 Norifumi Kawada
Norifumi Kawada- Department of Hepatology, Graduate School of Medicine, Osaka City University, Osaka, Japan
Myofibroblasts play important roles in inflammation, fibrosis and tumorigenesis in chronically inflamed liver. Liver myofibroblasts originate from hepatic stellate cells, portal fibroblasts or mesothelial cells, and they are localized in and around fibrotic septum and portal tracts. Liver myofibroblasts are the source of extracellular matrix materials, including type I collagen and multiple fibrogenic growth factors, such as transforming growth factor-β and vascular endothelial growth factor. Although a detailed characterization of the function of individual myofibroblasts has not been conducted, owing to the lack of appropriate cell markers, recent lineage-tracing technology has revealed the limited contribution of myofibroblasts that are derived from portal fibroblasts to various types of liver fibrosis, as compared with the contribution of hepatic stellate cells. In addition, cytoglobin, which is the fourth globin in mammals and function as a local gas sensor, provides a new perspective on the involvement of stellate cells in fibrosis and carcinogenesis, possibly through its anti-oxidative properties and is a promising new marker that discriminates between myofibroblasts derived from stellate cells and those from portal fibroblasts.
Introduction
The myofibroblast was originally described by Gabbiani in the granulation tissue of healing wounds as a cell type that had characteristics of fibroblasts and smooth muscle cells (Gabbiani et al., 1971).
In the liver, myofibroblasts were first identified in hepatic schistosomal fibrosis (Grimaud and Borojevic, 1977); this was followed by the identification of contractile fibroblasts (myofibroblasts) in chronic alcoholic cirrhosis (Rudolph et al., 1979).
Liver myofibroblasts are featured by the expression of α-smooth muscle actin (α-SMA), which is encoded by ACTA2 gene located in 10q22-q24 in the human genome (Schmitt-Gräff et al., 1991). The origin of liver myofibroblasts is complex: at least three cellular sources—hepatic stellate cells (HSCs), portal fibroblasts (PFs), and mesothelial cells (MCs)—are considered to transdifferentiate to liver myofibroblasts (Yin et al., 2013). The contribution of each type of myofibroblast to the development or resolution of liver fibrosis varies depending on the etiology of liver disease.
Conversely, cytoglobin (Cygb) belongs to the mammalian globin family, which consists of myoglobin, hemoglobin, neuroglobin, and androglobin (Burmester and Hankeln, 2014). Cygb was accidentally discovered in rat HSCs during the proteomics analysis of proteins that are expressed in HSCs. The amino acid sequence and gas-binding ability of Cygb are similar to those of myoglobin, thus suggesting that these two globins have the same primary function in pathophysiology (Kawada et al., 2001; Burmester et al., 2002; Trent and Hargrove, 2002). However, recent studies have suggested that Cygb is involved in the carcinogenesis of various organs, which is different from the function of myoglobin, a gas-binding sensor that affects muscle cell contraction (Shivapurkar et al., 2008; Shaw et al., 2009; Latina et al., 2015).
In this review, we will discuss the role of Cygb, which is uniquely expressed in HSCs in the liver, in the myofibroblastic transformation of HSCs and carcinogenesis in the liver.
Liver Myofibroblasts
In 2002, Cassiman et al. proposed the presence of at least four different subpopulations of myofibroblasts in human liver. Portal myofibroblasts and septal myofibroblasts are located in the extended collagenous area around portal tracts and in the internal region of fibrotic septa, respectively. These myofibroblasts are strongly positive for α-SMA, and they exhibit a variable positivity to glial fibrillar acidic protein (GFAP), brain-derived nerve growth factor (BDNF) and α-B-crystallin (ABCRYS). The third type of myofibroblasts is interface myofibroblasts, which present at the edge between fibrotic septa and the surrounding hepatic parenchyma, which are positive for α-SMA, GFAP, and ABCRYS as well as BDNF, neuronal cell adhesion molecule (N-CAM), neuronal growth factor (NGF) and neurotrophin 4 (NT-4). The fourth type is activated HSCs that are located in or around sinusoidal capillary and within pseudo-lobules and express intensively α-SMA and N-CAM and are positive for GFAP, NGF, BDNF, NT-3, and synaptophysin as well as NT-4, ABCRYS, p75 and the NT receptors tyrosine kinase A and B. Cassiman et al. proposed that portal myofibroblasts, in addition to activated HSCs, may originate from PFs (Cassiman et al., 2002). Recent cell-tracing technology has clarified the origin of myofibroblasts in further detail, as discussed below.
Hepatic Stellate Cells
HSCs reside in Disse's space in the hepatic sinusoid, a space between sinusoidal endothelial cells and hepatocytes (Wake, 1980). HSCs constitute approximately 10% of total liver cells in the adult healthy liver. In healthy situation, quiescent HSCs principally store vitamin A in droplets in their cytoplasm. It is calculated that HSCs in the liver store 45–72% of vitamin A in the body. Quiescent HSCs are positive for neural crest markers like GFAP and NTs, nerve growth factor receptor (p75), and desmin. Quiescent HSCs are the source of extracellular matrix materials (ECMs) and the component of basement membrane-like structure, such as laminin, proteoglycan, and type IV collagen (Friedman, 2008; Yin et al., 2013; Pellicoro et al., 2014). HSCs also function as liver-specific pericytes surrounding sinusoidal endothelial cells, and thus their contractility is regulated by the exposure to endothelin-1 (ET-1), angiotensin-II, and their relaxation by nitric oxide (NO) control the diameter of sinusoids and regulates the hepatic microcirculation (Kawada et al., 1993).
