 Rafaa Zeineddine
Rafaa Zeineddine Justin J. Yerbury
Justin J. Yerbury- 1Illawarra Health and Medical Research Institute, University of Wollongong, Wollongong, NSW, Australia
- 2Faculty of Science, Medicine and Health, School of Biological Sciences, University of Wollongong, Wollongong, NSW, Australia
With the onset of the rapidly aging population, the impact of age related neurodegenerative diseases is becoming a predominant health and economic concern. Neurodegenerative diseases such as Alzheimer's disease, Creutzfeldt-Jakob disease (CJD), Parkinson's disease, Huntington's disease, frontotemporal dementia (FTD), and amyotrophic lateral sclerosis (ALS) result from the loss of a specific subsets of neurons, which is closely associated with accumulation and deposition of specific protein aggregates. Protein aggregation, or fibril formation, is a well-studied phenomenon that occurs in a nucleation-dependent growth reaction. Recently, there has been a swell of literature implicating protein aggregation and its ability to propagate cell-to-cell in the rapid progression of these diseases. In order for protein aggregation to be kindled in recipient cells it is a requisite that aggregates must be able to be released from one cell and then taken up by others. In this article we will explore the relationship between protein aggregates, their propagation and the role of macropinocytosis in their uptake. We highlight the ability of neurons to undergo stimulated macropinocytosis and identify potential therapeutic targets.
Introduction
Neurodegenerative diseases are closely linked to the formation and deposition of protein aggregates, quite often fibrillar, that accumulate intracellularly, such as α-synuclein in Parkinson's disease (PD), or extracellularly, such as the amyloid-beta (Aβ) peptide plaques associated with Alzheimer's disease (AD) (Chiti and Dobson, 2006). Although the peptides and proteins that aggregate are seemingly unrelated in terms of primary or tertiary structure, the resulting deposits are remarkably similar, often sharing a rope-like fibrillar morphology, a common cross-β core structure, and the ability to bind specific dyes such as thioflavin T and Congo red (Dobson, 2003). It has been postulated that oligomeric species present in solution prior to the appearance of fibrils are more likely to be responsible for cellular toxicity (Kayed et al., 2003). However, it is probable that all aggregate species provoke some level of cellular stress, although how protein aggregates induce cell injury is not fully understood. One potential mechanism of toxicity is mediated through exposed hydrophobic residues found on protein aggregates that have been shown to interact with cell surface receptors and membranes (Stefani and Dobson, 2003; Bolognesi et al., 2010), leading to membrane disruption and inappropriate signaling cascades.
Protein aggregation or fibril formation can be described as a nucleated self-assembly reaction. In this context, misfolded monomeric proteins or peptides must first aggregate to form stable nuclei from which fibril growth can occur via addition of further monomers. In vitro, using bulk measurements such as light scattering or thioflavin T fluorescence, protein aggregation reactions display sigmoidal growth kinetics (Figure 1). Initially, there is a lag phase which is thought to reflect the time it takes for the nuclei to form, and where the formation of fibrils is below the threshold of detection (Serio et al., 2000; Pedersen et al., 2004). In solution, there are two predominant species; monomers and fibrils (Figure 1A). While oligomeric aggregates are thought to be present in small amounts (i.e., < 2% of total species). During the elongation phase, the concentration of fibrils increases dramatically as the monomer concentration decreases. This is thought to be due to increasing numbers of actively growing fibrils via fragmentation of fibrils creating new “growing ends” and/or secondary nucleation (Figure 1B; where available sites on existing fibrils catalyze the nucleation of new aggregates) (Chiti and Dobson, 2006; Wilson et al., 2008; Arosio et al., 2014; Knowles et al., 2014). The lag phase can be circumvented by the addition of exogenous nuclei or “seeds” in the form of preformed fibrils (Jarrett and Lansbury, 1993). As a consequence of seeding, the lag time is eliminated (Jarrett et al., 1993) resulting in a first-order growth polymerization (Figure 1C).
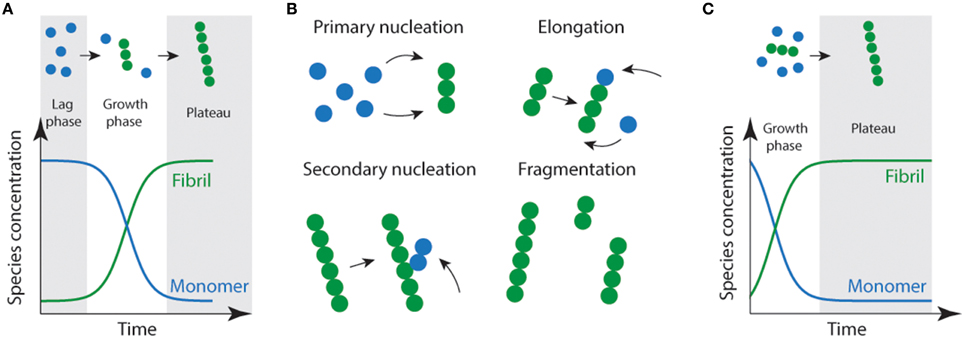
Figure 1. Schematic representation of amyloid fibril formation. (A) Fibril formation can be characterized by a lag phase where nucleation events occur, following critical nucleation a growth/elongation phase is observed which can proceed via primary (monomer addition) or secondary (fragmentation/secondary nucleation) events (B). During the latter stages, mature fibrils are formed which often display strong ThT emission signals. (C) Addition of fibrils or other functional seeds to the start of the reaction allows elongation to proceed without the requirement for primary nucleation removing the lag phase.
