
- Ottawa Hospital Research Institute, Ottawa, ON, Canada
The heart is a continually active pulsatile fluid pump. It generates appropriate forces by precisely timed and spaced engagement of its contractile machinery. Largely, it makes its own control signals, the most crucial of which are precisely timed and spaced fluxes of ions across the sarcolemma, achieved by the timely opening and closing of diverse voltage-gated channels (VGC). VGCs have four voltage sensors around a central ion-selective pore that opens and closes under the influence of membrane voltage. Operation of any VGC is secondarily tuned by the mechanical state (i.e., structure) of the bilayer in which it is embedded. Rates of opening and closing, in other words, vary with bilayer structure. Thus, in the intensely mechanical environment of the myocardium and its vasculature, VGCs kinetics might be routinely modulated by reversible and irreversible nano-scale changes in bilayer structure. If subtle bilayer deformations are routine in the pumping heart, VGCs could be subtly transducing bilayer mechanical signals, thereby tuning cardiac rhythmicity, collectively contributing to mechano-electric feedback. Reversible bilayer deformations would be expected with changing shear flows and tissue distension, while irreversible bilayer restructuring occurs with ischemia, inflammation, membrane remodeling, etc. I suggest that tools now available could be deployed to help probe whether/how the inherent mechanosensitivity of VGCs – an attribute substantially reflecting the dependence of voltage sensor stability on bilayer structure – contributes to cardiac rhythmicity. Chief among these tools are voltage sensor toxins (whose inhibitory efficacy varies with the mechanical state of bilayer) and arrhythmia-inducing VGC mutants with distinctive mechano-phenotypes.
Cardiac VGCs and Mechano-Physiology
Cardiac myocyte voltage-gated channels (VGCs) are abundant, ubiquitous, and diverse. Multiple isoforms and splice variants from the Kv, Nav, Cav, and HCN subfamilies work together to shape and propagate action potentials and to achieve electro-mechanical coupling. Knowing that VGC operation is inherently (Tabarean and Morris, 2002) mechanosensitive (MS; Figures 1–3), it is natural to pose the broad hypothesis that VGCs contribute to physiological and pathological mechano-electric feedback in the heart. Box 1 and 2 list classes of situations where VGC mechanosensitivity could potentially come into play in the myocardium. “VGC mechanosensitivity” is a robust, understandable, but highly nuanced phenomenon. Consult (Morris, 2011a) together with (Morris and Laitko, 2005) for a review of VGC mechanosensitivity as seen by biophysical studies of recombinant and native VGCs; in essence, voltage sensor repacking underlies VGC mechanosensitivity, and repacking plays out differently in different VGC species and different membrane environments. VGCs have complex kinetic schemes and their mechanosensitivity plays out against those schemes. Nav current is “inhibited by stretch” if one looks only on the fact that stretch accelerates fast inactivation. Nav current is “potentiated by stretch” if one looks only on the fact that stretch accelerates activation. And so it goes for all the VGCs, all of which are inherently MS voltage-and-time dependent conductances.
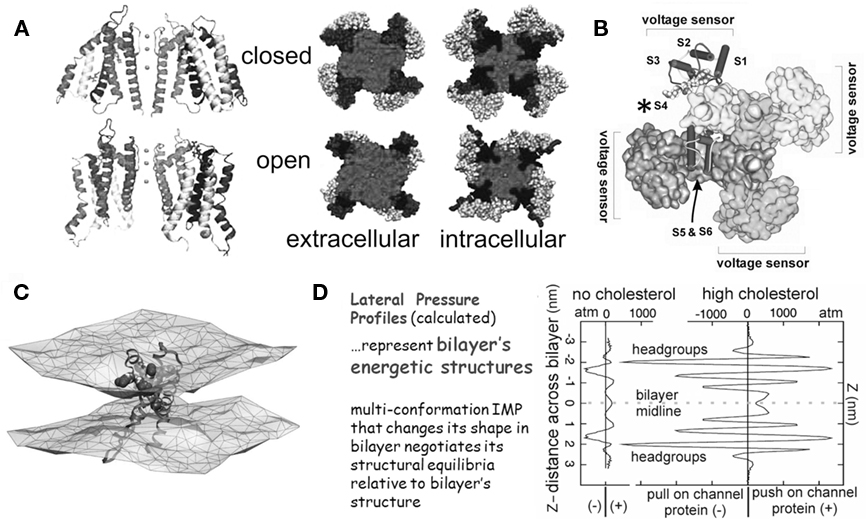
Figure 1. The structural basis of mechanosensitivity in VGCs. (A) Left, a Kv channel (Long et al., 2007) modeled in closed-like and open-like conformations and right (for intracellular and extracellular faces of the Kv) space-filling models of these states (modified from Chanda et al., 2005) and based on (Long et al., 2007) and refs therein). Due to its cruciform shape, the VGC’s lateral interactions with bilayer lipids are considerably more extensive than if the protein were cylindrical (the typical depiction before high resolution structures were available). Because of the substantial shape change of the closed-open transition, some of the free energy of the transition must be attributed to restructuring the lateral protein-lipid interface. (B) The 4 voltage sensors, whose gating charge is primarily in transmembrane segment S4 (asterisk), occupy the periphery (see Long et al., 2007). (C) A single activated-state voltage sensor domain modeled in lipid bilayer (the two water/lipid interfaces are depicted) showing that in its activated-state, the sensor locally deforms/thins the bilayer (modified from Krepkiy et al., 2009). (D) Bilayers, too have different structures (and hence energetics) that are dramatic and lipid-dependent, as seen from these lateral pressure profiles calculated for two simple bilayers, one without, one with cholesterol. Cholesterol thickens a bilayer (see Z axis) and increases its packing order. For any shape-changing integral membrane protein (IMP), the lateral pressure profile is an important component of the energetic landscape (for further explanation see Morris and Juranka, 2007b; Finol-Urdaneta et al., 2010; Morris, 2011a, b).
Box 1. Situations that might REVERSIBLY DEFORM VGC-bearing membranes in the heart
(1) Shear forces associated with blood flow. Endomyocardial cells, valve surfaces, papillary muscle surfaces, and the lumen of the cardiovascular system would all experience continually changing shear forces.
(2) Stretch/distension/twist of ventricles and atria associated with chamber filling. Sarcoplasmic membranes and, when present, t-tubular and intercalated disk membranes could all experience localized intermittent deformations. Likewise for the fine processes of intra-cardiac neurons embedded in the mechanically dynamic ventricular wall.
(3) Strains arising from respiratory motions and other external forces like chest thump.
Box 2. Situations that might IRREVERSIBLY RESTRUCTURE VGC-bearing membranes in the heart
Ischemia-reperfusion injury, cardiac inflammatory and sepsis conditions, diverse developmental changes and physiological adjustments, mutations or drugs that affect the composition of membrane lipids, mutations, or drugs that perturb membrane meso-structures such as caveolae, mutations that perturb normal membrane cortex structure (e.g., dystrophin-related genes), diet changes, temperature changes, presence of bilayer restructuring agents such as ethanol or of agents that modify cholesterol (packing and amounts), mechanical trauma severe enough to locally bleb the sarcolemma.
A commonly held view is that the only cardiac “stretch channel” of real interest is “the” stretch-activated cation channel. This idea needs to be dispelled before we proceed. First, stretch-activated cation channels that generate unitary current events occur only in atrial myocytes. In contrast, ventricular myocytes and sinoatrial node cells generate whole cell mechanically sensitive cation currents, but are devoid of unitary stretch-activated channel activity (Zeng et al., 2000; Cooper and Kohl, 2005; Morris and Laitko, 2005; Banderali et al., 2010). Second, as with cardiac VGCs (which are identified entities), it is still unknown whether atrial cation channels (which are unidentified entities) respond to stretch or shear forces in situ the way they do in vitro (Morris, 2011a, 2011b). Third, in biophysical tests (Figure 2), recombinant VGCs show the same level of sensitivity to membrane stretch as endogenous stretch-activated cation channels (Gu et al., 2001), raising the simple but telling question: if atrial stretch cation channels feel and respond to mechanical stimuli in situ (Bode et al., 2001) how do the more abundant and equally stretch-sensitive VGCs remain unaffected?
