 Joeri Lambrecht
Joeri Lambrecht Inge Mannaerts
Inge Mannaerts Leo A. van Grunsven
Leo A. van Grunsven- Liver Cell Biology Lab, Department of Biomedical Sciences, Vrije Universiteit Brussel, Brussels, Belgium
The progression of liver fibrosis and cirrhosis is associated with the persistence of an injury causing agent, leading to changes in the extracellular environment and a disruption of the cellular homeostasis of liver resident cells. Recruitment of inflammatory cells, apoptosis of hepatocytes, and changes in liver microvasculature are some examples of changing cellular environment that lead to the induction of stress responses in nearby cells. During liver fibrosis, the major stresses include hypoxia, oxidative stress, and endoplasmic reticulum stress. When hepatic stellate cells (HSCs) are subjected to such stress, they modulate fibrosis progression by induction of their activation toward a myofibroblastic phenotype, or by undergoing apoptosis, and thus helping fibrosis resolution. It is widely accepted that microRNAs are import regulators of gene expression, both during normal cellular homeostasis, as well as in pathologic conditions. MicroRNAs are short RNA sequences that regulate the gene expression by mRNA destabilization and inhibition of mRNA translation. Specific microRNAs have been identified to play a role in the activation process of HSCs on the one hand and in stress-responsive pathways on the other hand in other cell types (Table 2). However, so far there are no reports for the involvement of miRNAs in the different stress responses linked to HSC activation. Here, we review briefly the major stress response pathways and propose several miRNAs to be regulated by these stress responsive pathways in activating HSCs, and discuss their potential specific pro-or anti-fibrotic characteristics.
Introduction
Liver fibrosis is the pathological condition of the liver resulting from sustained wound healing in response to chronic liver injury. Multiple factors can lead to such injury, including genetic (the accumulation of misfolded alpha1-antitrypsin), cholestatic (sclerosing cholangitis), metabolic (non-alcoholic fatty liver disease and non-alcoholic steatohepatitis), drug induced (paracetamol-intoxication and alcohol) and viral diseases (hepatitis B and C) (Friedman, 2003; Wallace et al., 2008). Liver fibrosis can eventually progress toward cirrhosis, which is characterized by the loss of endothelial fenestrations, excessive scar formation in the space of Disse, and the presence of vascularized fibrotic septa. These distortions of liver architecture and subsequent cellular homeostasis lead to impaired organ function, ascites, encephalopathy, variceal hemorrhage, portal hypertension and the development of hepatocellular carcinoma (Schuppan and Afdhal, 2008).
Role of miRNAs during Hepatic Stellate Cell Activation
One of the key features in the development of liver fibrosis is the augmenting presence of myofibroblasts in the liver. Myofibroblasts are characterized by their stellate shape, the expression of some specific proteins, such as alpha-smooth muscle actin (α-SMA), and the excessive production of extracellular matrix proteins, including fibronectin and collagen type I, III, and IV. Hepatic stellate cells (HSCs) transdifferentiate upon injury into myofibroblasts, and can be considered as the major origin of myofibroblasts (Mederacke et al., 2013). During initiation and progression of the liver fibrosis process, the liver is subjected to various kinds of stress including hypoxia (Nath and Szabo, 2012), oxidative stress (Parola and Robino, 2001), and endoplasmic reticulum (ER) stress (Li et al., 2015). HSCs will respond by activating into myofibroblasts, which is characterized by a change in gene (Jiang et al., 2006; De Minicis et al., 2007) and microRNA expression (Guo et al., 2009a), as reviewed in He et al. (2012a); Huang et al. (2014a) and Coll et al. (2015). Numerous detailed reports on gene expression changes during HSC activation are available, but information regarding their regulation by specific miRNAs remains rather vague.
MiRNAs are short non-protein coding RNA sequences of 20–23 nucleotides that are evolutionary conserved and are encoded in the genome. The human genome is supposed to encode for approximately 1000 miRNAs, which can be expressed in an ubiquitous or a tissue/cell-type specific way (Lee, 2013), and each of these miRNAs is thought to have a great range of potential targets, thus indicating its importance in gene regulation (Bartel and Chen, 2004). MiRNA-encoding genes are transcribed by RNA polymerase II, with the generation of primary miRNA, which will then be processed in the nucleus by activity of a microprocessor complex, named Drosha. The activity of this Drosha containing complex leads to the production of a hairpin-shaped premature miRNA defined by a length of approximately 70 nucleotides and the presence of a stem-loop structure (Lee et al., 2003; Gregory et al., 2004). Correctly processed premature miRNAs are then bound by Exportin-5 in a Ran guanosine triphosphate (RanGTP)-dependent manner, leading to the transport of these pre-miRNAs toward the cytoplasm (Lund et al., 2004). In the cytoplasm, the pre-miRNAs undergo processing by Dicer, another ribonuclease III enzyme, resulting in the production of double stranded RNA (dsRNA) of 20–23 nucleotides (Bernstein et al., 2001). In this double stranded nucleotide-complex, a mature miRNA strand, known as the guide strand, and a miRNA* strand, known as the passenger strand can be identified. The mature miRNA strand will be loaded into the Argonaute 2 (Ago2)-containing RNA-induced silencing complex (RISC), which is the effector of miRNA-mediated activities (Gregory et al., 2005). It is believed that the RISC complex can cause down-regulation of gene expression through 2 mechanisms; by an inhibition of mRNA translation or by reducing the mRNA stability and thus facilitating the degradation (Figure 1) (Bagga et al., 2005; Orban and Izaurralde, 2005; Pillai et al., 2005).
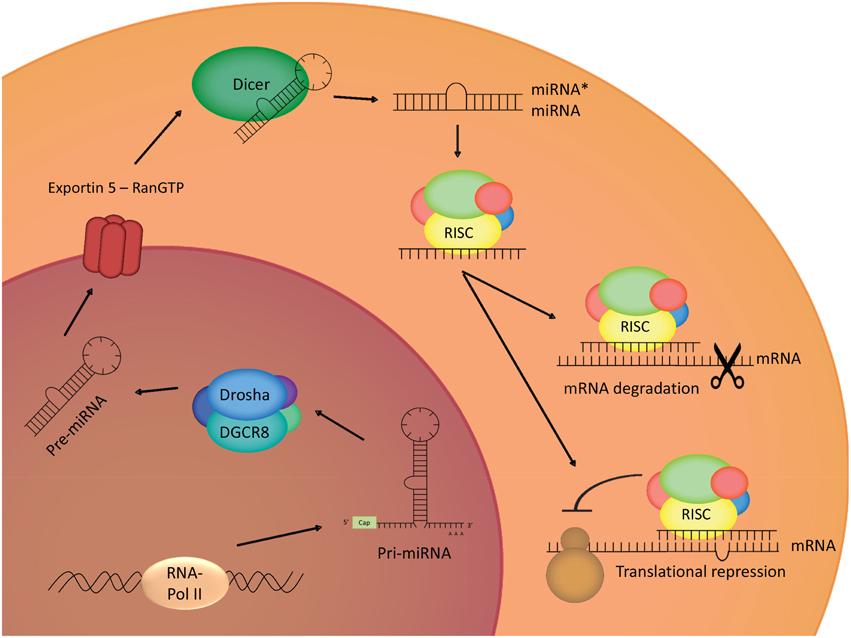
Figure 1. MiRNA biogenesis. Transcription of the genes coding for miRNAs leads to the generation of primary miRNAs, which will be cleaved in the nucleus by Drosha, a ribonuclease III complex. The produced ribonucleic structure is called premature miRNA, and will be transported to the cytoplasm by Exportin 5, where it will undergo cleaving by Dicer, another ribonuclease III enzyme. One strain of the double-stranded obtained structure will integrate in the RISC-complex, leading to translational repression, or degradation of the target mRNA.
Since the discovery of miRNAs in 1993 (Wightman et al., 1993), researchers continuously tried to evoke the role of miRNAs in cellular homeostasis and in development of pathological conditions, including liver fibrosis. There are many miRNAs expressed during, and described to be involved in, HSC activation (Table 1), making them the topic of concise reviews (He et al., 2012a; Huang et al., 2014a). Here, we only briefly highlight some key miRNAs to illustrate the possible roles a miRNA could have in quiescent or activated HSCs. When evaluating these miRNA-studies it is important to keep in mind that although many miRNAs are conserved among eukaryotic organisms, it is possible that they do not display the same expression patterns in specific (pathological) processes, and thus can display interspecies differences in expression (Ha et al., 2008).
