 Donna M. D'Souza
Donna M. D'Souza Dhuha Al-Sajee
Dhuha Al-Sajee Thomas J. Hawke
Thomas J. Hawke- Department of Pathology and Molecular Medicine, McMaster University, Hamilton, ON, Canada
Diabetes mellitus is defined as a group of metabolic diseases that are associated with the presence of a hyperglycemic state due to impairments in insulin release and/or function. While the development of each form of diabetes (Type 1 or Type 2) drastically differs, resultant pathologies often overlap. In each diabetic condition, a failure to maintain healthy muscle is often observed, and is termed diabetic myopathy. This significant, but often overlooked, complication is believed to contribute to the progression of additional diabetic complications due to the vital importance of skeletal muscle for our physical and metabolic well-being. While studies have investigated the link between changes to skeletal muscle metabolic health following diabetes mellitus onset (particularly Type 2 diabetes mellitus), few have examined the negative impact of diabetes mellitus on the growth and reparative capacities of skeletal muscle that often coincides with disease development. Importantly, evidence is accumulating that the muscle progenitor cell population (particularly the muscle satellite cell population) is also negatively affected by the diabetic environment, and as such, likely contributes to the declining skeletal muscle health observed in diabetes mellitus. In this review, we summarize the current knowledge surrounding the influence of diabetes mellitus on skeletal muscle growth and repair, with a particular emphasis on the impact of diabetes mellitus on skeletal muscle progenitor cell populations.
Skeletal Muscle and Muscle Progenitor Cells
Skeletal muscle is capable of adapting to numerous stimuli, with these responses manifested through changes in muscle size, fiber-type distribution, and/or metabolism. A critical component in skeletal muscle maintenance and plasticity is the presence of muscle progenitor cells. A complex network of intrinsic and extrinsic factors mediate changes to these progenitor cells, with such factors influenced by, and influential to, skeletal muscle health. Diseases that negatively impact muscle health, such as diabetes mellitus, may do so by negatively affecting progenitor cell quantity and/or functionality. As such, these cells (or a sub-population therein) function as a primary therapeutic target to attenuate deficits in muscle health with disease progression. While the most well defined of these progenitor cells is the satellite cell (SC; Hawke and Garry, 2001; Zammit and Relaix, 2012), evidence of a number of non-satellite cell progenitor populations contributing to the maintenance of skeletal muscle in health and disease has emerged in recent years (Pannérec et al., 2012). Throughout this brief review we will use the terms “satellite cells” or “SCs” to define this heterogeneous progenitor cell population, acknowledging that as our understanding of these other unique cell populations becomes clearer, we may revisit roles once allocated specifically to the muscle satellite cells.
Pathophysiology of Type 1 and 2 Diabetes Mellitus
The onset of Type 1 diabetes mellitus (T1DM) often occurs in childhood or adolescence and is characterized by the immune-mediated destruction of pancreatic β-cells leading to insulin deficiency. While T1DM accounts for only ~10% of diabetic cases, its prevalence over the past 30 years has increased worldwide (Onkamo et al., 1999; Gale, 2002). Adolescent muscles subjected to atrophic stimuli are more likely to endure irreversible changes (Darr and Schultz, 1989; Mozdziak et al., 2000), thus the presentation of T1DM during this critical growth period can detrimentally impact long-term muscle health. In contrast, Type 2 Diabetes Mellitus (T2DM) accounts for ~90% of diabetes mellitus cases (Masso-Gonzalez et al., 2009), and is expected to affect almost 8% of the worldwide population by 2030 (Shaw et al., 2010). Adverse health behaviors, particularly sedentary lifestyles and increased adiposity, have lead to a high incidence of insulin resistance and impaired fasting glucose (American Diabetes Association, 2006). Without therapeutic intervention, the insulin-resistant state often precipitates to pancreatic β-cell death and progression to insulin-dependent T2DM.
While the etiology of T1DM and T2DM are distinct, the end result is number of common co-morbidities including nephropathy, neuropathy, and cardiovascular disease. Diabetic myopathy, characterized by reduced physical capacity, strength, and muscle mass (Andersen et al., 1996, 1997, 2004, 2005), is a relatively understudied complication of diabetes mellitus, but is believed to directly influence the rate of co-morbidity development. This is based on the fact that skeletal muscle functions as the largest site for glucose uptake (DeFronzo et al., 1981), and therefore changes to skeletal muscle health can impact whole-body glucose homeostasis. A vital component to the maintenance of skeletal muscle is its SC population. As such, changes to SC functionality with diabetes mellitus would impact skeletal muscle health. Here we review the current state of knowledge on the relationship between skeletal muscle health and diabetes mellitus, with a particular focus on the fate and function of skeletal muscle progenitor cell populations.
Skeletal Muscle in Diabetes Mellitus
T1DM
Muscle growth and development is significantly impaired in T1DM, resulting in reduced muscle mass and myofiber size, poor metabolic control, and a switch to a glycolytic phenotype (Andersen et al., 1997, 2004; Crowther et al., 2003; Fritzsche et al., 2008; Krause et al., 2009, 2013). While initial studies in human T1DM reported no difference in capillary density (Leinonen et al., 1982), investigations in T1DM mice illustrate that the disease is associated with a decline in skeletal muscle capillarization and angiogenesis (Kivelä et al., 2006; Krause et al., 2009). These alterations to muscle structure and metabolism often are associated with reductions in muscle function, as previously demonstrated (Huttunen et al., 1984; Poortmans et al., 1986; Almeida et al., 2008; Gordon et al., 2010). In addition to growth and function, the capacity for repair from damage is also adversely affected by T1DM, as indicated by studies of muscle regeneration using chemical and genetic models of T1DM (Gulati and Swamy, 1991; Talesara and Vashishta, 2000; Vignaud et al., 2007; Krause et al., 2011, 2013). Collectively, these studies highlight the negative impact T1DM is having on skeletal muscle and its potential for growth, maintenance, and repair.