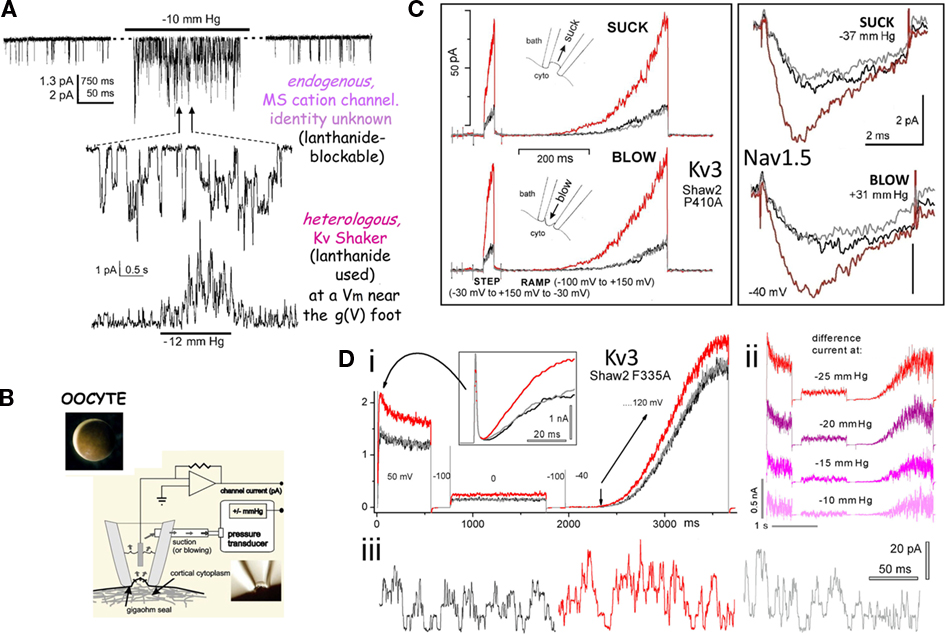
Figure 2. Use of Xenopus oocyte cell-attached patch recordings to study MS gating in VGCs. (A) Endogenous “stretch-activated cation channel” activity and heterologously expressed Kv channels (Shaker, fast inactivation removed) exhibiting what a naive observer could reasonably construe to be “stretch-activated K channel” activity. In reality it is stretch-modulated VGC current (Gu et al., 2001). (B) A Xenopus oocyte, and a cartoon of pipette aspiration as used for applying membrane stretch (inset, a cell-attached macropatch; typically, smaller patches are used but this allows for visualization of non-traumatized plasma membrane; inset is modified from Shcherbatko et al., 1999). As explained in refs (Morris et al., 2006) and (Wang et al., 2009), membrane trauma, when it happens, is submicroscopic. (C) During pipette aspiration, stretch increases membrane tension, and does so whether aspiration pressure is negative (“suck”) or positive (“blow”), as seen for two very different VGCs, a Kv3 homotetramer and a Nav1.5 pseudotetramer. Multivalent lanthanide ions included in the pipette inhibit endogenous stretch channel activity (and, as expected, right-shift VGC currents by tens of mV). Here, and throughout, black, red, gray traces signify before, during, after stretch. (Di,ii) Illustrates “stretch difference currents” obtained from before/during/after records (two step depolarizations and one ramp depolarization are used here) and demonstrates that the magnitude of the stretch difference currents increase with increasing stretch intensity, while (iii) shows that stretch increases unitary K currents frequency at 0 mV (which corresponds to the reversal potential of endogenous stretch-activated cation channels). These figures are modified from (Gu et al., 2001; Laitko et al., 2006; Morris and Juranka, 2007a).
Given that VGCs are molecularly identified and sensitive to a plethora of reasonably specific inhibitors, what has been the barrier to establishing if they do or do not contribute “mechano-modulated” components to myocardial currents? The difficulty is that the machinery of voltage sensing is the very machinery responsible for mechanosensitivity in these channels (Laitko and Morris, 2004; Krepkiy et al., 2009). Could one nevertheless recognize and dissect out MS contributions of VCGs to cardiac currents, and if so, by what means?
I discuss here two classes of ready-to-deploy molecular tools, namely voltage sensor toxins and arrhythmia-inducing voltage sensor mutations, that could help with rigorously posing the question: “Does VGC-X pass stretch-modulated current in the working myocardium?” Mechanical stimulation of soft tissues is notoriously hard to quantify; for this reason it is particularly tricky to characterize the modulation of VGCs by such stimuli (Bode et al., 2001; Morris et al., 2006; Huang et al., 2009). Unwavering insistence on blinded experimental and analysis procedures (see Wan et al., 1999; Gottlieb et al., 2008) will therefore be needed, plus inclusion of diverse controls for drug specificity, for the appropriateness of mechanical and voltage protocols, for time-effects, for reversibility, and so on. The pay-off will be an understanding of how nature either uses or, possibly, has managed to entirely bypass the inherent mechanosensitivity of the VGCs operating in the inherently and relentlessly electro-mechanical milieu of the heart.
Basic Operation of a VGC without/with Bilayer Mechanical Perturbations
Voltage-gated channels are either tetramers (Figure 1A) or pseudotetramers; evolution has evidently conjoined a voltage sensor protein (Figure 1C) to a pore-forming protein (Long et al., 2007), and then in various ways has multiplied this entity fourfold. The resulting VGCs differ in their selectivity for cations and their voltage-dependent gating has useful idiosynchrasies; depolarization promotes opening in Kv, Nav, and Cav channels, hyperpolarization promotes opening in HCN channels, repolarization lets HERG channels open. The molecular mechanisms by which voltage sensors respond to changing membrane voltage have become increasingly clear over the last decade. Gating current associated with movement of charged residues in the voltage sensor domains (Chanda et al., 2005) can be measured in the different VGC subtypes. Ionic or gating currents can be recorded while simultaneously monitoring the intensity of site-directed fluorescence signals embedded in or near the voltage sensor machinery (Mannuzzu et al., 1996; Horne and Fedida, 2009). Upon membrane depolarization, a transmembrane sequence (largely in S4) bearing positively charged arginine and lysine residues twists and repacks relative to the rest of the protein. The voltage-dependent repacking facilitates the opening or closing of various “gates” along the central ion permeation pathway.
By analogy to a hinged-and-bolted gate, outward movement the charge associated with S4 would be “bolt sliding.” “Unbolted” gates are free to open, but do so only under the influence of the ever-present jostling of thermal energy that makes them “swing open on their hinges.” The waiting (for appropriate random jostles) makes the overall gating process stochastic. VGCs, in other words, are voltage-and-time dependent devices and “VGC kinetic” parameters quantitatively summarize the interplay of electric and thermal energy. Even if membrane voltage changes instantaneously, as occurs in ideal voltage clamp, the individual VGCs in a population open with randomly distributed latencies or wait times. The resulting I(V, t) (“macroscopic” if from many channels simultaneously or “ensemble” if averaged from one channel tested multiple times) has a “characteristic time” for the opening and closing transitions at that voltage. The voltage at which being open or closed is equally likely is the midpoint (V0.5) of a voltage-dependent sigmoid curve, its “Boltzmann.” If some factor – a drug, an elevated temperature, a membrane fluidizer, hyperbaric pressures, membrane stretch – alters the characteristic waiting times and/or the V0.5, we say the VGC’s kinetics are modulated. In Kv1 and Nav channels, membrane stretch decreases the wait times without changing the voltage dependence (slope of the Boltzmann; Tabarean and Morris, 2002; Laitko and Morris, 2004; Banderali et al., 2010). In terms of our metaphor, stretch “lubricates the bolt” but makes it no smaller or bigger. In biophysical terms, stretch lowers the free energy barrier for voltage sensor movement without altering the quantity of gating charge that moves. A hyperpolarizing (“left”) shift of the Boltzmann means that voltage sensors move more easily, a depolarizing (“right”) shift means they move with greater difficulty.
