


- Danish National Research Foundation Centre for Cardiac Arrhythmia, Department of Biomedical Sciences, University of Copenhagen, Copenhagen, Denmark
Electrophysiological remodeling of cardiac potassium ion channels is important in the progression of heart failure. A reduction of the transient outward potassium current (Ito) in mammalian heart failure is consistent with a reduced expression of potassium channel interacting protein 2 (KChIP2, a KV4 subunit). Approaches have been made to investigate the role of KChIP2 in shaping cardiac Ito, including the use of transgenic KChIP2 deficient mice and viral overexpression of KChIP2. The interplay between Ito and myocardial calcium handling is pivotal in the development of heart failure, and is further strengthened by the dual role of KChIP2 as a functional subunit on both KV4 and CaV1.2. Moreover, the potential arrhythmogenic consequence of reduced Ito may contribute to the high relative incidence of sudden death in the early phases of human heart failure. With this review, we offer an overview of the insights into the physiological and pathological roles of KChIP2 and we discuss the limitations of translating the molecular basis of electrophysiological remodeling from animal models of heart failure to the clinical setting.
Introduction
The normal pumping action of the mammalian heart is critically dependent on the cardiac electrophysiological function. Within a heartbeat, the single cardiac cell generates an action potential in consequence of sequential depolarizing and repolarizing currents. The exact function of the multitude of ion channels and other transporters of electrical charges determines the morphology of the action potential. Owing to diversities in properties and expression profiles of ion channels in different regions in the heart, in different species and in different physiological and pathological settings, the action potential waveform is highly variable (Carmeliet, 1999; Nerbonne and Kass, 2005; Nattel et al., 2007; Ter Keurs and Boyden, 2007).
The human action potential has a spike-and-dome morphology, with a rapid early repolarizing phase or notch that reflects the transient outward potassium current, Ito. The magnitude of the notch determines the membrane potential for the plateau phase of the action potential, and thus establishes the driving force for Ca2+ entry through the L-type Ca2+ channel triggering calcium-induced calcium release and contraction of the myocyte (Greenstein et al., 2000; Sah et al., 2003). In rodents, Ito is the major repolarizing force abbreviating the action potential considerably in adaptation to the fast heart rates. Reduced Ito amplitude in non-rodent mammals is associated with desynchronized Ca2+ release from intracellular stores (Harris et al., 2005; Wasserstrom et al., 2009) and suboptimal cardiac performance secondary to uncoordinated calcium-induced calcium release and contraction (Sah et al., 2002, 2003; Cordeiro et al., 2004).
The landmark finding of reduced Ito in heart failure (Beuckelmann et al., 1993) spurred an intense interest into function and regulation of this current. An et al. (2000) reported how Ito was modulated by K+-channel interacting proteins (KChIPs). Since the initial characterization of KChIPs, several additional functions of KChIPs have been described, including tuning of several ionic currents and gene-transcription regulation (Buxbaum, 2004). In the present review, we outline the basic molecular biology and cardiac electrophysiology of KChIP2 and integrate these findings in a discussion of the role of KChIP2 in cardiac plasticity, hypertrophic remodeling, arrhythmia, and failure.
KChIP Structure and Expression
The KChIPs belong to the family of small cytosolic calcium-binding proteins and consists of four different KChIP isoforms ranging between 216 and 270 amino acids. They all have a reasonably conserved core region of 185 amino acids and a heterogeneous N-terminal part (An et al., 2000; Holmqvist et al., 2002). All KChIPs have calcium-binding EF hands which appear important for modulation KV-channels (An et al., 2000; Li et al., 2005) but not for CaV-channels (Thomsen et al., 2009c). Myristoylation of KChIP1 (Burgoyne and Weiss, 2001) and palmitoylation of KChIP2 (Takimoto et al., 2002) play important roles in post-translational modification to improve plasma membrane localization. It is well established that KChIP1–3 functionally increase peak KV4 current, slow channel inactivation and accelerate recovery from inactivation (An et al., 2000; Lundby et al., 2010); whereas KChIP4 eliminates inactivation (Holmqvist et al., 2002). Furthermore, KChIP2 has been reported to increase ICa,L (Thomsen et al., 2009c), and suggested to decrease KV1.5 cell surface expression (Li et al., 2005) and facilitate INa (Deschenes et al., 2008).
All four KChIP mRNAs are found in the brain; however only KChIP2 is expressed in the heart (An et al., 2000; Kuo et al., 2001; Rosati et al., 2001). KChIP expression in cardiac autonomic nerves has not been reported. The cardiac KChIP2 gene generates three protein isoforms of 220, 252, and 270 amino acids via alternative splicing of exons 2 and 3 (Kuo et al., 2001). Immunocytochemistry has localized KChIP2 to the sarcolemma and the nucleus (Deschenes et al., 2002). Cardiac expression of KChIP2 is absent in embryonic and neonatal stages; however developmental upregulation potentially suggests a physiological role for KChIP2 once a transmural gradient of Ito is appearing (Kuo et al., 2001; Plotnikov et al., 2004). KChIP2 is expressed heterogeneously across the canine and human left and right ventricular free wall with higher concentrations in epicardium compared to endocardium, whereas no transmural gradient of expression is found for KV4.3 (Rosati et al., 2001, 2003; Calloe et al., 2009b). This finding has led to the suggestion that KChIP2 is responsible for the Ito gradient; whereas other studies failed to show a transmural KChIP2 protein gradient in canine and human hearts (Deschenes et al., 2002), arguing against the role of KChIP2 in the generation of the transmural Ito gradient. More recently, studies of the human heart have shown strong KChIP2 expression in the epicardium based on both mRNA and protein levels (Gaborit et al., 2007; Soltysinska et al., 2009). In mice, transmural KV4.2 protein expression parallels the Ito,f gradient; whereas KV4.3 and KChIP2 protein levels were uniformly distributed in an early study (Guo et al., 2002). More recently, laser capture microdissection of the murine LV has revealed a transmural gradient of both KV4.2 and KChIP2 mRNA (Teutsch et al., 2007).
KChIP2 and Ion Channels
KV4
KChIP2 is a cytosolic protein when expressed in absence of KV4; however co-expression of KV4 and KChIP2 leads to co-localization at the cell surface (An et al., 2000; Bahring et al., 2001). Biochemically, KChIP2 co-immunoprecipitates with KV4.2 and KV4.3 in mouse ventricular homogenates (Guo et al., 2002). Crystallographic, electron microscopic and electrophysiological experiments have shown that KChIPs physically interact with the distal part of N-terminus of KV4 subunits, forming a cross-shaped octamer, where two interaction sites on each KV4 subunit are important for KChIP2-mediated channel trafficking and gating (Bahring et al., 2001; Kim et al., 2004; Pioletti et al., 2006). KChIP2 decreases the turnover rate of KV4.2 by stabilizing the KChIP2–KV4.2 complex at the cell surface. This stabilization is lost with deletion of the N-terminal part of KV4.2 (Foeger et al., 2010). KChIP2 protein levels are decreased in the absence of KV4.2, despite unaltered mRNA levels, suggesting post-translational downregulation of KChIP2 in the absence of the pore-forming subunit (Guo et al., 2005). In addition, both KV4.2 and KV4.3 mRNA and protein expression are augmented in hearts from KChIP2−/− mice as well as in cultured neonatal rat cardiomyocytes overexpressing KChIP2, suggesting regulation of the pore-forming subunits at gene and protein levels (Thomsen et al., 2009b; Jin et al., 2010).