Satellite Cells and T1DM
Satellite cells from streptozotocin (STZ)-treated diabetic mice fail to activate properly, resulting in failed regeneration following chemically induced muscle injury (Jeong et al., 2013). This extreme catabolic state has previously been shown to promote the fusion of SCs to adjacent muscle fibers in T1DM mice, as this is thought to promote the release of factors that function to sustain muscle integrity in this less than favorable metabolic condition (Brannon et al., 1989). Furthermore, Aragno et al. (2004) reported reduced myogenic regulatory factor expression and impaired differentiation in T1DM-derived myoblasts. Attenuated muscle repair has also been observed in T1DM mice (Krause et al., 2011, 2013). The impaired regeneration with diabetes was attributed to an elevation in plasma PAI-1 resulting from attenuated extracellular matrix (ECM) turnover. The delay in ECM turnover inhibited macrophage and SC migration into the damaged/necrotic regions of injured muscle. Interestingly, despite systemic increases in PAI-1, the impaired regeneration occurred in a muscle-specific pattern (Krause et al., 2013), indicating that muscles are intrinsically resistant to the T1DM environment. It is becoming increasingly clear that alterations to muscle protein turnover cannot, by itself, account for diabetic myopathy. Although studies investigating SCs in T1DM remains limited, evidence indicates that functionality is affected. Clinically, it is important to appreciate that T1DM-onset almost always occurs during childhood/adolescence, a period of extensive muscle growth. Thus, understanding alterations to the SC population in T1DM is essential for the development of therapeutic strategies to maximize muscle health during this vulnerable time.
T2DM
Similar to observations in T1DM, skeletal muscle of T2DM subjects exhibit increased glycolytic fiber number (Mårin et al., 1994; Nyholm et al., 1997), muscle atrophy (Huang et al., 2010), and decreases in capillary density (Prior et al., 2009). Perturbations to muscle metabolism in T2DM are common, resulting in decreased intermyofibrillar mitochondrial content and abnormal lipid deposition (Nielsen et al., 2010; Chomentowski et al., 2011). As a consequence of these unfavorable changes, the muscle becomes “metabolically inflexible” as it cannot easily switch between fat and carbohydrate oxidation in response to insulin (Kelley and Mandarino, 2000). Functional impairments are also evident, as demonstrated by a decline in muscle strength (Andersen et al., 2004; Park et al., 2006), a finding strongly correlated with intramuscular fat storage (Hilton et al., 2008). Studies of muscle regeneration in insulin-resistance/T2DM animal models (Nguyen et al., 2011) further identify attenuated skeletal muscle plasticity, with deleterious changes to SC function theorized as a central mechanism underlying the observed outcomes.
As discussed, diabetes mellitus impinges on skeletal muscle health. Studies have noted that the diabetic environment enhances protein degradation (Price et al., 1996; Lecker et al., 1999; Mitch et al., 1999; Mastrocola et al., 2008). While these studies are well conducted, it was not within their scope to investigate all components required for skeletal muscle growth and maintenance. Indeed, SCs are indispensable for such events (Zammit and Relaix, 2012), and thus a more complete understanding of the impact of the diabetic environment on SC function is needed.
Satellite Cells and T2DM
While studies directly assessing SC function with T2DM remain limited, a number of recent investigations have evaluated SC behavior with hyperglycemia and/or lipotoxicity. For instance, 3 weeks of a high fat feeding (HFF) affected SC content and functionality, with the latter classified as the quantity of regenerating fibers present following injury (Fitzpatrick et al., 2011). Hu et al. (2010) demonstrated reduced muscle regeneration after 8-months HFF that was attributed to a delay in myofiber maturation, rather than SC activation or proliferation. In vitro studies have also shown that SCs incubated in high glucose medium have an increased propensity to differentiate into adipocytes (Aguiari et al., 2008), suggesting that SC myogenic capacity may be impacted by uncontrolled diabetes. This is further substantiated through the use of genetic models of obesity and diabetes. The Obese Zucker Rat (OZR), a model for the metabolic syndrome, displays reduced SC proliferative capacity though quiescent SC percentages remain unchanged (Peterson et al., 2008); findings consistent with observed alterations to Akt signaling and myogenic regulatory factor expression (Peterson et al., 2008). Similar results were obtained in transgenic (ob/ob, db/db) models of T2DM. Specifically, impaired SC proliferation and activation were observed and were reflected in measurable impairments of muscle regeneration (Nguyen et al., 2011). A critical, but as of yet unanswered, question is the role of altered leptin signaling in mediating changes to SCs in these animal models. Interestingly, these authors found no difference in SC function or regenerative capacity in HFF mice (Nguyen et al., 2011).
In addition to altered myogenic potential, SCs derived from T2DM patients were found to retain a “diabetic phenotype” upon isolation and culturing. These T2DM-derived SCs displayed reduced lipid oxidation (Gaster et al., 2004), increased secretion of inflammatory markers leading to altered cell signaling (Green et al., 2011), impaired glucose transport (Gaster et al., 2002), and insulin-resistance (Scarda et al., 2010). These modifications were based on T2DM-induced epigenetic changes to muscle cell gene programming, modifying protein expression of factors essential to myogenesis, thereby permanently affecting muscle SCs (Broholm et al., 2012). Taken together, these findings suggest that the degree of T2DM disease severity (i.e., diet vs. genetic model) will differentially influence SC function. A more severe T2DM phenotype, as is found in genetic models of T2DM, results in impairments to the early stages of myogenesis (proliferation, activation), while the HFF models will alter the differentiation potential of the SCs. Finally, long-term exposure to T2DM may promote detrimental epigenetic changes to SCs that will inevitably affect their functionality, and ultimately, overall skeletal muscle health.
Mechanisms for Altered Satellite Cell Function in Diabetes Mellitus
The literature is clear that the uncontrolled diabetic environment is unfavorable for skeletal muscle growth and regeneration (Vignaud et al., 2007; Peterson et al., 2008; Krause et al., 2011; Nguyen et al., 2011). However, the molecular processes governing changes to the SCs are far from elucidated.