Kv3 and Cav channel responses to stretch show the same-fold increase in current at all voltages; their activation rates are unaffected and they show no Boltzmann shifts with stretch (Morris and Laitko, 2005; Laitko et al., 2006). Evidently, for these channels, the rate-limiting transition in the voltage-dependent activation path is not a stretch-sensitive transition (Laitko et al., 2006; Morris, 2011a). Cav and Kv3 channels behave as if stretch “promotes” channels into near-open states (probably by making an outward voltage sensor movement easier), so that when a slower voltage-dependent (and stretch-insensitive) transition occurs, more than the usual number of channels open (Figures 2C,D). Identifying the rate-limiting but stretch-insensitive transition in these channels would clearly be of interest for selective drug design.
Various physical and chemical deformations of the bilayer (Phillips et al., 2009) can modulate gating in VGCs (Figures 1C,D). Bilayer deformation could result from membrane stretch (or imposed change in local curvature), from hyperbaric pressure, from addition of a bilayer structure-distorting chemical reagent, from developmental processes that alter the bilayer’s cholesterol, fatty acid, sphingomyelin content, etc. Elastic membrane stretch thins and disorders the bilayer. Hyperbaric pressure, by increasing packing order, thickens and rigidifies it. Clearly, elastic membrane stretch can modulate VGC kinetics in biophysical recording contexts (Figures 2 and 3) for recombinant channels (see also Morris and Laitko, 2005; Morris, 2011a) but it is now up to cardiac physiologists to determine if stretch (e.g., Zabel et al., 1996) and other bilayer deformations modulate VGCs in situ.
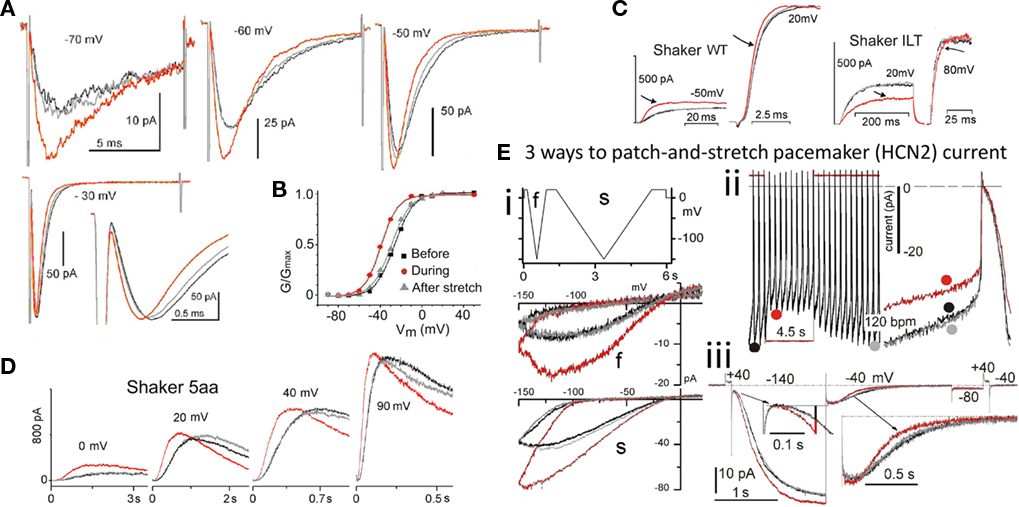
Figure 3. VGC operation is inherently mechanosensitive (MS). (A) Nav1.5 channel current accelerates reversibly with stretch; activation and inactivation rates both speed to the same extent (Gmax has been reached at −30 mV in this patch, so peak INa is unaffected by stretch, but activation and fast inactivation both reversibly accelerate; modified from Morris and Juranka, 2007a). (B) a Nav1.5 activation Boltzmann left-shifts reversibly with stretch. (C) Shaker (Kv1) and Shaker-ILT have different rate-limiting voltage transitions in the activation pathway. Stretch affects both, but it accelerates the (largely independent) movement of the Shaker voltage sensors, whereas it decelerates (the late concerted) movement of the voltage sensor in Shaker-ILT, as seen here for voltages near the foot and head of the respective G(V)s; in keeping with the acceleration vs deceleration, stretch left-shifts the Shaker Boltzmann and right-shifts the Shaker-ILT Boltzmann (not shown; see Reference Laitko et al., 2006). (D) In Shaker 5aa, voltage-dependent gating is two orders of magnitude slower than in Shaker (compare to C; note the different time scales), but since an independent (though sluggish) voltage-dependent transition is rate-limiting for activation, this mutant channel behaves like its “WT” counterpart (see Laitko and Morris, 2004). (E) HCN2 channels open with hyperpolarization, they close with depolarization, they have multiple open states and they exhibit pronounced hysteresis. Here HCN2 currents are studied by (i) ramp clamp, (ii) passive action potential clamp and (iii) classic step clamp (see Lin et al., 2007) for details. Upon a step depolarization (iii) from −140 to −40 mV, stretch accelerates channel closure. Gating currents show this transition to be analogous to the rate-limiting depolarization-induced “outward” voltage sensor motion in Kv Shaker.
Squid axon sodium and potassium currents have been voltage-clamped at hyperbaric pressures equivalent to the ocean depths and, of course, at atmospheric pressure (see Morris and Juranka, 2007a). The outcome? When large hyperbaric pressures are applied reversibly, both gating and ionic currents slow reversibly. Consider the giant squid, the diving whale, the elephant seal two, or more kilometers deep in the ocean. All their bilayers are thicker, more densely packed at depth than at the ocean surface. As they rise toward the surface, the bilayers again become thinner, more fluid. Collectively, the VGCs in these creatures unavoidably experience and accommodate bilayer mechanical modulations. VGCs have been at work in membranes since a time when all creatures were sea creatures. In terrestrial mammals such as ourselves, have VGCs learned to ignore the information available in bilayer structure? Probably not.
Both Elastic and Plastic Changes in Bilayer Structure Affect VGC Kinetics
Plasma membranes are principally defined by their lipid bilayer but also include intracellular and extracellular proteins (membrane skeleton and adherent extracellular matrix respectively). To a first approximation, our interest here is bilayer structure which is the molecular determinant of bilayer mechanics (Phillips et al., 2009) but with the proviso that bilayer structure and long-range organization are powerfully influenced by the state of the normally adherent and contractile actomyosin–spectrin based membrane cortex, by the extracellular matrix and by the membrane’s integral and peripheral membrane proteins (Charras and Paluch, 2008; Phillips et al., 2009; Lingwood and Simons, 2010; Morris, 2011b).
A VGC is reversibly “stretch-modulated” if it exhibits reversible changes in voltage-dependent current when subjected to reversible (elastic) membrane stretch. But membrane stretch can also impose irreversible (plastic) changes in membrane structure (Morris and Juranka, 2007b). Not surprisingly therefore, many channels, including VGCs, exhibit irreversible and reversible responses to experimental membrane stretch (Morris and Juranka, 2007b; Wang et al., 2009). In essence, abrupt irreversible changes reflect membrane trauma (see cartoon, Figure 4), typically due to membrane blebbing, the dissociation of bilayer from adherent membrane cortex proteins (Tabarean et al., 1999; Wang et al., 2009; Schmidt and MacKinnon, 2008). Irreversible changes in bilayer lipid composition/organization also occur physiologically, developmentally, with diet, and with various pathologies and these too modify VGC kinetics (e.g., Morris and Juranka, 2007b; Ben-Zeev et al., 2010).
