 Johannes Patzelt
Johannes Patzelt Admar Verschoor
Admar Verschoor Harald F. Langer
Harald F. Langer- 1University Clinic for Cardiovascular Medicine, University of Tuebingen, Tuebingen, Germany
- 2Institute for Medical Microbiology, Immunology and Hygiene, Technische Universität München, Munich, Germany
- 3Section for Cardioimmunology, Department of Cardiovascular Medicine, University of Tuebingen, Tuebingen, Germany
Atherosclerosis and its late sequels are still the number one cause of death in western societies. Platelets are a driving force not only during the genesis of atherosclerosis, but especially in its late stages, as evidenced by complications such as arterial thrombosis, myocardial infarction, and ischemic stroke. Atherosclerosis is increasingly recognized as an inflammatory disease, influenced by various immune mechanisms. The complement system is part of our innate immune system, and its diverse roles in atherosclerosis have become evident over the past years. In this review we identify points of intersection between platelets and the complement system and discuss their relevance for atherosclerosis. Specifically, we will focus on roles for platelets in the onset as well as progression of the disease, a possible dual role for complement in the genesis and development of atherosclerosis, and review emerging literature revealing previously unrecognized cross-talk between platelets and the complement system and discuss its possible impact for atherosclerosis. Finally, we identify limitations of current research approaches and discuss perspectives of complement modulation in the control of the disease.
Introduction
Despite remarkable progress in therapy, atherosclerosis and its associated complications such as coronary heart disease are still the leading cause of death in western societies (Ross, 1999). Atherosclerosis is defined as the process of plaque formation and stenosis in arterial vessels. Deleterious effects of plaque rupture include acute vessel occlusion and thromboembolic complications such as myocardial infarction resulting from thrombotic occlusion of an epicardial vessel or stroke resulting from carotid artery plaque formation.
A complex interplay between hemostatic and inflammatory cues is increasingly unraveled and recognized to drive the genesis and exacerbations of atherosclerosis. Platelets are thought to play a dual role here, mediating the often serious clinical effects of plaque rupture in established atherosclerosis, but also impacting the early development of atherosclerosis (Massberg et al., 2002). The classical function of platelets is coverage and closure of endothelial wounds, and contact between platelets and the subendothelial matrix triggers their activation and drives thrombus formation also during the early pathophysiological process of plaque formation (Figure 1). Moreover, platelets have been shown to also interact with intact endothelium and recruit leukocytes even before an atherosclerotic plaque has formed (Massberg et al., 2002).
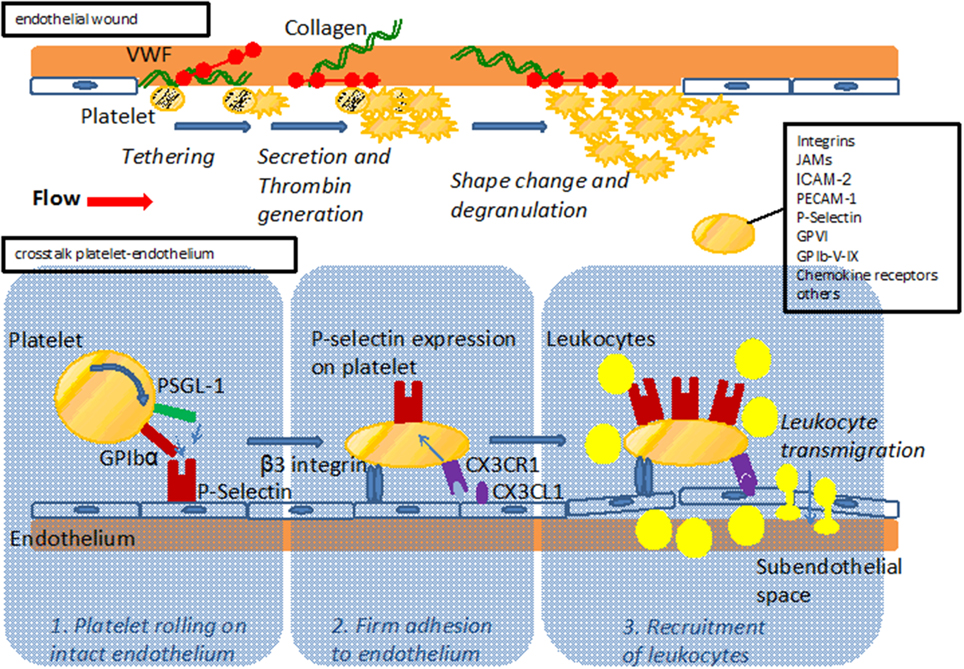
Figure 1. Upper part: Model of platelet interaction with the damaged vessel wall: exposure of subendothelial matrix after endothelial lesion leads to platelet tethering, activation and accumulation to provide sealing of the endothelial wound. Lower part: Model of platelet interaction with the endothelium. Platelets interact with the endothelium via adhesion receptors such as GPIbα and PSGL-1 promoting rolling and subsequent firm adhesion via β3 integrins. Interaction with endothelial bound chemokines such as Cx3CL1 (fractalkine) or CCL5 (Rantes) results in P-Selectin mediated recruitment of leukocytes to the vessel wall and subsequent transmigration. For both interaction with the subendothelial matix and the endothelium, major adhesion receptors expressed on platelets are listed.
On the other hand, atherosclerosis is recognized as an inflammatory disease (Ross, 1999), making the immune system a focus of interest. The complement system, as part of our immune system, has important protective functions in immune defense but can also be a driving force behind the pathological processes of chronic inflammatory diseases (Walport, 2001). The complement system consists of a series of plasmatic proteins (zymogens) that form enzymatic cascades, which produce a broad spectrum of immune-active molecules and pro-inflammatory mediators (Verschoor and Carroll, 2004). Besides these classical functions of immune defense, the complement system also directs central homeostatic and pathophysiological processes such as angiogenesis, tissue regeneration, the removal of immune complexes, apoptotic cells, and cellular debris (Ricklin et al., 2010). Thus, with respect to atherosclerosis complement may exert proatherogenic as well as atheroprotective effects, for which we will discuss the evidence below in detail.