Following a period of uncontrolled diabetes mellitus in humans (i.e., pre-diagnosis), insulin administration is the therapeutic standard, and is well studied in terms of regulating muscle protein turnover (Pain et al., 1983; Price et al., 1996; Charlton and Nair, 1998; Lee et al., 2004). What remains less known is the effect of insulin therapy on SCs. Insulin has been found to stimulate both proliferation and differentiation of SCs, with such evidence derived from a handful of in vitro studies (Ewton and Florini, 1981; Vandenburgh et al., 1991; Cassar-Malek et al., 1999). The paucity of data available from human diabetic muscle exposed to insulin treatment merits further consideration. In the absence of insulin, or poorly managed diabetic states, there may be a myriad of factors and processes stemming from the diabetic environment that have the potential to influence SC activity. After a review of many of these mechanisms, a select few are evident in both T1DM and T2DM. The precise modifications to skeletal muscle following diabetes onset is depicted along with the predicted mechanisms of action (Figure 1). These include, but are not limited to: oxidative stress, chronic low-grade inflammation, and impaired ECM remodeling. Though the impact of metabolic diseases on the changing metabolic needs of the muscle satellite cells as they move from quiescence through to differentiation is certainly of note, it is beyond the scope of this mini-review. We refer the readers to some excellent recent reviews (Fulco et al., 2008; Ryall, 2013) on this topic.
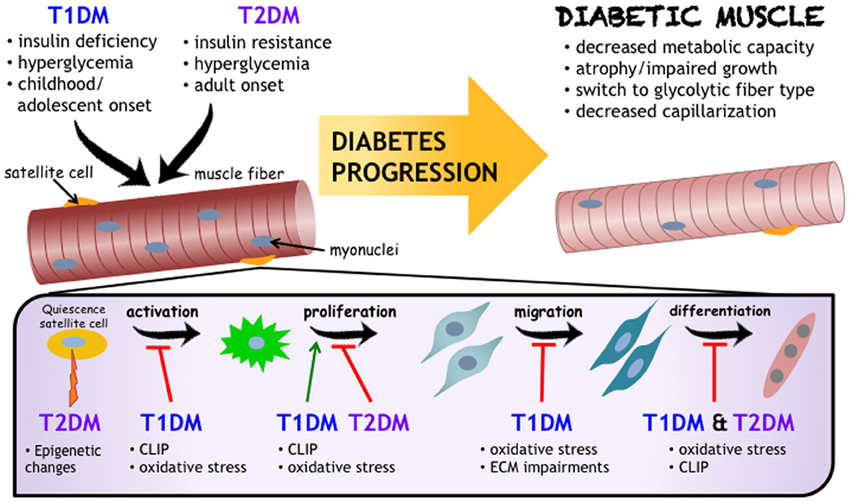
Figure 1. Impact of Diabetes Mellitus on Skeletal Muscle Health. While the etiology and progression for T1DM and T2DM development are distinct, both diseases negatively influence skeletal muscle (referred to as “Diabetic Muscle”) and their resident progenitor cell populations, including satellite cells. Satellite cells are critical to muscle health, and are affected by diabetes mellitus at varying stages of adult myogenesis. As outlined in this review, and schematized here, chronic low grade inflammation (also known as CLIP, or chronic low-grade inflammatory profile), oxidative stress, and impaired extracellular matrix remodeling are proposed to be common denominators for mechanisms underlying impairments to muscle health and decreased satellite cell functionality in diabetes mellitus.
Oxidative Stress
Oxidative stress is evident in both T1DM (Aragno et al., 2004) and T2DM (Henriksen et al., 2010), and has been directly associated with elevated glucose concentrations (Bonnefont-Rousselot, 2002). Dysregulation of nitric oxide (NO) production also occurs, as hyperglycemia promotes the formation of reactive nitrogen species (RNS) to further exacerbate levels of oxidative stress (Zou et al., 2002). A shift in pro-oxidant/antioxidant balance is regarded in the pathogenesis of diabetes and its complications (Evans et al., 2002). Although an emphasis of research relating oxidative stress to skeletal muscle health has been on its modulation of protein turnover (Li et al., 1998; Zhou et al., 2001; Aragno et al., 2004), it is speculated that the concomitant increase in ROS and decrease in NO hinders satellite cell function. In vitro work has found that acute treatment of human muscle SCs with the ROS-inducing agent hydrogen peroxide (H2O2) led to reduced cell viability, shortened lifespan, and decreased proliferative capacity (Renault et al., 2002). In support of oxidative stress impairing myogenesis, Aragno et al. (2004) found that in response to muscle damage, the expression of critical myogenic factors (MyoD, myogenin, and Jun D) was reduced in STZ-diabetic rodents compared to non-diabetic rodents. Muscle creatine kinase and myosin expression were also impaired, suggesting that defects in the early phases of regeneration (i.e., satellite cell functionality) led to a cascade of events to further hinder muscle repair. It is interesting to note that oxidative stress has been implicated in the adipogenic conversion of muscle SCs (Vettor et al., 2009). Now whether this occurs within diabetic muscle has yet to be defined, however, given that the demonstrated impairments in myogenesis with diabetes appear to be linked to oxidative stress, it is clear that this area requires further investigation.
Chronic Low-Grade Inflammatory Profile (CLIP)
With diabetes progression, a multitude of pro-inflammatory factors are elevated, constituting a state of chronic low-grade inflammation, or a chronic low-grade inflammatory profile (CLIP). The presence of this condition is evident in all forms of diabetes mellitus (Llauradó et al., 2012; Osborn and Olefsky, 2012) and is believed to occur as a result of the enhanced formation of advanced glycation end-products (AGEs; Tan et al., 2004; Ramasamy et al., 2005; Yan et al., 2008). The factors associated with CLIP can collectively and/or independently influence SC activity. While examination of each of these factors on SC function is beyond the breadth of this review, it is important to highlight a select few.
Chronic elevations of circulating Interleukin-6 (IL-6) are observed in T1DM and T2DM (Pradhan et al., 2001; Reis et al., 2012). While transient increases in IL-6 are associated with SC proliferation (Toth et al., 2011), chronically elevated IL-6 is correlated with significant decrements in muscle health (e.g., cancer cachexia; Roubenoff, 1997; McKay et al., 2013). Given the chronic elevations in IL-6 with diabetes, it is reasonable to surmise that impairments to SC functionality are occurring. Consistent with this hypothesis, obese diabetic individuals displayed significant impairments in IL-6 signaling within their skeletal muscle that persisted within the satellite cells even upon removal from the diabetic environment (Nielsen et al., 2012).
Akin to IL-6, tumor necrosis factor-α (TNF-α) functions as a key mediator of the inflammatory process. Not only is TNF-α correlated with diabetes progression (Csizuadia et al., 2012; Swaroop et al., 2012), it has also been found to alter insulin-mediated glucose uptake in muscle cells in vitro (Yoon et al., 2011), and has been implicated in the development of insulin resistance through studies knocking out its respective receptors (Uysal et al., 1997; Romanatto et al., 2009). With respect to SCs, TNF-α is thought to exert its effects through stimulation of factors that promote entry into the cell cycle (Li et al., 2003). In support of this, TNF-α treated myoblasts displayed an increased proliferative capacity while differentiation was hindered (Alter et al., 2008).