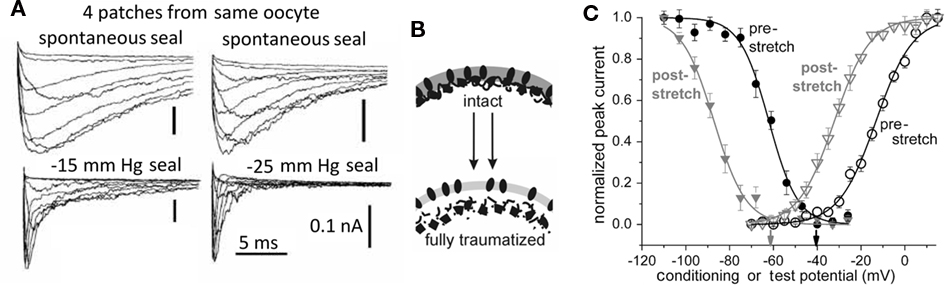
Figure 4. Membrane trauma can irreversibly left-shift Nav channel operation, producing “leaky” Nav channels, probably because the resident bilayer gets more bleb-like, i.e., thinner, more disordered. (A) This phenomenon was first discovered from accidental damage to membranes while making gigaohm seals on oocytes. Here, Nav1.4 (without the auxiliary beta subunit) kinetics recorded in 4 patches from the same oocyte look radically different depending on the extent to which the membrane was mechanically traumatized during seal formation. Modifed from (Tabarean et al., 1999). (B) Cartoon suggesting that bleb formation leaves channels (dark ovals) in a bilayer environment that is on average thinner and (not specifically depicted but indicated by the lighter color) more disordered/fluid/symmetric-across-leaflets. In oocytes, Nav1.5 channels behave as if they are trafficked to disordered membrane (Morris and Juranka, 2007a; Wang et al., 2009; Banderali et al., 2010) and so do not show irreversible kinetic changes with stretch, but in HEK cells, stretch is largely traumatic for Nav1.5 channels (Beyder et al., 2010). (C) The trauma phenomenon also occurs for Nav1.6 in oocyte membranes and is summarized by coupled left-shift of the activation and steady-state inactivation curves as shown here (see Wang et al., 2009).
Though reversible deformations of lipid bilayers are necessarily small (e.g., bilayers can stretch only ∼3–4% before rupture; Morris and Homann, 2001), reversible stretch modulation of VGC current can be dramatic, as in Figure 2. As a biological gauge, it is interesting to compare VGC mechanosensitivity (as in Figures 2 and 3) to that of MS prokaryotic channels which act as molecular osmotic emergency valves and open only at near-lytic membrane tensions. Walled prokaryotes, however, also have VGCs (e.g., Finol-Urdaneta et al., 2010; Irie et al., 2010). If voltage sensors first appeared in such cells, then they originated and evolved in high turgor cells, i.e., in cells where high curvature membranes subject to bilayer deformations and tension fluctuations were common. Prokaryote VGCs likely exhibit MS modulation more frequently than osmovalves exhibit stretch-activation (see Morris, 2002; Boucher et al., 2009). In animal cells, membrane tensions are well below the near-lytic range (Morris and Homann, 2001). Myocardial t-tubules and intra-cardiac neuron transmural neurites might well experience deformations, tension changes, osmotic dilations, and other mechanical perturbations (Dyachenko et al., 2008). Virtually nothing is known about this or about regular or intermittent bilayer deformation intensities in “flat” regions of membrane (sarcolemma proper, intercalated disk membrane) or in caveolin or amphiphysin-rich regions. Inherent biophysical mechanosensitivity is not sufficient evidence to say that VGCs are physiological mechano-transducers; experiments with native channels in their normal working environments are needed to determine if nature exploits the inherent mechanosensitivity of VGCs to monitor the mechanical (i.e., bilayer mechanical) status of various myocardial membranes, or alternatively, if nature has learned to circumvent this characteristic of VGCs.
Molecular Pharmacology Tools - Voltage Sensor Toxins and Their D-Enantiomers
Neither gadolinium nor the peptide GsMTx4 (a powerful inhibitor of stretch-induced atrial fibrillation) should be called “specific MS cation channel inhibitors.” Doing so has muddied the waters vis à vis both MS cation channels and the mechanosensitivity of VGCs (see Lin et al., 2007). Lanthanides (gadolinium, lanthanum) do indeed inhibit MS cation channels, but are entirely unsuitable in cell physiological settings since any lanthanide ions that manage to remain in solution (Caldwell et al., 1998; Morris et al., 2006) also modify gating of VGCs (Gu et al., 2001). That gadolinium-modified ventricular myocyte INa could be misconstrued as gadolinium-inhibition of “MS cation channels” has been meticulously demonstrated (Li and Baumgarten, 2001).
Whereas any physiological story that relies on gadolinium blockade of MS cation channels should be ignored, the actions of GsMTx4, an amphiphilic tarantula venom internal cysteine-knot (ICK) toxin, GsMTx4 (Suchyna et al., 2000; Jung et al., 2005) should be examined afresh in physiological settings. Along with similarly structured toxins that are well-established voltage sensor toxins, it offers fascinating promise as an in situ indicator of VGC involvement in mechano-electric feedback. GsMTx4 (see Figure 5) piques interest for at least three reasons: (1) it is an established inhibitor of cardiac phenomena relevant to mechano-electric feedback, since it powerfully inhibits, at biologically relevant concentrations, stretch-induced atrial fibrillation and swell-induced ventricular cation current (Suchyna et al., 2000; Bode et al., 2001), (2) by evolutionary pedigree, the tarantula venom peptide, GsMTx4, looks indistinguishable from known members the tarantula venom’s cabal of voltage sensor toxins, making it plausible that, like those toxins, it binds to sensors on one or several VGCs (Jung et al., 2005), (3) VsTx1, a related tarantula venom peptide, is a voltage sensor toxin and is demonstrably MS in its efficacy (Schmidt and MacKinnon, 2008) making it reasonable to anticipate that GsMTx4 might also be a MS voltage sensor toxin.
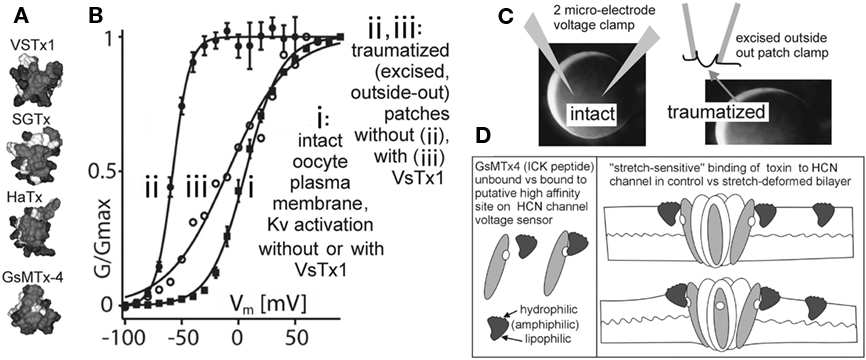
Figure 5. Are there more MS voltage sensor toxins out there?(A) A collection of ICK peptides purified from tarantula venom (modified from Jung et al., 2005). The top 3 are established voltage sensor toxins, and the bottom one, GsMTx4, might be too (micromolar level GsMTx4 inhibits unidentified MS cation channels in astrocytes, but that is not the effect of interest here. GsMTx4 has powerful cardiac actions at <200 nm; Bode et al., 2001). (B) The “silver bullet scenario” as described in the text. VsTx1 action is mechanosensitive in that it inhibits this species of Kv channel (right-shifts activation; ii → iii) only when bilayer organization has been disrupted. The irreversible left-shift (i → ii) upon excision/trauma is as seen for some Nav channels (Figure 4), and like the advent of toxin efficacy, is ascribed to, (C) the difference in bilayer mechanics between the two recording conditions. (D) how GsMTx4 might act as a stretch-sensitive toxin for HCN channel voltage sensors. The scenario at left mirrors known behavior of VsTx1 and SGTx on their targets (Kv voltage sensors) and that at right suggests how membrane stretch might increase the inhibitory efficacy of GsMtx4. It is also possible to imagine membrane deformations that decreased the affinity of amphiphilic voltage sensor toxins for their targets. HCN channels are mentioned here, but the idea would apply for any voltage sensor, even for stand-alone ones (see Figure 1C; proton channels, voltage sensitive phosphatases as described, e.g., in Krepkiy et al., 2009).