Platelets and Atherosclerosis
Platelets have a well-recognized contribution to the late thrombotic complications associated with atherosclerosis, making them a potent target for the development of diagnostic and therapeutic tools (Gawaz et al., 2008). In addition, platelets can also interact with endothelial cells before an atherosclerotic plaque has formed and may thus play a role in the genesis of atherosclerosis (Gawaz et al., 2005). Indeed, platelets of atherosclerotic apolipoprotein E deficient (ApoE–/–) mice were observed to adhere to intact endothelium via the von Willebrand factor receptor GPIbα and the fibrinogen receptor GPIIbIIIa well before atherosclerotic plaques had formed (Massberg et al., 2002; Huo et al., 2003; Gawaz et al., 2005). Still, absence of GPIIbIIIa alone does not fully protect from atherosclerosis, as is known from studies assessing patients with Glanzmann thrombasthenia whose platelets lack functional GPIIbIIIa. Indeed, ultrasound imaging of the carotid bifurcation of such patients revealed plaques in 4 of 7 individuals (Shpilberg et al., 2002). Therefore, platelet-vessel wall interactions via GPIIbIIIa may contribute to, but do not seem to be a prerequisite in the genesis of human atherosclerosis and may be functionally substituted by other platelet receptors. Mice with deficiency in GPIbα are not protected from atherosclerotic plaque formation (Strassel et al., 2009). On the other hand, platelet depletion with a GPIbα-specifc antibody in ApoE–/– mice leads to reduced leukocyte accumulation in the arterial intima and attenuated atherosclerotic plaque formation, importantly indicating that adhering platelets form a focal point for the immune-cell driven inflammation that typifies atherosclerosis (Massberg et al., 2002). Indeed, PSGL-1 or GPIbα allow platelets to interact with P-selectin expressed on intact endothelium. This initial “rolling” interaction is followed by β3 integrin–mediated firm “adhesion” of platelets to the vessel wall (Gawaz et al., 2005), and these events are seen as crucial steps in the initiation of atherosclerosis. Platelets express various inflammatory receptors, including fractalkine receptor (CX3CR1) which induces P-selectin on platelets upon binding to fractalkine (CX3CL1) expressed on inflamed endothelial cells (Schulz et al., 2007). In turn, P-selectin exposure initiates local accumulation of leukocytes, driven by arterial shear forces (Schulz et al., 2007) (Figure 1). The importance of P-selectin in the genesis of atherosclerosis is underlined by the finding of increased intima-media-thickness in human subjects presenting with high levels of platelet P-selectin (Koyama et al., 2003). Indeed, both platelet and endothelial P-selectin contributed to lesion formation in a mouse model of atherosclerosis that was based on the adoptive transfer of P-selectin positive or negative platelets (Burger and Wagner, 2003).
Whether platelet-adhesion to the intima mediates direct damage to the endothelial lining remains unclear, but several studies found how platelets contribute to vascular inflammation via their interaction with leukocytes (Santoso et al., 2002; Schober et al., 2002; Langer et al., 2007, 2012; Ley et al., 2007; Langer and Chavakis, 2009). Activated platelets were shown to exacerbate atherosclerosis in ApoE deficient mice via the recruitment of monocytes and other leukocytes (Wagner and Frenette, 2008; von Hundelshausen et al., 2009). In turn, the formation of platelet-leukocyte aggregates (PLA) facilitated the deposition of inflammatory platelet mediators on endothelial cells (Schober et al., 2002; Huo et al., 2003). Others found that the number of circulating PLAs is increased upon platelet activation (van Gils et al., 2009; Totani and Evangelista, 2010). As potential mechanisms underlying platelet-leukocyte crosstalk, several receptor/ligand pairs have been identified, including integrins or members of the JAM family of proteins (von Hundelshausen and Weber, 2007; Wagner and Frenette, 2008; von Hundelshausen et al., 2009). A list of platelet expressed receptors with potential relevance for artherosclerosis is given in Table 1. Finally, platelets may also contribute to vascular inflammation via release of active biomolecules from their granules (Langer and Gawaz, 2008; Patzelt and Langer, 2012). As prominent examples, the release of chemokines such as CCL5 or CXCL4 contributes to atherosclerosis in a P-selectin dependent manner (von Hundelshausen and Schmitt, 2014).
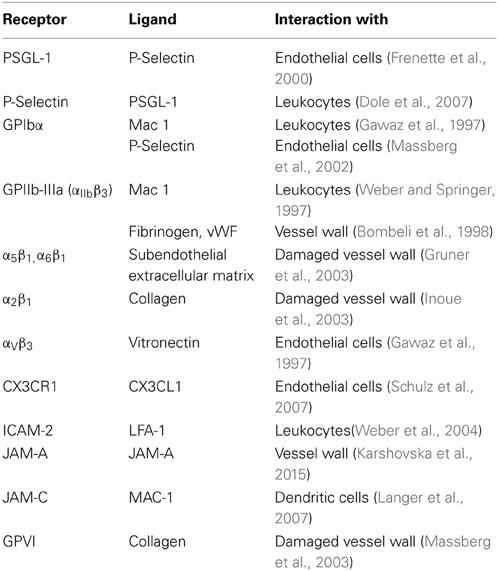
Table 1. Platelet receptors, their ligands, and their target structure contributing to vascular inflammation.
Given the relevance of fatty acids in the genesis of artherosclerosis, it should be noted that also oxidized LDL—one of the major initiators and drivers of atherosclerosis—can be bound by platelets and interactions with lipoproteins can change platelet function (Siegel-Axel et al., 2008; Stellos et al., 2012). In line with this, platelets of hypercholesterolemic patients show hyperaggregability in vitro and enhanced activity in vivo (Cipollone et al., 2002; Ferroni et al., 2006). In conclusion, platelet activation seems to confer proatherosclerotic effects, as well as effects of atheromodulation and tissue/vascular remodeling.
The Complement System and Atherosclerosis
As noted earlier, many cells and molecular mediators that were identified to modulate the development of artherosclerosis are components of the immune system. Complement, as part of the innate immune system, has a broad range of immune-modulatory effects, including the opsonization of microbial intruders with C1q or manose binding lectin (MBL), followed by the activation products of C2, C3, and C4 (including the opsonins C3b and C4b), the induction of mast cell degranulation via soluble anaphylatoxins C3a and C5a and the attraction of inflammatory cells (Verschoor and Carroll, 2004). Components C5b–C9 form the membrane attack complex (MAC), which mediates lysis of target cells. Beyond immune defense, the complement system directs central homeostatic and pathophysiological processes in tissue remodeling and the removal of immune complexes, apoptotic cells and cellular debris (Ricklin et al., 2010). Components of the classical pathway, including C1q, C2, and C4, are associated with the homeostatic control of such complexes, as their deficiency predisposes to diseases characterized by an impairment in the removal of cellular remnants, for example known in Systemic Lupus Erythomatosis (SLE) (Aggarwal et al., 2010). Apoptotic and necrotic cells also accumulate in atherosclerotic plaques, and a Swedish study identified a significant association between genetic C2 deficiency and atherosclerosis, including a higher rate of myocardial infarctions and stroke in a cohort of 40 patients (Jonsson et al., 2005). Furthermore, in patients with the premature atherosclerotic peripheral vascular disease C4 deficiency, a significant proportion of diseased patients revealed circulating immune complexes and their enhanced propensity to immune complex formation was associated with a higher prevalence of circulating immune complexes in atherosclerotic patients (Nityanand et al., 1999). A genome-wide analysis found a SNP of C1q receptor C1qRp (CD93) constituting a risk factor for coronary artery disease, which could be confirmed in patients with familial hypercholesteremia (van der Net et al., 2008) and polymorphisms for mannose binding lectin (MBL) with decreased levels of the protein went along with more coronary artery disease and increased carotid plaque (Madsen et al., 1998; Hegele et al., 2000; Best et al., 2004).
Complement activation also promotes inflammation, through the generation of anaphylatoxins. Analyzing circulating levels of such complement components demonstrated that patients with advanced atherosclerosis present with elevated levels of anaphylatoxin C5a, predictive of major cardiovascular events and independent of known risk markers such as C reactive protein (CRP) or fibrinogen (Speidl et al., 2005).
