

- 1 Program in Cell Biology, The Hospital for Sick Children, Biochemistry Department, University of Toronto, Toronto, ON, Canada
- 2 Department of Pharmacology and Toxicology, University of Lausanne, Lausanne, Switzerland
Nedd4-2 is a ubiquitin ligase previously demonstrated to regulate endocytosis and lysosomal degradation of the epithelial Na+ channel (ENaC) and other ion channels and transporters. Recent studies using Nedd4-2 knockout mice specifically in kidney or lung epithelia has revealed a critical role for this E3 ubiquitin ligase in regulating salt and fluid transport in these tissues/organs and in maintaining homeostasis of body blood pressure. Interestingly, the primary targets for Nedd4-2 may differ in these two organs: in the lung Nedd4-2 targets ENaC, and loss of Nedd4-2 leads to excessive ENaC function and to cystic fibrosis – like lung disease, whereas in the kidney, Nedd4-2 targets the Na+/Cl− cotransporter (NCC) in addition to targeting ENaC. In accord, loss of Nedd4-2 in the distal nephron leads to increased NCC abundance and function. The aldosterone-responsive kinase, Sgk1, appears to be involved in the regulation of NCC by Nedd4-2 in the kidney, similar to its regulation of ENaC. Collectively, these new findings underscore the physiological importance of Nedd4-2 in regulating epithelial salt and fluid transport and balance.
Introduction
Neuronal precursor cell expressed developmentally downregulated 4 (Nedd4) proteins are E3 ubiquitin ligases that belong to the HECT family. They are composed of an N-terminal C2 domain that directs subcellular localization or mediate intra-molecular inhibitory interactions, multiple WW domains (usually three or four) that recognize substrates by binding to PY motifs (PPxY or LPxY), and a ubiquitin ligase HECT domain (Rotin and Kumar, 2009). In mammals, there are eight or nine Nedd4 family members, including the closely related Nedd4 (Nedd4-1) and Nedd4-2 (NEDD4L; Yang and Kumar, 2010). While recent in vivo work has suggested that Nedd4 is involved in regulating cellular growth, differentiation and animal development (e.g., Cao et al., 2008; Yang et al., 2008; Liu et al., 2009; Fouladkou et al., 2010; Kawabe et al., 2010; Persaud et al., 2011), Nedd4-2 has been primarily demonstrated to regulate ion transport by controlling cellular trafficking/endocytosis and lysosomal degradation of ion channels and transporters. Of these, recent work on the regulation of epithelial salt and fluid transport by Nedd4-2 in the kidney and lung, using animal models, has received considerable attention, and is therefore the focus of this review.
Regulation of ENaC by Nedd4-2 in Kidney and Lung Epithelia
The amiloride-sensitive epithelial Na+ channel (ENaC) plays a critical role in regulating salt and fluid absorption in epithelia of the distal nephron, lung, distal colon, and other salt-absorbing tissues. In accord, loss of function (LOF) mutations in ENaC cause pseudohypoaldosteronism Type I (PHAI), and gain of function (GOF) mutations cause pseudoaldosteronism (Liddle syndrome, a hereditary hypertension), underscoring the importance of this channel for Na+ and fluid absorption in the kidney (Lifton et al., 2001). Similar diseases are also observed in knock in mice bearing the same mutations found in PHAI or Liddle syndrome patients, respectively; moreover, total ablation of ENaC in mice leads to severe pulmonary edema (Hummler and Vallon, 2005). ENaC function is up-regulated by the hormones aldosterone and vasopressin (ADH) in the kidney, and by glucocorticoids in the lung (Loffing and Korbmacher, 2009).
Epithelial Na+ channel is expressed in the distal convoluted tubules (DCT), connecting tubules (CNT), and cortical collecting ducts (CCD) of the distal nephron (Duc et al., 1994), and is composed of three subunits, αβγ (Canessa et al., 1994); A PY motif is present at the cytoplasmic C-terminal region of each ENaC subunit (Snyder et al., 1995; Schild et al., 1996; Staub et al., 1996). Liddle syndrome is caused by deletion or mutation of the PY motif of β or γ ENaC (Shimkets et al., 1994; Hansson et al., 1995a,b; Lifton et al., 2001). These PY motifs serve as binding sites for Nedd4 proteins (Staub et al., 1996), especially Nedd4-2 (Abriel et al., 1999; Harvey et al., 2001; Kamynina et al., 2001; Snyder et al., 2004b). Like other Nedd4 family members, Nedd4-2 is comprised of a C2-WW(x4)-HECT domain architecture. This E3 ligase binds, via it WW domain, to the PY motifs of ENaC, leading to channel ubiquitylation and endocytosis, thus suppressing ENaC function (Figure 1A); this suppression is alleviated in Liddle syndrome (PY motif) mutants, which cannot properly bind Nedd4-2 (Figure 1B) and hence are retained at the plasma membrane for prolonged times (Staub et al., 1996, 1997; Abriel et al., 1999; Kamynina et al., 2001; Lu et al., 2007; Wiemuth et al., 2007; Zhou et al., 2007). Such ENaC retention leads to excessive channel function and Na+ reabsorption, which significantly contributes to the development of hypertension in these Liddle patients. The PY motifs of ENaC also regulate its response to intracellular Na+ concentrations, because the normal feedback inhibition by Na+ seen in wildtype ENaC is inhibited in the PY motif mutants of this channel (Dinudom et al., 1998; Kellenberger et al., 1998; Palmer et al., 1998).