Liver trauma caused by hepatitis virus B or C infection, alcohol abuse, drug toxicity, autoimmunity, or steatohepatitis, triggers the activation of HSCs and their transdifferentiation from quiescent phenotype to myofibroblast-like one (Novo et al., 2014). The myofibroblast-like HSCs contain less amount of vitamin A droplets, express increased α–SMA and growth factor receptors, possess augmented contractility, and generate multiple ECMs, including type I and III collagens (Friedman, 2008; Yin et al., 2013; Pellicoro et al., 2014). This activation process of HSCs is initiated by paracrine stimulation by neighboring and activated sinusoidal endothelial cells, Kupffer cells (liver macrophages), hepatocytes and cholangiocytes, as well as platelets. When activated, HSCs synthesize transforming growth factor β (TGF-β), which stimulates HSCs themselves as an autocrine loop. Hepatic sinusoidal endothelial cells take part in the activation of TGF-β from its latency-associated peptide-binding form by its proteolytic cleavage (Schuppan and Kim, 2013). Cholangiocytes and platelets are also an important origin of growth factors, including platelet-derived growth factor (PDGF), TGF-β, and epidermal growth factor. Activation of HSCs is additionally initiated by damaged hepatocytes; they secrete fibrogenic lipid peroxides, and their apoptosis mediated by Fas and tumor-necrosis factor (TNF)-related apoptosis-inducing ligand initiates the activation of HSCs via damage-associated molecular pattern molecules including high-mobility group box 1, RNA and DNA, S100 molecules, and purine metabolites. In culture system, it was also demonstrated that apoptotic fragments derived from damaged hepatocytes stimulate HSC activation, and that fibrogenic activity of myofibroblasts are augmented by the phagocytosis of apoptotic hepatocytes due to NADPH oxidase 2 and the janus kinase/signal transducer and activator of transcription and phosphoinositide 3-kinase/Akt pathways (Wree et al., 2014). Activated HSCs also produce an increased amount of matrix metalloproteinases (MMPs), especially MMP13, and their inhibitors, tissue inhibitor of matrix metalloproteinases (TIMPs).
Transcription factors including activated protein-1, Jun D, Sp1, Kruppel-like factor 6, and nuclear factor kappa B, whose functions are strictly regulated by intracellular signaling molecules, such as Smad, Ras, Raf-1, and mitogen-activated protein kinase, control the activation of HSC, leading to the transcriptional upregulation of latent TGF-β (Mann and Smart, 2002; Lopez-Sanchez et al., 2014). Epigenetic regulation, specifically DNA methylation and histone modification, is also important in the regulation of the activation of HSCs. For example, the abnormal DNA methylation of phosphatase and tension homolog (PTEN) and MeCP2 has been identified in activated HSCs (Lee et al., 2015). Among epigenetic signals, microRNAs also participate in the activation of HSCs. The microRNAs that have been implicated in HSC activation include miR-199a,b, miR-221, miR-27, miR-21, miR-125, miR-195, miR-214, and miR-221/222 as profibrotic miRNAs, and miR-29, miR-15b, miR-200, miR-16, miR-133b, and miR-122 as anti-fibrotic miRNAs. Among these miRNAs, miR-29 has been the most intensively analyzed for its ability to suppress collagen 1A1 production in HSCs (Sekiya et al., 2011; Ogawa et al., 2012). In addittion, the augmented production of TIMPs impedes ECM degradation and triggers ECM accumulation in the damaged liver (Perugorria et al., 2013). It should be noted that leptin and other adipocytokines are involved in the process of HSC activation (Choi et al., 2010) and that activated HSCs are additionally characterized by augmented expression of receptors for PDGF, TGF-β, vascular endothelial growth factor (VEGF), angiotensin-II, and ET-1 (Novo et al., 2012). Through study of reporter Collagen α1(I)-GFP (Col-GFP) mice, GFP+Vitamin-A+Desmin+ activated HSCs have been shown to comprise more than 92% of myofibroblasts in the fibrotic liver induced by CCl4 intoxication (Scholten et al., 2010).
Hepatocellular carcinoma (HCC) is a consequence of chronic liver disease with many etiologies, and it develops after repetitive hepatocyte death and the development of fibrosis. In fact, it is thought that 80% of HCC develop in fibrotic and cirrhotic liver. Thus, fibrosis consisting of HSC-derived myofibroblasts may be involved in cancer development. Because activated HSCs are the source for growth factors and ECMs as mentioned above, they may contribute to the development of a microenvironment that is suitable for cancer growth. In fact, Coulouarn et al. demonstrated the importance of HSC-hepatocyte interaction due to the production of VEGFA and MMP-9 (Coulouarn et al., 2012). Very recently, Yoshimoto et al. elucidated that senescent HSCs activated by a bile acid derived from intestinal microbiota shows a senescent-associated secretary phenotype, in which a panel of cytokines, such as IL-1, IL-6, and GROα, are synthesized. This phenomenon is suggested in the central scenario for the development of liver cancer in obesity (Yoshimoto et al., 2013).
Portal Fibroblasts
PFs are the resident fibroblasts around the portal tract existing in the mesenchyme surrounding the bile ducts. PFs were first described more than 50 years ago in rat liver. It is considered that PFs in addition to HSC-derived myofibroblasts produce ECMs particularly in case of biliary fibrosis (Wells, 2014). Although it is considered that the septum tranversum-derived mesothelial cells are origin of PFs and HSCs, the definition of the roles of PFs in normal and injured liver has been unsatisfactory confirmed due to the lack of reliable markers to distinguish PFs from HSCs. PFs are generally positive for gremlin, Thy-1, fibulin 2, elastin, interleukin 6 (IL-6), cofilin, and the ectonucleotidase NTPDase 2. Lemoinne et al. demonstrated that COL15A1-positive portal and periportal cells give rise to portal myofibroblasts and release VEGFA-laden microparticles, thus promoting endothelial tubulogenesis (Lemoinne et al., 2015). Unlike HSCs, PFs lack desmin, cytoglobin, β2-macroglobulin, Hand2, GFAP, p75NGFr, and Vitamin A. Using a mouse model carrying a bacterial artificial chromosome with a Cre reporter derived from the lecithin-retinol acyltransferase, Mederacke et al. recently showed that HSCs are the source of 82–96% of myofibroblasts in seven models of fibrosis in mice, although they could not eliminate the possibility that PFs are required for biliary fibrosis. Particularly, bridging fibrosis occurs from the portal vein area to the central vein area where PFs and desmin–negative cells are localized (Mederacke et al., 2013). Iwaisako et al. showed the important role of myofibroblasts derived from PFs during the initial stages of bile-duct ligation-induced liver fibrosis by contributing more than 70% of myofibroblasts, although HSC-derived myofibroblasts become dominant at the last phase (Iwaisako et al., 2014). In their study, Iwaisako et al. also identified mesothelin as a useful marker to distinguish PF-myofibroblasts from HSC-myofibroblasts.
