 Jingzhen Yuan1*
Jingzhen Yuan1* Yannan Liu1,2
Yannan Liu1,2 Tanya Tan1 Sushovan Guha3 Ilya Gukovsky1 Anna Gukovskaya1
Tanya Tan1 Sushovan Guha3 Ilya Gukovsky1 Anna Gukovskaya1 Stephen J. Pandol1
Stephen J. Pandol1
- 1 South California Research Center for Alcoholic Liver and Pancreatic Diseases, Veterans Affairs Greater Los Angeles Healthcare System, University of California at Los Angeles, Los Angeles, CA, USA
- 2 Beijing Hospital, Beijing, China
- 3 Department of Gastroenterology, Hepatology and Nutrition, University of Texas M.D. Anderson Cancer Center, Houston, TX, USA
Inflammation and acinar cell necrosis are two major pathological responses of acute pancreatitis, a serious disorder with no current therapies directed to its molecular pathogenesis. Serine/threonine protein kinase D family, which includes PKD/PKD1, PKD2, and PKD3, has been increasingly implicated in the regulation of multiple physiological and pathophysiological effects. We recently reported that PKD/PKD1, the predominant PKD isoform expressed in rat pancreatic acinar cells, mediates early events of pancreatitis including NF-κB activation and inappropriate intracellular digestive enzyme activation. In current studies, we investigated the role and mechanisms of PKD/PKD1 in the regulation of necrosis in pancreatic acinar cells by using two novel small molecule PKD inhibitors CID755673 and CRT0066101 and molecular approaches in in vitro and in vivo experimental models of acute pancreatitis. Our results demonstrated that both CID755673 and CRT0066101 are PKD-specific inhibitors and that PKD/PKD1 inhibition by either the chemical inhibitors or specific PKD/PKD1 siRNAs attenuated necrosis while promoting apoptosis induced by pathological doses of cholecystokinin-octapeptide (CCK) in pancreatic acinar cells. Conversely, up-regulation of PKD expression in pancreatic acinar cells increased necrosis and decreased apoptosis. We further showed that PKD/PKD1 regulated several key cell death signals including inhibitors of apoptotic proteins, caspases, receptor-interacting protein kinase 1 to promote necrosis. PKD/PKD1 inhibition by CID755673 significantly ameliorated necrosis and severity of pancreatitis in an in vivo experimental model of acute pancreatitis. Thus, our studies indicate that PKD/PKD1 is a key mediator of necrosis in acute pancreatitis and that PKD/PKD1 may represent a potential therapeutic target in acute pancreatitis.
Introduction
Pancreatic acinar cell necrosis is a major pathological response of acute pancreatitis associated with significant morbidity and mortality from the disease (Gukovskaya and Pandol, 2004; Pandol et al., 2007). Currently, there are no therapies directed to the molecular pathogenesis of necrosis in pancreatitis (Pandol et al., 2007). Thus, elucidating the molecular signals that mediate acinar cell necrosis is important for developing new therapeutic strategies.
Apoptosis and necrosis are two major forms of acinar cell death in acute pancreatitis and associated with specific morphological and biochemical features (Gukovskaya and Pandol, 2004; Mareninova et al., 2006). Apoptosis is a tightly regulated process involving the caspase family of cysteine proteases and manifests as chromatin condensation, nuclear shrinkage, and reduction in cell volume but with intact plasma membranes, thus there is no leakage of cellular contents to the extracellular environment. Conversely, necrosis leads to early plasma membrane permeabilization and rupture, cytoplasmic swelling, and release of intracellular constituents to the extracellular space. There is no sign of nuclear shrinkage (Gukovskaya and Pandol, 2004; Pandol et al., 2007). Necrosis has been considered a passive, unregulated form of cell death; however, recent studies indicate that necrosis can occur in a tightly regulated fashion, so called as programmed necrosis (Edinger and Thompson, 2004; Mareninova et al., 2006; Krantic et al., 2007; He et al., 2009; Moquin and Chan, 2010).
Although the consequences of apoptosis and necrosis are distinct in pancreatitis, the mechanisms underlying these two types of cell death are interrelated (Kaiser et al., 1995; Bhatia et al., 1998; Bhatia, 2004a,b; Gukovskaya and Pandol, 2004; Pandol et al., 2007). Understanding the regulation of the two death pathways in acute pancreatitis is important because the severity of acute pancreatitis in both humans and in experimental models correlates with the extent of necrosis (Pandol et al., 2007). In animal models of pancreatitis the severity also inversely correlates with apoptosis (Kaiser et al., 1995; Bhatia et al., 1998; Bhatia, 2004a,b; Mareninova et al., 2006). That is, inhibition of apoptosis pathways leads to necrosis and increased severity of pancreatitis, whereas stimulation of apoptosis attenuates the severity of the disease (Kaiser et al., 1995; Bhatia et al., 1998; Bhatia, 2004a,b). Therefore, revealing the key signaling molecules that determine the pattern of pancreatic acinar cell death (i.e., apoptosis versus necrosis) in pancreatitis will provide potential molecular targets for therapy in this disease.
The family of caspases is a major mediator of apoptosis in pancreatic acinar cells (Pandol et al., 2007). Caspases are divided into two classifications. “Effector” caspases (caspase-3, -7) are responsible for the cleavages that disassemble the cell. “Initiator” caspases (caspase-8, -9) trigger cleavage and activation of the “effector” caspases. Activation of both initiator and effector caspases mediates cell apoptosis. Cholecystokinin-octapeptide (CCK-8 or CCK), at supraphysiologic doses which induces pancreatitis, has been shown to stimulate the initiator and the effector caspases-8, -9, and -3 and causes apoptosis of pancreatic acinar cells in vitro and in vivo experimental pancreatitis (Beil et al., 2002; Gukovskaya et al., 2002; Bhatia, 2004a; Mareninova et al., 2006; Sung et al., 2009). Interestingly, increasing evidence (Bhatia, 2004b; Mareninova et al., 2006) indicates that in addition to apoptosis, caspases also regulate other processes in pancreatitis; in particular, caspases negatively regulate necrosis. Thus, caspase activation may function as a critical point switching the cell death response toward apoptosis and away from necrosis.
NF-κB activation is a key intracellular event in acute pancreatitis (Pandol et al., 2007). NF-κB activation is known to increase the expression of the family of inhibitors of apoptosis proteins (IAPs; Stehlik et al., 1998; Deveraux and Reed, 1999; Pahl, 1999; Gukovskaya and Pandol, 2004; Zou et al., 2004; Kawakami et al., 2005; Kerbauy et al., 2005; Pandol et al., 2007), such as X-linked IAP (XIAP; Stehlik et al., 1998; Deveraux and Reed, 1999; Gukovskaya and Pandol, 2004) and survivin (Deveraux and Reed, 1999; Kawakami et al., 2005), and anti-apoptotic protein FLICE-inhibitory protein (c-FLIP; Kerbauy et al., 2005) that inhibit the caspase system, the essential mediator of apoptotic death pathways (Deveraux and Reed, 1999; Tang et al., 2000; Bratton et al., 2001). The importance of IAPs in regulating the type of death in pancreatitis has been reported by our group (Mareninova et al., 2006; Pandol et al., 2007). For example, blocking XIAP resulted in increased caspase activation and apoptosis while decreasing necrosis and the severity of pancreatitis.
The mechanisms underlying necrosis are beginning to be explored. A number of reports indicate that the programmed necrosis requires the receptor-interacting protein kinase 1 (RIP1; Lin et al., 1999; Chan et al., 2003; Meylan and Tschopp, 2005; Festjens et al., 2007; Galluzzi et al., 2009; He et al., 2009; Moquin and Chan, 2010; Trichonas et al., 2010). RIP1 forms a death-signaling complex with the Fas-associated death domain and caspases in response to death domain receptor stimulation (Lin et al., 1999; Chan et al., 2003; Meylan and Tschopp, 2005; Festjens et al., 2007; Trichonas et al., 2010). During apoptosis, RIP1 is cleaved and inactivated by caspase-3 and -8 (Lin et al., 1999; Chan et al., 2003; Moquin and Chan, 2010). The regulation of RIP1 by caspases has been suggested to be one of mechanisms underlying the protective role of caspases from necrosis in cerulein-induced pancreatitis (Mareninova et al., 2006; He et al., 2009).
Serine/threonine protein kinase D family, which includes PKD/PKD1, PKD2, and PKD3, has emerged as a major target in the signal transduction pathways induced by G protein coupled receptor (GPCR) agonists and polypeptide growth factors in a variety of cell types including pancreatic acinar cells (Berna et al., 2007; Yuan et al., 2008; Chen et al., 2009; Thrower et al., 2011). PKD family members are activated through PKC-dependent and -independent pathways (Matthews et al., 1999; Berna et al., 2007; Jacamo et al., 2008; Yuan et al., 2008; Chen et al., 2009; Rozengurt, 2011; Thrower et al., 2011) and have been increasingly implicated in the regulation of multiple cellular functions in health and disease (reviewed in Rozengurt, 2011), such as protein secretion and Golgi function, cell proliferation, and apoptosis, oxidative stress and inflammation, cardiac diseases and cancer (Trauzold et al., 2003; Storz et al., 2004; Chiu et al., 2007; Fielitz et al., 2008; Sharlow et al., 2008; Harikumar et al., 2010; Rozengurt, 2011). Particularly, PKD/PKD1, the predominant PKD isoform expressed in rat pancreatic acinar cells (Berna et al., 2007; Yuan et al., 2008) has been shown to mediate secretagogue-induced signal pathways (Berna et al., 2007; Yuan et al., 2008; Thrower et al., 2011). We recently reported that PKD/PKD1 mediates NF-κB activation and inappropriate intracellular digestive enzyme activation in pancreatitis (Yuan et al., 2008; Thrower et al., 2011).
Despite of these studies, the role of PKD/PKD1 in another critical pathological response in pancreatitis, acinar cell necrosis, has not been investigated. Our present study was performed to determine the role of PKD/PKD1 in cell death responses of acute pancreatitis. Our findings demonstrate that PKD/PKD1 promotes necrosis and inhibits apoptosis in pancreatitis through regulation of IAPs, caspases, and RIP1. In addition, PKD inhibitors significantly ameliorated necrosis and severity of pancreatitis. Thus, PKD/PKD1 is a potential therapeutic target in acute pancreatitis.
Experimental Procedures
Preparation and Treatments of Dispersed Pancreatic Acini
Pancreatic acini were prepared from male Sprague-Dawley rats (75–100 g) using a collagenase digestion method as described previously (Yuan et al., 2008; Thrower et al., 2011) and then incubated in medium 199 supplemented with 0.01% trypsin inhibitor (w/v), penicillin (100 U/ml), and streptomycin (0.1 mg/ml) at 37°C and 5% CO2-humidified atmosphere. For experimental purposes, the acini were pre-incubated in the medium 199 with or without inhibitors and then treated further with cholecystokinin-8 (CCK). Acini were collected and washed twice with cold PBS.
Experimental Pancreatitis
Pancreatitis was induced in male (120–150 g) Sprague-Dawley rats by up to four hourly intraperitoneal (IP) injections of 20 μg/kg cerulein, a CCK analog used for experimental pancreatitis models. Control animals received similar injections of physiologic saline. PKD inhibitor CID755673 (15 mg/kg) dissolved in 120–150 μl (15 mg/ml) vehicle (DMSO) or same volume DMSO was applied in rats as a single IP injection 60 min before cerulein treatment. The animals were euthanized by CO2-induced asphyxiation at 30 min, and 4 h after the first cerulein injection and then the blood and pancreas were harvested for measurements. Animal care and all procedures were approved by the Institutional Animal Care and Use Committees of the Veterans Affairs Greater Los Angeles Healthcare System.
Quantification of Necrosis
Necrosis in pancreatic acini and AR42J cells was determined by the release of lactate dehydrogenase (LDH) into the incubation medium, as previously described (Gukovskaya et al., 1997, 2002; Gukovskaya and Pandol, 2004; Sung et al., 2009), LDH activity was measured using Cytotoxicity Detection Kit (Roche Diagnostics, Indianapolis, IN, USA) according to the manufacturer’s protocol.
