 Rachel Lees-Green1*
Rachel Lees-Green1* Peng Du1 Gregory O’Grady1,2
Peng Du1 Gregory O’Grady1,2 Arthur Beyder3
Arthur Beyder3 Gianrico Farrugia3
Gianrico Farrugia3 Andrew J. Pullan1,4
Andrew J. Pullan1,4
- 1 Auckland Bioengineering Institute, The University of Auckland, Auckland, New Zealand
- 2 Department of Surgery, The University of Auckland, Auckland, New Zealand
- 3 Mayo Clinic, Rochester, MN, USA
- 4 Department of Engineering Science, The University of Auckland, Auckland, New Zealand
Gastrointestinal motility research is progressing rapidly, leading to significant advances in the last 15 years in understanding the cellular mechanisms underlying motility, following the discovery of the central role played by the interstitial cells of Cajal (ICC). As experimental knowledge of ICC physiology has expanded, biophysically based modeling has become a valuable tool for integrating experimental data, for testing hypotheses on ICC pacemaker mechanisms, and for applications in in silico studies including in multiscale models. This review is focused on the cellular electrophysiology of ICC. Recent evidence from both experimental and modeling domains have called aspects of the existing pacemaker theories into question. Therefore, current experimental knowledge of ICC pacemaker mechanisms is examined in depth, and current theories of ICC pacemaking are evaluated and further developed. Existing biophysically based ICC models and their physiological foundations are then critiqued in light of the recent advances in experimental knowledge, and opportunities to improve these models are identified. The review concludes by examining several potential clinical applications of biophysically based ICC modeling from the subcellular through to the organ level, including ion channelopathies and ICC network degradation.
Interstitial Cells of Cajal and Gastrointestinal Motility
The gastrointestinal (GI) system is a continuous tube from the mouth to the anus, formed into a series of discrete organs: the oropharynx, esophagus, stomach, small and large intestines. These organs have distinct functions that together encompass the purposes of storage, digestion, and absorption of nutrients, the excretion of waste, and protection from digestive agents and pathogens. Motility, being the movement of contents along and between gut segments as a consequence of coordinated smooth muscle contractions, is of fundamental importance to the health of all of these GI functions.
Motility is regulated by several cooperating mechanisms, involving bioelectrical, neural, and hormonal control systems (reviewed in Huizinga and Lammers, 2009). The focus of this review is on GI bioelectrical activity, and specifically the mechanisms of its generation within interstitial cells of Cajal (ICC).
Slow Wave Activity is Generated and Propagated by ICC
Among the mechanisms coordinating motility is a rhythmic electrical “slow wave” activity. Slow waves are cyclical oscillations in the ICC and smooth muscle cell (SMC) membrane potential (Vm) from a resting potential (RMP) of −80 to −55 mV to a peak potential of −25 to 0 mV in ICC and −40 to −25 mV in SMC, varying by region and species (Dickens et al., 1999; Kito and Suzuki, 2003). An example of ICC and SMC slow wave recordings is shown in Figure 1. Slow wave depolarizations shift SMC membrane potential from a region of low open probability for voltage-dependent calcium (Ca2+) channels to a potential with increased channel open probability, thereby influencing the timing and direction of contractions (Sanders et al., 2006). Although slow waves alone can cause sufficient calcium influx to initiate contractions (Ozaki et al., 1991), functionally significant contractions are a consequence of coregulatory conditions being met, as occurs following a meal (Huizinga and Lammers, 2009).
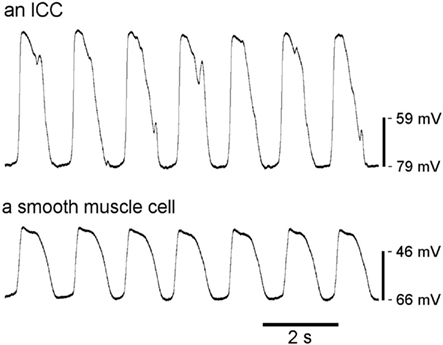
Figure 1. Typical slow wave recordings from an ICC and a SMC from murine small intestine. Slow waves from SMC have a slightly different morphology than those from ICC, including smaller amplitudes. (Courtesy of Dr. Lei Sha, Mayo Clinic).
It is widely accepted that ICC generate and propagate slow waves (reviewed previously e.g., in Sanders et al., 2006). Unlike ICC, SMC lack all the conductances and intracellular mechanisms responsible for generating slow wave activity, and cannot actively propagate slow waves (Koh et al., 2003; Ward et al., 2003). Moreover, neural input is not required for slow wave generation (Ward et al., 1999).
Slow Wave Entrainment, Propagation, and SMC Coupling
Individual ICC demonstrate spontaneous activity at intrinsic frequencies, and a declining gradient of slow wave frequencies is evident in the stomach and small intestine in the aboral direction (Code and Szurszewski, 1970; Kelly and Code, 1971). In intact tissue, ICC are organized in an electrical syncytium via gap junctions, and neighboring ICC become “entrained” to match the highest intrinsic frequency in the syncytium (Cousins et al., 2003; Koh et al., 2003). Electrical coupling between ICC and SMC is likely also achieved via small sparse gap junctions, enabling conduction of slow waves into the SMC syncytium, but without draining excessive current such that ongoing ICC entrainment would be prevented (Cousins et al., 2003).
The role of slow waves in GI motility is best understood in the stomach, where slow waves directly coordinate the phasic smooth contractions that underpin trituration of ingested food. In the healthy human stomach, slow waves originate in the corpus at 3 cycles per minute (cpm), and propagate through the corpus and antrum toward the pylorus (O’Grady et al., 2010). In the small intestine, slow waves have a higher frequency, occurring in humans at around 12 cpm in the proximal duodenum, with a declining frequency gradient to around 8–9 cpm distally in the ileum (Christensen et al., 1966). The organization of small intestine and colonic slow wave patterns and their role in motility is less well understood than in the stomach, and neural influences on some ICC populations adds complexity (Bayguinov et al., 2010).
Populations of ICC
Several types of ICC have been classified by location, as shown in Figure 2. ICC-MY (also called ICC-MP) are located in the region of the myenteric nerve plexus between the longitudinal and circular smooth muscle layers, although ICC-MY are not found in the gastric fundus of small animals (Hanani et al., 2005). ICC-IM are located intramuscularly, and are further subclassified as ICC-CM (in the circular muscle layer), and ICC-LM (in the longitudinal layer). ICC-LM are sparsely distributed in the small intestine, and ICC-CM are also sparse in the small intestine, particularly in small animals (Hanani et al., 2005). However, the small intestine contains an ICC network (ICC-DMP) in the deep muscular plexus between circular muscle layers (Hanani et al., 2005). A sub-population of ICC-IM are the ICC-SEP, identified in the septa between circular muscle bundles (Horiguchi et al., 2001). Submucosal ICC-SM are located at the submucosa-circular muscle border in the antrum (Horiguchi et al., 2001) and near the submucous plexus in the colon (often called ICC-SMP; Rumessen et al., 1993). Finally, ICC found in the subserosal layer have been called ICC-SS (Hanani et al., 2005).
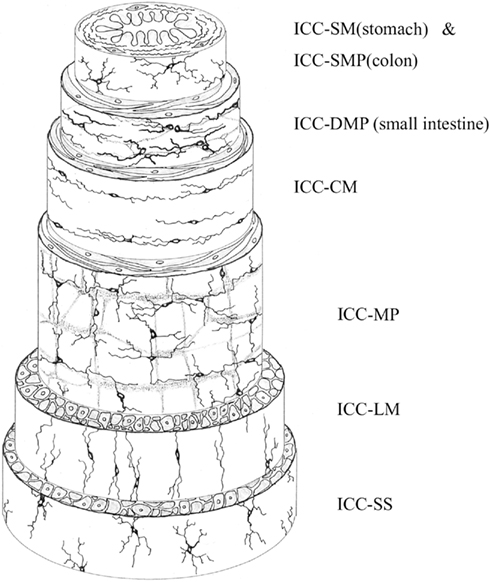
Figure 2. Schematic of the location of ICC within the different layers of the GI tract. ICC-SM and ICC-SMP are the innermost ICC, located at the border of the submucosa and circular muscle. ICC-DMP occur between circular muscle layers in the small intestine. ICC-CM and ICC-LM are located within circular and longitudinal muscle, respectively. ICC-MP (ICC-MY) are located between the circular and longitudinal muscle layers. ICC-SS occur within the subserosa. Reproduced from Hanani et al. (2005) with permission from Elsevier.
ICC-MY are generally held to be the primary pacemaker cells in the stomach and small intestine (Dickens et al., 1999), while ICC-SM are the primary pacemakers in the colon (Yoneda et al., 2002). ICC-CM actively regenerate slow waves in circular muscle (Dickens et al., 2001; Horiguchi et al., 2001), and ICC-SEP conduct slow waves from ICC-MY deep into circular muscle bundles in large animals (Lee et al., 2007). ICC-IM and ICC-DMP are responsible for mediating neural signals from the enteric nervous system, particularly cholinergic and nitrergic neurotransmission (Suzuki et al., 2003). Excitatory vagal stimulation can also lead ICC-IM to become primary pacemakers (Hirst et al., 2002b).
Slow waves generated by ICC-MY have been called pacemaker potentials or driving potentials in recognition of their role in initiating slow waves, and slow waves recorded from ICC-CM have been called regenerative potentials. This terminology can be used to differentiate between slow waves recorded directly from ICC and those recorded from SMC, but pacemaker potentials, regenerative potentials, and SMC slow waves all have the same electrical origins in ICC.
The Clinical Significance of ICC
Since the recognition that ICC play a critical role in normal motility, pathophysiological associations between ICC and dysmotility have been reported for all gut regions (reviewed in Farrugia, 2008 and Huizinga et al., 2009). Interpreting the functional significance of these defects is made difficult by the fact that other cell types, including enteric nerves, are also affected in many of these conditions. In particular, there is a strong association between loss of ICC and loss of enteric nerves in motility diseases. Further confounding the issue is the large variety of techniques used to identify ICC in human tissue and the quality of the tissue used (Garrity et al., 2009; Knowles et al., 2009). Most of the work done on normal tissue has used different fixation and visualization techniques from that done in pathological states, making comparisons hard. Additionally, human ICC numbers vary by site and decline markedly with age (Gomez-Pinilla et al., 2011) making use of proper controls fundamental.
Despite these issues, associations of likely functional significance are now established in several disorders, notably in diabetic gastroenteropathy (reviewed in Kashyap and Farrugia, 2010) and slow transit constipation (He et al., 2000), in both of which ICC network depletion is now regarded as a pathological hallmark. ICC defects have also been implicated in intestinal pseudo-obstruction (Isozaki et al., 1997), inflammatory conditions (Der et al., 2000), ileus (Zhou et al., 2011), and bowel obstruction (Chang et al., 2001). An absolute loss of ICC is not required to produce a functional defect because ICC act as a network to generate and propagate signals. Therefore, abnormal function or patchy loss can have significant effects even if total numbers are not markedly affected. A developing area of clinical interest is ICC ion channelopathies, which have been implicated in irritable bowel syndrome (Locke et al., 2006; Saito et al., 2009), which will be discussed in subsequent sections in relation to biophysically based modeling applications.
ICC Electrophysiology
Methods for Investigating the Electrical Activity of Gastrointestinal Tissues
The fundamental element of cellular electrical activity is the ion channel. Ion channels in the plasma membrane and in the membranes of cell organelles coordinate in setting the resting potential and responding to a variety of mechanical, chemical and electrical stimuli. In electrically active cells like ICC and SMC, specialized ion channels generate, amplify, and propagate electrical signals. Experimental tools have been developed to study the components of the GI electrical system from the single ion channel to the organ level.
The study of the bioelectrical activity requires, at minimum, the use of two electrodes: a recording electrode and a reference electrode. The voltage difference between these electrodes is the measured potential in volts. Typical recordings are performed in either current clamp or voltage clamp mode, with the design of the recording electrode depending on the sample of interest.
The activity of single ion channels or small populations of channels can be studied using patch clamp (Sakmann and Neher, 1995), which requires dissociation of ICC from tissue, or expression of the ion channels of interest in cell lines. However, in situ patch clamp recording is possible in special circumstances, and this has recently been done for ICC in situ (Wang et al., 2008). In a patch clamp setup a small area of cell membrane is aspirated into a capillary glass recording electrode filled with an electrolyte solution, forming a tight seal between the membrane and glass, which enables currents through single ion channels to be recorded (Sakmann and Neher, 1995).
To study the entire population of a cell’s ion channels, whole-cell voltage clamp may be employed (Strege et al., 2003). In this case, the seal is formed as above and then the membrane patch is ruptured so the patch electrode solution becomes continuous with the cell interior. In this mode of recording, single channels generally cannot be studied. However, the benefits of whole-cell recording include the ability to exchange the intracellular and extracellular solutions, and to study the effect of drug application on different ion currents.
If the electrical activity of intact tissue is of interest then sharp electrode intracellular recording or extracellular electrodes may be employed. Intracellular recording lacks the ability to record currents under voltage clamp, but a significant advantage is that complex autonomous electrical activity may be studied, and different cell types can be studied in their near-native environment.
Extracellular recordings, typically performed using surface contact electrodes, are a valuable recording technique for in vivo studies. Extracellular signals are generated by transmembrane currents arising due to the voltage gradient between excited and resting tissues, and correspond to the summated activity from a localized region of tissue around the electrode. Detailed spatiotemporal maps can be generated from multi-electrode recordings, describing propagation sequences in high-resolution (Lammers et al., 2005; O’Grady et al., 2010).
Model tissues
In the quest to understand the molecular mechanisms of gastrointestinal electrical activity, the functional details and arrangement of the ion channels and associated proteins needs to be understood. Opportunities to study human GI physiology are limited (Lee et al., 2007; O’Grady et al., 2010), so multiple animal models have been employed, including mice (e.g., Bayguinov et al., 2010), guinea-pigs (e.g., Dickens et al., 1999), dogs (e.g., Ward et al., 2004), and rats (e.g., Wang et al., 2008).
Interstitial cells of Cajal are difficult to study in culture because they change phenotype, quickly losing their automaticity and becoming more like SMCs (Epperson et al., 2000; Zhu et al., 2009). However, cultured ICC have often been used preferentially because freshly dissociated ICC are difficult to identify amongst the significantly more numerous SMC (Koh et al., 1998). In an effort to overcome some of these problems, a new mouse model has been developed with ICC expressing green fluorescent protein (GFP)-kit (Ro et al., 2010), which makes ICC identification easier (Zhu et al., 2009). Nevertheless, work performed in ex vivo biological systems requires great effort and patience.
Ion channels can be identified using an electrophysiological approach (patch or whole-cell clamp) or by molecular biology by amplifying genes or their products from tissues or single cells. In electrophysiology, electrical signals are frequently composites of multiple ion currents. Identification of contributing channels requires a decomposition of the current into individual components, by defining the channels’ ion selectivity, gating stimuli, and sensitivity to pharmacological agents.
Once candidate ion channels and modulating proteins are identified, these proteins can be expressed in heterologous cell systems, such as HEK cells, CHO cells, or Xenopus oocytes, so that electrophysiology of the protein can be studied in detail down to the single channel level. One way to classify ion channels is by gating sensitivity to voltage, ligands, mechanical stimuli, or ions. These channels gate from a closed to an open state over a narrow range of the specific stimulus, and gating sensitivity may often be described by a Boltzmann function (Dubois et al., 2009).
