 Kevin Danastas1*
Kevin Danastas1* Valery Combes2
Valery Combes2 Laura A. Lindsay1
Laura A. Lindsay1 Georges E. R. Grau2Michael B. Thompson3Christopher R. Murphy1
Georges E. R. Grau2Michael B. Thompson3Christopher R. Murphy1- 1Discipline of Anatomy and Histology, School of Medical Sciences, Bosch Institute, The University of Sydney, Sydney, NSW, Australia
- 2Discipline of Pathology, School of Medical Sciences, Bosch Institute, The University of Sydney, Sydney, NSW, Australia
- 3School of Biological Sciences, The University of Sydney, Sydney, NSW, Australia
Vascular endothelial growth factor is a secreted glycoprotein that acts on endothelial cells to induce developmental and physiological angiogenesis. It has also been implicated in angiogenesis occurring in several pathologies, most notably, cancer. Alternative splicing of VEGF mRNA transcripts results in several isoforms with distinct properties depending on their exon composition. Recently, a new isoform has been identified, VEGF111 with a unique exon composition responsible for its high angiogenic potential. In humans, the only known inducer of VEGF111 is DNA damage but its natural presence in the uterus of the viviparous lizard, Saiphos equalis, suggests other mechanisms of regulation. Most interestingly, the possible relationship between the evolution of viviparity and the associated increased risk in developing cancer may be important in understanding the mechanisms underlying tumor development.
Introduction
Angiogenesis is a crucial process during tumor growth, invasion and metastasis for the rapid development and maintenance of a blood supply to developing tumors (Weidner et al., 1991). Capillaries do not usually actively proliferate under normal conditions in adults, but tumors secrete several growth factors to stimulate surrounding endothelial cells (ECs) to invade, rapidly proliferate and develop a dedicated blood supply (Carmeliet and Jain, 2000).
One of the most important growth factors involved in angiogenesis is vascular endothelial growth factor (VEGF), a major mitogen that acts selectively on ECs to stimulate angiogenesis (Ferrara and Henzel, 1989; Keck et al., 1989). Also referred to as VEGF-A, vascular permeability factor and vasculotropin, VEGF belongs to a family of proteins along with VEGF-B, C, D and placenta growth factor (Ferrara, 2004).
VEGF is widely expressed in human fetal and adult organs, primarily in lung, kidney and spleen, and at lower concentrations in several other major organs. It is vital for both maintenance of the vasculature and stimulation of angiogenesis (Shifren et al., 1994). Cancers have developed strategies to utilize the angiogenic role of VEGF to grow and metastasize, but this rapid and uncontrolled angiogenesis results in the blood vessels within tumors often having abnormal characteristics including tortuosity, random branching and variable lumen size (Gimbrone et al., 1972; Langenkamp and Molema, 2009). A newly discovered splice variant of VEGF, known as VEGF111, is produced by human cells with DNA damage and potentially supports the development and metastasis of tumors due to its high angiogenic activity (Mineur et al., 2007) and may provide new insights into how cancers develop.
Background to a New Isoform: Biological Effects of VEGF
VEGF induces angiogenesis by selectively binding to two tyrosine kinase receptors, VEGFR-1 and VEGFR-2, as well as the neuropilin co-receptor, expressed on the surface of ECs (De-Vries et al., 1992; Terman et al., 1992). Both receptors are essential for normal vascular development, but VEGFR-2 is the major regulator of the biological effects of VEGF via downstream pathways (Shalaby et al., 1995; Olsson et al., 2006) (Figure 1). ECs within tumors are often hypoxic, which induces the overexpression of both VEGFR-1 and 2 on these ECs, contributing to uncontrolled angiogenesis (Veikkola et al., 2000).