Patterns of Neurodegenerative Pathology in Humans
In major neurodegenerative diseases, such as Amyotrophic Lateral Sclerosis (ALS), Frontotemporal Dementia (FTD), AD, PD, and Huntington's diseases (HD), pathological changes such as loss of neurons and presence of pathological protein aggregates, typically follow distinctive anatomical patterns. The observed patterns are consistent with a spreading of pathology from one part of the brain to another (Brundin et al., 2010). The progression of these disorders, which is also associated with increasing clinical severity, has enabled the development of several staging systems for a range of neurodegenerative diseases (Braak et al., 2003, 2006; Brettschneider et al., 2013, 2014). The resulting patterns suggest that pathology is not only propagated between nearby cells, but that it also remotely connects regions of the brain along axonal pathways (Brundin et al., 2010; Brettschneider et al., 2015).
Propagation of Protein Aggregation and Neurodegenerative Disease
The patterns identified in neuropathological studies are consistent with propagation of protein misfolding and aggregation reminiscent of the prion diseases (Aguzzi, 2009). Prion diseases include human disorders such as Kuru and Creutzfeldt-Jakob disease (CJD), but also exist in animal populations in the form of scrapie in sheep, and bovine spongiform encephalopathy (BSE) in cattle (and several other species have their own version of prion-opathies). Prion diseases are infectious neurodegenerative diseases where the pathogenic agent is a misfolded/aggregated form of the prion protein which can seed misfolding and aggregation of the normal cellular prion protein in naïve cells, and indeed in naïve hosts transmitting pathology between cells and between individuals (Prusiner, 1998). The neurodegenerative diseases described in this review differ from prion diseases in one crucial respect; they are not infectious. Importantly, most neurodegenerative diseases do not involve the prion protein but typically a disease specific misfolded protein. However, it is useful to model the progression of neurodegenerative diseases associated with protein aggregation on the propagation of misfolded prion protein within an individual host. It may help provide an explanation for the apparent patterns of pathology in ALS, PD, and AD.
It has been known for decades that fibril formation can be seeded in vitro through addition of preformed fibrils, removing the rate limiting lag phase (see Figure 1). Aβ from the brains of AD patients can seed amyloid formation in non-human primates (Baker et al., 1993) and in mice engineered to produce large amounts of APP (Kane et al., 2000). This propagation is easily understandable given that Aβ aggregates do not have to cross plasma membranes to seed further aggregation, as amyloid plaques are present outside of cells. One might imagine that it would be less likely that aggregates formed inside one cell could propagate aggregation in another. However, there is a wave of research that demonstrates that this is possible (Clavaguera et al., 2009; Desplats et al., 2009; Ren et al., 2009). Evidence suggests that large fibrillar aggregates of a range of proteins (tau, α-synuclein, polyglutamine repeats, SOD1) are able to gain access to the cytoplasmic compartment via an incompletely understood mechanism and propagate misfolding and aggregation (Clavaguera et al., 2009; Desplats et al., 2009; Ren et al., 2009; Grad et al., 2014). Injection of brain/spinal cord extracts from transgenic mice expressing human tau, SOD1 or α-synuclein can seed pathology in the sites of injection and spread to other regions of the nervous system (Clavaguera et al., 2009; Mougenot et al., 2012; Ayers et al., 2014). Further, cell culture experiments have shown that these neurodegenerative disease associated protein aggregates can propagate cell-to-cell in neuronal and other cell lines, and into primary neurons (Hansen et al., 2011; Münch et al., 2011; Volpicelli-Daley et al., 2011; Furukawa et al., 2013; Guo and Lee, 2013; Grad et al., 2014). Moreover, cell culture experiments also show that insoluble material from brain tissue can seed aggregation of neuropathological TDP-43, whose cytoplasmic accumulation is associated with ALS (Furukawa et al., 2011). It is clear that these disease-associated aggregates are able to spread to naïve cells in culture gaining access to the cytosol in an unknown manner linked with fluid phase endocytosis. It is vital then to understand this mechanism to identify targets that will halt passage of the “infectious” particle in a strategy analogous to drugs blocking viral entry. Logically there are two vital steps that must occur in order to propagate aggregation between cells; aggregates must first be released in to the extracellular space from the cells in which they are made, and then be taken up by nearby cells to seed further aggregation in the cytosol of naïve cells.
Aggregates can be Released by Neurons
The mechanism of release of protein aggregates is incompletely understood, and it has been proposed that both active and passive routes of escape may be responsible for aggregate release (Brundin et al., 2010). For example, cell death may promote the non-specific release of aggregates from the cell or there may be active mechanisms responsible for release through exocytosis pathways (Figure 2A). SOD1 aggregates are generally found to be intracellular in human SOD1-linked familial ALS patient samples, however, there is evidence from cell culture models that these aggregates can escape from neurons and come into contact with nearby cells (Münch et al., 2011; Grad et al., 2014). There is evidence of α-synuclein release from neurons into the extracellular space via brefeldin A-insensitive unconventional exocytosis (Lee et al., 2005; Jang et al., 2010). The released α-synuclein is thought to be misfolded or aggregated (Jang et al., 2010) and is consistent with β-sheet-rich α-synuclein oligomeric aggregates (Kim et al., 2013). Tau aggregates have also been shown to be able to transfer between cells (Frost et al., 2009; Frost and Diamond, 2010; Dujardin et al., 2014). As in the case of anti-SOD1 antibodies (Grad et al., 2014) the use of anti-tau antibodies can block this tau transfer (Yanamandra et al., 2013). While cell lysates from mutant huntingtin-expressing cells promote propagation of aggregation in naïve acceptor cells, huntingtin aggregate release from neurons has not been conclusively demonstrated (Ren et al., 2009). The mechanism underpinning misfolded SOD1 release in cell culture models has been shown to be linked to both the release of free aggregates associated with cell death and to active release via exosomes (Grad et al., 2014). Collectively these data leave open the possibility that both cell death and active secretion mechanisms are acting in concert to promote aggregate release in vivo.