Ion channels are also classified by their ion selectivity [e.g., anion, non-selective cation (NSC), sodium (Na+), potassium (K+), Ca2+, chloride (Cl−), etc.]. The reversal potential for a particular ion current, equal to the Nernst equilibrium potential, can be used to identify candidate ions. Individual ions can then be eliminated from the solutions, and subsequent loss of current indicates selectivity for the eliminated ion, which is significant progress toward ion channel identification. Additional functional description of ion channels may be done using pharmacologic modification.
Non-specific effects of pharmacological agents
A key issue in experimental studies investigating ICC pacemaker mechanisms has been the lack of specificity of almost all the pharmacological agents used to identify membrane ion currents and intracellular ion transfer mechanisms. The main agents employed to date are as follows:
2-aminoethoxydiphenyl borate (2-APB): blocks inositol trisphosphate (IP3)-mediated Ca2+ release from the endoplasmic reticulum (ER), but also inhibits store-operated Ca2+ entry, including entry through transient receptor potential (TRP) channels (Bootman et al., 2002), which often carry NSC currents (Parekh and Putney, 2005). 2-APB may also disrupt other intracellular Ca2+ transporters, including sarco-endoplasmic reticulum Ca2+ ATPase (SERCA) pumps and mitochondrial Na+/Ca2+ exchangers (Bootman et al., 2002).
Mibefradil: inhibits T-type Ca2+ channels when used in concentrations greater than 0.1 μM, but it also inhibits NaV1.5 sodium channels at 0.3 μM, and L-type Ca2+ channels at 1 μM (Strege et al., 2005).
Nickel ions (Ni2+): are also used to block T-type Ca2+ channels. However, the concentration of Ni2+ ([Ni2+]) needed to inhibit T-type current depends on the subtype of T-type channel present. CaV3.2 T-type channels are inhibited by 10–50 μM [Ni2+], whereas CaV3.1 and CaV3.3 channels require 10- to 20-fold greater [Ni2+] (Perez-Reyes, 2003).
Chloride channel blockers: A large number of pharmacological agents, including niflumic acid, flufenamic acid, DIDS, SITS, and 9-anthracene carboxylic acid (9-AC) are known blockers of Cl− channels, particularly Ca2+-activated Cl− channels (White and Aylwin, 1990; Dick et al., 1999; Park et al., 2005). However, these drugs have also been shown to inhibit other channels. NSC currents in ICC were inhibited by niflumic acid, DIDS, and 9-AC (Koh et al., 2002; Walker et al., 2002; Takeda et al., 2008), and NSC currents have also been inhibited by niflumic acid and flufenamic acid in pancreatic cells (Gögelein et al., 1990) and by DIDS in vascular smooth muscle (Welsh et al., 2000). Flufenamic acid and 9-AC inhibited L-type Ca2+ channels in rat cerebral arteries (Doughty et al., 1998), and DIDS inhibited L-type Ca2+ and delayed-rectifier K+ channels in canine colonic SMC (Dick et al., 1999). In addition, it has been shown that niflumic acid can act as an agonist to Ca2+-activated Cl− currents under the conditions of negative membrane potential and high intracellular Ca2+ concentration ([Ca2+]i; 250 and 500 nM), and that washout of niflumic acid can enhance Ca2+-activated Cl− currents (Piper et al., 2002). Physiologically, ICC always have negative membrane potential, and it is likely that local, transient increases of [Ca2+]i in ICC reach higher than 250 nM (Means and Sneyd, 2010).
Existing Theories of Pacemaker Mechanisms
Intracellular calcium handling and unitary potentials
Many aspects of pacemaker mechanisms are well established based on early work in the ICC field. These discoveries have been reviewed elsewhere (e.g., Sanders et al., 2006) but will be briefly covered here.
Uptake and release of Ca2+ from stores in the ER, causing localized changes in [Ca2+]i, is a key dynamic in ICC pacemaking. Pharmacological inhibition of IP3-mediated Ca2+ release and SERCA Ca2+ uptake has been shown to inhibit slow waves in mice and guinea-pigs (Suzuki and Hirst, 1999; Ward et al., 2000; Hirst and Edwards, 2001; Yoneda et al., 2002). The Ca2+ chelator BAPTA also blocked regenerative potentials in the guinea-pig stomach (Suzuki and Hirst, 1999).
Type 1 IP3R and type 2 IP3R are found in ICC from mouse stomach and intestine (Aoyama et al., 2004; Liu et al., 2005; Chen et al., 2007). Type 1 IP3R is likely to be the primary candidate for Ca2+ release in the pacemaker cycle, because gastric smooth muscle from mice without IP3R1 does not generate slow waves (Suzuki et al., 2000).
Mitochondria are also important for Ca2+ cycling. Mitochondrial uncouplers (Ward et al., 2000; Kito et al., 2002a; Yoneda et al., 2002), and drugs that inhibit the mitochondrial Ca2+ uniporter (Ward et al., 2000) and Na+/Ca2+ exchanger (Kim et al., 2006) were shown to inhibit slow waves and Ca2+ cycling.
Inhibition of Ca2+ cycling was often found to shorten the duration of slow waves (e.g., Kito et al., 2002a; Lee et al., 2007). Thus, the hypothesis arose that cycling of Ca2+ between the ER and mitochondria is particularly important for the plateau phase of slow wave production.
As well as producing slow waves, ICC also produce small transient depolarizations (up to 10 mV) called unitary potentials (Hirst and Edwards, 2001; Kito et al., 2002b). Unitary potentials are readily observed in ICC-IM (Edwards et al., 1999), and can also be seen between slow waves in ICC-MY (Kito et al., 2002b; Kito and Suzuki, 2003). Unitary potentials were demonstrated to be the fundamental pacemaker event, because slow waves from guinea-pig gastric ICC-CM and ICC-MY were shown to be generated by clustered unitary potentials (Edwards et al., 1999; Hirst and Edwards, 2001). Unitary potentials depend on increased [Ca2+]i and it was proposed that they are triggered by Ca2+ release from intracellular stores (Edwards et al., 1999).
Interstitial cells of Cajal have abundant mitochondria, and closely apposed sections of ER, mitochondria, and cell membrane (on the order of 10 nm apart) are thought to create pockets of cytoplasm that are partially isolated from the rest of the cell, in which localized [Ca2+]i dynamics can occur (Ward et al., 2000; Koh et al., 2002; Sanders et al., 2006). Sanders et al. (2006) called these structures pacemaker units (PMUs), and suggested that they are the elementary structural origin of unitary potentials, whereby the ion channels that generate unitary potentials are localized to PMUs and are activated by Ca2+ cycling between the mitochondria and ER.
Voltage-dependent entrainment of unitary potentials and slow waves
Unitary potentials are generated by Ca2+-dependent mechanisms, so a voltage-dependent mechanism is required to entrain multiple PMUs to fire in a coordinated manner to generate the slow wave plateau phase. IP3Rs can be opened by Ca2+, so Koh et al. (2003) proposed that voltage-dependent Ca2+ influx was responsible for entrainment. A candidate channel was the voltage-dependent, dihydropyridine (DHP)-resistant Ca2+ channel identified by Kim et al. (2002). This channel was inhibited by Ni2+ and mibefradil, which also inhibited the upstroke rate and propagation of slow waves, and is now believed to be a T-type Ca2+ channel (Lee et al., 2007).
The entrainment theory developed by Sanders and colleagues states that depolarization from several concurrent unitary potentials activates nearby T-type Ca2+ channels. The resultant influx of Ca2+ current is responsible for the upstroke depolarization, and causes an increase in [Ca2+]i in PMUs that have not yet fired, initiating new unitary potentials by stimulating IP3R-mediated Ca2+ release (Sanders et al., 2006; Lee et al., 2007). The same voltage-mediated entrainment mechanism is believed to be responsible for propagating slow waves through ICC networks, because slow waves propagate at least an order of magnitude faster than Ca2+ diffuses (Ward et al., 2003). It has been shown in canine gastric and colonic muscles that slow wave propagation requires Ca2+ entry and reactivation of the internal IP3R- and mitochondria-related pacemaker mechanism (Ward et al., 2003, 2004). Thus, T-type Ca2+ channels are responsible for entraining PMUs to generate the slow wave plateau phase and to propagate slow waves through ICC networks.
An alternative theory of entrainment has also been suggested, in which voltage-dependent oscillations in IP3 concentration ([IP3]) are responsible for entraining PMUs (Hirst et al., 2002a). This theory has been successfully implemented in the ICC modeling field (Du et al., 2010c). However, at present there is no experimental evidence in ICC that IP3R open probability or IP3 production or degradation is voltage-mediated.
The NSCC pacemaking hypothesis
The first attempt to assemble the above evidence into a comprehensive pacemaking hypothesis was made by Sanders et al. (2006), following the discovery of a Ca2+-inhibited NSC current in cultured murine intestinal ICC that oscillated at the frequency of slow waves and was activated by reductions in [Ca2+]i (Koh et al., 2002). This theory was called the “Sanders hypothesis” by Faville et al. (2008), but shall be referred to as the NSCC Hypothesis in this review. This Hypothesis informed the structure of two recent biophysical ICC slow wave models (Corrias and Buist, 2008; Faville et al., 2009). Recent experimental findings have led Sanders and colleagues (Hwang et al., 2009; Zhu et al., 2009) and others (Means and Sneyd, 2010) to question central aspects of the NSCC Hypothesis. Nevertheless, an understanding of the Hypothesis is necessary to appreciate the significance of these recent findings and their implications for future ICC models.
According to the NSCC Hypothesis, NSC channels (NSCC) and voltage-gated Ca2+ channels are localized to PMUs. The initiating action in the pacemaker cycle is Ca2+ release from IP3R-operated stores in the ER, which gates open Ca2+ uptake transporters in mitochondria. The mitochondrial Ca2+ uptake is sufficient to decrease [Ca2+]i within the PMU cytoplasm below the original level, thus activating Ca2+-inhibited NSCC. The resulting inward pacemaker current generates a unitary potential.
The pacemaker cycle relies on maintenance of intracellular Ca2+ stores, so Sanders et al. (2006) suggested that Ca2+ collected in the mitochondria is extruded to the cytoplasm by Na+/Ca2+ exchangers, and Ca2+ is taken back up into the ER by SERCA pumps. The theory that DHP-resistant Ca2+ channels facilitate slow wave entrainment and propagation was also incorporated into the NSCC Hypothesis.
Calcium-activated channels: an alternative to the NSCC Hypothesis
There has been evidence that Ca2+-activated Cl− currents, not Ca2+-inhibited NSC currents, may generate the primary pacemaker current in ICC (Hirst et al., 2002a; Huizinga et al., 2002). Cl− channel blockers were shown to inhibit slow wave activity (Hirst et al., 2002a; Kito et al., 2002a) and rhythmic Cl− currents were recorded in ICC by Huizinga et al. (2002). However, Cl− channel blockers were also shown to inhibit the NSC current described by Sanders and colleagues (Koh et al., 2002), and the Cl− currents observed in ICC did not appear to be Ca2+-dependent (Parsons and Sanders, 2008). Thus, until recently this theory never had enough supporting evidence to displace the NSCC Hypothesis.
There have also been suggestions that Ca2+-facilitated NSC currents may be responsible for primary pacemaker currents (Goto et al., 2004) or unitary potentials (Takeda et al., 2008).
Recent Developments
Critical challenges to the NSCC Hypothesis have come from both experimental and modeling studies. When the NSCC Hypothesis was developed, the Ca2+-inactivated NSCC seemed to generate a viable pacemaking current, based on experiments using cultured ICC. However, in the last two years a Ca2+-activated Cl− channel has been identified in ICC in situ (Gomez-Pinilla et al., 2009) and shown to be important for slow wave production (Hwang et al., 2009).
Means and Sneyd (2010) have recently developed a detailed mathematical model of Ca2+ transients in the PMU, which demonstrates that generating unitary potentials with the Ca2+-inactivated NSC current described by Sanders et al. (2006) is not feasible. Firstly, Means and Sneyd (2010) found that [Ca2+]i in the PMU could only be reduced below baseline levels under certain restrictive conditions. A pulse of IP3 could result in decreased [Ca2+]i only if the mitochondrial Ca2+ uptake transporter was implemented as a non-physiological, time-dependent uniporter. Alternatively, when [IP3] was held constant, the ER Ca2+ store was depleted after many seconds and [Ca2+]i could then decrease below baseline in the periods when IP3Rs were inactive, which suggests inhibiting IP3Rs should enhance pacemaker activity. Thus, the NSC current could only be activated under very specific conditions. Secondly, half-maximal inactivation of the NSC current occurs when [Ca2+]i is around 375 nM (Faville et al., 2008), which suggests resting [Ca2+]i in the PMU should be higher than this. Means and Sneyd (2010) showed that maintaining PMU [Ca2+]i at 400 nM caused [Ca2+]i in the rest of the cell to rise to levels that would be toxic to the cell. Therefore, the Ca2+-inhibited NSC current described by Sanders and colleagues cannot be the pacemaker current.
In light of these recent developments, the following section shall review current knowledge regarding ion channels in ICC, and propose how these channels may contribute to the pacemaker activity of ICC.
Ionic Conductances in ICC
Several different types of ion conductances are proposed to play a role in ICC slow wave production, including Ca2+, Na+, Cl−, K+, and NSCC.
Calcium channels
Two calcium channels are found in ICC: the transient, low-voltage-activated T-type channel, and the long-lasting, high-voltage-activated L-type channel.
L-type Ca2+ channels are abundantly expressed in both ICC-MY and ICC-DMP in the mouse small intestine (Cho and Daniel, 2005; Chen et al., 2007), and generate inward currents in cultured murine ICC (Kim et al., 2002). L-type channels are blocked by DHPs such as nifedipine and nicardipine. Nifedipine reduced the duration of the plateau potential in murine colon, suggesting L-type current may contribute to the plateau phase in the colon (Yoneda et al., 2002; Hotta et al., 2007). However, DHPs are routinely used to block SMC L-type channels to inhibit contractions in experiments on GI tissue, because movement can cause artifacts and damage microelectrodes, and several earlier studies showed that DHPs do not significantly affect slow waves (Dickens et al., 1999). Nifedipine and other L-type channel antagonists caused a small decrease in the amplitude of the slow wave plateau phase in canine gastric muscle (Ozaki et al., 1991) and reduced [Ca2+]i measured from smooth muscle strips but did not affect slow wave production by ICC (Dickens et al., 1999; Suzuki and Hirst, 1999). Therefore, L-type Ca2+ channels do not appear to play a significant role in slow wave generation.
The T-type Ca2+ channel blockers mibefradil and Ni2+ cause a decrease in the upstroke rate of slow waves (Kito et al., 2005), suggesting that T-type channels contribute to the upstroke phase (Kito and Suzuki, 2003; Hotta et al., 2007). T-type current during the upstroke is believed to contribute to unitary potential entrainment (Kito et al., 2005; Lee et al., 2007) and voltage-mediated slow wave propagation (Ward et al., 2004), as discussed above.