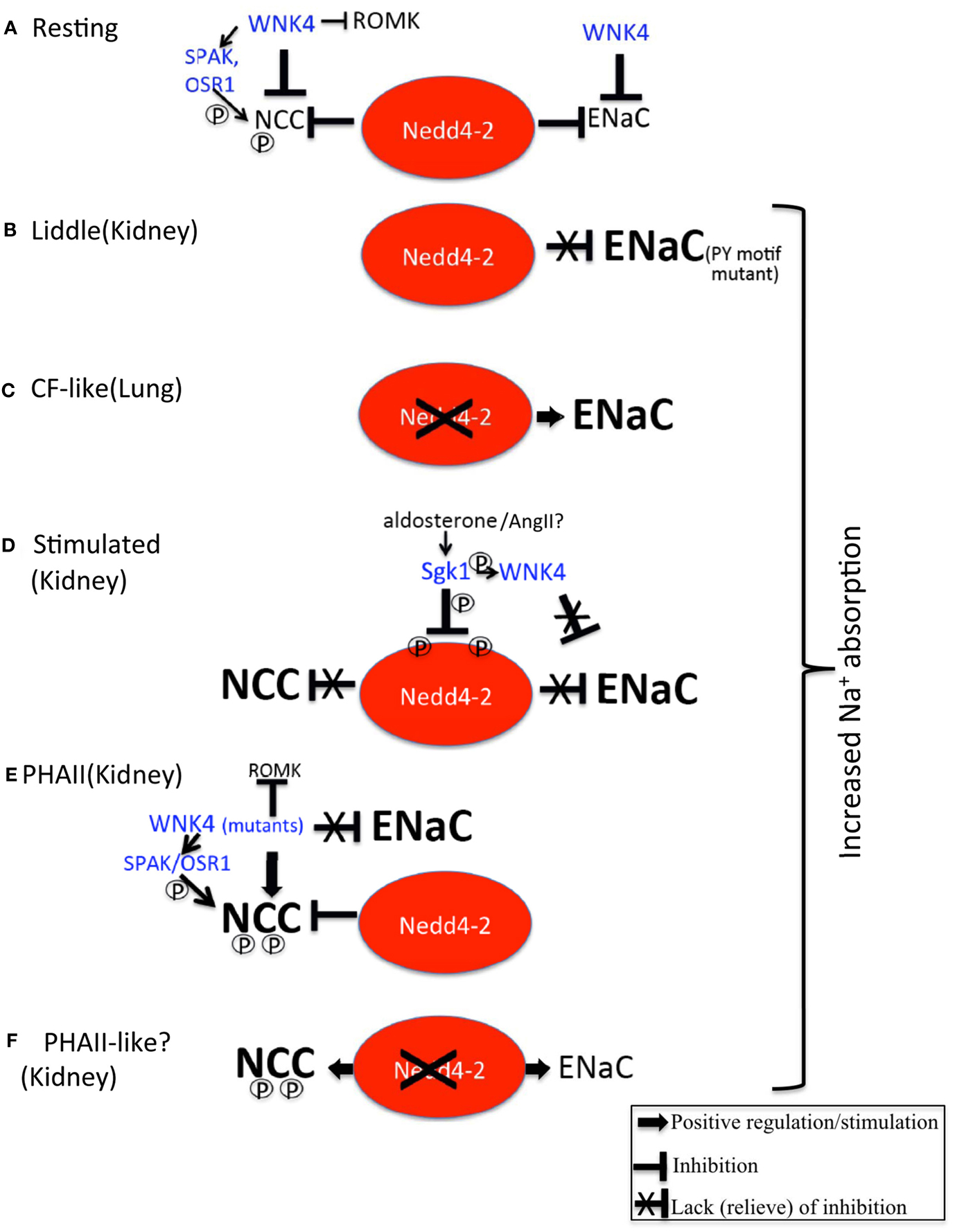
Figure 1. Regulation of ENaC and NCC by Nedd4-2. (A) Under resting conditions, Nedd4-2 inhibits ENaC and NCC, as does WNK4, ensuring no excess Na+ (and fluid) reabsorption takes place, especially in the kidney. An alternative model suggests that WNK4 activates NCC to a low degree by activating the kinases SPAK and OSR1, which then phosphorylate (P) NCC. WNK4 also inhibits the K+ channel ROMK, to restrain K+ excretion. (B) In Liddle syndrome, PY motif mutations in ENaC prevent Nedd4-2 from properly binding and suppressing ENaC, thus leading to increased ENaC abundance/function and excessive Na+ (and fluid) reabsorption in the kidney, causing hypertension. (C) Removal of Nedd4-2 in lung epithelia by gene knockout in mice leads to cystic fibrosis – like lung disease due to excessive abundance/function of ENaC. (D) Aldosterone (or Angiotensin II, AngII?) stimulation of Sgk1 leads to Sgk1-mediated phosphorylation of Nedd4-2 on two sites, thus preventing Nedd4-2 from suppressing NCC and ENaC. This results in increased abundance/function of these Na+ absorbing proteins. In addition, phosphorylation of WNK4 by Sgk1 interferes with the ability of WNK4 to inhibit ENaC. Collectively, this leads to aldosterone-mediated increased in Na+ and fluid reabsorption in the kidney, hence to elevated blood pressure. (E) In PHAII, WNK4 mutants either divert NCC to the recycling pathway instead of lysosomal degradation, and/or enhance phosphorylation (and activation) of SPAK/OSR1 and hence of NCC. Both of these effects, coupled with loss of inhibition of ENaC by WNK4, lead to increased Na+ reabsorption (and paracellular Cl− permeability) in the kidney, and hence to increased fluid absorption and hypertension. Mutant WNK4 also strongly suppresses ROMK, preventing K+ excretion and causing hyperkalemia. (F) Knockout of Nedd4-2 in the distal nephron leads to increased abundance and activation of NCC (and ENaC), causing hypertension and possibly mimicking features of PHAII.
Given the salt-induced hypertension, metabolic alkalosis and hypokalemia observed in Liddle patients and in mice bearing the Liddle syndrome mutations in βENaC (Pradervand et al., 1999), one would expect that ablation of Nedd4-2 would result in a similar phenotype. Indeed, Yang and colleagues have recently generated Nedd4-2 (total) knockout mice and showed that these mice exhibit salt-induced hypertension (although milder than in the βENaC mutant mice) and cardiac hypertrophy, which were inhibited by amiloride (Shi et al., 2008; see below).
The suppressive effect of Nedd4-2 on ENaC led to a search for mutations or polymorphism is NEDD4L (Nedd4-2) in hypertensive patients. Although to date no mutations in NEDD4L have been identified, polymorphisms in that gene that contribute to the regulation of blood pressure by less efficiently suppressing ENaC, have been described (Dunn et al., 2002; Fouladkou et al., 2004; Araki et al., 2008; Luo et al., 2009). These Nedd4L variants cause a rather mild effect on ENaC (e.g., Fouladkou et al., 2004) and it is not known if they also affect the Na+/Cl− cotransporter (NCC; see below).
As mentioned above, ENaC plays an important role in regulating salt/fluid absorption not only in the kidney, but also in the lung. Knockout mice that ablate αENaC (the key subunit in this channel) die at birth from an inability to clear their lungs of fluid (Hummler et al., 1996). In contrast, mice bearing a Liddle syndrome mutation demonstrate enhanced alveolar fluid clearance following pulmonary edema (Randrianarison et al., 2007).
An earlier study showed that (∼60%) knockdown of Nedd4-2 in rat lungs using RNA interference led to increased ENaC expression and to reduced extravascular lung fluid (Li et al., 2007). Indeed, a very recent study using genetic ablation of Nedd4-2 specifically in lung epithelia in mice has demonstrated elevated ENaC levels and function in lung epithelia harvested from these mice, leading to lung dehydration, massive sterile inflammation and mucus accumulation in the airways (Kimura et al., 2011); these features resemble those seen in the lungs of cystic fibrosis (CF) patients and also mimic the defects seen upon overexpression of βENaC in lung epithelia (Mall et al., 2004; see below). Moreover, they can be ameliorated by treatment of newborn knockout mice with the ENaC inhibitor amiloride (Kimura et al., 2011); These observations were also confirmed by a recent total knockout of Nedd4-2 in mice (Boase et al., 2011) using a different knockout approach than that described above by Shi et al. (2008), where a lung phenotype was not described). In the Boase et al. (2011) paper, systemic loss of Nedd4-2 led to neonatal death of many of the newborns, likely due to elevated ENaC activity (although no rescue studies were performed to confirm this), and surviving mice, on a different genetic background, exhibited a lung phenotype very similar to that observed by Kimura et al. (2011), described above. Collectively, these recent studies demonstrate that Nedd4-2 normally suppresses ENaC function in the lung (Figure 1C), a function that is important both at birth and at later stages of life (Boase et al., 2011; Kimura et al., 2011), to ensure normal pulmonary salt and fluid homeostasis. The inhibition of ENaC by Nedd4-2 in the lung resembles the suppression of ENaC by CFTR in that tissue, although the mechanism for this suppression is different.