Quantification of necrosis in in vivo pancreatitis was performed on pancreatic tissue (collected after four hourly cerulein injections) sections stained with H&E. Images from multiple random, non-overlapping sections were captured under a high-power field (×400-magnification, 6–10 random fields per section) with a Nikon Eclipse E600 microscope equipped with a digital camera using the SPOT imaging software (Diagnostic Instruments, Sterling Heights, MI, USA). Cells with swollen cytoplasm, loss of plasma membrane integrity, and leakage of organelles into interstitium were considered necrotic. A total of at least 2000 acinar cells were counted on tissue sections from each animal and three to five animals per condition were counted.
Quantification of Apoptosis
Apoptosis was quantified in pancreatic tissue sections (four hourly cerulein injections) stained with TdT-mediated dUTP nick end-labeling (TUNEL) to measure DNA breaks, as we described previously (Gukovskaya and Pandol, 2004; Mareninova et al., 2006) using DeadEnd™ Fluorometric TUNEL System kit (Promega, Madison, WI, USA). Briefly, tissue was fixed in 4% buffered formaldehyde, embedded in paraffin, and 5-μm-thick sections were adhered to glass slides. Sections were stained using terminal deoxynucleotidyl transferase and FITC-labeled dUTP according to the manufacturer’s protocol (Promega, Madison, WI, USA). Nuclei were counterstained with DAPI. Fluorescence images were obtained under a high-power field (×400-magnification with a fluorescence microscope (Nikon Eclipse E600) equipped with a monochrome camera (CoolSnap, 20 MHz; Roper Scientific, Germany) and the MetaMorph imaging system (Universal Imaging Corporation). Apoptosis was quantified by morphometric analysis of apoptotic acinar cells-stained with bright green relative to the number of total nuclei-stained with blue using ImageJ 1.42I software (from NIH). For these and other quantifications of histological measurements, a total of at least 2000 acinar cells were counted on tissue sections from each animal and three to five animals per condition were counted.
For the measurement of apoptosis in pancreatic acinar cells, the isolated pancreatic acinar cells were plated on polylysine-coated glass coverslips and fixed with methanol at −20°C for 10 min. The transfected AR42J cells growing on glass coverslips were fixed with 4% formaldehyde in PBS for 25 min at 4°C. Apoptosis of the acinar cells was quantified by use of TUNEL staining as described above. A total of at least 3000 acinar cells were counted on the cell smears for each condition.
Apoptosis of transfected AR42J cell culture was also quantified by measuring internucleosomal DNA fragmentation using Cell Death Detection ELISA Plus kit (Roche Molecular Biochemicals, Manheim, Germany) according to the manufacturer’s instructions.
Serum Amylase and Lipase Measurements
Animal serum samples were sent to Antech Diagnostics (Irvine, CA, USA) Custom Service for measurements. The serum amylase and lipase activities were determined in a Hitachi 707 analyzer using the company standard enzyme Kinetic methods (assay code T040 for amylase and T165 for lipase).
Histologic Analysis for Pancreas Inflammatory Cell Infiltration, Vacuolization, and Edema
Quantification of inflammatory cell infiltration and vacuolization was performed on H&E stained pancreatic tissue (four hourly cerulein injections) sections from at least four rats per group and expressed as the number of inflammatory cells or vacuoles per 100 acinar cells. Pancreatic edema grading was made from 0 to 3 according to the Schoenberg grading system (Schoenberg et al., 1990) as 0: no edema; 1: interlobular edema; 2: moderate interlobular and intra-acinar edema; and 3: severe interlobular and intra-acinar edema.
PKD/PKD1 Immunoprecipitation and in vitro Kinase Assay
PKD/PKD1 immunoprecipitation and in vitro kinase assay were performed by the procedures as described previously (Yuan et al., 2008; Thrower et al., 2011). Briefly, PKD/PKD1 in pancreatic acinar cell or pancreatic tissue lysates was immunoprecipitated at 4°C for 3 h with the PKD/PKD1 C-20 antibody (1:100) and protein-A-agarose. Exogenous substrate syntide-2 phosphorylation by immunoprecipitated PKD/PKD1 was carried out by mixing 20 μl of the washed immunocomplexes with 10 μl of a phosphorylation mixture containing 100 μM ATP (including [γ-32P] ATP at 2 μCi/assay or with specific activity, 400–600 cpm/pmol) and 2.5 mg/ml syntide-2 (PLARTLSVAGLPGKK) in kinase buffer. After 10 min of incubation at 30°C, the reaction was stopped by adding 100 μl of 75 mM H3PO4, and 75 μl of the supernatant was spotted on P-81 phosphocellulose paper. Free [γ-32P]ATP was separated from the labeled substrate by washing the P-81 paper four times for 5 min in 75 mM H3PO4. The papers were dried, and the radioactivity incorporated into syntide-2 was determined by Cerenkov counting.
Measurement of Caspase Activities
Caspase activities were measured using a fluorogenic assay with substrates specific for caspase-3 (Ac-DEVD-AMC), caspase-8 (Ac-IETD-AMC), or caspase-9 (Ac-LEHD-AMC) as we described previously (Mareninova et al., 2006; Sung et al., 2009). Pancreatic tissue or acinar cell samples were homogenized in a lysis buffer containing 150 mM NaCl, 50 mM Tris–HCl (pH 7.5), 0.5% Igepal CA-630, and 0.5 mM EDTA, centrifuged for 15 min at 16,000 g, and the supernatants were collected. Proteolytic reactions were carried out at 37°C in a buffer containing 25 mM HEPES (pH 7.5), 10% sucrose, 0.1% CHAPS, and 10 mM dithiothreitol, using specific substrates for each caspase. Cleavage of these substrates relieves 7-amino-4-methylcoumarin (AMC), which emits fluorescent signal with excitation at 380 nm and emission at 440 nm. Fluorescence was calibrated using a standard curve for AMC. The data are expressed as picomoles of AMC per milligram of protein per minute.
Cell Culture and Transient Transfection
AR42J cells were cultured in F-12K medium supplemented with 20% fetal bovine serum at 37°C with a humidified atmosphere containing 5% CO2. Transfection of AR42J cells with cDNA plasmids or siRNAs was done using Nucleofector™ II (Amaxa Inc., Gaithersburg, MD, USA) according to the manufacturer’s protocol.
The plasmid constructs encoding PKD/PKD1, PKD/PKD1 siRNAs Pool (pool of four duplexes; Yuan et al., 2008), and XIAP siRNAs (pool of three duplexes) were applied in the transfection. The sequences of the four PKD1 siRNAs (sense strands) are as follows: GAAGAGAUGUAGCUAUUAA; GAAAGAGUGUUUGUU-GUUA; CAUAAGAGAUGUGCAUUUA; and CAGCGAAUGUAGUGUAUUA. The sequences of the three XIAP siRNAs (sense strands) are GAAUGUUAAUGUUCGAAGUtt; GAAUUUCCCAAAUUCAACAtt; and GGAUAGAAAUAUUGCUAUAtt. The vector pcDNA3 and siCONTROL Non-Targeting siRNA Pool (pool of four duplexes) were used as controls. Three days after transfection, cells were used for experiments. The efficiency of transfection assessed by expression of the GFP plasmid was 70–80% in AR42J cells.
Preparation of Tissue and Cell Lysates and Western Blot Analysis
Portions of frozen pancreas tissue were homogenized and sonicated on ice in lysis buffer as described previously (Yuan et al., 2008). The tissue lysates were rotated for 40 min at 4°C and centrifuged at 4°C for 15 min at 16,000 g. The supernatants were collected and stored at −80°C. Aliquots of the acinar cells were resuspended in the lysis buffer and processed as described above for tissue samples. The protein concentration in supernatants was measured using the Bio-Rad protein assay reagent. Western blot analyses were performed as described previously (Mareninova et al., 2006; Yuan et al., 2008; Thrower et al., 2011). The membranes were blocked by 1–2 h incubation with 5% non-fat dried milk in Tris-buffered saline, pH 7.2, and probed overnight at 4°C with specific primary antibodies at a 1:500–1:1000 dilution in the Tris-buffered saline containing 3% non-fat dried milk. Then the membranes were incubated with secondary antibodies conjugated with horseradish peroxidase at 1:5000 dilution for 1 h at room temperature. Blots were developed by using the enhanced chemiluminescence detection kit (Pierce). When reprobing was necessary, the membrane was stripped of bound antibody by incubating in Re-Blot Plus Mild Solution (Millipore, Temecula, CA, USA) at room temperature for 20 min.
Statistical Analysis of Data
Results are means ± SE and represent data from at least three independent experiments. The experimental data was evaluated by the analysis of variance (ANOVA) followed by Bonferroni multiple comparison post hoc tests with the GraphPad Prism software (GraphPad Software Inc., La Jolla, California). T-tests were used to analyze differences between two groups. P < 0.05 was considered statistically significant.
Reagents
CCK-8 was from American Peptide (Sunnyvale, CA, USA); Cerulein was from Peninsula Laboratories (Belmont, CA, USA). Medium 199 was from GIBCO (Grand Island, NY, USA). AR42J cell line and medium F-12K were from the American Type Culture Collection (ATCC; Manassas, VA, USA). Mouse PKD/PKD1 siRNAs Pool (pool of 4 duplexes) and siCONTROL Non-Targeting siRNA Pool were from Dharmacon (Chicago, IL, USA). XIAP siRNAs were from Invitrogen Life Technologies (Carlsbad, CA, USA). CRT0066101 was obtained from Cancer Research Technology, London, UK. CID755673 was obtained from two sources: a custom made synthesis from Chem. Div. Res. Ins. (San Diego, CA, USA) and a commercially available source TOCRIS (MO, USA). [γ-32P] ATP was from Perkin Elmer (Torrance, CA, USA). Nitrocellulose membranes were from Schleicher and Schuell BioScience. Carbachol, GF1 (also known as GF 109203X or bisindolylmaleimide I) were from Calbiochem (La Jolla, CA, USA). Caspase fluorogenic substrates Ac-IETD-AMC, Ac-DEVD-AMC, and Ac-LEHD-AMC were from Biomol (Plymouth Meeting, PA, USA). Antibodies against PKD/PKD1 C-20, caspase-8, c-FLIP, survivin were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against phosphoserine 744/748 PKD (predominantly detects the phosphorylated state of Ser744), phosphoserine 916 PKD, phosphoserine 152/156-MARCKS, XIAP, Cleaved caspase-3, and GAPDH were obtained from Cell Signaling Technology (Beverly, MA, USA). phosphoserine 82 Hsp27 was obtained from ALEXIS Biochemicals (San Diego, CA, USA). Caspase-9 antibody was from Stressgen (San Diego, CA, USA). RIP1 antibody was from BD Science (San Diego, CA, USA). Protein-A-agarose was from Roche Applied Science (Mannheim, Germany) and PKD substrate, syntide-2, was from Bachem (Chicago, IL, USA). Other items were from standard suppliers or as indicated in text.
Results
CCK-Induced PKD/PKD1 Activation is Inhibited by PKD Inhibitors CID755673 and CRT0066101 in Pancreatic Acinar Cells
CID755673 and CRT0066101, two newly developed small molecule PKD inhibitors, have been demonstrated to inhibit PKD-mediated cancer growth and other cellular functions (Sharlow et al., 2008; Harikumar et al., 2010; Thrower et al., 2011). Both inhibitors were reported as selective PKD inhibitors (Sharlow et al., 2008; Harikumar et al., 2010; Thrower et al., 2011). For example, their specificity for PKD family members was confirmed in in vitro kinase assays comprising a panel of 14–90 protein kinases (Sharlow et al., 2008; Harikumar et al., 2010) including PLK1, CAK, AKT/PKB, CAMKII, MAPK, MAPKK, c-Raf, c-Src, and c-Abl (Sharlow et al., 2008; Harikumar et al., 2010). Importantly, both inhibitors were demonstrated to specifically block PKD activity but not suppress its upstream regulators, the PKCs including PKCα, PKCβ, PKCε, and PKCδ (Sharlow et al., 2008; Harikumar et al., 2010).