Voltage-gated channel mechanosensitivity is linked, in all likelihood, to the supposition, now experimentally justified (Krepkiy et al., 2009), that transition from resting to active states requires a voltage sensor to locally thin (i.e., deform) the bilayer (see Figure 1C). Reciprocally, therefore, any external agency that thins the bilayer will favor active states. By steric and thermodynamic arguments, it would be surprising if bilayer-resident amphiphilic voltage sensor toxins bound to their targets in an entirely mechano-in sensitive fashion. And indeed, empirical evidence supports this logic: simple but elegant experiments with VSTx1, a voltage sensor toxin for select Kv channels, show that inhibitory efficacy depends dramatically on the mechanical state of the bilayer (Schmidt and MacKinnon, 2008; see Figure 5). Moreover, efficacy and binding of this and of similar voltage sensor toxins to their Kv voltage sensor targets depends on bilayer lipid-species (Milescu et al., 2007, 2009; Schmidt and MacKinnon, 2008), in other words toxin-sensor actions depend on bilayer structure.
As implied above, I suspect that GsMTx4, like VSTx1, is a voltage sensor toxin whose actions are sensitive to the mechanical state of the bilayer. If so, the VGC target of GSMTx4 – cation-permeant “pacemaker channels” possibly? – have yet to be identified (Lin et al., 2007; Morris, 2011a). The argument to date has been, however, that this toxin evolved as a specific inhibitor of (non-VGC) MS cation channels (Suchyna et al., 2004) acting not as a specific ligand but as an amphiphilic perturbor of bilayer structure. This is thought to modulate MS transitions in unidentified voltage-insensitive cation channels. If, instead, the dramatic inhibitory effect reported for 170 nM GsMTx4 on atrial stretch-induced fibrillation (Bode et al., 2001) involves specific binding to voltage sensor, it can be expected to bind its target sensor(s) with greater avidity in some bilayer mechanical states than others. It nevertheless remains entirely unclear by what mechanism GsMTx4 in the submicromolar range inhibits stretch-induced atrial fibrillation (Bode et al., 2001) and swell-induced ventricular (inwardly rectifying) Ication (Suchyna et al., 2000). The possibility that specific binding of GsMTx4 to some species of cardiomyocyte VGC(s) is sensitive to bilayer stretch urgently needs to be tested. If GsMTx4 is behaving as a “stretch-sensitive voltage sensor toxin” (Figure 5) in the rabbit heart experiments mentioned, the biological implications are exciting. It would signify, for example, that bilayers of some of the VGC-bearing cardiomyocyte membranes of the atrium are deformed in a physiologically significant fashion by whole-tissue stretch.
Consideration of available data points to pacemaker cation channels as plausible VGC targets for GsMTx4. At 400 nM, GsMTx4 abolishes an unidentified swell-induced inwardly rectifying cation current (ventricular myocytes), and at 170 nM GsMTx4 it strongly inhibits stretch-induced atrial fibrillation. Atrial and ventricular myocytes and sinoatrial node cells all express several isoforms of HCN-type “pacemaker” channels, and this subfamily of VGCs precisely fits the description of an inwardly rectifying MS/swell-induced cation current” (with the caveat that only the HCN2 isoform has been tested; Calloe et al., 2005; Lin et al., 2007). If the idea embodied in Figure 5D (stretch-sensitive vs toxin) is broadly valid, then we have in hand a class of VGC-specific, discretely modifiable molecular tools for exploring the role of VGCs in cardiac mechano-electric feedback. Note that even if submicromolar GsMTx4 is found to modulate native HCN channels in stretched membranes, it might also act on additional VGC species in native membranes. Clearly, it will be important to test a range of different native VGCs for MS actions of GsMTx4.
HCN channel kinetics are challenging. Their hysteresis would make it difficult to seek evidence of reversible MS efficacy in a voltage sensor toxin. But the approach used by Schmidt and MacKinnon (2008) identifies irreversible MS changes in efficacy and this could make studies of HCN channel/toxin/stretch tractable. What should be sought? Large irreversible toxin-induced right-shifts in traumatized membrane patches, contrasting to “no-shift” in intact membrane. Use of sawtooth ramps at two speeds would give useful outputs (see Figure 2). GsMTx4 efficacy (or lack thereof) on HCN isoforms 1–4 would be determined by two microelectrode clamp (= intact membrane) then compared to efficacy in excised outside-out (= traumatized membrane) oocyte patches (refer to prototype in Figure 5; see figure legend). Other VGCs could also, of course, be tested with GsMTx4. The protocol needed to look for MS vs Toxins comes down to this: identical VGC protein, identical bilayer lipids, lipids structured differently (= different bilayer mechanics) in intact and traumatized (disorganized) membranes.
Exploiting the GsMTx4 Sensitivity of Stretch-Induced Fibrillation
Stretch-arrhythmia experiments like those done by Bode et al. (2001) with GsMTx4 should be undertaken to explore the in situ efficacy of a known MS voltage sensitive toxin. Does VSTx1 action on cardiac Kv channels change with tissue stretch? Response “polarity” (i.e., more vs less atrial arrhythmia) would depend on whether toxin binding increased or decreased with stretch. Either would indicate that tissue stretch was deforming cardiomyocyte bilayers that held Kv channels.
A program of blinded stretch-induced atrial fibrillation experiments involving 3–4 tarantula venom ICK peptides is needed. The peptides should include 2–3 well-characterized voltage sensor toxins (e.g., VSTx1) plus GsMTx4, all at, say, 200 nM, a concentration slightly above 170 nM used to such effect by Bode et al. (2001) in their atrial stretch experiments. Fully blinded no-voltage-sensor-toxin controls are not a problem; the d-amino acid enantiomer peptides of SGTx1 (Milescu et al., 2007) and of GsMTx4 (Suchyna et al., 2004; again, at 200 nM) would be ideal. d-enantiomer SGTx1 accumulates in the bilayer as effectively as its natural (WT) counterpart, but unlike the WT does not bind the Kv voltage sensor or right-shift Kv activation (Milescu et al., 2007). d-enantiomer GsMTx4 also would not act as voltage sensor toxin (one presumes!). What is claimed (Suchyna et al., 2004), however, is that d-enantiomer GsMTx4 inhibits stretch-activated cation channels as strongly as l-enantiomer described would rectify a major and surprising oversight: there has never been a test of prediction that l-enantiomer and d-enantiomers of GsMTx4 (at 170 nM) are equi-effective (Suchyna et al., 2004) as inhibitors of stretch-induced atrial fibrillation. If, as I suspect, the published 170 nM action of the l-enantiomer (Bode et al., 2001) reflects a stretch-sensitive voltage sensor toxin action (on a to-be-determined VGC), then 200 nm d-enantiomer GsMTx4 will not be effective as an inhibitor. The use of blinded procedures and double controls will obviate false negative problems.
Cardiac Arrhythmias in Ischemia – Targeting the “Leaky” VGCs of Damaged Bilayer
Mechanically induced irreversible changes (i.e., plastic change) in membrane nano-structure typically takes the form of blebbing – de-adhesion of membrane cortex from the bilayer, accompanied by loss of lipid order, including the loss of leaflet asymmetry. The operation of many VGCs, including Nav channels (Shcherbatko et al., 1999; Tabarean et al., 1999; Wang et al., 2009; Beyder et al., 2010), becomes irreversibly left-shifted in the mechanically disturbed bilayers of blebbed membrane. A key consequence: VGCs are inappropriately active near the resting potential. Kinetically speaking, the channels “leak.” For this reason, channels in mechanically or ischemically blebbed sarcolemma or in dilated t-tubule membranes would become foci of pathological channel activity (Wang et al., 2009).