The presence of CLIP, as found in diabetes, will undoubtedly alter skeletal muscle homeostasis. This emerging and exciting new area of interest, though still in its infancy, presents an intriguing avenue for further therapeutic investigations.
Impaired Extracellular Matrix (ECM) Remodeling
Studies have found that central constituents of the plasminogen system are required for normal growth and repair within a variety of tissues types, including skeletal muscle (Romer et al., 1996; Lluís et al., 2001; Shimizu et al., 2001). Within muscle, inhibition of PAI-1 (a critical inhibitor of the plasminogen system) was found to increase MyoD expression and accelerate muscle repair (Koh et al., 2005). Of particular note, elevated ECM levels have been demonstrated in a variety of diabetic tissues (Berria et al., 2006; Krause et al., 2011). Excessive ECM levels are likely the result of altered protein expression (Lecker et al., 2004), especially in regards to matrix metalloproteinases (Hopps and Caimi, 2012). The improper turnover of ECM proteins may also hinder growth factor signaling, further impeding myogenesis (Gopinath and Rando, 2008). While the aforementioned studies identify that aspects of muscle health are clearly subject to modification with diabetes mellitus, one must also account for diabetic-induced changes to the environment in which the muscle SCs reside. The adverse remodeling of the ECM in diabetic muscle, as evidenced by increased collagen presence, will inevitably affect SC functionality and its capacity to migrate within regenerating muscle (Krause et al., 2013). These defects are also prevalent in senescent skeletal muscle (Franceschi, 2007; Kurtz and Oh, 2012; Vasilaki and Jackson, 2013). Thus, potential therapies to attenuate negative alterations to SC behavior with diabetes onset may also function to mitigate sarcopenia.
Significance and Conclusions
Diabetes mellitus is a global health concern. While diabetes mellitus begins as a result of an impairment in insulin signaling (deficiency/resistance), numerous other factors quickly become altered making the pathogenesis of diabetic complications multi-faceted. Here we provide an overview of the importance of the muscle SC, the impact of T1DM and T2DM on this cell population, and potential “common” mechanisms for altered SC function. Based on the limited number of studies to date, it is evident that various stages of the myogenic process are affected by diabetes mellitus and impairments to SC function are occurring. Given the vital role of these cells in the lifelong maintenance of skeletal muscle, and the importance of a physically and metabolically healthy skeletal muscle mass in attenuating the morbidity and mortality associated with diabetes mellitus, a comprehensive understanding of SC in the diabetic environment is of fundamental significance. Identifying the proponents that attenuate normal SC function in diabetes mellitus will lead to the development of therapies that restore SC activity in order to sustain muscle health, and subsequently attenuate other diabetic complications.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aguiari, P., Leo, S., Zavan, B., Vindigni, V., Rimessi, A., Bianchi, K., et al. (2008). High glucose induces adipogenic differentiation of muscle-derived stem cells. Proc. Natl. Acad. Sci. U.S.A. 105, 1226–1231. doi: 10.1073/pnas.0711402105
Almeida, S., Riddell, M. C., and Cafarelli, E. (2008). Slower conduction velocity and motor unit discharge frequency are associated with muscle fatigue during isometric exercise in Type 1 diabetes mellitus. Muscle Nerve 37, 231–240. doi: 10.1002/mus.20919
Alter, J., Rozentzweig, D., and Bengal, E. (2008). Inhibition of myoblast differentiation by tumor necrosis factor alpha is mediated by c-Jun N-terminal kinase 1 and leukemia inhibitory factor. J. Biol. Chem. 283, 23224–23234. doi: 10.1074/jbc.M801379200
American Diabetes Association. (2006). Diagnosis and classification of diabetes mellitus. Diabetes Care 29, 43–48. doi: 10.2337/dc09-S062
Andersen, H., Gadeberg, P. C., Brock, B., and Jakobsen, J. (1997). Muscular atrophy in diabetic neuropathy: a stereological magnetic resonance imaging study. Diabetologia 40, 1062–1069. doi: 10.1007/s001250050788
Andersen, H., Gjerstad, M. D., and Jakobsen, J. (2004). Atrophy of foot muscles: a measure of diabetic neuropathy. Diabetes Care 27, 2382–2385. doi: 10.2337/diacare.27.10.2382
Andersen, H., Poulsen, P. L., Mogensen, C. E., and Jakobsen, J. (1996). Isokinetic muscle strength in long-term IDDM patients in relation to diabetic complications. Diabetes 45, 440–445. doi: 10.2337/diab.45.4.440
Andersen, H., Schmitz, O., and Nielsen, S. (2005). Decreased isometric muscle strength after acute hyperglycaemia in Type 1 diabetic patients. Diabet. Med. 22, 1401–1407. doi: 10.1111/j.1464-5491.2005.01649.x
Aragno, M., Mastrocola, R., Catalano, M. G., Brignardello, E., Danni, O., and Boccuzzi, G. (2004). Oxidative stress impairs skeletal muscle repair in diabetic rats. Diabetes 53, 1082–1088. doi: 10.2337/diabetes.53.4.1082
Berria, R., Wang, L., Richardson, D. K., Finlayson, J., Belfort, R., Pratipanawatr, T., et al. (2006). Increased collagen content in insulin-resistant skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 290, E560–E565. doi: 10.1152/ajpendo.00202.2005
Bonnefont-Rousselot, D. (2002). Glucose and reactive oxygen species. Curr. Opin. Clin. Nutr. Metab. Care 5, 561–568. doi: 10.1097/00075197-200209000-00016
Brannon, M. A., Dodson, M. V., Wheeler, B. A., Mathison, B. D., and Mathison, B. A. (1989). Satellite cells derived from streptozotocin-diabetic rats display altered fusion parameters in vitro. Metab. Clin. Exp. 38, 348–352. doi: 10.1016/0026-0495(89)90123-6
Broholm, C., Brandt, C., Schultz, N. S., Nielsen, A. R., Pedersen, B. K., and Scheele, C. (2012). Deficient leukemia inhibitory factor signaling in muscle precursor cells from patients with Type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 303, E283–E292. doi: 10.1152/ajpendo.00586.2011
Cassar-Malek, I., Langlois, N., Picard, B., and Geay, Y. (1999). Regulation of bovine satellite cell proliferation and differentiation by insulin and triiodothyronine. Domest. Anim. Endocrinol. 17, 373–388. doi: 10.1016/S0739-7240(99)00055-7
Charlton, M., and Nair, K. S. (1998). Protein metabolism in insulin-dependent diabetes mellitus. J. Nutr. 128, 323S–327S.