Complement factors have been detected in substantial amounts within atherosclerotic plaques (Laine et al., 2002; Speidl et al., 2011a). Under normal conditions, activated complement components are quickly cleared from the circulation. However, activated complement and the MAC were identified within fatty streaks (early stages of plaque formation, before the arrival of inflammatory cells) in cholesterol-fed rabbits (Seifert et al., 1989). This finding was confirmed by following studies, which demonstrated the presence of the terminal complement complex C5b–9 in human atherosclerotic arteries (Niculescu et al., 1985, 1987; Torzewski et al., 1998).
Such complement components may derive from the blood circulation (Vlaicu et al., 1985; Niculescu and Rus, 2004), but the presence of mRNA for several complement components (including C1r, C1s, C4, C7, and C8) indicates that these factors also may be produced locally within the plaque (Yasojima et al., 2001; Niculescu and Rus, 2004). Indeed, various studies identified the power of local complement production (as opposed to circulating systemic complement) in driving immune processes (Verschoor et al., 2001, 2003; Gadjeva et al., 2002; Li et al., 2007). Interestingly, complement activation differs between superficial and deeper layers of the atherosclerotic plaque: in the luminal layer, signs of classical and alternative, but not terminal, complement activation can be found, consistent with the local presence of complement regulators C4bp and fH (Oksjoki et al., 2003, 2007). In contrast, terminal complement complex deposition is detected in the deeper layers of the intima, associated with smooth muscle cells, cell debris, and extracellular lipids (Oksjoki et al., 2003, 2007) (Figure 2). In addition, C1q and the receptor for its globular domain (gC1q-R) are found in the necrotic core of advanced atherosclerotic lesions (Peerschke et al., 2004). Also C3b can be detected, with stronger deposition in ruptured compared to non-ruptured plaques of the same patients (Laine et al., 2002). Another sign of increased complement activation within ruptured plaques is the significantly higher C5a concentration associated with lipid-rich inflammatory lesions containing exposed cholesterol and necrotic cell debris than with stable plaques containing collagen- and elastin (Speidl et al., 2011b). Epidemiological data underline this notion and show increased C5a levels in patients with increased cardiovascular disease risk, independent of non-specific inflammatory markers such as C-reactive protein (CRP) or fibrinogen (Speidl et al., 2005). Also elevated C4 levels in the circulation are associated with severe atherosclerosis (Muscari et al., 1988). Still, mere detection of complement activation or deposition does not yet ascertain a deleterious or a protective role for the complement system in artherosclerosis. In fact, various studies underline a pro-atherosclerotic role of complement, while others suggest protective effects for complement. Already in the late 1970s Geertinger et al could show that C6 deficiency, for instance, protects cholesterol-fed rabbits from atherosclerosis (Geertinger and Sorensen, 1970) and inhibition of C5a or its receptor C5aR1 (CD88) reduces atherosclerosis in murine models (Shagdarsuren et al., 2010; Manthey et al., 2011). On the other hand, C1q deficiency leads to the development of significantly larger lesions in atherosclerotic low-density lipoprotein receptor deficient (LDLR–/–) mice compared to C1q-sufficient controls (Bhatia et al., 2007; Lewis et al., 2009). One explanation for this observation may be that C1q binds apoptotic or necrotic cells in plaques directly (or indirectly via IgM), facilitating their removal by macrophages by promoting classical pathway deposition of C3 activation products. Indeed, C3 deficiency promotes the development of larger abdominal and thoracic aorta lesions in atherosclerotic LDLR–/– mice than in C3-sufficient controls. The aortic root lesions of the C3-deficient mice showed increased lipid and macrophage deposition, combined with decreased collagen and smooth muscle cell content, indeed suggesting a net protective effect for C3 in this model (Buono et al., 2002). This notion is underlined by the fact that mice lacking both ApoE and LDLR show a dramatically increased aortic lesions load (+84%) when C3 is absent too (Persson et al., 2004). Given the ambiguous experimental findings regarding the role of complement in the genesis of artherosclerosis, the efficacy of targeting complement in coronary artery disease (CAD) was directly assessed in several clinical trials. Consistent with the protective effects seen in experimental studies interfering with C5 function (Manthey et al., 2011; Shagdarsuren et al., 2010), treatment with anti-C5 antibody (Pexelizumab) resulted in significantly reduced mortality in ST-elevated myocardial infarction (STEMI) patients (Granger et al., 2003). Other trials that examined complement inhibition in patients undergoing coronary artery bypass graft surgery found positive effects on morbidity and mortality (Testa et al., 2011). All in all, a heterogeneous picture emerges regarding the role of complement in atherosclerosis and also open questions remain (Figure 3), which will have to be addressed in future basic and clinical studies to further define the role of complement and its potential and perspective for targeted treatment of atherosclerosis patients.
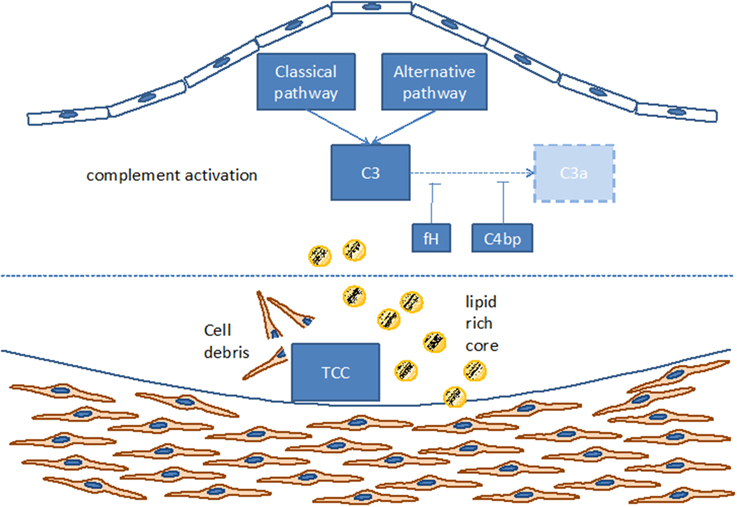
Figure 2. Complement activation within the atherosclerotic plaque. TCC, terminal complement complex; C3, C3a, C4bp, different components of the complement cascade; fH, factor H.
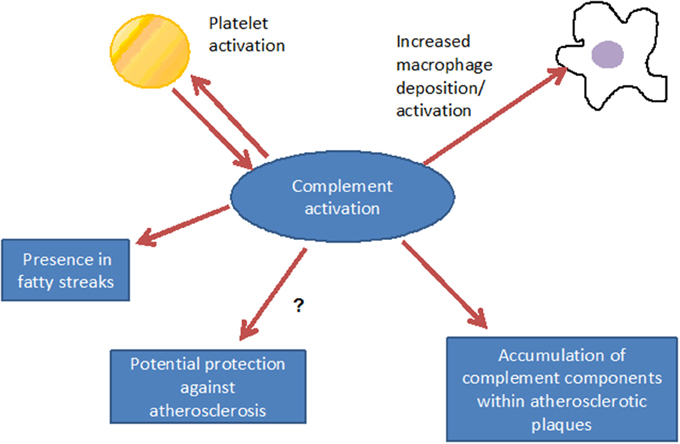
Figure 3. Consequences of complement activation: complement interacts with inflammatory cells, may, however, also have a functional relationship with platelet activation. Via promotion of inflammation, complement contributes to the formation of the fatty streak, under distinct conditions, however, atheroprotective effects can not be excluded.
