 Ben Ke
Ben Ke Afei Zhang
Afei Zhang Xianfeng Wu
Xianfeng Wu Xiangdong Fang
Xiangdong Fang- Department of Nephrology, Nanchang University School of Medicine, Second Affiliated Hospital to Nanchang University, Nanchang, China
Chronic kidney disease (CKD) caused by renal fibrosis is an important public health concern. It is therefore necessary to understand the molecular pathogenesis of renal fibrosis in order to develop novel therapeutic strategies. KLF4 is the most extensively studied factor among the various members of the Krüppel-like factor (KLF) family of zinc finger-containing transcription factors. Many studies have demonstrated that KLF4 inhibits the activation of myofibroblasts and exerts an inhibitory effect on fibrosis. However, other studies have indicated that KLF4 may promote renal fibrosis. These controversial results suggest that KLF4 may be crucially involved in the development of renal fibrosis, although the underlying mechanism(s) remain unclear. Here, we summarize the recent progress made in understanding the role of KLF4 in renal fibrosis. Together, these findings suggest that KLF4 may participate in the development of renal fibrosis, but that its inhibition of fibrosis is greater than its promotion of the condition, which suggests that KLF4 may serve as a novel therapeutic target for renal fibrosis.
Introduction
Renal fibrosis is characterized by excessive proliferation of fibroblasts and increased deposition of extracellular matrix (ECM), which together lead to extensive scarring (fibrotic tubular and glomerular sclerosis), renal artery stenosis and chronic inflammatory cell infiltration (Li et al., 2015). The development of renal fibrosis is a complex process that involves the activation of molecules both intrinsic to the kidney and the infiltrated cells, which results in the deposition of ECM, ultimately leading to the loss of renal function. Tubulointerstitial fibrosis represents the end stage of renal fibrosis.
The Krüppel-like factor (KLF) family consists of 17 zinc finger-containing transcription factors, among which KLF4 is the most extensively studied (Black et al., 2001). KLF4 is essential for development and regulates a variety of processes, such as cell proliferation and differentiation (Black et al., 2001). Many studies have reported that KLF4 inhibits the activation of myofibroblasts and exhibits an inhibitory effect on fibrosis (Yang et al., 2013; Gras et al., 2015). However, in animal models of cardiac hypertrophy, KLF4 promoted myocardial fibrosis (Liao et al., 2010; Zhang et al., 2013). The function of KLF4 in the kidney is still not well understood. In this review, we highlight recent findings on how KLF4 regulates renal fibrosis, with the aim of evaluating the potential of KLF4 as a novel therapeutic target for renal fibrosis.
The Biological Characteristics of KLF4
KLF4 was first described by Shields et al. (1996) after identification of the gene from a mouse NIH3T3 cell cDNA library. KLF4 was formerly known as gut-enriched KLF, or epithelial zinc finger KLF, and contains several functional domains, including an N-terminal transcription activation domain that interacts with other proteins, a C-terminal zinc finger structure that combines with the DNA binding domain, and a transcription inhibition zone close to the N-terminal zinc finger structure (Bieker, 2001). KLF4 is expressed in many tissues, tumors, vascular smooth muscle cells (VSMC), and monocytes/macrophages, but it has been reported to be most highly expressed in colon cancer, the lungs (Yoshida and Hayashi, 2014), and the kidneys (Hayashi et al., 2014).
After binding to its specific protein partners, KLF4 activates or inhibits the transcription of target genes. KLF4 thus regulates numerous cellular processes, including proliferation, differentiation, apoptosis, migration, and invasion (Wang et al., 2015). Furthermore, KLF4 plays a specific role in cell reprogramming and differentiation, transforming somatic cells into induced pluripotent stem cells (IPS; D'Anselmi et al., 2013; Yamaguchi et al., 2014).
KLF4 is abundantly expressed in kidney podocyte cells, and its expression is decreased in proteinuric states (Hayashi et al., 2014). In the diabetic kidney, the KLF4 mRNA level is remarkably reduced. In an animal model of diabetic nephropathy, KLF4 expression was significantly decreased in renal tubular cells (Mreich et al., 2015). Renal fibrosis is considered to be one of the major pathological changes that occur during the course of diabetic nephropathy (Arora and Singh, 2013). Together, these findings suggest that a change in the KLF4 level may be closely related to renal fibrosis.