PFs can be differentiated from HSCs-myofibroblasts by their production of elastin. As mentioned before, PFs were originally defined by their fibulin 2 expression (Knittel et al., 1999). Fibulin 2 is a linker protein that occupies the interface between microfibrils and their elastin core. Fibulin 2 mediates the progression of fibrosis by sequestering latent TGF-β binding proteins from TGF-β. In addition, PFs produce lysyl oxidase (LOX) and the related protein LOX-like 1, which cross-links elastin. Taken together, the function of PFs should be estimated regarding to the metabolism of elastin since elastin fibers are able to provide the bile ducts and the portal vasculature with mechanical stability (Perepelyuk et al., 2013).
The interaction of PFs with cholangiocytes is another important issue. Tanimizu et al. described that neurotrophin receptor p75-positive progenitors of PFs and cholangiocytes express laminin α1 and α5 subunits, respectively, which interact with the β1 integrin subunit to maintain the polarity and lumen of the bile ducts (Tanimizu et al., 2012). On the other hand, Jhandier et al. reported that PF-derived myofibroblasts do not express ectonucleotidase NTPDase2, which is equivalent to ecto-ATPase or CD39L1 and metabolizes extracellular nucleotides. This fact may explain how cholangiocyte proliferation is initiated by P2Y receptor activation in chronic cholangiopathies (Jhandier et al., 2005).
Mesodermal Mesenchymal Cells
Although myofibroblasts are able to be derived from epithelial cells like hepatocytes and cholangiocytes, during the epithelial-to-mesenchymal transition, recent genetic cell lineage tracing has clearly revealed that mesenchymal cells including HSCs, PFs, and vascular smooth muscle cells are derived from MCs in the mouse embryonic liver. In addition, in adult Wt1CreERT2; R26T/Gf mice after the induction of fibrosis by CCl4 or bile duct ligation, MCs that are positive for glycoprotein M6a, podoplanin and CD200 acquire mesenchymal characteristics—expression of collagen 1α1 and vimentin—but the epithelial cell markers are decreased. This phenomenon of the conversion of liver MCs to mesenchymal cells is called the mesothelial-mesenchymal transition. The canonical TGF-β pathway plays a central role in MMT via SMAD3 (Li et al., 2013; Yin et al., 2013; Lua et al., 2014).
Cytoglobin
Cygb belongs to the mammalian globin family including myoglobin in muscle cells, hemoglobin in red blood cells, neuroglobin in the nervous system, and androglobin in testis. Cygb, originally named stellate cell activation-associated protein by our group and histoglobin by Trent and Hagrove, was discovered in 2001 from cultured rat HSCs by proteomics analysis, namely the separation of proteins expressed in HSCs by 2D SDS-PAGE, followed by the identification of individual proteins by time of flight mass spectrometry. The molecular weight of Cygb is 21,496 Da, and it is composed of 190 amino acids (Kawada et al., 2001; Trent and Hargrove, 2002; Burmester and Hankeln, 2014).
Cygb is uniquely expressed in HSCs, but not in hepatocytes, endothelial cells, or Kupffer cells, in the liver (Figure 1). Cygb is also expressed in extrahepatic organs, including the pancreas, spleen, kidney, digestive tract, and heart. Cygb-positive cells usually localize proximally to capillaries, and they are considered to be pericytes that store vitamin A, produce collagen and closely correlate with vascular endothelium (Nakatani et al., 2004). In addition, Cygb is expressed in chondrocytes, osteoblasts and osteocytes (Schmidt et al., 2004). Furthermore, a recent report has indicated that Cygb is expressed in melanocytes and several types of melanoma, and it is involved in the melanocyte-to-melanoma transition through hyper-methylation in the promoter region of the CYGB gene (Fujita et al., 2014).
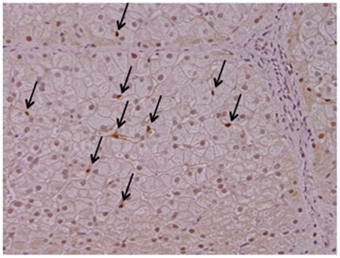
Figure 1. Cytoglobin expression in intact human liver. Cytoglobin (arrows) is localized along hepatic sinusoid, namely these cells are identical to hepatic stellate cells, but not hepatocytes. Intact human liver tissue was obtained at Osaka City University Hospital under the patient's consent (Motoyama et al., 2014). Written informed consent was obtained from the patient. NK contributed to the evaluation of diagnosis of human liver histopathology.
The 3D structure of Cygb is almost identical to that of myoglobin, and it has a three-over-three α-helical sandwich structure consisting of eight helices, whereas Cygb is a bis-histidyl hexa-coordinate globin similar to neuroglobin and different from myoglobin and hemoglobin, which are penta-coordinate globins. However, Cygb can bind gas molecules including oxygen, nitric oxide, and carbon monoxide with kinetic properties similar to those of myoglobin. Thus, Cygb is thought to function as a local gas-sensor (Makino et al., 2006, 2011). A recent report has indicated the role of Cygb as a nitric oxide dioxygenase [Cygb(Fe2+) → O2 → Cygb(Fe2+−O2) → NO → Cygb(Fe3+) + ] resulting in the regulation of the local concentration of nitric oxide and hence vascular tonus (Liu et al., 2012).