CFTR has been shown to inhibit ENaC function in the airways (Stutts et al., 1997; Boucher, 2007; although this notion has been debated; e.g., Itani et al., 2011). In CF, in which CFTR function (Cl− secretion) is impaired, Na+ and fluid absorption are increased. This results in airway dehydration, mucus accumulation and repeated bacterial infections (Boucher, 2007; Donaldson and Boucher, 2007). Mall, Boucher and co-workers have provided strong support for this model, by demonstrating that overexpression of ENaC (βENaC) in mouse airways leads to a CF-like lung phenotype, which was ameliorated by treatment of the mice with amiloride early in neonatal life (Mall et al., 2004; Zhou et al., 2008). An important question arising from these experiments was, therefore, why do Liddle syndrome patients, exhibiting systemic excessive ENaC activity, not develop CF-like lung disease? One possible explanation (although likely not the only one) lies in the difference between CFTR expression levels in the lower airways of mice vs. humans. Unlike in humans, CFTR is only weakly expressed in the distal airways of mice (Mall et al., 2010), thus it may not be able adequately suppress the elevated levels of ENaC obtained upon overexpression of its β subunit (Mall et al., 2004) or upon knockout of Nedd4-2 (Kimura et al., 2011).
Nedd4-2 Participates in the Hormonal Regulation of ENaC
Aldosterone and vasopressin are hormones well known to regulate Na+ balance and blood pressure, and both enhance ENaC function. Serum and glucocorticoid-regulated kinase 1 (Sgk1, a gene/protein related to Akt; Webster et al., 1993) is an aldosterone and glucocorticoid responsive gene. Sgk1 enhances ENaC function primarily by increasing its levels at the plasma membrane in response to aldosterone (Alvarez de la Rosa et al., 1999; Chen et al., 1999; Naray-Fejes-Toth et al., 1999; Shigaev et al., 2000; Loffing et al., 2001). This increase is achieved (at least in part) by Sgk1-mediated phosphorylation of Nedd4-2 (Debonneville et al., 2001; Snyder et al., 2002). Such phosphorylation leads to recruitment of specific 14-3-3 isoforms (themselves induced by aldosterone; Liang et al., 2006) to the phosphorylated Nedd4-2 (Bhalla et al., 2005; Ichimura et al., 2005), resulting in blockade of the Nedd4-2:ENaC interactions and loss of the ability of Nedd4-2 to suppress ENaC (Debonneville et al., 2001; Snyder et al., 2002; Flores et al., 2005; Figure 1D). Thus, this represents an important mechanism (although not the only one – see Alvarez de la Rosa et al., 1999; Shigaev et al., 2000; Auberson et al., 2003; Dahlmann et al., 2003; Diakov and Korbmacher, 2004; Fejes-Toth et al., 2008) by which aldosterone enhances ENaC function.
Much like Sgk1, PKA can also phosphorylate Nedd4-2 and prevent it from inhibiting ENaC (Snyder et al., 2004a). Since PKA is activated by cAMP, which is generated by vasopressin release and binding to its V2 receptor, it is likely that vasopressin, like aldosterone, can increase ENaC cell surface abundance by inhibiting Nedd4-2. In addition, the hormone insulin (Lee et al., 2007) and the kinases IKKB (Edinger et al., 2009) and with no lysine 1 (WNK1; Xu et al., 2005) were also shown to up-regulate ENaC via the Sgk1/Nedd4-2 pathway. In contrast, AMPK enhances the interactions between Nedd4-2 and ENaC, leading to increased ENaC endocytosis and channel downregulation under metabolic stress (Carattino et al., 2005; Bhalla et al., 2006; Almaca et al., 2009). Moreover, the WNK4 kinase inhibits ENaC following its (WNK4) phosphorylation by Sgk1 (Ring et al., 2007a,b; Figure 1D). Mutations in WNK1 and WNK4 cause Pseudohypoaldosteronism Type II (PHAII), as detailed below.
Regulation of NCC by Nedd4-2
The thiazide-sensitive NCC, encoded by the SLC12A3 gene, is expressed in the DCT of the nephron, and is known to play a critical role in the regulation Na+ and fluid transport (reviewed in Dimke, 2011). Ectopic expression of NCC in Xenopus oocytes and other cells leads to thiazide-sensitive Na+ uptake. This, together with the finding of expression of NCC in the DCT, suggests that this transporter plays an important role in regulation of salt and fluid balance. Indeed, Gitleman syndrome, a salt-losing disease exhibiting hypokalemic metabolic alkalosis, hypomagnesemia, and hypocalciuria, is caused by LOF mutations in NCC (SLC12A3; Simon et al., 1996).
NCC has been shown to be tightly regulated by several kinases, primarily by WNK 1 and 4, which regulate NCC trafficking and lysosomal sorting, phosphorylation, and function (Dimke, 2011; Figure 1A). Phosphorylation of several Ser/Thr residues on NCC appears to positively regulate its activity (Pacheco-Alvarez et al., 2006). Interestingly, a GOF mutation in WNK1, GOF or missense mutations in WNK4, all leading to elevated NCC function, cause PHAII (Figure 1E; Wilson et al., 2001; Lalioti et al., 2006; Kahle et al., 2008). PHAII is characterized by increased renal Na+ reabsorption (leading to hypertension), defective K+ secretion (hyperkalemia) and metabolic acidosis, exactly opposite to Gitleman syndrome. Treatment of PHAII patients with thiazide corrects these abnormalities, implicating excessive function of NCC as the culprit in this disease. Exactly how mutated WNK1 and WNK4 increase NCC function has been the subject of some debate. WNK4 was proposed to target NCC for lysosomal degradation (Subramanya et al., 2009), and accordingly, LOF mutations in WNK4 were shown to increase cell surface abundance of NCC (Wilson et al., 2003; Yang et al., 2003). Alternatively, it was proposed that WNK4 activates its substrates, the Ste20 kinases SPAK and OSR1, which in turn phosphorylate and activate NCC (Figure 1A). In GOF mutations in WNK4 that cause PHAII, this phosphorylation cascade is enhanced, leading to increased activation of NCC (Yang et al., 2007; Chiga et al., 2011; Figure 1E). Although this needs to be investigated further, it is possible that different mutations in WNK4 give rise to LOF or GOF, in either case leading to enhanced NCC function (by different routes; Figure 1E). Regarding WNK1, it was proposed to inhibit WNK4, hence a GOF WNK1 mutant found in PHAII was suggested to enhance suppression of WNK4, thus resulting in increased NCC function (Yang et al., 2003). Mutant WNK1 was also proposed to enhance phosphorylation of SPAK/OSR1, hence stimulating NCC phosphorylation and activation.