The α1H subunit for a T-type channel, CaV3.2 (encoded by cacna1h), has been found in mouse intestinal ICC (Gibbons et al., 2009). Gibbons et al. (2009) showed that mice homozygous for knockout of cacna1h produced slow waves with an abnormally slow upstroke rate and frequency, and that the majority of homozygous knockout mice (99%) died in utero, reinforcing the role of T-type Ca2+ channels in the upstroke phase and propagation.
Sodium channels
SCN5A encodes a voltage-dependent, mechanosensitive sodium channel, NaV1.5, which has been found in human intestinal ICC (Strege et al., 2003). In intracellular recordings from human jejunum circular muscle, removal of extracellular Na+ or inhibiting NaV1.5 with lidocaine and QX-314 resulted in hyperpolarization of the resting potential, and decreased slow wave upstroke rate and frequency, leading Strege et al. (2003) to propose that Na+ current contributes to the upstroke phase. The NaV1.5 current is mechanosensitive as it was shown to be activated by shear stress (Strege et al., 2003) and by direct stretch of patches containing NaV1.5 (Beyder et al., 2010).
Interestingly, expression of NaV1.5 in the gastrointestinal tract is not universal across species. Strege et al. (2007) found NaV1.5 in the jejunum circular smooth muscle layer from human and dog tissue, but not in those from pig or guinea-pig. Curiously, NaV1.5 was also expressed in mouse circular smooth muscle, but Na+ current was observed in only one out of 21 murine SMC. The expression of SCN5A in animal ICC has not yet been investigated.
Chloride channels
Three Cl− currents have been identified in ICC: a high-conductance Cl− current (Huizinga et al., 2002), a volume-activated Cl− current (Park et al., 2005), and a Ca2+-activated Cl− current attributed to the anoctamin 1 (ANO1) channel (Zhu et al., 2009).
ANO1 (previously called TMEM16A), recently identified as a Ca2+-activated Cl− channel (Caputo et al., 2008; Schroeder et al., 2008; Yang et al., 2008), is expressed in human and mouse ICC (Gomez-Pinilla et al., 2009) and appears to be essential for slow wave production (Hwang et al., 2009). ANO1 knockout mice have very low survival rates, and those that do survive display no slow wave activity (Hwang et al., 2009). The exact role of ANO1 in ICC is not yet clear, but it may generate a pacemaker current, as discussed below.
Zhu et al. (2009) investigated an inward current in ICC that they identified as being carried by Cl− ions. The current had a single channel conductance of 7.8 pS, close to the 8.3-pS conductance of ANO1 expressed in HEK 293 cells (Yang et al., 2008). The current activated at −72 mV, but the activation and inactivation kinetics suggested that the current was not directly voltage gated, but rather was dependent on [Ca2+]i (Zhu et al., 2009). However, the [Ca2+]i required to activate ANO1 does depend on membrane potential, and the voltage–current relationship of ANO1 is outwardly rectifying at low (<1 μM) [Ca2+]i (Caputo et al., 2008; Schroeder et al., 2008; Yang et al., 2008).
A high-conductance chloride current (HCCC) has been identified in mouse intestine ICC (Huizinga et al., 2002), which appears to be made up of strongly cooperating 30 or 60 pS subconductances (Zhu et al., 2005; Parsons and Sanders, 2008; Wang et al., 2008). However, the reported properties of this current have differed significantly between studies. Huizinga and colleagues reported that the HCCC is Ca2+-dependent (Huizinga et al., 2002; Zhu et al., 2005), that it is inwardly rectifying due to reduction in cooperation between subconductances (Zhu et al., 2005; Wang et al., 2008), and suggested that it may contribute directly to pacemaking and to stabilizing RMP. Conversely, Parsons and Sanders (2008) found that a HCCC was outwardly rectifying and Ca2+-independent, and they suggested the current may act to increase the excitability of ICC immediately after repolarization.
Finally, Park et al. (2005) identified a volume-activated Cl− current in ICC cultured from mouse small intestine. The whole-cell current was Ca2+-independent, outwardly rectifying, and appeared to contribute to RMP and increased excitability of the plateau phase during stretch.
Measurements of Cl− equilibrium potentials (ECl) and Cl− current reversal potentials in ICC from mouse small intestine have ranged from −27 mV in situ (Wang et al., 2008) to −63 mV in culture (Zhu et al., 2005). Zhu et al. (2010a) recently measured intracellular [Cl−] for varying extracellular [Cl−] in ICC-MY from mouse small intestine. They found that ECl was −47 mV to −52 mV in situ and −41 mV in culture (Zhu et al., 2010b). These values are close to many of the ECl values (−34 to −63 mV) previously measured in culture (Park et al., 2005; Zhu et al., 2005), and indicate that Cl− currents would be depolarizing at RMP but repolarizing during the slow wave plateau.
An important finding in the study by Zhu et al. (2010) was that ICC are able to modulate [Cl−]i to maintain ECl close to −50 mV in situ. In addition, rhythmic decreases in [Cl−]i were seen in cultured ICC networks, suggesting spontaneous activation of depolarizing Cl− currents, possibly as pacemaker currents.
Chloride channel blockers. Many studies other than the ones mentioned above have suggested that Ca2+-activated Cl− channels generate pacemaking currents in ICC, based on the inhibition of slow waves when applying Cl− channel blockers and modifying Cl− concentrations (e.g., Hirst et al., 2002a; Kito and Suzuki, 2003). However, all the Cl− channel blockers used have non-specific effects (as discussed earlier), and the effects of Cl− channel blockers (detailed in Table 1) vary depending on the tissue preparation and drug, making it difficult to determine how Cl− currents contribute to slow waves.
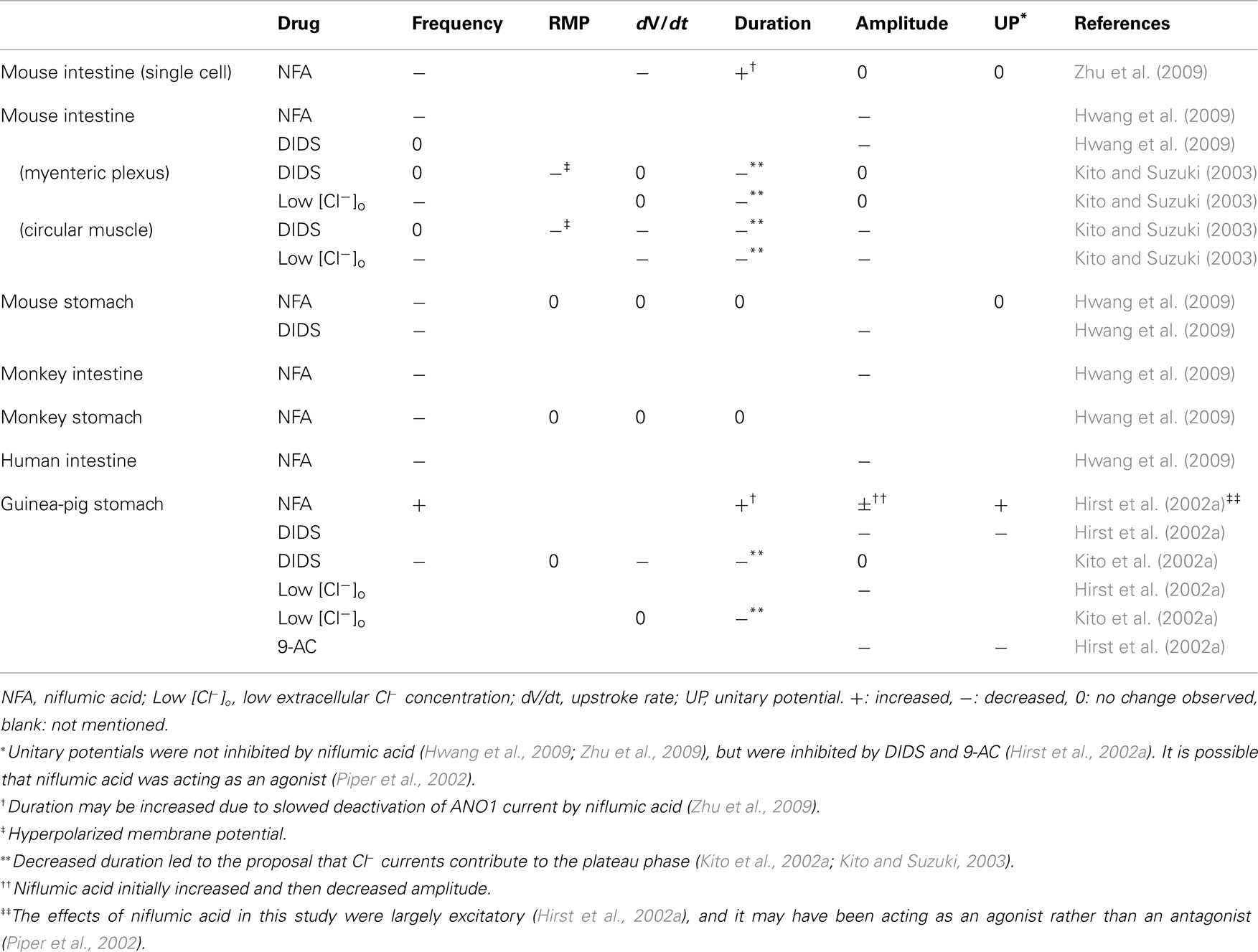
Table 1. Effects of Cl− channel blockers on slow waves recorded from isolated ICC (first row only) and GI tissue segments.
Nevertheless, Cl− channel blockers do inhibit Cl− currents in ICC. Niflumic acid has been shown to decrease the amplitude of the ANO1 current in isolated mouse intestine ICC, and to slow its deactivation (Zhu et al., 2009). The HCCC was reduced in amplitude by DIDS or 9-AC (Zhu et al., 2005) and blocked completely by DIDS or SITS (Huizinga et al., 2002) in cultured mouse intestine ICC. On the other hand, the volume-activated Cl− current was inhibited by DIDS at +80 mV membrane potential, but not at −80 mV (Park et al., 2005).
To summarize the results in Table 1, Cl− channel blockers and reduced [Cl−]o tended to reduce slow wave frequency (Kito et al., 2002a; Kito and Suzuki, 2003; Hwang et al., 2009; Zhu et al., 2009), with the exceptions that frequency changes were not observed in most experiments with guinea-pig gastric antrum (Hirst et al., 2002a) and DIDS did not reduce frequency in mouse small intestine tissue (Kito and Suzuki, 2003; Hwang et al., 2009). Cl− channel blockers and low [Cl−]o also tended to reduce slow wave amplitude (Hirst et al., 2002a; Kito and Suzuki, 2003; Hwang et al., 2009), with the caveat that reductions were often not seen until high drug concentrations were applied (Hwang et al., 2009). The effects of Cl− channel blockers on unitary potentials were inconclusive, although Sanders and colleagues suggested that ANO1 does not produce unitary potentials (Hwang et al., 2009; Zhu et al., 2009). Hwang et al. (2009) found that slow waves were completely absent in ANO1 knockout mice, but did not comment on either the absence or presence of unitary potentials. This information would be useful for determining whether ANO1 currents do generate unitary potentials.
The robustness of niflumic acid-inhibited currents or their role in ICC pacemaker mechanisms appears to be highly dependent on species, organ, and experimental preparation. Much higher concentrations of niflumic acid were required to inhibit slow waves in mouse small intestine tissue (Hwang et al., 2009) than in isolated mouse intestine cells (Zhu et al., 2009). Similarly, mouse small intestine slow waves were much less sensitive to niflumic acid than mouse gastric slow waves, and human intestine was much less sensitive than mouse intestine (Hwang et al., 2009).
Cl− channels, in particular ANO1, do seem to be important for slow wave production, but Cl− channel blocking drugs have too many varying and non-specific effects to aid in elucidating the exact role of Cl− currents in ICC.
Non-selective cation channels
Sanders and colleagues proposed that a Ca2+-inhibited NSC conductance generated unitary potentials (Sanders et al., 2006), but the discovery of ANO1 in particular raised questions about this NSCC Hypothesis (Hwang et al., 2009). Most electrophysiological recordings of NSC currents were made using cultured ICC (Koh et al., 1998, 2002; Torihashi et al., 2002), but phenotypic changes in ICC in culture may include changes in the expression of pacemaker conductances (Sanders et al., 2006; Zhu et al., 2009). Nevertheless, NSCC may still play an important role in ICC pacemaking.
Takeda et al. (2008) identified two different NSC currents in freshly dispersed ICC from mouse gastric antrum. A basally active, Ca2+-inhibited current was found in cells that resembled ICC-MY, while a Ca2+-facilitated conductance that was responsible for generating noisy spontaneous transient inward currents was found in cells that resembled ICC-IM. The Ca2+-inhibited current, putatively observed in ICC-MY, was a large basal inward current that decreased in magnitude when [Ca2+]i was increased secondary to depolarization, and this was shown to be mediated by calmodulin. The current was identified as a NSC current because it had a reversal potential near 0 mV and was inhibited by removing extracellular Na+ (Takeda et al., 2008).
The cells believed to be ICC-IM basally generated spontaneous transient inward currents, which appeared to summate during depolarization-induced increases in [Ca2+]i to produce the larger Ca2+-facilitated current in a calmodulin-dependent manner. The Ca2+-facilitated current was also identified as a NSC current and was inhibited by Cl− channel blockers (Takeda et al., 2008).
Ca2+-activated NSC currents have also been found in freshly dispersed ICC from murine small intestine (Goto et al., 2004). Goto et al. (2004) called this current an “autonomous inward current” because it was activated upon depolarization of the cell, and then followed a time course that was largely independent of the subsequent voltage steps applied. Goto et al. (2004) speculated that the autonomous inward NSC current was activated by increases in [Ca2+]i secondary to membrane depolarization, but did not provide direct evidence of Ca2+-dependence.
There were similarities between the autonomous inward current (Goto et al., 2004) and the Ca2+-facilitated current (Takeda et al., 2008). Both were activated by depolarization, which was believed to be due to depolarization-induced increases in [Ca2+]i. In addition, both the autonomous inward current and the Ca2+-facilitated current had linear conductance and demonstrated large tail currents on repolarization. Therefore, it is possible that these two currents were generated by the same type of NSCC.
The channels responsible for NSC currents have not been identified in ICC. A candidate for NSCC is the family of TRP homologs. At least four types of canonical TRP (TRPC) and two types of melastatin TRP (TRPM) are expressed in ICC from mouse small intestine and stomach (Epperson et al., 2000; Liu et al., 2005; Chen et al., 2007). TRPC4 (Torihashi et al., 2002; Walker et al., 2002) and TRPM7 (Kim et al., 2005, 2009) have both been suggested as generating a pacemaker current.
Sanders and colleagues proposed that NSC currents may still produce unitary potentials, because they found that Cl− channel blockers did not inhibit unitary potentials (Hwang et al., 2009; Zhu et al., 2009). However, earlier studies found that similar concentrations of Cl− channel blockers also inhibit NSC currents in ICC (Koh et al., 2002; Walker et al., 2002; Takeda et al., 2008), suggesting that NSC conductances may not generate unitary potentials. In addition, there is also previous evidence that Cl− channel blockers do inhibit unitary potentials (Hirst et al., 2002a; Suzuki et al., 2003). These competing studies and uncertainties are yet to be resolved.