Platelets and the Complement System
Given the association of platelets and the complement system with the genesis and late complications of atherosclerosis, as well as their intimate contact in the circulation, a closer review of their interactions is warranted. A wide variety of complement factors and receptors have been identified in platelet isolates (Hamad et al., 2010). We recently found that the expression of anaphylatoxin-receptors (C3aR and C5aR) and activation markers (i.e., P-selectin) on platelets correlates with CAD in patients (Patzelt et al., 2014). Others described a potentially self-reinforcing cycle, in which complement can activate platelets and, in turn, thrombin activated platelets are able to initiate the complement cascade (Hamad et al., 2008). In addition, CR4, also a receptor for iC3b, is expressed on platelets but so far with unclear function (Vik and Fearon, 1987). Besides the aforementioned C3-fragment receptors, platelet expressed C1q receptors gC1qR/p33 and cC1qR were shown to mediate platelet aggregating and activating effects (Wautier et al., 1977; Peerschke and Ghebrehiwet, 1997, 2001). Also receptors for anaphylatoxins C3a and C5a, which are generated further downstream in the complement activation cascade, are found on platelets. C3a and its derivative C3a-des-Arg induce platelet activation and aggregation in vitro (Polley and Nachman, 1983; Martel et al., 2011).
The intimate contact between platelets and plasma proteins, including complement, combined with the propensity of platelets to become activated by complement, requires that platelets actively counteract complement deposition onto their surface. To this end, platelets express and bind a variety of complement control proteins (CCPs) on their membrane (Verschoor and Langer, 2013). Indeed, absence or impairment of function of such CCPs is associated with platelet dysfunction, and activation thrombocytopenia, again underlining the potential of the complement system to influence platelet physiology and function (Verschoor and Langer, 2013). In atypical hemolytic uremic syndrome (aHUS), deficiencies or mutations in CCPs, frequently in factor H, may result in excessive complement activation on platelets, leading to thrombocytopenia and prothrombotic complications (Stahl et al., 2008). Another immune mediated pathology in which platelets are affected by overwhelming complement activation is paroxysmal nocturnal hemoglobinuria (PNH). Here, a mutation in the phosphatidylinositol glycan A (PIGA) enzyme prevents the effective anchoring of CCPs decay-accelerating factor (DAF, CD55) and protectin (CD59) to the platelet surface (Nicholson-Weller et al., 1982). With the advent of Eculizumab, a humanized version of the anti-C5 antibody h5G1.1, which was first described in 1996, both conditions can now be effectively treated. Eculizumab is a drug approved for the treatment of PNH. By binding directly to C5, this mAb prevents the cleavage of C5–C5b and, thus, suppresses the formation of the membrane damaging MAC (Thomas et al., 1996).
Finally, platelets may also interact with the complement system via proteins that are not considered classical complement receptors, such as P-selectin (Del Conde et al., 2005) or GP1bα (Verschoor et al., 2011). In the case of P-selectin it was observed that it can bind C3b and mediate the generation of C3a and MAC formation (Del Conde et al., 2005). As exemplified by the pathology and treatment of aHUS and PNH, platelet activation is enhanced by MAC formation causing a prothrombotic state (Sims and Wiedmer, 1991). In the case of GPIbα, bacterial infection studies in mice revealed that, upon systemic infection, C3b-opsonized bacteria form complexes with platelets in the bloodstream. Such complexes can only form in the presence of the alpha chain of GPIb on the platelet surface, strongly suggesting that GPIb directly or indirectly interacts with activated complement C3 (Verschoor et al., 2011). Interestingly, both GPIb and C3 deficient mice show prolonged bleeding times (Strassel et al., 2007; Gushiken et al., 2009), strengthening the notion that these molecules may synergize in physiological hemostatic processes.
Perspective and Outlook
Various clinical and experimental lines of evidence indicate that platelets and the complement system influence the initiation and pathogenesis of atherogenesis and modulate each other's function. While clinical evidence is mounting, most of our mechanistic understanding of atherosclerosis still derives from animal models which model some, but not all characteristics of human atherosclerosis. With non-invasive methods lacking, our ability to directly assess the mechanisms, pathophysiology and plaque burden of human atherogenesis remains limited and indirect parameters (i.e., intima-media thickness, CT-based coronary calcium, mortality) have to be used instead. While such parameters are important in terms of practical patient care, it is often difficult to assess, whether i.e., anti-platelet treatment works by reducing atherosclerotic lesions or by preventing the thrombotic complications of plaque rupture. Thus, such studies unfortunately remain limited in helping us understand the underlying mechanisms. We therefore believe that, in addition, a hypothesis-driven approach is needed to bring seemingly disparate mechanisms in atherosclerotic lesion development together, as we attempt here in this review with complement and platelets.
Several studies show that activated complement is more abundant in unstable plaques than in stable atherosclerotic lesions. Underlining the notion that complement may have long remained an underestimated factor in artherosclerosis is provided by recent clinical studies that show that pexelizumab, a humanized monoclonal antibody against C5, clearly reduced rates of acute major cardiac events after STEMI or coronary bypass surgery (Granger et al., 2003; Verrier et al., 2004). Nonetheless, the drug failed to reduce mortality or recurrent myocardial infarction in the large, randomized controlled APEX AMI trial (Investigators et al., 2007).
Thus, the mixed results highlight once more the complexity of the pathological mechanisms underlying artherosclerosis and the need for intensified clinical and experimental studies probing the interaction of complement and platelets in the context of atherosclerosis. It stresses the need for a comprehensive approach, combining both clinical and experimental research, to uncover and detail further links between platelets and complement with the aim to identify new and promising pharmacological targets in the treatment of atherosclerosis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
AV and HFL are funded by the Tuebingen platelet investigative consortium (TuePIC) funded by the German Research Council (Deutsche Forschungsgemeinschaft (DFG), KFO 274 – Platelets – basic mechanisms and clinical implications). Additionally, AV is funded by Deutsche Forschungsgemeinschaft (SFB 914, project B04) and HFL is supported by the Volkswagen Foundation (Lichtenberg program), German Heart Foundation and Wilhelm Sander Foundation, and the Juniorprofessorenprogramm of the county Baden-Wuerttemberg.