Chomentowski, P., Coen, P. M., Radiková, Z., Goodpaster, B. H., and Toledo, F. G. (2011). Skeletal muscle mitochondria in insulin resistance: differences in intermyofibrillar versus subsarcolemmal subpopulations and relationship to metabolic flexibility. J. Clin. Endocrinol. Metab. 96, 494–503. doi: 10.1210/jc.2010-0822
Crowther, G. J., Milstein, J. M., Jubrias, S. A., Kushmerick, M. J., Gronka, R. K., and Conley, K. E. (2003). Altered energetic properties in skeletal muscle of men with well-controlled insulin-dependent (type 1) diabetes. Am. J. Physiol. Endocrinol. Metab. 284, E655–E662. doi: 10.1152/ajpendo.00343.2002
Csizuadia, E., Qipo, A., Libermann, T. A., Strom, T. B., Bhasin, M., Awdeh, Z., et al. (2012). The role of TNF-α in mice with Type 1- and 2- diabetes. PLoS ONE 7:e33254. doi: 10.1371/journal.pone.0033254
Darr, K. C., and Schultz, E. (1989). Hindlimb suspension suppresses muscle growth and satellite cell proliferation. J. Appl. Physiol. 67, 1827–1834.
DeFronzo, R. A., Jacot, E., Jequier, E., Maeder, E., Wahren, J., and Felber, J. P. (1981). The effect of insulin on the disposal of intravenous glucose. results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes 30, 1000–1007. doi: 10.2337/diab.30.12.1000
Evans, J. L., Goldfine, I. D., Maddux, B. A., and Grodsky, G. M. (2002). Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of Type 2 diabetes. Endocr. Rev. 23, 599–622. doi: 10.1210/er.2001-0039
Ewton, Z., and Florini, R. (1981). Effects of the somatomedins differentiation and insulin on myoblast in vitro. Dev. Biol. 39, 31–39. doi: 10.1016/0012-1606(81)90312-2
Fitzpatrick, C., Wagers, A. J., Woo, M., Jimenez-Chillaron, J., Patti, M. E., Cerletti, M., et al. (2011). Early life nutrition modulates muscle stem cell number: implications for muscle mass and repair. Stem Cells Dev. 20, 1763–1769. doi: 10.1089/scd.2010.0349
Franceschi, C. (2007). Inflammaging as a major characteristic of old people: can it be prevented or cured? Nutr. Rev. 65, S173–S176. doi: 10.1301/nr.2007.dec.S173-S176
Fritzsche, K., Blüher, M., Schering, S., Buchwalow, I. B., Kern, M., Linke, A., et al. (2008). Metabolic profile and nitric oxide synthase expression of skeletal muscle fibers are altered in patients with Type 1 diabetes. Exp. Clin. Endocrinol. Diabetes 116, 606–613. doi: 10.1055/s-2008-1073126
Fulco, M., Cen, Y., Zhao, P., Hoffman, E. P., McBurney, M. W., Sauve, A. A., et al. (2008). Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev. Cell 14, 661–673. doi: 10.1016/j.devcel.2008.02.004
Gale, E. A. M. (2002). A missing link in the hygiene hypothesis? Diabetologia 45, 588–594. doi: 10.1007/s00125-002-0801-1
Gaster, M., Petersen, I., Højlund, K., Poulsen, P., and Beck-Nielsen, H. (2002). The diabetic phenotype is conserved in myotubes established from diabetic subjects: evidence for primary defects in glucose transport and glycogen synthase activity. Diabetes 51, 921–927. doi: 10.2337/diabetes.51.4.921
Gaster, M., Rustan, A. C., Aas, V., and Beck-Nielsen, H. (2004). Reduced lipid oxidation in skeletal muscle from Type 2 diabetic subjects may be of genetic origin: evidence from cultured myotubes. Diabetes 53, 542–548. doi: 10.2337/diabetes.53.3.542
Gopinath, S. D., and Rando, T. A. (2008). Stem cell review series: aging of the skeletal muscle stem cell niche. Aging Cell 7, 590–598. doi: 10.1111/j.1474-9726.2008.00399.x
Gordon, C. S., Serino, A. S., Krause, M. P., Campbell, J. E., Cafarelli, E., Adegoke, O. A., et al. (2010). Impaired growth and force production in skeletal muscles of young partially pancreatectomized rats: a model of adolescent Type 1 diabetic myopathy? PLoS ONE 5:e14032. doi: 10.1371/journal.pone.0014032
Green, C. J., Scheele, C., Pedersen, B. K., and Pedersen, M. (2011). Elevated NF- B activation is conserved in human myocytes cultured from obese Type 2 diabetic patients and attenuated by AMP-activated protein kinase. Diabetes 60, 2810–2819. doi: 10.2337/db11-0263
Gulati, A. K., and Swamy, M. S. (1991). Regeneration of skeletal muscle in streptozotocin-induced diabetic rats. Anat. Rec. 229, 298–304. doi: 10.1002/ar.1092290303
Hawke, T. J., and Garry, D. J. (2001). Myogenic satellite cells: physiology to molecular biology. J. Appl. Physiol. 91, 534–551. doi: 10.1063/1.3603452
Henriksen, E. J., Diamond-Stanic, M. K., and Marchionne, E. M. (2010). Oxidative stress and the etiology of insulin resistance and Type 2 diabetes. Free Radic. Biol. Med. 51, 993–999. doi: 10.1016/j.freeradbiomed.2010.12.005
Hilton, T. N., Tuttle, L. J., Bohnert, K. L., Mueller, M. J., and Sinacore, D. R. (2008). Excessive adipose tissue infiltration in skeletal muscle in individuals with obesity, diabetes mellitus, and peripheral neuropathy: association with performance and function. Phys. Ther. 88, 1336–1344. doi: 10.2522/ptj.20080079
Hopps, E., and Caimi, G. (2012). Matrix metalloproteinases in metabolic syndrome. Eur. J. Intern. Med. 23, 99–104. doi: 10.1016/j.ejim.2011.09.012
Hu, Z., Wang, H., Lee, I. H., Modi, S., Wang, X., Du, J., et al. (2010). PTEN inhibition improves muscle regeneration in mice fed a high-fat diet. Diabetes 59, 1312–1320. doi: 10.2337/db09-1155
Huang, B. K., Monu, J. U., and Doumanian, J. (2010). Diabetic myopathy: MRI patterns and current trends. Am. J. Roentgenol. 195, 198–204. doi: 10.2214/AJR.09.2494
Huttunen, N. P., Kaar, M. L., Knip, M., Mustonen, A., Puukka, R., and Akerblom, H. K. (1984). Physical fitness of children and adolescents with insulin-dependent diabetes mellitus. Ann. Clin. Res. 16, 1–5.