Re-imagine the scenario of Figure 5 (VsTx1, Kv, intact vs blebbed bilayer; Schmidt and MacKinnon, 2008) in a clinical context. The toxin is a “silver bullet” able to hit only “the bad guys” – the left-shifted, and hence leaky, channels in damaged bilayer. This scenario deserves attention. Wherever in cardiomyocytes, adhesions between bilayer and cortical cytoskeleton fail, membrane blebs form (in CNS subjected to traumatic brain injury, such blebbing occurs at Nav1.6-rich nodes of Ranvier (Wang et al., 2009). Ischemic and traumatic blebs with Nav channels are likely foci for ectopic excitation. Blebs are patches of disordered fluidized membrane which, for crawling motility and mitosis (Stewart et al., 2011) form reversibly, but which in trauma, ischemia, inflammation, apoptosis form irreversibly. Returning to the VSTx/Kv story, the “silver bullet” status of the toxin becomes possible because of the dramatically different bilayer structures in intact vs traumatized membranes. “[O]ne of the mechanisms by which riluzole exerts its neuroprotective action is to preferentially block the inactivated sodium channel of damaged or depolarized neurons under ischemic conditions,” Narahashi and colleagues concluded some years ago (Song et al., 1997), before it was understood that altered bilayer mechanics might underlie some of the differential action they reported. An important question that should be addressed is whether clinically important lipophilic Nav inhibitors (including ranolazine) are particularly useful post-ischemia (Dhalla et al., 2009) because they can preferentially target left-shifted (leaky) Navs in blebbed bilayer. If so, an elevated solubility for the drug in “fluidized” (blebbed) vs intact membrane might be partially responsible. Another possibility is that drug binding to slow-inactivated states is more stable in blebbed vs intact membrane. These possibilities are in no way mutually exclusive.
Since VGC gating and toxin binding to voltage sensors are both sensitive to bilayer conditions, differential efficacy is also likely for lipophilic reagents in different bilayer mechanical environments (see discussion in Reference Finol-Urdaneta et al., 2010). Structural tuning (which could be tested with a Schmidt and MacKinnon, 2008 approach) to amplify differential efficacy in traumatized vs intact membranes might bring lipophilic VGC drugs closer to “silver bullet” status.
Needed for Mechano-Electric Feedback Studies: A Nav1.5 “R1626P” Mouse
Voltage sensor mechanosensitivity renders all VGCs MS and makes it difficult to determine if VGCs contribute to the mechano-electrical life of the working myocardium. VGC-specific voltage sensor toxins could be one good approach. Another would be to exploit arrhythmia-associated disease mutants of VGCs that, at a biophysical level, show abnormal and distinctive mechano-phenotypes. While mechanical stimuli were applied (Huang et al., 2009) at varying frequencies and phases of the cardiac cycle, cardio-outputs could be followed in an animal (mouse, presumably) expressing the mutant VGC. Comparison of frequency domain data sets should help establish whether the WT vs mutant mechano-phenotypes affect stretch-arrhythmias. The power of such information is illustrated by a non-invasive experimental approach used in humans to seek evidence of cardiac mechano-electric feedback. Patients with atrial fibrillation and a variable secondary modulation (flutter) of their elevated beat frequency were monitored (electrocardiograms and respiratory motions) during metronomic breathing at various rates (Masé et al., 2008). Rhythmicity patterns were consistent with expectation from a coupled oscillator model (interacting cardiac and respiratory rhythms) of mechano-electric feedback. Mechano-electric feedback experiments with small mammals are challenging but doable (Huang et al., 2009), and provide diverse opportunities to probe with drugs (Fabritz, 2007), with rhythm resetting procedures, and so on (Morris, 2011a).
In a case of LQT1 syndrome in which the proband (and other family members) presented with late onset hypertension-induced atrial fibrillation, it has been suggested that the mutant KCNQ1 (delayed rectifier) channel is responsible for dangerously dysfunctional mechano-electric feedback (Otway et al., 2007). The mutation, in a region responsible for channel/actin interactions, renders the channel overly sensitive to hyposmotic swelling. It needs to be determined, however, whether the mutant KCNQ1 responses to swelling are (as implied; Otway et al., 2007) indeed due to membrane “stretch” (Calloe et al., 2005; Hammami et al., 2009).
Substandard mechano-electric feedback might add insult to injury in large enough measure to trigger potentially lethal arrhythmias in some LQT3 syndrome patients carrying Nav1.5 channel mutations. Of particular interest are mutations associated with sudden infant death syndrome (Banderali et al., 2010) and sudden unexplained nocturnal death syndrome. The latter is strongly associated in otherwise healthy adults with both sudden respiratory disturbances (Cheng et al., 2010) and with acute inflammatory conditions of the cardiovascular system, conditions that are blebbogenic (Nakajima et al., 2010). Whether large and sudden respiratory motions such as gasps, sneezes, snorts etc., or sudden respiratory rate changes can directly – that is, via mechano-electric feedback (Masé et al., 2008) – impact myocardial rhythmicity in humans needs further study.
Nav1.5 channel LQT3 mutations are pro-arrhythmic because they impair fast inactivation, engendering long Q–T intervals and fostering sudden re-entrant tachycardias when heart rate is low, as during sleep. We studied currents in two voltage sensor mutants involving domain-4 arginines residues (Banderali et al., 2010; the domain-4 voltage sensor governs the voltage dependence of inactivation; Figure 6). Both are associated with LQT3 syndrome: R1626P (identified and mexiletine-treated in a young adult; Ruan et al., 2007) and R1623Q (identified in a case of sudden infant death; see Banderali et al., 2010). Both showed the same abnormal mechano-phenotype: their inactivation rates, which have reduced or absent sensitivity to voltage, show significantly reduced sensitivity to stretch (Banderali et al., 2010; Figure 6B). The overall consequence is that in the mutants stretch does not speed up “INa turn off” as effectively as in WT. In situ, therefore, stretch would therefore be expected to be excessively pro-arrhythmic for the LQT3 mutants. I have suggested a transgenic mouse equivalent of human Nav1.5-R1626P as a good candidate for probing VGC involvement in mechano-electric feedback (see Morris, 2011a and Figure 6 legend). An “R1626P” mouse might help resolve the issue of why LQT3 mutations are suddenly lethal at time-X on night-Z as opposed to nights A-to-W. Even if the answer is “chaos” (Glass, 2009), chaotic storm systems are famously susceptible to being triggered by the beat of a butterfly wing. In sudden cardiac death, are minor stretch effects the butterfly kiss of death?
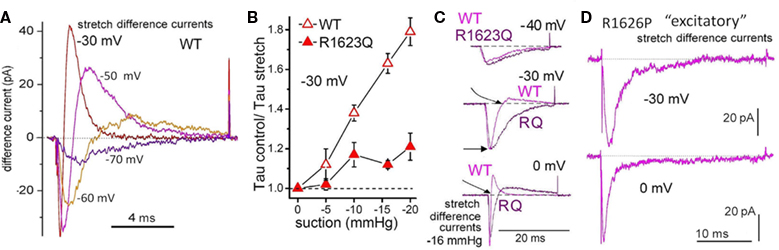
Figure 6. Two LQT3 channels: impaired domain-4 voltage sensors, impaired mechanosensitivity. (A) Stretch difference currents for WT Nav1.5 channels (see Figure 3A for INa before, during, after stretch; Morris and Juranka, 2007a). Because INa is inward over the entire physiological range, and because Nav channels activate then rapidly inactivate, stretch difference currents for Nav channels are more complex than stretch difference currents for Kv channels (Figure 2D). For Nav rule is: downward = more INa during stretch, upward = less INa during stretch. Based on the Nav stretch difference currents at the voltages illustrated, imagine what would happen during an action potential: briefly near the start, stretch would increase INa (downward) but quickly decreased INa would prevail. If this happens in situ, when there is stretch, the faster turn off of INa once an action potential was triggered would contribute in an anti-arrhythmic fashion to the overall mechano-electric feedback. The LQT3 mutations, R1623Q and R1626P, affect the Nav domain 4 voltage sensor, whose depolarization-induced repacking renders the inactivation rate (Tau−1) voltage dependent by stabilizing the inactivation particle in its bound state (see Banderali et al., 2010). Replacing arginine (positive) at R1623 with glutamine (neutral) impairs repacking of the voltage sensor protein and reduces the voltage sensitivity of Tau inactivation. Replacing arginine at 1626 with kink-inducing proline makes the inactivation rate entirely insensitive to voltage; at all voltages, R1626P has the same low-speed inactivation. The consequence: these LQT3 Nav channels remain open too long during the action potential; excess INa causes “torsades de pointes” arrhythmias. (B) Stretch (from pipette suction) is less effective at speeding the rate (Tau−1) of INa inactivation for R1623Q than for WT (likewise for R1626P, but not shown). (C) Overlaid stretch difference currents compare kinetics for WT and the LQT3 mutant. With increasing depolarization (imagine the action potential passing through −30 and 0 mV) WT INa turns off (goes upward) far sooner than for R1623Q. (D) For R1626P, stretch difference current always (even at 0 mV) represents excess (stretch-induced) inward INa. The R1626P response to stretch in situ, were it to occur, would be distinctly “excitatory” and pro-arrhythmic. Figures B–D are from Banderali et al. (2010).