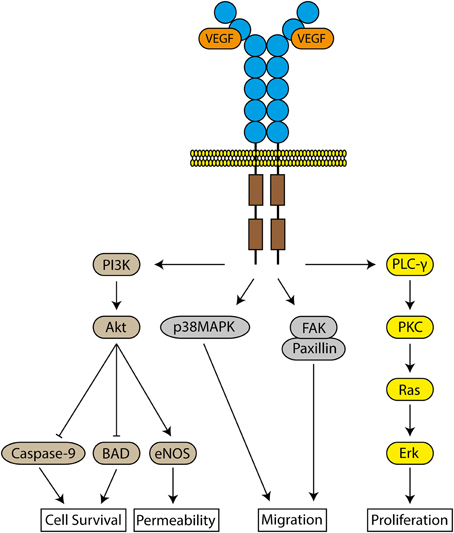
Figure 1. Signaling pathways of VEGFR-2. The binding of VEGF to VEGFR-2 results in the activation of several intracellular molecules to induce cell survival, increase vascular permeability, cellular migration and proliferation.
EC activation by VEGF induces the production of several proteins, including interstitial collagenase, and urokinase-type and tissue-type plasminogen activators, to facilitate EC invasion into the underlying basement membrane and extracellular matrix (Pepper et al., 1991; Unemori et al., 1992). There is also an increase in vascular permeability (Senger et al., 1983), leading to the extravasation of proteins into the surrounding tissue due to opening intercellular junctions and production of fenestrations between ECs. Tumors exhibit uncontrolled vascular permeability due to the increased incidence of these fenestrations (Roberts and Palade, 1995).
VEGF Gene and Isoforms
The gene for VEGF encodes 8 exons and 7 introns. Exons 1 and 2 encode for the signal peptide and exons 3 and 4 encode for the binding sites to VEGFR-1 and 2 respectively (Robinson and Stringer, 2001). The other exons may or may not be present, resulting in various isoforms. Exon splicing of the mRNA transcript results in four naturally expressed isoforms named after the number of amino acids encoded after signal sequence cleavage (Ferrara, 2004) (Figure 2).
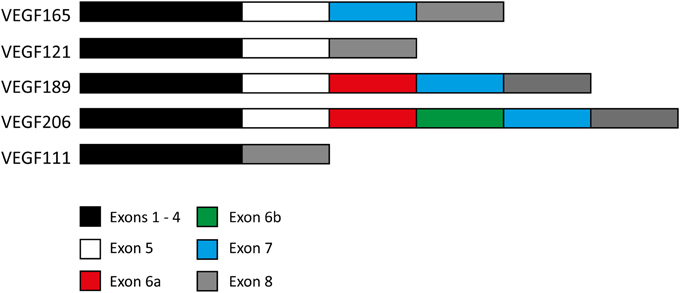
Figure 2. Exon composition of various VEGF isoforms. Exon composition of the four naturally expressed isoforms of VEGF in humans. Exons 6 and 7 encode for heparin and neuropilin binding sites. Note the 24 amino acid insertion within VEGF189 making up exon 6a, and also the same 24 amino acid insertion followed by a 17 amino acid insertion (exon 6b) in VEGF206. The exon composition of the VEGF111 isoform is also included for comparison.
The residues encoded by exons 6 and 7 contain heparin and neuropilin-binding sites, limiting the ability of VEGF189 and VEGF206, and to a lesser degree VEGF165, to diffuse freely upon secretion, resulting in a high proportion remaining bound to the cell surface or surrounding extracellular matrix (Leung et al., 1989). In contrast, VEGF121 lacks these binding domains making it freely diffusible (Houck et al., 1992). Whereas VEGF165, VEGF121, and VEGF189 are widely expressed in a range of normal and pathological tissues, VEGF206 is rare and has only been identified in the cDNA library of human fetal liver (Houck et al., 1991). Other isoforms have been identified in transformed cells but most recently, the discovery of the VEGF111 isoform has raised questions about its involvement in tumor development and metastasis (Mineur et al., 2007).