KLF4 and Renal Fibrosis
KLF4 and Kidney Inflammation
Inflammation is integral to the body's defense, although excessive inflammation is often considered to be the main driving force of fibrosis (Li et al., 2015). Inflammatory cells release large amounts of chemokines and vasoactive factors, such as monocyte chemotactic protein-1 (MCP-1) and angiotensin II, which contribute to the production of pro-fibrotic cytokines after kidney injury (Chung and Lan, 2011). At the site of injury, profibrogenic factors, such as interleukin (IL)-1, MCP-1, and macrophage migration inhibitory factor (MIF), stimulate the generation of myofibroblasts and the deposition of ECM, eventually leading to renal dysfunction (Mack and Yanagita, 2015). Therefore, kidney damage due to persistent inflammation is the originating factor for renal fibrosis (Meng et al., 2014).
KLF4 is involved in renal fibrosis by regulating inflammation (Mreich et al., 2015). MIF and MCP-1 are important inflammatory cytokines associated with kidney disease (Qi et al., 2006; Lan, 2008). It was recently demonstrated that KLF4 reduced inflammation by abrogating the transforming growth factor-β1 (TGF-β1)-induced production of MIF and MCP-1 in human renal tubular cells (Mreich et al., 2015). Consistently, in endothelial cells, KLF4 has shown anti-inflammatory effects by increasing the expression of endothelial nitric oxide synthase, decreasing inflammatory cell adhesion to the endothelial surface and prolonging the clotting time under inflammatory states (Hamik et al., 2007). In macrophages, however, the expression of TGF-β was shown to be regulated by the ratio of the M1 and M2 subtypes of macrophages (López-García et al., 2015), and KLF4 could activate epithelial factors and mediate proinflammatory signals, thereby exhibiting pro-inflammatory activity (Feinberg et al., 2005). The mechanism promoting such inflammation may be related to differentially regulated expression of KLF4 between the two subtypes of macrophages (M1/M2; Liao et al., 2011).
Nuclear factor-kappa B (NF-κB) is an important participant in a broad spectrum of inflammatory networks that regulate the cytokine activity in renal fibrosis (Wu et al., 2015). Upon NF-κB activation, KLF4 binds to the promoters of inflammatory cytokines, such as TNF-α and IL-6, to increase their transcription (Kaushik et al., 2010). In addition, KLF4 interacts with the p65 subunit of NF-κB to activate the transcription of proinflammatory genes and trigger inflammation (Autieri, 2008). KLF4 can also bind to high mobility group box-1 protein, an important mediator of systemic and local inflammatory responses, to achieve pro-inflammatory effects (Liu et al., 2008).
However, it remains unclear what function KLF4 plays at the onset of kidney inflammation. In particular, KLF4 generally shows anti-inflammatory effects in epithelial and endothelial cells, while it exhibits pro-inflammatory effects in other cell types (Figure 1).
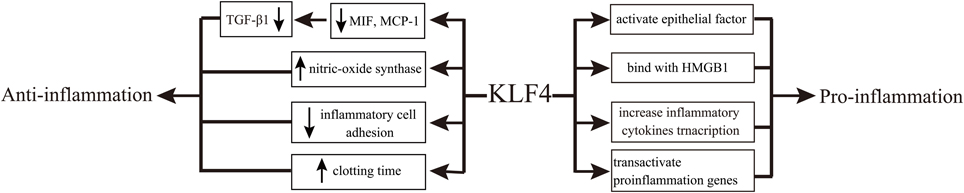
Figure 1. The role of KLF4 in kidney inflammation.
KLF4 and TGF-β
The TGF-β superfamily consists of three subtypes: TGF-β1, TGF-β2, and TGF-β3 (Piek et al., 1999). TGF-β plays important roles to promote cell proliferation and differentiation and induce the synthesis of the ECM. It is also known that TGF-β is a major cytokine/growth factor involved in renal fibrosis (Meng et al., 2012). Smad proteins, highly conserved transcription factors, are central to the signal transduction pathways that mediate the numerous effects of the TGF-β superfamily (Massagué, 2012). TGF-β1 strongly stimulates the renal tubular epithelial-to-mesenchymal transition (EMT), which is a crucial process in the development of tubulointerstitial fibrosis (Carew et al., 2012; Xiao et al., 2015).