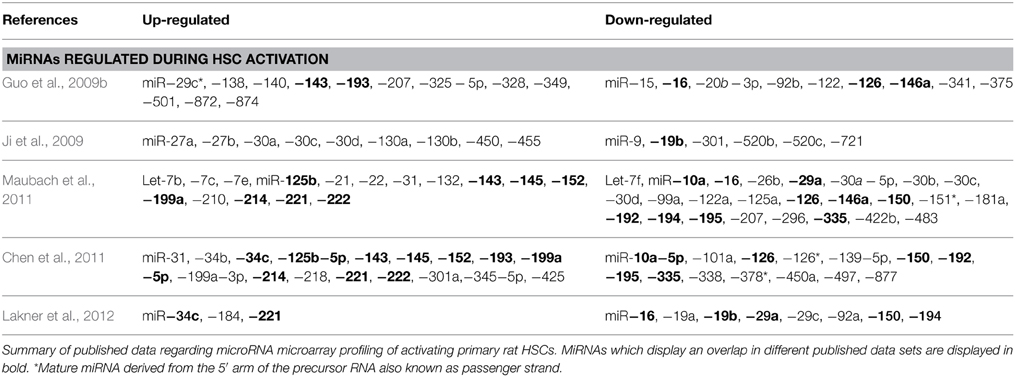
Table 1. Significantly regulated miRNAs during HSC-activation.
miR-29
miR-29 is the first and most thoroughly investigated miRNA-family in HSCs. miR-29a, miR-29b, and miR-29c are all down-regulated during the in vitro activation of isolated rat and mouse HSCs, and in liver biopsies from patients with advanced liver fibrosis. This down-regulation is promoted by transforming growth factor-β (TGF-β) and factors like inflammatory signals including lipopolysaccharide (LPS) and nuclear factor kappa B (NF-κB) (Roderburg et al., 2011). The miR-29 family is of importance for HSC activation, as they can bind to 3′-UTR collagen types I and IV (Kwiecinski et al., 2011). Consequently, miR-29 overexpression in HSCs reduces Collagen I and IV synthesis (Roderburg et al., 2011) and maintenance of the quiescent morphology (Sekiya et al., 2011). In addition to collagen targeting, PDGF-C and IGF-I are identified as targets of miR-29, with PDGF-C having pro-mitogenic and migratory capacities, and IGF-I being an important mitogenic factor when present in an autocrine manner in combination with PDGF-BB (Kwiecinski et al., 2012). In support with these findings, miR-29a/b levels were found to decrease in CCl4-treated male mice. Interestingly, female mice do not show this decrease, most likely due to differences in E2, which can induce miR-29a/b levels (Zhang et al., 2012). Not only collagen production, but also other aspects of HSC activation such as inflammatory response and cell proliferation can be regulated by miRNAs such as is the case for miR-146a and miR-16, respectively.
miR-146
miR-146 is also down-regulated during TGF-β-induced HSC activation (He et al., 2012b), while overexpression of miR-146a in HSCs leads to up-regulation of tissue inhibitor of metalloproteinase 3 (TIMP-3) and down-regulation of IL-6 mRNA (Maubach et al., 2011). In another study, overexpression of miR-146a lead to inhibition of proliferation of activated HSCs. This would be the result of direct binding to the promoter region of the SMAD4 mRNA, which regulates TGF-β1-mediated gene expression, thus leaving the cell insensitive to TGF-β1 stimulation (He et al., 2012b), demonstrating its importance in the inflammatory response, and its link with liver fibrosis. In addition, miR-146a is known to have a role in the inflammatory response during liver reperfusion injury, as it negatively regulates IL-1 receptor-associated kinase 1 (IRAK1) and Toll-like receptor-associated factor 6 (TRAF6), leading to a decrease in pro-inflammatory cytokine production, and by inhibiting the pro-inflammatory NF-κB pathway (Jiang et al., 2014). MiR-126 represents another miRNA that can regulate the NF-κB pathway by suppressing the expression of NF-κB inhibitor alpha (IκBα), thus leading to NF-κB activation (Feng et al., 2015).
MiR-16
miR-16 is another down-regulated miRNA during HSC activation. This miRNA has been shown to inhibit the expression of Cyclin D1, an important regulator of the cell cycle pathway. Expression levels of miR-16 and Cyclin D1 are inversely correlated in activating HSCs. Overexpression of this miRNA in activated HSCs leads to accumulation of the cells in the G0/G1-phase or G0/G1 to S-phase of cell cycle progression (Guo et al., 2009c). In HSCs, miR-16 also acts as an anti-apoptotic regulator in HSCs, by inhibition of B-cell lymphoma 2 (Bcl-2) translation, a known anti-apoptotic gene, leading to the enhanced expression levels of the underlying caspase-pathway consisting of caspases 3, 8, and 9, and thus induction of apoptosis (Guo et al., 2009b).
Function of Stress-Responsive Pathways and Possible Contribution of miRNAs during HSC Activation
As mentioned before, HSCs will undergo an activation process in the presence of different (fibrogenic) stimuli like liver injury, paracrine stimulation and autocrine regulation. This activation changes the quiescent fat storing cells into fibrogenic, proliferative and contractile myofibroblasts characterized by their expression of abundant intracellular filaments like α-SMA and vimentin, secretion of ECM including collagen type I and III and fibronectin and their high contractility (Kisseleva and Brenner, 2013). The contribution of stress response pathways in liver fibrosis, cirrhosis and to the HSC activation is generally accepted (Parola and Robino, 2001; Nath and Szabo, 2012; Li et al., 2015), but cannot be interpreted as a simple cause and consequence reaction. As literature mainly describes the contribution of hypoxia (Nath and Szabo, 2012), oxidative stress (Parola and Robino, 2001), and ER stress (Li et al., 2015) pathways during liver fibrosis and cirrhosis progression (Figure 2), we will focus on these three pathways.
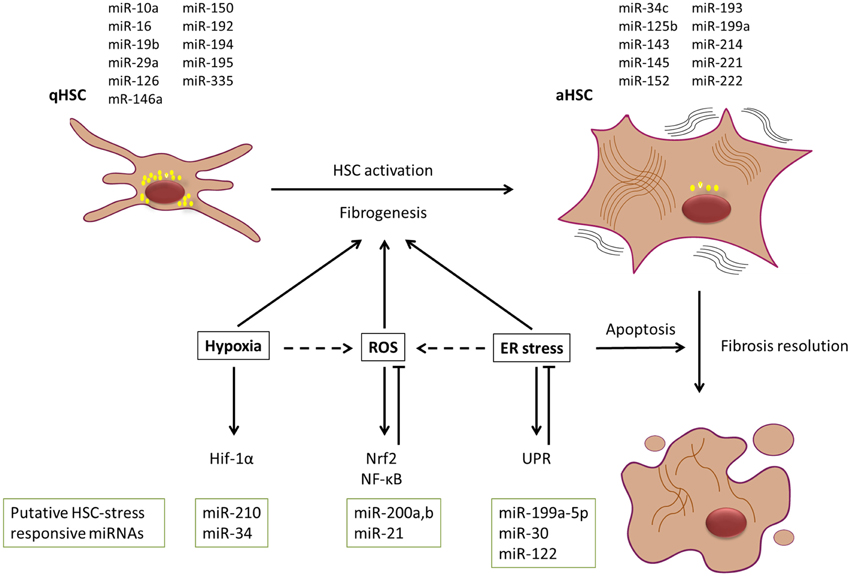
Figure 2. Dynamic contribution of stress stimuli and miRNAs to liver fibrosis progression and resolution. HSCs are major contributors to the myofibroblastic cell pool in the fibrotic liver. In the presence of various activation stimuli, HSCs will undergo a myofibroblastic transdifferentiation process toward an activated state, which is characterized by a change in miRNA and mRNA expression pattern. It is widely accepted that the presence of hypoxia, oxidative stress (ROS), and endoplasmic reticulum (ER) stress most likely supports this activation process. However, ER stress could have a potential dual role in the process, as it can also lead to induction of apoptosis in activated HSCs, and thus could contribute to resolution of fibrosis. Simplified representation of some of the signaling cascades and potential miRNAs involved in these stress responses are given. MiRNAs depicted above the HSCs have been reported to be enriched in either qHSC or aHSCs. Putative HSC-stress responsive miRNAs that are discussed in the text are depicted below the signaling cascades.
Specific stress-related genes can be quickly switched on and off in presence or absence of environmental stress-inducing factors and this can be mediated by miRNAs (Babar et al., 2008; Leung and Sharp, 2010) (Table 2, right panel). So far there are no reports describing the functionality of specific miRNAs in these stress response pathways of activating HSCs during liver fibrosis. However, assumptions about miRNAs forming the link in stress-responsive HSCs (Table 2) and their potential functions in these conditions can be made based on the available data and will be discussed here. We should keep in mind that the presence or lack of overlap in miRNA expression pattern can be due to cell-type and species-specificity and is no proof for actual involvement of the miRNA in stress responsive HSCs, and should be elucidated in future research.
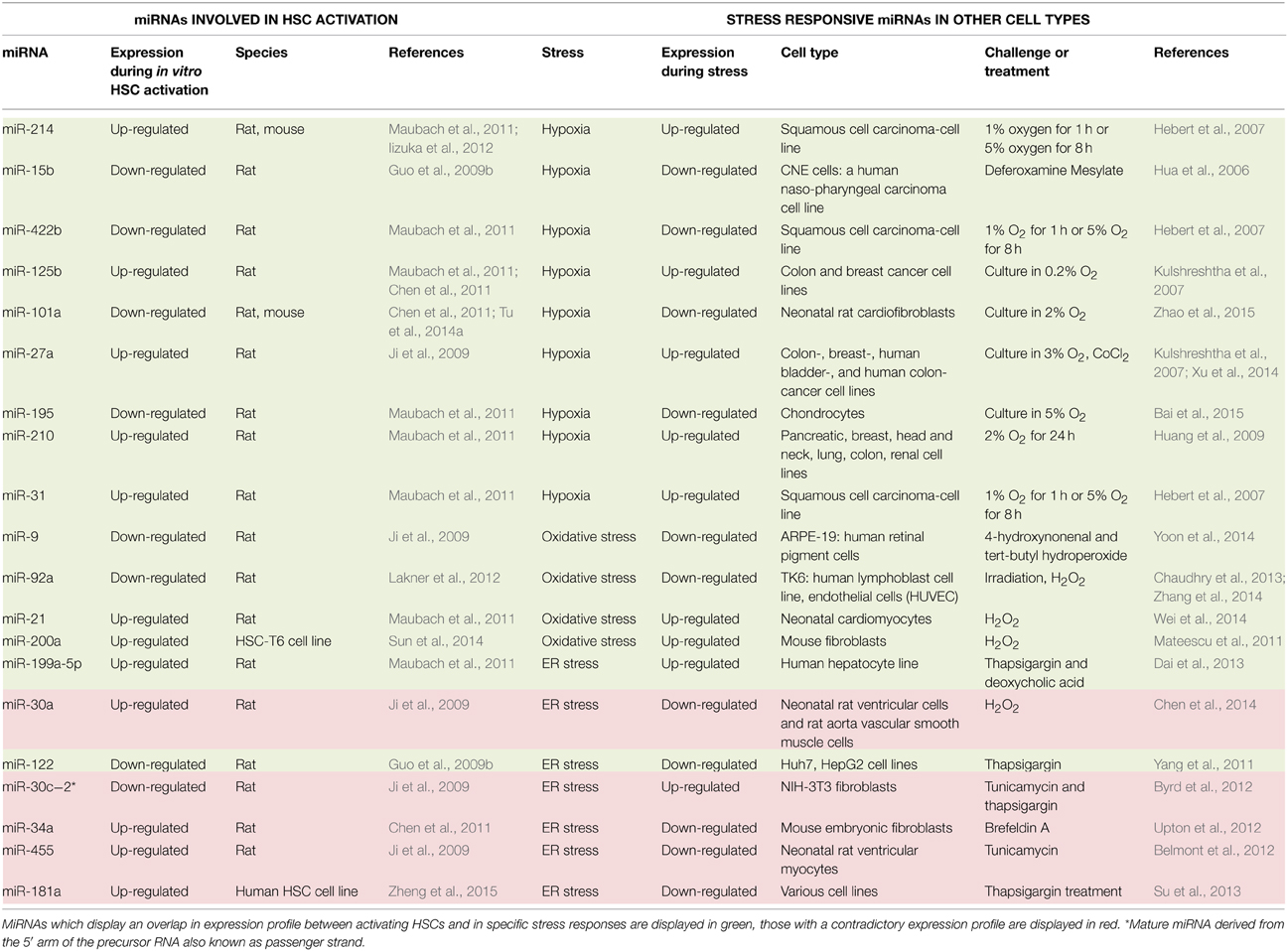
Table 2. Potential miRNAs involved in stress responsive HSC activation.
