 Yasmin L. Hashambhoy1,2*
Yasmin L. Hashambhoy1,2* John C. Chappell3,4
John C. Chappell3,4 Shayn M. Peirce5* Victoria L. Bautch3,4,6
Shayn M. Peirce5* Victoria L. Bautch3,4,6 Feilim Mac Gabhann1,2
Feilim Mac Gabhann1,2
- 1 Department of Biomedical Engineering, Johns Hopkins University, Baltimore, MD, USA
- 2 Institute for Computational Medicine, Johns Hopkins University, Baltimore, MD, USA
- 3 Department of Biology, The University of North Carolina at Chapel Hill, Chapel Hill, NC, USA
- 4 McAllister Heart Institute, The University of North Carolina at Chapel Hill, Chapel Hill, NC, USA
- 5 Department of Biomedical Engineering, University of Virginia, Charlottesville, VA, USA
- 6 Lineberger Comprehensive Cancer Center, The University of North Carolina at Chapel Hill, Chapel Hill, NC, USA
Experimental data indicates that soluble vascular endothelial growth factor (VEGF) receptor 1 (sFlt-1) modulates the guidance cues provided to sprouting blood vessels by VEGF-A. To better delineate the role of sFlt-1 in VEGF signaling, we have developed an experimentally based computational model. This model describes dynamic spatial transport of VEGF, and its binding to receptors Flt-1 and Flk-1, in a mouse embryonic stem cell model of vessel morphogenesis. The model represents the local environment of a single blood vessel. Our simulations predict that blood vessel secretion of sFlt-1 and increased local sFlt-1 sequestration of VEGF results in decreased VEGF–Flk-1 levels on the sprout surface. In addition, the model predicts that sFlt-1 secretion increases the relative gradient of VEGF–Flk-1 along the sprout surface, which could alter endothelial cell perception of directionality cues. We also show that the proximity of neighboring sprouts may alter VEGF gradients, VEGF receptor binding, and the directionality of sprout growth. As sprout distances decrease, the probability that the sprouts will move in divergent directions increases. This model is a useful tool for determining how local sFlt-1 and VEGF gradients contribute to the spatial distribution of VEGF receptor binding, and can be used in conjunction with experimental data to explore how multi-cellular interactions and relationships between local growth factor gradients drive angiogenesis.
Introduction
Angiogenesis is the growth of new blood vessels from an existing vascular network. While it has been studied extensively in pathological conditions such as cancer, angiogenesis is also essential for organ development and recovery of tissues from injury. This complex, multistep process is regulated, in part, by the extracellular ligands of the vascular endothelial growth factor (VEGF) family. VEGF ligands bind to and activate receptor tyrosine kinases on the endothelial cell surface, initiating downstream signaling that drives cellular behaviors such as proliferation and migration, culminating in angiogenesis (Keck et al., 1989; Leung et al., 1989; Olsson et al., 2006; Shibuya and Claesson-Welsh, 2006).
There are three known VEGF tyrosine kinase receptors: VEGFR-1 (Flt-1 in mice), VEGFR-2 (Flk-1 in mice), and VEGFR-3 (Flt-4 in mice), and each receptor binds one or more members of the VEGF ligand family to initiate different downstream signaling pathways. In addition to the membrane-bound form of Flt-1 (mFlt-1), there is a soluble version of the receptor (sFlt-1) that binds to and can sequester VEGF-A (VEGF) in the extracellular space (Nilsson et al., 2010). Flt-1 and Flk-1 are both essential for vascular development and viability, since genetic deletion of either gene in mice causes death in utero (Fong et al., 1995; Shalaby et al., 1995).
Vascular development is exquisitely sensitive to VEGF levels: embryonic lethality results from knockout of a single VEGF allele and from VEGF over-expression (Carmeliet et al., 1996; Ferrara et al., 1996; Miquerol et al., 2000). While VEGF concentrations are also important in angiogenesis for initiating new vessel sprouting, the spatial distribution of VEGF is an important regulator of blood vessel branching morphogenesis (Carmeliet et al., 1999; Ruhrberg et al., 2002; Stalmans et al., 2002). This has been demonstrated experimentally through the effects of different VEGF isoforms on vessel morphogenesis. These isoforms have varying heparin-binding affinities resulting in different local VEGF concentration gradients that are perceived by endothelial cells of nascent vessels.
The endothelial cells in vessel sprouts sense VEGF gradients, and they respond both by modifying their behavior and by communicating with their neighbors in order to influence their function. For example, Delta-like ligand 4 (Dll4) activates Notch signaling in adjacent cells to repress sprouting (Hellstrom et al., 2007), and Dll4 up-regulation is downstream of VEGF activation (Lawson et al., 2002). Endothelial tip cells, which are at the front of the growing blood vessel sprout, extend filopodia and migrate in response to VEGF gradients, whereas stalk cells, located behind the tip cell of the sprout, proliferate in response to VEGF levels (Gerhardt et al., 2003). Endothelial cells compete for the tip cell position, and during sprouting, cells that have higher levels of VEGF signaling are more likely to become tip cells (Jakobsson et al., 2010).
Over the past decade, studies have revealed mechanisms through which VEGF receptors modulate blood vessel formation. Flk-1 and mFlt-1 are monomeric VEGF receptors that have cytoplasmic tyrosine kinase domains. VEGF is bivalent and initiates signaling by binding to two receptor monomers. The two cytoplasmic receptor kinases in this complex phosphorylate each other, thus initiating downstream signaling (Mac Gabhann and Popel, 2008). If sFlt-1 replaces one of the receptors, then neither it nor the other receptor is activated, as sFlt-1 lacks the cytoplasmic kinase domain. In mouse embryonic development, Flt-1 and Flk-1 appear to compete for VEGF (Autiero et al., 2003). Flt-1 modulates the organization of embryonic vasculature (Fong et al., 1995); this activity appears to be through VEGF sequestration and seems to be independent of Flt-1 signaling (Hiratsuka et al., 1998). This suggests that during development, Flt-1–Flk-1 heterodimers also do not provide essential signaling. However, VEGF receptor heterodimers have been shown to exist and to be capable of signaling (Huang et al., 2001; Nilsson et al., 2010), although their particular role is still not clear. Similarly, analysis of ex vivo developmental systems, such as embryonic stem (ES) cell cultures that undergo differentiation to form primitive vessels, showed that elimination of all Flt-1 expression (both soluble and membrane-bound) caused vascular overgrowth (Kearney et al., 2002, 2004). This phenotype was partially rescued by Flk-1 inhibitors (Kearney et al., 2002; Roberts et al., 2004), exogenous Flt-1 (Roberts et al., 2004) and re-expression of sFlt-1 (Zeng et al., 2007). While sFlt-1 and mFlt-1 transgenes each partially rescued endothelial cell proliferation in flt-1−/− mutant vessels, only the sFlt-1 transgene rescued branching (Kappas et al., 2008). Furthermore, loss of Flt-1 activity disrupted local sprout guidance in ES-cell-derived vessels, which was partially rescued by sFlt-1 expression in endothelial cells in the lateral base of the sprout (Chappell et al., 2009).
Several groups have used computational models to describe processes involved in angiogenesis including endothelial cell migration (Stokes et al., 1991), branching (Sun et al., 2005; McDougall et al., 2006; Bauer et al., 2007), signaling (Levine et al., 2002; Bentley et al., 2008) and VEGF transport and binding (Mac Gabhann et al., 2006, 2007). Models of angiogenesis have been extensively reviewed (Peirce, 2008; Qutub et al., 2009). The model presented here focuses on how putative VEGF and sFlt-1 gradients affect vessel morphogenesis. Our previous studies regarding VEGF transport include analysis of sFlt-1 (Wu et al., 2010a,b) or receptor dimerization by VEGF (Mac Gabhann and Popel, 2007a), as well as simulations of 2- and 3-D VEGF diffusion gradients (Mac Gabhann et al., 2006, 2007; Ji et al., 2007; Vempati et al., 2010). However, this is the first simulation of sFlt-1 gradients, and the first simulation of sFlt-1 to include explicit receptor dimerization.