Cygb gene expression is regulated by hypoxia inducible factor-1 (HIF-1), which binds to the hypoxia response element of the Cygb gene promoter. In fact, exposure of HeLa cells to 1% O2 upregulates Cygb mRNA expression 1.7-fold (3 h) and 1.6-fold (6 h) in culture. HIF-1 and erythropoietin have 2 (-141 and -448) and 1 (-144) binding site, respectively, within the 5′-UTR lesion of the Cygb gene; these sites are important for Cygb gene transcription under hypoxia (Guo et al., 2007). The other transcription factors involved in Cygb gene expression include c-Ets-1, Sp1, nuclear factor of activated T cell and activator protein (Guo et al., 2006). In case of the liver, Cygb is induced during hypoxia (24 and 48 h at 7% O2) and reoxygenetion in mice (Fordel et al., 2007). Man et al. demonstrated that more than 3.5-fold upregulation of Cygb mRNA expression and the increase in the number of Cygb-expressing cells after the administration of CCl4 in mice (Man et al., 2008). Gninsky et al. reported the induction of Cygb in thioacetamide-intoxicated liver fibrosis model in rats, which was clearly suppressed by the treatment with halofuginone, an inhibitor of TGF-β-dependent Smad3 phosphorylation (Gnainsky et al., 2007). In culture studies, Cui et al. demonstrated the induction of Cygb in primary-cultured rat HSCs by arundic acid, an inhibitor of S100b synthesis in astrocytes (Cui et al., 2012). Stone et al. recently clarified the regulation of Cygb expression in HTC-T6 cells in culture; Cygb expression is upregulated by laminin via integrin α1β4 signaling while it is downregulated by type I collagen by phosphorylation of focal-adhesion kinase via integrin α2β1 signaling (Stone et al., 2015).
Although Cygb's function as a fas-binding molecule is well-established, its pathophysiological role has not been fully elucidated. Cygb protects SH-SY5Y neuroblastoma cells from apoptosis under exposure to 300 μM H2O2 (Fordel et al., 2006). Li et al. demonstrated that siCygb treatment facilitates N2a neuronal cell death under exposure to 500 μM H2O2 for 4 h (Li et al., 2007). These and other studies have revealed that Cygb is a possible cytoprotector against oxidative stress.
Recently, the role of Cygb in human carcinogenesis in various organs has been paid special attention. The downregulation of Cygb gene expression has been demonstrated in human esophageal cancer, head and neck cancer, lung cancer, ovarian cancer and prostatic cancer via the hyper-methylation of the promoter lesion of the Cygb gene (Shivapurkar et al., 2008; Shaw et al., 2009; Latina et al., 2015). In detail, Oleksiewicz et al. showed (1) marked down-regulation of Cygb protein in human lung cancer cell line compared to normal human epithelial cell lines and possible involvement of promoter methylation and (2) significantly reduced Cygb mRNA expression in non-small cell lung cancer tissues compared with normal lung tissue (Mann-Whitney test, P = 2.3 × 10−7), suggesting the possible tumor suppressor function of Cygb (Oleksiewicz et al., 2013). Based on these reports, Hubers et al. studied Cygb gene methylation in spontaneous sputum in order to utilize it for lung cancer diagnosis. As a result, they reported the sensitivity of Cygb for the diagnosis of lung cancer was 22% (Hubers et al., 2014). Except for the lung cancer, Wojnarowicz et al. demonstrated that Cygb gene expression was lowered in the majority of tumors with low malignant potential and cancer compared to benign tumors and normal ovarian surface epithelial cell samples (Wojnarowicz et al., 2012). Chen et al. also clarified the involvement of Cygb in ovarian cancer development at human tissue level and in cell culture models (Chen et al., 2014). Similar observations were additionally reported in human atrophy and adenocarcinoma of the prostate, suggesting that prostatic malignancy is accompanied with low level of Cygb, which play a role in protecting from oxidative damage (Mogal et al., 2012).
Using Cygb-knockout mice that were generated in our laboratory, we studied the role of Cygb in liver cancer development. First, mice were treated with 0.05 ppm diethylnitrosamine, an established carcinogen to the liver, for 36 weeks, thus resulting in the occurrence of liver tumors in 57.1% of the knockout mice compared with 0% of the wild-type mice. In this model, background liver tissues showed a marked development of liver fibrosis, augmented inflammatory reactions and overproduction of peroxynitrite (ONOO−) in knockout mice (Thuy le et al., 2011). Second, mice were given a choline-deficient L-amino acid-defined diet for 32 weeks to induce steatohepatitis. Unexpectedly, 100% of Cygb knockout mice developed multiple liver tumors compared with 0% of the wild type mice. Again, background liver tissues showed marked development of liver fibrosis and augmented inflammatory reactions, which were accompanied by DNA double strand breaks (γH2AX expression) in hepatocytes. These results show the protective role of Cygb against oxidative stress and liver fibrosis development under chronic inflammation, suggesting Cygb's role in tissue carcinogenesis (Thuy le et al., 2015). In accordance with these observations, previous two reports demonstrated the anti-fibrotic function of Cygb. Xu et al. reported that Cygb overexpression in the liver by recombinant adeno-associated virus-2 encoding full-length rat Cygb suppressed liver injury and fibrosis induced by CCl4 adminstration or bile-duct ligation in rats (Xu et al., 2006) and, furthermore, He at al. showed that administration of recombinant Cygb (10 mg/Kg) attenuates liver fibrosis in thioacetamide-induced liver fibrosis in rats (He et al., 2011), suggesting the hepato-protective role of Cygb.