Using inducible conditional knockout of Nedd4-2 specifically in renal tubules of mice, it was recently shown that deletion of Nedd4-2 in this region of the distal nephron leads, in addition to an increase in abundance of ENaC, to a dramatic elevation in NCC protein expression and a reduction in aldosterone levels (Arroyo et al., 2011; Figure 1F). Moreover, Nedd4-2 was demonstrated (in Hek293 cells) to bind NCC, likely indirectly, and to promote NCC ubiquitylation and downregulation of its cell surface pool (Arroyo et al., 2011). This suggests that in the DCT, NCC is a major substrate for Nedd4-2. It is not clear why this elevated NCC expression was not detected in the Nedd4-2 total knockout mice generated by Shi et al. (2008) using the same Nedd4-2 floxed mice for genetic crosses; possible explanations include differences in genetic background, differences in acute knockout (Arroyo et al., 2011) vs. chronic knockout (Shi et al., 2008), which could potentially allow compensation by other genes during development, or non-renal effects of total knockout of Nedd4-2 in the Shi et al. (2008) mice.
The Nedd4-2-mediated suppression of NCC in the kidney involves Sgk1, which phosphorylates Nedd4-2 in response to aldosterone stimulation at two sites, thus inhibiting the ability of this E3 ligase to suppress NCC (Arroyo et al., 2011; Figure 1D). In this regard, the Nedd4-2 mediated downregulation of NCC is similar to its downregulation of ENaC, although unlike ENaC, NCC does not contains classic PY motifs and likely does not interact directly with Nedd4-2.
Thus, it is clear that Nedd4-2 is able to regulate more than one salt-absorbing targets/pathways, and this regulation is tissue-specific. For example, in the distal nephron, Nedd4-2 regulates the stability of NCC and that of ENaC (Arroyo et al., 2011), whereas in the lung ENaC is a major target for Nedd4-2 regulation (Boase et al., 2011; Kimura et al., 2011), as described above. Putative Nedd4-2 targets in other salt-absorbing epithelia is/are still unknown.
It is also apparent from the above studies that Sgk1 plays a very important role in regulating ENaC and NCC, via phosphorylation and hence inhibition of Nedd4-2 function. To confirm its importance in vivo, several groups have generated Sgk1 knockout mice. Both Wulff et al. (2002) and Fejes-Toth et al. (2008) generated total Sgk1 knockout mice, which exhibited salt-wasting phenotype under low salt diet, as well as reduced ENaC expression. The latter study also showed weaker expression of NCC (Fejes-Toth et al., 2008). In support, kidney-specific knockout of Sgk1 resulted in a salt-losing phenotype, especially under low salt diet, with hyperkaliuria and low blood pressure, and with reduced phosphorylation of Nedd4-2, leading to decreased expression of NCC (and to a lesser extent reduced expression of ENaC; Faresse et al., 2012). These findings underscore the importance of Sgk1 in regulating the Nedd4-2:NCC interactions, as well as the previously described Nedd4-2:ENaC interactions, in the kidney.
Summary/Perspective
Recent knockout studies of Nedd4-2 in both the distal nephron and in lung epithelia have helped cement our understanding of the suppressive role of Nedd4-2 on ENaC, which was extensively studied earlier using cultured cells and Xenopus oocytes. Moreover, these in vivo knockout studies have uncovered a new unsuspected role for Nedd4-2 in suppressing NCC in the kidney. Collectively, these animal model studies have important implications to our understanding of human diseases, since total loss of Nedd4-2 leads to salt-induced hypertension, and its specific loss in lung epithelia results in CF-like lung disease. Additionally, given the large increase in NCC abundance in the kidney upon knockout Nedd4-2 specifically in the distal nephron, it may be speculated that such an increase could result in PHAII-like disease (Figure 1F). This needs to be confirmed experimentally. Identification of LOF mutations in NEDD4L in humans that are associated with PHAII (if ever identified) would further strengthen the current studies with knockout mouse models. Regardless, it is clear that Nedd4-2 plays a critical role in regulating salt and fluid balance in the body, by affecting Na+ and fluid absorption in organs such as the kidney and the lung. The very recent identification of mutations in the ubiquitin ligase KLHL3/CUL3 that cause PHAII (Boyden et al., 2012; Louis-Dit-Picard et al., 2012), which lead to hypertension by affecting NCC abundance/function, further underscores the importance of E3 ubiquitin ligases in the regulation of body salt and fluid homeostasis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abriel, H., Loffing, J., Rebhun, J. F., Pratt, J. H., Schild, L., Horisberger, J. D., Rotin, D., and Staub, O. (1999). Defective regulation of the epithelial Na+ channel by Nedd4 in Liddle’s syndrome. J. Clin. Invest. 103, 667–673.
Almaca, J., Kongsuphol, P., Hieke, B., Ousingsawat, J., Viollet, B., Schreiber, R., Amaral, M. D., and Kunzelmann, K. (2009). AMPK controls epithelial Na(+) channels through Nedd4-2 and causes an epithelial phenotype when mutated. Pflugers Arch. 458, 713–721.
Alvarez de la Rosa, D., Zhang, P., Naray-Fejes-Toth, A., Fejes-Toth, G., and Canessa, C. M. (1999). The serum and glucocorticoid kinase sgk increases the abundance of epithelial sodium channels in the plasma membrane of Xenopus oocytes. J. Biol. Chem. 274, 37834–37839.
Araki, N., Umemura, M., Miyagi, Y., Yabana, M., Miki, Y., Tamura, K., Uchino, K., Aoki, R., Goshima, Y., Umemura, S., and Ishigami, T. (2008). Expression, transcription, and possible antagonistic interaction of the human Nedd4L gene variant: implications for essential hypertension. Hypertension 51, 773–777.
Arroyo, J. P., Lagnaz, D., Ronzaud, C., Vazquez, N., Ko, B. S., Moddes, L., Ruffieux-Daidie, D., Hausel, P., Koesters, R., Yang, B., Stokes, J. B., Hoover, R. S., Gamba, G., and Staub, O. (2011). Nedd4-2 modulates renal Na+-Cl−cotransporter via the aldosterone-SGK1-Nedd4-2 pathway. J. Am. Soc. Nephrol. 22, 1707–1719.
Auberson, M., Hoffmann-Pochon, N., Vandewalle, A., Kellenberger, S., and Schild, L. (2003). Epithelial Na+ channel mutants causing Liddle’s syndrome retain ability to respond to aldosterone and vasopressin. Am. J. Physiol. Renal Physiol. 285, F459–F471.