A complex relationship exists between KLF4 and TGF-β, which plays an important role in the development of renal fibrosis. It was recently indicated that the regulation of cell proliferation and differentiation through TGF-β is mediated by KLF4, which binds Smad3 to modulate TGF-β-induced gene expression (Hu et al., 2007). Moreover, the overexpression of KLF4 significantly reduced the production of MIF and MCP-1, which are important mediators of TGF-β-induced renal fibrosis (Mreich et al., 2015). Surprisingly, in cardiac fibroblasts, KLF4 binds with TGF-β1 to upregulate the expression of TGF-β1, while this binding is suppressed by TGF-β signaling in macrophages (King et al., 2003; Figure 2).
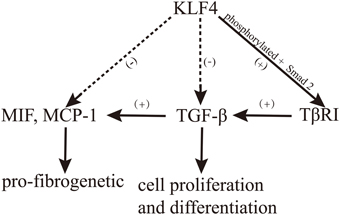
Figure 2. The role of KLF4 in TGF-β signaling.
TGF-β impacts KLF4 both directly and indirectly. In an indirect manner, TGF-β can quickly reduce the expression of KLF4 by inducing miRNA-143 and miRNA-145 (Davis-Dusenbery et al., 2011). In a direct manner, TGF-β1 induces KLF4 phosphorylation via the Smad and p38 MAPK signaling pathways, and phosphorylated KLF4 can, in turn, interact with Smad2 to cooperatively activate the promoter of TGF-β type I receptor (TβRI; Hu and Wan, 2011). Moreover, Cdh1-anaphase promoting complex (Cdh1/APC), a well-known regulator of mitosis (Peters, 2002), acts as a putative E3 ligase to control TGF-β-induced KLF4 degradation. Both depletion of Cdh1 by RNA interference and stabilization of KLF4 by disruption of its destruction box significantly attenuated the TGF-β-induced ubiquitylation and degradation of KLF4. Additionally, depletion of Cdh1 or stabilization of KLF4 antagonized the TGF-β-induced activation of target gene transcription (Hu and Wan, 2011). Meanwhile, KLF4 is degraded in response to TGF-β signaling through Cdh1/APC, which is catalyzed by KLF4 ubiquitylation (Hu and Wan, 2011). Taken together, these findings indicate that TGF-β plays an instrumental role in inducing KLF4 degradation (Figure 2). On the other hand, the overexpression of KLF4 inhibits the pro-fibrotic effects of TGF-β1 (Mreich et al., 2015).
EMT has been shown to play an active role in renal tubular fibrosis (Galichon and Hertig, 2011). DNA methylation, a type of epigenetic modification in mammals, plays a crucial role in the regulation of gene transcription, which is closely related to EMT (Carmona et al., 2014). DNA methyltransferase 1 (DNMT1) is responsible for the maintenance of pre-existing DNA methylation patterns after replication (Goll and Bestor, 2005). In in vivo and in vitro models of renal EMT, the KLF4 mRNA and protein levels were reduced compared with the control, and the downregulation of KLF4 was found to be due to DNMT1-mediated KLF4 promoter hypermethylation, which contributed to the progression of EMT in renal epithelial cells (Xiao et al., 2015). In tumor cells, KLF4 also demonstrated the ability to regulate EMT (Chen et al., 2014). In particular, KLF4 activated the transcription of the epithelial cell marker E-cadherin and repressed the expression of a mesenchymal cell marker, snail 2 (slug), by binding to their respective promoters.
KLF4 and Vascular Damage
In renal fibrosis, the significant changes in the structure and function of peritubular capillaries and the apoptosis of peritubular capillary endothelial cells give rise to capillary loss, tissue hypoxia and oxidative stress. At the same time, the loss of capillaries causes hypoxia or a reduction in the local nutrient supply, aggravating the renal fibrosis and resulting in kidney damage (Cao et al., 2012; Chapal et al., 2013). Moreover, changes in the renal tubular capillary structure and function lead to renal interstitial inflammation and a decrease or loss of renal tubular capillaries, eventually leading to further renal fibrosis. Thus, vascular damage is an essential factor contributing to renal fibrosis (Yamaguchi et al., 2012).