Hypoxia Regulated miRNAs
In the process of liver fibrosis and cirrhosis, hypoxia in the liver cells can be due to disruption of the normal hepatic blood flow, damage of the microvasculature, and excessive deposition of extracellular matrix in the sinusoidal space (Copple et al., 2006). Cellular hypoxia leads to the activation of several Hypoxia Inducible Factors (HIFs), a family of transcriptional factors that work as key regulators for the maintenance of cellular homeostasis when confronted with low oxygen levels (Paternostro et al., 2010). At normal cellular oxygen levels, the oxygen-dependent hypoxia inducible factor HIF-1α (HIF-1α) is hydroxylated by members of the prolyl hydroxylase family (PHD), leading to the rapid degradation of this protein. Decrease of the cellular oxygen levels leads to loss of function of PHD, and subsequent accumulation and translocation of HIF-1α/HIF-2α to the nucleus. In the nucleus, the functional HIF transcription factor complex is formed consisting of HIF-α, HIF-1β and some hypoxic responsive elements (Semenza, 2007). HIF regulates certain processes such as angiogenesis, iron metabolism, glycolysis, and pH control (Jiang et al., 1996; Rosmorduc et al., 1999; Moon et al., 2009). Hypoxic conditions lead to activation of the HSC cell line LX-2 as illustrated by an up-regulation of α-SMA and collagen I protein levels, possibly through activation of the Smad/TGF-β pathway (Shi et al., 2007). HIF is proposed as a main regulator of hypoxia-mediated HSC activation, since it can act as a regulator and stimulator of profibrogenic mediators such as platelet-derived growth factor (PDGF) A and B, plasminogen activator inhibitor-1, and vascular epithelial growth factor (VEGF) (Forsythe et al., 1996; Moon et al., 2009; Wang et al., 2013). The essential role of HIF-1α during hypoxia-induced HSC activation was confirmed in vitro by inhibition of HSC-activation due to silencing of HIF-1α (Wang et al., 2013), and the reduced expression of activation genes in HIF-1α-deficient HSCs undergoing hypoxia (Copple et al., 2011). In vivo experiments using bile duct ligated (BDL) Hif-1α-deficient and control mice, showed less fibrosis in Hif-1α-deficient mice, as observed by lower levels of α-SMA and type I collagen, thus further indicating its importance during liver fibrosis (Moon et al., 2009).
MiRNAs can act down-stream and up-stream of the HIF pathway. For example, miR-210 expression is directly regulated by HIF-1α as it can bind to the hypoxia responsive element (HRE) located up-stream of the transcription start site of miR-210, leading to its enhanced transcription (Huang et al., 2009). It is suggested, that HIF-2α would mediate miR-210 expression in the absence of HIF-1α, also by interaction with consensus HREs in the miR-210 promoter region (Zhang et al., 2009). MiR-210 effects a broad variety of cellular processes such as fine-tuning cell proliferation by targeting e2f transcription factor 3 (E2f3) (Giannakakis et al., 2008) and MNT, a known MYC antagonist, and a member of the Myc/Max/Mad network (Zhang et al., 2009) while regulating apoptosis by controlling expression of the pro-apoptotic FLICE-associated huge protein (FLASH)/caspase-8-associated protein 2 (Casp8ap2) (Kim et al., 2009). Genes such as Nptx1, Rad52, Acvr1b, Fgrl, Hoxa1, and Hoxa9 associated with pathways like angiogenesis, tumor invasion, regulation of the mitochondrial metabolism, and DNA damage repair were also found to be miR-210 targets (Fasanaro et al., 2009; Huang et al., 2009). The hypoxia-induced up-regulation of miR-210 in various cancer cell lines (Huang et al., 2009) displays an overlap with its enhanced expression during the activation process of HSCs, thus suggesting a potential role of this miRNA in hypoxia-mediated HSC activation.
Another potential link in hypoxia-mediated regulation of HSC activation is presented by miR-31. MiR-31 is up-regulated in both in vivo and in vitro activated rat HSCs (Maubach et al., 2011). This was confirmed in humans, where miR-31 was not changed in whole liver samples of fibrotic livers, but an increased expression of miR-31 was detected in HSCs during fibrogenesis. Functional studies showed repression of HSC activation by miR-31 inhibition, while miR-31 overexpression revealed its promoting role in cell migration (Hu et al., 2015). Interestingly it has been suggested that the biological function of miR-31 in activating HSCs would be obtained through its effect on Factor-inhibiting HIF-1(FIH) (Mahon et al., 2001; Hu et al., 2015). In head and neck carcinoma, miR-31 negatively regulates the expression of FIH and can thus regulate the expression of FIH in a hypoxia-independent manner (Liu et al., 2010). In cancer models this miRNA is up-regulated under hypoxic conditions (Hebert et al., 2007), suggesting a very complicated and diverse functionality of miR-31 during the reach for cellular homeostasis. Previous research identified a direct link between the increased nuclear levels of HIF-1α protein and an increased activated status of HSCs in a hypoxic environment. HIF-1α has an indirect activating effect on the expression of pro-fibrogenic genes such as TGF-β, IL-6 and CTGF (Copple et al., 2011; Wang et al., 2013). The exact role of miR-31 in this hypoxia-induced HSC activation remains to be elucidated. We speculate on two possible scenarios that are perhaps not exclusive. Due to pro-activating signals from surrounding liver cells, HSCs will up-regulate miR-31 expression, leading to inhibition of FIH function, and thus enhanced HIF-1α expression, thereby favoring HSC activation in normoxic conditions. Hypoxic regions appear in the liver due to injury, what could favor the (further) induction of miR-31 expression, boosting the already enhanced HIF-1α expression, further leading to progression or maintenance of HSC activation.
Oxidative Stress Regulated miRNAs
Cells in aerobic organisms have a continuous balance between the production of pro-oxidants, such as reactive oxygen species (ROS), and anti-oxidants. When a cell is subjected to oxidative stress, this normal balance fades by excessive production of pro-oxidants. Various types of ROS are known, such as the singlet molecular oxygen, hydrogen peroxide and the hydrogen radical, which all have a specific half-life and mechanism of action (Sies, 1991).
There are several possible sources of ROS in the cell. Mitochondria, the main site of oxygen consumption in aerobic cells, are the main producers of ROS derived mainly through the leakage of electrons and formation of superoxide (Guarente, 2008). Cytochrome P450 (CYP) acts in the detoxification of metabolic as well as xenobiotic compounds by means of oxidation (Aubert et al., 2011) making it also an important source of ROS. Specifically the form CYP2E1, which is highly expressed in hepatocytes, has been demonstrated to be a key source of ROS in the liver (Poli, 2000). Another major source of ROS in several cell types and HSCs is nicotinamide adenine dinucleotide phosphate-oxidase (NADPH oxidase) (De Minicis and Brenner, 2007; Sergey, 2011).
Oxidative stress and the subsequent decreased levels of anti-oxidants during liver fibrosis has been shown for a broad variety of etiologies (Poli, 2000). ROS are produced by various cell types, but it is thought that the major contributors of ROS production in this pathology are apoptotic hepatocytes. HSCs express a non-phagocytic form of NADPH oxidase, which presents a basal level of activity, producing constitutively low levels of ROS and increasing production upon different stimuli (Bataller et al., 2003). NADPH oxidase of HSCs is activated upon phagocytosis of these apoptotic bodies of hepatocytes (Shan-Shan et al., 2006). Furthermore, NADPH oxidase-generated ROS in HSCs is also induced by advanced glycation end-products (AGEs) which are products of a non-enzymatic reaction of sugars with molecules such as proteins, lipids and nucleic acids that accumulate in diseases related to the metabolic syndrome (Yan et al., 2010). Liver fibrosis is correlated with accumulation of systemic AGEs and ROS in HSCs has been show to participate during the development of liver diseases (Šebeková et al., 2002; Hyogo et al., 2007; Guimarães et al., 2010).