Bhalla, V., Daidie, D., Li, H., Pao, A. C., LaGrange, L. P., Wang, J., Vandewalle, A., Stockand, J. D., Staub, O., and Pearce, D. (2005). Serum- and glucocorticoid-regulated kinase 1 regulates ubiquitin ligase neural precursor cell-expressed, developmentally down-regulated protein 4-2 by inducing interaction with 14-3-3. Mol. Endocrinol. 19, 3073–3084.
Bhalla, V., Oyster, N. M., Fitch, A. C., Wijngaarden, M. A., Neumann, D., Schlattner, U., Pearce, D., and Hallows, K. R. (2006). AMP-activated kinase inhibits the epithelial Na+ channel through functional regulation of the ubiquitin ligase Nedd4-2. J. Biol. Chem. 281, 26159–26169.
Boase, N. A., Rychkov, G. Y., Townley, S. L., Dinudom, A., Candi, E., Voss, A. K., Tsoutsman, T., Semsarian, C., Melino, G., Koentgen, F., Cook, D. I., and Kumar, S. (2011). Respiratory distress and perinatal lethality in Nedd4-2-deficient mice. Nat. Commun. 2, 287.
Boucher, R. C. (2007). Airway surface dehydration in cystic fibrosis: pathogenesis and therapy. Annu. Rev. Med. 58, 157–170.
Boyden, L. M., Choi, M., Choate, K. A., Nelson-Williams, C. J., Farhi, A., Toka, H. R., Tikhonova, I. R., Bjornson, R., Mane, S. M., Colussi, G., Lebel, M., Gordon, R. D., Semmekrot, B. A., Poujol, A., Valimaki, M. J., De Ferrari, M. E., Sanjad, S. A., Gutkin, M., Karet, F. E., Tucci, J. R., Stockigt, J. R., Keppler-Noreuil K, M., Porter, C. C., Anand, S. K., Whiteford, M. L., Davis, I. D., Dewar, S. B., Bettinelli, A., Fadrowski, J. J., Belsha, C. W., Hunley, T. E., Nelson, R. D., Trachtman, H., Cole, T. R., Pinsk, M., Bockenhauer, D., Shenoy, M., Vaidyanathan, P., Foreman, J. W., Rasoulpour, M., Thameem, F., Al-Shahrouri, H. Z., Radhakrishnan, J., Gharavi, A. G., Goilav, B., and Lifton, R. P. (2012). Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature 482, 98–102.
Canessa, C. M., Schild, L., Buell, G., Thorens, B., Gautschi, I., Horisberger, J. D., and Rossier, B. C. (1994). Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature 367, 463–467.
Cao, X. R., Lill, N. L., Boase, N., Shi, P. P., Croucher, D. R., Shan, H., Qu, J., Sweezer, E. M., Place, T., Kirby, P. A., Daly, R. J., Kumar, S., and Yang, B. (2008). Nedd4 controls animal growth by regulating IGF-1 signaling. Sci. Signal. 1, ra5.
Carattino, M. D., Edinger, R. S., Grieser, H. J., Wise, R., Neumann, D., Schlattner, U., Johnson, J. P., Kleyman, T. R., and Hallows, K. R. (2005). Epithelial sodium channel inhibition by AMP-activated protein kinase in oocytes and polarized renal epithelial cells. J. Biol. Chem. 280, 17608–17616.
Chen, S. Y., Bhargava, A., Mastroberardino, L., Meijer, O. C., Wang, J., Buse, P., Firestone, G. L., Verrey, F., and Pearce, D. (1999). Epithelial sodium channel regulated by aldosterone-induced protein sgk. Proc. Natl. Acad. Sci. U.S.A. 96, 2514–2519.
Chiga, M., Rafiqi, F. H., Alessi, D. R., Sohara, E., Ohta, A., Rai, T., Sasaki, S., and Uchida, S. (2011). Phenotypes of pseudohypoaldosteronism type II caused by the WNK4 D561A missense mutation are dependent on the WNK-OSR1/SPAK kinase cascade. J. Cell. Sci. 124, 1391–1395.
Dahlmann, A., Pradervand, S., Hummler, E., Rossier, B. C., Frindt, G., and Palmer, L. G. (2003). Mineralocorticoid regulation of epithelial Na+ channels is maintained in a mouse model of Liddle’s syndrome. Am. J. Physiol. Renal Physiol. 285, F310–F318.
Debonneville, C., Flores, S. Y., Kamynina, E., Plant, P. J., Tauxe, C., Thomas, M. A., Munster, C., Chraibi, A., Pratt, J. H., Horisberger, J. D., Pearce, D., Loffing, J., and Staub, O. (2001). Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na(+) channel cell surface expression. EMBO J. 20, 7052–7059.
Diakov, A., and Korbmacher, C. (2004). A novel pathway of epithelial sodium channel activation involves a serum- and glucocorticoid-inducible kinase consensus motif in the C terminus of the channel’s alpha-subunit. J. Biol. Chem. 279, 38134–38142.
Dimke, H. (2011). Exploring the intricate regulatory network controlling the thiazide-sensitive NaCl cotransporter (NCC). Pflugers Arch. 462, 767–777.
Dinudom, A., Harvey, K. F., Komwatana, P., Young, J. A., Kumar, S., and Cook, D. I. (1998). Nedd4 mediates control of an epithelial Na+ channel in salivary duct cells by cytosolic Na+. Proc. Natl. Acad. Sci. U.S.A. 95, 7169–7173.
Donaldson, S. H., and Boucher, R. C. (2007). Sodium channels and cystic fibrosis. Chest 132, 1631–1636.
Duc, C., Farman, N., Canessa, C. M., Bonvalet, J. P., and Rossier, B. C. (1994). Cell-specific expression of epithelial sodium channel alpha, beta, and gamma subunits in aldosterone-responsive epithelia from the rat: localization by in situ hybridization and immunocytochemistry. J. Cell Biol. 127, 1907–1921.
Dunn, D. M., Ishigami, T., Pankow, J., von Niederhausern, A., Alder, J., Hunt, S. C., Leppert, M. F., Lalouel, J. M., and Weiss, R. B. (2002). Common variant of human NEDD4L activates a cryptic splice site to form a frameshifted transcript. J. Hum. Genet. 47, 665–676.
Edinger, R. S., Lebowitz, J., Li, H., Alzamora, R., Wang, H., Johnson, J. P., and Hallows, K. R. (2009). Functional regulation of the epithelial Na+ channel by IkappaB kinase-beta occurs via phosphorylation of the ubiquitin ligase Nedd4-2. J. Biol. Chem. 284, 150–157.
Faresse, N., Lagnaz, D., Debonneville, A., Ismailji, A., Maillard, M., Fejes-Toth, G., Naray-Fejes-Toth, A., and Staub, O. (2012). Inducible kidney specific Sgk1 knock-out mice show a salt losing phenotype. Am. J. Physiol. Renal Physiol. 302, F977–F985.