Jeong, J., Conboy, M. J., and Conboy, I. M. (2013). Pharmacological inhibition of myostatin/TGF-ß receptor/pSmad 3 signaling rescues muscle regenerative responses in mouse model of type 1 diabetes. Acta. Pharmacol. Sin. 34, 1052–1060. doi: 10.1038/aps.2013.67
Kelley, D. E., and Mandarino, L. J. (2000). Fuel selection in human skeletal muscle in insulin resistance: a reexamination. Diabetes 49, 677–683. doi: 10.2337/diabetes.49.5.677
Kivelä, R., Silvennoinen, M., Touvra, A. M., Lehti, T. M., Kainulainen, H., and Vihko, V. (2006). Effects of experimental Type 1 diabetes and exercise training on angiogenic gene expression and capillarization in skeletal muscle. FASEB J. 20, 1570–1572. doi: 10.1096/fj.05-4780fje
Koh, T. J., Bryer, S. C., Pucci, A. M., and Sisson, T. H. (2005). Mice deficient in plasminogen activator inhibitor-1 have improved skeletal muscle regeneration. Am. J. Physiol. Cell Physiol. 289, C217–C223. doi: 10.1152/ajpcell.00555.2004
Krause, M. P., Al-Sajee, D., D'Souza, D. M., Rebalka, I. A., Moradi, J., Riddell, M. C., et al. (2013). Impaired macrophage and satellite cell infiltration occurs in a muscle-specific fashion following injury in diabetic skeletal muscle. PLoS ONE 8:e70971. doi: 10.1371/journal.pone.0070971
Krause, M. P., Moradi, J., Nissar, A. A., Riddell, M. C., and Hawke, T. J. (2011). Inhibition of plasminogen activator inhibitor-1 restores skeletal muscle regeneration in untreated Type 1 diabetic mice. Diabetes 60, 1964–1972. doi: 10.2337/db11-0007
Krause, M. P., Riddell, M. C., Gordon, C. S., Imam, S. A., Cafarelli, E., and Hawke, T. J. (2009). Diabetic myopathy differs between Ins2Akita+/- and streptozotocin-induced Type 1 diabetic models. J. Appl. Physiol. 106, 1650–1659. doi: 10.1152/japplphysiol.91565.2008
Kurtz, A., and Oh, S. J. (2012). Age related changes of the extracellular matrix and stem cell maintenance. Prev. Med. Suppl:S50–S56. doi: 10.1016/j.ypmed.2012.01.003
Lecker, S. H., Jagoe, R. T., Gilbert, A., Gomes, M., Baracos, V., Bailey, J., et al. (2004). Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 18, 39–51. doi: 10.1096/fj.03-0610com
Lecker, S. H., Solomon, V., Price, S. R., Kwon, Y. T., Mitch, W. E., and Goldberg, A. L. (1999). Ubiquitin conjugation by the N-end rule pathway and mRNAs for its components increase in muscles of diabetic rats. J. Clin. Invest. 104, 1411–1420. doi: 10.1172/JCI7300
Lee, S. W., Dai, G., Hu, Z., Wang, X., Du, J., and Mitch, W. E. (2004). Regulation of muscle protein degradation: coordinated control of apoptotic and ubiquitin-proteasome systems by phosphatidylinositol 3 kinase. J. Am. Soc. Nephrol. 15, 1537–1545. doi: 10.1097/01.ASN.0000127211.86206.E1
Leinonen, H., Matikainen, E., and Juntunen, J. (1982). Permeability and morphology of skeletal muscle capillaries in Type 1 (insulin-dependent) diabetes mellitus. Diabetologia 22, 158–162. doi: 10.1007/BF00283744
Li, Y. P., Lecker, S. H., Chen, Y., Waddell, I. D., Goldberg, A. L., and Reid, M. B. (2003). TNF-alpha increases ubiquitin-conjugating activity in skeletal muscle by up-regulating UbcH2/E220k. FASEB J. 17, 1048–1057. doi: 10.1096/fj.02-0759com
Li, Y. P., Schwartz, R. J., Waddell, I. D., Holloway, B. R., and Reid, M. B. (1998). Skeletal muscle myocytes undergo protein loss and reactive oxygen-mediated NF-kappaB activation in response to tumor necrosis factor alpha. FASEB J. 12, 871–880.