Vascular endothelial growth factor secretion by parenchymal cells and binding to VEGF receptors on endothelial cells (see Figure 1) results in a VEGF gradient across the interstitial space. Experiments have predicted that sFlt-1 is intimately involved in endothelial sprouting processes; however, the existence of local sFlt-1 gradients and their role near vessel sprouts has yet to be measured or elucidated. sFlt-1 binds to VEGF, so it is possible that sFlt-1 secretion reduces available VEGF. We simulated the impact of opposing sFlt-1 and VEGF gradients on interstitial VEGF concentrations and distribution. In addition, the signaling of activated Flk-1 receptors can be affected to different extents by sFlt-1 in two ways: first, sFlt-1 sequesters VEGF to decrease free ligand concentrations available to bind cell surface receptors; second, heterodimerization of sFlt-1 to Flk-1 permits the formation of sFlt-1–VEGF–Flk-1 complexes, which are non-signaling but also sequester and internalize VEGF. By simulating sFlt-1 and VEGF transport and binding as well as receptor dimerization in a model that represents the local sprout environment, we can quantify the extent to which sFlt-1 sequestration vs. sFlt-1–(mFlt-1/Flk-1) heterodimerization affect Flk-1 activation. The model outputs the level and distribution of activated Flk-1 across the cell surface, and we interpret these as metrics for pro-angiogenic signaling.
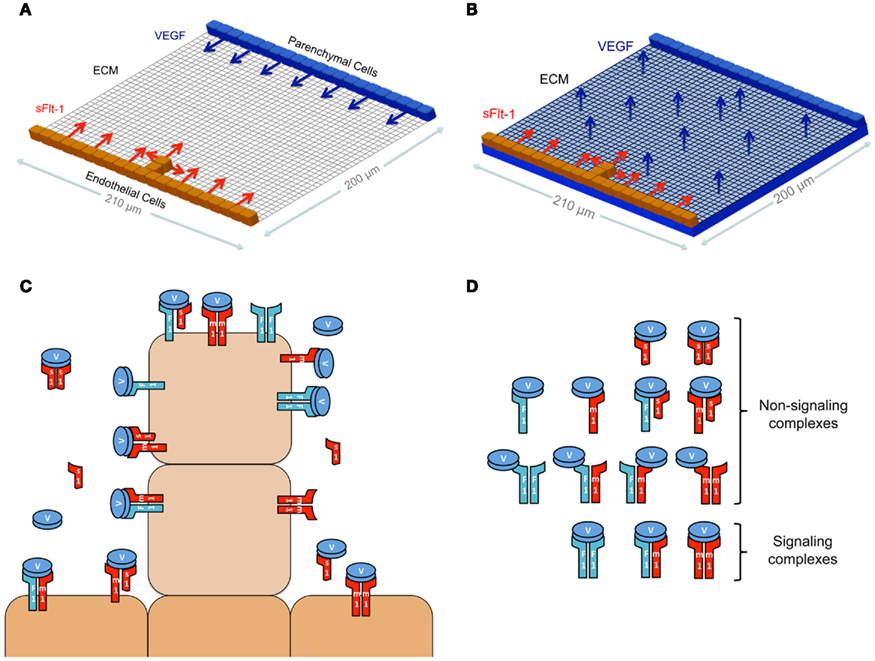
Figure 1. Schematics. (A,B) Schematics of the 2-D computational model space. In (A), sFlt-1 is secreted from an opposing wall of parenchymal cells. In (B), sFlt-1 is secreted from a layer of parenchymal cells below the interstitial space. Diffusion in the z direction is assumed to be instantaneous, and both cases (A,B) are essentially 2-D models. (C) Schematic of the molecular species and the cell surface signaling and non-signaling receptor complexes. (D) Schematic showing non-signaling (top) and signaling (bottom) receptor complexes.
To quantify the extent to which sFlt-1 alters VEGF signaling, we have developed an experimentally based computational model describing dynamic spatial transport of VEGF and its receptors in the mouse ES-cell culture system. Briefly, in the experiments simulated by our computational model, ES-cells undergo a programmed differentiation to produce multiple cell types including vascular endothelial cells, which give rise to primitive blood vessels via angiogenic sprouting in a context similar to the developing embryo and yolk sac (Zeng and Bautch, 2009). Our computational model system represents the local environment of a blood vessel sprout and nearby mesendoderm tissue at the leading edge of developing blood vessel networks. In the model, cell positions are static, but we simulate dynamic VEGF and sFlt-1 transport and binding. Our simulations suggest that sFlt-1 sequestration of VEGF in the interstitial space significantly alters VEGF–Flk-1 levels on the cell surface, thus influencing directionality of future sprout growth. We also predict that the proximity of neighboring sprouts to one another may alter local VEGF gradients, VEGF receptor binding, and the directionality of sprout growth.
Materials and Methods
Simulation Environment
We present a computational model that represents the local environment of a single blood vessel within a biological system with several cell types and a source of VEGF. The environment has the dimensions 210 μm × 200 μm × 10 μm (Figure 1A), and includes endothelial cells and parenchymal cells (source of VEGF), which are separated by interstitial space (see Figure 1 in Chappell et al., 2009 for images of ES-cell-derived vessels). In our model, the interstitial space is filled with extracellular matrix (ECM). This is treated as a homogenous matrix, representing the amalgamation of various proteins that are present in the microenvironment; in the future, we will expand the model to include other cell types in this space. In this model, cells do not move (their position is static) or proliferate, but parenchymal cells dynamically secrete VEGF, and endothelial cells dynamically bind VEGF, secrete sFlt-1 and internalize mFlt-1 and Flk-1. There are a number of splice variants of mouse (and human) VEGF-A; in this study, we have modeled the most common isoform, VEGF164 (hereafter referred to as VEGF). This splice variant has the ability to reversibly bind to ECM and to cell surface receptors.
Figure 1A illustrates the configuration that was simulated to explore the gradients that are shown in Figure 2. At the initiation of the simulation, a sprout has emerged from the base vessel. The base vessel is composed of multiple endothelial cells. The sprout protrudes out of the middle of the base, with dimensions 10 μm × 20 μm, which are consistent with experimentally observed values (Kearney et al., 2004; Chappell et al., 2009). A line of parenchymal cells 200 μm from the base vessel secretes VEGF into the interstitial space. Figures 3–5 show results from simulations in which VEGF is secreted by parenchymal cells below the ECM (as represented in the schematic in Figure 1B). In all simulations, endothelial cells are capable of secreting sFlt-1 into the interstitial space. In ES-cell cultures, day 3 embryoid bodies are seeded so that upon differentiation (day 8) they cover the bottom of a 24-well dish (Kearney and Bautch, 2003). The simulation area is ∼0.02% of the area of one of these wells.