References
Aggarwal, R., Sestak, A. L., D'Sousa, A., Dillon, S. P., Namjou, B., and Scofield, R. H. (2010). Complete complement deficiency in a large cohort of familial systemic lupus erythematosus. Lupus 19, 52–57. doi: 10.1177/0961203309346508
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Best, L. G., Davidson, M., North, K. E., MacCluer, J. W., Zhang, Y., Lee, E. T., et al. (2004). Prospective analysis of mannose-binding lectin genotypes and coronary artery disease in American Indians: the strong heart study. Circulation 109, 471–475. doi: 10.1161/01.CIR.0000109757.95461.10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bhatia, V. K., Yun, S., Leung, V., Grimsditch, D. C., Benson, G. M., Botto, M. B., et al. (2007). Complement C1q reduces early atherosclerosis in low-density lipoprotein receptor-deficient mice. Am. J. Pathol. 170, 416–426. doi: 10.2353/ajpath.2007.060406
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bombeli, T., Schwartz, B. R., and Harlan, J. M. (1998). Adhesion of activated platelets to endothelial cells: evidence for a GPIIbIIIa-dependent bridging mechanism and novel roles for endothelial intercellular adhesion molecule 1 (ICAM-1), alphavbeta3 integrin, and GPIbalpha. J. Exp. Med. 187, 329–339. doi: 10.1084/jem.187.3.329
Buono, C., Come, C. E., Witztum, J. L., Maguire, G. F., Connelly, P. W., Carroll, M., et al. (2002). Influence of C3 deficiency on atherosclerosis. Circulation 105, 3025–3031. doi: 10.1161/01.CIR.0000019584.04929.83
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Burger, P. C., and Wagner, D. D. (2003). Platelet P-selectin facilitates atherosclerotic lesion development. Blood 101, 2661–2666. doi: 10.1182/blood-2002-07-2209
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cipollone, F., Mezzetti, A., Porreca, E., Di Febbo, C., Nutini, M., Fazia, M., et al. (2002). Association between enhanced soluble CD40L and prothrombotic state in hypercholesterolemia: effects of statin therapy. Circulation 106, 399–402. doi: 10.1161/01.CIR.0000025419.95769.F0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Del Conde, I., Cruz, M. A., Zhang, H., Lopez, J. A., and Afshar-Kharghan, V. (2005). Platelet activation leads to activation and propagation of the complement system. J. Exp. Med. 201, 871–879. doi: 10.1084/jem.20041497
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dole, V. S., Bergmeier, W., Patten, I. S., Hirahashi, J., Mayadas, T. N., and Wagner, D. D. (2007). PSGL-1 regulates platelet P-selectin-mediated endothelial activation and shedding of P-selectin from activated platelets. Thromb. Haemost. 98, 806–812.
Ferroni, P., Basili, S., Santilli, F., and Davi, G. (2006). Low-density lipoprotein-lowering medication and platelet function. Pathophysiol. Haemost. Thromb. 35, 346–354. doi: 10.1159/000093226
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Frenette, P. S., Denis, C. V., Weiss, L., Jurk, K., Subbarao, S., Kehrel, B., et al. (2000). P-Selectin glycoprotein ligand 1 (PSGL-1) is expressed on platelets and can mediate platelet-endothelial interactions in vivo. J. Exp. Med. 191, 1413–1422. doi: 10.1084/jem.191.8.1413
Gadjeva, M., Verschoor, A., Brockman, M. A., Jezak, H., Shen, L. M., Knipe, D. M., et al. (2002). Macrophage-derived complement component C4 can restore humoral immunity in C4-deficient mice. J. Immunol. 169, 5489–5495. doi: 10.4049/jimmunol.169.10.5489
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gawaz, M., Langer, H., and May, A. E. (2005). Platelets in inflammation and atherogenesis. J. Clin. Invest. 115, 3378–3384. doi: 10.1172/JCI27196
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gawaz, M., Neumann, F. J., Dickfeld, T., Reininger, A., Adelsberger, H., Gebhardt, A., et al. (1997). Vitronectin receptor (alpha(v)beta3) mediates platelet adhesion to the luminal aspect of endothelial cells: implications for reperfusion in acute myocardial infarction. Circulation 96, 1809–1818. doi: 10.1161/01.CIR.96.6.1809
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gawaz, M., Stellos, K., and Langer, H. F. (2008). Platelets modulate atherogenesis and progression of atherosclerotic plaques via interaction with progenitor and dendritic cells. J. Thromb. Haemost. 6, 235–242. doi: 10.1111/j.1538-7836.2007.02867.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Geertinger, P., and Sorensen, H. (1970). Complement as a factor in arteriosclerosis. Acta Pathol. Microbiol. Scand. A 78, 284–288.
Granger, C. B., Mahaffey, K. W., Weaver, W. D., Theroux, P., Hochman, J. S., Filloon, T. G., et al. (2003). Pexelizumab, an anti-C5 complement antibody, as adjunctive therapy to primary percutaneous coronary intervention in acute myocardial infarction: the COMplement inhibition in Myocardial infarction treated with Angioplasty (COMMA) trial. Circulation 108, 1184–1190. doi: 10.1161/01.CIR.0000087447.12918.85
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gruner, S., Prostredna, M., Schulte, V., Krieg, T., Eckes, B., Brakebusch, C., et al. (2003). Multiple integrin-ligand interactions synergize in shear-resistant platelet adhesion at sites of arterial injury in vivo. Blood 102, 4021–4027. doi: 10.1182/blood-2003-05-1391
Gushiken, F. C., Han, H., Li, J., Rumbaut, R. E., and Afshar-Kharghan, V. (2009). Abnormal platelet function in C3-deficient mice. J. Thromb. Haemost. 7, 865–870. doi: 10.1111/j.1538-7836.2009.03334.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hamad, O. A., Ekdahl, K. N., Nilsson, P. H., Andersson, J., Magotti, P., Lambris, J. D., et al. (2008). Complement activation triggered by chondroitin sulfate released by thrombin receptor-activated platelets. J. Thromb. Haemost. 6, 1413–1421. doi: 10.1111/j.1538-7836.2008.03034.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hamad, O. A., Nilsson, P. H., Wouters, D., Lambris, J. D., Ekdahl, K. N., and Nilsson, B. (2010). Complement component C3 binds to activated normal platelets without preceding proteolytic activation and promotes binding to complement receptor 1. J. Immunol. 184, 2686–2692. doi: 10.4049/jimmunol.0902810
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hegele, R. A., Ban, M. R., Anderson, C. M., and Spence, J. D. (2000). Infection-susceptibility alleles of mannose-binding lectin are associated with increased carotid plaque area. J. Investig. Med. 48, 198–202.