The New Isoform: VEGF111 Discovery and Identification
VEGF111 is encoded by exons 1–4 and 8 (Figure 2) and is induced by DNA damage to human cells caused by ultraviolet B (UV-B) radiation and genotoxic drugs (Mineur et al., 2007) as well as mild hypothermia (Neutelings et al., 2013). The total VEGF mRNA levels are not increased, but rather the proportions of the other VEGF isoforms are altered to accommodate VEGF111 production. This is primarily at the expense of VEGF165 (Mineur et al., 2007). The concentration of VEGF111 also increases with increasing UV-B intensity, presumably because there is a higher rate of genetic damage occurring, in this case the formation of pyrimidine dimers. Genotoxic agents that result in genetic damage through double strand breaks, including captothecin, mimosin and mitomycin C, also induce VEGF111 formation.
The only documented evidence of the natural expression of VEGF111 is in the uterine wall, testes and kidneys of Saiphos equalis, a viviparous lizard from eastern Australia. VEGF111 is absent from any other somatic organ tested (Murphy et al., 2010). Uterine expression of VEGF111 increases during pregnancy in S. equalis. Other isoforms involved in uterine angiogenesis are also expressed within the uterus. Important changes occur in uterine microvascular architecture in S. equalis to support the developing embryo, with increased vessel density evident particularly in late stage pregnancy (Parker et al., 2010). No VEGF111 mRNA transcripts have yet been identified in any other species (Murphy et al., 2010).
The role of VEGF111 in the uterus of S. equalis is currently unknown, but may be involved in the rapid angiogenesis of both the uterus and chorioallantoic membrane of the embryo (Murphy et al., 2010). The significance of the natural production of VEGF111 in S. equalis suggests there are means of regulating its production, other than genetic damage. VEGF111 may be under hormonal regulation, possibly by estrogen or progesterone, but the exact nature of VEGF111 regulation is unknown.
Properties of VEGF111
VEGF111 is angiogenic and will induce vascularization in vivo and in vitro (Mineur et al., 2007) resulting in the formation of a functional vasculature (Delcombel et al., 2013). VEGF111 is unique in that it is resistant to proteolytic cleavage and retains its complete biological activity upon exposure to plasmin, due to not encoding for exon 5, which contains the residues Arg110-Ala111, the site of plasmin cleavage (Keyt et al., 1996). All other isoforms encode for this cleavage site and their biological activity is decreased upon exposure to plasmin (Houck et al., 1992). Like VEGF121, VEGF111 lacks extracellular matrix binding regions and thus is also freely diffusible (Mineur et al., 2007), which is evident from the widespread vascular permeability induced by VEGF111 in comparison to VEGF165 (Delcombel et al., 2013). These unique characteristics result in its high angiogenic activity compared to other VEGF isoforms as shown by the ability of VEGF111 to promote early blood vessel recruitment and reduce the effects of ischemia and hypoxia in newly grafted tissue (Labied et al., 2013) and improve healing in tendon injuries (Kaux et al., 2014) compared to other isoforms. The high angiogenic activity, and the fact that it is induced by genetic damage, allows VEGF111 to support the development of a blood supply to tumors (Mineur et al., 2007; Delcombel et al., 2013).
VEGF111 is fully glycosylated, unlike VEGF165 and VEGF121, which are only partially glycosylated. Glycosylation is important in efficient protein secretion, which occurs with VEGF111, but does not affect its biological activity (Mineur et al., 2007). VEGF111 also stimulates the migration of ECs but not monocytes. As monocyte chemotaxis is primarily mediated via VEGFR-1, the chemotactic signals stimulated by VEGF111 are primarily mediated via VEGFR-2 (Mineur et al., 2007). VEGF111 binds to VEGFR-2 with similar affinity to the other isoforms and along with VEGF121, is the strongest inducer of VEGFR-2 phosphorylation in the absence of the neuropilin co-receptor, which is needed for complete activation by VEGF165 (Delcombel et al., 2013). VEGF111 is unable to bind this neuropilin co-receptor due to lacking the important binding domains, but the lack of binding has no impact on its biological activity (Delcombel et al., 2013).