Fejes-Toth, G., Frindt, G., Naray-Fejes-Toth, A., and Palmer, L. G. (2008). Epithelial Na+ channel activation and processing in mice lacking SGK1. Am. J. Physiol. Renal Physiol. 294, F1298–F1305.
Flores, S. Y., Loffing-Cueni, D., Kamynina, E., Daidie, D., Gerbex, C., Chabanel, S., Dudler, J., Loffing, J., and Staub, O. (2005). Aldosterone-induced serum and glucocorticoid-induced kinase 1 expression is accompanied by Nedd4-2 phosphorylation and increased Na+ transport in cortical collecting duct cells. J. Am. Soc. Nephrol. 16, 2279–2287.
Fouladkou, F., Alikhani-Koopaei, R., Vogt, B., Flores, S. Y., Malbert-Colas, L., Lecomte, M. C., Loffing, J., Frey, F. J., Frey, B. M., and Staub, O. (2004). A naturally occurring human Nedd4-2 variant displays impaired ENaC regulation in Xenopus laevis oocytes. Am. J. Physiol. Renal Physiol. 287, F550–F561.
Fouladkou, F., Lu, C., Jiang, C., Zhou, L., She, Y., Walls, J. R., Kawabe, H., Brose, N., Henkelman, R. M., Huang, A., Bruneau, B. G., and Rotin, D. (2010). The ubiquitin ligase Nedd4-1 is required for heart development and is a suppressor of thrombospondin-1. J. Biol. Chem. 285, 6770–6780.
Hansson, J. H., Nelson-Williams, C., Suzuki, H., Schild, L., Shimkets, R., Lu, Y., Canessa, C., Iwasaki, T., Rossier, B., and Lifton, R. P. (1995a). Hypertension caused by a truncated epithelial sodium channel gamma subunit: genetic heterogeneity of Liddle syndrome. Nat. Genet. 11, 76–82.
Hansson, J. H., Schild, L., Lu, Y., Wilson, T. A., Gautschi, I., Shimkets, R., Nelson-Williams, C., Rossier, B. C., and Lifton, R. P. (1995b). A de novo missense mutation of the beta subunit of the epithelial sodium channel causes hypertension and Liddle syndrome, identifying a proline-rich segment critical for regulation of channel activity. Proc. Natl. Acad. Sci. U.S.A. 92, 11495–11499.
Harvey, K. F., Dinudom, A., Cook, D. I., and Kumar, S. (2001). The Nedd4-like protein KIAA0439 is a potential regulator of the epithelial sodium channel. J. Biol. Chem. 276, 8597–8601.
Hummler, E., Barker, P., Gatzy, J., Beermann, F., Verdumo, C., Schmidt, A., Boucher, R., and Rossier, B. C. (1996). Early death due to defective neonatal lung liquid clearance in alpha-ENaC-deficient mice. Nat. Genet. 12, 325–328.
Hummler, E., and Vallon, V. (2005). Lessons from mouse mutants of epithelial sodium channel and its regulatory proteins. J. Am. Soc. Nephrol. 16, 3160–3166.
Ichimura, T., Yamamura, H., Sasamoto, K., Tominaga, Y., Taoka, M., Kakiuchi, K., Shinkawa, T., Takahashi, N., Shimada, S., and Isobe, T. (2005). 14-3-3 proteins modulate the expression of epithelial Na+ channels by phosphorylation-dependent interaction with Nedd4-2 ubiquitin ligase. J. Biol. Chem. 280, 13187–13194.
Itani, O. A., Chen, J. H., Karp, P. H., Ernat, S., Keshavjee, S., Parkeh, K., Klesney-Tate, Z. J., and Welsh, M. J. (2011). Human cystic fibrosis airway epithelia have reduced Cl- conductance but not increased Na+ conductance. Proc. Natl. Acad. Sci. U.S.A. 108, 10260–10265.
Kahle, K. T., Ring, A. M., and Lifton, R. P. (2008). Molecular physiology of the WNK kinases. Annu. Rev. Physiol. 70, 329–355.
Kamynina, E., Debonneville, C., Bens, M., Vandewalle, A., and Staub, O. (2001). A novel mouse Nedd4 protein suppresses the activity of the epithelial Na+ channel. FASEB J. 15, 204–214.
Kawabe, H., Neeb, A., Dimova, K., Young, S. M. Jr., Takeda, M., Katsurabayashi, S., Mitkovski, M., Malakhova, O. A., Zhang, D. E., Umikawa, M., Kariya, K., Goebbels, S., Nave, K. A., Rosenmund, C., Jahn, O., Rhee, J., and Brose, N. (2010). Regulation of Rap2A by the ubiquitin ligase Nedd4-1 controls neurite development. Neuron 65, 358–372.
Kellenberger, S., Gautschi, I., Rossier, B. C., and Schild, L. (1998). Mutations causing Liddle syndrome reduce sodium-dependent downregulation of the epithelial sodium channel in the Xenopus oocyte expression system. J. Clin. Invest. 101, 2741–2750.
Kimura, T., Kawabe, H., Jiang, C., Zhang, W., Xiang, Y. Y., Lu, C., Salter, M. W., Brose, N., Lu, W. Y., and Rotin, D. (2011). Deletion of the ubiquitin ligase Nedd4L in lung epithelia causes cystic fibrosis-like disease. Proc. Natl. Acad. Sci. U.S.A. 108, 3216–3221.
Lalioti, M. D., Zhang, J., Volkman, H. M., Kahle, K. T., Hoffmann, K. E., Toka, H. R., Nelson-Williams, C., Ellison, D. H., Flavell, R., Booth, C. J., Lu, Y., Geller, D. S., and Lifton, R. P. (2006). Wnk4 controls blood pressure and potassium homeostasis via regulation of mass and activity of the distal convoluted tubule. Nat. Genet. 38, 1124–1132.
Lee, I. H., Dinudom, A., Sanchez-Perez, A., Kumar, S., and Cook, D. I. (2007). Akt mediates the effect of insulin on epithelial sodium channels by inhibiting Nedd4-2. J. Biol. Chem. 282, 29866–29873.
Li, T., Koshy, S., and Folkesson, H. G. (2007). Involvement of {alpha}ENaC and Nedd4-2 in the conversion from lung fluid secretion to fluid absorption at birth in the rat as assayed by RNA interference analysis. Am. J. Physiol. Lung Cell Mol. Physiol. 293, L1069–L1078.
Liang, X., Peters, K. W., Butterworth, M. B., and Frizzell, R. A. (2006). 14-3-3 isoforms are induced by aldosterone and participate in its regulation of epithelial sodium channels. J. Biol. Chem. 281, 16323–16332.
Lifton, R. P., Gharavi, A. G., and Geller, D. S. (2001). Molecular mechanisms of human hypertension. Cell 104, 545–556.