Huo, Y., Schober, A., Forlow, S. B., Smith, D. F., Hyman, M. C., Jung, S., et al. (2003). Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat. Med. 9, 61–67. doi: 10.1038/nm810
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Inoue, O., Suzuki-Inoue, K., Dean, W. L., Frampton, J., and Watson, S. P. (2003). Integrin alpha2beta1 mediates outside-in regulation of platelet spreading on collagen through activation of Src kinases and PLCgamma2. J. Cell Biol. 160, 769–780. doi: 10.1083/jcb.200208043
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Investigators, A. A., Armstrong, P. W., Granger, C. B., Adams, P. X., Hamm, C., Holmes, D., et al. (2007). Pexelizumab for acute ST-elevation myocardial infarction in patients undergoing primary percutaneous coronary intervention: a randomized controlled trial. JAMA 297, 43–51. doi: 10.1001/jama.297.1.43
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jonsson, G., Truedsson, L., Sturfelt, G., Oxelius, V. A., Braconier, J. H., and Sjoholm, A. G. (2005). Hereditary C2 deficiency in Sweden: frequent occurrence of invasive infection, atherosclerosis, and rheumatic disease. Medicine (Baltimore). 84, 23–34. doi: 10.1097/01.md.0000152371.22747.1e
Karshovska, E., Zhao, Z., Blanchet, X., Schmitt, M. M., Bidzhekov, K., Soehnlein, O., et al. (2015). Hyperreactivity of junctional adhesion molecule a-deficient platelets accelerates atherosclerosis in hyperlipidemic mice. Circ. Res. 116, 587–599. doi: 10.1161/CIRCRESAHA.116.304035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Koyama, H., Maeno, T., Fukumoto, S., Shoji, T., Yamane, T., Yokoyama, H., et al. (2003). Platelet P-selectin expression is associated with atherosclerotic wall thickness in carotid artery in humans. Circulation 108, 524–529. doi: 10.1161/01.CIR.0000081765.88440.51
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Laine, P., Pentikainen, M. O., Wurzner, R., Penttila, A., Paavonen, T., Meri, S., et al. (2002). Evidence for complement activation in ruptured coronary plaques in acute myocardial infarction. Am. J. Cardiol. 90, 404–408. doi: 10.1016/S0002-9149(02)02498-0
Langer, H. F., and Chavakis, T. (2009). Leukocyte-endothelial interactions in inflammation. J. Cell. Mol. Med. 13, 1211–1220. doi: 10.1111/j.1582-4934.2009.00811.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Langer, H. F., and Gawaz, M. (2008). Platelet-vessel wall interactions in atherosclerotic disease. Thromb. Haemost. 99, 480–486. doi: 10.1160/TH07-11-0685
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Langer, H. F., Choi, E. Y., Zhou, H., Schleicher, R., Chung, K. J., Tang, Z., et al. (2012). Platelets contribute to the pathogenesis of experimental autoimmune encephalomyelitis. Circ. Res. 110, 1202–1210. doi: 10.1161/CIRCRESAHA.111.256370
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Langer, H. F., Daub, K., Braun, G., Schonberger, T., May, A. E., Schaller, M., et al. (2007). Platelets recruit human dendritic cells via Mac-1/JAM-C interaction and modulate dendritic cell function in vitro. Arterioscler. Thromb. Vasc. Biol. 27, 1463–1470. doi: 10.1161/ATVBAHA.107.141515
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lewis, M. J., Malik, T. H., Ehrenstein, M. R., Boyle, J. J., Botto, M., and Haskard, D. O. (2009). Immunoglobulin M is required for protection against atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation 120, 417–426. doi: 10.1161/CIRCULATIONAHA.109.868158
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ley, K., Laudanna, C., Cybulsky, M. I., and Nourshargh, S. (2007). Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat. Rev. Immunol. 7, 678–689. doi: 10.1038/nri2156
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, K., Sacks, S. H., and Zhou, W. (2007). The relative importance of local and systemic complement production in ischaemia, transplantation and other pathologies. Mol. Immunol. 44, 3866–3874. doi: 10.1016/j.molimm.2007.06.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Madsen, H. O., Videm, V., Svejgaard, A., Svennevig, J. L., and Garred, P. (1998). Association of mannose-binding-lectin deficiency with severe atherosclerosis. Lancet 352, 959–960. doi: 10.1016/S0140-6736(05)61513-9
Manthey, H. D., Thomas, A. C., Shiels, I. A., Zernecke, A., Woodruff, T. M., Rolfe, B., et al. (2011). Complement C5a inhibition reduces atherosclerosis in ApoE-/- mice. FASEB J. 25, 2447–2455. doi: 10.1096/fj.10-174284
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martel, C., Cointe, S., Maurice, P., Matar, S., Ghitescu, M., Theroux, P., et al. (2011). Requirements for membrane attack complex formation and anaphylatoxins binding to collagen-activated platelets. PLoS ONE 6:e18812. doi: 10.1371/journal.pone.0018812
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Massberg, S., Brand, K., Gruner, S., Page, S., Muller, E., Muller, I., et al. (2002). A critical role of platelet adhesion in the initiation of atherosclerotic lesion formation. J. Exp. Med. 196, 887–896. doi: 10.1084/jem.20012044
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Massberg, S., Gawaz, M., Gruner, S., Schulte, V., Konrad, I., Zohlnhofer, D., et al. (2003). A crucial role of glycoprotein VI for platelet recruitment to the injured arterial wall in vivo. J. Exp. Med. 197, 41–49. doi: 10.1084/jem.20020945
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Muscari, A., Bozzoli, C., Gerratana, C., Zaca, F., Rovinetti, C., Zauli, D., et al. (1988). Association of serum IgA and C4 with severe atherosclerosis. Atherosclerosis 74, 179–186. doi: 10.1016/0021-9150(88)90204-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nicholson-Weller, A., Burge, J., Fearon, D. T., Weller, P. F., and Austen, K. F. (1982). Isolation of a human erythrocyte membrane glycoprotein with decay-accelerating activity for C3 convertases of the complement system. J. Immunol. 129, 184–189.
Niculescu, F., and Rus, H. (2004). The role of complement activation in atherosclerosis. Immunol. Res. 30, 73–80. doi: 10.1385/IR:30:1:073
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Niculescu, F., Rus, H. G., and Vlaicu, R. (1987). Immunohistochemical localization of C5b-9, S-protein, C3d and apolipoprotein B in human arterial tissues with atherosclerosis. Atherosclerosis 65, 1–11. doi: 10.1016/0021-9150(87)90002-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Niculescu, F., Rus, H., Cristea, A., and Vlaicu, R. (1985). Localization of the terminal C5b-9 complement complex in the human aortic atherosclerotic wall. Immunol. Lett. 10, 109–114. doi: 10.1016/0165-2478(85)90185-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nityanand, S., Truedsson, L., Mustafa, A., Bergmark, C., and Lefvert, A. K. (1999). Circulating immune complexes and complement C4 null alleles in patients in patients operated on for premature atherosclerotic peripheral vascular disease. J. Clin. Immunol. 19, 406–413. doi: 10.1023/A:1020506901117
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Oksjoki, R., Jarva, H., Kovanen, P. T., Laine, P., Meri, S., and Pentikainen, M. O. (2003). Association between complement factor H and proteoglycans in early human coronary atherosclerotic lesions: implications for local regulation of complement activation. Arterioscler. Thromb. Vasc. Biol. 23, 630–636. doi: 10.1161/01.ATV.0000057808.91263.A4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Oksjoki, R., Kovanen, P. T., Mayranpaa, M. I., Laine, P., Blom, A. M., Meri, S., et al. (2007). Complement regulation in human atherosclerotic coronary lesions. Immunohistochemical evidence that C4b-binding protein negatively regulates the classical complement pathway, and that C5b-9 is formed via the alternative complement pathway. Atherosclerosis 192, 40–48. doi: 10.1016/j.atherosclerosis.2006.06.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Patzelt, J., and Langer, H. F. (2012). Platelets in angiogenesis. Curr. Vasc. Pharmacol. 10, 570–577. doi: 10.2174/157016112801784648
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Patzelt, J., Mueller, K. A., Breuning, S., Karathanos, A., Schleicher, R., Seizer, P., et al. (2014). Expression of anaphylatoxin receptors on platelets in patients with coronary heart disease. Atherosclerosis 238, 289–295. doi: 10.1016/j.atherosclerosis.2014.12.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Peerschke, E. I., and Ghebrehiwet, B. (1997). C1q augments platelet activation in response to aggregated Ig. J. Immunol. 159, 5594–5598.