To demonstrate effects of these two PKD inhibitors in pancreas, we first examined their specificity for PKD inhibition in an in vitro experimental pancreatitis model of isolated pancreatic acini (Figure 1). Isolated pancreatic acini were pretreated with CID755673 or CRT0066101 and then stimulated with a supramaximal dose (100 nM) of the pancreatic secretagogue CCK which is known to cause cell pathologies of pancreatitis in vitro (Gukovskaya and Pandol, 2004). We applied 10 μM CRT0066101 and 25 μM CID755673 for maximal inhibition of PKD activation based on previous reports including our own measurements (Sharlow et al., 2008; Harikumar et al., 2010; Thrower et al., 2011).
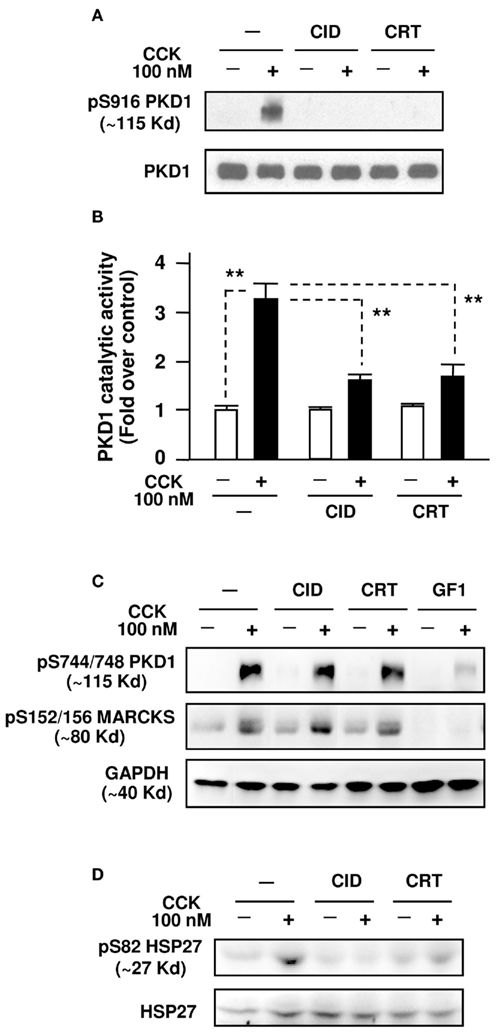
Figure 1. CID755673 and CRT0066101 selectively inhibit supramaximal dose CCK-induced PKD/PKD1 activation in pancreatic acinar cells. Isolated pancreatic acini were pre-incubated for 3 h with CID755673 (CID), CRT0066101 (CRT), or GF 109203X (GF1), followed by incubation with 100 nM CCK for 30 min. The acini were collected and lysed. (A) PKD/PKD1 autophosphorylation at its Ser916 (pS916 PKD1) was determined by Western blot analysis of the acinar lysates using anti-phospho-Ser916 PKD/PKD1 antibody, as described in Section “Experimental Procedures.” Blots were stripped and re-probed with PKD1 C-20 antibody for loading control. Shown are representative blots from at least three independent experiments. (B) PKD/PKD1 was immunoprecipitated from the acini lysate with PKD1 C-20 antibody and the PKD/PKD1 catalytic activity was measured with in vitro kinase assay as described in Section “Experimental Procedures.” Activity values were normalized by the control without CCK stimulation. Results are means ± SE (n = 3) for each condition. **P < 0.01 comparing the control versus CCK alone without inhibitor pretreatments, and comparing CID + CCK or CRT + CCK versus CCK alone, as indicated with dash lines. (C) Western blot analyses of the acini lysates with anti-phospho-Ser744/748 PKD or anti-phospho-Ser152/156-MARCKS antibodies. Blots were stripped and re-probed with GAPDH antibody for loading controls. (D) Western blot analyses of the acinar lysates with anti-phospho-Ser82 Hsp27 antibodies. Blots were stripped and re-probed with Hsp27 antibody for loading controls.
For the current studies, our results showed that CCK-induced PKD/PKD1 activation dramatically/significantly (∼3- to 4-fold over the control) measured by both autophosphorylation at PKD/PKD1 Ser916 with Western blot and an in vitro kinase assay (Figures 1A,B). Pre-incubation with either 25 μM CID755673 or 10 μM CRT0066101 almost completely blocked PKD/PKD1 activation (Figures 1A,B). Of note, the PKD/PKD1 inhibitors did not affect CCK-induced PKD/PKD1 activation loop (Ser744/748) phosphorylation (Figure 1C) which is known to result from PKC-dependent (predominant) and -independent (minor) pathways in pancreatic acinar cells (Berna et al., 2007; Yuan et al., 2008). But, the Ser744/748 phosphorylation was largely inhibited by GF109203X (GF1, 5 μM), a conventional and novel PKC inhibitor (Figure 1C). These results demonstrated the selectivity of our inhibitors for PKD/PKD1 without an effect on PKC-dependent or -independent pathways of PKD activation.
Furthermore, we showed that neither CID755673 nor CRT0066101 blocked phosphorylation of myristolated alanine-rich C kinase substrate (pS152/156-MARCKS), a known PKC substrate (Heemskerk et al., 1993; Ramsden, 2000), whereas the PKC inhibitor GF1 potently blocked the phosphorylation of MARCKS (Figure 1C). Conversely, both CID755673 and CRT0066101 abrogated PKD-dependent phosphorylation of heat shock protein 27 (pS82-Hsp27), a physiologic substrate of PKD/PKD1 (Doppler et al., 2005; Yuan and Rozengurt, 2008), in pancreatic acini (Figure 1D). All these results confirmed that both CID755673 and CRT0066101 are PKD-specific inhibitors without an effect on activities of the PKCs.
PKD/PKD1 Regulates Cell Death Pathways in Pancreatic Acinar Cells – Promoting Cell Necrosis and Suppressing Apoptosis and Caspase Activation
We next investigated the role of PKD/PKD1 in death pathways by examining the effect of the two PKD inhibitors on cell necrosis and apoptosis death pathways in pancreatic acinar cells stimulated with a supramaximal dose of CCK, which serves as an in vitro experimental pancreatitis model (Figure 2). Isolated pancreatic acini were pretreated with CID755673 or CRT0066101 for 1 h and then further incubated with 100 nM CCK for 4 h.
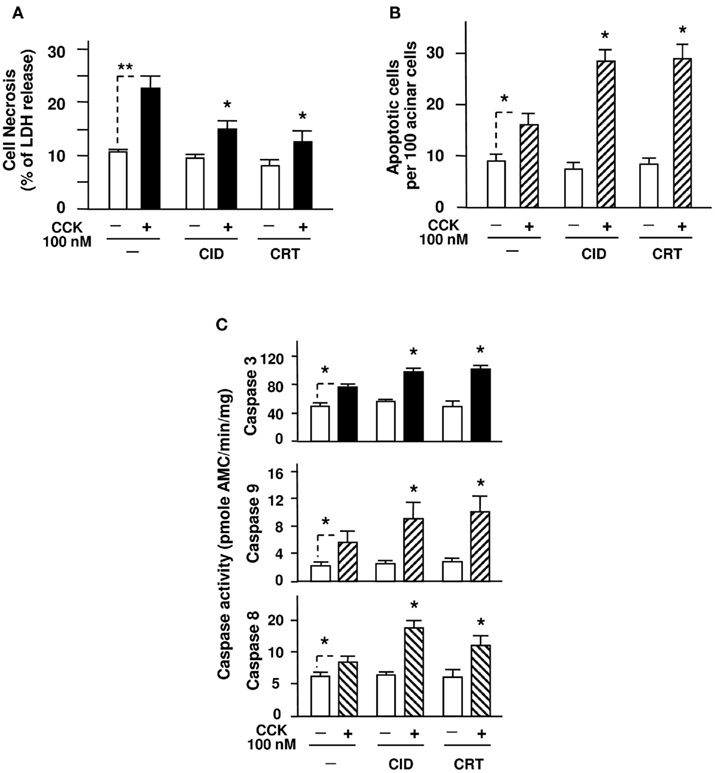
Figure 2. PKD inhibitors CID755673 and CRT0066101 decrease necrosis and enhance apoptosis in pancreatic acini induced by supramaximal dose CCK. Rat pancreatic acini were pre-incubated for 1 h with CID755673 (CID, 25 μM) or CRT0066101 (CRT, 10 μM), followed by incubation with 100 nM CCK for 4 h. (A) Acinar cell necrosis was measured by the percentage of total cellular LDH released into the medium, as described in Section “Experimental Procedures.” (B) Apoptosis was measured by the percentage of TUNEL staining positive cells. For each condition, at least 3000 cells were counted. (C) Caspase activities were measured as described in Section “Experimental Procedures” in the lysates of isolated pancreatic acini. Graphs (A–C) show mean ± SE (n = 3–5) for each condition. **P < 0.01 comparing the control versus CCK alone without inhibitor pretreatments as indicated with dash lines; *P < 0.05 comparing CID + CCK or CRT + CCK versus CCK alone without inhibitor.
To assess necrosis in pancreatic acini, we measured LDH release into the extracellular medium. Necrosis is characterized by progressive cell swelling, rupture of the plasma membrane and, hence, release of cytoplasmic constituents, particularly, the small molecule protein LDH, in the incubation medium. Thus, measurement of LDH release into the extracellular medium has been used for measuring necrosis of isolated pancreatic acini in multiple studies of pancreatitis (Gukovskaya et al., 1997, 2002; Gukovskaya and Pandol, 2004; Sung et al., 2009). LDH levels in both cell lysate and medium samples were assayed and the results were expressed as the percent of cellular LDH released into the medium.
Our results showed that either CID755673 or CRT0066101 alone did not effect on the basal level of LDH release, which was in accordance with our previous report (Thrower et al., 2011). Importantly, pretreatment of the acinar cells with CID755673 or CRT0066101 significantly reduced supramaximal dose of CCK-induced necrosis after 4 h stimulation (Figure 2A). Of note, our previous results from 2 h of incubation of CCK in PKD/PKD1 inhibitor CRT0066101-treated pancreatic acini already showed a small decrease in LDH release (Thrower et al., 2011). The difference with the present results from 4 h of incubation with CCK suggests that the necrosis effect of CCK is time-dependent, thus the inhibitors show greater benefit with a longer duration of incubation.
Next, we determined the effect of the two PKD inhibitors on apoptosis of the isolated acini. Distinct from necrosis, there is no leakage of cellular contents to the extracellular environment in apoptosis. The features of morphologic changes in apoptosis include shrinkage of the cell and its organelles and condensation of nuclear chromatin followed by intranucleosomal DNA cleavage. Thus, we measured CCK-stimulated acinar cell apoptosis by measuring DNA breaks, a biochemical hallmark of apoptosis, with a TUNEL assay. Our results showed that in contrast with their suppressing effect on CCK-induced necrosis, the two PKD inhibitors both promoted CCK-stimulated acinar cell apoptosis (Figure 2B) but had no effect on the basal apoptosis.
Apoptosis in pancreatic acinar cells is mediated mainly by activation of caspases. Supramaximal dose (100 nM) of CCK has been known to activate initiator and effector caspases-8, -9, and -3 and cause apoptosis of pancreatic acinar cells in pancreatitis (Beil et al., 2002; Gukovskaya et al., 2002; Bhatia, 2004a; Mareninova et al., 2006; Sung et al., 2009). We here determined whether the two PKD inhibitors promote apoptosis through effect on caspase activation. Similarly to the previous results (Beil et al., 2002; Gukovskaya et al., 2002; Mareninova et al., 2006; Sung et al., 2009), we also observed that 100 nM CCK caused activation of caspases-8, -9, and -3 in pancreatic acinar cells (Figure 2C). More importantly, in the acini pre-incubated with either PKD inhibitor, CID755673 and CRT0066101 both exerted a proapoptotic effect, i.e., enhanced the activation of these caspases induced by a supramaximal dose CCK (Figure 2C).