The rodent outward potassium currents (Figure 1) involved in action potential repolarization can broadly be divided into transient outward (Ito) and delayed rectifier (IK) currents (for a review, see Nerbonne et al., 2001), which can be further separated into four components, Ito,fast (Ito,f, KV4.2/KV4.3 + KChIP2), Ito,slow (Ito,s, KV1.4), IK,slow1 (IKur, KV1.5), and Ik,slow2 (ISS, KV2.1). In order to dissect these current components, several studies have used electrophysiology combined with pharmacology on cardiomyocytes of wild-type (WT) or transgenic mice (Himmel et al., 1999; Brouillette et al., 2004; Brunet et al., 2004; Liu et al., 2011). Ito,f and Ito,s both have a fast activation and a fast inactivation and are under normal conditions very similar, however they can be distinguished by the rate of recovery from inactivation (Wickenden et al., 1999a; Patel and Campbell, 2005). Ito,f recovers from inactivation in the order of milliseconds, whereas Ito,s recovers much slower. In order to inhibit Ito,f, an inactivating pre-pulse to −40 mV for 50 ms can be applied, which captures Ito,f in a closed state (Brouillette et al., 2004); alternatively Heteropoda toxin can be used, which selectively blocks KV4 channels in low-micromolar concentrations (Sanguinetti et al., 1997; DeSimone et al., 2011; Liu et al., 2011). For evaluating IK,slow1, the sensitivity toward 4-aminopyridine can be exploited, because IK,slow1 is sensitive in the micromolar range while Ito,f, Ito,s, and IK,slow2 are sensitive in the millimolar range (Guo et al., 1999). Furthermore, in order to dissect IK,slow2, an inactivating pre-pulse in combination with micromolar 4-aminopyridine can be used (Brouillette et al., 2004). Several studies have failed to find Ito,s in the free wall of the mouse ventricle and only located it in myocytes from the interventricular septum (London et al., 1998; Guo et al., 1999).
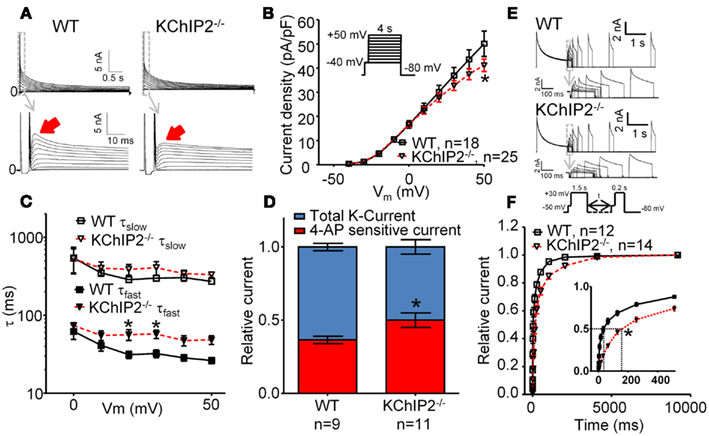
Figure 1. Whole-cell voltage-clamp recordings of outward K-currents recorded in disaggregated left ventricular cardiomyocytes from wild-type (WT) versus KChIP2−/− mice. Cardiomyocyte isolation and patch-clamp solutions (37°C) were made according to (Brouillette et al., 2004) with minor modifications. Cadmium (300 μM) was added to inhibit L-type Ca2+ currents. (A) Representative current traces showing that KChIP2−/− cardiomyocytes lack a fast decaying component of the outward K-current (solid arrows). (B) Peak current density as a function of test potential, however, was not significantly changed at any voltage (inset, voltage protocol). (C) The decay of the outward current (0–700 ms) was fitted to a second-order exponential function to obtain time-constants (τ) of inactivation for positive test potentials. No differences between WT and KChIP2−/− were found for the slow component; however, the fast component of the current decay was significantly slower for KChIP2−/− myocytes, confirming the loss of a transient component. (D) In order to dissect IK,slow1 from Ito,f, the current sensitive to 100 μM 4-AP was determined. The 4-AP sensitive fraction (red) of the peak current (blue, normalized to 1) was increased in KChIP2−/− compared to WT, indicating increased IK,slow1. (E) Recovery from inactivation was addressed by a double-pulse voltage-clamp protocol with varying interpulse intervals (see inset). Representative current traces from WT and KChIP2−/− cardiomyocytes are presented. (F) The mean recovery from inactivation is illustrated as the peak current during the 0.2-s test pulse relative to the 1.5-s base pulse. Recovery from inactivation was significantly slowed in KChIP2−/− compared to WT in interpulse intervals between 1 and 4000 ms. This is compatible with a larger fraction on the outward potassium currents attributable to IK,slow relative to Ito,f in KChIP2−/− cardiomyocytes. Data are reported as mean ± SEM. Statistical significance was evaluated by ANOVA followed by Newman–Keuls’ post hoc test where appropriate. *p < 0.05. Comparable data have been published previously by Thomsen et al. (2009b).
Electrophysiologically, KChIP2 increases KV4-mediated current amplitude and modulates channel gating by slowing the fast time constant of inactivation at depolarizing potentials and accelerating recovery from inactivation (Decher et al., 2001; Deschenes et al., 2002; Patel et al., 2002). The binding of KChIP2 to KV4 is calcium-independent; however the effect of KChIP2 on slowing the fast time constant of inactivation at depolarizing potentials is dependent on calcium-binding to three of the four EF hands on KChIP2 (Deschenes et al., 2002; Patel et al., 2002). Interestingly, when KV4 and KChIP2 are co-expressed in HEK293 the current inactivation is much slower than the native Ito in human and canine cardiomyocytes (Deschenes et al., 2002), which could be explained by the presence of additional subunits in the native cardiac cell. Dipeptidyl-peptidase-like proteins (DPPX) have been suggested as part of the missing link since a ternary complex of KV4.3, KChIP2, and DPPX produced currents closely resembling Ito,f in mammalian expression systems (Radicke et al., 2005; Lundby et al., 2010). Additionally, KCNE2–4 have all been implicated in regulation of the KV4-mediated current, suggesting a diverse macromolecular complex governing Ito (Zhang et al., 2001; Lundby and Olesen, 2006; Lundby et al., 2010).
The development of the KChIP2 deficient mouse provided the first clues to the role of KChIP2 in generating the native Ito (Kuo et al., 2001). Voltage-clamp recordings of KChIP2−/− cardiomyocytes revealed a complete loss of Ito,f defined as the portion of the potassium currents sensitive to an inactivating voltage pre-pulse to −40 mV. Moreover, administration of Heteropoda toxin to KChIP2−/− cardiomyocytes had no effect on potassium current amplitude (Thomsen et al., 2009b). In vivo, the loss of Ito,f translated to an elevated and earlier J wave of the murine ECG, normal QT intervals and ventricular effective refractory periods, and susceptibility to pacing-induced polymorphic ventricular tachyarrhythmias (Kuo et al., 2001). Furthermore, we showed that transmembrane action potentials were comparable in ventricular multicellular preparations from KChIP2−/− and WT control mice paced at 2 Hz (Thomsen et al., 2009c), which was presumably secondary to an upregulation of a 4-aminopyridine-sensitive current that restored peak outward potassium current density in KChIP2−/− mice (Figure 1). Action potentials recorded from disaggregated right ventricular myocytes from KChIP2−/− mice are longer than those from WT mice and show reverse rate dependency (Kuo et al., 2001), potentially consequential of the slowed recovery from inactivation of the remaining outward potassium currents (Figure 1).