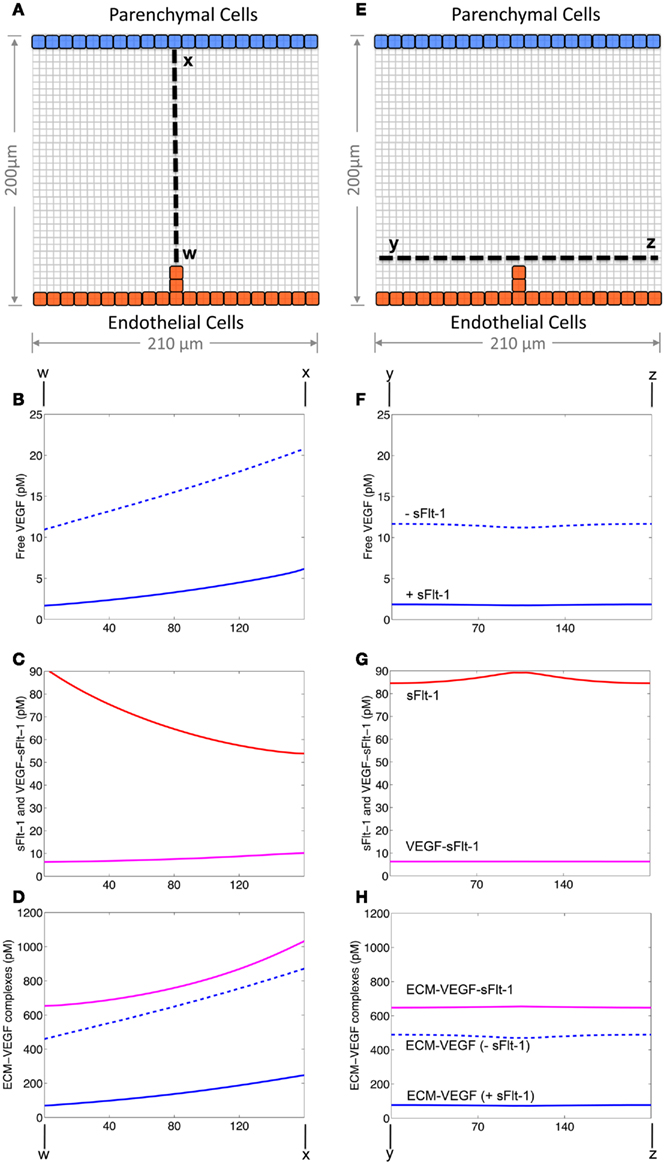
Figure 2. Volumetric Gradients of VEGF and sFlt-1. (A–D) Gradients of VEGF, sFlt-1, and their complexes along a line from the sprout leading edge to the parenchymal cell wall. (E–H) Gradients of VEGF, sFlt-1, and their complexes along a transverse line in front of the sprout leading edge. (B,F) VEGF gradients in the absence (dotted line) and presence (solid line) of sFlt-1 (C,G), sFlt-1 (red) and VEGF–sFlt-1 complex (purple) gradients, (D,H), ECM-bound gradients: ECM–VEGF in the absence (dotted blue line) and presence (solid blue line) of sFlt-1 and ECM–VEGF–sFlt-1 (purple line). All results are from 10 h after the start of simulation.
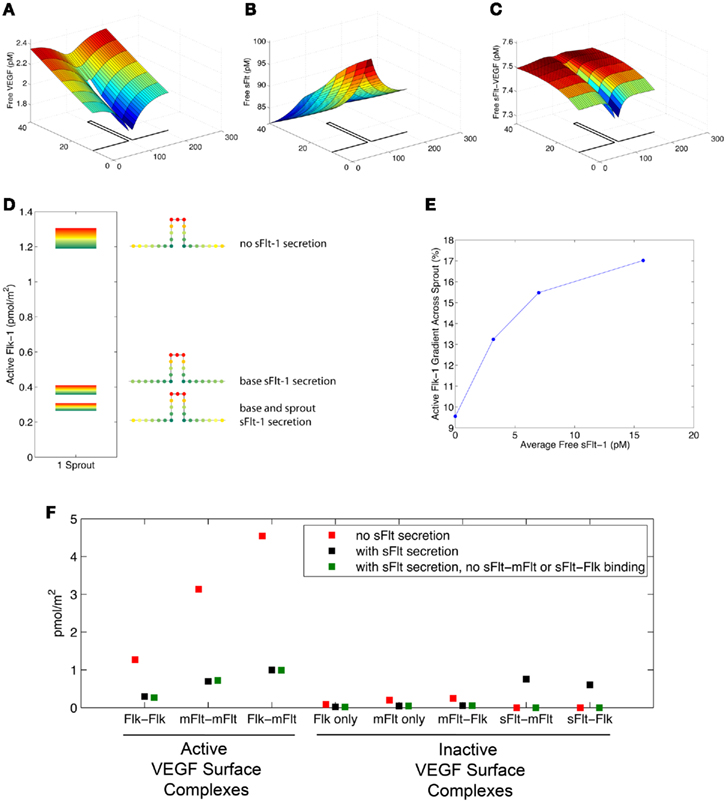
Figure 3. sFlt-1 can alter both absolute and relative levels of active Flk-1 on the sprout. (A–C) local VEGF and sFlt-1 gradients near the sprout for the scenario with sFlt-1 secretion from both the base vessel and the sprout. Note that the y-axis is stretched compared to the x-axis to emphasize the area local to the sprout. See the text and Figure 2 for more on the gradients in the x- and y-directions. (D) Active distribution across the endothelial surface. Flk-1 is considered active if both subunits in a dimerized Flk-1 complex are bound to VEGF. The results of three different scenarios are displayed: no sFlt-1 secretion from the sprout; sFlt-1 secretion only from the endothelial cells making up the base vessel; sFlt-1 secretion from both vessel and sprout. sFlt-1 secretion decreases the absolute amount and relative gradient of Flk-1 activation on the surface of sprouts. (E) As free sFlt-1 increases (due to increased sFlt-1 secretion) so does relative Flk-1 activation along the sprout. (F) Average receptor complex concentrations along the sprout for three cases: no sFlt-1 secretion (red), base and sprout sFlt-1 secretion (black), and base and sprout sFlt-1 secretion without any sFlt-1 binding to surface receptors mFlt-1 or Flk-1 (green). Active VEGF surface complexes are shown on the left, and inactive surface complexes are on the right. The removal of sFlt-1 heterodimerization barely affects active VEGF surface receptor complex levels.
The reaction–diffusion equations on which this model is based are similar to those that have been published previously (Mac Gabhann et al., 2006; Mac Gabhann and Popel, 2007a; Wu et al., 2010b). The model includes a number of physiological processes involved in sprout development, including: VEGF and sFlt-1 secretion by parenchymal and endothelial cells, respectively; VEGF and sFlt-1 diffusion, degradation and binding to ECM in the interstitial space; VEGF binding to sFlt-1 and surface receptors mFlt-1 and Flk-1; dimerization of sFlt-1, mFlt-1, and Flk-1 by VEGF; insertion of mFlt-1 and Flk-1 in the endothelial cell membrane; and internalization and degradation of receptors. Table 1 lists the kinetic parameters of the system.

Table 1. Kinetic parameters of the VEGF system.
Diffusion
Vascular endothelial growth factor, sFlt-1 and the VEGF–sFlt-1 complexes diffuse through the interstitial space. These species degrade at constant rates, and bind to other volumetric as well as membrane-bound species. Diffusion in the z direction is assumed to be very fast, and therefore is not explicitly modeled, however we include a defined thickness to allow a non-zero sprout edge surface area for binding and secretion. The top surface of cells is not considered in this model. An example of a reaction–diffusion equation is shown below (for VEGF):
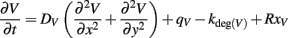
where V is the concentration of VEGF; DV is the diffusivity of VEGF; q is the secretion rate; kdeg is the degradation rate; and RxV refers to binding/unbinding reactions for VEGF. The model does not allow for degradation of ECM-bound complexes. The diffusivity constants for sFlt-1 and VEGF-bound sFlt-1 complexes were calculated by using the ratios of molecular weights between these molecules and VEGF and the calculated diffusivity of VEGF (Vempati et al., 2010).
Binding
The reaction term (Rx) includes ligand binding to soluble receptors, cell surface receptors, and glycoprotein sites in the ECM, along with the dimerization of receptors (Figure 1B). For example, for VEGF:
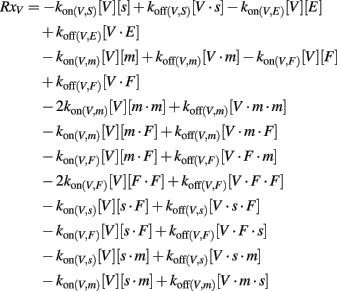
where E is ECM, s is sFlt-1, m is mFlt-1, and F is Flk-1; kon and koff are the kinetic rates of binding and unbinding, respectively.