Activated Kupffer cells and neutrophils are also described as important producers of ROS during early stages of liver fibrosis (Kisseleva and Brenner, 2007). The most important result of oxidative stress is lipid peroxidation. As example, liver fibrosis caused by excessive alcohol intake leads to injury of the different liver cell types and consecutive excessive oxidation of polyunsaturated membrane lipids due to enhanced generation of ROS due to the elevated levels of cytochrome CYP2E1 (Nieto et al., 1999). The products of such lipid peroxidation could further catalyze the progression of fibrosis by activation of the production of collagen α2 (I) in HSCs in a paracrine manner (Bedossa et al., 1994). Furthermore, exposure of HSCs to ROS can promote their proliferation and invasiveness. It is thought that it would obtain these effects by an induction of MMP-2 expression, and the enhancement of MT1-MMP and TIMP-2 protein levels, in an ERK1/2 and PI3K dependent manner (Galli et al., 2005).
Several miRNAs have already been linked to the regulation of the oxidative stress pathway, including members of the miR-200 family. From this miRNA-family, especially miR-200c has been shown to display an increased expression after cellular exposure to H2O2. This miRNA would lead to down-regulation of zinc finger E-box binding homeobox 1 (Zfhx1a, aka Zeb1, or TCF8), a transcriptional repressor, both on mRNA and protein level, leading to cellular senescence and inhibition of cell proliferation. Interestingly, an inhibitory loop was found between miR-200c and Zeb1, as the promoter region of miR-200c contains two conserved Zeb1 binding sites (Magenta et al., 2011). MiR-200c can also regulate apoptosis, as it inhibits the translation of FAS associated phosphatase (FAP-1) mRNA. Decreased expression of FAP-1 leads to a greater sensitivity to CD95-mediated apoptosis (Schickel et al., 2010). Some of the other identified targets of miR-200c include Moesin (MSN), Fibronectin 1 (FN1), and Rho GTPase activating protein 19 (ARHGAP19), important regulators of the migratory and invasive capacity of cancer cells (Howe et al., 2011). Another miRNA associated with oxidative stress is miR-21. Cells exposed to ROS would up-regulate miR-21, which can directly interact with the 3′UTR of the programmed cell death 4 (PDCD4) gene, a known tumor suppressor and apoptosis-regulator, thereby preventing cell death. Oxidative stress mediated up-regulation of miR-21 can be induced by NF-κB activation through five NF-κB binding sites in the 5' miR-21 promoter region (Tu et al., 2014b; Wei et al., 2014). Up-regulation of miR-21 would be a down-stream effect of NADPH oxidase activity (Dattaroy et al., 2015), as this induces NF-κB translocation to the nucleus (Yao et al., 2007) and its subsequent binding to the miR-21 promotor (Sheedy et al., 2010). This enhanced expression of miR-21 also leads to a suppression of SMAD7 expression and therefore favors assembly of SMAD2/3-SMAD4 heterodimers, a crucial event in the pro-fibrogenic TGF-β signaling pathway (Dattaroy et al., 2015).
A potential link in oxidative stress-induced HSC activation could be represented by miR-200a, which is down-regulated during the process of liver fibrosis in rat, and in TGF-β1-mediated activation of a rat HSC cell line (Sun et al., 2014). MiR-200a also regulates proliferation of these activating HSCs, shown by an accumulation of cells in the G0/G1 phase upon miR-200a overexpression. Targets of miR-200a include pro-fibrogenic factors TGF-β2 and β-catenin (Sun et al., 2014). Another important miRNA-200a target gene is Kelch-like ECH-associated protein 1 (Keap1), which negatively regulates the stability of nuclear factor-erythroid-2-related factor 2 (Nrf2), a known regulator of the expression of antioxidants involved in the protection against oxidative damage (Yang et al., 2014). While no information is available for miR-200c, in rat, miR-200a seems to be down-regulated upon HSC activation while during liver fibrosis progression in human and mouse, miR-200a and miR-200b undergo a significant up-regulation (Murakami et al., 2011). This is in line with the up-regulation in expression of the miR-200 family after induction of oxidative stress in mouse fibroblasts where miR-200a can target p38α mitogen-activated protein kinase (MAPK) (Mateescu et al., 2011), which is downstream of the oxidative stress stimulus, and leads to an inhibition of cell division (Kurata, 2000). Despite opposing expression patterns observed in different species, the involvement of miR-200a in both HSC activation and oxidative stress response is clear. It is therefore tempting to speculate that miR-200a could participate in the anti-oxidant response of HSCs during liver injury.
Endoplasmic Reticulum Stress Regulated miRNAs
The generation of mediators that lead to a perturbation of the ER homeostasis can be evoked by various stimuli associated with the initiation or progression of the liver fibrosis process, such as repeated cycles of ischemia and reperfusion due to distorted hepatic flow, genetic mutations of proteins involved in ER constitution and function, excessive exposure to certain drugs (paracetamol, ethanol), obesity-linked enhanced presence of lipids, and viral infections (HCV, HBV). These stimuli can lead to oxidative stress, formation of protein aggregates, altered membrane lipid-composition, and hyperhomocysteinemia with resulting N-homocysteinylation, all leading to the dysfunction of the ER, and accumulation of unfolded and misfolded proteins (Malhi and Kaufman, 2011). Cells will try to counteract this accumulation of misfolded proteins by diverse mechanisms such as the unfolded protein response (UPR). The activation of the UPR pathway, due to ER-resident stress sensors such as ATF-6, IRE1, and PERK (Asselah et al., 2010), will lead to an enhanced and more stringent folding and degradation of proteins in the ER, and an overall diminishment of protein synthesis. When the UPR fails to diminish the ER stress, the cells go into apoptosis. Persistent ER stress has several consequences including the excessive energy depletion due to the enhanced utilization of energy for translocation of misfolded proteins; ASK1/JNK mediated signaling leading to activation of caspases, and the activation of the pro-apoptotic pathway of CHOP/GADD153 transcription factor, which all direct the cell toward apoptosis (Xu et al., 2005). It will also lead to the release of the stored calcium in the ER, which affects mitochondria; moreover it will lead to the induction of oxidative stress, activation of the pro-inflammatory NF-κB pathway and apoptosis of the cell. ER stress will also lead to translocation and activation of SREBP, causing an enhanced synthesis of lipids such as fatty acids and cholesterol, and an enhanced cellular uptake of lipoproteins (Ji and Kaplowitz, 2006; Ji, 2008).
Cultured HSCs, which are known to be relatively apoptosis-insensitive, have been shown to undergo apoptosis in response to persistent ER stress due to an increase of the amount of intracellular calcium, and activation of JNK/p38 MAPK and Calpain/Caspase pathways (Huang et al., 2014b). Activation of the latter pathway can be explained by the decrease of Calpastatin expression, which works as an inhibitor of the pro-apoptotic Calpain. During the activation of HSCs, Calpastatin levels become elevated, leading to the desensitization of the HSCs toward apoptotic stimuli. ER-stress mediated decrease of Calpastatin expression can thus lead to higher Calpain levels, and consequent sensitization toward apoptotic stimuli (De Minicis et al., 2012). The fibrosis counteracting effect of ER stress was further supported by the decrease in α-SMA and Col1a1-expression in ER-stress responsive activating HSCs (Huang et al., 2014b). However, it is found that when HSCs are exposed to oxidative stress-induced ER stress, the UPR will lead to the up-regulation of different pathways leading to enhanced autophagy and consequent HSC activation in vitro (Hernandez-Gea et al., 2013). All described ER stress could thus be considered as a complex mechanism of fibrosis regulation, with a possible stimulatory role in HSC activation and a possible role in fibrosis resolution due to its pro-apoptotic effects in activated HSCs.
The role of miRNAs during ER-stress remains largely unknown. One of the miRNAs that has been studied in this process is miR-199a-5p, which displays an up-regulation in hepatocytes undergoing ER stress. This miRNA would have several ER-stress related targets including the chaperone protein GRP78 (which is also known as Bip and HSPA5), activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1α (IRE1α), with the latter two being UPR transducers. As IRE1α activated ER stress can induce cell death, activation of miR-199a-5p, and thus subsequent down-regulation of IRE1α, would work as a rescue mechanism to prevent the induction of apoptosis. In silico target prediction identified DNA-damage regulated autophagy modulator 1 (DRAM1) and cyclin-dependent kinase inhibitor 1B (p27), both pro-apoptotic genes, as additional potential targets of miR-199a-5p, thus further underlining its pro-survival role (Dai et al., 2013). miR-199a-5p could also have some effect on cell proliferation, as it has been shown to target frizzles type 7 receptor (FZD7), and thus regulates the expression of its downstream genes including β-catenin, Jun, Cyclin D1, and Myc (Song et al., 2014). A second class of miRNAs linked with ER stress includes members of the miR-30 family, which are being down-regulated due to this specific stress responsive pathway. This miRNA family contains six members (from a to e), which contain all an identical seed sequence motif, but are located at different sites of the genome. GRP78 is targeted by miR-30a, which further underlines the importance of this miRNA in this stress response. Knockdown of miR-30 in cardiac cells identified ATF6, CHOP, and caspase-12 as indirect targets of this miRNA, thus revealing its role in regulation of cell death (Chen et al., 2014).