Great efforts have been made to understand the role of KLF4 in VSMC, and KLF4 is known to inhibit VSMC differentiation and proliferation through multiple pathways (Shi and Chen, 2014). For example, it has been shown that TGF-β control element (TCE), a smooth muscle (SM) alpha-actin promoter required for the TGF-β inducibility of SM alpha-actin in SM cells, is needed for gene transcription in VSMC (Liu et al., 2003). KLF4 is a trans-acting factor with TCE (Adam et al., 2000). Specificity protein (Sp) 1, a ubiquitous transcription factor that mediates the transcription of ECM genes (Verrecchia et al., 2001), forms a complex with KLF4 and binds TCE at the promoter of the angiotensin II type 1 receptor (AT1R) in the absence of TGF-β to maintain a basal expression level of AT1R in VSMC. Upon TGF-β activation, the expression of AT1R is suppressed through TGF-β1-mediated dissociation of the KLF4-Sp1 complex from TCE at the AT1R promoter (Zhang et al., 2012). Moreover, in VSMC, phosphorylated KLF4 can activate the TGF-β1 receptor promoter by interacting with Smad2 (Zhang et al., 2012). Furthermore, KLF4 exhibited an inhibitory effect on the proliferation of VSMC via binding to p53 (Yoshida et al., 2008).
Serum response factor (SRF) regulates the expression of VSMC differentiation markers by cooperating with its co-activator, myocardin, or its co-repressor, phosphorylated KLF4 (Wang et al., 2001). KLF4 also suppresses the expression of VSMC contractile markers by interacting with SRF (Hu et al., 2014). Therefore, KLF4 plays a catalytic role in renal fibrosis by inhibiting the differentiation and proliferation of VSMC.
KLF4 in Renal Fibrosis
Recently, Chen et al. showed that KLF4 was indeed involved in the regulation of renal physiological functions and the progression of fibrosis (Chen et al., 2015). In two in vivo models of unilateral ureteral obstruction, a decrease in KLF4 expression was observed (Chen et al., 2015) and (Xiao et al., 2015), indicating that KLF4 has anti-fibrotic action in the kidney. Nevertheless, the role of KLF4 in renal fibrosis is still not clear, there is a need for further study.
Conclusions and Perspectives
The role of KLF4 in renal fibrosis is complex. For instance, KLF4 can induce the transformation of primary fibroblasts (Rowland et al., 2005) but can also induce fibroblasts to become pluripotent stem cells in mice (Takahashi and Yamanaka, 2006; Okita et al., 2007; Wernig et al., 2007). These findings make the function of KLF4 in renal fibrosis confusing; nevertheless, the findings described above indicate that KLF4 participates in the development of renal fibrosis and that its inhibition of fibrosis is greater than its promotion of the condition.
Because KLF4 is expressed in kidney cells (Hayashi et al., 2014), the ability to reprogramming somatic cells into IPS cells makes it possible to utilize KLF4 as a novel therapeutic target for Chronic kidney disease (CKD; D'Anselmi et al., 2013; Yamaguchi et al., 2014). Moreover, epigenetic modifications are known to play an essential role in kidney function and development (Dressler, 2008), and overexpression of KLF4 can reduce DNA methylation at the nephrin promoter, increasing the expression of nephrin and reducing the expression of mesenchymal genes (Hayashi et al., 2014). KLF4 and epigenetic modulation can thus be targeted as part of an intervention to treat proteinuria (Hayashi and Itoh, 2015). In addition, KLF4 can reduce the inflammation stimulated by TGF-β1 in cases of renal fibrosis caused by diabetic nephropathy (Mreich et al., 2015). Taken together, these findings strongly suggest that KLF4 represents a potential therapeutic target for renal fibrosis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank XW for guidance on this article. This work was supported by grants from the National Natural Science Foundation of China (General Program 81460142).