For mFlt-1 and Flk-1, which are anchored to the cell membrane, we do not include diffusion along the cell surface, and thus cell surface molecule concentrations are calculated using equations of the general form:
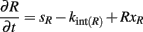
where sR is the insertion rate of new receptors into the membrane, kint is the internalization and Rx includes ligand binding and receptor dimerization reactions. The values of the binding affinities and kinetic rates are similar to our previous models (Mac Gabhann and Popel, 2007a; Wu et al., 2010b), and derived from empirical data. This assumes that kinetic parameters, such as binding and unbinding rates, are similar among different experimental systems, e.g., in vitro cell culture and endothelial cells in developing vasculature. sFlt-1–VEGF binding and dimerization parameters were constrained to produce observed Kd values for VEGF–sFlt-1 binding close to those that were measured assuming 1:1 binding (Tanaka et al., 1997). We assumed that the affinity of sFlt-1 dimerization with mFlt-1 was similar to that of VEGF binding mFlt-1. We also assumed that sFlt-1 and VEGF bind ECM with the same affinity, since both proteins have heparin-binding properties (Park and Lee, 1999; Wu et al., 2010b). We did not include binding between cell surface receptors and ECM-bound ligands. Surface edges have a thickness of 10 μm, corresponding to the thickness of the simulation environment. Endothelial cells express membrane-bound receptors mFlt-1 and Flk-1 on their surfaces at a density of 6.64 × 10−6 nmol/cm2 or 40 receptors/μm2, which is consistent with expression levels in our previous models (Mac Gabhann and Popel, 2007a). In the model, sFlt-1 can also heterodimerize with Flk-1; determining the extent to which sFlt-1–Flk-1 binding has an effect on Flk-1 signaling was a secondary goal of this study.
Ligand Secretion
Vascular endothelial growth factor secretion was constrained in order for simulation mean concentrations in the tissue to match steady state interstitial concentrations in previous models (Wu et al., 2010b). Kappas et al. (2008) observed that in ES-cell-derived vessels, the relative proportions of sFlt-1 and mFlt-1 RNA levels are roughly equal. Therefore, in the model, the sFlt-1 secretion rate was fixed so that it matched the mFlt-1 production rate. Each cell has a fixed secretion rate, and the amount of VEGF or sFlt-1 secreted from a patch of the cell’s membrane is a function of the cell’s secretion rate and the surface area of the patch that is exposed to interstitial space. Following secretion, the ligands diffuse through the ECM and form a concentration gradient driven by degradation and receptor binding (Mac Gabhann et al., 2006, 2007; Ji et al., 2007; Vempati et al., 2010).
Model Simulation
The simulation space is divided into a 2-D 43 × 41 grid, and partial differential equations are solved using the central difference method in space and the forward difference method in time. An odd number of grid points allows us to place the sprout symmetrically. The grid size was chosen to be smaller than the cells size so that receptor activation could be visualized across the surface of each cell. Each grid size represents 5 μm. A no-flux boundary condition is used at the edges of the grid. At the start of each simulation, it is assumed that there is an average of 1 pM of free VEGF, 1 nM of ECM, and 100 pM of ECM-bound VEGF in the interstitial space. The simulations were coded and run using Fortran.
Results
SFlt-1 Secretion Lowers Available VEGF Concentration
Figure 2 shows the predicted gradients of soluble VEGF, sFlt-1, VEGF–sFlt-1, and ECM-bound VEGF complexes along straight lines across the interstitial space (dashed lines in Figures 2A,E) after 10 h of sFlt-1 secretion. Free VEGF concentrations are higher in the absence of endothelial cell sFlt-1 secretion (compare solid (+sFlt) vs. dashed (−sFlt) lines in Figures 2B,F).
We quantified the relative gradients over the straight line across the interstitial space (dashed line in Figure 2A). The gradient was calculated by dividing the difference between the maximum and minimum concentration by the concentration at the midpoint, and normalizing to 20 μm, the typical length of a tip cell. In the presence of sFlt-1 secretion, free VEGF ranges from 1.7 pM at the sprout leading edge to 6.1 pM at the parenchymal cell wall (an average gradient of 17.0% per 20 μm). In the absence of sFlt-1 secretion, the concentration ranges from 11 to 20.8 pM (an average gradient of 7.9% per 20 μm). ECM-bound VEGF concentrations display relative gradients similar to those of free VEGF over the length of the sprout in the presence or absence of sFlt-1 (16.2 and 8.0%, respectively). Free sFlt-1 concentrations range from 54 to 91 pM (an average gradient of 7.3% per 20 μm). ECM-bound sFlt ranges from 2.24 to 3.81 nM, a gradient of 7.3% along the length of the sprout.
Local VEGF gradients are close to constant across the interstitial space, ranging from 18.6% near the sprout to 17.0% near the parenchymal cells. In contrast, the local sFlt-1 gradients are much steeper closer to the sprout (10.4% per 20 μm) than near the parenchymal cells (2.3% per 20 μm).
Note that the total amount of sFlt-1 in the interstitial space is over 100-fold higher than the total amount of mFlt-1 on the vessels; this is in broad agreement with placental data showing a 50-fold excess of sFlt-1 protein (Carmeliet et al., 2001), and occurs despite the 50:50 ratio of sFlt-1 and mFlt-1 expression, primarily due to sFlt-1 binding to ECM.
When endothelial cells secrete sFlt-1, sFlt-1 is highest where VEGF is lowest – near the endothelial cells (Figures 2C,G, red lines). Free sFlt-1 binds and forms complexes with VEGF (Figures 2C,G, purple lines), thus reducing the concentration of free VEGF available for receptor binding. Perhaps more significantly, ECM-bound sFlt-1 binds VEGF (Figures 2D,H, purple lines), which reduces free VEGF degradation and increases total VEGF levels in the interstitial space. However, the sFlt-1 sequestration of VEGF is high enough that free VEGF and ECM-bound VEGF levels are reduced.
SFlt-1 Secretion Alters Absolute and Relative Flk-1 Activation on Sprout Surfaces
We investigated the hypothesis that sFlt-1 sequesters VEGF, altering VEGF gradients, and influencing sprout morphogenesis (Chappell et al., 2009). It is well established that VEGF signaling through Flk-1 leads to cell proliferation and migration (Waltenberger et al., 1994; Gerber et al., 1998). Based on VEGF isoform-specific experiments in mouse embryos (Ruhrberg et al., 2002), it is hypothesized that endothelial cells proliferate in response to the perceived VEGF concentration, and they sprout and migrate in response to gradients of VEGF. Therefore in analyzing the simulation results, we present both the amount and the spatial organization of VEGF-activated Flk-1 as indicators of the strength of VEGF-induced cell signaling. In this set of simulations, VEGF is secreted by a layer of parenchymal cells below the ECM (as shown in Figure 1B). This scenario more closely mimics the sprouting environment in ES-cell cultures, since the underlying endoderm is the primary source of VEGF ligand (Bautch et al., 2000). Figures 3A–C show local free VEGF, sFlt-1, and VEGF–sFlt-1 levels around the sprout. In the previous set of simulations, the absence of sFlt-1 allows more free VEGF to be available for membrane receptor binding and activation (Figures 2B,F). Therefore we predict that there is increased active Flk-1 over the entire sprout surface, and simulations confirm that this is the case (Figure 3D).
Chappell et al. (2009) observed that in ES-cell-derived vessels, the flt-1 gene is more highly activated in base vessel cells, just lateral to the sprout, than in sprout cells. In our simulations, all the endothelial cells of the base vessel secrete sFlt-1, and the soluble receptor binds to VEGF, thus reducing the amount of free VEGF available for Flk-1 binding, which results in decreased active Flk-1 levels. Active Flk-1 concentration is reduced further when both the base and the nascent sprout secrete sFlt-1 (Figure 3D).