Cytoglobin as a Marker of Hepatic Stellate Cell-derived Myofibroblasts
As mentioned above, Cygb was initially discovered from rat HSCs in primary culture (Kawada et al., 2001). Cygb is expressed in mouse and human HSCs (Figure 1). Thus, whether Cygb can be utilized as a specific marker to distinguish myofibroblasts derived from HSCs from those derived from PFs is an interesting issue. Schmidt et al. first provided a detailed description of Cygb-expressing cells in vivo in mice, and they demonstrated that, in the liver, Cygb is expressed in HSCs and fibroblasts that surround the portal vein, but not in hepatocytes (Schmidt et al., 2004). However, in this study, fibroblasts around the portal vein were insufficiently characterized. Using fibulin 2 as a PF marker, Tateaki et al. demonstrated that there are several types of fibroblastic cells in the liver, namely fibulin 2+/Cygb+, fibulin 2+/Cygb−, fibulin 2−/Cygb+, fibulin 2−/Cygb−, and they concluded that fibulin 2+/Cygb+ and fibulin 2−/Cygb+ are identical to portal myofibroblasts and activated HSCs, respectively. A limitation of this study is that the identification of individual cell types was performed only by immunofluorescence staining (Tateaki et al., 2004). Ogawa et al. isolated the mouse liver non-parenchymal cell fraction by the Nycodenz density gradient method and further separated the cells in the fraction on the basis of vitamin A autofluorescence with FACS. UV+ cells had minimum growth potential, expressed desmin and Cygb, but not αSMA and fibulin 2 at 1 day in culture, and later became positive for desmin, Cygb, and αSMA at 7 day in culture. Hence, UV+ cells (vitamin A-storing cells) are identical to HSCs. Conversely, UV− cells showed high proliferation activity and expressed αSMA, fibulin 2, and Gremlin, but not desmin and Cygb, thus indicating that UV− cells are derived from portal fibroblasts (Ogawa et al., 2007). Bosselut at al. demonstrated that isolated rat portal myofibroblasts do not express Cygb, and thus are different from rat HSCs, as determined by 2-D MS/MS (Bosselut et al., 2010). Recent study by Fausther et al. described that rat portal fibroblasts and portal myofibroblast cell lines, RGF and RGF-N2, express Cygb at least in mRNA level in culture model (Fausther et al., 2015). In contrast, Motoyama et al. demonstrated that in human liver damaged by chronic hepatitis C, Cygb- and cellular retinol-binding protein-positive cells localize along hepatic sinusoids in liver parenchyma and at the margin of fibrotic septum, and they are not identical to αSMA, fibulin 2, and Thy-1-positive myofibroblasts (Motoyama et al., 2014; Figure 2). In summary, in mouse, rat, and human liver, Cygb is expressed in quiescent HSCs, and its expression level increases in activated HSCs that also express αSMA. In contrast, myofibroblasts derived from PFs are positive for fibulin 2 and αSMA, and they are negative for Cygb in human, while in rodent Cygb expression in PF-derived myofibroblasts needs to be characterized further by, for example, using genetic cell-labeling technology.
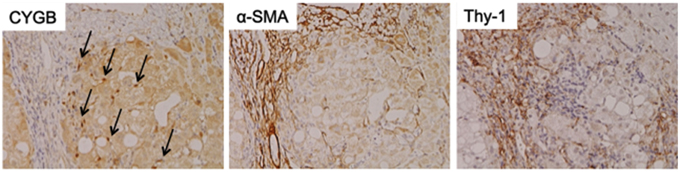
Figure 2. Cytoglobin expression in fibrotic human liver. Cytoglobin (arrows) is localized along hepatic sinusoid, while α-SMA and Thy-1, myofibroblast markers, were strongly positive at fibrotic septum. Human liver tissue with fibrosis caused by chronic hepatitis C was obtained at Osaka City University Hospital under the patient's consent (Motoyama et al., 2014). Written informed consent was obtained from the patients. NK contributed to the evaluation of diagnosis of human liver histopathology.
Author Contributions
NK contributed to the evaluation of diagnosis of human liver histopathology.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
NK was supported by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS) through Grant No. 25293177 (2013-present) and Research on Hepatitis and BSE, the Ministry of Health, Labor and Welfare (2013-present).
Abbreviations
ABCRYS, α-B-crystallin; α-SMA, α-smooth muscle actin; BDNF, brain-derived nerve growth factor; Cygb, cytoglobin; ET-1, endothelin-1; ECM, extracellular matrix material; GFAP, glial fibrillar acidic protein; HSC, hepatic stellate cell; HIF, hypoxia inducible factor; LOX, lysyl oxidase; MMP, matrix metalloproteinase; MC mesothelial cell; NGF, neuronal growth factor; NT, neurotrophin; NO, nitric oxide; PDGF, platelet-derived growth factor; PF, portal fibroblast; TIMP, tissue inhibitor of matrix metalloproteinases; TGF-β, transforming growth factor β; TNF, tumor-necrosis factor; VEGF, vascular endothelial growth factor.
References
Bosselut, N., Housset, C., Marcelo, P., Rey, C., Burmester, T., Vinh, J., et al. (2010). Distinct proteomic features of two fibrogenic liver cell populations: hepatic stellate cells and portal myofibroblasts. Proteomics 10, 1017–1028. doi: 10.1002/pmic.200900257
Burmester, T., Ebner, B., Weich, B., and Hankeln, T. (2002). Cytoglobin: a novel globin type ubiquitously expressed in vertebrate tissues. Mol. Biol. Evol. 19, 416–421. doi: 10.1093/oxfordjournals.molbev.a004096
Burmester, T., and Hankeln, T. (2014). Function and evolution of vertebrate globins. Acta Physiol. 211, 501–514. doi: 10.1111/apha.12312