MiR-122 could perhaps represent a regulator of ER-stress-modulated HSC activation. MiR-122 is described as liver-specific and the most abundant miRNA in the liver (Lagos-Quintana et al., 2002). It has been shown that miR-122 is down-regulated in total liver samples during the progression of liver disease in mouse, rat (Li et al., 2013) and human (Padgett et al., 2009), and this down-regulation was furthermore observed in activating HSCs (Li et al., 2013). Overexpression of this miRNA in LX-2 cells leads to a decrease in cell proliferation and maturation of Col1a1, most likely through regulation of P4HA1 by miR-122. The expression of P4HA1 is up-regulated during fibrosis progression, and encodes a component of prolyl 4-hydroxylase, which is necessary for collagen maturation (Li et al., 2013). Overexpression of miR-122 in LX2 further identified FN1, which is involved in the assembly of collagen fibrils, and serum response factor (SRF) as direct targets, and confirmed its inhibitory effect on TGF-β-induced HSC activation (Zeng et al., 2015). Further target identification studies in hepatocytes identified mitogen-activated protein kinase kinase kinase 3 (MAP3K3), which plays a role in cell survival and proliferation, the intermediate filament vimentin, and HIF-1α (Csak et al., 2015). MiR-122 inhibition in hepatoma cells suggests a role in the UPR. Moreover its inhibition leads to an up-regulation of the 26S proteasome non-ATPase regulatory subunit 10 (PMSD10), which can enhance the protein folding-capacity and thus promoting recovery, by up-regulation of GRP78. MiR-122 would have this effect on PMSD10 in an indirect manner through targeting of cyclin dependent kinase 4 (CDk4) which interacts with PMSD10. Other miR-122 targets include the ER stress chaperones calreticulin (CALR), ER protein 29 (ERP29) and SET nuclear oncogene (SET), which help in the correct folding of malfunctional proteins (Yang et al., 2011). Taken together, even though miR-122 is not abundantly expressed in HSCs, it is tempting to speculate that down-regulation of miR-122 is involved in the UPR in HSCs.
Discussion
MiRNAs have been proposed as key regulators of gene expression and dysregulated patterns of miRNA expression were observed in various diseases (Tufekci et al., 2014), including the progression of liver fibrosis and cirrhosis (Wang et al., 2012; Xin et al., 2014). Studying miRNAs is very popular and raised a lot of expectations in their use as biomarkers for diseases and therapeutic interventions using miRNA mimics and antagomirs. Unfortunately, so far this has not turned out to be easy, partly because of their cell type-specific and species-specific activity and wide range of targets.
Diagnosis of liver fibrosis could be facilitated by identification of blood-circulating biomarkers representative for HSC activation, as the current golden standard for diagnosis remains the invasive and harmful liver biopsy (Piccinino et al., 1986; Friedman, 2003). Circulating miRNAs, both protein-bound and packaged into extracellular vesicles (Turchinovich et al., 2011), have been proposed as such a potential biomarker, and various research groups already tried to identify circulating miRNAs that could be linked with progression and regression of liver disease (Roderburg and Luedde, 2014). To date, this has not yet led to a diagnostic protocol that is used in clinic. It is tempting to speculate that perhaps stress-responsive miRNAs of activating HSCs secreted in the blood could also be used as a liquid biopsy to document the stress present in the liver.
We discussed several miRNAs with a potential role in stress-mediated regulation of HSC activation. Experimental validation of these suggested links between stress-related miRNAs and HSCs should address a number of issues. First, are specific miRNAs dysregulated in HSCs in response to specific stress signals and does this lead to an imbalance of the cellular homeostasis and consequent HSC apoptosis or activation? In vivo, paracrine stimulation of quiescent HSCs by stress-undergoing surrounding cells is likely to create a warning for the quiescent cell, leading to its activation and reducing its responsiveness to more stress-signals. Secondly, responding to stress is necessary to counteract short term challenges to restore cell homeostasis. Thus the question is, whether there are miRNAs that specifically respond to prolonged stresses present in the fibrotic liver, and if so, could a targeted mimic/antagomir approach inhibit HSC activation or promote HSC apoptosis or inactivation?
In conclusion, HSC activation in vivo can be seen as a very complicated and multifactorial process in which hypoxia (Cannito et al., 2014), oxidative stress (Poli, 2000), and ER stress (Malhi and Kaufman, 2011) are surely involved. This suggests a potential role for stress-related miRNAs during HSC activation and disease development and opens perspectives for new therapeutic approaches.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
IM is supported by a Fund of Scientific Research Flanders FWO-V post-doctoral fellowships (12N5415N LV) and JL and LV are supported by the Vrije Universiteit Brussel (GOA78, OZR1930).
References
Asselah, T., Bieche, I., Mansouri, A., Laurendeau, I., Cazals-Hatem, D., Feldmann, G., et al. (2010). In vivo hepatic endoplasmic reticulum stress in patients with chronic hepatitis C. J. Pathol. 221, 264–274. doi: 10.1002/path.2703
Aubert, J., Begriche, K., Knockaert, L., Robin, M. A., and Fromenty, B. (2011). Increased expression of cytochrome P450 2E1 in nonalcoholic fatty liver disease: mechanisms and pathophysiological role. Clin. Res. Hepatol. Gastroenterol. 35, 630–637. doi: 10.1016/j.clinre.2011.04.015
Babar, I. A., Slack, F. J., and Weidhaas, J. B. (2008). miRNA modulation of the cellular stress response. Future Oncol. 4, 289–298. doi: 10.2217/14796694.4.2.289
Bagga, S., Bracht, J., Hunter, S., Massirer, K., Holtz, J., Eachus, R., et al. (2005). Regulation by let-7 and lin-4 miRNAs results in target mRNA degradation. Cell 122, 553–563. doi: 10.1016/j.cell.2005.07.031
Bai, R., Zhao, A. Q., Zhao, Z. Q., Liu, W. L., and Jian, D. M. (2015). MicroRNA-195 induced apoptosis in hypoxic chondrocytes by targeting hypoxia-inducible factor 1 alpha. Eur. Rev. Med. Pharmacol. Sci. 19, 545–551.
Bartel, D. P., and Chen, C. Z. (2004). Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat. Rev. Genet. 5, 396–400. doi: 10.1038/nrg1328
Bataller, R., Schwabe, R. F., Choi, Y. H., Yang, L., Paik, Y. H., Lindquist, J., et al. (2003). NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J. Clin. Invest. 112, 1383–1394. doi: 10.1172/JCI18212
Bedossa, P., Houglum, K., Trautwein, C., Holstege, A., and Chojkier, M. (1994). Stimulation of collagen alpha 1(I) gene expression is associated with lipid peroxidation in hepatocellular injury: a link to tissue fibrosis? Hepatology 19, 1262–1271. doi: 10.1002/hep.1840190527
Belmont, P. J., Chen, W. J., Thuerauf, D. J., and Glembotski, C. C. (2012). Regulation of microRNA expression in the heart by the ATF6 branch of the ER stress response. J. Mol. Cell. Cardiol. 52, 1176–1182. doi: 10.1016/j.yjmcc.2012.01.017
Bernstein, E., Caudy, A. A., Hammond, S. M., and Hannon, G. J. (2001). Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409, 363–366. doi: 10.1038/35053110
Byrd, A. E., Aragon, I. V., and Brewer, J. W. (2012). MicroRNA-30c-2* limits expression of proadaptive factor XBP1 in the unfolded protein response. J. Cell Biol. 196, 689–698. doi: 10.1083/jcb.201201077
Cannito, S., Paternostro, C., Busletta, C., Bocca, C., Colombatto, S., Miglietta, A., et al. (2014). Hypoxia, hypoxia-inducible factors and fibrogenesis in chronic liver diseases. Histol. Histopathol. 29, 33–44.
Chaudhry, M. A., Omaruddin, R. A., Brumbaugh, C. D., Tariq, M. A., and Pourmand, N. (2013). Identification of radiation-induced microRNA transcriptome by next-generation massively parallel sequencing. J. Radiat. Res. 54, 808–822. doi: 10.1093/jrr/rrt014
Chen, C., Wu, C. Q., Zhang, Z. Q., Yao, D. K., and Zhu, L. (2011). Loss of expression of miR-335 is implicated in hepatic stellate cell migration and activation. Exp. Cell Res. 317, 1714–1725. doi: 10.1016/j.yexcr.2011.05.001
Chen, M., Ma, G., Yue, Y., Wei, Y., Li, Q., Tong, Z., et al. (2014). Downregulation of the miR-30 family microRNAs contributes to endoplasmic reticulum stress in cardiac muscle and vascular smooth muscle cells. Int. J. Cardiol. 173, 65–73. doi: 10.1016/j.ijcard.2014.02.007
Coll, M., Taghdouini, A. E., Perea, L., Mannaerts, I., Vila-Casadesús, M., Blaya, D., et al. (2015). Integrative miRNA and gene expression profiling analysis of human quiescent hepatic stellate cells. Sci. Rep. 5:11549. doi: 10.1038/srep11549
Copple, B. L., Bai, S., Burgoon, L. D., and Moon, J. O. (2011). Hypoxia-inducible factor-1alpha regulates the expression of genes in hypoxic hepatic stellate cells important for collagen deposition and angiogenesis. Liver Int. 31, 230–244. doi: 10.1111/j.1478-3231.2010.02347.x
Copple, B. L., Roth, R. A., and Ganey, P. E. (2006). Anticoagulation and inhibition of nitric oxide synthase influence hepatic hypoxia after monocrotaline exposure. Toxicology 225, 128–137. doi: 10.1016/j.tox.2006.05.016
Csak, T., Bala, S., Lippai, D., Satishchandran, A., Catalano, D., Kodys, K., et al. (2015). microRNA-122 regulates hypoxia-inducible factor-1 and vimentin in hepatocytes and correlates with fibrosis in diet-induced steatohepatitis. Liver Int. 35, 532–541. doi: 10.1111/liv.12633
Dai, B. H., Geng, L., Wang, Y., Sui, C. J., Xie, F., Shen, R. X., et al. (2013). microRNA-199a-5p protects hepatocytes from bile acid-induced sustained endoplasmic reticulum stress. Cell Death Dis. 4, e604. doi: 10.1038/cddis.2013.134
Dattaroy, D., Pourhoseini, S., Das, S., Alhasson, F., Seth, R. K., Nagarkatti, M., et al. (2015). Micro-RNA 21 inhibition of SMAD7 enhances fibrogenesis via leptin-mediated NADPH oxidase in experimental and human nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 308, G298–G312. doi: 10.1152/ajpgi.00346.2014
De Minicis, S., and Brenner, D. A. (2007). NOX in liver fibrosis. Arch. Biochem. Biophys. 462, 266–272. doi: 10.1016/j.abb.2007.04.016
De Minicis, S., Candelaresi, C., Agostinelli, L., Taffetani, S., Saccomanno, S., Rychlicki, C., et al. (2012). Endoplasmic Reticulum stress induces hepatic stellate cell apoptosis and contributes to fibrosis resolution. Liver Int. 32, 1574–1584. doi: 10.1111/j.1478-3231.2012.02860.x
De Minicis, S., Seki, E., Uchinami, H., Kluwe, J., Zhang, Y., Brenner, D. A., et al. (2007). Gene expression profiles during hepatic stellate cell activation in culture and in vivo. Gastroenterology 132, 1937–1946. doi: 10.1053/j.gastro.2007.02.033
Fasanaro, P., Greco, S., Lorenzi, M., Pescatori, M., Brioschi, M., Kulshreshtha, R., et al. (2009). An integrated approach for experimental target identification of hypoxia-induced miR-210. J. Biol. Chem. 284, 35134–35143. doi: 10.1074/jbc.M109.052779
Feng, X., Tan, W., Cheng, S., Wang, H., Ye, S., Yu, C., et al. (2015). Upregulation of microRNA-126 in hepatic stellate cells may affect pathogenesis of liver fibrosis through the NF-kappaB pathway. DNA Cell Biol. 34, 470–480. doi: 10.1089/dna.2014.2760
Forsythe, J. A., Jiang, B. H., Iyer, N. V., Agani, F., Leung, S. W., Koos, R. D., et al. (1996). Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 16, 4604–4613.