CaV1.2
KChIP2 binds to the N-terminal cytosolic domain of CaV1.2 in a calcium-independent manner. In heterologous expression systems, KChIP2 co-expression causes an increase in current mediated by CaV1.2, comparable to the effect of the calcium channel auxiliary β2 subunit; however KChIP2 was not able to replace the β2-mediated macromolecular ion channel trafficking to the cell membrane (Thomsen et al., 2009c). Also this functional modulation was independent of calcium-binding to the EF hands of KChIP2. Furthermore, the L-type calcium current is significantly smaller in disaggregated cardiomyocytes from KChIP2−/− mice compared to WT controls (Figure 2). This decrease in current density was observed in the presence of an intact trafficking pathway of CaV1.2 and increased transcriptional activity of the β2 subunit (Thomsen et al., 2009a,c). Protein levels of CaV1.2 in the KChIP2−/− mice are increased, whereas acute gene silencing of KChIP2 in neonatal rat cardiomyocytes does not affect CaV1.2 protein expression (Deschenes et al., 2008). In addition, in neurons KChIP3 has been described as part of a macromolecular signaling complex together with CaV3 and KV4 that confer physiological calcium modulation of KV4-mediated currents (Anderson et al., 2010).
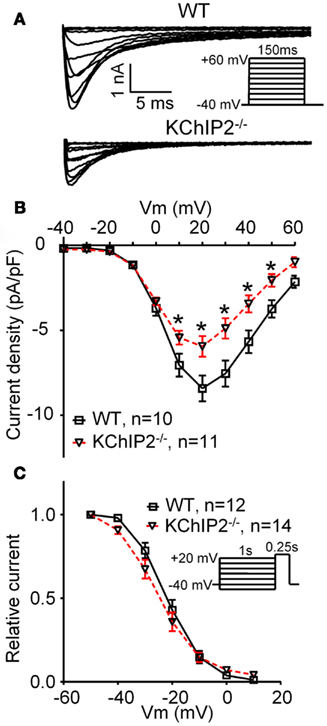
Figure 2. L-type Ca2+ current in disaggregated cardiomyocytes from wild-type (WT) versus KChIP2−/−. ICa,L was recorded at 37°C in left ventricular cardiomyocytes as the current fraction sensitive to 300 μM Cd2+, as described by Sah et al. (2002) with minor modifications. (A) Representative current traces of WT and KChIP2−/− (inset, voltage protocol). (B) Mean peak current densities show a decreased ICa,L in KChIP2−/− cardiomyocytes. (C) No changes were found in the steady-state inactivation of ICa,L in WT versus KChIP2−/− cardiomyocytes (inset, voltage protocol). Data are reported as mean ± SEM. Statistical significance was evaluated by ANOVA followed by Newman–Keuls’ post hoc test where appropriate. *p < 0.05. Comparable data have been published previously by Thomsen et al. (2009c).
NaV1.5
A structural and functional association of Ito and INa has been suggested based on data from neonatal rat cardiomyocytes (Deschenes et al., 2008). Genetic silencing of KChIP2 caused a decrease in protein contents of KV4.2 and KV4.3, presumably due to a trafficking defect. Interestingly, the authors showed a biochemical association between KV4 and the auxiliary β1 subunit of the sodium channel, and went on to demonstrate that KChIP2 silencing also dramatically reduced mRNA and protein levels of NaV1.5 and the sodium channel β1 subunit to a point where no INa could be recorded and no action potentials could be elicited (Deschenes et al., 2008). Reversibly, co-expression of KChIP2 with NaV1.5 in a mammalian cell line augmented the sodium current, further substantiating the functional effect of KChIP2 on INa (Deschenes et al., 2008), suggesting a close structural and functional link of Ito and INa. These findings are not reproduced in mice with complete deletion of KChIP2 as they do not show embryonic lethality which would be expected if INa was reduced to the point the hearts were inexcitable (Kuo et al., 2001). The KChIP2−/− mice show a small, but statistically not significant, reduction of the upstroke velocity of action potentials, generally accepted to be an indication of INa availability (Thomsen et al., 2009b). Furthermore, mRNA levels for NaV1.5 and β1 are comparable in KChIP2−/− mice and WT controls (M Thomsen, unpublished data). The reason underlying the discrepant results is currently unknown, but could include an unidentified compensatory adaptation in the embryogenic development of the knockout mouse to counteract a reduction in INa.
KV1.5
KV1.5 governs the murine IK,slow and the human atrial-specific IKur. In HEK-293 cells, KChIP2 has been reported to reduce KV1.5 current density without modifying the kinetics of the current (Li et al., 2005). Immunocytochemistry suggested that KChIP2 attenuate trafficking of KV1.5 to the plasma membrane and EF-hand mutations abolished the effect of KChIP2. In the same study, mouse ventricular KV1.5 and KChIP2 were found to co-immunoprecipitate (Li et al., 2005). However, co-expression of KV1.5 with KChIP2 in Xenopus laevis oocytes revealed no difference in current levels in absence or presence of KChIP2 (Lundby et al., 2010). Additionally, both groups found no effect of KChIP2 on Kv1.4 currents (Li et al., 2005; Lundby et al., 2010). In KChIP2−/− mice, KV1.5 mRNA levels were elevated in KChIP2−/− hearts, suggesting a potential KChIP2-mediated regulation of Kv1.5 on a transcriptional level.
KChIP2 and Arrhythmias
Spontaneous ventricular arrhythmias are rarely seen in mice with genetic deletion of single potassium currents (Nerbonne et al., 2001). For example, the KV4.2−/− mouse show complete elimination of Ito,f but normal action potentials and ECG and no spontaneous arrhythmia (Guo et al., 2005). On the other hand, prolonged action potentials and QT intervals were reported in another mouse model with total elimination of Ito,f (KV4.2-W362F; Barry et al., 1998). Interestingly, overexpression of a dominant-negative KV4.2 fragment generated a mouse line with reduced Ito,f, prolonged action potentials and hypercontractility that developed into heart failure within 3 months of age (Wickenden et al., 1999b).
The KChIP2−/− mouse had no spontaneous arrhythmia during continuous monitoring of conscious mice; however, it was possible to induce cardiac tachyarrhythmia by pacing the ventricles (Kuo et al., 2001). Presently, it is unclear whether the increased susceptibility to pacing-induced arrhythmias is secondary to decreased repolarizing potassium currents or decreased depolarizing calcium currents, or a combination of the two.
KChIP2 and Remodeling
Cardiac Memory
Cardiac memory describe a special form of cardiac plasticity observed as an altered T wave on the surface ECG (Rosenbaum et al., 1982). After a period of altered ventricular activation, due to pacing or arrhythmias, the T wave “remembers” the QRS complex from the paced or arrhythmic phase. The electrocardiographic T wave vector during sinus rhythm reflects the QRS complex vector during the preceding period of ventricular activation (Rosen, 2001).