Potassium channels
A vast range of K+ channels are expressed in excitable cells, and many types have been found in ICC, including Ca2+-activated K+ channels, inward-rectifier K+ channels, and a delayed-rectifier K+ channel. K+ channels generate outward current at physiological potentials, and can contribute to slow wave repolarization, balancing inward current during the plateau phase, or maintaining the RMP to control ICC excitability.
Small (SK), intermediate (IK), and large (BK) conductance Ca2+-activated K+ channels have been found in ICC. BK channel proteins (Cho and Daniel, 2005; Zhu and Huizinga, 2008) and RNA (Chen et al., 2007) have been identified in ICC from murine small intestine, and a large conductance (218 pS) Ca2+-activated K+ current was recorded from canine colonic ICC (Langton et al., 1989). Zhu and Huizinga (2008) proposed that the primary role of BK channels may be to regulate ICC excitability, particularly during the plateau phase, in response to neurotransmitters and other chemical stimuli because nitrergic innervation appears to inhibit ICC by activating BK channels.
An IK current with 38 pS single channel conductance was identified in murine jejunal ICC in situ and in culture (Zhu et al., 2007). This study suggested that IK may regulate ICC excitability, because blocking the current depolarized RMP and increased slow wave duration. The current was enhanced by nitric oxide, so it may also play a role in mediating nitrergic innervation (Zhu et al., 2007). An SK channel, SK3, is expressed in ICC-MY and ICC-IM in the stomach, small intestine and colon of rats (Fujita et al., 2001).
KV1.1 channels generate a delayed-rectifier K+ current. KV1.1 is expressed in ICC from dog, guinea-pig, and mouse, and a KV1.1 blocker, dendrotoxin-K, blocked part of a delayed-rectifier outward current in ICC cultured from mouse gastric fundus (Hatton et al., 2001). KV1.1 channels may contribute to repolarization and ICC excitability (Huizinga et al., 2004).
Ether-a-go-go related gene (ERG) K+ channels are found in ICC. ERG1 was abundantly expressed in murine small intestine ICC-MY (Chen et al., 2007), while ERG3 has been found in the same cell type in culture (White et al., 2008). ERG current in cultured murine small intestine ICC was found to activate on depolarization then rapidly inactivate to a smaller sustained current, and to reactivate upon repolarization (McKay et al., 2006). ERG currents act as inward rectifiers on hyperpolarization from potentials positive to RMP (Zhu et al., 2003). ERG currents contribute to the slow wave plateau, because the ERG window current is maximal around −30 mV (McKay et al., 2006), and blocking ERG with E4031 leads to increased slow wave duration (Zhu et al., 2003; McKay et al., 2006; White et al., 2008). ERG current also regulates RMP because E4031 depolarizes RMP (Zhu et al., 2003; White et al., 2008).
Another inward-rectifier K+ current thought to be present in ICC is an ATP-sensitive K+ current. This current is activated by phentolamine (Ahn et al., 2010), pinacidil (Choi et al., 2006), and hydrogen peroxide (Choi et al., 2010). These drugs have been shown to inhibit pacemaker currents, increase outward currents, cause hyperpolarization, and decrease slow wave frequency in ICC from mice and guinea-pigs (Kito et al., 2002b, 2005; Choi et al., 2006, 2010; Ahn et al., 2010). These actions were blocked by glibenclamide, which inhibits ATP-sensitive K+ currents (Ahn et al., 2010; Choi et al., 2010), indicating that ATP-sensitive K+ channels are present in the cell membrane. However, the frequency and amplitude of unitary potentials was increased by pinacidil (Kito et al., 2002b), and glibenclamide blocked internal Ca2+ cycling (Fukuta et al., 2002), suggesting an important role for ATP-sensitive K+ channels in the mitochondrial membrane.
KCNK3, which encodes an outwardly rectifying background K+ channel, is abundantly expressed in ICC-MY (Chen et al., 2007), and probably contributes an outward leak current to maintain RMP (Goldstein et al., 2005).
Finally, Parsons and Huizinga (2010) recently identified a transient outward K+ current that activated immediately on depolarization from RMP in ICC cultured from murine small intestine and proposed that it may play a role in regulating the slow wave upstroke phase.
An Updated Theory of ICC Pacemaker Mechanisms
For reasons detailed above, serious reservations now exist regarding the previously dominant NSCC Hypothesis for ICC pacemaking, although the intracellular pacemaking processes remain largely unchanged. There are three major contentions for resolution: (i) What are the specific membrane conductances involved in ICC pacemaking?; (ii) Are pacemaker conductances activated by the rising or falling phase of the internal Ca2+ oscillations?; and (iii) How do conductances other than the pacemaker conductance contribute to slow wave production and propagation?
Based on the evidence outlined above, slow waves are generated by the following process (summarized in Figure 3): the pacemaker cycle is initiated by stochastic Ca2+ release into the PMU cytoplasm via IP3 receptors on the ER (Suzuki et al., 2000; Ward et al., 2000; Hirst and Edwards, 2001), resulting in a localized [Ca2+]i increase that activates a transient inward pacemaker current, producing a unitary potential (Edwards et al., 1999). Ca2+ influx through the basally active Ca2+-inhibited NSC current described by Takeda et al. (2008) may enhance IP3R open probability in ICC-MY.
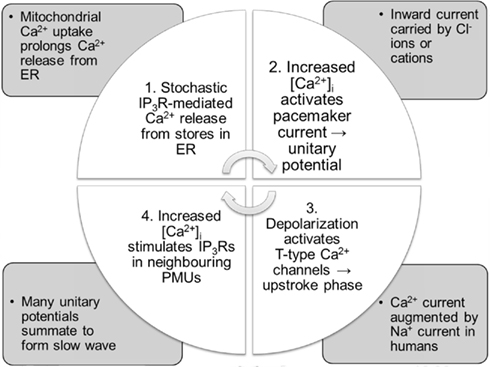
Figure 3. Summary of the updated pacemaker cycle. The mechanisms involved are described in more detail in the text.
Increased [Ca2+]i could stimulate mitochondrial Ca2+ uptake, but the role of mitochondria is no longer as clear as it was in the NSCC Hypothesis. In cardiac myocytes, the main role for mitochondria is ATP synthesis to power contraction and SERCA pumps (Lukyanenko et al., 2009), and mitochondrial Ca2+ uptake and extrusion mechanisms were traditionally considered too slow to contribute to pacemaker activity (Griffiths et al., 2010). However, interactions between Ca2+ and mitochondria are important for regulating ATP supply and modulating cellular Ca2+ signaling, and more recent evidence suggests that mitochondria proximal to Ca2+ stores can also contribute directly to [Ca2+]i transients during excitation–contraction coupling (Griffiths et al., 2010). Mitochondria may similarly have multiple important roles in ICC. Mitochondria can indirectly contribute to Ca2+ cycling by generating ATP to power the SERCA pump, and by contributing to Ca2+ homeostasis through slow mitochondrial Ca2+ uptake and release processes. At the same time, faster mitochondrial Ca2+ cycling may have a direct effect on [Ca2+]i transients during the pacemaker cycle, as previously suggested. In SMC, mitochondrial Ca2+ uptake helps to reduce the inhibitory effect of high [Ca2+]i (>500 nM) on IP3Rs (Chalmers and McCarron, 2009; Olson et al., 2010), and it may have a similar effect in ICC, prolonging ER Ca2+ release and the duration of the pacemaker current, although further definitive research on the role of mitochondria in ICC Ca2+ cycling is still required.
SERCA pumps (Ward et al., 2000) and mitochondrial Na+/Ca2+ exchangers (Kim et al., 2006) are important for slow wave activity. The intracellular calcium cycle is thought to be completed by Ca2+ efflux from the mitochondria via the Na+/Ca2+ exchanger and calcium uptake into the ER by the SERCA pump. TRP channels acting as Ca2+-inhibited NSCC may also play a role in ER replenishment, thus helping to set the pace of slow wave generation (Means and Sneyd, 2010).
As detailed above, both Ca2+-activated Cl− currents (Zhu et al., 2009) and NSC currents (Takeda et al., 2008) have been implicated as the pacemaker current in ICC (Hirst et al., 2002a; Koh et al., 2002). Modeling studies have clarified that the pacemaker current is most likely Ca2+-activated, not Ca2+-inhibited (Means and Sneyd, 2010). Unfortunately, pharmacological studies cannot differentiate between Cl− and NSC currents (Gögelein et al., 1990; White and Aylwin, 1990), so there is no evidence that conclusively shows either one is exclusively responsible for generating unitary potentials. It is possible that NSCCs and ANO1 Cl− channels both contribute as pacemaker currents. Wang et al. (2008) observed that the reversal potential of spontaneous inward currents in mouse and rat ICC pointed to a role for both Cl− channels and NSCC in pacemaking, and Kito and Suzuki (2003) suggested that the initial and plateau components of slow waves may be produced by two different populations of PMUs. It is likely that ANO1 and NSCC are activated by different levels of Ca2+, so one may initiate slow waves while the other may contribute only to unitary potentials that shape the plateau phase. Having two ion channels capable of generating unitary potentials may be a built-in redundancy that enables ICC to generate slow waves under a wide variety of conditions.
ANO1 current was found to activate at −72 mV in murine intestinal ICC (Zhu et al., 2009), and thus is likely to contribute as a pacemaker current at RMP. Spontaneous transient inward NSC currents were observed in ICC-IM but not ICC-MY from mouse stomach (Takeda et al., 2008). Therefore, the initial pacemaker current in ICC-MY may be a Ca2+-activated Cl− current carried by ANO1 channels, whereas unitary potentials in ICC-IM may be generated at RMP by both ANO1 and NSCC.
The small depolarization from the pacemaker current activates nearby T-type Ca2+ channels (Lee et al., 2007), resulting in a large influx of Ca2+ and a rapid depolarization forming the slow wave upstroke phase. Some T-type channels may be localized within PMUs (Sanders et al., 2006), and Ca2+ influx during the upstroke phase entrains previously quiescent PMUs by opening IP3Rs in the ER, thus triggering more unitary potentials. The summation of many unitary potentials maintains the slow wave plateau (Edwards et al., 1999). CaV3.2 T-type channels are more abundant in murine intestinal ICC-MY than ICC-DMP (Chen et al., 2007). If a similar disparity between T-type channel expression exists in guinea-pig gastric ICC, it may help explain why unitary potentials produced by ICC-IM are less likely than unitary potentials in ICC-MY to entrain to produce slow waves (Edwards et al., 1999; Kito et al., 2002b).
NaV1.5 channels are also likely to contribute to the upstroke depolarization in human ICC (Strege et al., 2003). Mibefradil slows the rise rate of slow waves (Kito et al., 2005), and inhibits both T-type Ca2+ and NaV1.5 currents (Strege et al., 2005). Both T-type Ca2+ channels (Perez-Reyes, 2003) and NaV1.5 channels (Strege et al., 2003) activate around −60 to −50 mV, peak around −30 to −20 mV, and are fast-inactivating, making them prime candidates for producing the upstroke phase.
A lower concentration of Ca2+ is required to activate ANO1 current at more positive potentials (Yang et al., 2008). This finding suggests that depolarization-dependent entrainment may be in part due to an increase in the Ca2+ sensitivity of ANO1 channels.
Depolarizing unitary potentials during the plateau phase are generated by NSCC. Cl− channels generate outward, repolarizing currents at membrane potentials positive to −50 mV (Zhu et al., 2010b). It is likely that ANO1 pacemaker currents do activate during the plateau phase, and the balance of inward NSC currents and outward Cl− currents may help to maintain the plateau potential. ERG K+ channels (McKay et al., 2006), BK channels and other Ca2+-activated K+ channels (Zhu and Huizinga, 2008) may also contribute an outward current during the plateau phase.
Slow wave repolarization occurs by inactivation of inward currents and activation of outward K+ currents toward the end of the plateau phase. Delayed-rectifier KV1.1 channels in particular probably contribute to repolarization (Huizinga et al., 2004).
RMP in ICC is typically close to the equilibrium potential for K+ ions because of background K+ channels (Goldstein et al., 2005; Chen et al., 2007). Inward-rectifier ERG K+ currents (Zhu et al., 2003) and high-conductance Cl− currents (Zhu et al., 2005) may also contribute to maintaining RMP.
The pacemaker mechanism described here is based on fairly consistent experimental evidence across different types of ICC, and the ion channels thought to contribute to each phase of the slow wave are summarized in Figure 4. However, tissue-specific NaV1.5 expression in the GI tract (Strege et al., 2007) suggests that the slow wave mechanisms from different species, organs, and layers of the GI tract may utilize different subsets of ion channels for slow wave generation and propagation. Additionally, more experimental work is needed to confirm or refute this proposed pacemaker mechanism.
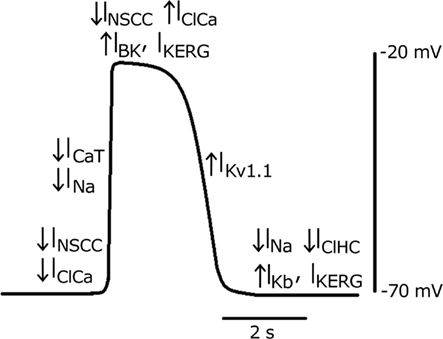
Figure 4. A theoretical slow wave trace from human small intestine, with the ion currents contributing to the initial unitary potential, upstroke phase, plateau phase, repolarization, and resting potential. A downward arrow indicates an inward current, while an upward arrow indicates an outward current. INSCC: Ca2+-activated NSC current, IClCa: Ca2+-activated Cl− current, INa: NaV1.5 current, ICaT: T-type Ca2+ current, IBK: Ca2+-activated K+ current, IKERG: ERG K+ current, IKv1.1: delayed-rectifier K+ current, IKb: background K+ current, IClHC: high-conductance Cl− current.
Existing Mathematical ICC Models
As experimental data on ICC electrophysiology continues to accumulate, it is increasingly important to coherently integrate this knowledge in order to form, test and extend hypotheses on whole-cell function. Biophysically based models provide an ideal platform to achieve this integration. Cell models can be employed in virtual studies, whereby appropriate parameter values are adjusted to simulate experimental conditions. Such in silico studies offer broad potential, for example in evaluating physical variables not easily assessed experimentally, quantitatively relating structure with function, and potentially reducing experimental animal usage (e.g., Du et al., 2010a,2010c).
Mathematical cell models can be classified roughly into two categories: phenomenological and biophysical. Phenomenological slow wave models generally include a system of ordinary differential equations that produce oscillatory patterns to match the periodicity of slow waves in different regions of the GI tract. Phenomenological models had a significant impact on early theories of slow wave propagation, but they have now been superseded by more sophisticated modeling methods.
In 1968, Nelsen and Becker published one of the earliest simulations of slow wave activity of the small intestine, consisting of a series of coupled “relaxation oscillators.” This model applied a generalized version of van der Pol’s equation, transformed into a system of two first order differential equations with a stimulus term added to one equation (Nelsen and Becker, 1968). The morphology and frequency of the simulated slow waves could be controlled by adjusting the parameter values to match experimental data. Sarna et al. (1971) expanded on the concept of coupled oscillators adopted by Nelsen and Becker (1968), and demonstrated entrainment of slow waves to an “intact frequency” in a linear network of coupled oscillators by incorporating forward, backward, and phase-shifted backward couplings. However, the applications of relaxation oscillator slow wave models were ultimately limited by their absence of intracellular details. Consequently, Publicover and Sanders (1989) discussed the inability of the relaxation oscillator based models to represent the effects of pharmacological agents on slow wave activity, and the mismatches between the morphologies of simulated slow wave activity and intracellular slow wave recordings.