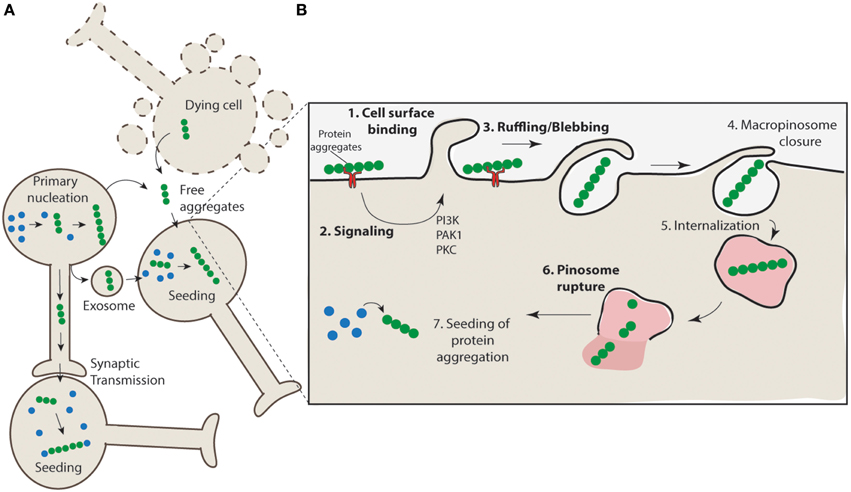
Figure 2. Propagation of aggregation and a proposed mechanism for aggregate uptake via macropinocytosis. (A) Protein aggregates form in neurons (primary nucleation) and have the potential to further nucleate the aggregation of other proteins. These protein aggregates can transfer directly from cell-to-cell through synaptic transfer, or be actively released via secretion mechanisms (e.g., exosomes) or in their naked form, either actively released from live cells or non-specifically released from dying cells, to neighboring interconnecting neurons. The uptake of such aggregates nucleates aggregation in naïve cells. (B) Aggregates may interact with cell surface receptors (such as HSG) (1) and promote the clustering and activation of signaling receptors such as receptor tyrosine kinases. This may result in the activation of signaling pathways such as those regulated by PAK1 and PKC (2) and subsequent mobilization of actin and formation of ruffles/blebs (3). Upon macropinosome closure (4) the structure is internalized (5). Given the unstructured nature of the macropinosomes it is likely that rigid aggregate structures may cause rupture (6) and allow access of the aggregates to the cytosol where nucleation of aggregation can proceed (7).
Aggregates can be Taken up by Neurons
In order to facilitate the propagation of intracellular aggregation in neurodegenerative diseases, such as ALS, PD, AD, and HD, active aggregate nuclei or seeds must gain access to the cytosol of naïve cells. Active nuclei could be large aggregates such as those that are macroscopically visible and accumulating in neurons (>2 microns in size), or small soluble oligomeric aggregates that might diffuse between cells. Fibrillar aggregates are generally 1–20 nm across and can be hundreds of nm long (Dobson, 2003), large aggregates that accumulate many fibrils in cells can be several μm in diameter (Farrawell et al., 2015). In this light, and given that neurons are not professional phagocytes it would be reasonable to assume that endocytosis of protein aggregates by neurons would be almost impossible. However, previous work has shown that exogenously applied SOD1 aggregates associated with ALS can be taken up efficiently and rapidly into living cells (Furukawa et al., 2013), such as neuronal cells (Neuro2a; N2a, NSC-34, and SH-SY5Y), in a time dependent manner (Münch et al., 2011; Sundaramoorthy et al., 2013).
In the context of PD, direct cell-to-cell transfer of aggregated α-synuclein has been demonstrated in humans, cell culture and animal models (Desplats et al., 2009; Lee et al., 2010). Post mortem studies were able to show that approximately 2–5% of normal embryonic neurons transplanted in the brains of PD patients acquired α-synuclein rich Lewy bodies over a period of 5 years (Brundin et al., 2008; Hansen et al., 2011). This phenomenon was also demonstrated in mice, where α-synuclein was shown to propagate from mouse host brain cells to grafted dopaminergic neurons (Hansen et al., 2011). Cell culture studies have shown that extracellular α-synuclein in various forms (fibrils, oligomers, and monomers) can be internalized by cultured neuronal cells (Lee et al., 2008). Furthermore, mouse cortical neuronal stem cells were shown to internalize extracellular aggregated α-synuclein either applied as recombinantly produced protein aggregates or from co-culture with cells overexpressing and subsequently releasing α-synuclein aggregates (Desplats et al., 2009). Together, these findings provide evidence that neurons are capable of engulfing even large α-synuclein structures. Tau aggregates, associated with AD and tauopathies such as FTD, have also been shown to be taken up into neurons. A recent study has shown that recombinant synthetic tau fibrils have been internalized in primary neurons derived from embryonic mouse hippocampus, and this triggered robust aggregation of endogenous soluble protein (Guo and Lee, 2013). Similarly, cell culture studies have shown that fibrillar polyQ aggregates (K2Q44K2), a model for polyQ expansion in huntingtin associated with HD, can efficiently enter into N2A cells and gain access to the cytosol to potentially nucleate the aggregation of otherwise soluble proteins (Ren et al., 2009). Collectively these data show that, despite their size, aggregates from a range of neurodegenerative diseases can be efficiently taken up by cells and most importantly this can occur in neurons. Given the similar structure and large size of these aggregates it is likely that similar mechanisms are used by subsets of neurons to engulf such large aggregates. Intuitively, the large size of the protein aggregates argues against neuronal entry by caveolae (generally used for particle sizes from 50 to 100 nm) (Richter et al., 2008) or clathrin-coated pits (for particle sizes < 200 nm; Traub, 2009). One potential mechanism that may explain the uptake of such large structures is the process of macropinocytosis.