Thus, with the two PKD/PKD1 inhibitors we demonstrated that PKD/PKD1 regulates cell death in pancreatic acinar cells by promoting cell necrosis while suppressing apoptosis/caspase activation. These findings prompted us to evaluate the potential anti-necrotic effect of PKD/PKD1 inhibitors in vivo. Due to its availability, we choose CID755673 for our in vivo experimental pancreatitis model.
CID755673 Ameliorates Necrosis and Other Pancreatitis Parameters in Cerulein-induced Pancreatitis
We first tested dose- and time-dependency of the effect of CID755673 pretreatment on PKD/PKD1 catalytic activation in a CCK analog (cerulein)-induced pancreatitis to find an optimal dose of the inhibitor and the interval time between the inhibitor injection and cerulein injection for maximum inhibition of PKD/PKD1. Three doses of the inhibitor (10, 15, or 20 mg/kg) at different pretreatment lasting time (30, 45, or 60 min) were tested. Rats first received an IP injection of CID755673 at a certain dose (10, 15, or 20 mg/kg). The control animals received same volume of vehicle. After a different waiting time (30, 45, or 60 min), the inhibitor- or vehicle-pretreated animals were given an IP injection of cerulein or saline. Each combination of the inhibitor pretreatment (dose and time) was performed on three rats. The pancreas was collected in 30 min after the cerulein injection.
PKD/PKD1 catalytic activity in the pancreatic tissue lysate was measured by in vitro kinase assays. Figure 3A showed that ∼50–60% of cerulein-induced PKD/PKD1 activation was inhibited by 30 min-pretreatment of the animals with CID 755673 at a dose of 15 mg/kg. PKD/PKD1 activation was almost abolished with 60 min of pretreatment (close to basal level) at this dose. Similar results were also observed in animals pretreated with CID 755673 at a dose of 20 mg/kg, but less than 50% inhibition of PKD/PKD1 activity was achieved with a lower dose (10 mg/kg) of CID755673 at this time point (data not shown). Thus, we decided to use 15 mg/kg CID755673 and the time (60 min) for animal pretreatment with the inhibitor prior to cerulein injection throughout this study.
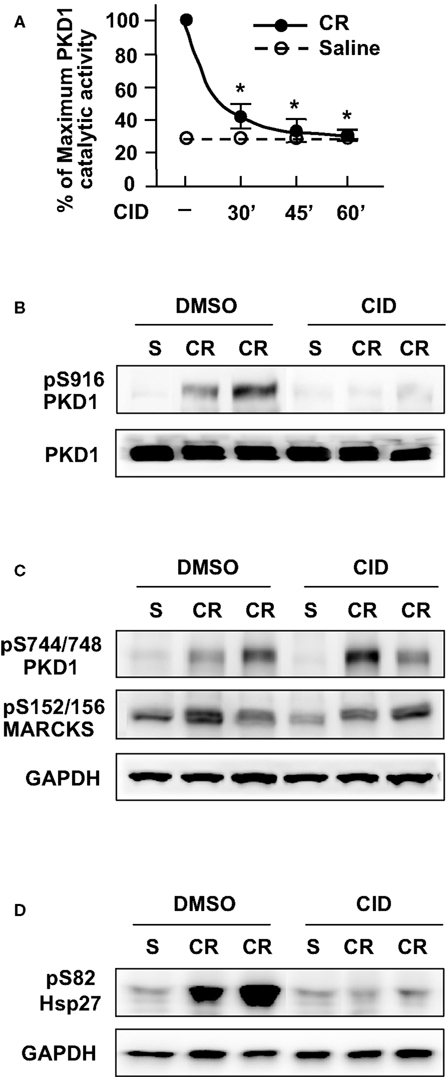
Figure 3. CID755673 pretreatment inhibits PKD/PKD1 activation in pancreatic tissue in cerulein-induced pancreatitis. (A) Rats received intraperitoneal (IP) injection of CID755673 (CID, 15 mg/kg) or same volume of vehicle DMSO. After 30, 45, or 60 min waiting time, the inhibitor- or vehicle-pretreated animals were given an IP injection of cerulean (CR) or saline (S). The pancreas was collected in 30 min after the cerulein or saline injection. The pancreas tissue lysates were immunoprecipitated with PKD/PKD1 C-20 antibody and PKD1 activity in the immunocomplexes was determined by in vitro kinase assay. PKD/PKD1 catalytic activities were expressed as percentage of maximum activity in the animals injected with cerulein without inhibitor pretreatment. Results are means ± SE (n = 3) for each condition. *P < 0.05 versus cerulein alone without inhibitor pretreatments. (B–D) The pancreas tissue was collected in 30 min after cerulein injection in rats pretreated for 60 min with CID755673 as described above and the tissue lysates were analyzed by Western blot using antibodies against phospho-Ser916 PKD1 (B), phospho-Ser744/748 PKD1 or phospho-Ser152/156-MARCKS (C), or phospho-Ser82 Hsp27 antibodies (D). The blots were then reblotted with antibodies against PKD1 C-20 or GAPDH to verify equal protein loading.
Corresponding to its inhibition to PKD/PKD1 catalytic activity, pretreatment of rats with CID755673 for 60 min also blocked PKD/PKD1 Ser916 autophosphorylation (Figure 3B). Of note, similar to the results from in vitro experiments, in vivo treatment with CID755673 had no effect on PKD/PKD1 Ser744/748 phosphorylation which was predominantly PKC-dependent and on phosphorylation of PKC substrate (pS152/156-MARCKS) in pancreatitis (Figure 3C) while CID755673 treatment blocked phosphorylation of PKD/PKD1 substrate (pS82-Hsp27) in pancreatitis (Figure 3D), confirming the specificity of CID755673 targeting PKD/PKD1 in vivo.
Next, we evaluated the effect of PKD/PKD1 inhibition on the parameters of cerulein-induced pancreatitis. After 1 h pretreatment of CID755673 (15 mg/kg) or vehicle as described above, the rats received up to four hourly IP injections of cerulein or saline. The animals were sacrificed at 30 min and 4 h after the first cerulein injection. As illustrated in Figure 4A, cerulein caused striking increases of blood amylase and lipase after four hourly IP injections of cerulein, which were significantly attenuated in CID755673-treated rats. Furthermore, CID755673 treatment dramatically ameliorated the histological damage in cerulein-induced pancreatitis (Figure 4B). The salient feature of pancreas histological changes in CID755673-pretreated animals was that cerulein-induced severe acinar cell necrosis was strikingly attenuated and almost prevented (Figure 4C). Other histopathologic features of pancreatitis including interstitial edema, accumulation of cytoplasmic vacuoles, and inflammatory cell infiltration were also greatly attenuated by CID755673 treatment (Figures 4D–F). These results from the in vivo pancreatitis model demonstrated that PKD/PKD1 inhibition blocks necrosis and ameliorates severity of acute pancreatitis.
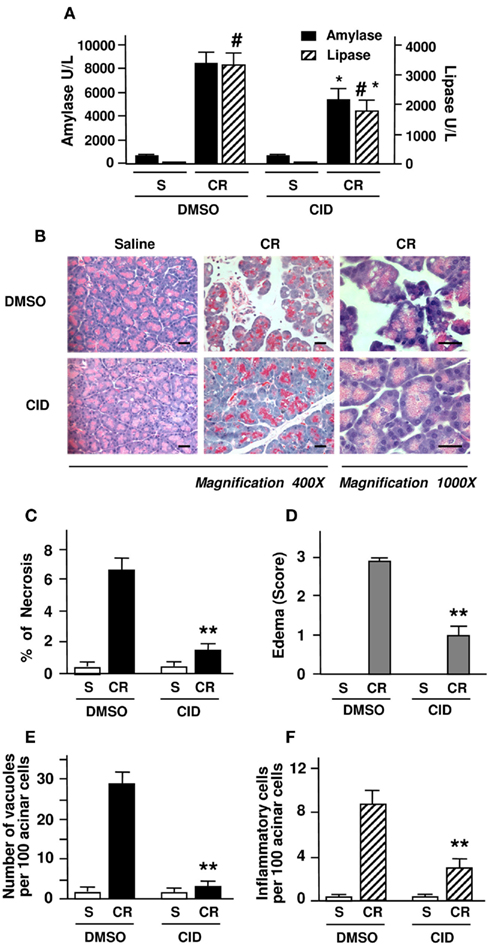
Figure 4. PKD/PKD1 inhibition by CID755673 ameliorates necrosis and other pancreatitis parameters in cerulein-induced pancreatitis. Rats received intraperitoneal (IP) injection of CID755673 (CID, 15 mg/kg) or same volume of vehicle DMSO. At 60 min after the pretreatment, pancreatitis was induced by four hourly IP injections of cerulein (CR), as described in Section “Experimental Procedures.” Control rats received saline (S). The blood and pancreas were harvested in 4 h after the first injection of cerulein for the following measurements. (A) Serum amylase and lipase levels were measured. (B) H&E staining of pancreatic tissue sections from the experiments. Each image is representative of at least 6 rats with similar results at each condition. Bars represent 20 μm. (C) Necrosis was measured on H&E stained pancreatic tissue sections. Cells with swollen cytoplasm, loss of plasma membrane integrity, and leakage of organelles into interstitium were considered necrotic. For necrosis measurement and the quantifications of following other histological measurements, a total of at least 2000 acinar cells were counted on tissue sections from each animal and three to five animals each condition were counted. (D) Pancreatic edema grading was made from 1 to 3 on the H&E stained tissue sections with Schoenberg grading system as described in Section “Experimental Procedures.” (E,F) Number of vacuole and inflammatory cell infiltration were counted on these H&E stained pancreatic tissue sections and expressed as percentage of total acinar cells. Graphs (A,C–F) show means ± SE (n = 4) for each condition. **P < 0.01 or *P < 0.05 versus cerulein alone without inhibitor pretreatments as indicated.
CID755673 Increases Apoptosis and Caspase Activities in Cerulein-induced Pancreatitis
We then investigated the effects of CID755673 on apoptosis in cerulein-induced pancreatitis (Figure 5A). The in situ TUNEL assays showed that cerulein-induced apoptosis increased two fold in CID755673-pretreated rats compared with the controls (Figure 5B). This data confirmed our in vitro experimental results demonstrating the proapoptotic effect of CID755673 on acinar cell death in pancreatitis. Combination of the results of the CID755673 effects on the two death pathways (necrosis versus apoptosis) shows that the ratio of necrosis/apoptosis in cerulein-induced pancreatitis was dramatically decreased (∼10 times less) in the CID755673-treated rats (Figure 5C).