Responding to the need for a class of more physiologically realistic cell model, an attempt was made by Aliev et al. (2000) to add more meaningful parameters to the oscillator models. This model was applied in multiscale modeling (reviewed in Du et al., 2010b), but it was evident that phenomenological cell models had reached their limit in the face of new discoveries about the electrophysiology of ICC and SMC.
Biophysically Based ICC Models
Biophysical cell models are based on the mathematical approach developed by Hodgkin and Huxley (1952), allowing individual ion currents to be quantitatively evaluated under effects of parameters with meaningful physical quantities, such as temperature, ion concentration, and voltage. For more detail on the Hodgkin–Huxley approach, see the review by Du et al. (2010b). A primary focus of biophysical ICC models is to quantitatively understand the signaling pathways that give rise to slow wave activity. The development of biophysical cell models of ICC is at relatively nascent stage, with the first full biophysical ICC model created in 2006 (Youm et al., 2006), and two further biophysical ICC models (Corrias and Buist, 2008; Faville et al., 2009) published more recently. The following section details each of these models and discusses their respective merits, as well as potential areas for improvement.
The ICC model developed by Youm et al.
The ICC model by Youm et al. (2006), partially based on cardiac cell models, was the first biophysical cell model to include a detailed description of the ion conductances and intracellular Ca2+ transients that were thought to contribute to slow wave activity. Figure 5A shows slow wave activity simulated using the Youm et al. (2006) model. The model included four ion conductances: an inward-rectifier K+ current (IK1), the L-type Ca2+ current (ICaL), a voltage-dependent DHP-resistant current (IVDDR), and a Ca2+-activated autonomous inward current (IAI) carried by K+, Ca2+, and Na+ ions; and three ion transporters: a Na+/K+ pump (INaK), a plasmalemmal Ca2+ pump (IPMCA), and a Na+/Ca2+ exchanger (INaCa). The time-dependent membrane potential is described by the following expression,
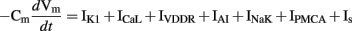
where Cm denotes the membrane capacitance of an ICC isolated from mouse small intestine (25 pF; Koh et al., 2002; Youm et al., 2006), and Is is a stimulus current.
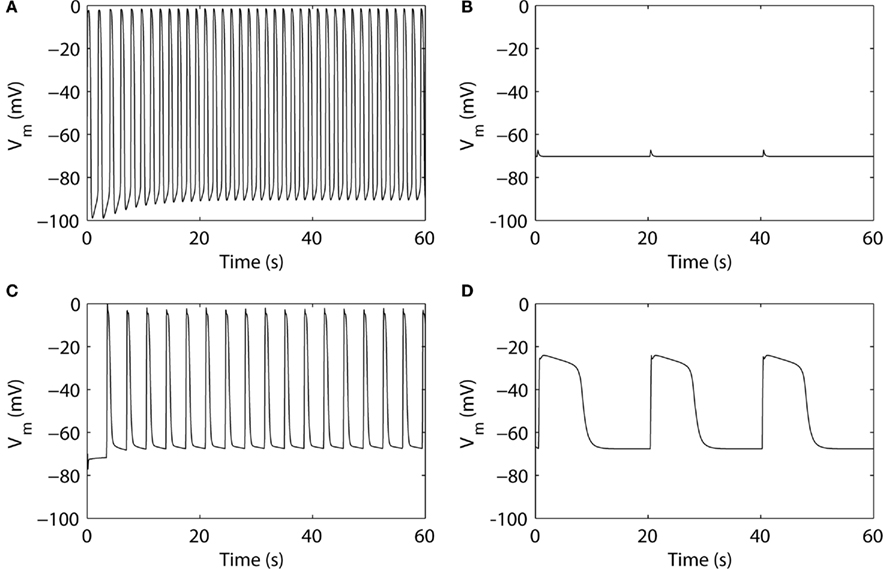
Figure 5. Simulated slow wave activities. (A) The ICC model by Youm et al. (2006). (B) The pacemaker unit model by Faville et al. (2008) at gastric slow wave frequency. (C) The bulk cytoplasm ICC model by Faville et al. (2009) at intestinal slow wave frequency. (D) The ICC model by Corrias and Buist (2008).
The intracellular calcium transient in the ICC model by Youm et al. (2006) is governed by four conductances: an ER (called sarcoplasmic reticulum, SR, in the original literature) uptake current (Iup), IP3-mediated Ca2+ release from the ER (IIP3R), a diffusive Ca2+ leak current from the ER (Ileak), and the net sum of ICaL, IAI, IVDDR, IPMCA, and INaCa. The metabolism of IP3 was governed by a three-state model of PIP2, IP3, and IP4 production and degradation, with voltage- and Ca2+-dependent rate constants. The synthesis of IP3 controls the conductance of IIP3R. The ER was modeled with separate sites for calcium uptake from and release to the cytoplasm. ER calcium uptake is governed by three currents: Iup, Ileak, and a transfer current between the uptake and release sites on the ER (Itr). Calcium release at the ER release site is governed by Itr and IIP3R. The time-dependent intracellular calcium transient is described by the following expression,
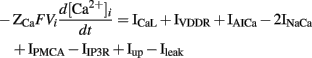
where ZCa denotes valence of Ca2+, F denotes Faraday’s constant (96.4867 C mmol-1), Vi denotes the cell volume (712.5 μm3), and IAICa is the Ca2+ component of IAI (Youm et al., 2006).
The ICC model developed by Corrias and Buist
The ICC model by Corrias and Buist (2008) was principally based on the NSCC Hypothesis. The model ICC contained a single PMU with ER, mitochondria, and a small cytosolic subspace, representing the aggregate of all the PMUs in an ICC. The PMU contained a Ca2+-inhibited NSC current, INSCC. Because the PMUs were represented by a single bulk PMU, the simulated slow wave was not generated by unitary potential summation, but by current flow through many ion conductances.
The model included nine types of ion conductances in the bulk cytoplasm: the delayed-rectifier KV1.1 current (IKv1.1), the ether-a-go-go K+ current (IK(ERG)), a Ca2+-activated K+ conductance (IBK), a background K+ leak current (IK(B), termed IKb in the original literature), ICaL (termed IL-type in the original literature), IVDDR, the voltage-dependent Na+ current (INa), a Ca2+-dependent chloride channel (IClCa), and a Ca2+ extrusion mechanism (JCaEXT). Figure 5D shows slow wave activity simulated using the Corrias and Buist ICC model. The time-dependent membrane potential is described by the following expression,
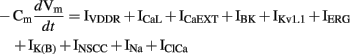
Corrias and Buist (2008) adapted an extensive description of intracellular Ca2+ dynamics from Fall and Keizer (2001) to represent Ca2+ handling in the PMU. Ca2+ flux between the PMU cytosolic subspace and the greater cytoplasm is governed by a passive diffusive current (Jleak). The Ca2+ handling dynamics of the mitochondria and ER induce Ca2+ oscillations in the cytosolic subspace, from which Ca2+ diffuses into the bulk cytoplasm through Jleak. This leads to a global increase in [Ca2+]i, which in turn activates the Ca2+-dependent ion conductances in the cell model: ICaL, IBK, and IClCa. Conversely, INSCC is activated by the falling phase of the Ca2+ oscillations within the PMU. The time-dependent intracellular calcium transient is described by the following expression,
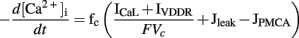
where fc denotes the cytosolic free Ca2+ proportion (set to 0.01), F denotes Faraday’s constant, and Vc denotes the cytosolic volume (7 × 10−7 mm3).
Entrainment of ICC slow wave activity has been modeled in two independent studies using the Corrias and Buist ICC model. Du et al. (2010c) adapted an existing voltage-dependent IP3 pathway from Imtiaz et al. (2002) and incorporated it into the Corrias and Buist (2008) ICC model. Voltage-dependent IP3 synthesis enables intracellular Ca2+ release in response to changes in the membrane potential, such as the depolarization of another ICC, thus entraining the slow wave activity of one model ICC to other surrounding ICC. The voltage-dependent IP3 mechanism is described by the following equation,
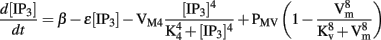
where parameter β denotes IP3 production in response to a chemical stimulus agent (such as acetylcholine), ε is the rate constant for linear IP3 degradation, VM4 is the rate constant for non-linear IP3 degradation, PMV denotes the maximal rate of voltage-dependent IP3 synthesis, and K4 and Kv are the half-saturation constants for non-linear IP3 degradation and voltage-dependent IP3 synthesis, respectively.
The other entrainment modeling study was conducted by Buist et al. (2010), who added an additional IVDDR (denoted IVDDRPU) into the PMU, along with a Ca2+ extrusion current (ICaEXTPU) to maintain homeostasis. The expression for IVDDRPU is as follows,
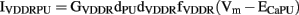
where GVDDRdPU denotes the maximum conductance of IVDDRPU, dVDDR, and fVDDR represent the activation gate and inactivation gate, respectively, and ECaPU is the Nernst potential of Ca2+ in the PMU. IVDDRPU added a voltage-dependent pathway for modulating [Ca2+]i in the PMU to the original Corrias and Buist (2008) ICC model, enabling entrainment of the pacemaker mechanism.
The ICC model developed by Faville et al.
The ICC model by Faville et al. includes two major components; a PMU model (Faville et al., 2008) and a bulk cytoplasm cell model (Faville et al., 2009). Like the Corrias and Buist ICC model, the Faville model was formulated based on the NSCC Hypothesis, but it is the only biophysical model to incorporate multiple PMUs.
Faville et al. (2008) modeled the PMU using a compartmentalized approach. Figure 5B shows a unitary potential simulated using the Faville et al. (2008) PMU model. The PMU intracellular space was further divided into four subspaces: the ER, the mitochondria, the main cytoplasmic subspace (S1), and a smaller cytoplasmic subspace between the ER and mitochondria (S2).
The membrane conductance in the PMU model is governed by two ion conductances: an inward Ca2+ current (ICa) and a Ca2+-inhibited NSC conductance (INSCC); and two ion transporters: a plasma membrane Ca2+-ATPase (IPM) and an outward Na pump (INaP). Faville et al. (2008) adapted an IP3 model by Sneyd et al. (2000) to represent the Ca2+ release from the ER into the cytoplasm (JIPR). In addition, four more intracellular Ca2+ fluxes were also defined: a SERCA pump (JSERCA), a mitochondrial Ca2+ uniporter (JMCU), a mitochondrial Na+/Ca2+ exchanger (JNCX), and an inter-cytosolic subspace Ca2+ flux (JS1S2). These ion conductances, pumps, and fluxes govern the changes in [Ca2+] in each of the four PMU subspaces. Specifically, the Ca2+ transient in S1 is governed by: JS1S2, JNCX, ICa, JSERCA; the Ca2+ transient in S2 is governed by: JIPR, JS1S2, JMCU; the Ca2+ transient in the ER is governed by: JSERCA and JIPR; and the Ca2+ transient in the mitochondria is governed by JMCU and JNCX. The time-dependent unitary potential is described by the following expression,
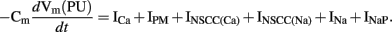
The full ICC model by Faville et al. (2009) contains multiple PMUs along with ion conductances in the bulk cytoplasm, including the DHP-resistant T-type Ca2+ current (ICa(T)), IK(ERG), IKv1.1, IK(B), a small non-selective inward leak current (IL), and a Ca2+ extrusion pump (ICa(Ext)). Using 10 PMUs, Faville et al. (2009) demonstrated intracellular entrainment of unitary potentials and simulated intestinal slow waves at 17.4 cpm (Figure 5C). The time-dependent membrane potential is described by the following expression,
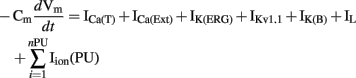
where Iion(PU) is the net current from one PMU, and nPU is the number of PMUs.
Comparison of the ICC models
Each of the biophysical ICC models outlined above are compartmental models, but differ in their approach to modeling the intracellular Ca2+ transient and pacemaker activity. The Corrias and Buist ICC model (Corrias and Buist, 2008) and the Faville ICC model (Faville et al., 2009) contained similar ion conductances, whereas the ICC model by Youm et al. (2006) included a set of different ion conductances, as detailed in Table 2. Youm et al. (2006) included a NSC current (IAI), but it was Ca2+-activated, unlike the Ca2+-inhibited INSCC used by Faville et al. (2009) and Corrias and Buist (2008). The major omissions from the Faville ICC model were the Ca2+-activated Cl− conductance (IClCa), which has since been identified to have a critical role in the generation of slow wave activity (Hwang et al., 2009; Zhu et al., 2009), and the voltage-dependent Na+ channel (INa(V)), which is now believed to play an important role in mechanotransduction (Beyder et al., 2010) and GI pathologies (Saito et al., 2009).
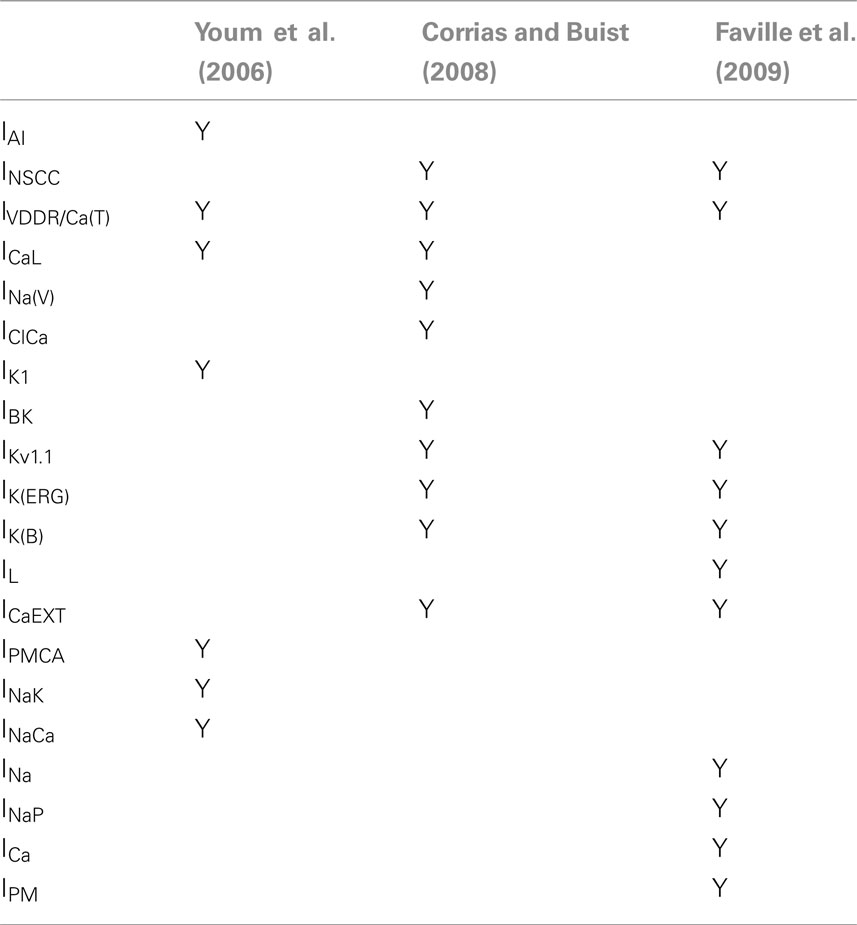
Table 2. A comparison of the different types of ion conductances and intracellular components in the three ICC models (Youm et al., 2006; Corrias and Buist, 2008; Faville et al., 2009).