Cardiac memory in dogs is associated with a decrease in Ito and a reduction in KV4.3 mRNA and KChIP2 mRNA and protein (Yu et al., 1999; Patberg et al., 2003). KV4.2, KChIP2, and the angiotensin-1 receptor co-localize and immunoprecipitate together in heart tissue (Doronin et al., 2004). Furthermore, when ventricular pacing alters cardiac stretch, angiotensin-II is released (Sadoshima and Izumo, 1993). Activation of the angiotensin-1 receptor results in an internalization of the macromolecular complex and a decrease in Ito density. Secondly, during prolonged periods of ventricular pacing inducing cardiac memory, cyclic AMP response element binding protein (CREB) is downregulated via ubiquitination and proteosomal degradation (Ozgen et al., 2010). Transcriptional activity of KChIP2 is reduced by low levels of CREB as this fails to associate with cyclic AMP response elements in the KChIP2-gene promotor region, indirectly causing a further reduction in Ito (Patberg et al., 2005). In summary, altered ventricular pacing causes a rapid internalization if the KV4 ion channel complex and a reduction in KChIP2 levels further reducing Ito (Rosen and Cohen, 2006). Angiotensin-1 receptor blockade do not suppress the latter pathway, whereas inhibition of ICa,L attenuates cardiac memory and Ito downregulation, suggesting a role central for calcium handling in the development of cardiac memory (Plotnikov et al., 2003).
Hypertrophy
Hypertrophic enlargement of the ventricles involves complex cellular processes, and is typically divided into concentric hypertrophy due to pressure overload and eccentric hypertrophy due to volume overload. Hypertrophy is associated with growth of the individual cardiomyocyte which adds sarcomeres to the existing contractile apparatus, thus responding to the increased demand on the ventricle. In contrast, the failing heart cannot compensate sufficiently, and is impaired in its ability to sustain an adequate blood flow to the body. Among a plethora of changes, cardiac hypertrophy is accompanied by electrophysiological remodeling causing prolonged action potential durations (Gaughan et al., 1998; Kaab et al., 1998; Marionneau et al., 2008). Ion channel remodeling in cardiac hypertrophy and failure has been described recently in excellent reviews (Nattel et al., 2007; Nass et al., 2008; Aiba and Tomaselli, 2010). Here, we focus on KChIP2.
Modulating Ito affects action potential duration, intracellular calcium load and contractile force in a complex manner. In larger mammals, pharmacological activation of Ito lead to prolongation of the action potential in a concentration-dependent manner (Calloe et al., 2009a). Moreover, large increases in Ito to a point where CaV1.2 fails to activate, action potentials show a dramatic shortening (Greenstein et al., 2000; Sah et al., 2003; Calloe et al., 2011). Selective pharmacological inhibition of Ito,f in species with a spike-and-dome morphology action potential is yet to be studied.
In rodents, Ito is part of the terminal repolarization and action potential prolongation is consistently seen in the absence of the current (Nerbonne et al., 2001). This action potential prolongation elevates cytosolic calcium levels and improves excitation–contraction coupling by mechanisms distinct from the effects of Ito modulation in larger mammals (Sah et al., 2003). Furthermore, an elevated calcium level activates the calcium-sensitive phosphatase calcineurin and initiates hypertrophic signaling (Kassiri et al., 2002; Zobel et al., 2002; Lebeche et al., 2004). Transcriptional regulation of many hypertrophic genes is regulated via NFAT by the activity of calcineurin (Gaughan et al., 1998; Zobel et al., 2002; Gong et al., 2006; Jin et al., 2010). It appears that an increased cytosolic calcium concentration during adrenergically induced hypercontractility activates calcineurin initiating the hypertrophic response. Hypertrophy is not observed in models with genetically reduced Ito,f in the absence of action potential prolongation, potentially due to chronic upregulation of other repolarizing currents (Kuo et al., 2001; Guo et al., 2005). Interestingly, concentric hypertrophy in WT mice secondary to aortic banding is associated with prolonged action potentials, decreased Ito,f and an increased protein level of the ion channel underlying IK,slow1 (Marionneau et al., 2008), whereas angiotensin-II mediated hypertension causing cardiac hypertrophy leaves Ito,f density and KV4.2, KV4.3, and KChIP2 protein contents unaltered.
Using KChIP2 overexpression to successfully prevent hypertrophy in rats showed that a KChIP2-mediated upregulation of Ito shortened action potentials and reduced myocardial shortening (Jin et al., 2010). In the same study, the authors showed that KChIP2 overexpression decreased calcineurin expression in vitro, suggesting that action potential shortening abrogates the hypertrophic response via the calcineurin pathway. Moreover, simultaneous overexpression of KV4.3 and induction of concentric hypertrophy in rat hearts showed a robust increase in Ito, action potential abbreviation, reduced calcineurin expression and an attenuated hypertrophic response (Lebeche et al., 2004).
Transgenic mice overexpressing calcineurin have a reduced Ito,f, prolonged action potential duration and display cardiac hypertrophy, which could all be reversed by pharmacological inhibition of calcineurin (Dong et al., 2003, 2010). In young mice overexpressing calcineurin, a paradoxical increased Ito,f was measured, secondary to elevated KV4.2 expression (Gong et al., 2006). It was hypothesized that two regulatory pathways operated in parallel, one leading to positive regulation of KV4.2 transcription via activation of calcineurin and NFAT translocation (Gong et al., 2006); and one that via NF-κB phosphorylation reduces Ito,f via downregulation of KChIP2 and KV4.3 (Panama et al., 2011).
Heart Failure
Heart failure is the condition, where the ability of the heart to adequately supply blood flow to meet the demand of the body is impaired. Whereas the underlying index events are often manageable if identified early, treatment options for heart failure are mainly palliative or merely slowing the relentless progression of symptoms (Dickstein et al., 2008). At end-stage heart failure, therapeutic options are heart transplantation or mechanical support devices. The lifetime risk of acquiring heart failure is estimated to 20% (Lloyd-Jones et al., 2002) and the 5-year mortality rate after diagnosis is 50% (Stewart et al., 2001).
Functional reduction of Ito has been observed in many animal models of heart failure (Nass et al., 2008) in addition to human samples from terminal heart failure transplants (Beuckelmann et al., 1993; Wettwer et al., 1994; Nabauer et al., 1996). KV4.3 mRNA is reduced in human (Kaab et al., 1998) and canine heart failure (Akar et al., 2005). KChIP2 mRNA and protein is downregulated in human heart failure (Radicke et al., 2006; Soltysinska et al., 2009), although reports of unchanged KChIP2 levels are also available (Zicha et al., 2004). Heart failure secondary to pressure overload reduces peak potassium currents in mice, but does not completely abolish Ito,f (Wang et al., 2007; Grubb et al., 2011). Furthermore, preliminary studies show a reduction of non-Ito,f currents in KChIP2−/− mice with heart failure, adding another layer of complexity to the electrophysiological remodeling seen in heart failure (Grubb et al., 2011).
Limitations
In order to translate findings in experimental models to useful information in a clinical setting, a clear understanding of species dependent differences is electrophysiology of rodents and larger mammals is imperative. Compatible with having a higher heart rate than larger mammals, rodents have a much shorter action potential and Ito is involved with total repolarization, whereas it constitutes the notch between the spike-and-dome in the action potential of larger mammals (for review, see Nerbonne et al., 2001). In rodents, a loss of Ito leads to a prolonged APD which results in a loss of temporal synchronization of the Ca2+ entry through ICa,L due to channel inactivation instead of deactivation causing a longer Ca2+ entry and a stronger but less synchronized SR Ca2+ release. In larger mammals, instead of being responsible for ICa,L deactivation, Ito provides a driving force for Ca2+ entry, and a decreased Ito therefore leads to an ICa,L reduction and a desynchronized intracellular Ca2+-transient (for review, see Sah et al., 2003). This is an important species dependent difference, and should be considered when using rodents as a model of hypertrophy or heart failure. Moreover, extrapolation of physiological data from rodent studies to clinical practice should always be performed with outmost caution and only with supporting results from additional experimental models and human trials.