Hijacking Macropinocytosis for Aggregate Entry into Cells
Macropinocytosis is a transient, actin-dependent process that leads to the internalization of fluid, membrane and other particles into large vacuoles (up to 5 μm). It is generally triggered by growth factors but can be triggered by a variety of particles such as bacteria, apoptotic bodies, necrotic cells and viruses (Mercer and Helenius, 2012). The activation of macropinocytosis induces membrane ruffling (or membrane extensions or even blebbing) that can fold back on to the cell, fusing with the plasma membrane and forming large fluid filled randomly sized vacuoles that lack supporting coating molecules (Meier et al., 2002). Several studies now suggest that macropinocytosis may be involved in the uptake of protein aggregates associated with various neurodegenerative diseases (Münch et al., 2011; Holmes et al., 2013; Sundaramoorthy et al., 2013; Grad et al., 2014; Tang et al., 2015).
Studies on the propagation of SOD1 aggregation by Münch et al. were the first to suggest that mutant SOD1 ALS protein aggregates enter into N2A cells via macropinocytosis (Münch et al., 2011). The work used a large panel of inhibitors of a range of cellular functions to systematically rule out specific pathways of endocytosis such as caveolin and clatherin-dependent endocytosis (Münch et al., 2011). Subsequently, these results were confirmed by a study that showed that uptake of both extracellular wild-type and mutant SOD1 soluble forms into NSC-34 cells can be inhibited by small molecule inhibitors of macropinocytosis EIPA and rottlerin (Sundaramoorthy et al., 2013). Similarly, EIPA was shown to inhibit the uptake of human wild-type SOD1 aggregates into NSC34 cells (Grad et al., 2014) consistent with macropinocytosis. EIPA (ethylisopropylamiloride) is an analog of amiloride that is an inhibitor of Na+/H+ exchangers. It is thought to be specific to macropinocytosis due to the susceptibility of GTPases such as Rac1 to pH changes (Koivusalo et al., 2010).
More broadly, uptake and cell-to-cell transmission of α-synuclein into neurons has been shown to be mediated via an unconventional endocytosis pathway (Lee et al., 2008; Desplats et al., 2009). Also, while initially synthetic polyglutamine fibrils were proposed to enter cells via direct penetration of the lipid bilayer (Ren et al., 2009) cell surfaces structures have been recently implicated in the binding and internalization of both synthetic polyglutamine (K2Q44K2) and huntingtin exon 1 (Htt exon1Q4444) fibrils suggesting a role for endocytosis mediated uptake (Trevino et al., 2012). A recent study was also able to show that the amyloid precursor protein (APP) can be rapidly internalized from the cell surface to lysosomes, bypassing early and late endosomes via macropinosome-like structures. This process was found to be mediated by Arf6, a regulator of macropinocytosis. A dominant negative mutant version of Arf6 inhibits transport of APP to lysosomes, and therefore reduces the secretion of Aβ (Tang et al., 2015). Further, exogenously added AD associated-recombinant tau fibrils have also been shown to be taken up by cultured cells in a process consistent with pinocytosis suggested by the co-localization of tau aggregates with Dextran, a marker of fluid phase endocytosis (Frost et al., 2009). In support of these findings, a recent study was able to show that small misfolded tau species are also internalized through the process of bulk endocytosis (Wu et al., 2013). Further to this, uptake of fibrillar tau into C17.2 cells (mouse neural stem cells) could be inhibited by both amiloride and rottlerin consistent with macropinocytosis (Holmes et al., 2013). The same study demonstrates that tau fibrils could be observed in vacuoles and invaginations with diameters of approximately 5 μm. Lastly, in a result that suggests stimulated macropinocytosis has been activated, the uptake of the fluid phase marker dextran was increased with increased doses of tau fibrils (Holmes et al., 2013). This uptake could be inhibited by suppressing cell binding of tau fibrils by blocking binding to heparan sulfate proteoglycans, indeed this also blocked uptake when aggregates were injected in to the brains of mice (Holmes et al., 2013). This was also shown in a more recent study where recombinant Tau assemblies (trimers) bound heparan sulfate proteoglycans on the cell surface to mediated Tau uptake and seeding into primary cortical neurons and HEK293 cells (Mirbaha et al., 2015). This binding stimulated macropinocytosis. However, how the binding to heparan sulfate proteoglycans on the cell surface relate to activation of macropinocytosis remains unclear.
In the context of cell-to-cell transfer of misfolded or aggregated protein, it is interesting to note that exosomes can contain misfolded and aggregated protein (Bellingham et al., 2012), and further, that exosomes can enter cells via macropinocytosis (Fitzner et al., 2011). However, additional work needs to be performed to confirm a role of macropinocytosis in exosome associated protein misfolded propagation.