Conclusion
The key messages from this essay have both biological and a pathological implications: (1) we now have molecular tools – peptide toxins and LQT3 syndrome VGCs – that can help in asking whether VGCs in the mechanically active myocardium are routinely modulated by bilayer mechanics, and (2) VGCs in mechanically or ischemically blebbed membrane, as compared with intact healthy membrane, are embedded in mechanically abnormal bilayers where voltage sensors move too readily and where the channels interact differently with amphiphilic drugs than they do in intact membrane. As an example, the kinetically “leaky” Nav channels of blebbed membrane would foster ectopic excitation but would also be especially susceptible to appropriately designed “silver bullet” lipophilic Nav channel inhibitors.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Thanks to Chris Miller, Bob French, and Ruben Coronel for critiquing the manuscript and providing helpful and intriguing comments.
References
Banderali, U., Juranka, P. F., Clark, R. B., Giles, W. R., and Morris, C. E. (2010). Impaired stretch modulation in potentially lethal cardiac sodium channel mutants. Channels 4, 12–21.
Ben-Zeev, G., Telias, M., and Nussinovitch, I. (2010). Lysophospholipids modulate voltage-gated calcium channel currents in pituitary cells; effects of lipid stress. Cell Calcium 47, 514–524.
Beyder, A., Rae, J. L., Bernard, C., Strege, P. R., Sachs, F., and Farrugia, G. (2010). Mechanosensitivity of Nav1.5, a voltage-sensitive sodium channel. J. Physiol. 588, 4969–4985.
Bode, F., Sachs, F., and Franz, M. R. (2001). Tarantula peptide inhibits atrial fibrillation. Nature 409, 35–36.
Boucher, P. A., Morris, C. E., and Joós, B. (2009). Mechanosensitive closed-closed transitions in large membrane proteins: osmoprotection and tension damping. Biophys. J. 97, 2761–2770.
Caldwell, R. A., Clemo, H. F., and Baumgarten, C. M. (1998). Using gadolinium to identify stretch-activated channels: technical considerations. Am. J. Physiol. 275, C619–C621.
Calloe, K., Elmedyb, P., Olesen, S. P., Jorgensen, N. K., and Grunnet, M. (2005). Hypoosmotic cell swelling as a novel mechanism for modulation of cloned HCN2 channels. Biophys. J. 89, 2159–2169.
Chanda, B., Asamoah, O. K., Blunck, R., Roux, B., and Bezanilla, F. (2005). Gating charge displacement in voltage-gated ion channels involves limited transmembrane movement. Nature 436, 852–856.
Charras, G., and Paluch, E. (2008). Blebs lead the way: how to migrate without lamellipodia. Nat. Rev. Mol. Cell Biol. 9, 730–736.
Cheng, J., Makielski, J. C., Yuan, P., Shi, N., Zhou, F., Ye, B., Tan, B. H., and Kroboth, S. (2010). Sudden unexplained nocturnal death syndrome in Southern China: an epidemiological survey and SCN5A gene screening. Am. J. Forensic Med. Pathol. PMID: 20110800. [Epub ahead of print].
Cooper, P. J., and Kohl, P. (2005). Species- and preparation-dependence of stretch effects on sino-atrial node pacemaking. Ann. N. Y. Acad. Sci. 1047, 324–335.
Dhalla, A. K., Wang, W. Q., Dow, J., Shryock, J. C., Belardinelli, L., Bhandari, A., and Kloner, R. A. (2009). Ranolazine, an antianginal agent, markedly reduces ventricular arrhythmias induced by ischemia and iscehmia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 297, H1923–H1929.
Dyachenko, V., Christ, A., Gubanov, R., and Isenberg, G. (2008). Bending of z-lines by mechanical stimuli: an input signal for integrin dependent modulation of ion channels? Prog. Biophys. Mol. Biol. 97, 196–216.
Fabritz, L. (2007). Drug-induced torsades de pointes – a form of mechano-electric feedback? Cardiovasc. Res. 76, 202–203.
Finol-Urdaneta, R. K., Juranka, P. F., French, R. J., and Morris, C. E. (2010). Modulation of KvAP unitary conductance and gating by 1-alkanols and other surface active agents. Biophys. J. 98, 762–772.
Glass, L. (2009). Introduction to controversial topics in nonlinear science: is the normal heart rate chaotic? Chaos 19, 028501.
Gottlieb, P., Folgering, J., Maroto, R., Raso, A., Wood, T. G., Kurosky, A., Bowman, C., Bichet, D., Patel, A., Sachs, F., Martinac, B., Hamill, O. P., and Honoré, E. (2008). Revisiting TRPC1 and TRPC6 mechanosensitivity. Pflugers Arch. 455, 1097–1103.
Gu, C. X., Juranka, P. F., and Morris, C. E. (2001). Stretch-activation and stretch-inactivation of Shaker-IR, a voltage-gated K+ channel. Biophys. J. 80, 2678–2693.
Hammami, S., Willumsen, N. J., Olsen, H. L., Morera, F. J., Latorre, R., and Klaerke, D. A. (2009). Cell volume and membrane stretch independently control K+ channel activity. J. Physiol. 587, 2225–2231.
Horne, A. J., and Fedida, D. (2009). Use of voltage clamp fluorimetry in understanding potassium channel gating: a review of Shaker fluorescence data. Can. J. Physiol. Pharmacol. 87, 411–418.
Huang, H., Wei, H., Liu, P., Wang, W., Sachs, F., and Niu, W. (2009). A simple automated stimulator of mechanically induced arrhythmias in the isolated rat heart. Exp. Physiol. 94, 1054–1061.
Irie, K., Kitagawa, K., Nagura, H., Imai, T., Shimomura, T., and Fujiyoshi, Y. (2010). Comparative study of the gating motif and C-type inactivation in prokaryotic voltage-gated sodium channels. J. Biol. Chem. 285, 3685–3694.
Jung, H. J., Lee, J. Y., Kim, S. H., Eu, Y. J., Shin, S. Y., Milescu, M., Swartz, K. J., and Kim, J. I. (2005). Solution structure and lipid membrane partitioning of VSTx1, an inhibitor of the KvAP potassium channel. Biochemistry 44, 6015–6023.
Krepkiy, D., Mihailescu, M., Freites, J. A., Schow, E. V., Worcester, D. L., Gawrisch, K., Tobias, D. J., White, S. H., and Swartz, K. J. (2009). Structure and hydration of membranes embedded with voltage-sensing domains. Nature 462, 473–479.
Laitko, U., Juranka, P. F., and Morris, C. E. (2006). Membrane stretch slows the concerted step prior to opening in a Kv channel. J. Gen. Physiol. 127, 687–701.