Peerschke, E. I., and Ghebrehiwet, B. (2001). Human blood platelet gC1qR/p33. Immunol. Rev. 180, 56–64. doi: 10.1034/j.1600-065X.2001.1800105.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Peerschke, E. I., Minta, J. O., Zhou, S. Z., Bini, A., Gotlieb, A., Colman, R. W., et al. (2004). Expression of gC1q-R/p33 and its major ligands in human atherosclerotic lesions. Mol. Immunol. 41, 759–766. doi: 10.1016/j.molimm.2004.04.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Persson, L., Boren, J., Robertson, A. K., Wallenius, V., Hansson, G. K., and Pekna, M. (2004). Lack of complement factor C3, but not factor B, increases hyperlipidemia and atherosclerosis in apolipoprotein E-/- low-density lipoprotein receptor-/- mice. Arterioscler. Thromb. Vasc. Biol. 24, 1062–1067. doi: 10.1161/01.ATV.0000127302.24266.40
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Polley, M. J., and Nachman, R. L. (1983). Human platelet activation by C3a and C3a des-arg. J. Exp. Med. 158, 603–615. doi: 10.1084/jem.158.2.603
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ricklin, D., Hajishengallis, G., Yang, K., and Lambris, J. D. (2010). Complement: a key system for immune surveillance and homeostasis. Nat. Immunol. 11, 785–797. doi: 10.1038/ni.1923
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ross, R. (1999). Atherosclerosis–an inflammatory disease. N. Engl. J. Med. 340, 115–126. doi: 10.1056/NEJM199901143400207
Santoso, S., Sachs, U. J., Kroll, H., Linder, M., Ruf, A., Preissner, K. T., et al. (2002). The junctional adhesion molecule 3 (JAM-3) on human platelets is a counterreceptor for the leukocyte integrin Mac-1. J. Exp. Med. 196, 679–691. doi: 10.1084/jem.20020267
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schober, A., Manka, D., von Hundelshausen, P., Huo, Y., Hanrath, P., Sarembock, I. J., et al. (2002). Deposition of platelet RANTES triggering monocyte recruitment requires P-selectin and is involved in neointima formation after arterial injury. Circulation 106, 1523–1529. doi: 10.1161/01.CIR.0000028590.02477.6F
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schulz, C., Schafer, A., Stolla, M., Kerstan, S., Lorenz, M., von Bruhl, M. L., et al. (2007). Chemokine fractalkine mediates leukocyte recruitment to inflammatory endothelial cells in flowing whole blood: a critical role for P-selectin expressed on activated platelets. Circulation 116, 764–773. doi: 10.1161/CIRCULATIONAHA.107.695189
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Seifert, P. S., Hugo, F., Hansson, G. K., and Bhakdi, S. (1989). Prelesional complement activation in experimental atherosclerosis. Terminal C5b-9 complement deposition coincides with cholesterol accumulation in the aortic intima of hypercholesterolemic rabbits. Lab. Invest. 60, 747–754.
Shagdarsuren, E., Bidzhekov, K., Mause, S. F., Simsekyilmaz, S., Polakowski, T., Hawlisch, H., et al. (2010). C5a receptor targeting in neointima formation after arterial injury in atherosclerosis-prone mice. Circulation 122, 1026–1036. doi: 10.1161/CIRCULATIONAHA.110.954370
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shpilberg, O., Rabi, I., Schiller, K., Walden, R., Harats, D., Tyrrell, K. S., et al. (2002). Patients with Glanzmann thrombasthenia lacking platelet glycoprotein alpha(IIb)beta(3) (GPIIb/IIIa) and alpha(v)beta(3) receptors are not protected from atherosclerosis. Circulation 105, 1044–1048. doi: 10.1161/hc0902.104676
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Siegel-Axel, D., Daub, K., Seizer, P., Lindemann, S., and Gawaz, M. (2008). Platelet lipoprotein interplay: trigger of foam cell formation and driver of atherosclerosis. Cardiovasc. Res. 78, 8–17. doi: 10.1093/cvr/cvn015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sims, P. J., and Wiedmer, T. (1991). The response of human platelets to activated components of the complement system. Immunol. Today 12, 338–342. doi: 10.1016/0167-5699(91)90012-I
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Speidl, W. S., Exner, M., Amighi, J., Kastl, S. P., Zorn, G., Maurer, G., et al. (2005). Complement component C5a predicts future cardiovascular events in patients with advanced atherosclerosis. Eur. Heart J. 26, 2294–2299. doi: 10.1093/eurheartj/ehi339
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Speidl, W. S., Kastl, S. P., Huber, K., and Wojta, J. (2011a). Complement in atherosclerosis: friend or foe? J. Thromb. Haemost. 9, 428–440. doi: 10.1111/j.1538-7836.2010.04172.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Speidl, W. S., Kastl, S. P., Hutter, R., Katsaros, K. M., Kaun, C., Bauriedel, G., et al. (2011b). The complement component C5a is present in human coronary lesions in vivo and induces the expression of MMP-1 and MMP-9 in human macrophages in vitro. FASEB J. 25, 35–44. doi: 10.1096/fj.10-156083
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stahl, A. L., Vaziri-Sani, F., Heinen, S., Kristoffersson, A. C., Gydell, K. H., Raafat, R., et al. (2008). Factor H dysfunction in patients with atypical hemolytic uremic syndrome contributes to complement deposition on platelets and their activation. Blood 111, 5307–5315. doi: 10.1182/blood-2007-08-106153
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stellos, K., Sauter, R., Fahrleitner, M., Grimm, J., Stakos, D., Emschermann, F., et al. (2012). Binding of oxidized low-density lipoprotein on circulating platelets is increased in patients with acute coronary syndromes and induces platelet adhesion to vascular wall in vivo–brief report. Arterioscler. Thromb. Vasc. Biol. 32, 2017–2020. doi: 10.1161/ATVBAHA.111.244707
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Strassel, C., Hechler, B., Bull, A., Gachet, C., and Lanza, F. (2009). Studies of mice lacking the GPIb-V-IX complex question the role of this receptor in atherosclerosis. J. Thromb. Haemost. 7, 1935–1938. doi: 10.1111/j.1538-7836.2009.03608.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Strassel, C., Nonne, C., Eckly, A., David, T., Leon, C., Freund, M., et al. (2007). Decreased thrombotic tendency in mouse models of the Bernard-Soulier syndrome. Arterioscler. Thromb. Vasc. Biol. 27, 241–247. doi: 10.1161/01.ATV.0000251992.47053.75
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Testa, L., Meco, M., Cirri, S., and Bedogni, F. (2011). Pexelizumab and survival in cardiac surgery. HSR Proc. Intensive Care Cardiovasc. Anesth. 3, 23–24.