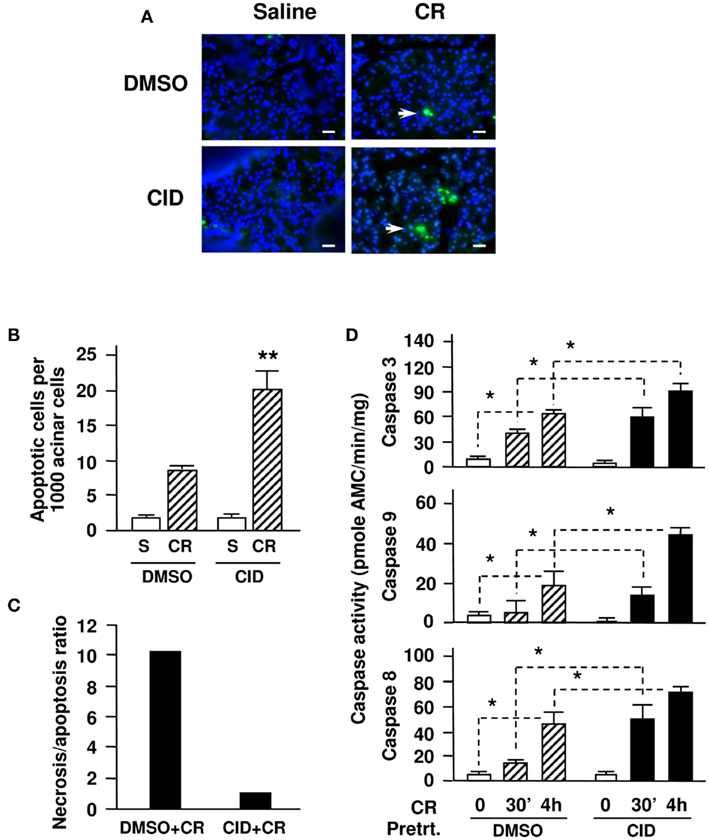
Figure 5. CID755673 enhances apoptosis and caspase activation in cerulein-induced pancreatitis. (A) In situ TUNEL staining of apoptotic acinar cells (green staining, pointed with arrows) in pancreatic tissue sections from the experiments. Nuclei were counterstained with DAPI (blue staining). Fluorescence images were obtained under a high-power field (×400-magnification with a fluorescence microscope (Nikon Eclipse E600) equipped with a monochrome camera (CoolSnap, 20 MHz; Roper Scientific, Germany) and the MetaMorph imaging system (Universal Imaging Corporation). Each image is representative of at least 6 rats with similar results at each condition. Bars represent 20 μm. (B) Apoptosis were quantified by morphometric analysis of apoptotic acinar cells-stained with bright green relative to the total nuclei number (blue) using ImageJ 1.42I software (from NIH) on pancreatic tissue sections from rats with 4 h injections of cerulein (CR) and saline (S). A total of at least 2000 acinar cells were counted on tissue sections from each animal and 3–5 animals per condition were counted. (C) Necrosis/apoptosis ratios were compared in the groups of rats with and without CID pretreatment. The ratio calculation was based on the percentage of necrotic cells versus percentage of apoptotic cells using the data from (A) and Figure 4C. (D) Caspase activities at the indicated time points were measured in pancreatic tissue homogenates. Graphs (B,D) show means ± SE (n = 3). **P < 0.01 or *P < 0.05 versus cerulein alone without inhibitor pretreatments (CR + DMSO) or versus control without cerulein (DMSO) as indicated.
Next, we measured caspase activities in pancreas tissue homogenates. The activities of caspase -3, -9, and -8 were all significantly increased in CID755673-treated rats at either early stage (30 min) or late stage (4 h) of cerulein-induced pancreatitis compared with the non-inhibitor-treated controls (Figure 5D). These results indicated that the PKD inhibitor shifts necrosis to apoptosis in pancreatitis by removing the blockade of PKD/PKD1 on caspase activation.
Genetic Regulation of PKD/PKD1 Expression Alters Cell Death Pathways in Pancreatic Acinar Cells
Although the combination of our in vitro and in vivo experiments with the two pharmacological inhibitors of PKD shows that PKD/PKD1 plays a critical role in regulation of cell death in pancreatitis, we further validate the function of PKD/PKD1 by altering PKD/PKD1 expression with molecular approaches in the cell culture model of pancreatic acinar AR42J cells (Piiper et al., 2001). AR42J cells were transfected with pooled PKD1siRNAs (four duplexes) or pcDNA3-PKD1 vector. Control cells received non-target (NT) siRNAs or empty vector (pcDNA3). After 3 day incubation in cell culture medium, PKD/PKD1 protein expression and CCK-induced PKD/PKD1 activation was analyzed with Western blot analysis of the transfected cell lysates. The results confirmed that PKD/PKD1 expression was down-regulated in PKD1siRNAs-transfected cells but dramatically increased in pcDNA-PKD1-transfected cells (Figure 6A). Similarly to our previously reported results (Yuan et al., 2008), CCK-induced PKD1 activation was correlated with its expression levels. That is, CCK-induced PKD/PKD1 activation was markedly enhanced in cells overexpressing PKD1 but was dramatically decreased in cells with PKD1 knocked down (data not shown).
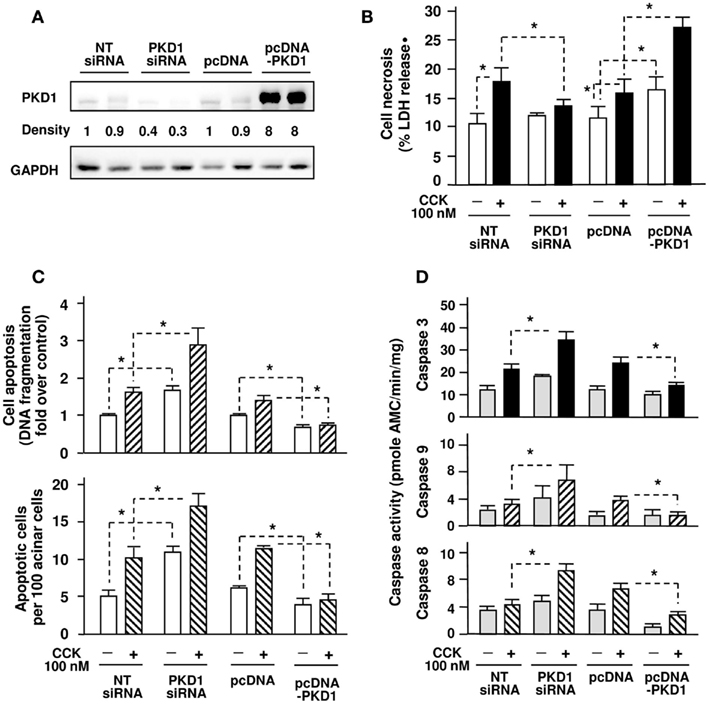
Figure 6. Effects of genetic down- or up-regulation of PKD/PKD1 expression on cell necrosis and apoptosis/caspase activation in AR42J cells induced by supramaximal dose CCK. AR42J cells were transfected with pooled four PKD1 siRNA duplexes or pcDNA3-PKD1 vector. Control cells received non-target (NT) siRNAs or empty vector (pcDNA3). (A) Western blot analysis of the transfected AR42J cell lysates using PKD1 C-20 antibody to measure PKD/PKD1 expression and using GAPDH antibody to verify equal loading. Shown are representative blots from at least 3 independent experiments. The band densities were normalized with corresponding controls (PKD1 siRNA versus NT siRNAs; pcDNA-PKD1 versus pcDNA). (B) The transfected cells were incubated in serum-free F-12K medium for 1 h and then incubated for 5 h with or without 100 nM CCK. Cell necrosis was measured by the percentage of total cellular LDH released into the extracellular medium. Results are expressed as the percentage of total cellular LDH released into the medium. (C) Apoptosis was quantified by two methods: Upper panel, measuring internucleosomal DNA fragmentation using Cell Death Detection ELISA Plus kit according to the manufacturer’s instructions. Results are expressed as increase fold over control in DNA fragmentation; Lower panel, the transfected AR42J cells growing on glass coverslips were fixed and apoptosis was measured by the percentage of TUNEL staining positive cells. For each condition, at least 3000 cells were counted. (D) Caspase activities were measured as described in Section “Experimental Procedures.” Graphs (B–D) show means ± SE (n = 3–5) for each condition. *P < 0.05 versus corresponding controls (NT siRNA or pcDNA3) as indicated.
We then measured cell necrosis and apoptosis after the transfected cells were incubated for 5 h with 100 nM CCK. As shown in Figure 6B, cell incubation with CCK caused an increase in cellular LDH release (a measure of necrosis) in control cells (NTsiRNAs, pcDNA cells). Importantly, inhibition of PKD/PKD1 expression by PKD1siRNAs attenuated CCK-induced cell necrosis. Conversely, CCK-induced cell necrosis was significantly enhanced in PKD/PKD1-overexpressing cells. In contrast to its stimulating effect on necrosis, increased PKD/PKD1 expression resulted in inhibition of CCK-stimulated apoptosis while PKD/PKD1 down-regulation caused dramatically increased CCK-induced apoptosis determined by measuring internucleosomal DNA fragmentation with ELISA and by cell staining with TUNEL assay (Figure 6C). Interestingly, PKD/PKD1 overexpression or knockdown also affected the basal necrosis and apoptosis rates of the transfected cells.
Furthermore, we found that PKD/PKD1 down-regulation by PKD1siRNAs promoted CCK-induced activation of caspase-3, -9, -8, whereas PKD/PKD1 overexpression markedly inhibited these caspases (Figure 6D). Thus, with molecular approaches we confirmed that PKD/PKD1 promotes cell necrosis while suppresses apoptosis and caspase activation in pancreatic acinar cells.
PKD/PKD1 Inhibition Decreases Anti-Apoptotic Protein Levels in Cerulein-Induced Pancreatitis
Inhibitors of apoptosis proteins are potent endogenous suppressors of apoptosis and function by directly inhibiting the activity of caspases (Stehlik et al., 1998; Deveraux and Reed, 1999; Gukovskaya and Pandol, 2004; Kawakami et al., 2005; Kerbauy et al., 2005; Mareninova et al., 2006; Pandol et al., 2007). A wealth of evidence indicates that IAPs, such as XIAP and survivin, play a pivotal role in regulation of cell death (Stehlik et al., 1998; Deveraux and Reed, 1999; Pahl, 1999; Zou et al., 2004; Kawakami et al., 2005; Kerbauy et al., 2005; Harikumar et al., 2010). To further explore the signal pathways mediating the regulation of PKD/PKD1 on cell death, we hypothesized that PKD/PKD1 blocks caspase activation through regulating IAPs in pancreatitis.
We first determined whether PKD/PKD1 regulates XIAP, the most potent among the eight mammalian IAPs (Stehlik et al., 1998; Zou et al., 2004) and an endogenous inhibitor of caspase-9 and -3, in in vivo experimental pancreatitis. Our previous report (Mareninova et al., 2006) showed that rate of XIAP degradation during cerulein-induced pancreatitis is a key determinant of the type of cell death and that pharmacological inhibition of XIAP ameliorated necrosis and severity of cerulein-induced pancreatitis. Here we found that PKD1 inhibition by CID755673 altered protein levels of XIAP in cerulein-induced pancreatitis (Figure 7A). Although both non-inhibitor-treated and inhibitor-treated groups exhibited XIAP degradation in pancreatitis after four hourly cerulein injections, of importance, XIAP in CID755673-treated rats was degraded earlier (in 30 min after first cerulein injection) and to a much greater extent than in non-inhibitor-treated animals, indicating that the PKD/PKD1 inhibition promotes degradation of XIAP in pancreatitis.
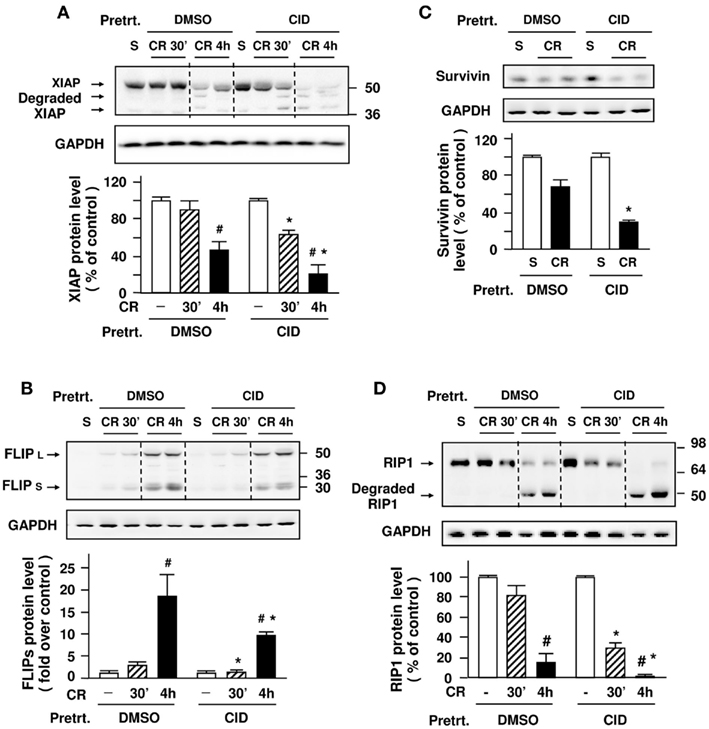
Figure 7. Effect of CID755673 on anti-apoptotic proteins levels and degradation of RIP1 in cerulein-induced pancreatitis. Western blot analysis of pancreas tissue lysate using antibodies against XIAP (A), cFLIP (B), survivin (C), and RIP1 (D) to measure the protein levels of these proteins at early (30 min) and late (4 h) stages of cerulein (CR)-induced pancreatitis. Blots were re-probed for GAPDH to confirm equal protein loading. The bar figures are the quantification of these protein band intensities that were normalized on the band intensity in control animals injected with saline (S). Graphs show means ± SE (n = 4) for each condition. *P < 0.05 versus cerulein alone (DMSO + CR) in first injection and fourth injection (#).