Liu, Y., Oppenheim, R. W., Sugiura, Y., and Lin, W. (2009). Abnormal development of the neuromuscular junction in Nedd4-deficient mice. Dev. Biol. 330, 153–166.
Loffing, J., and Korbmacher, C. (2009). Regulated sodium transport in the renal connecting tubule (CNT) via the epithelial sodium channel (ENaC). Pflugers Arch. 458, 111–135.
Loffing, J., Zecevic, M., Feraille, E., Kaissling, B., Asher, C., Rossier, B. C., Firestone, G. L., Pearce, D., and Verrey, F. (2001). Aldosterone induces rapid apical translocation of ENaC in early portion of renal collecting system: possible role of SGK. Am. J. Physiol. Renal Physiol. 280, F675–F682.
Louis-Dit-Picard, H., Barc, J., Trujillano, D., Miserey-Lenkei, S., Bouatia-Naji, N., Pylypenko, O., Beaurain, G., Bonnefond, A., Sand, O., Simian, C., Vidal-Petiot, E., Soukaseum, C., Mandet, C., Broux, F., Chabre, O., Delahousse, M., Esnault, V., Fiquet, B., Houillier, P., Bagnis, C. I., Koenig, J., Konrad, M., Landais, P., Mourani, C., Niaudet, P., Probst, V., Thauvin, C., Unwin, R. J., Soroka, S. D., Ehret, G., Ossowski, S., Caulfield, M., International Consortium For Blood Pressure Icbp, Bruneval, P., Estivill, X., Froguel, P., Hadchouel, J., Schott, J. J., and Jeunemaitre, X. (2012). KLHL3 mutations cause familial hyperkalemic hypertension by impairing ion transport in the distal nephron. Nat. Genet. 44, 456–460.
Lu, C., Pribanic, S., Debonneville, A., Jiang, C., and Rotin, D. (2007). The PY motif of ENaC, mutated in Liddle syndrome, regulates channel internalization, sorting and mobilization from subapical pool. Traffic 8, 1246–1264.
Luo, F., Wang, Y., Wang, X., Sun, K., Zhou, X., and Hui, R. (2009). A functional variant of NEDD4L is associated with hypertension, antihypertensive response, and orthostatic hypotension. Hypertension 54, 796–801.
Mall, M., Grubb, B. R., Harkema, J. R., O’Neal, W. K., and Boucher, R. C. (2004). Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat. Med. 10, 487–493.
Mall, M. A., Button, B., Johannesson, B., Zhou, Z., Livraghi, A., Caldwell, R. A., Schubert, S. C., Schultz, C., O’Neal, W. K., Pradervand, S., Hummler, E., Rossier, B. C., Grubb, B. R., and Boucher, R. C. (2010). Airway surface liquid volume regulation determines different airway phenotypes in liddle compared with betaENaC-overexpressing mice. J. Biol. Chem. 285, 26945–26955.
Naray-Fejes-Toth, A., Canessa, C., Cleaveland, E. S., Aldrich, G., and Fejes-Toth, G. (1999). sgk is an aldosterone-induced kinase in the renal collecting duct. Effects on epithelial Na+ channels. J. Biol. Chem. 274, 16973–16978.
Pacheco-Alvarez, D., Cristobal, P. S., Meade, P., Moreno, E., Vazquez, N., Munoz, E., Diaz, A., Juarez, M. E., Gimenez, I., and Gamba, G. (2006). The Na+:Cl− cotransporter is activated and phosphorylated at the amino-terminal domain upon intracellular chloride depletion. J. Biol. Chem. 281, 28755–28763.
Palmer, L. G., Sackin, H., and Frindt, G. (1998). Regulation of Na+ channels by luminal Na+ in rat cortical collecting tubule. J. Physiol. 509(Pt 1), 151–162.
Persaud, A., Alberts, P., Hayes, M., Guettler, S., Clarke, I., Sicheri, F., Dirks, P., Ciruna, B., and Rotin, D. (2011). Nedd4-1 binds and ubiquitylates activated FGFR1 to control its endocytosis and function. EMBO J. 30, 3259–3273.
Pradervand, S., Wang, Q., Burnier, M., Beermann, F., Horisberger, J. D., Hummler, E., and Rossier, B. C. (1999). A mouse model for Liddle’s syndrome. J. Am. Soc. Nephrol. 10, 2527–2533.
Randrianarison, N., Escoubet, B., Ferreira, C., Fontayne, A., Fowler-Jaeger, N., Clerici, C., Hummler, E., Rossier, B. C., and Planes, C. (2007). beta-Liddle mutation of the epithelial sodium channel increases alveolar fluid clearance and reduces the severity of hydrostatic pulmonary oedema in mice. J. Physiol. 582, 777–788.
Ring, A. M., Cheng, S. X., Leng, Q., Kahle, K. T., Rinehart, J., Lalioti, M. D., Volkman, H. M., Wilson, F. H., Hebert, S. C., and Lifton, R. P. (2007a). WNK4 regulates activity of the epithelial Na+ channel in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 104, 4020–4024.
Ring, A. M., Leng, Q., Rinehart, J., Wilson, F. H., Kahle, K. T., Hebert, S. C., and Lifton, R. P. (2007b). An SGK1 site in WNK4 regulates Na+ channel and K+ channel activity and has implications for aldosterone signaling and K+ homeostasis. Proc. Natl. Acad. Sci. U.S.A. 104, 4025–4029.
Rotin, D., and Kumar, S. (2009). Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 10, 398–409.
Schild, L., Lu, Y., Gautschi, I., Schneeberger, E., Lifton, R. P., and Rossier, B. C. (1996). Identification of a PY motif in the epithelial Na channel subunits as a target sequence for mutations causing channel activation found in Liddle syndrome. EMBO J. 15, 2381–2387.
Shi, P. P., Cao, X. R., Sweezer, E. M., Kinney, T. S., Williams, N. R., Husted, R. F., Nair, R., Weiss, R. M., Williamson, R. A., Sigmund, C. D., Snyder, P. M., Staub, O., Stokes, J. B., and Yang, B. (2008). Salt-sensitive hypertension and cardiac hypertrophy in mice deficient in the ubiquitin ligase Nedd4-2. Am. J. Physiol. Renal Physiol. 295, F462–F470.
Shigaev, A., Asher, C., Latter, H., Garty, H., and Reuveny, E. (2000). Regulation of sgk by aldosterone and its effects on the epithelial Na(+) channel. Am. J. Physiol. Renal Physiol. 278, F613–F619.
Shimkets, R. A., Warnock, D. G., Bositis, C. M., Nelson-Williams, C., Hansson, J. H., Schambelan, M., Gill, J. R. Jr., Ulick, S., Milora, R. V., Findling, J. W., Canessa, C. M., Rossier, B. C., and Lifton, R. P. (1994). Liddle’s syndrome: heritable human hypertension caused by mutations in the beta subunit of the epithelial sodium channel. Cell 79, 407–414.