Laitko, U., and Morris, C. E. (2004). Membrane tension accelerates rate-limiting voltage-dependent activation and slow inactivation steps in a Shaker channel. J. Gen. Physiol. 123, 135–154.
Li, G. R., and Baumgarten, C. M. (2001). Modulation of cardiac Na(+) current by gadolinium, a blocker of stretch-induced arrhythmias. Am. J. Physiol. Heart Circ. Physiol. 280, H272–H279.
Lin, W., Laitko, U., Juranka, P. F., and Morris, C. E. (2007). Dual stretch responses of mHCN2 pacemaker channels: accelerated activation, accelerated deactivation. Biophys. J. 92, 1559–1572.
Lingwood, D., and Simons, K. (2010). Lipid rafts as a membrane-organizing principle. Science 327, 46–50.
Long, S. B., Tao, X., Campbell, E. B., and MacKinnon, R. (2007). Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 450, 376–382.
Mannuzzu, L. M., Moronne, M. M., and Isacoff, E. Y. (1996). Direct physical measure of conformational rearrangement underlying potassium channel gating. Science 271, 213–216.
Masé, M., Glass, L., and Ravelli, F. (2008). A model for mechano-electrical feedback effects on atrial flutter interval variability. Bull. Math. Biol. 70, 1326–1347.
Milescu, M., Bosmans, F., Lee, S., Alabi, A. A., Kim, J. I., and Swartz, K. J. (2009). Interactions between lipids and voltage sensor paddles detected with tarantula toxins. Nat. Struct. Mol. Biol. 16, 1080–1085.
Milescu, M., Vobecky, J., Roh, S. H., Kim, S. H., Jung, H. J., Kim, J. I., and Swartz, K. J. (2007). Tarantula toxins interact with voltage sensors within lipid membranes. J. Gen. Physiol. 130, 497–511.
Morris, C. E. (2011a). “Pacemaker, potassium, calcium, sodium: stretch modulation of voltage-gated channels,” in Cardiac Mechano-Electric Feedback and Arrhythmias, Chapter 6, 2nd Edn, eds, P. Kohl, F. Sachs, and M. Franz (Philadelphia, PA: Elsevier Saunders), 42–49.
Morris, C. E. (2011b). “Why are so many ion channels mechanosensitive?” in Cell Physiology Source Book, 4th Edn, ed. N. Sperelakis (Elsevier). (in press).
Morris, C. E., and Homann, U. (2001). Cell surface area regulation and membrane tension. J. Membr. Biol. 179, 79–102.
Morris, C. E., and Juranka, P. F. (2007a). Nav channel mechanosensitivity: activation and inactivation accelerate reversibly with stretch. Biophys. J. 93, 822–833.
Morris, C. E., and Juranka, P. F. (2007b). “Lipid stress at play: mechanosensitivity of voltage-gated channels,” in Mechanosensitive Ion Channels, B. Current Topics in Membranes, Vol. 59, eds O. Hamill, S. Simon, and D. Benos (San Digeo, CA: Academic Press), 297–337.
Morris, C. E., Juranka, P. F., Lin, W., Morris, T. J., and Laitko, U. (2006). Studying the mechanosensitivity of voltage-gated channels using oocyte patches. Methods Mol. Biol. 322, 315–329.
Morris, C. E., and Laitko, U. (2005). “The mechanosensitivity of voltage-gated channels may contribute to cardiac mechanoelectric feedback,” in Cardiac Mechano-Electric Feedback and Arrhythmias, eds, P. Kohl, F. Sachs, and M. R. Franz (Philadelphia, PA: Elsevier Saunders), 33–41.
Nakajima, K., Takeichi, S., Nakajima, Y., and Fujita, M. Q. (2010). Pokkuri death syndrome: sudden cardiac death cases without coronary atherosclerosis in South Asian young males. Forensic Sci. Int. 207, 6–13.
Otway, R., Vandenberg, J. I., Guo, G., Varghese, A., Castro, M. L., Liu, J., Zhao, J., Bursill, J. A., Wyse, K. R., Crotty, H., Baddeley, O., Walker, B., Kuchar, D., Thorburn, C., and Fatkin, D. (2007). Stretch-sensitive KCNQ1 mutation A link between genetic and environmental factors in the pathogenesis of atrial fibrillation? J. Am. Coll. Cardiol. 49, 578–586.
Phillips, R., Ursell, T., Wiggins, P., and Sens, P. (2009). Emerging roles for lipids in shaping membrane-protein function. Nature 459, 379–385.
Ruan, Y., Liu, N., Bloise, R., Napolitano, C., and Priori, S. G. (2007). Gating properties of SCN5A mutations and the response to mexiletine in long-QT syndrome type 3 patients. Circulation 116, 1137–1144.
Schmidt, D., and MacKinnon, R. (2008). Voltage-dependent K+ channel gating and voltage sensor toxin sensitivity depend on the mechanical state of the lipid membrane. Proc. Natl. Acad. Sci. U.S.A 105, 19276–19281.
Shcherbatko, A., Ono, F., Mandel, G., and Brehm, P. (1999). Voltage-dependent sodium channel function is regulated through membrane mechanics. Biophys. J. 77, 1945–1959.
Song, J. H., Huang, C. S., Nagata, K., Yeh, J. Z., and Narahashi, T. (1997). Differential action of riluzole on tetrodotoxin-sensitive and tetrodotoxin-resistant sodium channels. J. Pharmacol. Exp. Ther. 282, 707–714.
Stewart, M. P., Helenius, J., Toyoda, Y., Ramanathan, S. P., Muller, D. J., and Hyman, A. A. (2011). Hydrostatic pressure and the actomyosin cortex drive mitotic cell rounding. Nature 469, 226–230.
Suchyna, T. M., Johnson, J. H., Hamer, K., Leykam, J. F., Gage, D. A., Clemo, H. F., Baumgarten, C. M., and Sachs, F. (2000). Identification of a peptide toxin from Grammostola spatulata spider venom that blocks cation-selective stretch-activated channels. J. Gen. Physiol. 115, 583–598.
Suchyna, T. M., Tape, S. E., Koeppe, R. E. II, Andersen, O. S., Sachs, F., and Gottlieb, P. A. (2004). Bilayer-dependent inhibition of mechanosensitive channels by neuroactive peptide enantiomers. Nature 430, 235–240.
Tabarean, I. V., Juranka, P., and Morris, C. E. (1999). Membrane stretch affects gating modes of a skeletal muscle sodium channel. Biophys. J. 77, 758–774.
Tabarean, I. V., and Morris, C. E. (2002). Membrane stretch accelerates activation and slow inactivation in Shaker channels with S3-S4 linker deletions. Biophys. J. 82, 2982–2994.
Wan, X., Juranka, P., and Morris, C. E. (1999). Activation of mechanosensitive currents in traumatized membrane. Am. J. Physiol. 276, C318–C327.
Wang, J. A., Lin, W., Morris, T., Banderali, U., Juranka, P. F., and Morris, C. E. (2009). Membrane trauma and Na(-leak from Nav1.6 channels. Am. J. Physiol. 297, C823–C834.
Keywords: stretch, mechano-electric feedback, arrhythmias, ectopic excitation, LQT3, pacemaker, sodium channel, bleb
Citation: Morris CE (2011) Voltage-gated channel mechanosensitivity: fact or friction?. Front. Physio. 2:25. doi: 10.3389/fphys.2011.00025
Received: 14 March 2011;
Paper pending published: 27 March 2011;
Accepted: 18 May 2011;
Published online: 31 May 2011.
Edited by:
Zhilin Qu, University of California Los Angeles, USAReviewed by:
Lai-Hua Xie, University of Medicine and Dentistry of New Jersey– New Jersey Medical School, USAArthur Beyder, Mayo Clinic, USA; Riccardo Olcese, University of California Los Angeles, USA
Copyright: © 2011 Morris. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Catherine E. Morris, Ottawa Hospital Research Institute, 725 Parkdale Avenue, Ottawa, ON, Canada K1Y 4E9. e-mail:Y21vcnJpc0B1b3R0YXdhLmNh