Macropinocytosis in Neurons
The fact that neurons can engulf large particles such as protein aggregates at all seems counterintuitive. Endocytosis of large particles is usually thought of as restricted to professional phagocytes such as macrophages and microglia (Swanson, 2008; Mercer and Helenius, 2012). This is useful for engulfment of viral particles, bacteria, and fragments of dying cells in order to dispose of them. Without extensive degradation machinery, such as in macrophages, neurons are seemingly left vulnerable with the ability to take up large particles whilst not built to remove them. So why is it that neurons have the capacity to perform such a function? It is unlikely that neurons have evolved to specifically endocytose pathogens and large protein aggregates. Although, several viruses are known to enter neurons via macropinocytosis (Talekar et al., 2011; Hollidge et al., 2012; Kalia et al., 2013), this is likely due to the hijacking of machinery involved in macropinocytosis evolved for other purposes. Macropinocytosis, or closely related processes, are thought to regulate growth cone membrane recycling and are an integral part of growth cone collapse, axon retraction and turning during development and injury (Jurney et al., 2002; Tom et al., 2004; Kolpak et al., 2009; Kabayama et al., 2011). The membrane recycling process in growth cones is associated with actin-dependent membrane ruffles that fuse back on the plasma membrane creating large pinosomes dependent on PI3K and Rac1; all characteristic of macropinocytosis. Upon synaptogenesis this process is suppressed, consistent with the idea that mature neurons do not undergo bulk changes in the membrane via macropinocytosis (Bonanomi et al., 2008). However, axonal injury in in vitro and in vivo models triggers axonal remodeling in adult neurons that is associated with large amounts of fluid uptake (Tom et al., 2004) consistent with macropinocytosis. Interestingly, some disease associated mutations such as those in ALS2 (in ALS) and γPKC (in spinocerebellar ataxia 14) result in dysregulation of macropinocytosis (Otomo et al., 2008; Yamamoto et al., 2014) suggesting dysfunction of these processes are detrimental to large neurons.
Therapeutic Targeting of Macropinocytosis for Neurodegenerative Diseases
Collectively, the data summarized above suggests that macropinocytosis plays a role in allowing the passage of protein aggregates into naïve cells. Understanding neuron specific macropinocytosis mechanisms will be vital in identifying a target to slow disease progression. In particular, the intracellular pathways that result in activation of macropinocytosis, the formation and closure of macropinosomes, and importantly potential disintegration of macropinosomes must be examined. In an analogous situation, mechanisms underpinning Ebola virus entry via macropinocytosis are being scrutinized, with promising compounds targeting endocytosis and escape of viral particles from endosomes proving successful in mice (Sakurai et al., 2015). Macropinocytosis may be a viable target given that other cell types such as microglia that clear protein aggregates appear to be via different pathways (Roberts et al., 2013). It may be possible then to redirect aggregates from entering neurons by suppressing macropinocytosis in pathological conditions while maintaining receptor mediated phagocytic pathways utilized by microglia to engulf protein aggregates.
One potential target could be the cell surface receptor triggering activation of macropinocytosis (Figure 2B). Recent work suggests heparan sulfate proteoglycans are involved in the entry of tau aggregates, but how this relates to activation of macropinocytosis and to entry of other neurodegenerative disease associated aggregates is unclear (Holmes et al., 2013). Although there is no evidence in the context of aggregate activated macropinocytosis, previous work would suggest that receptor tyrosine kinases are involved in endocytosis via macropinocytosis (Kerr and Teasdale, 2009). Activation of receptor tyrosine kinases causes an increase in actin polymerization at the cell surface, resulting in an elevation in actin-mediated ruffling and therefore an increase in macropinosome formation, which is the mechanism distinguishing it from other endocytic pathways (Kerr and Teasdale, 2009). In addition to this, inhibition of RAC1 activity inhibits ruffle formation (Figure 2B) irrespective of receptor tyrosine kinase signaling (Lanzetti et al., 2004). These are two examples of pathways that could potentially be exploited to slow or stop the progression of toxic protein aggregates that enter cells via macropinocytosis.
Conclusions
Propagation of protein aggregation is implicated in the progressive nature of several neurodegenerative diseases such as AD, PD, and ALS. This may be explained by the nucleation of aggregation in nearby or connected neurons. In the cases where protein aggregates are intracellular, in order for this propagation to occur aggregates must be able to gain access to the cytosol of naïve neurons. Evidence is accumulating that implicates neuronal macropinocytosis in the uptake of protein aggregates. This may provide rational therapeutic targets that could stop the spread of pathology and halt these progressive neurodegenerative diseases in their tracks.
Funding
JJY is supported by a NHMRC career development fellowship 1084144.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aguzzi, A. (2009). Cell biology: beyond the prion principle. Nature 459, 924–925. doi: 10.1038/459924a
Arosio, P., Vendruscolo, M., Dobson, C. M., and Knowles, T. P. (2014). Chemical kinetics for drug discovery to combat protein aggregation diseases. Trends Pharmacol. Sci. 35, 127–135. doi: 10.1016/j.tips.2013.12.005
Ayers, J. I., Fromholt, S., Koch, M., deBosier, A., McMahon, B., Xu, G., et al. (2014). Experimental transmissibility of mutant SOD1 motor neuron disease. Acta Neuropathol. 128, 791–803. doi: 10.1007/s00401-014-1342-7
Baker, H. F., Ridley, R. M., Duchen, L. W., Crow, T. J., and Bruton, C. J. (1993). Evidence for the experimental transmission of cerebral beta-amyloidosis to primates. Int. J. Exp. Pathol. 74, 441–454.