Faville et al. (2008, 2009) implemented unitary potential summation using multiple PMUs, giving the model 41 subspaces in total (four per PMU plus one bulk cytoplasm), with a simple model of Ca2+ cycling between the ER and mitochondria. On the other hand, Corrias and Buist (2008) modeled bulk slow wave activity using a single PMU, including a complex model of mitochondrial function (Fall and Keizer, 2001), giving the ICC model four subspaces. It is interesting to note that Youm et al. (2006) did not consider the role of mitochondria in intracellular Ca2+ handling, so their model contained just the ER and the bulk cytoplasm.
A more comprehensive spatial PMU model specific to GI ICC has recently been published (Means and Sneyd, 2010). As outlined above, Means and Sneyd (2010) demonstrated that the NSCC Hypothesis, as adopted by Faville et al. (2008), theoretically does not enable PMUs to generate a [Ca2+]i profile capable of invoking unitary potentials. Even though the underlying basis of the PMU mechanism in the Faville ICC model is now in question (Hwang et al., 2009; Zhu et al., 2009; Means and Sneyd, 2010), the model nevertheless holds merit in its multiple intracellular compartments approach, and its focus on describing the unitary potentials that are thought to be responsible for generating whole-cell slow wave activity. Additionally, the two cytoplasmic subspaces within the PMU are consistent with evidence that mitochondrial uniporters are highly localized within IP3R clusters in guinea-pig colon SMC (Olson et al., 2010).
Both of the approaches applied to model the entrainment of slow wave activity using the Corrias and Buist ICC model aimed to induce a voltage-dependent component to the PMU Ca2+ transient (Buist et al., 2010; Du et al., 2010c). Du et al. (2010c) adopted the voltage-dependent [IP3] approach, which altered the probability of Ca2+ release at the IP3Rs in the presence of changes in the membrane potential. Buist et al. (2010) incorporated additional voltage-gated Ca2+ conductances to directly couple [Ca2+] in the PMU to the membrane potential. Even though the two approaches were different in their implementation, the common concept of perturbing Ca2+ concentration in the PMU to achieve entrainment is particularly noteworthy.
Computational efficiency of cell models is an important consideration, especially for large scale simulation of electrophysiology (Du et al., 2010b). A detailed comparison of the simulated slow wave outputs and computational efficiency in the three biophysical ICC models was conducted (Table 3). The ICC models by Faville et al. (2009) and Youm et al. (2006) both produced electrical activity in the frequency range of intestinal slow waves, whereas the ICC model by Corrias and Buist (2008) produced gastric slow wave activity with a more depolarized RMP and a lower amplitude. In terms of computational efficiency, the ICC model by Faville et al. (2009) took the longest time to solve when the same solution method was used to solve each model, which could be attributed to the large number of subspaces and parameters in the Faville ICC model.
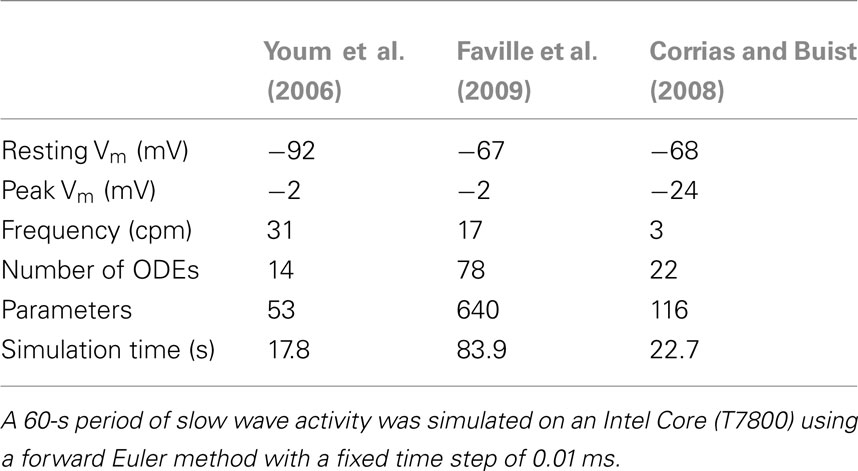
Table 3. Comparison of ICC models.
Future Directions
GI electrophysiological modeling is still in a relatively embryonic state, and the potential to further develop and apply GI models is vast. To realize the full potential of cell modeling, it will be essential to move beyond reproducing experimental results to predicting behaviors, in silico testing of the physiological likelihood of competing hypotheses, and ultimately informing new targeted experiments. In this section, discussion of future potential is focused on areas of current significance, or where modeling applications are currently being pursued.
Updating the Pacemaker Unit Model
An updated PMU model is needed, as the pacemaker conductances implemented by Corrias and Buist (2008) and Faville et al. (2009) were based on the NSCC Hypothesis. As discussed above, an impediment to progress is that it is not yet clear exactly which mechanisms and components play significant roles in generating slow waves in ICC. However, this knowledge gap presents the opportunity for modeling to inform which pacemaking theories are likely to be practicable, as was commendably shown by Means and Sneyd (2010). By creating an updated PMU model with both Cl− and NSC currents, it will be possible to test whether both or only one of these channels contributes to pacemaker activity in ICC, and this work is currently being undertaken.
The Ca2+-inhibited NSCC used in existing model PMUs should be replaced with Ca2+-activated Cl− channels and Ca2+-activated NSCC, either within the same or separate PMUs. In addition, a voltage-dependent T-type Ca2+ channel should be incorporated into the PMU to allow voltage-dependent entrainment of the pacemaker process, as implemented by Buist et al. (2010).
The cycling of Ca2+ between ER and mitochondria that was implemented in the existing models (Corrias and Buist, 2008; Faville et al., 2009) can be retained, pending further investigation into the role of mitochondria in Ca2+ cycling.
Subcellular Modeling Applications: the NaV1.5 Channel
The bulk cytoplasm ion conductances implemented by Corrias and Buist (2008) and Faville et al. (2009) are generally consistent with our current experimental understanding of ICC conductances. A key exception is the mechanosensitive NaV1.5 current that is found in human intestinal ICC (Strege et al., 2003). A voltage-gated Na+ current was incorporated into the gastric Corrias and Buist (2008) model based on the study by Strege et al. (2003), although NaV1.5 has not yet been described in gastric ICC, but Na+ current was excluded from the Faville ICC model. More information on the electrophysiology of NaV1.5 in the GI tract has recently been published, enabling model improvements (Saito et al., 2009; Beyder et al., 2010).
The NaV1.5 channel is of clinical interest, because of developing research indicating a role for channelopathies related to SCN5A mutations in the pathophysiology of irritable bowel syndrome (Saito et al., 2009). In the heart, SCN5A mutations sometimes cause lethal cardiac arrhythmias, such as type 3 long-QT syndrome or Brugada syndrome, the pathophysiology of which has been clarified by modeling (Clancy and Rudy, 1999). In a survey of patients with long-QT syndrome and their families, 50% of those with an SCN5A mutation had abdominal pain, and 65% had a GI symptom complex (Locke et al., 2006). Investigation of 49 subjects who had been diagnosed with IBS found that one patient had a loss-of-function missense mutation in SCN5A. When this mutated gene was expressed in HEK 293 cells, there was a decrease in whole-cell Na+ current, delayed channel activation and decreased mechanosensitivity compared with control SCN5A transcripts (Saito et al., 2009). Recent data show that the voltage sensitivity of NaV1.5 is also significantly modulated by mechanical forces (Beyder et al., 2010).
ICC models provide a useful tool to investigate how changes in subcellular components affect slow wave generation, for example, to explore how the mechanosensitivity of NaV1.5 may modulate slow wave production (Beyder et al., 2010), or how NaV1.5 channelopathies may impair normal GI motility (Saito et al., 2009). Both L-type Ca2+ channels (Lyford et al., 2002) and NaV1.5 channels (Beyder et al., 2010) have demonstrated mechanosensitivity in GI smooth muscle. Given the difficulties in studying electromechanical coupling experimentally in intact GI tissues, in silico investigations provide the ideal platform for predicting the effect of mechanical modulation of voltage-dependent currents on slow wave physiology. As an example, the phenomenological inward leak current, IL, in the Faville et al. (2009) ICC model could be replaced with a simple steady-state kinetic model of INav1.5 using parameters from the electrophysiological data of Beyder et al. (2010), and the stretch-dependent changes in these parameters could then be applied to predict the effects on slow wave production.
Specificity and Diversity of ICC Models
The pacemaker mechanisms within all types of ICC are believed to be similar, but key differences do exist. The frequency of slow waves varies markedly in the different GI organs, and also differs between species. Gene expression is known to differ between ICC-MY and ICC-DMP (Chen et al., 2007), and across species, with a notable example being NaV1.5 channels (Strege et al., 2007). There are also differences in ion channel function between organs, as L-type Ca2+ channels appear to play an important role in slow wave generation in the colon (Yoneda et al., 2002), but not the stomach (Dickens et al., 1999).
The existing biophysical ICC models are specific to either gastric (Corrias and Buist, 2008) or intestinal (Youm et al., 2006; Faville et al., 2009) slow wave frequencies. As our understanding of ICC physiology grows, we may also need to develop specific mathematical models to study ICC from both human and animal models. Separate cell models to represent the pacemaking properties of ICC-MY, and the roles of the various intramuscular ICC in slow wave propagation and neuromediation, may also become valuable.
Multiscale Modeling
Biophysically based cell models are actively being employed to study slow wave behavior at the tissue, whole-organ, and body scales. These “multiscale models” seek to provide an integrative and quantified understanding of the role of slow wave activity in motility and dysmotility. Multiscale GI electrophysiological modeling was recently reviewed in Du et al. (2010b), and is only discussed in brief here.
Achieving reliable predictions of behaviors across multiple biophysical scales and structures is a complex task, requiring accurate representations of biochemical, biophysical, and anatomical details (Hunter and Borg, 2003). More experimental data regarding all of these elements is needed before GI multiscale modeling can reach its full potential, however many worthwhile applications are currently available.
One example is the use of multiscale modeling to investigate the complex interactions between subcellular components and organ-level function. For instance, a logical next step to follow on from modeling the NaV1.5-mediated stretch effects on a single ICC is to model how these effects translate to the tissue or organ scale, including in the presence of modeled channelopathies. In a recent study by Du et al. (2010c), slow wave propagation was simulated over anatomically realistic ICC structures digitized from ICC-MY network images from wild-type and 5HT2B-knockout (ICC-depleted) mouse models, in order to demonstrate physiological consequences of ICC loss. This work is now being expanded to look at how varying degrees of ICC loss affects entrainment. Another recent application has been to generate multiscale models of whole stomach slow wave function (Buist et al., 2010; Du et al., 2010a). One of these models, spanning physiological scales from the subcellular to whole body levels, demonstrated an improved theory of the electrophysiological basis of the human electrogastrogram (Du et al., 2010a).
Special consideration must be given to the design of biophysically based models employed in multiscale applications. The finer details of subcellular functions, which are critical for reliably modeling ICC pacemaker mechanisms, may be neither necessary nor useful for understanding or investigating functions at higher scales in the physiological hierarchy (Crampin et al., 2004). Model reduction and simplification is desirable to achieve computational efficiency in multiscale simulations, and it is partly for this reason that the Corrias and Buist ICC model has been preferred over the Faville model in multiscale applications to date, due to its lower complexity (Du et al., 2010a,2010c). As ICC models are updated, consideration must therefore be given to appropriate reductions for multiscale modeling applications.
In conclusion, this review has examined the current evidence regarding the cellular processes responsible for generating slow waves in ICC, and has proposed an update to the NSCC Hypothesis that takes recent advances into account. Specifically, it appears that the pacemaker current is carried by Ca2+-activated Cl− currents and Ca2+-activated NSC currents, but not Ca2+-inactivated NSC currents. Existing biophysically based ICC models need to be modified to bring them in line with current physiological knowledge. The updated models can then be applied to investigate in silico how GI motility is affected by changes in ICC function on all levels from subcellular components to organs.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ahn, S. W., Kim, S. H., Kim, J. H., Choi, S., Yeum, C. H., Wie, H. W., Sun, J. M., So, I., and Jun, J. Y. (2010). Phentolamine inhibits the pacemaker activity of mouse interstitial cells of Cajal by activating ATP-sensitive K+ channels. Arch. Pharm. Res. 33, 479–489.
Aliev, R. R., Richards, W., and Wikswo, J. P. (2000). A simple nonlinear model of electrical activity in the intestine. J. Theor. Biol. 204, 21–28.
Aoyama, M., Yamada, A., Wang, J., Ohya, S., Furuzono, S., Goto, T., Hotta, S., Ito, Y., Matsubara, T., Shimokata, K., Chen, S. R. W., Imaizumi, Y., and Nakayama, S. (2004). Requirement of ryanodine receptors for pacemaker Ca 2+ activity in ICC and HEK293 cells. J. Cell Sci. 117, 2813–2825.
Bayguinov, P. O., Hennig, G. W., and Smith, T. K. (2010). Ca2+ imaging of activity in ICC-MY during local mucosal reflexes and the colonic migrating motor complex in the murine large intestine. J. Physiol. 22, 4453–4474.
Beyder, A., Rae, J. L., Bernard, C. E., Strege, P. R., Sachs, F., and Farrugia, G. (2010). Mechanosensitivity of Nav1.5, a voltage-sensitive sodium channel. J. Physiol. 588, 4969–4985.
Bootman, M. D., Collins, T. J., Mackenzie, L., Roderick, H. L., Berridge, M. J., and Peppiatt, C. M. (2002). 2-Aminoethoxydiphenyl borate (2-APB) is a reliable blocker of store-operated Ca2+ entry but an inconsistent inhibitor of InsP3-induced Ca2+ release. FASEB J. 16, 1145–1150.
Buist, M. L., Corrias, A., and Poh, Y. C. (2010). A model of slow wave propagation and entrainment along the stomach. Ann. Biomed. Eng. 38, 3022–3030.
Caputo, A., Caci, E., Ferrera, L., Pedemonte, N., Barsanti, C., Sondo, E., Pfeffer, U., Ravazzolo, R., Zegarra-Moran, O., and Galietta, L. J. V. (2008). TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science 322, 590–594.
Chalmers, S., and McCarron, J. G. (2009). Inhibition of mitochondrial calcium uptake rather than efflux impedes calcium release by inositol-1,4,5-trisphosphate-sensitive receptors. Cell Calcium 46, 107–113.
Chang, I. Y., Glasgow, N. J., Takayama, I., Horiguchi, K., Sanders, K. M., and Ward, S. M. (2001). Loss of interstitial cells of Cajal and development of electrical dysfunction in murine small bowel obstruction. J. Physiol. 536, 555–568.