We next examined whether c-FLIP and survivin, the other two anti-apoptotic proteins (Kawakami et al., 2005; Kerbauy et al., 2005), are also altered in pancreatitis. c-FLIP, an important endogenous protein inhibitor of caspase-8, has two splice isoforms: c-FLIPS (short) and c-FLIPL (long). c-FLIPS completely inhibits caspase-8 processing, whereas c-FLIPL allows partial processing of caspase-8 (Krueger et al., 2001; Thome and Tschopp, 2001; Peter, 2004). We found that both c-FLIPS and c-FLIPL were markedly up-regulated in pancreatitis (Figure 7B). There was no significant difference in the magnitude of the up-regulation of c-FLIPL between non-inhibitor and inhibitor-treated groups. However, the up-regulation of c-FLIPS, which can completely block caspase-8 processing, was much earlier (in 30 min after first cerulein injection) and to a greater extent in non-inhibitor-treated rats than in inhibitor-treated rats (Figure 7B), indicating that the PKD/PKD1 inhibitor CID755673 suppressed the up-regulation of c-FLIPS in pancreatitis. Further, we found a significantly decreased protein level of survivin, an endogenous inhibitor of caspase-3, after four hourly cerulein injections in CID755673-treated rats (Figure 7C).
Thus, our in vivo experimental results indicate that inhibition of PKD/PKD1 has significant effects on these endogenous protein inhibitors of caspases, including XIAP, c-FLIPs, and survivin, accounting for the effects observed on caspase activation and apoptosis in vivo and indicating that CID755673 ameliorated necrosis in pancreatitis probably through suppressing the levels of these IAP proteins.
PKD/PKD1 Inhibition Promotes Degradation of RIP1 in Cerulein-Induced Pancreatitis
Receptor-interacting protein kinase has recently emerged as a key mediator of programmed necrosis in acute pancreatitis (Mareninova et al., 2006; He et al., 2009) and other diseases (Lin et al., 1999; Chan et al., 2003; Meylan and Tschopp, 2005; Festjens et al., 2007; Galluzzi et al., 2009; Moquin and Chan, 2010; Trichonas et al., 2010). RIP1 forms a death-signaling complex with the Fas-associated death domain and caspases in response to death domain receptor stimulation (Lin et al., 1999; Chan et al., 2003; Meylan and Tschopp, 2005; Festjens et al., 2007; Trichonas et al., 2010). During apoptosis, RIP1 is cleaved and inactivated by caspase-3 and -8 (Lin et al., 1999; Chan et al., 2003; Moquin and Chan, 2010). The regulation of RIP1 by caspases has been suggested to be one of mechanisms underlying the protective role of caspases from necrosis in cerulein-induced pancreatitis (Mareninova et al., 2006; He et al., 2009).
For the present study, we determined the effect of the inhibition of PKD/PKD1 on RIP1 degradation. In agreement to our previous findings (Mareninova et al., 2006), RIP1 underwent cleavage to a 47-kDa product after four hourly cerulein injections (Figure 7D). The salient feature of the present results was that the cleavage of RIP1 started earlier (in first cerulein injection) and progressed to a greater extent in inhibitor-treated rats than in non-inhibitor-treated animals. The results with RIP1 during PKD/PKD1 inhibition correlated with the changes in caspase activities (Figure 5D) and cleavage of IAPs (Figures 7A–C). These findings suggest that PKD/PKD1-regulated IAPs/caspases pathways play a role in RIP1 cleavage/inactivation in cerulein-induced pancreatitis.
PKD/PKD1Regulates Protein Levels of IAPs and RIP1 in Pancreatic Acinar Cells
To provide insights into the mechanism of effects of PKD/PKD1 on IAPs and RIP1, we examined whether genetic regulation of PKD/PKD1 expression alters protein levels of IAPs and RIP1 in pancreatic acinar cells. We found that cells transfected with PKD1 siRNAs markedly down-regulated PKD/PKD1 expression, which resulted in dramatically decreased protein levels of XIAP, survivin, and FILPs (Figure 8A). Conversely, overexpression of PKD/PKD1 enhanced the levels of these IAPs (Figure 8B). Furthermore, up-regulation of PKD1 expression promoted RIP1 production and inhibited CCK-induced RIP1 cleavage/inactivation (Figure 8C) whereas knockdown PKD1 promoted RIP1 cleavage/inactivation (Figure 8D). These findings confirmed that PKD/PKD1 regulates IAPs and RIP1 protein levels, as observed in cerulein-induced pancreatitis.
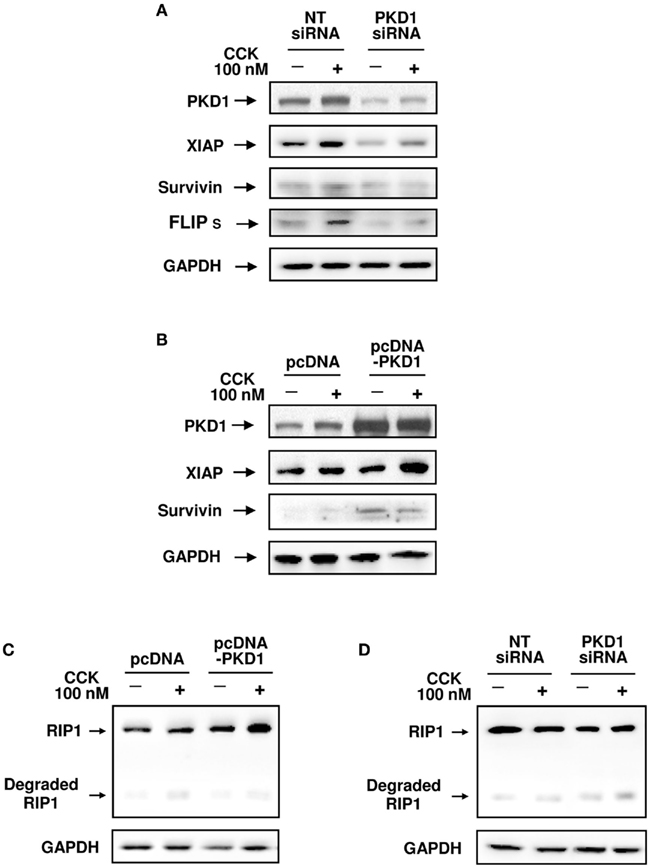
Figure 8. Effects of genetic down- or up-regulation of PKD/PKD1 expression on protein levels of IAPs and RIP1. AR42J cells were transfected with pooled four PKD1 siRNA duplexes or pcDNA3-PKD1 vector. Control cells received non-target (NT) siRNAs or empty vector (pcDNA3). Three days after transfection, the cells were incubated in serum-free F-12K medium for 1 h and then incubated for 5 h with or without 100 nM CCK. (A,B) Western blot analysis of the transfected AR42J cell lysates using antibodies against PKD1 C-20, XIAP, survivin, and FLIP to show effects of down- or up-regulation of PKD/PKD1 expression on the protein levels of these IAPs. Blots were re-probed for GAPDH to confirm equal protein loading. Shown are representative blots from at least three independent experiments. (C,D) Western blot analysis of the cell lysates using RIP1 antibody to detect the effect of up-regulation or knockdown of PKD/PKD1 expression on RIP1 expression and cleavage. The membranes were reblotted with GAPDH antibody to verify equal loading. Shown are representative blots from three independent experiments.
Thus, with pharmacologic and genetic approaches, we have demonstrated that PKD/PKD1 mediates cell necrosis in pancreatitis through regulating IAPs, caspases, and RIP1.
Genetic Inhibition of XIAP Stimulates Apoptosis and Caspase Activation While Promoting RIP1 Degradation and Attenuating Necrosis
Next, to further confirm the pivotal role of IAPs in cell death in pancreatitis, we assessed whether the down-regulation of XIAP, the major IAP, affects cell death pathways and RIP1 inactivation. We transfected AR42J cells with pooled XIAP siRNAs (three siRNA duplexes) to suppress XIAP protein expression, as confirmed with Western blot analysis (Figure 9A). Interestingly, specific inhibition of the expression of this endogenous inhibitor of caspases resulted in not only increased apoptosis and caspase activation (Figure 9B) but also a dramatically attenuated CCK-induced cell necrosis (Figure 9C). These results support our previous findings that caspases protect from necrosis in pancreatitis (Mareninova et al., 2006) and support the importance of PKD/PKD1-regulated IAPs/caspases pathways in mediating necrotic death pathways in pancreatitis.
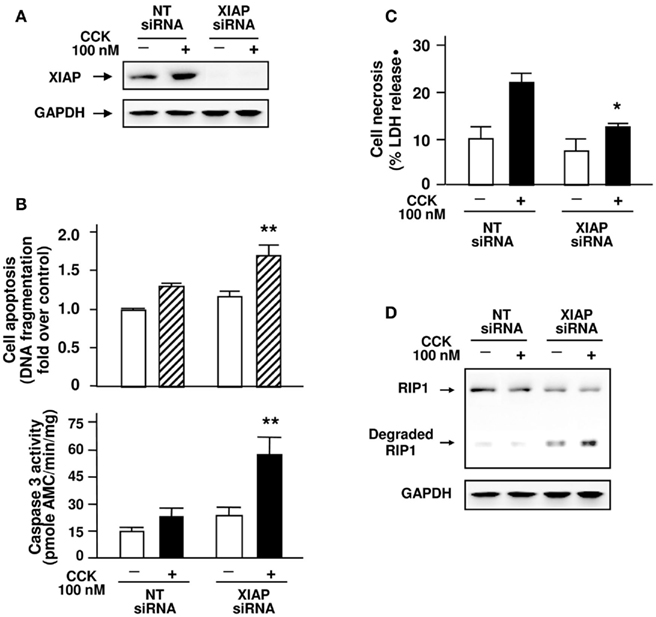
Figure 9. Inhibition of XIAP expression stimulates apoptosis and caspase activation while promoting RIP1 degradation and attenuating necrosis induced by CCK. AR42J cells were transfected with non-target (NT) or pooled XIAP siRNAs (three siRNA duplexes) as indicated. The transfected cells were incubated in serum-free F-12K medium for 1 h and then incubated for 5 h with or without 100 nM CCK. (A) Western blot analysis of the cell lysates using XIAP antibody for XIAP expression and using GAPDH antibody to verify equal loading. Shown are representative blots from at least three independent experiments. (B) Apoptosis quantified by measuring internucleosomal DNA fragmentation, as described in Section “Experimental Procedures.” Results are expressed as increased fold over control. Caspase activities were measured as described in Section “Experimental Procedures” for the lysates of transfected AR42J cells. (C) Cell necrosis was measured by the percentage of total cellular LDH released into the extracellular medium. Graphs (B,C) show means ± SE (n = 3 to 4) for each condition. **P < 0.01 or *P < 0.05 versus each corresponding NTsiRNA control with CCK-treated, as indicated. (D) Western blot analysis of the cell lysates using RIP1 antibody for RIP1 protein level and degradation and using GAPDH antibody to verify equal loading. Shown are representative blots from three independent experiments.