References
Adam, P. J., Regan, C. P., Hautmann, M. B., and Owens, G. K. (2000). Positive- and negative-acting Kruppel-like transcription factors bind a transforming growth factor beta control element required for expression of the smooth muscle cell differentiation marker SM22 alpha in vivo. J. Biol. Chem. 275, 37798–37806. doi: 10.1074/jbc.M006323200
Arora, M. K., and Singh, U. K. (2013). Molecular mechanisms in the pathogenesis of diabetic nephropathy: an update. Vascul. Pharmacol. 58, 259–271. doi: 10.1016/j.vph.2013.01.001
Autieri, M. V. (2008). Kruppel-like factor 4: transcriptional regulator of proliferation, or inflammation, or differentiation, or all three? Circ. Res. 102, 1455–1457. doi: 10.1161/CIRCRESAHA.108.178954
Bieker, J. J. (2001). Kruppel-like factors: three fingers in many pies. J. Biol. Chem. 276, 34355–34358. doi: 10.1074/jbc.R100043200
Black, A. R., Black, J. D., and Azizkhan-Clifford, J. (2001). Sp1 and kruppel-like factor family of transcription factors in cell growth regulation and cancer. J. Cell. Physiol. 188, 143–160. doi: 10.1002/jcp.1111
Cao, N., Feng, J., Bai, J., Sun, L., Li, S., Ma, J., et al. (2012). Netrin-1 attenuates the progression of renal dysfunction by inhibiting peritubular capillary loss and hypoxia in 5/6 nephrectomized rats. Kidney Blood Press. Res. 36, 209–219. doi: 10.1159/000343410
Carew, R. M., Wang, B., and Kantharidis, P. (2012). The role of EMT in renal fibrosis. Cell Tissue Res. 347, 103–116. doi: 10.1007/s00441-011-1227-1
Carmona, F. J., Davalos, V., Vidal, E., Gomez, A., Heyn, H., Hashimoto, Y., et al. (2014). A comprehensive DNA methylation profile of epithelial-to-mesenchymal transition. Cancer Res. 74, 5608–5619. doi: 10.1158/0008-5472.can-13-3659
Chapal, M., Néel, M., Le Borgne, F., Meffray, E., Carceles, O., Hourmant, M., et al. (2013). Increased soluble Flt-1 correlates with delayed graft function and early loss of peritubular capillaries in the kidney graft. Transplantation 96, 739–744. doi: 10.1097/TP.0b013e31829f4772
Chen, W.-C., Lin, H.-H., and Tang, M.-J. (2015). Matrix-stiffness–regulated inverse expression of Krüppel-like factor 5 and Krüppel-like factor 4 in the pathogenesis of renal fibrosis. Am. J. Pathol. 185, 2468–2481. doi: 10.1016/j.ajpath.2015.05.019
Chen, Z., Wang, Y., Liu, W., Zhao, G., Lee, S., Balogh, A., et al. (2014). Doxycycline inducible Kruppel-like factor 4 lentiviral vector mediates mesenchymal to epithelial transition in ovarian cancer cells. PLoS ONE 9:e105331. doi: 10.1371/journal.pone.0105331
Chung, A. C. K., and Lan, H. Y. (2011). Chemokines in renal injury. J. Am. Soc. Nephrol. 22, 802–809. doi: 10.1681/asn.2010050510
D'Anselmi, F., Masiello, M. G., Cucina, A., Proietti, S., Dinicola, S., Pasqualato, A., et al. (2013). Microenvironment promotes tumor cell reprogramming in human breast cancer cell lines. PLoS ONE 8:e83770. doi: 10.1371/journal.pone.0083770
Davis-Dusenbery, B. N., Chan, M. C., Reno, K. E., Weisman, A. S., Layne, M. D., Lagna, G., et al. (2011). Down-regulation of Kruppel-Like Factor-4 (KLF4) by MicroRNA-143/145 is critical for modulation of vascular smooth muscle cell phenotype by transforming growth factor-beta and bone morphogenetic protein 4. J. Biol. Chem. 286, 28097–28110. doi: 10.1074/jbc.M111.236950
Dressler, G. R. (2008). Epigenetics, development, and the kidney. J. Am. Soc. Nephrol. 19, 2060–2067. doi: 10.1681/asn.2008010119