Friedman, S. L. (2003). Liver fibrosis – from bench to bedside. J. Hepatol. 38, 38–53. doi: 10.1016/S0168-8278(02)00429-4
Galli, A., Svegliati-Baroni, G., Ceni, E., Milani, S., Ridolfi, F., Salzano, R., et al. (2005). Oxidative stress stimulates proliferation and invasiveness of hepatic stellate cells via a MMP2-mediated mechanism. Hepatology 41, 1074–1084. doi: 10.1002/hep.20683
Giannakakis, A., Sandaltzopoulos, R., Greshock, J., Liang, S., Huang, J., Hasegawa, K., et al. (2008). miR-210 links hypoxia with cell cycle regulation and is deleted in human epithelial ovarian cancer. Cancer Biol. Ther. 7, 255–264. doi: 10.4161/cbt.7.2.5297
Gregory, R. I., Chendrimada, T. P., Cooch, N., and Shiekhattar, R. (2005). Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell 123, 631–640. doi: 10.1016/j.cell.2005.10.022
Gregory, R. I., Yan, K. P., Amuthan, G., Chendrimada, T., Doratotaj, B., Cooch, N., et al. (2004). The microprocessor complex mediates the genesis of microRNAs. Nature 432, 235–240. doi: 10.1038/nature03120
Guarente, L. (2008). Mitochondria–a nexus for aging, calorie restriction, and sirtuins? Cell 132, 171–176. doi: 10.1016/j.cell.2008.01.007
Guimarães, E. L. M., Empsen, C., Geerts, A., and van Grunsven, L. A. (2010). Advanced glycation end products induce production of reactive oxygen species via the activation of NADPH oxidase in murine hepatic stellate cells. J. Hepatol. 52, 389–397. doi: 10.1016/j.jhep.2009.12.007
Guo, C. J., Pan, Q., Cheng, T., Jiang, B., Chen, G. Y., and Li, D. G. (2009a). Changes in microRNAs associated with hepatic stellate cell activation status identify signaling pathways. FEBS J. 276, 5163–5176. doi: 10.1111/j.1742-4658.2009.07213.x
Guo, C. J., Pan, Q., Jiang, B., Chen, G. Y., and Li, D. G. (2009c). Effects of upregulated expression of microRNA-16 on biological properties of culture-activated hepatic stellate cells. Apoptosis 14, 1331–1340. doi: 10.1007/s10495-009-0401-3
Guo, C. J., Pan, Q., Li, D. G., Sun, H., and Liu, B. W. (2009b). miR-15b and miR-16 are implicated in activation of the rat hepatic stellate cell: an essential role for apoptosis. J. Hepatol. 50, 766–778. doi: 10.1016/j.jhep.2008.11.025
Ha, M., Pang, M., Agarwal, V., and Chen, Z. J. (2008). Interspecies regulation of microRNAs and their targets. Biochim. Biophys. Acta 1779, 735–742. doi: 10.1016/j.bbagrm.2008.03.004
He, Y., Huang, C., Sun, X., Long, X. R., Lv, X. W., and Li, J. (2012b). MicroRNA-146a modulates TGF-beta1-induced hepatic stellate cell proliferation by targeting SMAD4. Cell. Signal. 24, 1923–1930. doi: 10.1016/j.cellsig.2012.06.003
He, Y., Huang, C., Zhang, S.-P., Sun, X., Long, X.-R., and Li, J. (2012a). The potential of microRNAs in liver fibrosis. Cell. Signal. 24, 2268–2272. doi: 10.1016/j.cellsig.2012.07.023
Hebert, C., Norris, K., Scheper, M. A., Nikitakis, N., and Sauk, J. J. (2007). High mobility group A2 is a target for miRNA-98 in head and neck squamous cell carcinoma. Mol. Cancer 6, 5. doi: 10.1186/1476-4598-6-5
Hernandez-Gea, V., Hilscher, M., Rozenfeld, R., Lim, M. P., Nieto, N., Werner, S., et al. (2013). Endoplasmic reticulum stress induces fibrogenic activity in hepatic stellate cells through autophagy. J. Hepatol. 59, 98–104. doi: 10.1016/j.jhep.2013.02.016
Howe, E. N., Cochrane, D. R., and Richer, J. K. (2011). Targets of miR-200c mediate suppression of cell motility and anoikis resistance. Breast Cancer Res. 13, R45. doi: 10.1186/bcr2867
Hu, J., Chen, C., Liu, Q., Liu, B., Song, C., Zhu, S., et al. (2015). The role of miR-31/FIH1 pathway in TGFbeta-induced liver fibrosis. Clin. Sci. 129, 305–317. doi: 10.1042/CS20140012
Hua, Z., Lv, Q., Ye, W., Wong, C. K., Cai, G., Gu, D., et al. (2006). MiRNA-directed regulation of VEGF and other angiogenic factors under hypoxia. PLoS ONE 1:e116. doi: 10.1371/journal.pone.0000116
Huang, J., Yu, X., Fries, J. W., Zhang, L., and Odenthal, M. (2014a). MicroRNA function in the profibrogenic interplay upon chronic liver disease. Int. J. Mol. Sci. 15, 9360–9371. doi: 10.3390/ijms15069360
Huang, X., Ding, L., Bennewith, K. L., Tong, R. T., Welford, S. M., Ang, K. K., et al. (2009). Hypoxia-inducible mir-210 regulates normoxic gene expression involved in tumor initiation. Mol. Cell 35, 856–867. doi: 10.1016/j.molcel.2009.09.006
Huang, Y., Li, X., Wang, Y., Wang, H., Huang, C., and Li, J. (2014b). Endoplasmic reticulum stress-induced hepatic stellate cell apoptosis through calcium-mediated JNK/P38 MAPK and Calpain/Caspase-12 pathways. Mol. Cell. Biochem. 394, 1–12. doi: 10.1007/s11010-014-2073-8
Hyogo, H., Yamagishi, S.-i., Iwamoto, K., Arihiro, K., Takeuchi, M., Sato, T., et al. and Tazuma, S. (2007). Elevated levels of serum advanced glycation end products in patients with non-alcoholic steatohepatitis. J. Gastroenterol. Hepatol. 22, 1112–1119. doi: 10.1111/j.1440-1746.2007.04943.x
Iizuka, M., Ogawa, T., Enomoto, M., Motoyama, H., Yoshizato, K., Ikeda, K., et al. (2012). Induction of microRNA-214-5p in human and rodent liver fibrosis. Fibrogenesis Tissue Repair 5, 12. doi: 10.1186/1755-1536-5-12
Ji, C. (2008). Dissection of endoplasmic reticulum stress signaling in alcoholic and non-alcoholic liver injury. J. Gastroenterol. Hepatol. 23(Suppl. 1), S16–S24. doi: 10.1111/j.1440-1746.2007.05276.x
Ji, C., and Kaplowitz, N. (2006). ER stress: can the liver cope? J. Hepatol. 45, 321–333. doi: 10.1016/j.jhep.2006.06.004
Ji, J., Zhang, J., Huang, G., Qian, J., Wang, X., and Mei, S. (2009). Over-expressed microRNA-27a and 27b influence fat accumulation and cell proliferation during rat hepatic stellate cell activation. FEBS Lett. 583, 759–766. doi: 10.1016/j.febslet.2009.01.034
Jiang, B. H., Semenza, G. L., Bauer, C., and Marti, H. H. (1996). Hypoxia-inducible factor 1 levels vary exponentially over a physiologically relevant range of O2 tension. Am. J. Physiol. 271, C1172–C1180.