We then examined whether XIAP regulates RIP1 cleavage. Inhibition of XIAP expression with specific siRNAs markedly increased RIP1 cleavage/inactivation in the transfected cells (Figure 9D). These results indicate that similar to PKD/PKD1, XIAP negatively regulates RIP1 cleavage and inactivation. The negative regulation of RIP1 cleavage or inactivation by both PKD/PKD1 and XIAP suggest that one mechanism by which PKD1/IAPs signaling pathway promotes necrotic cell death in acute pancreatitis is through prevention of RIP1 cleavage/inactivation. We summarize the signal pathways in Figure 10.
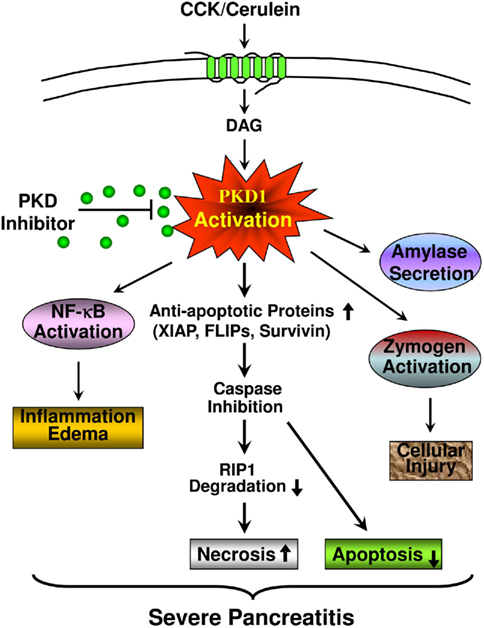
Figure 10. A scheme summarizing the role of PKD/PKD1 in multiple pathological processes of pancreatitis and the mechanisms of PKD activation mediating cell death in pancreatitis. CCK/cerulein-induced PKD activation increases the levels of anti-apoptotic proteins (XIAP, c-FLIPs, and survivin). The increased anti-apoptotic proteins suppress caspase activation and apoptosis. Caspases inhibition also results in decreased cleavage/inactivation of RIP1, thus, leading to increased necrosis and the severity of pancreatitis. This cell necrotic death response is blocked when PKD is inhibited. PKD inhibition promotes apoptosis and decreases necrosis. Thus, PKD inhibition will switch cell death away from necrosis. As we previously reported, PKD/PKD1 also mediates other pathological processes of pancreatitis, including NF-κB activation and inflammatory response, zymogen activation, and amylase secretion. All these effects indicate that PKD/PKD1 may represent a potential therapeutic target in pancreatitis. PKD inhibition decreases the severity of pancreatitis.
Discussion
PKD/PKD1 has been shown to mediate NF-κB activation and inappropriate intracellular digestive enzyme activation in pancreatitis (Yuan et al., 2008; Thrower et al., 2011), but its role in regulation of cell necrosis in pancreatitis has not been investigated. The studies presented here demonstrate that PKD/PKD1 activation promotes necrosis and inhibits apoptosis in both in vitro and in vivo models of pancreatitis. In vivo treatment of animals with PKD/PKD1 inhibitor CID755673 significantly ameliorated necrosis and the severity of pancreatitis.
Apoptosis in pancreatic acinar cells is mediated mainly by activation of caspases. Of importance, it has been increasingly recognized that caspases not only mediate apoptosis but also protect from necrosis and decrease the severity of pancreatitis (Bhatia, 2004a; Gukovskaya and Pandol, 2004; Mareninova et al., 2006; Pandol et al., 2007). We found that up-regulation of PKD/PKD1 expression prevented CCK/cerulein-induced activation of caspase-9, -8, and -3 and promoted necrosis. On the other hand, inhibition of PKD/PKD1 enhanced the activation of caspases in both in vitro and in vivo models of pancreatitis, resulting in decreased necrosis and the severity of pancreatitis. Therefore, the pro-necrosis effect of PKD/PKD1 activation in pancreatitis is mediated through inhibition of caspase activation.
NF-κB-dependent anti-apoptotic gene products are an important class of endogenous protein inhibitors of caspase activation (Stehlik et al., 1998; Deveraux and Reed, 1999; Pahl, 1999; Bratton et al., 2001; Zou et al., 2004; Kawakami et al., 2005; Kerbauy et al., 2005; Pandol et al., 2007). Transcriptional activation of XIAP, survivin, and c-FLIP through NF-κB pathways has been demonstrated to be crucial in regulating cell death (Stehlik et al., 1998; Deveraux and Reed, 1999; Pahl, 1999; Bratton et al., 2001; Zou et al., 2004; Kawakami et al., 2005; Kerbauy et al., 2005; Mareninova et al., 2006; Pandol et al., 2007), including pancreatitis (Mareninova et al., 2006; Pandol et al., 2007). We previously demonstrated that PKD/PKD1 is required for NF-κB activation in pancreatic acinar cells (Yuan et al., 2008) and that CCK-induced NF-κB activation is significantly decreased by PKD/PKD1 inhibition during experimental pancreatitis (Thrower et al., 2011). Here we show that PKD/PKD1 inhibition in cerulein-induced pancreatitis results in a more rapid and greater extent of decrease in the protein levels of XIAP, c-FLIPs, and survivin. Using cell culture approaches, we found that down-regulation of PKD/PKD1 decreased protein levels of XIAP, c-FLIPs, and survivin whereas up-regulation of PKD/PKD1 expression enhanced protein levels of these IAPs, supporting a role for PKD/PKD1 in the regulation of the IAPs. We also showed that inhibition of XIAP expression (as a representative of IAPs) significantly reduced necrosis while enhancing apoptosis and caspase activation, further supporting a mechanism by which PKD/PKD1 regulates cell death pathways through its effects on protein levels of IAPs. Although NF-κB is responsible for at least some of changes in IAPs levels caused by PKD inhibition observed in the in vivo pancreatitis experiments, the complete mechanism of reduction in IAPs with PKD inhibition may include changes in degradation rates as well. Further studies will be needed to investigate whether PKD regulates rates of degradation of IAPs.
Receptor-interacting protein kinase 1, a key regulator of programmed necrosis in many diseases including acute pancreatitis (Lin et al., 1999; Chan et al., 2003; Meylan and Tschopp, 2005; Festjens et al., 2007; Galluzzi et al., 2009; He et al., 2009; Moquin and Chan, 2010; Trichonas et al., 2010), has been known to be cleaved/inactivated by caspase-3 and -8. Our studies indicate that PKD1/IAPs signaling pathway regulates the expression and inactivation of RIP1 in pancreatitis. For example, (i) pharmacological inhibition of PKD/PKD1 led to earlier and greater degradation/inactivation of RIP1 in cerulein-induced pancreatitis. (ii) Up-regulation of PKD1 expression, which enhanced protein levels of XIAP and survivin, resulted in increased RIP1 production as well as decreased degradation. (iii) Down-regulation of PKD1 with PKDsiRNAs, which decreased the protein levels of these IAPs, promoted RIP1 cleavage/inactivation. (iv) Genetic inhibition of XIAP expression markedly increased RIP1 cleavage/inactivation in the pancreatic acinar cells. These results indicate that PKD/PKD1 regulates RIP1 expression and inactivation through the IAPs.
Development of specific PKD inhibitors has been of intense interest during recent years following the recognition of multiple physiological and pathophysiological effects of PKDs in human. Of note, although PKD/PKD1 is activated in many cases through PKCs (Berna et al., 2007; Yuan et al., 2008; Chen et al., 2009; Rozengurt, 2011; Thrower et al., 2011), selectively targeting each PKC isotype is difficult in pancreatitis and other disorders because of the number of PKC isotypes activated and the lack of isoform-specific PKC inhibitors. Our previous studies demonstrated that at least four PKC isotypes (α, δ, ε, ζ) are activated during pancreatitis. PKD/PKD1, as a convergent point, integrates the signaling from these PKCs to mediate zymogen activation, amylase secretion, and NF-κB activation (Yuan et al., 2008; Thrower et al., 2011), which are key signals involved in pancreatic injury and the inflammatory response of pancreatitis. The findings presented here show that PKD/PKD1 plays a critical role in the mechanisms of cell death in pancreatitis. Thus, PKD/PKD1 inhibition will have beneficial effects for preventing multiple pathological processes of pancreatitis including inappropriate intracellular digestive enzyme activation, inflammation, and cell necrotic death response, as summarized in Figure 10.
In summary, our studies demonstrate that PKD/PKD1 is a key mediator of necrosis in acute pancreatitis through its effects on IAPs, caspases, and RIP1. The mechanisms are described in Figure 10. PKD activation in pancreatitis increases the levels of anti-apoptotic proteins, XIAP, c-FLIPs, and survivin, which suppress caspase activation and inhibit apoptosis. Caspase inhibition also results in decreased cleavage/inactivation of RIP1, thus, leading to increased necrosis and severity of pancreatitis. This cell necrotic death response is blocked when PKD is inhibited. PKD inhibition promotes apoptosis and decreases necrosis. Thus, PKD inhibition switches cell death away from necrosis and ameliorates the severity of pancreatitis. The critical role of PKD/PKD1 in necrotic cell death and in other pathological processes of pancreatitis, as we previously identified, all indicates that PKD/PKD1 may represent a potential therapeutic target in pancreatitis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Dr. Aurelia Lugea for discussion and help in animal experiment design and TUNEL staining, Dr. Olga Mareninova for discussion of acinar cell staining. The authors also thank Dr. Edwin Thrower (Yale University School of Medicine) and Dr. Christopher R. Ireson and his group at the Cancer Research Technology Discovery Laboratories, London, UK for providing us with CRT0066101. This study was supported by the Department of Veterans Affairs Merits Grant and the Southern California Research Center for Alcoholic Liver and Pancreatic Diseases (NIH Grant P50-A11999); UCLA Center of Excellence in Pancreatic Diseases (NIH Grant 1P01AT0003960-01); and Lee Summer Student Research Award from Southern California Research Center for Liver and Pancreatic Diseases (to Tanya Tan).
References
Beil, M., Leser, J., Lutz, M. P., Gukovskaya, A., Seufferlein, T., Lynch, G., Pandol, S. J., and Adler, G. (2002). Caspase 8-mediated cleavage of plectin precedes F-actin breakdown in acinar cells during pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 282, G450–G460.
Berna, M. J., Hoffmann, K. M., Tapia, J. A., Thill, M., Pace, A., Mantey, S. A., and Jensen, R. T. (2007). CCK causes PKD1 activation in pancreatic acini by signaling through PKC-delta and PKC-independent pathways. Biochim. Biophys. Acta 1773, 483–501.
Bhatia, M. (2004a). Apoptosis of pancreatic acinar cells in acute pancreatitis: is it good or bad? J. Cell. Mol. Med. 8, 402–409.
Bhatia, M. (2004b). Apoptosis versus necrosis in acute pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 286, G189–G196.
Bhatia, M., Wallig, M. A., Hofbauer, B., Lee, H. S., Frossard, J. L., Steer, M. L., and Saluja, A. K. (1998). Induction of apoptosis in pancreatic acinar cells reduces the severity of acute pancreatitis. Biochem. Biophys. Res. Commun. 246, 476–483.
Bratton, S. B., Walker, G., Srinivasula, S. M., Sun, X. M., Butterworth, M., Alnemri, E. S., and Cohen, G. M. (2001). Recruitment, activation and retention of caspases-9 and -3 by Apaf-1 apoptosome and associated XIAP complexes. EMBO J. 20, 998–1009.
Chan, F. K., Shisler, J., Bixby, J. G., Felices, M., Zheng, L., Appel, M., Orenstein, J., Moss, B., and Lenardo, M. J. (2003). A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J. Biol. Chem. 278, 51613–51621.