The KChIP2−/− mouse does not show any apparent pathology; however, it is not uncommon that knockout animals lack phenotypic changes which may be due to a redundancy of genes that can compensate for the reduced repolarization reserve. The KChIP2−/− mouse is not an organ-specific knockout, which may have unspecific, peripheral effects compared to a cardiac-specific knockout mice, since many genes are expressed in several organs, including KChIP2 (An et al., 2000). Furthermore, the knockout of KChIP2 is congenital and non-inducible, which means that remodeling in the knockout animal could happen during development. This could be avoided by using an inducible organ-specific knockout or a tissue specific gene transfer by viral infection, which would also be preferable for studying the remodeling in hypertrophy or heart failure, since no pre-existing remodeling would exist.
Summary and Concluding Remarks
Substantial progress has been made in the mapping of the multiple characteristics and roles of KChIP2 in the heart. KChIP2 have been reported to modulate KV1.5, KV4.2, KV4.3, CaV1.2, and NaV1.5. Both KV4.2 and KV4.3 are intimately regulated by KChIP2, where peak current is increased, inactivation kinetics are slowed and recovery from inactivation is accelerated by the presence of KChIP2 (An et al., 2000). CaV1.2-conducted current is increased by KChIP2 via a potential interaction at the amino-terminal inhibitory module on the α1C subunit of the L-type Ca2+ channel (Thomsen et al., 2009c). Conflicting reports regarding KChIP2-mediated modulation of KV1.5 and NaV1.5 exists and points toward areas where intensified research is required to resolve discrepancies and clarify our understanding of cardiac electrophysiology. Cardiac memory, hypertrophy, and failure are all associated with reductions in KChIP2 and it appears that overexpression of KChIP2 can prevent the development of hypertrophy in rats (Jin et al., 2010).
Several aspects remains poorly understood, however. Given the promiscuity of KChIP2, it is possible that additional binding partners will be identified, potentially in non-cardiac tissue. The physiological relevance of the many individual interactions needs to be tested and compared in health and disease. The finding that KChIP2 overexpression can prevent the development of the hypertrophic response may have implications for future clinical approaches to heart disease, although extrapolation from rodent to patient should be done with outmost caution. Excessive KChIP2 overexpression increasing Ito may in spite of an augmented inward ICa,L lead to an early repolarization in mammals associated with a Brugada-like phenotype (Calloe et al., 2009a). Presently it is unknown whether KChIP2 holds regulatory control in the development of heart failure similar to its central role in the progress of cardiac hypertrophy. Furthermore, the antiarrhythmic properties of KChIP2 are still ill described, notwithstanding its initial report (Kuo et al., 2001). Reduced KChIP2 levels confer smaller ICa,L potentially reducing the amplitude of early afterdepolarizations in events of current reactivation at long action potentials, reducing the risk of triggering ventricular arrhythmias. However, optimal KChIP2 levels contribute to maintaining minimal regional repolarization dispersion, thus reducing the substrate for arrhythmias. Nonetheless, KChIP2 is a multipronged modifier of a series of voltage-gated cardiac ion channels and has important physiological functions in health and in heart disease.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the Danish Agency for Science, Technology and Innovation, Medical Research Council (to Morten B. Thomsen) and from the Danish National Research Foundation Centre for Cardiac Arrhythmia (to Kirstine Calloe).
References
Aiba, T., and Tomaselli, G. F. (2010). Electrical remodeling in the failing heart. Curr. Opin. Cardiol. 25, 29–36.
Akar, F. G., Wu, R. C., Juang, G. J., Tian, Y., Burysek, M., Disilvestre, D., Xiong, W., Armoundas, A. A., and Tomaselli, G. F. (2005). Molecular mechanisms underlying K+ current downregulation in canine tachycardia-induced heart failure. Am. J. Physiol. Heart Circ. Physiol. 288, H2887–H2896.
An, W. F., Bowlby, M. R., Betty, M., Cao, J., Ling, H. P., Mendoza, G., Hinson, J. W., Mattsson, K. I., Strassle, B. W., Trimmer, J. S., and Rhodes, K. J. (2000). Modulation of A-type potassium channels by a family of calcium sensors. Nature 403, 553–556.
Anderson, D., Mehaffey, W. H., Iftinca, M., Rehak, R., Engbers, J. D., Hameed, S., Zamponi, G. W., and Turner, R. W. (2010). Regulation of neuronal activity by Cav3–Kv4 channel signaling complexes. Nat. Neurosci. 13, 333–337.
Bahring, R., Dannenberg, J., Peters, H. C., Leicher, T., Pongs, O., and Isbrandt, D. (2001). Conserved Kv4 N-terminal domain critical for effects of Kv channel-interacting protein 2.2 on channel expression and gating. J. Biol. Chem. 276, 23888–23894.
Barry, D. M., Xu, H., Schuessler, R. B., and Nerbonne, J. M. (1998). Functional knockout of the transient outward current, long-QT syndrome, and cardiac remodeling in mice expressing a dominant-negative Kv4 alpha subunit. Circ. Res. 83, 560–567.
Beuckelmann, D. J., Nabauer, M., and Erdmann, E. (1993). Alterations of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circ. Res. 73, 379–385.
Brouillette, J., Clark, R. B., Giles, W. R., and Fiset, C. (2004). Functional properties of K+ currents in adult mouse ventricular myocytes. J. Physiol. (Lond.) 559, 777–798.
Brunet, S., Aimond, F., Li, H., Guo, W., Eldstrom, J., Fedida, D., Yamada, K. A., and Nerbonne, J. M. (2004). Heterogeneous expression of repolarizing, voltage-gated K+ currents in adult mouse ventricles. J. Physiol. (Lond.) 559, 103–120.
Burgoyne, R. D., and Weiss, J. L. (2001). The neuronal calcium sensor family of Ca2+-binding proteins. Biochem. J. 353, 1–12.
Buxbaum, J. D. (2004). A role for calsenilin and related proteins in multiple aspects of neuronal function. Biochem. Biophys. Res. Commun. 322, 1140–1144.
Calloe, K., Cordeiro, J. M., Di Diego, J. M., Hansen, R. S., Grunnet, M., Olesen, S. P., and Antzelevitch, C. (2009a). A transient outward potassium current activator recapitulates the electrocardiographic manifestations of Brugada syndrome. Cardiovasc. Res. 81, 686–694.
Calloe, K., Soltysinska, E., Jespersen, T., Lundby, A., Antzelevitch, C., Olesen, S. P., and Cordeiro, J. M. (2009b). Differential effects of the transient outward K+ current activator NS5806 in the canine left ventricle. J. Mol. Cell. Cardiol. 48, 191–200.
Calloe, K., Nof, E., Jespersen, T., Di Diego, J. M., Chlus, N., Olesen, S. P., Antzelevitch, C., and Cordeiro, J. M. (2011). Comparison of the effects of a transient outward potassium channel activator on currents recorded from atrial and ventricular cardiomyocytes. J. Cardiovasc. Electrophysiol. 22, 1057–1066.
Carmeliet, E. (1999). Cardiac ionic currents and acute ischemia: from channels to arrhythmias. Physiol. Rev. 79, 917–1017.