Bellingham, S. A., Guo, B. B., Coleman, B. M., and Hill, A. F. (2012). Exosomes: vehicles for the transfer of toxic proteins associated with neurodegenerative diseases? Front. Physiol. 3:124. doi: 10.3389/fphys.2012.00124
Bolognesi, B., Kumita, J. R., Barros, T. P., Esbjorner, E. K., Luheshi, L. M., Crowther, D. C., et al. (2010). ANS binding reveals common features of cytotoxic amyloid species. ACS Chem. Biol. 5, 735–740. doi: 10.1021/cb1001203
Bonanomi, D., Fornasiero, E. F., Valdez, G., Halegoua, S., Benfenati, F., Menegon, A., et al. (2008). Identification of a developmentally regulated pathway of membrane retrieval in neuronal growth cones. J. Cell Sci. 121(Pt 22), 3757–3769. doi: 10.1242/jcs.033803
Braak, H., Alafuzoff, I., Arzberger, T., Kretzschmar, H., and Del Tredici, K. (2006). Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 112, 389–404. doi: 10.1007/s00401-006-0127-z
Braak, H., Del Tredici, K., Rub, U., de Vos, R. A., Jansen Steur, E. N., and Braak, E. (2003). Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol. Aging 24, 197–211. doi: 10.1016/S0197-4580(02)00065-9
Brettschneider, J., Del Tredici, K., Irwin, D. J., Grossman, M., Robinson, J. L., Toledo, J. B., et al. (2014). Sequential distribution of pTDP-43 pathology in behavioral variant frontotemporal dementia (bvFTD). Acta Neuropathol. 127, 423–439. doi: 10.1007/s00401-013-1238-y
Brettschneider, J., Del Tredici, K., Lee, V. M., and Trojanowski, J. Q. (2015). Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat. Rev. Neurosci. 16, 109–120. doi: 10.1038/nrn3887
Brettschneider, J., Del Tredici, K., Toledo, J. B., Robinson, J. L., Irwin, D. J., Grossman, M., et al. (2013). Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann. Neurol. 74, 20–38. doi: 10.1002/ana.23937
Brundin, P., Li, J. Y., Holton, J., Lindvall, O., and Revesz, T. (2008). Research in motion: the enigma of Parkinson's disease pathology spread. Nat. Rev. Neurosci. 9, 741–745. doi: 10.1038/nrn2477
Brundin, P., Melki, R., and Kopito, R. (2010). Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat. Rev. Mol. Cell Biol. 11, 301–307. doi: 10.1038/nrm2873
Chiti, F., and Dobson, C. M. (2006). Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366. doi: 10.1146/annurev.biochem.75.101304.123901
Clavaguera, F., Bolmont, T., Crowther, R. A., Abramowski, D., Frank, S., Probst, A., et al. (2009). Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 11, 909–913. doi: 10.1038/ncb1901
Desplats, P., Lee, H. J., Bae, E. J., Patrick, C., Rockenstein, E., Crews, L., et al. (2009). Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc. Natl. Acad. Sci. U.S.A. 106, 13010–13015. doi: 10.1073/pnas.0903691106
Dujardin, S., Lecolle, K., Caillierez, R., Begard, S., Zommer, N., Lachaud, C., et al. (2014). Neuron-to-neuron wild-type Tau protein transfer through a trans-synaptic mechanism: relevance to sporadic tauopathies. Acta Neuropathol. Commun. 2:14. doi: 10.1186/2051-5960-2-14
Farrawell, N. E., Lambert-Smith, I. A., Warraich, S. T., Blair, I. P., Saunders, D. N., Hatters, D. M., et al. (2015). Distinct partitioning of ALS associated TDP-43, FUS and SOD1 mutants into cellular inclusions. Sci. Rep. 5:13416. doi: 10.1038/srep13416
Fitzner, D., Schnaars, M., van Rossum, D., Krishnamoorthy, G., Dibaj, P., Bakhti, M., et al. (2011). Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis. J. Cell Sci. 124(Pt 3), 447–458. doi: 10.1242/jcs.074088
Frost, B., and Diamond, M. I. (2010). Prion-like mechanisms in neurodegenerative diseases. Nat. Rev. Neurosci. 11, 155–159. doi: 10.1038/nrn2786
Frost, B., Jacks, R. L., and Diamond, M. I. (2009). Propagation of tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 284, 12845–12852. doi: 10.1074/jbc.M808759200
Furukawa, Y., Kaneko, K., Watanabe, S., Yamanaka, K., and Nukina, N. (2011). A seeding reaction recapitulates intracellular formation of Sarkosyl-insoluble transactivation response element (TAR) DNA-binding protein-43 inclusions. J. Biol. Chem. 286, 18664–18672. doi: 10.1074/jbc.M111.231209
Furukawa, Y., Kaneko, K., Watanabe, S., Yamanaka, K., and Nukina, N. (2013). Intracellular seeded aggregation of mutant Cu, Zn-superoxide dismutase associated with amyotrophic lateral sclerosis. FEBS Lett. 587, 2500–2505. doi: 10.1016/j.febslet.2013.06.046
Grad, L. I., Yerbury, J. J., Turner, B. J., Guest, W. C., Pokrishevsky, E., O'Neill, M. A., et al. (2014). Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. U.S.A. 111, 3620–3625. doi: 10.1073/pnas.1312245111
Guo, J. L., and Lee, V. M. (2013). Neurofibrillary tangle-like tau pathology induced by synthetic tau fibrils in primary neurons over-expressing mutant tau. FEBS Lett. 587, 717–723. doi: 10.1016/j.febslet.2013.01.051
Hansen, C., Angot, E., Bergstrom, A. L., Steiner, J. A., Pieri, L., Paul, G., et al. (2011). α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Invest. 121, 715–725. doi: 10.1172/JCI43366
Hollidge, B. S., Nedelsky, N. B., Salzano, M. V., Fraser, J. W., Gonzalez-Scarano, F., and Soldan, S. S. (2012). Orthobunyavirus entry into neurons and other mammalian cells occurs via clathrin-mediated endocytosis and requires trafficking into early endosomes. J. Virol. 86, 7988–8001. doi: 10.1128/JVI.00140-12
Holmes, B. B., DeVos, S. L., Kfoury, N., Li, M., Jacks, R., Yanamandra, K., et al. (2013). Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. U.S.A. 110, E3138–E3147. doi: 10.1073/pnas.1301440110
Jang, A., Lee, H. J., Suk, J. E., Jung, J. W., Kim, K. P., and Lee, S. J. (2010). Non-classical exocytosis of alpha-synuclein is sensitive to folding states and promoted under stress conditions. J. Neurochem. 113, 1263–1274. doi: 10.1111/j.1471-4159.2010.06695.x
Jarrett, J. T., Berger, E. P., and Lansbury, P. T. Jr. (1993). The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease. Biochemistry 32, 4693–4697. doi: 10.1021/bi00069a001
Jarrett, J. T., and Lansbury, P. T. Jr. (1993). Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell 73, 1055–1058. doi: 10.1016/0092-8674(93)90635-4
Jurney, W. M., Gallo, G., Letourneau, P. C., and McLoon, S. C. (2002). Rac1-mediated endocytosis during ephrin-A2- and semaphorin 3A-induced growth cone collapse. J. Neurosci. 22, 6019–6028.