Chen, L. A., Li, J., Silva, S. R., Jackson, L. N., Zhou, Y., Watanabe, H., Ives, K. L., Hellmich, M. R., and Evers, B. M. (2009). PKD3 is the predominant protein kinase D isoform in mouse exocrine pancreas and promotes hormone-induced amylase secretion. J. Biol. Chem. 284, 2459–2471.
Chiu, T. T., Leung, W. Y., Moyer, M. P., Strieter, R. M., and Rozengurt, E. (2007). Protein kinase D2 mediates lysophosphatidic acid-induced interleukin 8 production in nontransformed human colonic epithelial cells through NF-kappaB. Am. J. Physiol. Cell Physiol. 292, C767–C277.
Deveraux, Q. L., and Reed, J. C. (1999). IAP family proteins-suppressors of apoptosis. Genes Dev. 13, 239–252.
Doppler, H., Storz, P., Li, J., Comb, M. J., and Toker, A. (2005). A phosphorylation state-specific antibody recognizes Hsp27, a novel substrate of protein kinase D. J. Biol. Chem. 280, 15013–15019.
Edinger, A. L., and Thompson, C. B. (2004). Death by design: apoptosis, necrosis and autophagy. Curr. Opin. Cell Biol. 16, 663–669.
Festjens, N., Berghe, T. V., Cornelis, S., and Vandenabeele, P. (2007). RIP1, a kinase on the crossroads of a cell’s decision to live or die. Cell Death Differ. 14, 400–410.
Fielitz, J., Kim, M. S., Shelton, J. M., Qi, X., Hill, J. A., Richardson, J. A., Bassel-Duby, R., and Olson, E. N. (2008). Proc. Natl. Acad. Sci. U. S. A. 105, 3059–3063.
Galluzzi, L., Kepp, O., and Kroemer, G. I. (2009). RIP kinases initiate programmed necrosis. J. Mol. Cell. Biol. 1, 8–10.
Gukovskaya, A. S., Gukovsky, I., Jung, Y., Mouria, M., and Pandol, S. J. (2002). Cholecystokinin induces caspase activation and mitochondrial dysfunction in pancreatic acinar cells. roles in cell injury processes of pancreatitis. J. Biol. Chem. 277, 22595–22604.
Gukovskaya, A. S., Gukovsky, I., Zaninovic, V., Song, M., Sandoval, D., Gukovsky, S., and Pandol, S. J. (1997). Pancreatic acinar cells produce, release, and respond to tumor necrosis factor-alpha. role in regulating cell death and pancreatitis. J. Clin. Invest. 100, 1853–1862.
Gukovskaya, A. S., and Pandol, S. J. (2004). Cell death pathways in pancreatitis and pancreatic cancer. Pancreatology 4, 567–586.
Harikumar, K. B., Kunnumakkara, A. B., Ochi, N., Tong, Z., Deorukhkar, A., Sung, B., Kelland, L., Jamieson, S., Sutherland, R., Raynham, T., Charles, M., Bagherzadeh, A., Foxton, C., Boakes, A., Farooq, M., Maru, D., Diagaradjane, P., Matsuo, Y., Sinnett-Smith, J., Gelovani, J., Krishnan, S., Aggarwal, B. B., Rozengurt, E., Ireson, C. R., and Guha, S. (2010). A novel small molecule inhibitor of protein kinase D blocks pancreatic cancer growth in vitro and in vivo. Mol. Cancer Ther. 9, 1136–1146.
He, S., Wang, L., Miao, L., Wang, T., Du, F., and Wang, X. (2009). Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137, 1100–1111.
Heemskerk, F. M., Chen, H. C., and Huang, F. L. (1993). Protein kinase C phosphorylates Ser152, Ser156 and Ser163 but not Ser160 of MARCKS in rat brain. Biochem. Biophys. Res. Commun. 190, 236–241.
Jacamo, R., Sinnett-Smith, J., Rey, O., Waldron, R. T., and Rozengurt, E. (2008). Sequential protein kinase C (PKC)-dependent and PKC-independent protein kinase D catalytic activation via Gq-coupled receptors: differential regulation of activation loop SER(744) and SER(748) phosphorylation. J. Biol. Chem. 283, 12877–12887.
Kaiser, A. M., Saluja, A. K., Sengupta, A., Saluja, M., and Steer, M. L. (1995). Relationship between severity, necrosis, and apoptosis in five models of experimental acute pancreatitis. Am. J. Physiol. 269, C1295–C1304.
Kawakami, H., Tomita, M., Matsuda, T., Ohta, T., Tanaka, Y., Fujii, M., Hatano, M., Tokuhisa, T., and Mori, N. (2005). Transcriptional activation of survivin through the NF-kappaB pathway by human T-cell leukemia virus type I tax. Int. J. Cancer 115, 967–974.
Kerbauy, D. M., Lesnikov, V., Abbasi, N., Seal, S., Scott, B., and Deeg, H. J. (2005). NF-kappaB and FLIP in arsenic trioxide (ATO)-induced apoptosis in myelodysplastic syndromes (MDSs). Blood 106, 3917–3925.
Krantic, S., Mechawar, N., Reix, S., and Quirion, R. (2007). Apoptosis-inducing factor: a matter of neuron life and death. Prog. Neurobiol. 81, 179–196.
Krueger, A., Schmitz, I., Baumann, S., Krammer, P. H., and Kirchhoff, S. (2001). Cellular FLICE-inhibitory protein splice variants inhibit different steps of caspase-8 activation at the CD95 death-inducing signaling complex. J. Cell Biochem. 276, 20633–20640.
Lin, Y., Devin, A., Rodriguez, Y., and Liu, Z. G. (1999). Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 13, 2514–2526.
Mareninova, O. A., Sung, K. F., Hong, P., Lugea, A., Pandol, S. J., Gukovsky, I., and Gukovskaya, A. S. (2006). Cell death in pancreatitis: caspases protect from necrotizing pancreatitis. J. Biol. Chem. 281, 3370–3381.
Matthews, S. A., Rozengurt, E., and Cantrell, D. (1999). Characterization of serine 916 as an in vivo autophosphorylation site for protein kinase D/Protein kinase Cmu. J. Biol. Chem. 274, 26543–26549.
Meylan, E., and Tschopp, J. (2005). The RIP kinases: crucial integrators of cellular stress. Trends Biochem. Sci. 30, 151–159.
Moquin, D., and Chan, F. K. (2010). The molecular regulation of programmed necrotic cell injury. Trends Biochem. Sci. 35, 434–441.
Pahl, H. L. (1999). Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 18, 6853–6866.
Pandol, S. J., Saluja, A. K., Imrie, C. W., and Banks, P. A. (2007). Acute pancreatitis: bench to the bedside. Gastroenterology 132, 1127–11251.
Piiper, A., Leser, J., Lutz, M. P., Beil, M., and Zeuzem, S. (2001). Subcellular distribution and function of Rab3A-D in pancreatic acinar AR42J cells. Biochem. Biophys. Res. Commun. 287, 746–751.
Ramsden, J. J. (2000). MARCKS: a case of molecular exaptation? Int. J. Biochem. Cell Biol. 32, 475–479.
Rozengurt, E. (2011). Protein kinase D signaling: multiple biological functions in health and disease. Physiology 26, 23–33.
Schoenberg, M. H., Büchler, M., Gaspar, M., Stinner, A., Younes, M., Melzner, I., Bültmann, B., and Beger, H. G. (1990). Oxygen free radicals in acute pancreatitis of the rat. Gut 31, 1138–1143.
Sharlow, E. R., Giridhar, K. V., LaValle, C. R., Chen, J., Leimgruber, S., Barrett, R., Bravo-Altamirano, K., Wipf, P., Lazo, J. S., and Wang, Q. J. (2008). Potent and selective disruption of protein kinase D functionality by a benzoxoloazepinolone. J. Biol. Chem. 283, 33516–33526.
Stehlik, C., Martin, R., Kumabashiri, I., Schmid, J. A., Binder, B. R., and Lipp, J. (1998). NF-κB regulated X-chromosome-linked IAP gene expression protects endothelial cells from TNF-α induced apoptosis. J. Exp. Med. 188, 211–216.
Storz, P., Doppler, H., and Toker, A. (2004). Protein kinase Cdelta selectively regulates protein kinase D-dependent activation of NF-kappaB in oxidative stress signaling. Mol. Cell Biol. 24, 2614–2626.
Sung, K. F., Odinokova, I. V., Mareninova, O. A., Rakonczay, Z. Jr., Hegyi, P., Pandol, S. J., Gukovsky, I., and Gukovskaya, A. S. (2009). Prosurvival Bcl-2 proteins stabilize pancreatic mitochondria and protect against necrosis in experimental pancreatitis. Exp. Cell Res. 315, 1975–1989.
Tang, D., Lahti, J. M., and Kidd, V. J. (2000). Caspase-8 activation and bid cleavage contribute to MCF7 cellular execution in a caspase-3-dependent manner during staurosporine-mediated apoptosis. J. Biol. Chem. 275, 9303–9307.
Thome, M., and Tschopp, J. (2001). Regulation of lymphocyte proliferation and death by FLIP. Nat. Rev. Immunol. 1, 50–58.
Thrower, E. C., Yuan, J., Usmani, A., Liu, Y., Jones, C., Samantha, N., Minervini, S. N., Alexandre, M., Pandol, S. J., and Guha, S. (2011). A novel protein kinase D inhibitor attenuates early events of experimental pancreatitis in isolated rat acini. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G120–G129.
Trauzold, A., Schmiedel, S., Sipos, B., Wermann, H., Westphal, S., Roder, C., Klapper, W., Arlt, A., Lehnert, L., Ungefroren, H., Johannes, F. J., and Kalthoff, H. (2003). PKCmu prevents CD95-mediated apoptosis and enhances proliferation in pancreatic tumour cells. Oncogene 22, 8939–8947.
Trichonas, G., Murakami, Y., Thanos, A., Morizane, Y., Kayamaa, M., Debouckc, C. M., Hisatomi, T., Miller, J. W., and Vavvas, D. G. (2010). Receptor interacting protein kinases mediate retinal detachment-induced photoreceptor necrosis and compensate for inhibition of apoptosis. Proc. Natl. Acad. Sci. U.S.A. 107, 21695–21700.
Yuan, J., Lugea, A., Zheng, L., Gukovsky, I., Edderkaoui, M., Rozengurt, E., and Pandol, S. J. (2008). Protein kinase D1 mediates NF-κB activation induced by cholecystokinin and cholinergic signaling in pancreatic acinar cells. (2008). Am. J. Physiol. Gastrointest. Liver Physiol. 295, G1190–G1201.
Yuan, J., and Rozengurt, E. (2008). PKD, PKD2, and p38 MAPK mediate Hsp27 serine-82 phosphorylation induced by neurotensin in pancreatic cancer PANC-1 cells. J. Cell Biochem. 103, 648–662.
Keywords: pancreatic acinar cells, CCK, CID755673, CRT0066101, apoptosis, necrosis
Citation: Yuan J, Liu Y, Tan T, Guha S, Gukovsky I, Gukovskaya A and Pandol SJ (2012) Protein kinase D regulates cell death pathways in experimental pancreatitis. Front. Physio. 3:60. doi: 10.3389/fphys.2012.00060
Received: 30 January 2012; Paper pending published: 13 February 2012;
Accepted: 29 February 2012; Published online: 27 March 2012.
Edited by:
Peter Hegyi, University of Szeged, HungaryReviewed by:
Julia Gerasimenko, Cardiff University, UKZoltan Rakonczay Jr., University of Szeged, Hungary
Copyright: © 2012 Yuan, Liu, Tan, Guha, Gukovsky, Gukovskaya and Pandol. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Jingzhen Yuan, South California Research Center for Alcoholic Liver and Pancreatic Diseases, Veterans Affairs Greater Los Angeles Healthcare System, West Los Angeles VA Healthcare Center, University of California at Los Angeles, 11301 Wilshire Blvd, Building 258, Room 340, Los Angeles, CA 90073, USA. e-mail:anp5dWFuQHVjbGEuZWR1.