While hypoxia and hypoglycemia induce VEGF expression by increasing the stability of the mRNA transcript (Stein et al., 1995), mammalian cells do not produce VEGF111 in these conditions. Apoptosis and reactive oxygen species also do not induce VEGF111 expression (Mineur et al., 2007). While inducers of VEGF111 expression remain unclear, caffeine, epigallocatechin gallate (an antioxidant extracted from tea) and resveratrol (a phenol produced by some plants) inhibit VEGF111 production (Munaut et al., 2010). Thus, the only known inducer of VEGF111 in human cells is genetic damage (Mineur et al., 2007; Neutelings et al., 2013).
Role of VEGF in Cancer
Angiogenesis is critical in the growth of solid tumors, as a tumor cannot grow beyond a size of 2 mm without its own vascular support (Gimbrone et al., 1972). VEGF mRNA is expressed in several types of tumors and inhibiting VEGF inhibits tumor growth in vivo, indicating its importance in tumor angiogenesis. Several anti-VEGF therapies have been developed as cancer treatments, some showing success in improving survival rates (review by Jain et al., 2006).
An issue that has recently emerged is the detrimental effects of VEGF inhibitors on treatment progression, particularly regarding increased malignancy and metastasis (Paez-Ribes et al., 2009). The normalization of the vessels, which involves blocking VEGF signaling to repair vascular abnormalities, may be a better treatment option than eliminating the tumor vasculature, reducing metastasis and improving chemotherapeutic treatments (Mazzone et al., 2009).
Genetic mutations, such as those caused by UV-B radiation and genotoxic drugs, are a major cause of cancer, resulting in abnormal cellular functions (Brash et al., 1991). Thus, it is possible that cancers induced by genotoxic actions may produce VEGF111. Genotoxic drugs such as campothecin and mitomycin C, both of which induce VEGF111 production, are common chemotherapeutics, and thus it is possible that the production of VEGF111 during cancer therapy increases drug resistance (Mineur et al., 2007). Therefore, examining the role of VEGF111 in angiogenesis and EC activation may provide a significant contribution to understanding this process.
When nude mice are injected with cells expressing equal concentrations of different VEGF isoforms, the vascular structures differ. Tumors expressing VEGF111 are poorly vascularized in comparison to tumors expressing other isoforms. However, the vessel density in the surrounding tissue is highest in the VEGF111 tumors compared to all other isoforms tested, which have significantly lower vessel density (Mineur et al., 2007; Delcombel et al., 2013). This result stems from the ability of VEGF111 to freely diffuse from its point of secretion because it is not restricted by extracellular matrix binding. VEGF111 has a longer half-life compared to VEGF165 and so its actions remain active for longer (Mineur et al., 2007). Thus, a tumor with the ability to secrete several different isoforms, including VEGF111, would result in both a highly vascularized tumor and surrounding tissue, potentially increasing the ability to metastasize.
Other VEGF isoforms have been identified in transformed cells including VEGF145 (Poltorak et al., 1997) and VEGF162 (Lange et al., 2003). VEGF145 lacks exons 6b and 7, and is secreted primarily from cancer cell lines derived from the female reproductive system (Poltorak et al., 1997), whereas VEGF162 only lacks exon 7 and is secreted by ovarian carcinoma cells (Lange et al., 2003). Both isoforms are angiogenic and will induce angiogenesis in vitro and in vivo and their biological activity is similar to the other isoforms (Lange et al., 2003). The distinct difference to VEGF111 however, is that they still encode the site of plasmin cleavage, and thus lose their biological activity upon exposure to plasmin. Further investigation in mammalian cancers is necessary, however the presence of VEGF111 primarily in reproductive tissues of S. equalis raises questions whether this isoform may also be present in human reproductive tract cancers.