Simon, D. B., Nelson-Williams, C., Bia, M. J., Ellison, D., Karet, F. E., Molina, A. M., Vaara, I., Iwata, F., Cushner, H. M., Koolen, M., Gainza, F. J., Gitleman, H. J., and Lifton, R. P. (1996). Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat. Genet. 12, 24–30.
Snyder, P. M., Olson, D. R., Kabra, R., Zhou, R., and Steines, J. C. (2004a). cAMP and serum and glucocorticoid-inducible kinase (SGK) regulate the epithelial Na(+) channel through convergent phosphorylation of Nedd4-2. J. Biol. Chem. 279, 45753–45758.
Snyder, P. M., Steines, J. C., and Olson, D. R. (2004b). Relative contribution of Nedd4 and Nedd4-2 to ENaC regulation in epithelia determined by RNA interference. J. Biol. Chem. 279, 5042–5046.
Snyder, P. M., Olson, D. R., and Thomas, B. C. (2002). Serum and glucocorticoid-regulated kinase modulates Nedd4-2-mediated inhibition of the epithelial Na+ channel. J. Biol. Chem. 277, 5–8.
Snyder, P. M., Price, M. P., McDonald, F. J., Adams, C. M., Volk, K. A., Zeiher, B. G., Stokes, J. B., and Welsh, M. J. (1995). Mechanism by which Liddle’s syndrome mutations increase activity of a human epithelial Na+ channel. Cell 83, 969–978.
Staub, O., Dho, S., Henry, P., Correa, J., Ishikawa, T., McGlade, J., and Rotin, D. (1996). WW domains of Nedd4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle’s syndrome. EMBO J. 15, 2371–2380.
Staub, O., Gautschi, I., Ishikawa, T., Breitschopf, K., Ciechanover, A., Schild, L., and Rotin, D. (1997). Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J. 16, 6325–6336.
Stutts, M. J., Rossier, B. C., and Boucher, R. C. (1997). Cystic fibrosis transmembrane conductance regulator inverts protein kinase A-mediated regulation of epithelial sodium channel single channel kinetics. J. Biol. Chem. 272, 14037–14040.
Subramanya, A. R., Liu, J., Ellison, D. H., Wade, J. B., and Welling, P. A. (2009). WNK4 diverts the thiazide-sensitive NaCl cotransporter to the lysosome and stimulates AP-3 interaction. J. Biol. Chem. 284, 18471–18480.
Webster, M. K., Goya, L., Ge, Y., Maiyar, A. C., and Firestone, G. L. (1993). Characterization of sgk, a novel member of the serine/threonine protein kinase gene family which is transcriptionally induced by glucocorticoids and serum. Mol. Cell. Biol. 13, 2031–2040.
Wiemuth, D., Ke, Y., Rohlfs, M., and McDonald, F. J. (2007). Epithelial sodium channel (ENaC) is multi-ubiquitinated at the cell surface. Biochem. J. 405, 147–155.
Wilson, F. H., Disse-Nicodeme, S., Choate, K. A., Ishikawa, K., Nelson-Williams, C., Desitter, I., Gunel, M., Milford, D. V., Lipkin, G. W., Achard, J. M., Feely, M. P., Dussol, B., Berland, Y., Unwin, R. J., Mayan, H., Simon, D. B., Farfel, Z., Jeunemaitre, X., and Lifton, R. P. (2001). Human hypertension caused by mutations in WNK kinases. Science 293, 1107–1112.
Wilson, F. H., Kahle, K. T., Sabath, E., Lalioti, M. D., Rapson, A. K., Hoover, R. S., Hebert, S. C., Gamba, G., and Lifton, R. P. (2003). Molecular pathogenesis of inherited hypertension with hyperkalemia: the Na-Cl cotransporter is inhibited by wild-type but not mutant WNK4. Proc. Natl. Acad. Sci. U.S.A. 100, 680–684.
Wulff, P., Vallon, V., Huang, D. Y., Volkl, H., Yu, F., Richter, K., Jansen, M., Schlunz, M., Klingel, K., Loffing, J., Kauselmann, G., Bosl, M. R., Lang, F., and Kuhl, D. (2002). Impaired renal Na(+) retention in the sgk1-knockout mouse. J. Clin. Invest. 110, 1263–1268.
Xu, B. E., Stippec, S., Chu, P. Y., Lazrak, A., Li, X. J., Lee, B. H., English, J. M., Ortega, B., Huang, C. L., and Cobb, M. H. (2005). WNK1 activates SGK1 to regulate the epithelial sodium channel. Proc. Natl. Acad. Sci. U.S.A. 102, 10315–10320.
Yang, B., Gay, D. L., MacLeod, M. K., Cao, X., Hala, T., Sweezer, E. M., Kappler, J., Marrack, P., and Oliver, P. M. (2008). Nedd4 augments the adaptive immune response by promoting ubiquitin-mediated degradation of Cbl-b in activated T cells. Nat. Immunol. 9, 1356–1363.
Yang, B., and Kumar, S. (2010). Nedd4 and Nedd4-2: closely related ubiquitin-protein ligases with distinct physiological functions. Cell Death Differ. 17, 68–77.
Yang, C. L., Angell, J., Mitchell, R., and Ellison, D. H. (2003). WNK kinases regulate thiazide-sensitive Na-Cl cotransport. J. Clin. Invest. 111, 1039–1045.
Yang, S. S., Morimoto, T., Rai, T., Chiga, M., Sohara, E., Ohno, M., Uchida, K., Lin, S. H., Moriguchi, T., Shibuya, H., Kondo, Y., Sasaki, S., and Uchida, S. (2007). Molecular pathogenesis of pseudohypoaldosteronism type II: generation and analysis of a Wnk4(D561A/+) knockin mouse model. Cell Metab. 5, 331–344.
Zhou, R., Patel, S. V., and Snyder, P. M. (2007). Nedd4-2 catalyzes ubiquitination and degradation of cell surface ENaC. J. Biol. Chem. 282, 20207–20212.
Keywords: ubiquitin, Nedd4, ENaC, NCC, Sgk1, WNK, hypertension, cystic fibrosis
Citation: Rotin D and Staub O (2012) Nedd4-2 and the regulation of epithelial sodium transport. Front. Physio. 3:212. doi: 10.3389/fphys.2012.00212
Received: 24 April 2012; Paper pending published: 11 May 2012;
Accepted: 30 May 2012; Published online: 21 June 2012.
Edited by:
Kenneth R. Hallows, University of Pittsburgh School of Medicine, USAReviewed by:
Vivek Bhalla, Stanford University School of Medicine, USAFiona McDonald, University of Otago Dunedin, New Zealand
Copyright: © 2012 Rotin and Staub. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Daniela Rotin, The Hospital for Sick Children, TMDT-MaRS, Room 11-305, 101 College Street, Toronto, ON, Canada M5G 1L7. e-mail:ZHJvdGluQHNpY2traWRzLmNh