Cordeiro, J. M., Greene, L., Heilmann, C., Antzelevitch, D., and Antzelevitch, C. (2004). Transmural heterogeneity of calcium activity and mechanical function in the canine left ventricle. Am. J. Physiol. Heart Circ. Physiol. 286, H1471–H1479.
Decher, N., Uyguner, O., Scherer, C. R., Karaman, B., Yuksel-Apak, M., Busch, A. E., Steinmeyer, K., and Wollnik, B. (2001). HKChIP2 is a functional modifier of hKv4.3 potassium channels: cloning and expression of a short hKChIP2 splice variant. Cardiovasc. Res. 52, 255–264.
Deschenes, I., Armoundas, A. A., Jones, S. P., and Tomaselli, G. F. (2008). Post-transcriptional gene silencing of KChIP2 and Navbeta1 in neonatal rat cardiac myocytes reveals a functional association between Na and Ito currents. J. Mol. Cell. Cardiol. 45, 336–346.
Deschenes, I., Disilvestre, D., Juang, G. J., Wu, R. C., An, W. F., and Tomaselli, G. F. (2002). Regulation of Kv4.3 current by KChIP2 splice variants: a component of native cardiac I(to)? Circulation 106, 423–429.
DeSimone, C. V., Zarayskiy, V. V., Bondarenko, V. E., and Morales, M. J. (2011). Heteropoda toxin 2 interaction with Kv4.3 and Kv4.1 reveals differences in gating modification. Mol. Pharmacol. 80, 345–355.
Dickstein, K., Cohen-Solal, A., Filippatos, G., Mcmurray, J. J., Ponikowski, P., Poole-Wilson, P. A., Stromberg, A., Van Veldhuisen, D. J., Atar, D., Hoes, A. W., Keren, A., Mebazaa, A., Nieminen, M., Priori, S. G., Swedberg, K., Vahanian, A., Camm, J., De Caterina, R., Dean, V., Funck-Brentano, C., Hellemans, I., Kristensen, S. D., Mcgregor, K., Sechtem, U., Silber, S., Tendera, M., Widimsky, P., and Zamorano, J. L. (2008). ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2008: the Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2008 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association of the ESC (HFA) and endorsed by the European Society of Intensive Care Medicine (ESICM). Eur. Heart J. 29, 2388–2442.
Dong, D., Duan, Y., Guo, J., Roach, D. E., Swirp, S. L., Wang, L., Lees-Miller, J. P., Sheldon, R. S., Molkentin, J. D., and Duff, H. J. (2003). Overexpression of calcineurin in mouse causes sudden cardiac death associated with decreased density of K+ channels. Cardiovasc. Res. 57, 320–332.
Dong, M., Yan, S., Chen, Y., Niklewski, P. J., Sun, X., Chenault, K., and Wang, H. S. (2010). Role of the transient outward current in regulating mechanical properties of canine ventricular myocytes. J. Cardiovasc. Electrophysiol. 21, 697–703.
Doronin, S. V., Potapova, I. A., Lu, Z. J., and Cohen, I. S. (2004). Angiotensin receptor type 1 forms a complex with the transient outward potassium channel Kv4.3 and regulates its gating properties and intracellular localization. J. Biol. Chem. 279, 48231–48237.
Foeger, N. C., Marionneau, C., and Nerbonne, J. M. (2010). Co-assembly of Kv4 {alpha} subunits with K+ channel-interacting protein 2 stabilizes protein expression and promotes surface retention of channel complexes. J. Biol. Chem. 285, 33413–33422.
Gaborit, N., Le Bouter, S., Szuts, V., Varro, A., Escande, D., Nattel, S., and Demolombe, S. (2007). Regional and tissue specific transcript signatures of ion channel genes in the non-diseased human heart. J. Physiol. (Lond.) 582, 675–693.
Gaughan, J. P., Hefner, C. A., and Houser, S. R. (1998). Electrophysiological properties of neonatal rat ventricular myocytes with alpha1-adrenergic-induced hypertrophy. Am. J. Physiol. 275, H577–H590.
Gong, N. L., Bodi, I., Zobel, C., Schwartz, A., Molkentin, J. D., and Backx, P. H. (2006). Calcineurin increases cardiac transient outward K+ currents via transcriptional up-regulation of Kv4.2 channel subunits. J. Biol. Chem. 281, 38498–38506.
Greenstein, J. L., Wu, R., Po, S., Tomaselli, G. F., and Winslow, R. L. (2000). Role of the calcium-independent transient outward current I(to1) in shaping action potential morphology and duration. Circ. Res. 87, 1026–1033.
Grubb, S., Speerschneider, T., Olesen, S. P., Thomsen, M. B., and Calloe, K. (2011). Reduction of potassium currents in heart failure is accentuated in K+ channel interacting protein 2 deficient mice. Heart Rhythm 8, S459.
Guo, W., Jung, W. E., Marionneau, C., Aimond, F., Xu, H., Yamada, K. A., Schwarz, T. L., Demolombe, S., and Nerbonne, J. M. (2005). Targeted deletion of Kv4.2 eliminates I(to,f) and results in electrical and molecular remodeling, with no evidence of ventricular hypertrophy or myocardial dysfunction. Circ. Res. 97, 1342–1350.
Guo, W., Xu, H., London, B., and Nerbonne, J. M. (1999). Molecular basis of transient outward K+ current diversity in mouse ventricular myocytes. J. Physiol. (Lond.) 521(Pt 3), 587–599.
Guo, W. N., Li, H. L., Aimond, F., Johns, D. C., Rhodes, K. J., Trimmer, J. S., and Nerbonne, J. M. (2002). Role of heteromultimers in the generation of myocardial transient outward K+ currents. Circ. Res. 90, 586–593.
Harris, D. M., Mills, G. D., Chen, X., Kubo, H., Berretta, R. M., Votaw, V. S., Santana, L. F., and Houser, S. R. (2005). Alterations in early action potential repolarization causes localized failure of sarcoplasmic reticulum Ca2+ release. Circ. Res. 96, 543–550.
Himmel, H. M., Wettwer, E., Li, Q., and Ravens, U. (1999). Four different components contribute to outward current in rat ventricular myocytes. Am. J. Physiol. 277, H107–H118.
Holmqvist, M. H., Cao, J., Hernandez-Pineda, R., Jacobson, M. D., Carroll, K. I., Sung, M. A., Betty, M., Ge, P., Gilbride, K. J., Brown, M. E., Jurman, M. E., Lawson, D., Silos-Santiago, I., Xie, Y., Covarrubias, M., Rhodes, K. J., Distefano, P. S., and An, W. F. (2002). Elimination of fast inactivation in Kv4 A-type potassium channels by an auxiliary subunit domain. Proc. Natl. Acad. Sci. U.S.A. 99, 1035–1040.
Jin, H., Hadri, L., Palomeque, J., Morel, C., Karakikes, I., Kaprielian, R., Hajjar, R., and Lebeche, D. (2010). KChIP2 attenuates cardiac hypertrophy through regulation of Ito and intracellular calcium signaling. J. Mol. Cell. Cardiol. 48, 1169–1179.
Kaab, S., Dixon, J., Duc, J., Ashen, D., Nabauer, M., Beuckelmann, D. J., Steinbeck, G., Mckinnon, D., and Tomaselli, G. F. (1998). Molecular basis of transient outward potassium current downregulation in human heart failure: a decrease in Kv4.3 mRNA correlates with a reduction in current density. Circulation 98, 1383–1393.