Llauradó, G., Gallart, L., Tirado, R., Megia, A., Simón, I., Caixàs, A., et al. (2012). Insulin resistance, low-grade inflammation and Type 1 diabetes mellitus. Acta Diabetol. 49, 33–39. doi: 10.1007/s00592-011-0257-1
Lluís, F., Roma, J., Suelves, M., Parra, M., Aniorte, G., Gallardo, E., et al. (2001). Urokinase-dependent plasminogen activation is required for efficient skeletal muscle regeneration in vivo. Blood 97, 1703–1711. doi: 10.1182/blood.V97.6.1703
Mårin, P., Andersson, B., Krotkiewski, M., and Björntorp, P. (1994). Muscle fiber composition and capillary density in women and men with NIDDM. Diabetes Care 17, 382–386. doi: 10.2337/diacare.17.5.382
Masso-Gonzalez, E., Johansson, S., Wallander, M.-A., and Garcia-Rodriguez, L. (2009). Trends in the prevalence and incidence of diabetes in the UK - 1996 to 2005. J. Epidemiol. Community Health 63, 332–336. doi: 10.1136/jech.2008.080382
Mastrocola, R., Reffo, P., Penna, F., Tomasinelli, C. E., Boccuzzi, G., Baccino, F. M., et al. (2008). Muscle wasting in diabetic and in tumor-bearing rats: role of oxidative stress. Free Radic. Biol. Med. 44, 584–593. doi: 10.1016/j.freeradbiomed.2007.10.047
McKay, B. R., Ogborn, D. I., Baker, J. M., Toth, K. G., Tarnopolsky, M. A., and Parise, G. (2013). Elevated SOCS3 and altered IL-6 signaling is associated with age-related human muscle stem cell dysfunction. Am. J. Physiol. Cell Physiol. 304, C717–C728. doi: 10.1152/ajpcell.00305.2012
Mitch, W. E., Bailey, J. L., Wang, X., Jurkovitz, C., Newby, D., and Price, S. R. (1999). Evaluation of signals activating ubiquitin-proteasome proteolysis in a model of muscle wasting. Am. J. Physiol. 276, C1132–C1138.
Mozdziak, P. E., Pulvermacher, P. M., and Schultz, E. (2000). Unloading of juvenile muscle results in a reduced muscle size 9 wk after reloading. J. Appl. Physiol. 88, 158–164.
Nguyen, M.-H., Cheng, M., and Koh, T. J. (2011). Impaired muscle regeneration in Ob/ob and Db/db mice. ScientificWorldJournal 11, 1525–1535. doi: 10.1100/tsw.2011.137
Nielsen, A. R., Kelly, M., Fischer, C. P., Pedersen, M., Broholm, C., Taudorf, S., et al. (2012). Satellite cells derived from obese humans with Type 2 diabetes and differentiated into myocytes in vitro exhibit abnormal response to IL-6. PLoS ONE 7:0039657. doi: 10.1371/journal.pone.0039657
Nielsen, J., Mogensen, M., Vind, B. F., Sahlin, K., Højlund, K., Schrøder, H. D., et al. (2010). Increased subsarcolemmal lipids in Type 2 diabetes: effect of training on localization of lipids, mitochondria, and glycogen in sedentary human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 298, E706–E713. doi: 10.1152/ajpendo.00692.2009
Nyholm, B., Qu, Z., Kaal, A., Pedersen, S. B., Gravholt, C. H., Andersen, J. L., et al. (1997). Evidence of an increased number of Type IIb muscle fibers in insulin-resistant first-degree relatives of patients with NIDDM. Diabetes 46, 1822–1828. doi: 10.2337/diab.46.11.1822
Onkamo, P., Väänänen, S., Karvonen, M., and Tuomilehto, J. (1999). Worldwide increase in incidence of Type I diabetes–the analysis of the data on published incidence trends. Diabetologia 42, 1395–1403. doi: 10.1007/s001250051309
Osborn, O., and Olefsky, J. M. (2012). The cellular and signaling networks linking the immune system and metabolism in disease. Nat. Med. 18, 363–374. doi: 10.1038/nm.2627
Pain, V. M., Albertse, E. C., and Garlick, P. J. (1983). Protein metabolism in skeletal muscle, diaphragm, and heart of diabetic rats. Am. J. Physiol. 245, E604–E610.
Pannérec, A., Marazzi, G., and Sassoon, D. (2012). Stem cells in the hood: the skeletal muscle niche. Trends Mol. Med. 18, 599–606. doi: 10.1016/j.molmed.2012.07.004
Park, S. W., Goodpaster, B. H., Strotmeyer, E. S., de Rekeneire, N., Harris, T. B., Schwartz, A. V., et al. (2006). Decreased muscle strength and quality in older adults with Type 2 diabetes: the health, aging, and body composition study. Diabetes 55, 1813–1818. doi: 10.2337/db05-1183
Peterson, J. M., Bryner, R. W., and Alway, S. E. (2008). Satellite cell proliferation is reduced in muscles of Obese Zucker Rats but restored with loading. Am. J. Physiol. Cell Physiol. 295, C521–C528. doi: 10.1152/ajpcell.00073.2008
Poortmans, J. R., Saerens, P., Edelman, R., Vertongen, F., and Dorchy, H. (1986). Influence of the degree of metabolic control on physical fitness in Type I diabetic adolescents. Int. J. Sports Med. 7, 232–235. doi: 10.1055/s-2008-1025765
Pradhan, A. D., Manson, J. E., Rifai, N., Buring, J. E., and Ridker, P. M. (2001). C-reactive protein, interleukin 6, and risk of developing Type 2 diabetes mellitus. J. Am. Med. Assoc. 286, 327–334. doi: 10.1001/jama.286.3.327
Price, S. R., Bailey, J. L., Wang, X., Jurkovitz, C., England, B. K., Ding, X., et al. (1996). Muscle wasting in insulinopenic rats results from activation of the ATP-dependent, ubiquitin-proteasome proteolytic pathway by a mechanism including gene transcription. J. Clin. Invest. 98, 1703–1708. doi: 10.1172/JCI118968
Prior, S. J., McKenzie, M. J., Joseph, L. J., Ivey, F. M., Macko, R. F., Hafer-Macko, C. E., et al. (2009). Reduced skeletal muscle capillarization and glucose intolerance. Microcirculation 16, 203–212. doi: 10.1080/10739680802502423
Ramasamy, R., Vannucci, S. J., Yan, S. S., Herold, K., Yan, S. F., and Schmidt, A. M. (2005). Advanced glycation end products and RAGE: a common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology 15, 16R–28R. doi: 10.1093/glycob/cwi053
Renault, V., Thornell, L. E., Eriksson, P. O., Butler-Browne, G., and Mouly, V. (2002). Regenerative potential of human skeletal muscle during aging. Aging Cell 1, 132–139. doi: 10.1046/j.1474-9728.2002.00017.x