Jiang, F., Parsons, C. J., and Stefanovic, B. (2006). Gene expression profile of quiescent and activated rat hepatic stellate cells implicates Wnt signaling pathway in activation. J. Hepatol. 45, 401–409. doi: 10.1016/j.jhep.2006.03.016
Jiang, W., Kong, L., Ni, Q., Lu, Y., Ding, W., Liu, G., et al. (2014). miR-146a ameliorates liver ischemia/reperfusion injury by suppressing IRAK1 and TRAF6. PLoS ONE 9:e101530. doi: 10.1371/journal.pone.0101530
Kim, H. W., Haider, H. K., Jiang, S., and Ashraf, M. (2009). Ischemic preconditioning augments survival of stem cells via miR-210 expression by targeting caspase-8-associated protein 2. J. Biol. Chem. 284, 33161–33168. doi: 10.1074/jbc.M109.020925
Kisseleva, T., and Brenner, D. A. (2007). Role of hepatic stellate cells in fibrogenesis and the reversal of fibrosis. J. Gastroenterol. Hepatol. 22(Suppl. 1), S73–S78. doi: 10.1111/j.1440-1746.2006.04658.x
Kisseleva, T., and Brenner, D. A. (2013). Inactivation of myofibroblasts during regression of liver fibrosis. Cell Cycle 12, 381–382. doi: 10.4161/cc.23549
Kulshreshtha, R., Ferracin, M., Wojcik, S. E., Garzon, R., Alder, H., Agosto-Perez, F. J., et al. (2007). A microRNA signature of hypoxia. Mol. Cell. Biol. 27, 1859–1867. doi: 10.1128/MCB.01395-06
Šebeková, K. N., Kupèová, V., Schinzel, R., and Heidland, A. (2002). Markedly elevated levels of plasma advanced glycation end products in patients with liver cirrhosis – amelioration by liver transplantation. J. Hepatol. 36, 66–71. doi: 10.1016/S0168-8278(01)00232-X
Kurata, S. (2000). Selective activation of p38 MAPK cascade and mitotic arrest caused by low level oxidative stress. J. Biol. Chem. 275, 23413–23416. doi: 10.1074/jbc.C000308200
Kwiecinski, M., Elfimova, N., Noetel, A., Tox, U., Steffen, H. M., Hacker, U., et al. (2012). Expression of platelet-derived growth factor-C and insulin-like growth factor I in hepatic stellate cells is inhibited by miR-29. Lab. Invest. 92, 978–987. doi: 10.1038/labinvest.2012.70
Kwiecinski, M., Noetel, A., Elfimova, N., Trebicka, J., Schievenbusch, S., Strack, I., et al. (2011). Hepatocyte growth factor (HGF) inhibits collagen I and IV synthesis in hepatic stellate cells by miRNA-29 induction. PLoS ONE 6:e24568. doi: 10.1371/journal.pone.0024568
Lagos-Quintana, M., Rauhut, R., Yalcin, A., Meyer, J., Lendeckel, W., and Tuschl, T. (2002). Identification of tissue-specific microRNAs from mouse. Curr. Biol. 12, 735–739. doi: 10.1016/S0960-9822(02)00809-6
Lakner, A. M., Steuerwald, N. M., Walling, T. L., Ghosh, S., Li, T., McKillop, I. H., et al. (2012). Inhibitory effects of microRNA 19b in hepatic stellate cell-mediated fibrogenesis. Hepatology 56, 300–310. doi: 10.1002/hep.25613
Lee, H. J. (2013). Exceptional stories of microRNAs. Exp. Biol. Med. 238, 339–343. doi: 10.1258/ebm.2012.012251
Lee, Y., Ahn, C., Han, J., Choi, H., Kim, J., Yim, J., et al. (2003). The nuclear RNase III Drosha initiates microRNA processing. Nature 425, 415–419. doi: 10.1038/nature01957
Leung, A. K., and Sharp, P. A. (2010). MicroRNA functions in stress responses. Mol. Cell 40, 205–215. doi: 10.1016/j.molcel.2010.09.027
Li, J., Ghazwani, M., Zhang, Y., Lu, J., Li, J., Fan, J., et al. (2013). miR-122 regulates collagen production via targeting hepatic stellate cells and suppressing P4HA1 expression. J. Hepatol. 58, 522–528. doi: 10.1016/j.jhep.2012.11.011
Li, X., Wang, Y., Wang, H., Huang, C., Huang, Y., and Li, J. (2015). Endoplasmic reticulum stress is the crossroads of autophagy, inflammation, and apoptosis signaling pathways and participates in liver fibrosis. Inflamm. Res. 64, 1–7. doi: 10.1007/s00011-014-0772-y
Liu, C. J., Tsai, M. M., Hung, P. S., Kao, S. Y., Liu, T. Y., Wu, K. J., et al. (2010). miR-31 ablates expression of the HIF regulatory factor FIH to activate the HIF pathway in head and neck carcinoma. Cancer Res. 70, 1635–1644. doi: 10.1158/0008-5472.CAN-09-2291
Lund, E., Guttinger, S., Calado, A., Dahlberg, J. E., and Kutay, U. (2004). Nuclear export of microRNA precursors. Science 303, 95–98. doi: 10.1126/science.1090599
Magenta, A., Cencioni, C., Fasanaro, P., Zaccagnini, G., Greco, S., Sarra-Ferraris, G., et al. (2011). miR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via ZEB1 inhibition. Cell Death Differ. 18, 1628–1639. doi: 10.1038/cdd.2011.42
Mahon, P. C., Hirota, K., and Semenza, G. L. (2001). FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 15, 2675–2686. doi: 10.1101/gad.924501
Malhi, H., and Kaufman, R. J. (2011). Endoplasmic reticulum stress in liver disease. J. Hepatol. 54, 795–809. doi: 10.1016/j.jhep.2010.11.005
Mateescu, B., Batista, L., Cardon, M., Gruosso, T., de Feraudy, Y., Mariani, O., et al. (2011). miR-141 and miR-200a act on ovarian tumorigenesis by controlling oxidative stress response. Nat. Med. 17, 1627–1635. doi: 10.1038/nm.2512
Maubach, G., Lim, M. C., Chen, J., Yang, H., and Zhuo, L. (2011). miRNA studies in in vitro and in vivo activated hepatic stellate cells. World J. Gastroenterol. 17, 2748–2773. doi: 10.3748/wjg.v17.i22
Mederacke, I., Hsu, C. C., Troeger, J. S., Huebener, P., Mu, X., Dapito, D. H., et al. (2013). Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun. 4, 2823. doi: 10.1038/ncomms3823
Moon, J. O., Welch, T. P., Gonzalez, F. J., and Copple, B. L. (2009). Reduced liver fibrosis in hypoxia-inducible factor-1alpha-deficient mice. Am. J. Physiol. Gastrointest. Liver Physiol. 296, G582–G592. doi: 10.1152/ajpgi.90368.2008
Murakami, Y., Toyoda, H., Tanaka, M., Kuroda, M., Harada, Y., Matsuda, F., et al. (2011). The progression of liver fibrosis is related with overexpression of the miR-199 and 200 families. PLoS ONE 6:e16081. doi: 10.1371/journal.pone.0016081
Nath, B., and Szabo, G. (2012). Hypoxia and hypoxia inducible factors: diverse roles in liver diseases. Hepatology 55, 622–633. doi: 10.1002/hep.25497
Nieto, N., Friedman, S. L., Greenwel, P., and Cederbaum, A. I. (1999). CYP2E1-mediated oxidative stress induces collagen type, I., expression in rat hepatic stellate cells. Hepatology 30, 987–996. doi: 10.1002/hep.510300433
Orban, T. I., and Izaurralde, E. (2005). Decay of mRNAs targeted by RISC requires XRN1, the Ski complex, and the exosome. RNA 11, 459–469. doi: 10.1261/rna.7231505
Padgett, K. A., Lan, R. Y., Leung, P. C., Lleo, A., Dawson, K., Pfeiff, J., et al. (2009). Primary biliary cirrhosis is associated with altered hepatic microRNA expression. J. Autoimmun. 32, 246–253. doi: 10.1016/j.jaut.2009.02.022
Parola, M., and Robino, G. (2001). Oxidative stress-related molecules and liver fibrosis. J. Hepatol. 35, 297–306. doi: 10.1016/S0168-8278(01)00142-8
Paternostro, C., David, E., Novo, E., and Parola, M. (2010). Hypoxia, angiogenesis and liver fibrogenesis in the progression of chronic liver diseases. World J. Gastroenterol. 16, 281–288. doi: 10.3748/wjg.v16.i3.281
Piccinino, F., Sagnelli, E., Pasquale, G., and Giusti, G. (1986). Complications following percutaneous liver biopsy. A multicentre retrospective study on 68,276 biopsies. J. Hepatol. 2, 165–173. doi: 10.1016/S0168-8278(86)80075-7
Pillai, R. S., Bhattacharyya, S. N., Artus, C. G., Zoller, T., Cougot, N., Basyuk, E., et al. (2005). Inhibition of translational initiation by Let-7 MicroRNA in human cells. Science 309, 1573–1576. doi: 10.1126/science.1115079
Poli, G. (2000). Pathogenesis of liver fibrosis: role of oxidative stress. Mol. Aspects Med. 21, 49–98. doi: 10.1016/S0098-2997(00)00004-2
Roderburg, C., and Luedde, T. (2014). Circulating microRNAs as markers of liver inflammation, fibrosis and cancer. J. Hepatol. 61, 1434–1437. doi: 10.1016/j.jhep.2014.07.017
Roderburg, C., Urban, G. W., Bettermann, K., Vucur, M., Zimmermann, H., Schmidt, S., et al. (2011). Micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology 53, 209–218. doi: 10.1002/hep.23922
Rosmorduc, O., Wendum, D., Corpechot, C., Galy, B., Sebbagh, N., Raleigh, J., et al. (1999). Hepatocellular hypoxia-induced vascular endothelial growth factor expression and angiogenesis in experimental biliary cirrhosis. Am. J. Pathol. 155, 1065–1073. doi: 10.1016/S0002-9440(10)65209-1