Cassiman, D., Libbrecht, L., Desmet, V., Denef, C., and Roskams, T. (2002). Hepatic stellate cell/myofibroblast subpopulations in fibrotic human and rat livers. J. Hepatol. 36, 200–209. doi: 10.1016/S0168-8278(01)00260-4
Chen, H., Zhao, X., and Meng, T. (2014). Expression and biological role of cytoglobin in human ovarian cancer. Tumour Biol. 35, 6933–6939. doi: 10.1007/s13277-014-1941-x
Choi, S. S., Syn, W. K., Karaca, G. F., Omenetti, A., Moylan, C. A., Witek, R. P., et al. (2010). Leptin promotes the myofibroblastic phenotype in hepatic stellate cells by activating the hedgehog pathway. J. Biol. Chem. 285, 36551–35560. doi: 10.1074/jbc.M110.168542
Coulouarn, C., Corlu, A., Glaise, D., Guénon, I., Thorgeirsson, S. S., and Clément, B. (2012). Hepatocyte-stellate cell cross-talk in the liver engenders a permissive inflammatory microenvironment that drives progression in hepatocellular carcinoma. Cancer Res. 72, 2533–2542. doi: 10.1158/0008-5472.CAN-11-3317
Cui, W., Wang, M., Maegawa, H., Teranishi, Y., and Kawada, N. (2012). Inhibition of the activation of hepatic stellate cells by arundic acid via the induction of cytoglobin. Biochem. Biophys. Res. Commun. 425, 642–648. doi: 10.1016/j.bbrc.2012.07.126
Fausther, M., Goree, J. R., Lavoie, É. G., Graham, A. L., Sévigny, J., and Dranoff, J. A. (2015). Establishment and characterization of rat portal myofibroblast cell lines. PLoS ONE 10:e0121161. doi: 10.1371/journal.pone.0121161
Fordel, E., Thijs, L., Martinet, W., Lenjou, M., Laufs, T., Van Bockstaele, D., et al. (2006). Neuroglobin and cytoglobin overexpression protects human SH-SY5Y neuroblastoma cells against oxidative stress-induced cell death. Neurosci. Lett. 410, 146–151. doi: 10.1016/j.neulet.2006.09.027
Fordel, E., Thijs, L., Moens, L., and Dewilde, S. (2007). Neuroglobin and cytoglobin expression in mice. Evidence for a correlation with reactive oxygen species scavenging. FEBS J. 274, 1312–1317. doi: 10.1111/j.1742-4658.2007.05679.x
Friedman, S. L. (2008). Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 88, 125–172. doi: 10.1152/physrev.00013.2007
Fujita, Y., Koinuma, S., De Velasco, M. A., Bolz, J., Togashi, Y., Terashima, M., et al. (2014). Melanoma transition is frequently accompanied by a loss of cytoglobin expression in melanocytes: a novel expression site of cytoglobin. PLoS ONE 9:e94772. doi: 10.1371/journal.pone.0094772
Gabbiani, G., Ryan, G. B., and Majno, G. (1971). Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia 27, 549–550. doi: 10.1007/BF02147594
Gnainsky, Y., Kushnirsky, Z., Bilu, G., Hagai, Y., Genina, O., Volpin, H., et al. (2007). Gene expression during chemically induced liver fibrosis: effect of halofuginone on TGF-beta signaling. Cell Tissue Res. 328, 153–166. doi: 10.1007/s00441-006-0330-1
Grimaud, J. A., and Borojevic, R. (1977). Myofibroblasts in hepatic schistosomal fibrosis. Experientia 33, 890–892. doi: 10.1007/BF01951264
Guo, X., Philipsen, S., and Tan-Un, K. C. (2007). Study of the hypoxia-dependent regulation of human CYGB gene. Biochem. Biophys. Res. Commun. 364, 145–150. doi: 10.1016/j.bbrc.2007.09.108
Guo, X., Philipsen, S., and Tan-Un, K. C. (2006). Characterization of human cytoglobin gene promoter region. Biochim. Biophys. Acta 1759, 208–215. doi: 10.1016/j.bbaexp.2006.04.002
He, X., Lv, R., Wang, K., Huang, X., Wu, W., Yin, L., et al. (2011). Cytoglobin exhibits anti-fibrosis activity on liver in vivo and in vitro. Protein J. 30, 437–446. doi: 10.1007/s10930-011-9340-2
Hubers, A. J., van der Drift, M. A., Prinsen, C. F., Witte, B. I., Wang, Y., Shivapurkar, N., et al. (2014). Methylation analysis in spontaneous sputum for lung cancer diagnosis. Lung Cancer 84, 127–133. doi: 10.1016/j.lungcan.2014.01.019
Iwaisako, K., Jiang, C., Zhang, M., Cong, M., Moore-Morris, T. J., Park, T. J., et al. (2014). Origin of myofibroblasts in the fibrotic liver in mice. Proc. Natl. Acad. Sci. U.S.A. 111, E3297–E3305. doi: 10.1073/pnas.1400062111
Jhandier, M. N., Kruglov, E. A., Lavoie, E. G., Sévigny, J., and Dranoff, J. A. (2005). Portal fibroblasts regulate the proliferation of bile duct epithelia via expression of NTPDase2. J. Biol. Chem. 280, 22986–22992. doi: 10.1074/jbc.M412371200
Kawada, N., Kristensen, D. B., Asahina, K., Nakatani, K., Minamiyama, Y., Seki, S., et al. (2001). Characterization of a stellate cell activation-associated protein (STAP) with peroxidase activity found in rat hepatic stellate cells. J. Biol. Chem. 276, 25318–25323. doi: 10.1074/jbc.M102630200
Kawada, N., Tran-Thi, T. A., Klein, H., and Decker, K. (1993). The contraction of hepatic stellate (Ito) cells stimulated with vasoactive substances. Possible involvement of endothelin 1 and nitric oxide in the regulation of the sinusoidal tonus. Eur. J. Biochem. 213, 815–823.
Knittel, T., Kobold, D., Saile, B., Grundmann, A., Neubauer, K., Piscaglia, F., et al. (1999). Rat liver myofibroblasts and hepatic stellate cells: different cell populations of the fibroblast lineage with fibrogenic potential. Gastroenterology 117, 1205–1221. doi: 10.1016/S0016-5085(99)70407-5
Latina, A., Viticchiè, G., Lena, A. M., Piro, M. C., Annicchiarico-Petruzzelli, M., Melino, G., et al. (2015). ΔNp63 targets cytoglobin to inhibit oxidative stress-induced apoptosis in keratinocytes and lung cancer. Oncogene. doi: 10.1038/onc.2015.222. [Epub ahead of print].