Feinberg, M. W., Cao, Z., Wara, A. K., Lebedeva, M. A., Senbanerjee, S., and Jain, M. K. (2005). Kruppel-like factor 4 is a mediator of proinflammatory signaling in macrophages. J. Biol. Chem. 280, 38247–38258. doi: 10.1074/jbc.M509378200
Galichon, P., and Hertig, A. (2011). Epithelial to mesenchymal transition as a biomarker in renal fibrosis: are we ready for the bedside? Fibrogenesis Tissue Repair 4:11. doi: 10.1186/1755-1536-4-11
Goll, M. G., and Bestor, T. H. (2005). Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 74, 481–514. doi: 10.1146/annurev.biochem.74.010904.153721
Gras, C., Ratuszny, D., Hadamitzky, C., Zhang, H., Blasczyk, R., and Figueiredo, C. (2015). miR-145 contributes to hypertrophic scarring of the skin by inducing myofibroblast activity. Mol. Med. 21, 296–304. doi: 10.2119/molmed.2014.00172
Hamik, A., Lin, Z., Kumar, A., Balcells, M., Sinha, S., Katz, J., et al. (2007). Kruppel-like factor 4 regulates endothelial inflammation. J. Biol. Chem. 282, 13769–13779. doi: 10.1074/jbc.M700078200
Hayashi, K., and Itoh, H. (2015). Transcription factors and epigenetic modulation: its therapeutic implication in chronic kidney disease. Arch. Immunol. Ther. Exp. (Warsz.) 63, 193–196. doi: 10.1007/s00005-014-0326-6
Hayashi, K., Sasamura, H., Nakamura, M., Azegami, T., Oguchi, H., Sakamaki, Y., et al. (2014). KLF4-dependent epigenetic remodeling modulates podocyte phenotypes and attenuates proteinuria. J. Clin. Invest. 124, 2523–2537. doi: 10.1172/jci69557
Hu, B., Song, J. T., Qu, H. Y., Bi, C. L., Huang, X. Z., Liu, X. X., et al. (2014). Mechanical stretch suppresses microRNA-145 expression by activating extracellular signal-regulated kinase 1/2 and upregulating angiotensin-converting enzyme to alter vascular smooth muscle cell phenotype. PLoS ONE 9:e96338. doi: 10.1371/journal.pone.0096338
Hu, B., Wu, Z., Liu, T., Ullenbruch, M. R., Jin, H., and Phan, S. H. (2007). Gut-enriched Krüppel-like factor interaction with Smad3 inhibits myofibroblast differentiation. Am. J. Respir. Cell Mol. Biol. 36, 78–84. doi: 10.1165/rcmb.2006-0043OC
Hu, D., and Wan, Y. (2011). Regulation of Kruppel-like factor 4 by the anaphase promoting complex pathway is involved in TGF-beta signaling. J. Biol. Chem. 286, 6890–6901. doi: 10.1074/jbc.M110.179952
Kaushik, D. K., Gupta, M., Das, S., and Basu, A. (2010). Kruppel-like factor 4, a novel transcription factor regulates microglial activation and subsequent neuroinflammation. J. Neuroinflammation 7:68. doi: 10.1186/1742-2094-7-68
King, K. E., Iyemere, V. P., Weissberg, P. L., and Shanahan, C. M. (2003). Kruppel-like factor 4 (KLF4/GKLF) is a target of bone morphogenetic proteins and transforming growth factor beta 1 in the regulation of vascular smooth muscle cell phenotype. J. Biol. Chem. 278, 11661–11669. doi: 10.1074/jbc.M211337200
Lan, H. Y. (2008). Role of macrophage migration inhibition factor in kidney disease. Nephron. Exp. Nephrol. 109, e79–e83. doi: 10.1159/000145463
Li, R. X., Yiu, W. H., and Tang, S. C. W. (2015). Role of bone morphogenetic protein-7 in renal fibrosis. Front. Physiol. 6:114. doi: 10.3389/fphys.2015.00114
Liao, X., Haldar, S. M., Lu, Y., Jeyaraj, D., Paruchuri, K., Nahori, M., et al. (2010). Kruppel-like factor 4 regulates pressure-induced cardiac hypertrophy. J. Mol. Cell. Cardiol. 49, 334–338. doi: 10.1016/j.yjmcc.2010.04.008