Kabayama, H., Takeuchi, M., Taniguchi, M., Tokushige, N., Kozaki, S., Mizutani, A., et al. (2011). Syntaxin 1B suppresses macropinocytosis and semaphorin 3A-induced growth cone collapse. J. Neurosci. 31, 7357–7364. doi: 10.1523/JNEUROSCI.2718-10.2011
Kalia, M., Khasa, R., Sharma, M., Nain, M., and Vrati, S. (2013). Japanese encephalitis virus infects neuronal cells through a clathrin-independent endocytic mechanism. J. Virol. 87, 148–162. doi: 10.1128/JVI.01399-12
Kane, M. D., Lipinski, W. J., Callahan, M. J., Bian, F., Durham, R. A., Schwarz, R. D., et al. (2000). Evidence for seeding of beta -amyloid by intracerebral infusion of Alzheimer brain extracts in beta -amyloid precursor protein-transgenic mice. J. Neurosci. 20, 3606–3611.
Kayed, R., Head, E., Thompson, J. L., McIntire, T. M., Milton, S. C., Cotman, C. W., et al. (2003). Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489. doi: 10.1126/science.1079469
Kerr, M. C., and Teasdale, R. D. (2009). Defining macropinocytosis. Traffic 10, 364–371. doi: 10.1111/j.1600-0854.2009.00878.x
Kim, C., Ho, D. H., Suk, J. E., You, S., Michael, S., Kang, J., et al. (2013). Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 4, 1562. doi: 10.1038/ncomms2534
Knowles, T. P., Vendruscolo, M., and Dobson, C. M. (2014). The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 15, 384–396. doi: 10.1038/nrm3810
Koivusalo, M., Welch, C., Hayashi, H., Scott, C. C., Kim, M., Alexander, T., et al. (2010). Amiloride inhibits macropinocytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. J. Cell Biol. 188, 547–563. doi: 10.1083/jcb.200908086
Kolpak, A. L., Jiang, J., Guo, D., Standley, C., Bellve, K., Fogarty, K., et al. (2009). Negative guidance factor-induced macropinocytosis in the growth cone plays a critical role in repulsive axon turning. J. Neurosci. 29, 10488–10498. doi: 10.1523/JNEUROSCI.2355-09.2009
Lanzetti, L., Palamidessi, A., Areces, L., Scita, G., and Fiore, P. P. D. (2004). Rab5 is a signalling GTPase involved in actin remodelling by receptor tyrosine kinases. Nature 20, 309–314. doi: 10.1038/nature02542
Lee, H., Suk, J., Bae, E., Lee, J., Paik, S., and Lee, S. (2008). Assembly-dependent endocytosis and clearance of extracellular alphasynuclein. Int. J. Biochem. Cell Biol. 40, 1835–1849. doi: 10.1016/j.biocel.2008.01.017
Lee, H. J., Patel, S., and Lee, S. J. (2005). Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J. Neurosci. 25, 6016–6024. doi: 10.1523/JNEUROSCI.0692-05.2005
Lee, H. J., Suk, J. E., Patrick, C., Bae, E. J., Cho, J. H., Rho, S., et al. (2010). Direct transfer of α-Synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem. 285, 9262–9272. doi: 10.1074/jbc.M109.081125
Meier, O., Boucke, K., Hammer, S. V., Keller, S., Stidwill, R. P., Hemmi, S., et al. (2002). Adenovirus triggers macropinocytosis and endosomal leakage together with its clathrin-mediated uptake. J. Cell Biol. 158, 1119–1131. doi: 10.1083/jcb.200112067
Mercer, J., and Helenius, A. (2012). Gulping rather than sipping: macropinocytosis as a way of virus entry. Curr. Opin. Microbiol. 15, 490–499. doi: 10.1016/j.mib.2012.05.016
Mirbaha, H., Holmes, B. B., Sanders, D. W., Bieschke, J., and Diamond, M. I. (2015). Tau trimers are the minimal propagation unit spontaneously internalized to seed intracellular aggregation. J. Biol. Chem. 290, 14893–14903. doi: 10.1074/jbc.M115.652693
Mougenot, A. L., Nicot, S., Bencsik, A., Morignat, E., Verchere, J., Lakhdar, L., et al. (2012). Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol. Aging 33, 2225–2228. doi: 10.1016/j.neurobiolaging.2011.06.022
Münch, C., O'Brien, J., and Bertolotti, A. (2011). Prion-like propagation of mutant superoxide dismutase1 misfolding in neuronal cells. Proc. Natl. Acad. Sci. U.S.A. 108, 3548–3553. doi: 10.1073/pnas.1017275108
Otomo, A., Kunita, R., Suzuki-Utsunomiya, K., Mizumura, H., Onoe, K., Osuga, H., et al. (2008). ALS2/alsin deficiency in neurons leads to mild defects in macropinocytosis and axonal growth. Biochem. Biophys. Res. Commun. 370, 87–92. doi: 10.1016/j.bbrc.2008.01.177
Pedersen, J. S., Christensen, G., and Otzen, D. E. (2004). Modulation of S6 fibrillation by unfolding rates and gatekeeper residues. J. Mol. Biol. 341, 575–588. doi: 10.1016/j.jmb.2004.06.020
Prusiner, S. B. (1998). Prions. Proc. Natl. Acad. Sci. U.S.A. 95, 13363–13383. doi: 10.1073/pnas.95.23.13363
Ren, P. H., Lauckner, J. E., Kachirskaia, I., Heuser, J. E., Melki, R., and Kopito, R. R. (2009). Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat. Cell Biol. 11, 219–225. doi: 10.1038/ncb1830