Similarities of Cancer to Invasive Placentation
There are similar molecular mechanisms between how tumors invade and metastasize compared to the trophoblastic invasion in hemochorial placentation that occurs in humans, particularly regarding angiogenesis (Murray and Lessey, 1999; Hayakawa, 2006). It has been proposed that there is a positive correlation between the degree of placental invasiveness and the ability of metastatic tumors to develop (D'Souza and Wagner, 2014). Mammals that have evolved to develop the less invasive epitheliochorial and endotheliochorial placentas have a lower observed rate of cancer malignancies. In less invasive forms of placentation, the maternal endometrium limits the invasion of the trophoblast cells, and it is hypothesized that this may correlate with a global suppression of tumor metastasis (Priester and Mantel, 1971; D'Souza and Wagner, 2014).
The discovery of VEGF111 as both a potential stimulator of angiogenesis in mammalian tumors (Mineur et al., 2007; Delcombel et al., 2013) and in the uterus of S. equalis during pregnancy (Murphy et al., 2010) further supports a connection between placentation and cancer. The fact that S. equalis is transitioning between egg laying and giving live birth with a placenta (Stewart et al., 2000) also suggests that this evolutionary process could contribute to an increased susceptibility to cancer. Therefore, an understanding into the role of VEGF111 in EC biology may lead to a better understanding of this susceptibility.
VEGF111 as a Potential Therapeutic Agent
The high angiogenic activity of VEGF111 could also be used as a potential therapeutic agent to increase vascularization in ischemic diseases and lesions. The presence of plasmin in chronic wounds results in rapid VEGF cleavage (Lauer et al., 2000), but the ability of VEGF111 to resist this degradation has recently been exploited to improve ischemia and wound healing (Labied et al., 2013; Kaux et al., 2014). This resistance, as well being freely diffusible, allows VEGF111 to induce rapid angiogenesis and rapid EC proliferation compared to the other isoforms, which results in the earlier formation of a functional vasculature and blood supply to the ischemic region (Labied et al., 2013). Further investigation into the clinical applications of VEGF111 is necessary, but the initial data are promising.
Conclusion
VEGF plays a crucial role in normal developmental and embryological angiogenesis as well as contributing to the progression of several pathological conditions. The discovery of VEGF111 is an important advance in understanding the mechanisms by which tumors develop their dedicated blood supply and is critical in their ability to grow and metastasize rapidly. While there are several isoforms of VEGF, VEGF111 is the only isoform to resist proteolytic degradation and may provide further insights into the pathogenesis and treatment of diseases that are dependent on angiogenesis. Future work on this molecule should be directed at understanding the mechanisms underlying angiogenesis and has great promise in developing new therapeutics to target tumorigenesis and to treat ischemic diseases. This should involve examining the actions of VEGF111 on EC biology and its capacity to facilitate the formation of functional vessels in several models to specifically address its role in angiogenesis. This should encompass its actions in cancer, both as a potential contributor to metastasis during disease progression, and drug resistance during disease treatment. Further investigation should also expand on the previous work that focused on VEGF111 as a therapeutic agent. Its presence in the uterus of pregnant lizards provides a unique way to understand those mechanisms.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by an Australian Research Council Discovery Grant to Michael B. Thompson, Christopher R. Murphy, and Georges E. R. Grau and by funds from the Murphy laboratory.