Liao, X., Sharma, N., Kapadia, F., Zhou, G., Lu, Y., Hong, H., et al. (2011). Kruppel-like factor 4 regulates macrophage polarization. J. Clin. Invest. 121, 2736–2749. doi: 10.1172/jci45444
Liu, J., Liu, Y., Zhang, H., Chen, G., Wang, K., and Xiao, X. (2008). KLF4 promotes the expression, translocation, and release of HMGB1 IN RAW264.7 macrophages in response to LPS. Shock 30, 260–266. doi: 10.1097/SHK.0b013e318162bef7
Liu, Y., Sinha, S., and Owens, G. (2003). A transforming growth factor-beta control element required for SM alpha-actin expression in vivo also partially mediates GKLF-dependent transcriptional repression. J. Biol. Chem. 278, 48004–48011. doi: 10.1074/jbc.M301902200
López-Garcia, S., Castañed-Sanchéz, J. I., Jiménez-Arellanes, A., Domínguez-López, L., Castro-Mussot, M. E., Hernández-Sanchéz, J., et al. (2015). Macrophage activation by ursolic and oleanolic acids during mycobacterial infection. Molecules 20, 14348–14364. doi: 10.3390/molecules200814348
Mack, M., and Yanagita, M. (2015). Origin of myofibroblasts and cellular events triggering fibrosis. Kidney Int. 87, 297–307. doi: 10.1038/ki.2014.287
Massagué, J. (2012). TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 13, 2105–2124. doi: 10.1038/nrm3434
Meng, X.-M., Huang, X. R., Xiao, J., Chung, A. C. K., Qin, W., Chen, H.-Y., et al. (2012). Disruption of Smad4 impairs TGF-beta/Smad3 and Smad7 transcriptional regulation during renal inflammation and fibrosis in vivo and in vitro. Kidney Int. 81, 266–279. doi: 10.1038/ki.2011.327
Meng, X.-M., Nikolic-Paterson, D. J., and Lan, H. Y. (2014). Inflammatory processes in renal fibrosis. Nat. Rev. Nephrol. 10, 493–503. doi: 10.1038/nrneph.2014.114
Mreich, E., Chen, X.-M., Zaky, A., Pollock, C. A., and Saad, S. (2015). The role of Kruppel-like factor 4 in transforming growth factor-beta-induced inflammatory and fibrotic responses in human proximal tubule cells. Clin. Exp. Pharmacol. Physiol. 42, 680–686. doi: 10.1111/1440-1681.12405
Okita, K., Ichisaka, T., and Yamanaka, S. (2007). Generation of germline-competent induced pluripotent stem cells. Nature 448, 313–317. doi: 10.1038/nature05934
Peters, J. M. (2002). The anaphase-promoting complex: proteolysis in mitosis and beyond. Mol. Cell 9, 931–943. doi: 10.1016/S1097-2765(02)00540-3
Piek, E., Heldin, C. H., and Ten, D. P. (1999). Specificity, diversity, and regulation in TGF-beta superfamily signaling. FASEB J. 13, 2105–2124.
Qi, W., Chen, X., Holian, J., Mreich, E., Twigg, S., Gilbert, R. E., et al. (2006). Transforming growth actor-beta1 differentially mediates fibronectin and inflammatory cytokine expression in kedney tubular cells. Am. J. Physiol. Renal Physiol. 291, 1070–1077. doi: 10.1152/ajprenal.00013.2006
Rowland, B. D., Bernards, R., and Peeper, D. S. (2005). The KLF4 tumour suppressor is a transcriptional repressor of p53 that acts as a context-dependent oncogene. Nat. Cell Biol. 7, 1074–1082. doi: 10.1038/ncb1314
Shi, N., and Chen, S.-Y. (2014). Mechanisms simultaneously regulate smooth muscle proliferation and differentiation. J. Biomed. Res. 28, 40–46. doi: 10.7555/jbr.28.20130130
Shields, J. M., Christy, R. J., and Yang, V. W. (1996). Identification and characterization of a gene encoding a gut-enriched Kruppel-like factor expressed during growth arrest. J. Biol. Chem. 271, 20009–20017.