Kassiri, Z., Zobel, C., Nguyen, T. T., Molkentin, J. D., and Backx, P. H. (2002). Reduction of I(to) causes hypertrophy in neonatal rat ventricular myocytes. Circ. Res. 90, 578–585.
Kim, L. A., Furst, J., Butler, M. H., Xu, S. H., Grigorieff, N., and Goldstein, S. A. N. (2004). I-to channels are octomeric complexes with four subunits of each Kv4.2 and K+ channel-interacting protein 2. J. Biol. Chem. 279, 5549–5554.
Kuo, H. C., Cheng, C. F., Clark, R. B., Lin, J. J., Lin, J. L., Hoshijima, M., Nguyen-Tran, V. T., Gu, Y., Ikeda, Y., Chu, P. H., Ross, J., Giles, W. R., and Chien, K. R. (2001). A defect in the Kv channel-interacting protein 2 (KChIP2) gene leads to a complete loss of I(to) and confers susceptibility to ventricular tachycardia. Cell 107, 801–813.
Lebeche, D., Kaprielian, R., Del Monte, F., Tomaselli, G., Gwathmey, J. K., Schwartz, A., and Hajjar, R. J. (2004). In vivo cardiac gene transfer of Kv4.3 abrogates the hypertrophic response in rats after aortic stenosis. Circulation 110, 3435–3443.
Li, H. L., Guo, W. N., Mellor, R. L., and Nerbonne, J. M. (2005). KChIP2 modulates the cell surface expression of Kv1.5-encoded K+ channels. J. Mol. Cell. Cardiol. 39, 121–132.
Liu, J., Kim, K. H., London, B., Morales, M. J., and Backx, P. H. (2011). Dissection of the voltage-activated potassium outward currents in adult mouse ventricular myocytes: I(to,f), I(to,s), I(K,slow1), I(K,slow2), and I(ss). Basic Res. Cardiol. 106, 189–204.
Lloyd-Jones, D. M., Larson, M. G., Leip, E. P., Beiser, A., D’Agostino, R. B., Kannel, W. B., Murabito, J. M., Vasan, R. S., Benjamin, E. J., and Levy, D. (2002). Lifetime risk for developing congestive heart failure: the Framingham Heart Study. Circulation 106, 3068–3072.
London, B., Wang, D. W., Hill, J. A., and Bennett, P. B. (1998). The transient outward current in mice lacking the potassium channel gene Kv1.4. J. Physiol. 509(Pt 1), 171–182.
Lundby, A., Jespersen, T., Schmitt, N., Grunnet, M., Olesen, S. P., Cordeiro, J. M., and Calloe, K. (2010). Effect of the I(to) activator NS5806 on cloned K(v)4 channels depends on the accessory protein KChIP2. Br. J. Pharmacol. 160, 2028–2044.
Lundby, A., and Olesen, S. P. (2006). KCNE3 is an inhibitory subunit of the Kv4.3 potassium channel. Biochem. Biophys. Res. Commun. 346, 958–967.
Marionneau, C., Brunet, S., Flagg, T. P., Pilgram, T. K., Demolombe, S., and Nerbonne, J. M. (2008). Distinct cellular and molecular mechanisms underlie functional remodeling of repolarizing K+ currents with left ventricular hypertrophy. Circ. Res. 102, 1406–1415.
Nabauer, M., Beuckelmann, D. J., Uberfuhr, P., and Steinbeck, G. (1996). Regional differences in current density and rate-dependent properties of the transient outward current in subepicardial and subendocardial myocytes of human left ventricle. Circulation 93, 168–177.
Nass, R. D., Aiba, T., Tomaselli, G. F., and Akar, F. G. (2008). Mechanisms of disease: ion channel remodeling in the failing ventricle. Nat. Clin. Pract. Cardiovasc. Med. 5, 196–207.
Nattel, S., Maguy, A., Le Bouter, S., and Yeh, Y. H. (2007). Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol. Rev. 87, 425–456.
Nerbonne, J. M., and Kass, R. S. (2005). Molecular physiology of cardiac repolarization. Physiol. Rev. 85, 1205–1253.
Nerbonne, J. M., Nichols, C. G., Schwarz, T. L., and Escande, D. (2001). Genetic manipulation of cardiac K+ channel function in mice: what have we learned, and where do we go from here? Circ. Res. 89, 944–956.
Ozgen, N., Lau, D. H., Shlapakova, I. N., Sherman, W., Feinmark, S. J., Danilo, P. Jr., and Rosen, M. R. (2010). Determinants of CREB degradation and KChIP2 gene transcription in cardiac memory. Heart Rhythm 7, 964–970.
Panama, B. K., Latour-Villamil, D., Farman, G. P., Zhao, D., Bolz, S. S., Kirshenbaum, L. A., and Backx, P. H. (2011). Nuclear factor kappaB downregulates the transient outward potassium current I(to,f) through control of KChIP2 expression. Circ. Res. 108, 537–543.
Patberg, K. W., Obreztchikova, M. N., Giardina, S. F., Symes, A. J., Plotnikov, A. N., Qu, J. H., Chandra, P., Mckinnon, D., Liou, S. R., Rybin, A. V., Shlapakova, I., Danilo, P., Yang, J., and Rosen, M. R. (2005). The cAMP response element binding protein modulates expression of the transient outward current: implications for cardiac memory. Cardiovasc. Res. 68, 259–267.
Patberg, K. W., Plotnikov, A. N., Quamina, A., Gainullin, R. Z., Rybin, A., Danilo, P. Jr., Sun, L. S., and Rosen, M. R. (2003). Cardiac memory is associated with decreased levels of the transcriptional factor CREB modulated by angiotensin II and calcium. Circ. Res. 93, 472–478.
Patel, S. P., and Campbell, D. L. (2005). Transient outward potassium current, ‘I-to’, phenotypes in the mammalian left ventricle: underlying molecular, cellular and biophysical mechanisms. J. Physiol. (Lond.) 569, 7–39.
Patel, S. P., Campbell, D. L., and Strauss, H. C. (2002). Elucidating KChIP effects on Kv4.3 inactivation and recovery kinetics with a minimal KChIP2 isoform. J. Physiol. (Lond.) 545, 5–11.
Pioletti, M., Findeisen, F., Hura, G. L., and Minor, D. L. (2006). Three-dimensional structure of the KChIP1–Kv4.3 T1 complex reveals a cross-shaped octamer. Nat. Struct. Mol. Biol. 13, 987–995.
Plotnikov, A. N., Sosunov, E. A., Patberg, K. W., Anyukhovsky, E. P., Gainullin, R. Z., Shlapakova, I. N., Krishnamurthy, G., Danilo, P. Jr., and Rosen, M. R. (2004). Cardiac memory evolves with age in association with development of the transient outward current. Circulation 110, 489–495.
Plotnikov, A. N., Yu, H., Geller, J. C., Gainullin, R. Z., Chandra, P., Patberg, K. W., Friezema, S., Danilo, P. Jr., Cohen, I. S., Feinmark, S. J., and Rosen, M. R. (2003). Role of L-type calcium channels in pacing-induced short-term and long-term cardiac memory in canine heart. Circulation 107, 2844–2849.
Radicke, S., Cotella, D., Graf, E. M., Banse, U., Jost, N., Varro, A., Tseng, G. N., Ravens, U., and Wettwer, E. (2006). Functional modulation of the transient outward current Ito by KCNE beta-subunits and regional distribution in human non-failing and failing hearts. Cardiovasc. Res. 71, 695–703.