References
Brash, D. E., Rudolph, J. A., Simon, J. A., Lin, A., McKenna, G. J., Baden, H. P., et al. (1991). A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma. Proc. Natl. Acad. Sci. U.S.A. 88, 10124–10128. doi: 10.1073/pnas.88.22.10124
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Carmeliet, P., and Jain, R. K. (2000). Angiogenesis in cancer and other diseases. Nature 407, 249–257. doi: 10.1038/35025220
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Delcombel, R., Janssen, L., Vassy, R., Gammons, M., Haddad, O., Richard, B., et al. (2013). New prospects in the roles of the C-terminal domains of VEGF-A and their cooperation for ligand binding, cellular signaling and vessels formation. Angiogenesis 16, 353–371. doi: 10.1007/s10456-012-9320-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
De-Vries, C., Escobedo, J. A., Ueno, H., Houck, K., Ferrara, N., and Williams, L. T. (1992). The fms tyrosine-like kinase, a receptor for vascular endothelial growth factor. Science 255, 989–991. doi: 10.1126/science.1312256
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
D'Souza, A. W., and Wagner, G. P. (2014). Malignant cancer and invasive placentation: a case for positive pleiotropy between endometrial and malignancy phenotypes. Evol. Med. Public Health 2014, 136–145. doi: 10.1093/emph/eou022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ferrara, N. (2004). Vascular endothelial growth factor: basic science and clinical progress. Endocr Revs. 25, 581–611. doi: 10.1210/er.2003-0027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ferrara, N., and Henzel, W. J. (1989). Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem. Biophys. Res. Commun. 161, 851–858. doi: 10.1016/0006-291X(89)92678-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gimbrone, M. A., Leapman, S. B., Cotran, R. S., and Folkman, J. (1972). Tumor dormancy in vivo by prevention of neovascularization. J. Exp. Med. 136, 261–276. doi: 10.1084/jem.136.2.261
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hayakawa, S. (2006). No cancer in cancers: evolutionary trade-off between successful viviparity and tumor escape from the adaptive immune system. Med. Hypotheses 66, 888–897. doi: 10.1016/j.mehy.2005.12.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Houck, K. A., Ferrara, N., Winer, J., Cachianes, G., Li, B., and Leung, D. (1991). The vascular endothelial growth factor family: identification of a fourth molecular species and characterization of alternative splicing RNA. Mol. Endocrinol. 5, 1806–1814. doi: 10.1210/mend-5-12-1806
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Houck, K. A., Leung, D. W., Rowland, A. M., Winer, J., and Ferrara, N. (1992). Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J. Biol. Chem. 267, 26031–26037.
Jain, R. K., Duda, D. G., Clark, J. W., and Loeffler, J. S. (2006). Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat. Clin. Pract. Oncol. 3, 24–40. doi: 10.1038/ncponc0403
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kaux, J. F., Janssen, L., Drion, P., Nusgens, B., Libertiaux, V., Pascon, F., et al. (2014). Vascular Endothelial Growth Factor-111 (VEGF-111) and tendon healing: preliminary results in a rat model of tendon injury. Muscles Ligaments Tendons J. 4, 24–28. doi: 10.11138/mltj/2014.4.1.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keck, P. J., Hauser, S. D., Krivi, G., Sanzo, K., Warren, T., Feder, J., et al. (1989). Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science 246, 1309–1312. doi: 10.1126/science.2479987
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keyt, B. A., Berleau, L. T., Nguyen, H. V., Chen, H., Heinsohn, H., Vandlen, R., et al. (1996). The carboxyl-terminal domain (111-165) of vascular endothelial growth factor is critical for its mitogenic potency. J. Biol. Chem. 271, 7788–7795. doi: 10.1074/jbc.271.13.7788