Takahashi, K., and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. doi: 10.1016/j.cell.2006.07.024
Verrecchia, F., Rossert, J., and Mauviel, A. (2001). Blocking Sp1 transcription factor broadly inhibits extracellular matrix gene expression in vitro and in vivo: implications for the treatment of tissue fibrosis. J. Invest. Dermatol. 116, 755–763. doi: 10.1046/j.1523-1747.2001.01326.x
Wang, B., Zhao, M.-Z., Cui, N.-P., Lin, D.-D., Zhang, A.-Y., Qin, Y., et al. (2015). Kruppel-like factor 4 induces apoptosis and inhibits tumorigenic progression in SK-BR-3 breast cancer cells. FEBS Open Bio. 5, 147–154. doi: 10.1016/j.fob.2015.02.003
Wang, D. Z., Chang, P. S., Wang, Z. G., Sutherland, L., Richardson, J. A., Small, E., et al. (2001). Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell 105, 851–862. doi: 10.1016/s0092-8674(01)00404-4
Wernig, M., Meissner, A., Foreman, R., Brambrink, T., Ku, M., Hochedlinger, K., et al. (2007). In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature 448, 318–324. doi: 10.1038/nature05944
Wu, J. S., Shi, R., Lu, X., Ma, Y. M., and Cheng, N. N. (2015). Combination of active components of Xiexin decoction ameliorates renal fibrosis through the inhibition of NF-kappaB and TGF-beta1/Smad pathways in db/db diabetic mice. PLoS ONE 10:e0122661. doi: 10.1371/journal.pone.0122661
Xiao, X., Tang, W., Yuan, Q., Peng, L., and Yu, P. (2015). Epigenetic repression of Krupple-like factor 4 through Dnmt1 contributes to EMT in renal fibrosis. Int. J. Mol. Med. 35, 1596–1602. doi: 10.3892/ijmm.2015.2189
Yamaguchi, I., Tchao, B. N., Burger, M. L., Yamada, M., Hyodo, T., Giampietro, C., et al. (2012). Vascular endothelial cadherin modulates renal interstitial fibrosis. Nephron. Exp. Nephrol. 120, E20–E31. doi: 10.1159/000332026
Yamaguchi, S., Marumoto, T., Nii, T., Kawano, H., Liao, J., Nagai, Y., et al. (2014). Characterization of common marmoset dysgerminoma-like tumor induced by the lentiviral expression of reprogramming factors. Cancer Sci. 105, 402–408. doi: 10.1111/cas.12367
Yang, S., Cui, H., Xie, N., Icyuz, M., Banerjee, S., Antony, V. B., et al. (2013). miR-145 regulates myofibroblast differentiation and lung fibrosis. FASEB J. 27, 2382–2391. doi: 10.1096/fj.12-219493
Yoshida, T., and Hayashi, M. (2014). Role of Kruppel-like factor 4 and its binding proteins in vascular disease. J. Atheroscler. Thromb. 21, 402–413. doi: 10.5551/jat.23044
Yoshida, T., Kaestner, K. H., and Owens, G. K. (2008). Conditional deletion of Kruppel-like factor 4 delays downregulation of smooth muscle cell differentiation markers but accelerates neointimal formation following vascular injury. Circ. Res. 102, 1548–1557. doi: 10.1161/circresaha.108.176974
Zhang, X.-H., Zheng, B., Gu, C., Fu, J.-R., and Wen, J.-K. (2012). TGF-beta 1 downregulates AT1 receptor expression via PKC-delta-mediated Sp1 dissociation from KLF4 and Smad-mediated PPAR-gamma association With KLF4. Arterioscl. Throm. Vas. 32, U1015–U1405. doi: 10.1161/atvbaha.111.244962
Keywords: KLF4, renal fibrosis, inflammation, TGF-β, vascular disease
Citation: Ke B, Zhang A, Wu X and Fang X (2015) The Role of Krüppel-like Factor 4 in Renal Fibrosis. Front. Physiol. 6:327. doi: 10.3389/fphys.2015.00327
Received: 05 August 2015; Accepted: 29 October 2015;
Published: 12 November 2015.
Edited by:
Christine Kranz, University of Ulm, GermanyReviewed by:
Roger Evans, Monash University, AustraliaHui Y. Lan, The Chinese University of Hong Kong, China
Copyright © 2015 Ke, Zhang, Wu and Fang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiangdong Fang, eGlhbmdkb25nZmFuZzgxOEBzaW5hLmNvbQ==