Chen, H., Ördög, T., Chen, J., Young, D. L., Bardsley, M. R., Redelman, D., Ward, S. M., and Sanders, K. M. (2007). Differential gene expression in functional classes of interstitial cells of Cajal in murine small intestine. Physiol. Genomics 31, 492–509.
Cho, W. J., and Daniel, E. E. (2005). Proteins of interstitial cells of Cajal and intestinal smooth muscle, colocalized with caveolin-1. Am. J. Physiol. Gastrointest. Liver Physiol. 288, G571–G585.
Choi, S., Park, C. G., Kim, M. Y., Lim, G. H., Kim, J. H., Yeum, C. H., Yoon, P. J., So, I., Kim, K. W., and Jun, J. Y. (2006). Action of imipramine on activated ATP-sensitive K+ channels in interstitial cells of Cajal from murine small intestine. Life Sci. 78, 2322–2328.
Choi, S., Yeum, C. H., Kim, Y. D., Park, C. G., Kim, M. Y., Park, J.-S., Jeong, H.-S., Kim, B. J., So, I., Kim, K. W., and Jun, J. Y. (2010). Receptor tyrosine and MAP kinase are involved in effects of H2O2 on interstitial cells of Cajal in murine intestine. J. Cell. Mol. Med. 14, 257–266.
Christensen, J., Schedl, H. P., and Clifton, J. A. (1966). The small intestinal basic electrical rhythm (slow wave) frequency gradient in normal men and in patients with variety of diseases. Gastroenterology 50, 309–315.
Clancy, C. E., and Rudy, Y. (1999). Linking a genetic defect to its cellular phenotype in a cardiac arrhythmia. Nature 400, 566–569.
Code, C. F., and Szurszewski, J. H. (1970). The effect of duodenal and mid small bowel transection on the frequency gradient of the pacesetter potential in the canine small intestine. J. Physiol. 207, 281–289.
Corrias, A., and Buist, M. L. (2008). Quantitative cellular description of gastric slow wave activity. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G989–G995.
Cousins, H. M., Edwards, F. R., Hickey, H., Hill, C. E., and Hirst, G. D. S. (2003). Electrical coupling between the myenteric interstitial cells of Cajal and adjacent muscle layers in the guinea-pig gastric antrum. J. Physiol. 550, 829–844.
Crampin, E. J., Smith, N. P., and Hunter, P. J. (2004). Multi-scale modelling and the IUPS physiome project. J. Mol. Histol. 35, 707–714.
Der, T., Bercik, P., Donnelly, G., Jackson, T., Berezin, I., Collins, S. M., and Huizinga, J. D. (2000). Interstitial Cells of Cajal and Inflammation- Induced Motor Dysfunction in the Mouse Small Intestine. Gastroenterology 119, 1590–1599.
Dick, G. M., Kong, I. D., and Sanders, K. M. (1999). Effects of anion channel antagonists in canine colonic myocytes: comparative pharmacology of Cl−, Ca2+ and K+ currents. Br. J. Pharmacol. 127, 1819–1831.
Dickens, E. J., Edwards, F. R., and Hirst, G. D. S. (2001). Selective knockout of intramuscular interstitial cells reveals their role in the generation of slow waves in mouse stomach. J. Physiol. 531, 827–833.
Dickens, E. J., Hirst, G. D. S., and Tomita, T. (1999). Identification of rhythmically active cells in guinea-pig stomach. J. Physiol. 514, 515–531.
Doughty, J. M., Miller, A. L., and Langton, P. D. (1998). Non-specificity of chloride channel blockers in rat cerebral arteries: block of the L-type calcium channel. J. Physiol. 507, 433–439.
Du, P., O’Grady, G., Cheng, L. K., and Pullan, A. J. (2010a). A multiscale model of the electrophysiological basis of the human electrogastrogram. Biophys. J. 99, 2784–2792.
Du, P., O’Grady, G., Davidson, J. B., Cheng, L. K., and Pullan, A. J. (2010b). Multiscale modeling of gastrointestinal electrophysiology and experimental validation. Crit. Rev. Biomed. Eng. 38, 225–254.
Du, P., O’Grady, G., Gibbons, S. J., Yassi, R., Lees-Green, R., Farrugia, G., Cheng, L. K., and Pullan, A. J. (2010c). Tissue-specific mathematical models of slow wave entrainment in wild-type and 5-HT(2B) knockout mice with altered interstitial cells of Cajal networks. Biophys. J. 98, 1772–1781.
Dubois, J.-M., Ouanounou, G., and Rouzaire-Dubois, B. (2009). The Boltzmann equation in molecular biology. Prog. Biophys. Mol. Biol. 99, 87–93.
Edwards, F. R., Hirst, G. D. S., and Suzuki, H. (1999). Unitary nature of regenerative potentials recorded from circular smooth muscle of guinea-pig antrum. J. Physiol. 519, 235–250.
Epperson, A., Hatton, W. J., Callaghan, B., Doherty, P., Walker, R. L., Sanders, K. M., Ward, S. M., and Horowitz, B. (2000). Molecular markers expressed in cultured and freshly isolated interstitial cells of Cajal. Am. J. Physiol. Cell Physiol. 279, C529–C539.
Fall, C. P., and Keizer, J. E. (2001). Mitochondrial modulation of intracellular Ca(2+) signaling. J. Theor. Biol. 210, 151–165.
Farrugia, G. (2008). Interstitial cells of Cajal in health and disease. Neurogastroent. Motil. 20(Suppl. 1), 54–63.
Faville, R. A., Pullan, A. J., Sanders, K. M., Koh, S. D., Lloyd, C. M., and Smith, N. P. (2009). Biophysically based mathematical modeling of interstitial cells of Cajal slow wave activity generated from a discrete unitary potential basis. Biophys. J. 96, 4834–4852.
Faville, R. A., Pullan, A. J., Sanders, K. M., and Smith, N. P. (2008). A biophysically based mathematical model of unitary potential activity in interstitial cells of Cajal. Biophys. J. 95, 88–104.
Fujita, A., Takeuchi, T., Saitoh, N., Hanai, J., and Hata, F. (2001). Expression of Ca(2+)-activated K(+) channels, SK3, in the interstitial cells of Cajal in the gastrointestinal tract. Am. J. Physiol. Cell Physiol. 281, C1727–C1733.
Fukuta, H., Kito, Y., and Suzuki, H. (2002). Spontaneous electrical activity and associated changes in calcium concentration in guinea-pig gastric smooth muscle. J. Physiol. 540, 249–260.
Garrity, M. M., Gibbons, S. J., Smyrk, T. C., Vanderwinden, J. M., Gomez-Pinilla, P. J., Nehra, A., Borg, M., and Farrugia, G. (2009). Diagnostic challenges of motility disorders: optimal detection of CD117+ interstitial cells of Cajal. Histopathology 54, 286–294.
Gibbons, S. J., Strege, P. R., Sha, L., Roeder, J. L., Mazzone, A., Ou, Y., Rich, A., and Farrugia, G. (2009). The alpha1H Ca2+ channel subunit is expressed in mouse jejunal interstitial cells of Cajal and myocytes. J. Cell. Mol. Med. 13, 4422–4431.
Gögelein, H., Dahlem, D., Englert, H. C., and Lang, H. J. (1990). Flufenamic acid, mefenamic acid and niflumic acid inhibit single nonselective cation channels in the rat exocrine pancreas. FEBS Lett. 268, 79–82.
Goldstein, S. A. N., Bayliss, D. A., Kim, D., Lesage, F., and Plant, L. D. (2005). International union of pharmacology. LV. Nomenclature and molecular relationships of two-P potassium channels. Pharmacol. Rev. 57, 527–540.
Gomez-Pinilla, P. J., Gibbons, S. J., Bardsley, M. R., Lorincz, A., Pozo, M. J., Pasricha, P. J., Van de Rijn, M., West, R. B., Sarr, M. G., Kendrick, M. L., Cima, R. R., Dozois, E. J., Larson, D. W., Ördög, T., and Farrugia, G. (2009). Ano1 is a selective marker of interstitial cells of Cajal in the human and mouse gastrointestinal tract. Am. J. Physiol. Gastrointest. Liver Physiol. 296, G1370–G1381.
Gomez-Pinilla, P. J., Gibbons, S. J., Sarr, M. G., Kendrick, M. L., Robert Shen, K., Cima, R. R., Dozois, E. J., Larson, D. W., Ördög, T., Pozo, M. J., and Farrugia, G. (2011). Changes in interstitial cells of Cajal with age in the human stomach and colon. Neurogastroent. Motil. 23, 36–44.
Goto, K., Matsuoka, S., and Noma, A. (2004). Two types of spontaneous depolarizations in the interstitial cells freshly prepared from the murine small intestine. J. Physiol. 559, 411–422.
Griffiths, E. J., Balaska, D., and Cheng, W. H. Y. (2010). The ups and downs of mitochondrial calcium signalling in the heart. Biochim. Biophys. Acta 1797, 865–864.
Hanani, M., Farrugia, G., and Komuro, T. (2005). Intercellular coupling of interstitial cells of Cajal in the digestive tract. Int. Rev. Cytol. 242, 249–282.
Hatton, W. J., Mason, H. S., Carl, A., Doherty, P., Latten, M. J., Kenyon, J. L., Sanders, K. M., and Horowitz, B. (2001). Functional and molecular expression of a voltage-dependent K(+) channel (Kv1.1) in interstitial cells of Cajal. J. Physiol. 533, 315–327.
He, C.-L., Burgart, L., Wang, L., Pemberton, J., Young-Fadok, T., Szurszewski, J. H., and Farrugia, G. (2000). Decreased interstitial cell of Cajal volume in patients with slow-transit constipation. Gastroenterology 118, 14–21.
Hirst, G. D. S., Bramich, N. J., Teramoto, N., Suzuki, H., and Edwards, F. R. (2002a). Regenerative component of slow waves in the guinea-pig gastric antrum involves a delayed increase in [Ca2+]i and Cl− channels. J. Physiol. 540, 907–919.
Hirst, G. D. S., Dickens, E. J., and Edwards, F. R. (2002b). Pacemaker shift in the gastric antrum of guinea-pigs produced by excitatory vagal stimulation involves intramuscular interstitial cells. J. Physiol. 541, 917–928.
Hirst, G. D. S., and Edwards, F. R. (2001). Generation of slow waves in the antral region of guinea-pig stomach–a stochastic process. J. Physiol. 535, 165–180.
Hodgkin, A. L., and Huxley, A. F. (1952). A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 117, 500–544.
Horiguchi, K., Semple, G. S. A., Sanders, K. M., and Ward, S. M. (2001). Distribution of pacemaker function through the tunica muscularis of the canine gastric antrum. J. Physiol. 537, 237–250.
Hotta, A., Okada, N., and Suzuki, H. (2007). Mibefradil-sensitive component involved in the plateau potential in submucosal interstitial cells of the murine proximal colon. Biochem. Biophys. Res. Commun. 353, 170–176.
Huizinga, J. D., Golden, C. M., Zhu, Y., and White, E. J. (2004). Ion channels in interstitial cells of Cajal as targets for neurotransmitter action. Neurogastroenterol. Motil. 16(Suppl. 1), 106–111.
Huizinga, J. D., and Lammers, W. J. E. P. (2009). Gut peristalsis is governed by a multitude of cooperating mechanisms. Am. J. Physiol. Gastrointest. Liver Physiol. 296, G1–G8.
Huizinga, J. D., Zarate, N., and Farrugia, G. (2009). Physiology, injury, and recovery of interstitial cells of Cajal: basic and clinical science. Gastroenterology 137, 1548–1556.
Huizinga, J. D., Zhu, Y., Ye, J., and Molleman, A. (2002). High-conductance chloride channels generate pacemaker currents in interstitial cells of Cajal. Gastroenterology 123, 1627–1636.
Hunter, P. J., and Borg, T. K. (2003). Integration from proteins to organs: the Physiome Project. Nat. Rev. Mol. Cell Biol. 4, 237–243.
Hwang, S. J., Blair, P. J. A., Britton, F. C., O’Driscoll, K. E., Hennig, G. W., Bayguinov, Y. R., Rock, J. R., Harfe, B. D., Sanders, K. M., and Ward, S. M. (2009). Expression of anoctamin 1/TMEM16A by interstitial cells of Cajal is fundamental for slow wave activity in gastrointestinal muscles. J. Physiol. 587, 4887–4904.
Imtiaz, M. S., Smith, D. W., and van Helden, D. F. (2002). A theoretical model of slow wave regulation using voltage-dependent synthesis of inositol 1,4,5-trisphosphate. Biophys. J. 83, 1877–1890.
Isozaki, K., Hirota, S., Miyagawa, J., Taniguchi, M., Shinomura, Y., and Matsuzawa, Y. (1997). Deficiency of c-kit+ cells in patients with a myopathic form of chronic idiopathic intestinal pseudo-obstruction. Am. J. Gastroenterol. 92, 332–334.
Kashyap, P., and Farrugia, G. (2010). Diabetic gastroparesis: what we have learned and had to unlearn in the past 5 years. Gut 59, 1716–1726.
Kim, B. J., Jun, J. Y., So, I., and Kim, K. W. (2006). Involvement of mitochondrial Na+-Ca 2+ exchange in intestinal pacemaking activity. World J. Gastroenterol. 12, 796–799.
Kim, B. J., Lim, H.-H., Yang, D. K., Jun, J. Y., Chang, I. Y., Park, C.-S., So, I., Stanfield, P. R., and Kim, K. W. (2005). Melastatin-type transient receptor potential channel 7 is required for intestinal pacemaking activity. Gastroenterology 129, 1504–1517.
Kim, B. J., Park, K. J., Kim, H. W., Choi, S., Jun, J. Y., Chang, I. Y., Jeon, J.-H., So, I., and Kim, S. J. (2009). Identification of TRPM7 channels in human intestinal interstitial cells of Cajal. World J. Gastroenterol. 15, 5799–5804.
Kim, Y. C., Koh, S. D., and Sanders, K. M. (2002). Voltage-dependent inward currents of interstitial cells of Cajal from murine colon and small intestine. J. Physiol. 541, 797–810.
Kito, Y., Fukuta, H., and Suzuki, H. (2002a). Components of pacemaker potentials recorded from the guinea pig stomach antrum. Pflügers Arch. 445, 202–217.
Kito, Y., Suzuki, H., and Edwards, F. R. (2002b). Properties of unitary potentials recorded from myenteric interstitial cells of Cajal distributed in the guinea-pig gastric antrum. J. Smooth Muscle Res. 38, 165–179.
Kito, Y., and Suzuki, H. (2003). Properties of pacemaker potentials recorded from myenteric interstitial cells of Cajal distributed in the mouse small intestine. J. Physiol. 553, 803–818.
Kito, Y., Ward, S. M., and Sanders, K. M. (2005). Pacemaker potentials generated by interstitial cells of Cajal in the murine intestine. Am. J. Physiol. Cell Physiol. 288, C710–C720.
Knowles, C. H., De Giorgio, R., Kapur, R. P., Bruder, E., Farrugia, G., Geboes, K., Gershon, M. D., Hutson, J., Lindberg, G., Martin, J. E., Meier-Ruge, W. A., Milla, P. J., Smith, V. V., Vandervinden, J. M., Veress, B., and Wedel, T. (2009). Gastrointestinal neuromuscular pathology: guidelines for histological techniques and reporting on behalf of the Gastro 2009 International Working Group. Acta Neuropathol. 118, 271–301.