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Labied, S., Delforge, Y., Munaut, C., Blacher, S., Colige, A., Delcombel, R., et al. (2013). Isoform 111 of vascular endothelial growth factor (VEGF111) improves angiogenesis of ovarian tissue xenotransplantation. Transplantation 95, 426–433. doi: 10.1097/TP.0b013e318279965c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lange, T., Guttmann-Raviv, N., Baruch, L., Machluf, M., and Neufeld, G. (2003). VEGF162, a new heparin-binding vascular endothelial growth factor splice form that is expressed in transformed human cells. J. Biol. Chem. 278, 17164–17169. doi: 10.1074/jbc.M212224200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Langenkamp, E., and Molema, G. (2009). Microvascular endothelial cell heterogeneity: general concepts and pharmacological consequences for anti-angiogenic therapy of cancer. Cell Tissue Res. 335, 205–222. doi: 10.1007/s00441-008-0642-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lauer, G., Sollberg, S., Cole, M., Flamme, I., Sturzebecher, J., Mann, K., et al. (2000). Expression and proteolysis of vascular endothelial growth factor is increased in chronic wounds. J. Invest. Dermatol. 115, 12–18. doi: 10.1046/j.1523-1747.2000.00036.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leung, D. W., Cachianes, G., Kuang, W. J., Goeddel, D. V., and Ferrara, N. (1989). Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 246, 1306–1309. doi: 10.1126/science.2479986
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mazzone, M., Dettori, D., Leite de Oliveira, R., Loges, S., Schmidt, T., Jonckx, B., et al. (2009). Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell 136, 839–851. doi: 10.1016/j.cell.2009.01.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mineur, P., Colige, A. C., Deroanne, C. F., Dubail, J., Kesteloot, F., Habraken, Y., et al. (2007). Newly identified biologically active and proteolysis resistant VEGF-A isoform VEGF111 is induced by genotoxic agents. J. Cell Biol. 179, 1261–1273. doi: 10.1083/jcb.200703052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Munaut, C., Colige, A. C., and Lambert, C. A. (2010). Alternative splicing: a promising target for pharmaceutical inhibition of pathological angiogenesis? Curr. Pharm. Des. 16, 3864–3876. doi: 10.2174/138161210794455012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Murphy, B. F., Belov, K., and Thompson, M. B. (2010). Evolution of viviparity and uterine angiogenesis: vascular endothelial growth factor (VEGF) in oviparous and viviparous skinks. J. Exp. Zool. B Mol. Dev. Evol. 314, 148–156. doi: 10.1002/jez.b.21317
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Murray, M. J., and Lessey, B. A. (1999). Embryo implantation and tumor metastasis: common pathways of invasion and angiogenesis. Semin. Reprod. Endocrinol. 17, 275–290. doi: 10.1055/s-2007-1016235
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Neutelings, T., Lambert, C. A., Nusgens, B. V., and Colige, A. C. (2013). Effects of mild cold shock (25°C) followed by warming up at 37°C on the cellular stress response. PLoS ONE 8:e69687. doi: 10.1371/journal.pone.0069687
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Olsson, A. K., Dimberg, A., Kreuger, J., and Claesson-Welsh, L. (2006). VEGF receptor signaling–in control of vascular function. Nat. Rev. Mol. Cell Biol. 7, 359–371. doi: 10.1038/nrm1911
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Paez-Ribes, M., Allen, E., Hudock, J., Takeda, T., Okuyama, H., Vinals, F., et al. (2009). Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 15, 220–231. doi: 10.1016/j.ccr.2009.01.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Parker, S. L., Maconi, F., Murphy, C. R., and Thompson, M. B. (2010). Uterine and placental angiogenesis in the Australian skinks, Ctenotus taeniolatus, and Saiphos equalis. Anat. Rec. 293, 829–838. doi: 10.1002/ar.21052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pepper, M. S., Ferrara, N., Orci, L., and Montesano, R. (1991). Vascular endothelial growth factor (VEGF) induces plasminogen activators and plasminogen activator inhibitor-1 in microvascular endothelial cells. Biochem. Biophys. Res. Commun. 181, 902–906. doi: 10.1016/0006-291X(91)91276-I
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Poltorak, Z., Cohen, T., Sivan, R., Kandelis, Y., Spira, G., Vlodavsky, I., et al. (1997). VEGF145, a secreted vascular endothelial growth factor isoform that binds to extracellular matrix. J. Biol. Chem. 272, 7151–7158. doi: 10.1074/jbc.272.11.7151
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Priester, W. A., and Mantel, N. (1971). Occurrence of tumors in domestic animals. Data from 12 United States and Canadian colleges of veterinary medicine. J. Natl. Cancer Inst. 47, 1333–1344.