Lee, Y. A., Wallace, M. C., and Friedman, S. L. (2015). Pathobiology of liver fibrosis: a translational success story. Gut 64, 830–841. doi: 10.1136/gutjnl-2014-306842
Lemoinne, S., Cadoret, A., Rautou, P. E., El Mourabit, H., Ratziu, V., Corpechot, C., et al. (2015). Portal myofibroblasts promote vascular remodeling underlying cirrhosis formation through the release of microparticles. Hepatology 61, 1041–1055. doi: 10.1002/hep.27318
Li, D., Chen, X. Q., Li, W. J., Yang, Y. H., Wang, J. Z., and Yu, A. C. (2007). Cytoglobin up-regulated by hydrogen peroxide plays a protective role in oxidative stress. Neurochem. Res. 32, 1375–1380. doi: 10.1007/s11064-007-9317-x
Li, Y., Wang, J., and Asahina, K. (2013). Mesothelial cells give rise to hepatic stellate cells and myofibroblasts via mesothelial-mesenchymal transition in liver injury. Proc. Natl. Acad. Sci. U.S.A. 110, 2324–2329. doi: 10.1073/pnas.1214136110
Liu, X., Follmer, D., Zweier, J. R., Huang, X., Hemann, C., Liu, K., et al. (2012). Characterization of the function of cytoglobin as an oxygen-dependent regulator of nitric oxide concentration. Biochemistry 51, 5072–5082. doi: 10.1021/bi300291h
Lopez-Sanchez, I., Dunkel, Y., Roh, Y. S., Mittal, Y., De Minicis, S., Muranyi, A., et al. (2014). GIV/Girdin is a central hub for profibrogenic signalling networks during liver fibrosis. Nat. Commun. 5, 4451. doi: 10.1038/ncomms5451
Lua, I., James, D., Wang, J., Wang, K. S., and Asahina, K. (2014). Mesodermal mesenchymal cells give rise to myofibroblasts, but not epithelial cells, in mouse liver injury. Hepatology 60, 311–322. doi: 10.1002/hep.27035
Makino, M., Sawai, H., Shiro, Y., and Sugimoto, H. (2011). Crystal structure of the carbon monoxide complex of human cytoglobin. Proteins 79, 1143–1153. doi: 10.1002/prot.22950
Makino, M., Sugimoto, H., Sawai, H., Kawada, N., Yoshizato, K., and Shiro, Y. (2006). High-resolution structure of human cytoglobin: identification of extra N- and C-termini and a new dimerization mode. Acta Crystallogr. D Biol. Crystallogr. 62, 671–677. doi: 10.1107/S0907444906013813
Man, K. N., Philipsen, S., and Tan-Un, K. C. (2008). Localization and expression pattern of cytoglobin in carbon tetrachloride-induced liver fibrosis. Toxicol. Lett. 183, 36–44. doi: 10.1016/j.toxlet.2008.09.015
Mann, D. A., and Smart, D. E. (2002). Transcriptional regulation of hepatic stellate cell activation. Gut 50, 891–896. doi: 10.1136/gut.50.6.891
Mederacke, I., Hsu, C. C., Troeger, J. S., Huebener, P., Mu, X., Dapito, D. H., et al. (2013). Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun. 4, 2823. doi: 10.1038/ncomms3823
Mogal, A. P., Watson, M. A., Ozsolak, F., Salavaggione, L., and Humphrey, P. A. (2012). Gene expression profiles and differential cytoglobin expression in atrophy and adenocarcinoma of the prostate. Prostate 72, 931–937. doi: 10.1002/pros.21494
Motoyama, H., Komiya, T., Thuy le, T. T., Tamori, A., Enomoto, M., Morikawa, H., et al. (2014). Cytoglobin is expressed in hepatic stellate cells, but not in myofibroblasts, in normal and fibrotic human liver. Lab. Invest. 94, 192–207. doi: 10.1038/labinvest.2013.135
Nakatani, K., Okuyama, H., Shimahara, Y., Saeki, S., Kim, D. H., Nakajima, Y., et al. (2004). Cytoglobin/STAP, its unique localization in splanchnic fibroblast-like cells and function in organ fibrogenesis. Lab. Invest. 84, 91–101. doi: 10.1038/labinvest.3700013
Novo, E., Cannito, S., Paternostro, C., Bocca, C., Miglietta, A., and Parola, M. (2014). Cellular and molecular mechanisms in liver fibrogenesis. Arch. Biochem. Biophys. 548, 20–37. doi: 10.1016/j.abb.2014.02.015
Novo, E., Povero, D., Busletta, C., Paternostro, C., di Bonzo, L. V., Cannito, S., et al. (2012). The biphasic nature of hypoxia-induced directional migration of activated human hepatic stellate cells. J. Pathol. 226, 588–597. doi: 10.1002/path.3005
Ogawa, T., Enomoto, M., Fujii, H., Sekiya, Y., Yoshizato, K., Ikeda, K., et al. (2012). MicroRNA-221/222 upregulation indicates the activation of stellate cells and the progression of liver fibrosis. Gut 61, 1600–1609. doi: 10.1136/gutjnl-2011-300717
Ogawa, T., Tateno, C., Asahina, K., Fujii, H., Kawada, N., Obara, M., et al. (2007). Identification of vitamin A-free cells in a stellate cell-enriched fraction of normal rat liver as myofibroblasts. Histochem. Cell Biol. 127, 161–174. doi: 10.1007/s00418-006-0237-7
Oleksiewicz, U., Liloglou, T., Tasopoulou, K. M., Daskoulidou, N., Bryan, J., Gosney, J. R., et al. (2013). Cytoglobin has bimodal: tumour suppressor and oncogene functions in lung cancer cell lines. Hum. Mol. Genet. 22, 3207–3217. doi: 10.1093/hmg/ddt174
Pellicoro, A., Ramachandran, P., Iredale, J. P., and Fallowfield, J. A. (2014). Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 14, 181–194. doi: 10.1038/nri3623
Perepelyuk, M., Terajima, M., Wang, A. Y., Georges, P. C., Janmey, P. A., Yamauchi, M., et al. (2013). Hepatic stellate cells and portal fibroblasts are the major cellular sources of collagens and lysyl oxidases in normal liver and early after injury. Am. J. Physiol. Gastrointest. Liver Physiol. 304, G605–G614. doi: 10.1152/ajpgi.00222.2012
Perugorria, M. J., Murphy, L. B., Fullard, N., Chakraborty, J. B., Vyrla, D., Wilson, C. L., et al. (2013). Tumor progression locus 2/Cot is required for activation of extracellular regulated kinase in liver injury and toll-like receptor-induced TIMP-1 gene transcription in hepatic stellate cells in mice. Hepatology 57, 1238–1249. doi: 10.1002/hep.26108
Rudolph, R., McClure, W. J., and Woodward, M. (1979). Contractile fibroblasts in chronic alcoholic cirrhosis. Gastroenterology 76, 704–709.