Reis, J. S., Amaral, C. A., Volpe, C. M., Fernandes, J. S., Borges, E. A., Isoni, C. A., et al. (2012). Oxidative stress and interleukin-6 secretion during the progression of type 1 diabetes. Arg. Bras. Endocrinol. Metabol. 56, 441–448. doi: 10.1590/S0004-27302012000700006
Romanatto, T., Roman, E. A., Arruda, A. P., Denis, R. G., Solon, C., Milanski, M., et al. (2009). Deletion of tumor necrosis factor-alpha receptor 1 (TNFR1) protects against diet-induced obesity by means of increased thermogenesis. J. Biol. Chem. 284, 36213–36222. doi: 10.1074/jbc.M109.030874
Romer, J., Bugge, T. H., Pyke, C., Lund, L. R., Flick, M. J., Degen, J. L., et al. (1996). Impaired wound healing in mice with a disrupted plasminogen gene. Nat. Med. 2, 287–292. doi: 10.1038/nm0396-287
Ryall, J. G. (2013). Metabolic reprogramming as a novel regulator of skeletal muscle development and regeneration. FEBS J. 280, 4004–4013. doi: 10.1111/febs.12189
Scarda, A., Franzin, C., Milan, G., Sanna, M., Dal Prà, C., Pagano, C., et al. (2010). Increased adipogenic conversion of muscle satellite cells in Obese Zucker Rats. Int. J. Obes. 34, 1319–1327. doi: 10.1038/ijo.2010.47
Shaw, J. E., Sicree, R. A., and Zimmet, P. Z. (2010). Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res. Clin. Pract. 87, 4–14. doi: 10.1016/j.diabres.2009.10.007
Shimizu, M., Hara, A., Okuno, M., Matsuno, H., Okada, K., Ueshima, S., et al. (2001). Mechanism of retarded liver regeneration in plasminogen activator-deficient mice: impaired activation of hepatocyte growth factor after fas-mediated massive hepatic apoptosis. Hepatology 33, 569–576. doi: 10.1053/jhep.2001.22650
Swaroop, J. J., Rajarajeswari, D., and Naidu, J. N. (2012). Association of TNF-α with insulin resistance in Type 2 diabetes mellitus. Indian J. Med. Res. 135, 127–130. doi: 10.4103/0971-5916.93435
Talesara, C. L., and Vashishta, N. (2000). Regenerative ability of gastrocnemius muscle under diabetic condition with special reference to SDH & m-ATPase. Indian J. Exp. Biol. 38, 567–574.
Tan, K. C., Chow, W. S., Tam, S., Bucala, R., and Betteridge, J. (2004). Association between acute-phase reactants and advanced glycation end products in Type 2 diabetes. Diabetes Care 27, 223–228. doi: 10.2337/diacare.27.1.223
Toth, K. G., McKay, B. R., De Lisio, M., Little, J. P., Tarnopolsky, M. A., and Parise, G. (2011). IL-6 induced STAT3 signalling is associated with the proliferation of human muscle satellite cells following acute muscle damage. PLoS ONE 6:0017392. doi: 10.1371/journal.pone.0017392
Uysal, K. T., Wiesbrock, S. M., Marino, M. W., and Hotamisligil, G. S. (1997). Protection from obesity-induced insulin-resistance in mice lacking Tnf- alpha function. Nature 389, 610–614. doi: 10.1038/39335
Vandenburgh, H. H., Karlisch, P., Shansky, J., and Feldstein, R. (1991). Insulin and IGF-I induce pronounced hypertrophy of skeletal myofibers in tissue culture. Am. J. Physiol. 260, C475–C484.
Vasilaki, A., and Jackson, M. J. (2013). Role of reactive oxygen species in the defective regeneration seen in aging muscle. Free Radic. Biol. Med. 65, 317–323. doi: 10.1016/j.freeradbiomed.2013.07.008
Vettor, R., Milan, G., Franzin, C., Sanna, M., De Coppi, P., Rizzuto, R., et al. (2009). The origin of intermuscular adipose tissue and its pathophysiological implications. Am. J. Physiol. Endocrinol. Metab. 297, E987–E998. doi: 10.1152/ajpendo.00229.2009
Vignaud, A., Ramond, F., Hourdé, C., Keller, A., Butler-Browne, G., and Ferry, A. (2007). Diabetes provides an unfavorable environment for muscle mass and function after muscle injury in mice. Pathobiology 74, 291–300. doi: 10.1159/000105812
Yan, S. F., Ramasamy, R., and Schmidt, A. M. (2008). Mechanisms of disease: advanced glycation end-products and their receptor in inflammation and diabetes complications. Nat. Clin. Pract. Endocrinol. Metab. 4, 285–293. doi: 10.1038/ncpendmet0786
Yoon, J. H., Song, P., Jang, J. H., Kim, D. K., Choi, S., Kim, J., et al. (2011). Proteomic analysis of tumor necrosis factor-alpha (TNF-alpha)-induced L6 myotube secretome reveals novel TNF-alpha-dependent myokines in diabetic skeletal muscle. J. Proteome Res. 10, 5315–5325. doi: 10.1021/pr200573b
Zammit, P. S., and Relaix, F. (2012). Satellite cells are essential for skeletal muscle regeneration: the cell on the edge returns centre stage. Development 139, 2845–2856. doi: 10.1242/dev.069088
Zhou, L. Z., Johnson, A. P., and Rando, T. A. (2001). NF Kappa B and AP-1 mediate transcriptional responses to oxidative stress in skeletal muscle cells. Free Radic. Biol. Med. 31, 1405–1416. doi: 10.1016/S0891-5849(01)00719-5
Keywords: diabetes mellitus, muscle satellite cells, PICs, skeletal muscle, muscle regeneration, Type 1 diabetes mellitus, Type 2 diabetes mellitus
Citation: D'Souza DM, Al-Sajee D and Hawke TJ (2013) Diabetic myopathy: impact of diabetes mellitus on skeletal muscle progenitor cells. Front. Physiol. 4:379. doi: 10.3389/fphys.2013.00379
Received: 31 October 2013; Paper pending published: 20 November 2013;
Accepted: 04 December 2013; Published online: 20 December 2013.
Edited by:
Lucas Guimarães-Ferreira, Federal University of Espirito Santo, BrazilReviewed by:
Espen Spangenburg, University of Maryland, USACarlos Hermano J. Pinheiro, University of São Paulo, Brazil
Rebecca Berdeaux, University of Texas Health Science Center at Houston, USA
Copyright © 2013 D'Souza, Al-Sajee and Hawke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas J. Hawke, Department of Pathology and Molecular Medicine, McMaster University, 1280 Main Street West, Hamilton, ON L8S 4L8, Canada e-mail:aGF3a2VAbWNtYXN0ZXIuY2E=