Roberts, W. G., and Palade, G. E. (1995). Increased microvascular permeability and endothelial fenestration induced by vascular endothelial growth factor. J. Cell Sci. 108, 2369–2379.
Robinson, C. J., and Stringer, S. E. (2001). The splice variants of vascular endothelial growth factor (VEGF) and their receptors. J. Cell Sci. 114, 853–865.
Senger, D. R., Galli, S. J., Dvorak, A. M., Perruzzi, C. A., Harvey, V. S., and Dvorak, H. F. (1983). Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 219, 983–985. doi: 10.1126/science.6823562
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shalaby, F., Rossant, J., Yamaguchi, T. P., Gertsenstein, M., Wu, X. F., Breitman, M. L., et al. (1995). Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 376, 62–66. doi: 10.1038/376062a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shifren, J. L., Doldi, N., Ferrara, N., Mesiano, S., and Jaffe, R. B. (1994). In the human fetus, vascular endothelial growth factor is expressed in epithelial cells and myocytes, but not vascular endothelium: implications for mode of action. J. Clin. Endocrinol. Metab. 79, 316–322.
Stein, I., Neeman, M., Shweiki, D., Itin, A., and Keshet, E. (1995). Stabilization of vascular endothelial growth factor mRNA by hypoxia and hypoglycemia and coregulation with other ischemia-induced genes. Mol. Cell. Biol. 15, 5363–5368.
Stewart, J. R., Mathieson, A. N., Ecay, T. W., Herbert, J. F., Parker, S. L., and Thompson, M. B. (2000). Uterine and eggshell structure and histochemistry in a lizard with prolonged uterine egg retention (Lacertilia, Scincidae, Saiphos). J. Morphol. 271, 1342–1351. doi: 10.1002/jmor.10877
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Terman, B. I., Doughervermazen, M., Carrion, M. E., Dimitrov, D., Armellino, D. C., Gospodarowics, D., et al. (1992). Identification of the KDR tyrosine kinase as a receptor for vascular endothelial growth factor. Biochem. Biophys. Res. Commun. 187, 1579–1586. doi: 10.1016/0006-291X(92)90483-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Unemori, E. N., Ferrara, N., Bauer, E. A., and Amento, E. P. (1992). Vascular endothelial growth factor induces interstitial collagenase expression in human endothelial cells. J. Cell. Physiol. 153, 557–562. doi: 10.1002/jcp.1041530317
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Veikkola, T., Karkkainen, M., Claesson-Welsh, L., and Alitalo, K. (2000). Regulation of angiogenesis via vascular endothelial growth factor receptors. Cancer Res. 60, 203–212.
Weidner, N., Semple, J. P., Welch, W. R., and Folkman, J. (1991). Tumor angiogenesis and metastasis–correlation in invasive breast carcinoma. N. Engl. J. Med. 324, 1–8. doi: 10.1056/NEJM199101033240101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: vascular endothelial growth factor, angiogenesis, cancer, metastasis, tumorigenesis, placentation
Citation: Danastas K, Combes V, Lindsay LA, Grau GER, Thompson MB and Murphy CR (2015) VEGF111: new insights in tissue invasion. Front. Physiol. 6:2. doi: 10.3389/fphys.2015.00002
Received: 05 December 2014; Accepted: 05 January 2015;
Published online: 22 January 2015.
Edited by:
John D. Imig, Medical College of Wisconsin, USAReviewed by:
Brenda Lilly, The Ohio State University, USAGeorge Wilkinson, Concordia University of Wisconsin, USA
Copyright © 2015 Danastas, Combes, Lindsay, Grau, Thompson and Murphy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kevin Danastas, Cell and Reproductive Biology Laboratory, Discipline of Anatomy and Histology, The University of Sydney, Rm N364, Anderson Stuart Building (F13), Sydney, NSW 2006, Australia e-mail:a2RhbjY0OTdAdW5pLnN5ZG5leS5lZHUuYXU=