Plots of the spatial distribution of active Flk-1 along the sprout surface at different scales (Figure 3D) suggest that sFlt-1 secretion alters the absolute levels of Flk-1 activation, but it does not alter the surface pattern of active Flk-1. That is, the highest activation is always at the leading edge of the tip cell, and the lowest active Flk-1 levels are at the base of the sprout. Relative active Flk-1 gradients are calculated as the difference between the average active Flk-1 values on the leading and trailing edges, divided by the active Flk-1 midway along the cell. The relative activation profile of Flk-1 on the sprout is affected by sFlt-1 secretion. The active Flk-1 gradient along the sprout is 14.6% when there is sFlt-1 secretion from the base and the sprout compared to 13.4% when there is sFlt-1 secretion only from the base and 9.1% when there is no sFlt-1 secretion. These differences in the distribution of Flk-1 activation are similar to the change in interstitial VEGF gradients. Higher Flk-1 activation gradients accompanied lower absolute active Flk-1 levels, suggesting that small changes in activation could magnify the relative difference along the sprout. Simulations predict that when sFlt-1 secretion (and therefore free sFlt-1 level) is increased, the active Flk-1 gradient increases as well (Figure 3E).
Figure 3F illustrates the average magnitude of VEGF-bound receptors on the sprout for three scenarios: no sFlt-1 secretion (red), base and sprout sFlt-1 secretion (black), and base and sprout sFlt-1 secretion in the absence of sFlt-1 heterodimerization (green). In the base and sprout secretion case (black points), the number of inactive VEGF-bound sFlt-1 heterodimers is on the same order of magnitude as the active VEGF-bound membrane receptors. Based on these results alone, we would predict that sFlt-1 binding to mFlt-1 and Flk-1 might have a large influence on Flk-1 and mFlt-1 activation. However, calculations show that there is 2.8 × 10−10 nmol of VEGF-bound sFlt-1 that is unbound to the vessel, compared to 8.3 × 10−14 that is bound to mFlt-1 or Flk-1; simulations in the absence of sFlt-1 heterodimerization (green points) show that binding of sFlt-1 to membrane receptors has very little effect on absolute receptor activation. In addition, the Flk-1 gradient is only affected by a small amount (it decreases from 15.5% with heterodimerization to 14.5% without dimerization, data not shown). Therefore, it appears that sFlt-1 sequestration of VEGF away from the vessel surface has a larger effect on Flk-1 activation than sFlt-1 heterodimerization with surface receptors.
In addition, we investigated the possible role that sprout length plays in sFlt-1 modulation of Flk-1 activation (Figure 4). We simulated shorter sprouts (protruding out of the base by 10 μm), and longer sprouts (protruding out of the base by 30 μm). The active Flk-1 gradient along the (10 μm long) side of the tip cell increases with sprout length. The active Flk-1 gradient along the tip cell on the short sprout is 7.2%, on the medium (20 μm long) sprout it is 8.8%, and on the long sprout it is 9.0%. Jakobsson et al. (2010) showed that cells with lower Flt-1 expression dominate the tip cell position. Therefore, for the short and long sprout cases, we also included a scenario in which tip cell sFlt-1 secretion is an order of magnitude lower than base vessel sFlt-1 secretion. With lower tip cell secretion, average active Flk-1 levels on the tip cell increase by 19% from 0.32 to 0.38 pmol/m2 (shown in Figure 4A) for the short sprout and these active receptor levels are higher than those on the cells near the base of the sprout, suggesting that the leading edge of the growing sprout is the most active local site of VEGFR signaling. When tip cell secretion is the same as that of the base vessel, Flk-1 activation on base vessel cells 50 μm away from the sprout is at similar levels to the tip of the sprout, suggesting that new sprouts may form near to the existing sprout. Thus, sFlt-1 expression modulation in the tip cell may aid in nearby sprout repression. When we simulated longer sprouts with lower tip cell secretion, average Flk-1 levels on the tip cell increased by 17% from 0.27 to 0.32 pmol/m2 (shown in Figure 4B). However, Flk-1 activation on the nearby endothelial cells along the base vessel does not reach the same levels as that on the tip of the sprout, even when sFlt-1 secretion by the tip cell is reduced, which suggests that longer sprouts may repress nearby sprouting regardless of sFlt-1 modulation. These results indicate that reduced tip cell sFlt-1 secretion may play a role in determining the location from which a sprout emerges from a parent vessel, and is most critical at the very early stage of sprout development.
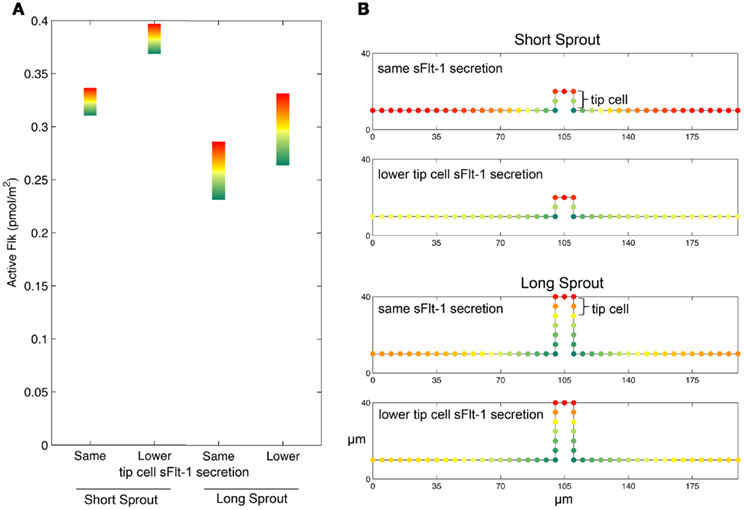
Figure 4. Decreased tip cell sFlt-1 secretion results in increased Flk-1 activation on the sprout. (A,B) Active Flk-1 levels on 10 μm long (A) or 30 μm long (B) sprouts in which tip cell sFlt-1 secretion is the same or lower than that of base vessel cells. The difference in Flk-1 activation on the leading edge of the sprout (due to decreased sFlt-1 secretion) compared to Flk-1 activation on the base vessel is more pronounced in short sprouts than in longer sprouts.
Nearby Sprouts Influence Perceived VEGF Gradient
The correct assembly of vessels into a functional vascular network requires that new vessels have cues for correct patterning. Just as the Dll4-Notch system prevents over-sprouting, new sprouts grow preferentially perpendicular to the base vessel, apparently in response to sFlt-mediated cues. We simulated a pair of sprouts protruding from the same vessel, and determined the effects they had on each other via their sFlt-1 production as a function of the distance between them. In this set of simulations, the vessel base and sprouts secrete sFlt-1 into the interstitial space.
Figures 5A,B illustrate the distribution of activated Flk-1 on cell surfaces after 10 h of sFlt-1 secretion from the base vessel and sprouts. When the sprouts are closest to each other (10 μm apart, Figure 5A), activated Flk-1 levels are highest (dark orange) at the corners of the sprout tip that are furthest away from the neighboring sprout. Active Flk-1 levels are lowest on the cell surfaces of the vessel base between the sprouts. If active Flk-1 is a predictor of cell proliferation and/or migration, this result suggests that sprouts that are in close proximity tend to diverge. This may prevent the formation of sprout anastamoses too close to the parent vessel. However, when sprouts develop further apart (150 μm, Figure 5B), active Flk-1 levels are more uniform along the tip cells, indicating a lack of lateral directional cues; therefore, simulation results suggest that they are likely to migrate straight ahead. When sprouts are close together, free VEGF is higher near the outside edges of the sprouts (Figure 5C), while free sFlt-1 is highest in the space between the sprouts (Figure 5D), where it sequesters VEGF. These findings suggest that sequestration of VEGF by sFlt-1 results in local activated Flk-1 relative distribution patterns that influence directionality. Note, in particular, that the concentration of activated Flk-1 between the two close sprouts is significantly lower than the concentration on the other side of the endothelial cells (Figure 5A). These simulation results lead us to predict that, based on active Flk-1 profiles, nearby sprouts will tend to diverge. Interestingly, the simulation predicts that when sFlt-1 secretion is absent, sprouts display qualitatively similar activated Flk-1 profiles as those shown in Figure 5A (data not shown). However, the results differ quantitatively when sFlt-1 secretion is present. Namely, with the addition of sFlt-1 secretion, Flk-1 activation is 40% higher at the leading outside edge of the sprout compared to the inside edge, whereas in the absence of sFlt-1, this increase is only 26%. These predictions suggest that VEGF sequestration by sFlt-1 magnifies the lateral cues for sprout divergence that result from spatial barriers to diffusion caused by the endothelial cells.