Koh, S. D., Jun, J. Y., Kim, T. W., and Sanders, K. M. (2002). A Ca2+-inhibited non-selective cation conductance contributes to pacemaker currents in mouse interstitial cell of Cajal. J. Physiol. 540, 803–814.
Koh, S. D., Sanders, K. M., and Ward, S. M. (1998). Spontaneous electrical rhythmicity in cultured interstitial cells of Cajal from the murine small intestine. J. Physiol. 513, 203–213.
Koh, S. D., Ward, S. M., Ördög, T., Sanders, K. M., and Horowitz, B. (2003). Conductances responsible for slow wave generation and propagation in interstitial cells of Cajal. Curr. Opin. Pharmacol. 3, 579–582.
Lammers, W. J. E. P., Donck, L. V., Schuurkes, J. A. J., and Stephen, B. (2005). Peripheral pacemakers and patterns of slow wave propagation in the canine small intestine in vivo. Can. J. Physiol. Pharmacol. 83, 1031–1043.
Langton, P., Ward, S. M., Carl, A., Norell, M. A., and Sanders, K. M. (1989). Spontaneous electrical activity of interstitial cells of Cajal isolated from canine proximal colon. Proc. Natl. Acad. Sci. U.S.A. 86, 7280–7284.
Lee, H.-T., Hennig, G. W., Fleming, N. W., Keef, K. D., Spencer, N. J., Ward, S. M, Sanders, K. M., and Smith, T. K. (2007). The mechanism and spread of pacemaker activity through myenteric interstitial cells of Cajal in human small intestine. Gastroenterology 132, 1852–1865.
Liu, H.-N., Ohya, S., Furuzono, S., Wang, J., Imaizumi, Y., and Nakayama, S. (2005). Co-contribution of IP3R and Ca2+ influx pathways to pacemaker Ca2+ activity in stomach ICC. J. Biol. Rhythms 20, 15–26.
Locke, G. R., Ackerman, M. J., Zinsmeister, A. R., Thapa, P., and Farrugia, G. (2006). Gastrointestinal symptoms in families of patients with an SCN5A-encoded cardiac channelopathy: evidence of an intestinal channelopathy. Am. J. Gastroenterol. 101, 1299–1304.
Lukyanenko, V., Chikando, A., and Lederer, W. J. (2009). Mitochondria in cardiomyocyte Ca2+ signaling. Int. J. Biochem. Cell Biol. 41, 1957–1971.
Lyford, G. L., Strege, P. R., Shepard, A., Ou, Y., Ermilov, L., Miller, S. M., Gibbons, S. J., Rae, J. L., Szurszewski, J. H., and Farrugia, G. (2002). alpha(1C) (Ca(V)1.2) L-type calcium channel mediates mechanosensitive calcium regulation. Am. J. Physiol. Cell Physiol. 283, C1001–C1008.
McKay, C. M., Ye, J., and Huizinga, J. D. (2006). Characterization of depolarization-evoked ERG K currents in interstitial cells of Cajal. Neurogastroenterol. Motil. 18, 324–333.
Means, S. A., and Sneyd, J. (2010). Spatio-temporal calcium dynamics in pacemaking units of the interstitial cells of Cajal. J. Theor. Biol. 267, 137–152.
Nelsen, T., and Becker, J. (1968). Simulation of the electrical and mechanical gradient of the small intestine. Am. J. Physiol. 214, 749–757.
O’Grady, G., Du, P., Cheng, L. K., Egbuji, J. U., Lammers, W. J. E. P., Windsor, J. A., and Pullan, A. J. (2010). Origin and propagation of human gastric slow-wave activity defined by high-resolution mapping. Am. J. Physiol. Gastrointest. Liver Physiol. 299, G585–G592.
Olson, M. L., Chalmers, S., and McCarron, J. G. (2010). Mitochondrial Ca2+ uptake increases Ca2+ release from inositol 1,4,5-trisphosphate receptor clusters in smooth muscle cells. J. Biol. Chem. 285, 2040–2050.
Ozaki, H., Stevens, R. J., Blondfield, D. P., Publicover, N. G., and Sanders, K. M. (1991). Simultaneous measurement of membrane potential, cytosolic Ca2+, and tension in intact smooth muscles. Am. J. Physiol. Cell Physiol. 260, C917–C925.
Parekh, A. B., and Putney, J. W. (2005). Store-operated calcium channels. Physiol. Rev. 85, 757–810.
Park, S. J., McKay, C. M., Zhu, Y., and Huizinga, J. D. (2005). Volume-activated chloride currents in interstitial cells of Cajal. Am. J. Physiol. Gastrointest. Liver Physiol. 289, G791–G797.
Parsons, S. P., and Huizinga, J. D. (2010). Transient outward potassium current in ICC. Am. J. Physiol. Gastrointest. Liver Physiol. 298, G456–G466.
Parsons, S. P., and Sanders, K. M. (2008). An outwardly rectifying and deactivating chloride channel expressed by interstitial cells of Cajal from the murine small intestine. J. Membr. Biol. 221, 123–132.
Perez-Reyes, E. (2003). Molecular physiology of low-voltage-activated T-type calcium channels. Physiol. Rev. 83, 117–161.
Piper, A. S., Greenwood, I. A., and Large, W. A. (2002). Dual effect of blocking agents on Ca2+-activated Cl− currents in rabbit pulmonary artery smooth muscle cells. J. Physiol. 539, 119–131.
Publicover, N. G., and Sanders, K. M. (1989). Are relaxation oscillators an appropriate model of gastrointestinal electrical activity? Am. J. Physiol. 256, G265–G274.
Ro, S., Park, C., Jin, J., Zheng, H., Blair, P. J. A., Redelman, D., Ward, S. M., Yan, W., and Sanders, K. M. (2010). A model to study the phenotypic changes of interstitial cells of Cajal in gastrointestinal diseases. Gastroenterology 138, 1068–1078.
Rumessen, J. J., Peters, S., and Thuneberg, L. (1993). Light- and electron microscopical studies of interstitial cells of Cajal and muscle cells at the submucosal border of human colon. Lab. Invest. 68, 481–495.
Saito, Y. A., Strege, P. R., Tester, D. J., Locke, G. R., Talley, N. J., Bernard, C. E., Rae, J. L., Makielski, J. C., Ackerman, M. J., and Farrugia, G. (2009). Sodium channel mutation in irritable bowel syndrome: evidence for an ion channelopathy. Am. J. Physiol. Gastrointest. Liver Physiol. 296, G211–G218.
Sakmann, B., and Neher, E. (eds). (1995). Single-Channel Recording, 2nd Edn. New York: Plenum Press.
Sanders, K. M., Koh, S. D., and Ward, S. M. (2006). Interstitial cells of Cajal as pacemakers in the gastrointestinal tract. Annu. Rev. Physiol. 68, 307–343.
Sarna, S. K., Daniel, E. E., and Kingma, Y. J. (1971). Simulation of small of slow-wave intestine electrical activity. Am. J. Physiol. 221, 166–175.
Schroeder, B. C., Cheng, T., Jan, Y. N., and Jan, L. Y. (2008). Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell 134, 1019–1029.
Sneyd, J., LeBeau, A., and Yule, D. (2000). Traveling waves of calcium in pancreatic acinar cells: model construction and bifurcation analysis. Physica D 145, 158–179.
Strege, P. R., Bernard, C. E., Ou, Y., Gibbons, S. J., and Farrugia, G. (2005). Effect of mibefradil on sodium and calcium currents. Am. J. Physiol. Gastrointest. Liver Physiol. 289, G249–G253.
Strege, P. R., Mazzone, A., Kraichely, R. E., Sha, L., Holm, A. N., Ou, Y., Lim, I., Gibbons, S. J., Sarr, M. G., and Farrugia, G. (2007). Species dependent expression of intestinal smooth muscle mechanosensitive sodium channels. Neurogastroenterol. Motil. 19, 135–143.
Strege, P. R., Ou, Y., Sha, L., Rich, A., Gibbons, S. J., Szurszewski, J. H., Sarr, M. G., and Farrugia, G. (2003). Sodium current in human intestinal interstitial cells of Cajal. Am. J. Physiol. Gastrointest. Liver Physiol. 285, G1111–G1121.
Suzuki, H., and Hirst, G. D. S. (1999). Regenerative potentials evoked in circular smooth muscle of the antral region of guinea-pig stomach. J. Physiol. 517, 563–573.
Suzuki, H., Takano, H., Yamamoto, Y., Komuro, T., Saito, M., Kato, K., and Mikoshiba, K. (2000). Properties of gastric smooth muscles obtained from mice which lack inositol trisphosphate receptor. J. Physiol. 525, 105–111.
Suzuki, H., Ward, S. M., Bayguinov, Y. R., Edwards, F. R., and Hirst, G. D. S. (2003). Involvement of intramuscular interstitial cells in nitrergic inhibition in the mouse gastric antrum. J. Physiol. 546, 751–763.
Takeda, Y., Koh, S. D., Sanders, K. M., and Ward, S. M. (2008). Differential expression of ionic conductances in interstitial cells of Cajal in the murine gastric antrum. J. Physiol. 586, 859–873.
Torihashi, S., Fujimoto, T., Trost, C., and Nakayama, S. (2002). Calcium oscillation linked to pacemaking of interstitial cells of Cajal: requirement of calcium influx and localization of TRP4 in caveolae. J. Biol. Chem. 277, 19191–19197.
Walker, R. L., Koh, S. D., Sergeant, G. P., Sanders, K. M., and Horowitz, B. (2002). TRPC4 currents have properties similar to the pacemaker current in interstitial cells of Cajal. Am. J. Physiol. Cell Physiol. 283, C1637–C1645.
Wang, B., Kunze, W. A., Zhu, Y., and Huizinga, J. D. (2008). In situ recording from gut pacemaker cells. Pflügers Arch. 457, 243–251.
Ward, S. M., Abu Baker, S., de Faoite, A., and Sanders, K. M. (2003). Propagation of slow waves requires IP3 receptors and mitochondrial Ca2+ uptake in canine colonic muscles. J. Physiol. 549, 207–218.
Ward, S. M., Dixon, R. E., de Faoite, A., and Sanders, K. M. (2004). Voltage-dependent calcium entry underlies propagation of slow waves in canine gastric antrum. J. Physiol. 561, 793–810.
Ward, S. M., Ordög, T., Bayguinov, J. R., Horowitz, B., Epperson, A., Shen, L., Westphal, H., and Sanders, K. M. (1999). Development of interstitial cells of Cajal and pacemaking in mice lacking enteric nerves. Gastroenterology 117, 584–594.
Ward, S. M., Ördög, T., Koh, S. D., Abu Baker, S., Jun, J. Y., Amberg, G., Monaghan, K., and Sanders, K. M. (2000). Pacemaking in interstitial cells of Cajal depends upon calcium handling by endoplasmic reticulum and mitochondria. J. Physiol. 525, 355–361.
Welsh, D. G., Nelson, M. T., Eckman, D. M., and Brayden, J. E. (2000). Swelling-activated cation channels mediate depolarization of rat cerebrovascular smooth muscle by hyposmolarity and intravascular pressure. J. Physiol. 527, 139–148.
White, E. J., Park, S. J., Foster, J. A., and Huizinga, J. D. (2008). Ether-a-go-go-related gene 3 is the main candidate for the E-4031-sensitive potassium current in the pacemaker interstitial cells of Cajal. Am. J. Physiol. Gastrointest. Liver Physiol. 295, G691–G699.
White, M. M., and Aylwin, M. (1990). Niflumic and flufenamic acids are potent reversible blockers of Ca2+-activated Cl( channels in Xenopus oocytes. Mol. Pharmacol. 37, 720–724.
Yang, Y. D., Cho, H., Koo, J. Y., Tak, M. H., Cho, Y., Shim, W.-S., Park, S. P., Lee, J., Lee, B., Kim, B.-M., Raouf, R., Shin, Y. K., and Oh, U. (2008). TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 455, 1210–1215.
Yoneda, S., Takano, H., Takaki, M., and Suzuki, H. (2002). Properties of spontaneously active cells distributed in the submucosal layer of mouse proximal colon. J. Physiol. 542, 887–897.
Youm, J. B., Kim, N., Han, J., Kim, E., Joo, H., Leem, C. H., Goto, G., Noma, A., and Earm, Y. E. (2006). A mathematical model of pacemaker activity recorded from mouse small intestine. Philos. Transact. A Math. Phys. Eng. Sci. 364, 1135–1154.
Zhou, H., Liu, L., Bai, Y., Wu, W., Li, G., Li, J., Zou, D., Gao, J., and Li, Z. (2011). Damage of the interstitial cells of Cajal and myenteric neurons causing ileus in acute necrotizing pancreatitis rats. Surgery 149, 262–275.
Zhu, M. H., Kim, T. W., Ro, S., Yan, W., Ward, S. M., Koh, S. D., and Sanders, K. M. (2009). A Ca(2+)-activated Cl(() conductance in interstitial cells of Cajal linked to slow wave currents and pacemaker activity. J. Physiol. 587, 4905–4918.
Zhu, Y., Golden, C. M., Ye, J., Wang, X.-Y., Akbarali, H. I., and Huizinga, J. D. (2003). ERG K+ currents regulate pacemaker activity in ICC. Am. J. Physiol. Gastrointest. Liver Physiol. 285, G1249–G1258.
Zhu, Y., and Huizinga, J. D. (2008). Nitric oxide decreases the excitability of interstitial cells of Cajal through activation of the BK channel. J. Cell. Mol. Med. 12, 1718–1727.
Zhu, Y., Mucci, A., and Huizinga, J. D. (2005). Inwardly rectifying chloride channel activity in intestinal pacemaker cells. Am. J. Physiol. Gastrointest. Liver Physiol. 288, G809–G821.
Zhu, Y., Parsons, S. P., and Huizinga, J. D. (2010a). Measurement of intracellular chloride ion concentration in ICC in situ and in explant culture. Neurogastroenterol. Motil. 22, 704–709.
Zhu, Y., Parsons, S. P., and Huizinga, J. D. (2010b). Corrigendum. Neurogastroent. Motil. 22, 1042–1042.
Keywords: electrophysiology, interstitial cells of Cajal, gastrointestinal motility, pacemaker, ion channels, mathematical models, physiome
Citation: Lees-Green R, Du P, O’Grady G, Beyder A, Farrugia G and Pullan AJ (2011) Biophysically based modeling of the interstitial cells of Cajal: current status and future perspectives. Front. Physio. 2:29. doi: 10.3389/fphys.2011.00029
Received: 31 January 2011;
Paper pending published: 25 April 2011;
Accepted: 13 June 2011;
Published online: 04 July 2011.
Edited by:
Joseph L. Greenstein, The Johns Hopkins University, USAReviewed by:
Bradley John Roth, Oakland University, USAYung E. Earm, Seoul National University College of Medicine, South Korea
Copyright: © 2011 Lees-Green, Du, O’Grady, Beyder, Farrugia and Pullan. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Rachel Lees-Green, Auckland Bioengineering Institute, The University of Auckland, 70 Symonds Street, Auckland 1010, New Zealand. e-mail:cmxlZTA3NEBhdWNrbGFuZHVuaS5hYy5ueg==