Schickel, R., Park, S. M., Murmann, A. E., and Peter, M. E. (2010). miR-200c regulates induction of apoptosis through CD95 by targeting FAP-1. Mol. Cell 38, 908–915. doi: 10.1016/j.molcel.2010.05.018
Schuppan, D., and Afdhal, N. H. (2008). Liver cirrhosis. Lancet 371, 838–851. doi: 10.1016/S0140-6736(08)60383-9
Sekiya, Y., Ogawa, T., Yoshizato, K., Ikeda, K., and Kawada, N. (2011). Suppression of hepatic stellate cell activation by microRNA-29b. Biochem. Biophys. Res. Commun. 412, 74–79. doi: 10.1016/j.bbrc.2011.07.041
Semenza, G. L. (2007). Hypoxia-inducible factor 1 (HIF-1) pathway. Sci. STKE 2007:cm8. doi: 10.1126/stke.4072007cm8
Sergey, D. (2011). Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 51, 1289–1301. doi: 10.1016/j.freeradbiomed.2011.06.033
Shan-Shan, Z., Joy, X. J., Jian, W., Charles, H., Scott, L. F., Mark, A. Z., et al. (2006). Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology 43, 435–443. doi: 10.1002/hep.21093
Sheedy, F. J., E., Palsson-McDermott, Hennessy, E. J., Martin, C., O'Leary, J. J., Ruan, Q., et al. (2010). Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat. Immunol. 11, 141–147. doi: 10.1038/ni.1828
Shi, Y. F., Fong, C. C., Zhang, Q., Cheung, P. Y., Tzang, C. H., Wu, R. S., et al. (2007). Hypoxia induces the activation of human hepatic stellate cells LX-2 through TGF-beta signaling pathway. FEBS Lett. 581, 203–210. doi: 10.1016/j.febslet.2006.12.010
Sies, H. (1991). Oxidative stress: from basic research to clinical application. Am. J. Med. 91, 31S–38S. doi: 10.1016/0002-9343(91)90281-2
Song, J., Gao, L., Yang, G., Tang, S., Xie, H., Wang, Y., et al. (2014). MiR-199a regulates cell proliferation and survival by targeting FZD7. PLoS ONE 9:e110074. doi: 10.1371/journal.pone.0110074
Su, S. F., Chang, Y. W., Andreu-Vieyra, C., Fang, J. Y., Yang, Z., Han, B., et al. (2013). miR-30d, miR-181a and miR-199a-5p cooperatively suppress the endoplasmic reticulum chaperone and signaling regulator GRP78 in cancer. Oncogene 32, 4694–4701. doi: 10.1038/onc.2012.483
Sun, X., He, Y., Ma, T. T., Huang, C., Zhang, L., and Li, J. (2014). Participation of miR-200a in TGF-beta1-mediated hepatic stellate cell activation. Mol. Cell. Biochem. 388, 11–23. doi: 10.1007/s11010-013-1895-0
Tu, H., Sun, H., Lin, Y., Ding, J., Nan, K., Li, Z., et al. (2014b). Oxidative stress upregulates PDCD4 expression in patients with gastric cancer via miR-21. Curr. Pharm. Des. 20, 1917–1923. doi: 10.2174/13816128113199990547
Tu, X., Zhang, H., Zhang, J., Zhao, S., Zheng, X., Zhang, Z., et al. (2014a). MicroRNA-101 suppresses liver fibrosis by targeting the TGFbeta signalling pathway. J. Pathol. 234, 46–59. doi: 10.1002/path.4373
Tufekci, K. U., Oner, M. G., Meuwissen, R. L., and Genc, S. (2014). The role of microRNAs in human diseases. Methods Mol. Biol. 1107, 33–50. doi: 10.1007/978-1-62703-748-8_3
Turchinovich, A., Weiz, L., Langheinz, A., and Burwinkel, B. (2011). Characterization of extracellular circulating microRNA. Nucleic Acids Res. 39, 7223–7233. doi: 10.1093/nar/gkr254
Upton, J. P., Wang, L., Han, D., Wang, E. S., Huskey, N. E., Lim, L., et al. (2012). IRE1alpha cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science 338, 818–822. doi: 10.1126/science.1226191
Wallace, K., Burt, A. D., and Wright, M. C. (2008). Liver fibrosis. Biochem. J. 411, 1–18. doi: 10.1042/BJ20071570
Wang, X. W., Heegaard, N. H., and Orum, H. (2012). MicroRNAs in liver disease. Gastroenterology 142, 1431–1443. doi: 10.1053/j.gastro.2012.04.007
Wang, Y., Huang, Y., Guan, F., Xiao, Y., Deng, J., Chen, H., et al. (2013). Hypoxia-inducible factor-1alpha and MAPK co-regulate activation of hepatic stellate cells upon hypoxia stimulation. PLoS ONE 8:e74051. doi: 10.1371/journal.pone.0074051
Wei, C., Li, L., Kim, I. K., Sun, P., and Gupta, S. (2014). NF-kappaB mediated miR-21 regulation in cardiomyocytes apoptosis under oxidative stress. Free Radic. Res. 48, 282–291. doi: 10.3109/10715762.2013.865839
Wightman, B., Ha, I., and Ruvkun, G. (1993). Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 75, 855–862. doi: 10.1016/0092-8674(93)90530-4
Xin, X., Zhang, Y., Liu, X., Xin, H., Cao, Y., and Geng, M. (2014). MicroRNA in hepatic fibrosis and cirrhosis. Front. Biosci. 19, 1418–1424. doi: 10.2741/4292
Xu, C., Bailly-Maitre, B., and Reed, J. C. (2005). Endoplasmic reticulum stress: cell life and death decisions. J. Clin. Invest. 115, 2656–2664. doi: 10.1172/JCI26373
Xu, Y., Zhou, M., Wang, J., Zhao, Y., Li, S., Zhou, B., et al. (2014). Role of microRNA-27a in down-regulation of angiogenic factor AGGF1 under hypoxia associated with high-grade bladder urothelial carcinoma. Biochim. Biophys. Acta 1842, 712–725. doi: 10.1016/j.bbadis.2014.01.007
Yan, S. F., Ramasamy, R., and Schmidt, A. M. (2010). The RAGE axis: a fundamental mechanism signaling danger to the vulnerable vasculature. Circ. Res. 106, 842–853. doi: 10.1161/CIRCRESAHA.109.212217
Yang, F., Zhang, L., Wang, F., Wang, Y., Huo, X. S., Yin, Y. X., et al. (2011). Modulation of the unfolded protein response is the core of microRNA-122-involved sensitivity to chemotherapy in hepatocellular carcinoma. Neoplasia 13, 590–600. doi: 10.1593/neo.11422
Yang, J. J., Tao, H., Hu, W., Liu, L. P., Shi, K. H., Deng, Z. Y., et al. (2014). MicroRNA-200a controls Nrf2 activation by target Keap1 in hepatic stellate cell proliferation and fibrosis. Cell. Signal. 26, 2381–2389. doi: 10.1016/j.cellsig.2014.07.016
Yao, H., Yang, S. R., Kode, A., Rajendrasozhan, S., Caito, S., Adenuga, D., et al. (2007). Redox regulation of lung inflammation: role of NADPH oxidase and NF-kappaB signalling. Biochem. Soc. Trans. 35, 1151–1155. doi: 10.1042/BST0351151
Yoon, C., Kim, D., Kim, S., Park, G. B., Hur, D. Y., Yang, J. W., et al. (2014). MiR-9 regulates the post-transcriptional level of VEGF165a by targeting SRPK-1 in ARPE-19 cells. Graefes Arch. Clin. Exp. Ophthalmol. 252, 1369–1376. doi: 10.1007/s00417-014-2698-z
Zeng, C., Wang, Y. L., Xie, C., Sang, Y., Li, T. J., Zhang, M., et al. (2015). Identification of a novel TGF-beta-miR-122-fibronectin 1/serum response factor signaling cascade and its implication in hepatic fibrogenesis. Oncotarget 6, 12224–12233. doi: 10.1074/jbc.M111.314922
Zhang, L., Zhou, M., Qin, G., Weintraub, N. L., and Tang, Y. (2014). MiR-92a regulates viability and angiogenesis of endothelial cells under oxidative stress. Biochem. Biophys. Res. Commun. 446, 952–958. doi: 10.1016/j.bbrc.2014.03.035
Zhang, Y., Wu, L., Wang, Y., Zhang, M., Li, L., Zhu, D., et al. (2012). Protective role of estrogen-induced miRNA-29 expression in carbon tetrachloride-induced mouse liver injury. J. Biol. Chem. 287, 14851–14862. doi: 10.1074/jbc.M111.314922
Zhang, Z., Sun, H., Dai, H., Walsh, R. M., Imakura, M., Schelter, J., et al. (2009). MicroRNA miR-210 modulates cellular response to hypoxia through the MYC antagonist MNT. Cell Cycle 8, 2756–2768. doi: 10.4161/cc.8.17.9387
Zhao, X., Wang, K., Liao, Y., Zeng, Q., Li, Y., Hu, F., et al. (2015). MicroRNA-101a inhibits cardiac fibrosis induced by hypoxia via targeting TGFbetaRI on cardiac fibroblasts. Cell. Physiol. Biochem. 35, 213–226. doi: 10.1159/000369689
Keywords: miRNAs, hepatic stellate cells, fibrosis, ER stress, hypoxia, oxidative stress
Citation: Lambrecht J, Mannaerts I and van Grunsven LA (2015) The role of miRNAs in stress-responsive hepatic stellate cells during liver fibrosis. Front. Physiol. 6:209. doi: 10.3389/fphys.2015.00209
Received: 06 May 2015; Accepted: 13 July 2015;
Published: 28 July 2015.
Edited by:
Atsushi Masamune, Tohoku University Graduate School of Medicine, JapanReviewed by:
Kyoko Shimizu, Tokyo Women's Medical University, JapanStephen J. Pandol, University of California, Los Angeles, USA
Copyright © 2015 Lambrecht, Mannaerts and van Grunsven. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Leo A. van Grunsven, Liver Cell Biology Lab, Department of Biomedical Sciences, Vrije Universiteit Brussel, Laarbeeklaan 103, Fac. G&F, Brussel B-1090, Belgium,bGVvLnZhbi5ncnVuc3ZlbkB2dWIuYWMuYmU=