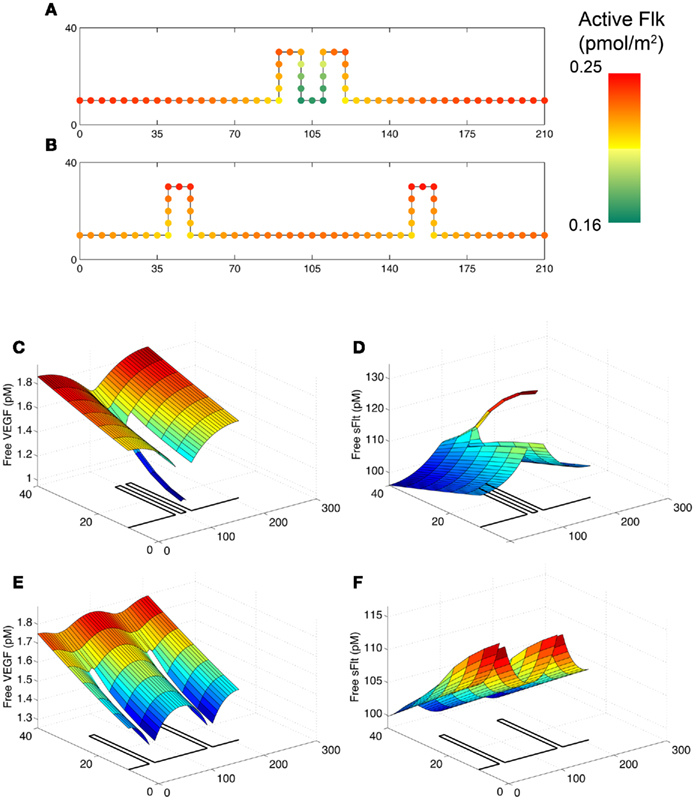
Figure 5. Active Flk-1 levels show that sprout directionality is partially governed by proximity to neighboring sprouts. All panels are for the scenario with both base and sprout sFlt-1 secretion. (A,B) Active Flk-1 levels on sprouts that are closer to (A) or farther away from (B) each other. (C) Free VEGF and (D), free sFlt-1 in the vicinity of the pair of sprouts that are close together. (E) Free VEGF and (F), free sFlt-1 in the vicinity of the pair of sprouts that are farther apart. Note that for panels (C–F), the y-axis is stretched compared to the x-axis, and the geometry of the sprouts is outlined on the x–y plane.
Discussion and Conclusion
Studies in differentiated ES-cell cultures have shown that flt-1−/− ES-cell-derived blood vessels display significant dysmorphogenesis (Kearney et al., 2002, 2004). Vessel branching is perturbed, and flt-1 mutant endothelial cells proliferate more than their wild-type counterparts. These properties are partially rescued by expression of a sFlt-1 transgene (Kappas et al., 2008). To quantify the extent to which sFlt-1 alters VEGF signaling in a biological context, we have developed an experimentally based computational model describing dynamic spatial transport of VEGF and its receptors, based on blood vessel morphogenesis during mouse ES-cell differentiation. One of the goals of this study is to use this model to further understand how sFlt-1 modulates vessel morphogenesis.
Simulation predictions indicate that sFlt-1 sequesters enough VEGF as to reduce free VEGF in the interstitial space (Figure 2) and to decrease active Flk-1 levels on the endothelial cell surface (Figure 3). Since Flk-1 activation promotes endothelial cell division and migration, our results suggest that sFlt-1 reduces the probability of proliferation along the whole vessel. This is in agreement with the observations of Kappas et al. (2008), that sFlt-1 partially rescues the increased mitotic index displayed by flt-1−/− cells. The simulations also predict that sFlt-1 decreases absolute active Flk-1 and creates differences in relative spatial distribution of Flk-1 activation (Figure 3) and that it does so primarily through sequestration of VEGF in the ECM (rather than through heterodimerization of receptors). This supports the hypothesis of Chappell et al. (2009) that sFlt-1 sequestration of VEGF increases relative VEGF gradients around the sprout, which results in guidance cues and rescue of vessel dysmorphogenesis.
There is an experimentally observed link between sprout proximity and sprout angle in flt-1−/− ES-cell cultures (Chappell et al., 2009). Compared to wild-type cells, sprouts from flt-1−/− ES-cell-derived vessels develop closer to each other, and grow at angles more acute to the base vessel. While our simulations are static and not dynamic, the results suggest that sprout proximity may be a contributory factor to the morphological outcome of sprouting vessels. The model results suggest that, as the distance between sprouts decreases, lateral directionality cues become more pronounced, possibly to prevent early anastamoses (too close to the base vessel). This is only part of the story, however, as sFlt-1 clearly influences sprout angle independent of sprout proximity (Chappell et al., 2009).
One of the inherent limitations of the model is that it does not represent the 3-D nature of the experimental system. Thus, it cannot account for diffusion above or below cells. However, even when we simulate VEGF secretion from cells below the surface, we see that there is a diffusion barrier created by nearby sprouts that drives lateral directionality cues. These cues are magnified in the presence of sFlt-1 secretion. This may explain why sFlt-1 rescues vessel dysmorphogenesis experimentally (Chappell et al., 2009). We are developing models to incorporate 3-D cell and tissue geometry. The model currently represents the environment surrounding blood vessels in a static system (i.e., cells do not migrate or proliferate). To dynamically simulate growing sprouts, which would permit the emergence of non-perpendicular vessels and other features, we plan to incorporate an agent-based model (Peirce et al., 2004; Bailey et al., 2007, 2009; Robertson et al., 2007). The model does not include neuropilin-1, a membrane-bound receptor found on the surface of endothelial cells. Neuropilin-1 binds to mFlt-1, Flk-1, and VEGF, thus affecting the distribution of activated VEGF receptors (Mac Gabhann and Popel, 2005, 2007b). We will include this receptor in future simulations. We are also working on decreasing the computational time required for the simulations, which would allow us to perform more detailed parameter sensitivity analyses and further investigate how different sprout configurations, secretion rates, and VEGF and sFlt-1 gradients contribute to vessel morphogenesis.
The multi-scale model we have introduced allows us to isolate at high resolution how sFlt-1 and VEGF modulate guidance cues and direct sprout growth. It has been constrained by kinetic data, and supports sprout morphogenesis observations in ES-cell culture systems. It is a useful tool that can be used as a foundation upon which to explore multi-scale interactions and relationships between local growth factor gradients driving angiogenesis behaviors in this unique experimental model.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Autiero, M., Waltenberger, J., Communi, D., Kranz, A., Moons, L., Lambrechts, D., Kroll, J., Plaisance, S., De Mol, M., Bono, F., Kliche, S., Fellbrich, G., Ballmer-Hofer, K., Maglione, D., Mayr-Beyrle, U., Dewerchin, M., Dombrowski, S., Stanimirovic, D., Van Hummelen, P., Dehio, C., Hicklin, D. J., Persico, G., Herbert, J. M., Communi, D., Shibuya, M., Collen, D., Conway, E. M., and Carmeliet, P. (2003). Role of PlGF in the intra- and intermolecular cross talk between the VEGF receptors Flt1 and Flk1. Nat. Med. 9, 936–943.