Thomas, T. C., Rollins, S. A., Rother, R. P., Giannoni, M. A., Hartman, S. L., Elliott, E. A., et al. (1996). Inhibition of complement activity by humanized anti-C5 antibody and single-chain Fv. Mol. Immunol. 33, 1389–1401. doi: 10.1016/S0161-5890(96)00078-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Torzewski, M., Klouche, M., Hock, J., Messner, M., Dorweiler, B., Torzewski, J., et al. (1998). Immunohistochemical demonstration of enzymatically modified human LDL and its colocalization with the terminal complement complex in the early atherosclerotic lesion. Arterioscler. Thromb. Vasc. Biol. 18, 369–378. doi: 10.1161/01.ATV.18.3.369
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Totani, L., and Evangelista, V. (2010). Platelet-leukocyte interactions in cardiovascular disease and beyond. Arterioscler. Thromb. Vasc. Biol. 30, 2357–2361. doi: 10.1161/ATVBAHA.110.207480
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
van der Net, J. B., Oosterveer, D. M., Versmissen, J., Defesche, J. C., Yazdanpanah, M., Aouizerat, B. E., et al. (2008). Replication study of 10 genetic polymorphisms associated with coronary heart disease in a specific high-risk population with familial hypercholesterolemia. Eur. Heart J. 29, 2195–2201. doi: 10.1093/eurheartj/ehn303
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
van Gils, J. M., Zwaginga, J. J., and Hordijk, P. L. (2009). Molecular and functional interactions among monocytes, platelets, and endothelial cells and their relevance for cardiovascular diseases. J. Leukoc. Biol. 85, 195–204. doi: 10.1189/jlb.0708400
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Verrier, E. D., Shernan, S. K., Taylor, K. M., Van de Werf, F., Newman, M. F., Chen, J. C., et al. (2004). Terminal complement blockade with pexelizumab during coronary artery bypass graft surgery requiring cardiopulmonary bypass: a randomized trial. JAMA 291, 2319–2327. doi: 10.1001/jama.291.19.2319
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Verschoor, A., Brockman, M. A., Gadjeva, M., Knipe, D. M., and Carroll, M. C. (2003). Myeloid C3 determines induction of humoral responses to peripheral herpes simplex virus infection. J. Immunol. 171, 5363–5371. doi: 10.4049/jimmunol.171.10.5363
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Verschoor, A., Brockman, M. A., Knipe, D. M., and Carroll, M. C. (2001). Cutting edge: myeloid complement C3 enhances the humoral response to peripheral viral infection. J. Immunol. 167, 2446–2451. doi: 10.4049/jimmunol.167.5.2446
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Verschoor, A., and Carroll, M. C. (2004). “Complement and its receptors in infection,” in The Innate Immune Response to Infection, eds S. H. E. Kaufmann, R. Medzhitov and S. Gordon (Washington, DC: American Society for Microbiology Press), 219–240. doi: 10.1128/9781555817671.ch11
Verschoor, A., and Langer, H. F. (2013). Crosstalk between platelets and the complement system in immune protection and disease. Thromb. Haemost. 110, 910–919. doi: 10.1160/TH13-02-0102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Verschoor, A., Neuenhahn, M., Navarini, A. A., Graef, P., Plaumann, A., Seidlmeier, A., et al. (2011). A platelet-mediated system for shuttling blood-borne bacteria to CD8alpha+ dendritic cells depends on glycoprotein GPIb and complement C3. Nat. Immunol. 12, 1194–1201. doi: 10.1038/ni.2140
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vik, D. P., and Fearon, D. T. (1987). Cellular distribution of complement receptor type 4 (CR4): expression on human platelets. J. Immunol. 138, 254–258.
Vlaicu, R., Rus, H. G., Niculescu, F., and Cristea, A. (1985). Quantitative determinations of immunoglobulins and complement components in human aortic atherosclerotic wall. Med. Interne 23, 29–35.
von Hundelshausen, P., and Schmitt, M. M. (2014). Platelets and their chemokines in atherosclerosis-clinical applications. Front. Physiol. 5:294. doi: 10.3389/fphys.2014.00294
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
von Hundelshausen, P., and Weber, C. (2007). Platelets as immune cells: bridging inflammation and cardiovascular disease. Circ. Res. 100, 27–40. doi: 10.1161/01.RES.0000252802.25497.b7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
von Hundelshausen, P., Koenen, R. R., and Weber, C. (2009). Platelet-mediated enhancement of leukocyte adhesion. Microcirculation 16, 84–96. doi: 10.1080/10739680802564787
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wagner, D. D., and Frenette, P. S. (2008). The vessel wall and its interactions. Blood 111, 5271–5281. doi: 10.1182/blood-2008-01-078204
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Walport, M. J. (2001). Complement. First of two parts. N. Engl. J. Med. 344, 1058–1066. doi: 10.1056/NEJM200104053441406
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wautier, J. L., Souchon, H., Reid, K. B., Peltier, A. P., and Caen, J. P. (1977). Studies on the mode of reaction of the first component of complement with platelets: interaction between the collagen-like portion of C1q and platelets. Immunochemistry 14, 763–766. doi: 10.1016/0019-2791(77)90351-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weber, C., and Springer, T. A. (1997). Neutrophil accumulation on activated, surface-adherent platelets in flow is mediated by interaction of Mac-1 with fibrinogen bound to alphaIIbbeta3 and stimulated by platelet-activating factor. J. Clin. Invest. 100, 2085–2093. doi: 10.1172/JCI119742
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weber, K. S., Alon, R., and Klickstein, L. B. (2004). Sialylation of ICAM-2 on platelets impairs adhesion of leukocytes via LFA-1 and DC-SIGN. Inflammation 28, 177–188. doi: 10.1023/B:IFLA.0000049042.73926.eb
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yasojima, K., Schwab, C., McGeer, E. G., and McGeer, P. L. (2001). Complement components, but not complement inhibitors, are upregulated in atherosclerotic plaques. Arterioscler. Thromb. Vasc. Biol. 21, 1214–1219. doi: 10.1161/hq0701.092160
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: atherosclerosis, platelets, complement system proteins, innate immunity, inflammation
Citation: Patzelt J, Verschoor A and Langer HF (2015) Platelets and the complement cascade in atherosclerosis. Front. Physiol. 6:49. doi: 10.3389/fphys.2015.00049
Received: 18 November 2014; Accepted: 04 February 2015;
Published online: 02 March 2015.
Edited by:
Christian A. Gleissner, University of Heidelberg, GermanyReviewed by:
Klaus Ley, La Jolla Institute for Allergy and Immunology, USAIrena Levitan, University of Illinois at Chicago, USA
Copyright © 2015 Patzelt, Verschoor and Langer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Admar Verschoor, Institute for Medical Microbiology, Immunology and Hygiene, Technische Universität München, Trogerstr. 30, 81675 Munich, Germany e-mail:YWRtYXIudmVyc2Nob29yQHR1bS5kZQ==;
Harald F. Langer, Medizinische Klinik III, Kardiologie und Kreislauferkrankungen, Eberhard Karls-Universität Tübingen, Otfried-Müllerstr. 10, 72076 Tübingen, Germany e-mail:aGFyYWxkLmxhbmdlckBtZWQudW5pLXR1ZWJpbmdlbi5kZQ==