Schmidt, M., Gerlach, F., Avivi, A., Laufs, T., Wystub, S., Simpson, J. C., et al. (2004). Cytoglobin is a respiratory protein in connective tissue and neurons, which is up-regulated by hypoxia. J. Biol. Chem. 279, 8063–8069. doi: 10.1074/jbc.M310540200
Schmitt-Gräff, A., Krüger, S., Bochard, F., Gabbiani, G., and Denk, H. (1991). Modulation of alpha smooth muscle actin and desmin expression in perisinusoidal cells of normal and diseased human livers. Am. J. Pathol. 138, 1233–1242.
Scholten, D., Osterreicher, C. H., Scholten, A., Iwaisako, K., Gu, G., Brenner, D. A., et al. (2010). Genetic labeling does not detect epithelial-to-mesenchymal transition of cholangiocytes in liver fibrosis in mice. Gastroenterology 139, 987–998. doi: 10.1053/j.gastro.2010.05.005
Schuppan, D., and Kim, Y. O. (2013). Evolving therapies for liver fibrosis. J. Clin. Invest. 123, 1887–1901. doi: 10.1172/JCI66028
Sekiya, Y., Ogawa, T., Yoshizato, K., Ikeda, K., and Kawada, N. (2011). Suppression of hepatic stellate cell activation by microRNA-29b. Biochem. Biophys. Res. Commun. 412, 74–79. doi: 10.1016/j.bbrc.2011.07.041
Shaw, R. J., Omar, M. M., Rokadiya, S., Kogera, F. A., Lowe, D., Hall, G. L., et al. (2009). Cytoglobin is upregulated by tumour hypoxia and silenced by promoter hypermethylation in head and neck cancer. Br. J. Cancer 101, 139–144. doi: 10.1038/sj.bjc.6605121
Shivapurkar, N., Stastny, V., Okumura, N., Girard, L., Xie, Y., Prinsen, C., et al. (2008). Cytoglobin, the newest member of the globin family, functions as a tumor suppressor gene. Cancer Res. 68, 7448–7456. doi: 10.1158/0008-5472.CAN-08-0565
Stone, L. C., Thorne, L. S., Weston, C. J., Graham, M., and Hodges, N. J. (2015). Cytoglobin expression in the hepatic stellate cell line HSC-T6 is regulated by extracellular matrix proteins dependent on FAK-signalling. Fibrogenesis Tissue Repair 8, 15. doi: 10.1186/s13069-015-0032-y
Tanimizu, N., Kikkawa, Y., Mitaka, T., and Miyajima, A. (2012). α1- and α5-containing laminins regulate the development of bile ducts via β1 integrin signals. J. Biol. Chem. 287, 28586–28597. doi: 10.1074/jbc.M112.350488
Tateaki, Y., Ogawa, T., Kawada, N., Kohashi, T., Arihiro, K., Tateno, C., et al. (2004). Typing of hepatic nonparenchymal cells using fibulin-2 and cytoglobin/STAP as liver fibrogenesis-related markers. Histochem. Cell Biol. 122, 41–49. doi: 10.1007/s00418-004-0666-0
Thuy le, T. T., Matsumoto, Y., Thuy, T. T., Hai, H., Suoh, M., Urahara, Y., et al. (2015). Cytoglobin deficiency promotes liver cancer development from hepatosteatosis through activation of the oxidative stress pathway. Am. J. Pathol. 185, 1045–1060. doi: 10.1016/j.ajpath.2014.12.017
Thuy le, T. T., Morita, T., Yoshida, K., Wakasa, K., Iizuka, M., Ogawa, T., et al. (2011). Promotion of liver and lung tumorigenesis in DEN-treated cytoglobin-deficient mice. Am. J. Pathol. 179, 1050–1060. doi: 10.1016/j.ajpath.2011.05.006
Trent, J. T. III, and Hargrove, M. S. (2002). A ubiquitously expressed human hexacoordinate hemoglobin. J. Biol. Chem. 277, 19538–19545. doi: 10.1074/jbc.M201934200
Wake, K. (1980). Perisinusoidal stellate cells (fat-storing cells, interstitial cells, lipocytes), their related structure in and around the liver sinusoids, and vitamin A-storing cells in extrahepatic organs. Int. Rev. Cytol. 66, 303–353. doi: 10.1016/S0074-7696(08)61977-4
Wells, R. G. (2014). The portal fibroblast: not just a poor man's stellate cell. Gastroenterology 147, 41–47. doi: 10.1053/j.gastro.2014.05.001
Wojnarowicz, P. M., Provencher, D. M., Mes-Masson, A. M., and Tonin, P. N. (2012). Chromosome 17q25 genes, RHBDF2 and CYGB, in ovarian cancer. Int. J. Oncol. 40, 1865–1880. doi: 10.3892/ijo.2012.1371
Wree, A., Eguchi, A., McGeough, M. D., Pena, C. A., Johnson, C. D., Canbay, A., et al. (2014). NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 59, 898–910. doi: 10.1002/hep.26592
Xu, R., Harrison, P. M., Chen, M., Li, L., Tsui, T. Y., Fung, P. C., et al. (2006). Cytoglobin overexpression protects against damage-induced fibrosis. Mol. Ther. 13, 1093–1100. doi: 10.1016/j.ymthe.2005.11.027
Yin, C., Evason, K. J., Asahina, K., and Stainier, D. Y. (2013). Hepatic stellate cells in liver development, regeneration, and cancer. J. Clin. Invest. 123, 1902–1910. doi: 10.1172/JCI66369
Keywords: stellate cell, cytoglobin, myofibroblast, liver fibrosis, hypoxia
Citation: Kawada N (2015) Cytoglobin as a Marker of Hepatic Stellate Cell-derived Myofibroblasts. Front. Physiol. 6:329. doi: 10.3389/fphys.2015.00329
Received: 29 July 2015; Accepted: 30 October 2015;
Published: 13 November 2015.
Edited by:
Jiri Kanta, Charles University, Czech RepublicReviewed by:
Charles Wang, Beckman Research Institute of City of Hope, USAMichel Fausther, University of Arkansas for Medical Sciences, USA
Copyright © 2015 Kawada. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Norifumi Kawada, a2F3YWRhbm9yaUBnbWFpbC5jb20=