Bailey, A. M., Lawrence, M. B., Shang, H., Katz, A. J., and Peirce, S. M. (2009). Agent-based model of therapeutic adipose-derived stromal cell trafficking during ischemia predicts ability to roll on P-selectin. PLoS Comput. Biol. 5, e1000294. doi: 10.1371/journal.pcbi.1000294
Bailey, A. M., Thorne, B. C., and Peirce, S. M. (2007). Multi-cell agent-based simulation of the microvasculature to study the dynamics of circulating inflammatory cell trafficking. Ann. Biomed. Eng. 35, 916–936.
Bauer, A. L., Jackson, T. L., and Jiang, Y. (2007). A cell-based model exhibiting branching and anastomosis during tumor-induced angiogenesis. Biophys. J. 92, 3105–3121.
Bautch, V.L., Redick, S. D., Scalia, A., Harmaty, M., Carmeliet, P., and Rapoport, R. (2000). Characterization of the vasculogenic block in the absence of vascular endothelial growth factor-A. Blood 95, 1979–1987.
Bentley, K., Gerhardt, H., and Bates, P. A. (2008). Agent-based simulation of notch-mediated tip cell selection in angiogenic sprout initialisation. J. Theor. Biol. 250, 25–36.
Carmeliet, P., Ferreira, V., Breier, G., Pollefeyt, S., Kieckens, L., Gertsenstein, M., Fahrig, M., Vandenhoeck, A., Harpal, K., Eberhardt, C., Declercq, C., Pawling, J., Moons, L., Collen, D., Risau, W., and Nagy, A. (1996). Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 380, 435–439.
Carmeliet, P., Moons, L., Luttun, A., Vincenti, V., Compernolle, V., De Mol, M., Wu, Y., Bono, F., Devy, L., Beck, H., Scholz, D., Acker, T., DiPalma, T., Dewerchin, M., Noel, A., Stalmans, I., Barra, A., Blacher, S., Vandendriessche, T., Ponten, A., Eriksson, U., Plate, K. H., Foidart, J. M., Schaper, W., Charnock-Jones, D. S., Hicklin, D. J., Herbert, J. M., Collen, D., and Persico, M. G. (2001). Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat. Med. 7, 575–783.
Carmeliet, P., Ng, Y. S., Nuyens, D., Theilmeier, G., Brusselmans, K., Cornelissen, I., Ehler, E., Kakkar, V. V., Stalmans, I., Mattot, V., Perriard, J. C., Dewerchin, M., Flameng, W., Nagy, A., Lupu, F., Moons, L., Collen, D., D’Amore, P. A., and Shima, D. T. (1999). Impaired myocardial angiogenesis and ischemic cardiomyopathy in mice lacking the vascular endothelial growth factor isoforms VEGF164 and VEGF188. Nat. Med. 5, 495–502.
Chappell, J. C., Taylor, S. M., Ferrara, N., and Bautch, V. L. (2009). Local guidance of emerging vessel sprouts requires soluble Flt-1. Dev. Cell 17, 377–386.
Ferrara, N., Carver-Moore, K., Chen, H., Dowd, M., Lu, L., O’Shea, K. S., Powell-Braxton, L., Hillan, K. J., and Moore, M. W. (1996). Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 380, 439–442.
Fong, G. H., Rossant, J., Gertsenstein, M., and Breitman, M. L. (1995). Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 376, 66–70.
Gerber, H. P., McMurtrey, A., Kowalski, J., Yan, M., Keyt, B. A., Dixit, V., and Ferrara, N. (1998). Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3’-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J. Biol. Chem. 273, 30336–30343.
Gerhardt, H., Golding, M., Fruttiger, M., Ruhrberg, C., Lundkvist, A., Abramsson, A., Jeltsch, M., Mitchell, C., Alitalo, K., Shima, D., and Betsholtz, C. (2003). VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 161, 1163–1177.
Hellstrom, M., Phng, L. K., Hofmann, J. J., Wallgard, E., Coultas, L., Lindblom, P., Alva, J., Nilsson, A. K., Karlsson, L., Gaiano, N., Yoon, K., Rossant, J., Iruela-Arispe, M. L., Kalén, M., Gerhardt, H., and Betsholtz, C. (2007). Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 445, 776–780.
Hiratsuka, S., Minowa, O., Kuno, J., Noda, T., and Shibuya, M. (1998). Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc. Natl. Acad. Sci. U.S.A. 95, 9349–9354.
Huang, K., Andersson, C., Roomans, G. M., Ito, N., and Claesson-Welsh, L. (2001). Signaling properties of VEGF receptor-1 and -2 homo- and heterodimers. Int. J. Biochem. Cell Biol. 33, 315–324.
Jakobsson, L., Franco, C. A., Bentley, K., Collins, R. T., Ponsioen, B., Aspalter, I. M., Rosewell, I., Busse, M., Thurston, G., Medvinsky, A., Schulte-Merker, S., and Gerhardt, H. (2010). Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat. Cell Biol. 12, 943–953.
Ji, J. W., Mac Gabhann, F., and Popel, A. S. (2007). Skeletal muscle VEGF gradients in peripheral arterial disease: simulations of rest and exercise. Am. J. Physiol. Heart Circ. Physiol. 293, H3740–H3749.
Kappas, N.C., Zeng, G., Chappell, J. C., Kearney, J. B., Hazarika, S., Kallianos, K. G., Patterson, C., Annex, B. H., and Bautch, V. L. (2008). The VEGF receptor Flt-1 spatially modulates Flk-1 signaling and blood vessel branching. J. Cell Biol. 181, 847–858.
Kearney, J. B., Ambler, C. A., Monaco, K. A., Johnson, N., Rapoport, R. G., and Bautch, V. L. (2002). Vascular endothelial growth factor receptor Flt-1 negatively regulates developmental blood vessel formation by modulating endothelial cell division. Blood 99, 2397–2407.
Kearney, J. B., and Bautch, V. L. (2003). In vitro differentiation of mouse ES cells: hematopoietic and vascular development. Meth. Enzymol. 365, 83–98.
Kearney, J. B., Kappas, N. C., Ellerstrom, C., DiPaola, F. W., and Bautch, V. L. (2004). The VEGF receptor flt-1 (VEGFR-1) is a positive modulator of vascular sprout formation and branching morphogenesis. Blood 103, 4527–4535.
Keck, P. J., Hauser, S. D., Krivi, G., Sanzo, K., Warren, T., Feder, J., and Connolly, D. T. (1989). Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science 246, 1309–1312.
Lawson, N. D., Vogel, A. M., and Weinstein, B. M. (2002). Sonic hedgehog and vascular endothelial growth factor act upstream of the Notch pathway during arterial endothelial differentiation. Dev. Cell 3, 127–136.
Leung, D. W., Cachianes, G., Kuang, W. J., Goeddel, D. V., and Ferrara, N. (1989). Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 246, 1306–1309.
Levine, H. A., Tucker, A. L., and Nilsen-Hamilton, M. (2002). A mathematical model for the role of cell signal transduction in the initiation and inhibition of angiogenesis. Growth Factors 20, 155–175.
Mac Gabhann, F., Ji, J. W., and Popel, A. S. (2006). Computational model of vascular endothelial growth factor spatial distribution in muscle and pro-angiogenic cell therapy. PLoS Comput. Biol. 2, e127. doi: 10.1371/journal.pcbi.0020127
Mac Gabhann, F., Ji, J. W., and Popel, A. S. (2007). VEGF gradients, receptor activation, and sprout guidance in resting and exercising skeletal muscle. J. Appl. Physiol. 102, 722–734.
Mac Gabhann, F., and Popel, A. S. (2005). Differential binding of VEGF isoforms to VEGF receptor 2 in the presence of neuropilin-1: a computational model. Am. J. Physiol. Heart Circ. Physiol. 288, H2851–H2860.
Mac Gabhann, F., and Popel, A. S. (2007a). Dimerization of VEGF receptors and implications for signal transduction: a computational study. Biophys. Chem. 128, 125–139.
Mac Gabhann, F., and Popel, A. S. (2007b). Interactions of VEGF isoforms with VEGFR-1, VEGFR-2, and neuropilin in vivo: a computational model of human skeletal muscle. Am. J. Physiol. Heart Circ. Physiol. 292, H459–H474.