Richter, T., Floetenmeyer, M., Ferguson, C., Galea, J., Goh, J., Lindsay, M. R., et al. (2008). High-resolution 3D quantitative analysis of caveolar ultrastructure and caveola-cytoskeleton interactions. Traffic 9, 893–909. doi: 10.1111/j.1600-0854.2008.00733.x
Roberts, K., Zeineddine, R., Corcoran, L., Li, W., Campbell, I. L., and Yerbury, J. J. (2013). Extracellular aggregated Cu/Zn superoxide dismutase activates microglia to give a cytotoxic phenotype. Glia 61, 409–419. doi: 10.1002/glia.22444
Sakurai, Y., Kolokoltsov, A. A., Chen, C. C., Tidwell, M. W., Bauta, W. E., Klugbauer, N., et al. (2015). Ebola virus. Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science 347, 995–998. doi: 10.1126/science.1258758
Serio, T. R., Cashikar, A. G., Kowal, A. S., Sawicki, G. J., Moslehi, J. J., Serpell, L., et al. (2000). Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science 289, 1317–1321. doi: 10.1126/science.289.5483.1317
Stefani, M., and Dobson, C. M. (2003). Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 81, 678–699. doi: 10.1007/s00109-003-0464-5
Sundaramoorthy, V., Walker, A. K., Yerbury, J., Soo, K., Farg, M. A., Hoang, V., et al. (2013). Extracellular wildtype and mutant SOD1 induces ER-Golgi pathology characteristic of amyotrophic lateral sclerosis in neuronal cells. Cell. Mol. Life Sci. 70, 4181–4195. doi: 10.1007/s00018-013-1385-2
Swanson, J. A. (2008). Shaping cups into phagosomes and macropinosomes. Nat. Rev. Mol. Cell Biol. 9, 639–649. doi: 10.1038/nrm2447
Talekar, A., Pessi, A., and Porotto, M. (2011). Infection of primary neurons mediated by nipah virus envelope proteins: role of host target cells in antiviral action. J. Virol. 85, 8422–8426. doi: 10.1128/JVI.00452-11
Tang, W., Tam, J. H., Seah, C., Chiu, J., Tyrer, A., Cregan, S. P., et al. (2015). Arf6 controls beta-amyloid production by regulating macropinocytosis of the Amyloid Precursor Protein to lysosomes. Mol. Brain 8, 41. doi: 10.1186/s13041-015-0129-7
Tom, V. J., Steinmetz, M. P., Miller, J. H., Doller, C. M., and Silver, J. (2004). Studies on the development and behavior of the dystrophic growth cone, the hallmark of regeneration failure, in an in vitro model of the glial scar and after spinal cord injury. J. Neurosci. 24, 6531–6539. doi: 10.1523/JNEUROSCI.0994-04.2004
Traub, L. M. (2009). Clathrin couture: fashioning distinctive membrane coats at the cell surface. PLoS Biol. 7:e1000192. doi: 10.1371/journal.pbio.1000192
Trevino, R. S., Lauckner, J. E., Sourigues, Y., Pearce, M. M., Bousset, L., Melki, R., et al. (2012). Fibrillar structure and charge determine the interaction of polyglutamine protein aggregates with the cell surface. J. Biol. Chem. 287, 29722–29728. doi: 10.1074/jbc.M112.372474
Volpicelli-Daley, L. A., Luk, K. C., Patel, T. P., Tanik, S. A., Riddle, D. M., Stieber, A., et al. (2011). Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72, 57–71. doi: 10.1016/j.neuron.2011.08.033
Wilson, M. R., Yerbury, J. J., and Poon, S. (2008). Potential roles of abundant extracellular chaperones in the control of amyloid formation and toxicity. Mol. Biosyst. 4, 42–52. doi: 10.1039/B712728F
Wu, J. W., Herman, M., Liu, L., Simoes, S., Acker, C. M., Figueroa, H., et al. (2013). Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J. Biol. Chem. 288, 1856–1870. doi: 10.1074/jbc.M112.394528
Yamamoto, K., Seki, T., Yamamoto, H., Adachi, N., Tanaka, S., Hide, I., et al. (2014). Deregulation of the actin cytoskeleton and macropinocytosis in response to phorbol ester by the mutant protein kinase C gamma that causes spinocerebellar ataxia type 14. Front. Physiol. 5:126. doi: 10.3389/fphys.2014.00126
Keywords: protein aggregation, amyloid fibrils, macropinocytosis, endocytosis, propagation
Citation: Zeineddine R and Yerbury JJ (2015) The role of macropinocytosis in the propagation of protein aggregation associated with neurodegenerative diseases. Front. Physiol. 6:277. doi: 10.3389/fphys.2015.00277
Received: 17 July 2015; Accepted: 18 September 2015;
Published: 16 October 2015.
Edited by:
Katsuhiko Mikoshiba, RIKEN Brain Science Institute, JapanReviewed by:
Alexi Alekov, Medizinische Hochschule Hannover, GermanyKoji Yamanaka, Nagoya University, Japan
Copyright © 2015 Zeineddine and Yerbury. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Justin J. Yerbury, Faculty of Science, Medicine and Health, School of Biological Sciences, Illawarra Health and Medical Research Institute, Northfields Ave., Wollongong, NSW 2522, Australia,anllcmJ1cnlAdW93LmVkdS5hdQ==