Radicke, S., Cotella, D., Graf, E. M., Ravens, U., and Wettwer, E. (2005). Expression and function of dipeptidyl-aminopeptidase-like protein 6 as a putative beta-subunit of human cardiac transient outward current encoded by Kv4.3. J. Physiol. (Lond.) 565, 751–756.
Rosati, B., Grau, F., Rodriguez, S., Li, H. L., Nerbonne, J. M., and Mckinnon, D. (2003). Concordant expression of KChIP2 mRNA, protein and transient outward current throughout the canine ventricle. J. Physiol. (Lond.) 548, 815–822.
Rosati, B., Pan, Z. M., Lypen, S., Wang, H. S., Cohen, I., Dixon, J. E., and Mckinnon, D. (2001). Regulation of KChIP2 potassium channel beta subunit gene expression underlies the gradient of transient outward current in canine and human ventricle. J. Physiol. (Lond.) 533, 119–125.
Rosen, M. R., and Cohen, I. S. (2006). Cardiac memory… new insights into molecular mechanisms. J. Physiol. 570, 209–218.
Rosenbaum, M. B., Blanco, H. H., Elizari, M. V., Lazzari, J. O., and Davidenko, J. M. (1982). Electrotonic modulation of the T wave and cardiac memory. Am. J. Cardiol. 50, 213–222.
Sadoshima, J., and Izumo, S. (1993). Molecular characterization of angiotensin II-induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts. Critical role of the AT1 receptor subtype. Circ. Res. 73, 413–423.
Sah, R., Ramirez, R. J., and Backx, P. H. (2002). Modulation of Ca(2+) release in cardiac myocytes by changes in repolarization rate: role of phase-1 action potential repolarization in excitation–contraction coupling. Circ. Res. 90, 165–173.
Sah, R., Ramirez, R. J., Oudit, G. Y., Gidrewicz, D., Trivieri, M. G., Zobel, C., and Backx, P. H. (2003). Regulation of cardiac excitation–contraction coupling by action potential repolarization: role of the transient outward potassium current (I(to)). J. Physiol. (Lond.) 546, 5–18.
Sanguinetti, M. C., Johnson, J. H., Hammerland, L. G., Kelbaugh, P. R., Volkmann, R. A., Saccomano, N. A., and Mueller, A. L. (1997). Heteropodatoxins: peptides isolated from spider venom that block Kv4.2 potassium channels. Mol. Pharmacol. 51, 491–498.
Soltysinska, E., Olesen, S. P., Christ, T., Wettwer, E., Varro, A., Grunnet, M., and Jespersen, T. (2009). Transmural expression of ion channels and transporters in human nondiseased and end-stage failing hearts. Pflügers Arch. 459, 11–23.
Stewart, S., Macintyre, K., Hole, D. J., Capewell, S., and Mcmurray, J. J. (2001). More ‘malignant’ than cancer? Five-year survival following a first admission for heart failure. Eur. J. Heart Fail. 3, 315–322.
Takimoto, K., Yang, E. K., and Conforti, L. (2002). Palmitoylation of KChIP splicing variants is required for efficient cell surface expression of Kv4.3 channels. J. Biol. Chem. 277, 26904–26911.
Ter Keurs, H. E., and Boyden, P. A. (2007). Calcium and arrhythmogenesis. Physiol. Rev. 87, 457–506.
Teutsch, C., Kondo, R. P., Dederko, D. A., Chrast, J., Chien, K. R., and Giles, W. R. (2007). Spatial distributions of Kv4 channels and KChip2 isoforms in the murine heart based on laser capture microdissection. Cardiovasc. Res. 73, 739–749.
Thomsen, M. B., Foster, E., Nguyen, K. H., and Sosunov, E. A. (2009a). Transcriptional and electrophysiological consequences of KChIP2-mediated regulation of CaV1.2. Channels (Austin) 3, 308–310.
Thomsen, M. B., Sosunov, E. A., Anyukhovsky, E. P., Özgen, N., Boyden, P. A., and Rosen, M. R. (2009b). Deleting the accessory subunit KChIP2 results in loss of Ito,f and increased IK,slow that maintains normal action potential configuration. Heart Rhythm 6, 370–377.
Thomsen, M. B., Wang, C., Ozgen, N., Wang, H. G., Rosen, M. R., and Pitt, G. S. (2009c). Accessory subunit KChIP2 modulates the cardiac L-type calcium current. Circ. Res. 104, 1382–1389.
Wang, Y. G., Cheng, J., Chen, G. H., Rob, F., Naseem, R. H., Nguyen, L., Johnstone, J. L., and Hill, J. A. (2007). Remodeling of outward K+ currents in pressure-overload heart failure. J. Cardiovasc. Electrophysiol. 18, 869–875.
Wasserstrom, J. A., Sharma, R., Kapur, S., Kelly, J. E., Kadish, A. H., Balke, C. W., and Aistrup, G. L. (2009). Multiple defects in intracellular calcium cycling in whole failing rat heart. Circ. Heart Fail 2, 223–232.
Wettwer, E., Amos, G. J., Posival, H., and Ravens, U. (1994). Transient outward current in human ventricular myocytes of subepicardial and subendocardial origin. Circ. Res. 75, 473–482.
Wickenden, A. D., Jegla, T. J., Kaprielian, R., and Backx, P. H. (1999a). Regional contributions of Kv1.4, Kv4.2, and Kv4.3 to transient outward K+ current in rat ventricle. Am. J. Physiol. 276, H1599–H1607.
Wickenden, A. D., Lee, P., Sah, R., Huang, Q., Fishman, G. I., and Backx, P. H. (1999b). Targeted expression of a dominant-negative K(v)4.2 K+ channel subunit in the mouse heart. Circ. Res. 85, 1067–1076.
Yu, H., Mckinnon, D., Dixon, J. E., Gao, J., Wymore, R., Cohen, I. S., Danilo, P. Jr., Shvilkin, A., Anyukhovsky, E. P., Sosunov, E. A., Hara, M., and Rosen, M. R. (1999). Transient outward current, Ito1, is altered in cardiac memory. Circulation 99, 1898–1905.
Zhang, M., Jiang, M., and Tseng, G. N. (2001). minK-related peptide 1 associates with Kv4.2 and modulates its gating function: potential role as beta subunit of cardiac transient outward channel? Circ. Res. 88, 1012–1019.
Zicha, S., Xiao, L., Stafford, S., Cha, T. J., Han, W., Varro, A., and Nattel, S. (2004). Transmural expression of transient outward potassium current subunits in normal and failing canine and human hearts. J. Physiol. 561(Pt 3), 735–748.
Keywords: potassium, ion channels, heart failure, ventricular arrhythmia, knockout mice, action potential, repolarization, sudden death
Citation: Grubb S, Calloe K and Thomsen MB (2012) Impact of KChIP2 on cardiac electrophysiology and the progression of heart failure. Front. Physio. 3:118. doi: 10.3389/fphys.2012.00118
Received: 27 February 2012; Paper pending published: 03 April 2012;
Accepted: 11 April 2012; Published online: 04 May 2012.
Edited by:
Carol Ann Remme, University of Amsterdam, NetherlandsCopyright: © 2012 Grubb, Calloe and Thomsen. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Morten B. Thomsen, Danish National Research Foundation Centre for Cardiac Arrhythmia, Department of Biomedical Sciences, University of Copenhagen, Blegdamsvej 3b, Building 12.5.36, DK-2200 Copenhagen, Denmark. e-mail:bWJ0aG9tQHN1bmQua3UuZGs=