Mac Gabhann, F., and Popel, A. S. (2008). Systems biology of vascular endothelial growth factors. Microcirculation 15, 715–738.
McDougall, S. R., Anderson, A. R., and Chaplain, M. A. (2006). Mathematical modelling of dynamic adaptive tumour-induced angiogenesis: clinical implications and therapeutic targeting strategies. J. Theor. Biol. 241, 564–589.
Miquerol, L., Langille, B. L., and Nagy, A. (2000). Embryonic development is disrupted by modest increases in vascular endothelial growth factor gene expression. Development 127, 3941–3946.
Nilsson, I., Bahram, F., Li, X., Gualandi, L., Koch, S., Jarvius, M., Söderberg, O., Anisimov, A., Kholová, I., Pytowski, B., Baldwin, M., Ylä-Herttuala, S., Alitalo, K., Kreuger, J., and Claesson-Welsh, L. (2010). VEGF receptor 2/-3 heterodimers detected in situ by proximity ligation on angiogenic sprouts. EMBO J. 29, 1377–1388.
Olsson, A.-K., Dimberg, A., Kreuger, J., and Claesson-Welsh, L. (2006). VEGF receptor signalling? In control of vascular function. Nat. Rev. Mol. Cell Biol. 7, 359–371.
Park, M., and Lee, S. T. (1999). The fourth immunoglobulin-like loop in the extracellular domain of FLT-1, a VEGF receptor, includes a major heparin-binding site. Biochem. Biophys. Res. Commun. 264, 730–734.
Peirce, S. M. (2008). Computational and mathematical modeling of angiogenesis. Microcirculation 15, 739–751.
Peirce, S. M., Van Gieson, E. J., and Skalak, T. C. (2004). Multicellular simulation predicts microvascular patterning and in silico tissue assembly. FASEB J. 18, 731–733.
Qutub, A. A., Mac Gabhann, F., Karagiannis, E. D., Vempati, P., and Popel, A. S. (2009). Multiscale models of angiogenesis. IEEE Eng. Med. Biol. Mag. 28, 14–31.
Roberts, D. M., Kearney, J. B., Johnson, J. H., Rosenberg, M. P., Kumar, R., and Bautch, V. L. (2004). The vascular endothelial growth factor (VEGF) receptor Flt-1 (VEGFR-1) modulates Flk-1 (VEGFR-2) signaling during blood vessel formation. Am. J. Pathol. 164, 1531–1535.
Robertson, S. H., Smith, C. K., Langhans, A. L., McLinden, S. E., Oberhardt, M. A., Jakab, K. R., Dzamba, B., DeSimone, D. W., Papin, J. A., and Peirce, S. M. (2007). Multiscale computational analysis of Xenopus laevis morphogenesis reveals key insights of systems-level behavior. BMC Syst. Biol. 1, 46. doi: 10.1186/1752-0509-1-46
Ruhrberg, C., Gerhardt, H., Golding, M., Watson, R., Ioannidou, S., Fujisawa, H., Betsholtz, C., and Shima, D. T. (2002). Spatially restricted patterning cues provided by heparin-binding VEGF-A control blood vessel branching morphogenesis. Genes Dev. 16, 2684–2698.
Shalaby, F., Rossant, J., Yamaguchi, T. P., Gertsenstein, M., Wu, X. F., Breitman, M. L., and Schuh, A. C. (1995). Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 376, 62–66.
Shibuya, M., and Claesson-Welsh, L. (2006). Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp. Cell Res. 312, 549–560.
Stalmans, I., Ng, Y. S., Rohan, R., Fruttiger, M., Bouché, A., Yuce, A., Fujisawa, H., Hermans, B., Shani, M., Jansen, S., Hicklin, D., Anderson, D. J., Gardiner, T., Hammes, H. P., Moons, L., Dewerchin, M., Collen, D., Carmeliet, P., and D’Amore, P. A. (2002). Arteriolar and venular patterning in retinas of mice selectively expressing VEGF isoforms. J. Clin. Invest. 109, 327–336.
Stokes, C. L., Lauffenburger, D. A., and Williams, S. K. (1991). Migration of individual microvessel endothelial cells: stochastic model and parameter measurement. J. Cell. Sci. 99(Pt 2), 419–430.
Storkebaum, E., Lambrechts, D., Dewerchin, M., Moreno-Murciano, M. P., Appelmans, S., Oh, H., Van Damme, P., Rutten, B., Man, W. Y., De Mol, M., Wyns, S., Manka, D., Vermeulen, K., Van Den Bosch, L., Mertens, N., Schmitz, C., Robberecht, W., Conway, E. M., Collen, D., Moons, L., and Carmeliet, P. (2005). Treatment of motoneuron degeneration by intracerebroventricular delivery of VEGF in a rat model of ALS. Nat. Neurosci. 8, 85–92.
Sun, S., Wheeler, M. F., Obeyesekere, M., and Patrick, C. W. Jr. (2005). A deterministic model of growth factor-induced angiogenesis. Bull. Math. Biol. 67, 313–337.
Tanaka, K., Yamaguchi, S., Sawano, A., and Shibuya, M. (1997). Characterization of the extracellular domain in vascular endothelial growth factor receptor-1 (Flt-1 tyrosine kinase). Jpn. J. Cancer Res. 88, 867–876.
Vempati, P., Mac Gabhann, F., and Popel, A. S. (2010). Quantifying the proteolytic release of extracellular matrix-sequestered VEGF with a computational model. PLoS ONE 5, e11860. doi: 10.1371/journal.pone.0011860
Waltenberger, J., Claesson-Welsh, L., Siegbahn, A., Shibuya, M., and Heldin, C. H. (1994). Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J. Biol. Chem. 269, 26988–26995.
Wu, F. T., Stefanini, M. O., Mac Gabhann, F., Kontos, C. D., Annex, B. H., and Popel, A. S. (2010a). A systems biology perspective on sVEGFR1: its biological function, pathogenic role and therapeutic use. J. Cell. Mol. Med. 14, 528–552.
Wu, F. T., Stefanini, M. O., Mac Gabhann, F., Kontos, C. D., Annex, B. H., and Popel, A. S. (2010b). VEGF and soluble VEGF receptor-1 (sFlt-1) distributions in peripheral arterial disease: an in silico model. Am. J. Physiol. Heart Circ. Physiol. 298, H2174–H2191.
Zeng, G., and Bautch, V. L. (2009). Differentiation and dynamic analysis of primitive vessels from embryonic stem cells. Methods Mol. Biol. 482, 333–344.
Keywords: angiogenesis, vascular development, computational model, mathematical model, sFlt-1, VEGF, capillary sprouting
Citation: Hashambhoy YL, Chappell JC, Peirce SM, Bautch VL and Mac Gabhann F (2011) Computational modeling of interacting VEGF and soluble VEGF receptor concentration gradients. Front. Physio. 2:62. doi: 10.3389/fphys.2011.00062
Received: 16 February 2011; Accepted: 30 August 2011; Published online: 04 October 2011.
Edited by:
Timothy W. Secomb, University of Arizona, USAReviewed by:
Katie Bentley, Cancer Research UK, UKYi Jiang, Los Alamos National Laboratory, USA
Alexander R. A. Anderson, Moffitt Cancer Center and Research Institute, USA
Copyright: © 2011 Hashambhoy, Chappell, Peirce, Bautch and Mac Gabhann. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Yasmin L. Hashambhoy, Institute for Computational Medicine, Johns Hopkins University, 3400 North Charles Street, Hackerman Hall 316A, Baltimore, MD 21218, USA. e-mail:eWhhc2hhbTJAamhtaS5lZHU=; Shayn M. Peirce, Department of Biomedical Engineering, University of Virginia, Box 800759, Health System, Charlottesville, VA 22908. e-mail:c21wNnBAdmlyZ2luaWEuZWR1