacher, S., Hortmann, M., Baldus, S., Gori, T., Brandes, R. P., Munzel, T., and Daiber, A. (2008). First evidence for a crosstalk between mitochondrial and NADPH oxidase-derived reactive oxygen species in nitroglycerin-triggered vascular dysfunction. Antioxid. Redox Signal. 10, 1435–1447.
Whitmer, J. T., Idell-Wenger, J. A., Rovetto, M. J., and Neely, J. R. (1978). Control of fatty acid metabolism in ischemic and hypoxic hearts. J. Biol. Chem. 253, 4305–4309.
Williamson, C. L., Dabkowski, E. R., Baseler, W. A., Croston, T. L., Alway, S. E., and Hollander, J. M. (2010). Enhanced apoptotic propensity in diabetic cardiac mitochondria: influence of subcellular spatial location. Am. J. Physiol. Heart Circ. Physiol. 298, H633–H642.
Williamson, J. R. (1965). Glycolytic control mechanisms. I. Inhibition of glycolysis by acetate and pyruvate in the isolated, perfused rat heart. J. Biol. Chem. 240, 2308–2321.
Williamson, J. R. (1966). Glycolytic control mechanisms. II. Kinetics of intermediate changes during the aerobic-anoxic transition in perfused rat heart. J. Biol. Chem. 241, 5026–5036.
Williamson, J. R., Ford, C., Illingworth, J., and Safer, B. (1976). Coordination of citric acid cycle activity with electron transport flux. Circ. Res. 38(Suppl. 1), I39–I51.
Wojtczak, L., and Zablocki, K. (2008). Mitochondria in cell life, death and disease. Postepy Biochem. 54, 129–141.
Wojtovich, A. P., and Brookes, P. S. (2009). The complex II inhibitor atpenin A5 protects against cardiac ischemia-reperfusion injury via activation of mitochondrial KATP channels. Basic Res. Cardiol. 104, 121–129.
Wolf, H. P. (1992). Possible new therapeutic approach in diabetes mellitus by inhibition of carnitine palmitoyltransferase 1 (CPT1). Horm. Metab. Res. Suppl. 26, 62–67.
Wong-Riley, M. T., Liang, H. L., Eells, J. T., Chance, B., Henry, M. M., Buchmann, E., Kane, M., and Whelan, H. T. (2005). Photobiomodulation directly benefits primary neurons functionally inactivated by toxins: role of cytochrome c oxidase. J. Biol. Chem. 280, 4761–4771.
Wood, K. C., and Gladwin, M. T. (2007). The hydrogen highway to reperfusion therapy. Nat. Med. 13, 673–674.
Wu, F., Yang, F., Vinnakota, K. C., and Beard, D. A. (2007). Computer modeling of mitochondrial tricarboxylic acid cycle, oxidative phosphorylation, metabolite transport, and electrophysiology. J. Biol. Chem. 282, 24525–24537.
Wu, F., Zhang, E. Y., Zhang, J., Bache, R. J., and Beard, D. A. (2008). Phosphate metabolite concentrations and ATP hydrolysis potential in normal and ischaemic hearts. J. Physiol. 586(Pt 17), 4193–4208.
Wyatt, K. M., Skene, C., Veitch, K., Hue, L., and McCormack, J. G. (1995). The antianginal agent ranolazine is a weak inhibitor of the respiratory complex I, but with greater potency in broken or uncoupled than in coupled mitochondria. Biochem. Pharmacol. 50, 1599–1606.
Xu, W., Liu, Y., Wang, S., McDonald, T., Van Eyk, J. E., Sidor, A., and O’Rourke, B. (2002). Cytoprotective role of Ca2+-activated K+ channels in the cardiac inner mitochondrial membrane. Science 298, 1029–1033.
Yu, T., Sheu, S. S., Robotham, J. L., and Yoon, Y. (2008). Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc. Res. 79, 341–351.
Zhang, J., Gong, G., Ye, Y., Guo, T., Mansoor, A., Hu, Q., Ochiai, K., Liu, J., Wang, X., Cheng, Y., Iverson, N., Lee, J., From, A. H., Ugurbil, K., and Bache, R. J. (2005). Nitric oxide regulation of myocardial O2 consumption and HEP metabolism. Am. J. Physiol. Heart Circ. Physiol. 288, H310–H316.
Zhang, R., Mio, Y., Pratt, P. F., Lohr, N., Warltier, D. C., Whelan, H. T., Zhu, D., Jacobs, E. R., Medhora, M., and Bienengraeber, M. (2009). Near infrared light protects cardiomyocytes from hypoxia and reoxygenation injury by a nitric oxide dependent mechanism. J. Mol. Cell. Cardiol. 46, 4–14.
Zhang, W. H., Wang, H., Wang, X., Narayanan, M. V., Stavrovskaya, I. G., Kristal, B. S., and Friedlander, R. M. (2008). Nortriptyline protects mitochondria and reduces cerebral ischemia/hypoxia injury. Stroke 39, 455–462.
Zhao, Y., Wang, Z. B., and Xu, J. X. (2003). Effect of cytochrome c on the generation and elimination of O2o− and H2O2 in mitochondria. J. Biol. Chem. 278, 2356–2360.
Zmijewski, J. W., Lorne, E., Zhao, X., Tsuruta, Y., Sha, Y., Liu, G., Siegal, G. P., and Abraham, E. (2008). Mitochondrial respiratory complex I regulates neutrophil activation and severity of lung injury. Am. J. Respir. Crit. Care Med. 178, 168–179.
Zoccarato, F., Cavallini, L., Bortolami, S., and Alexandre, A. (2007). Succinate modulation of H2O2 release at NADH:ubiquinone oxidoreductase (Complex I) in brain mitochondria. Biochem. J. 406, 125–129.
Zorov, D. B., Filburn, C. R., Klotz, L. O., Zweier, J. L., and Sollott, S. J. (2000). Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 192, 1001–1014.
Zorov, D. B., Kobrinsky, E., Juhaszova, M., and Sollott, S. J. (2004). Examining intracellular organelle function using fluorescent probes: from animalcules to quantum dots. Circ. Res. 95, 239–252.
Zuckerbraun, B. S., Chin, B. Y., Bilban, M., d’Avila, J. C., Rao, J., Billiar, T. R., and Otterbein, L. E. (2007). Carbon monoxide signals via inhibition of cytochrome c oxidase and generation of mitochondrial reactive oxygen species. FASEB J. 21, 1099–1106.
Keywords: mitochondria, cardiac ischemia/reperfusion, cardiac metabolism, reactive oxygen species, nitric oxide, peroxynitrite, mitochondrial Ca2+, therapy
Citation: Camara AKS, Bienengraeber M and Stowe DF (2011) Mitochondrial approaches to protect against cardiac ischemia and reperfusion injury. Front. Physio. 2:13. doi: 10.3389/fphys.2011.00013
Received: 27 October 2010; Accepted: 24 March 2011;
Published online: 12 April 2011.
Edited by:
Miguel A. Aon, Johns Hopkins University School of Medicine, USAReviewed by:
Valdur Saks, National Institute of Chemical Physics and Biophysics, EstoniaNazareno Paolocci, Johns Hopkins University, USA
Shey-Shing Sheu, University of Rochester, USA
Copyright: © 2011 Camara, Bienengraeber and Stowe. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Amadou K. S. Camara, Medical College of Wisconsin, M4240, 8701 Watertown Plank Road, Milwaukee, WI 53226, USA. e-mail:YWtzY0BtY3cuZWR1