 Janelle Geist
Janelle Geist Aikaterini Kontrogianni-Konstantopoulos
Aikaterini Kontrogianni-Konstantopoulos- Department of Biochemistry and Molecular Biology, University of Maryland School of Medicine, Baltimore, MD, USA
Myosin Binding Protein-C (MyBP-C) comprises a family of accessory proteins that includes the cardiac, slow skeletal, and fast skeletal isoforms. The three isoforms share structural and sequence homology, and localize at the C-zone of the sarcomeric A-band where they interact with thick and thin filaments to regulate the cycling of actomyosin crossbridges. The cardiac isoform, encoded by MYBPC3, has been extensively studied over the last several decades due to its high mutational rate in congenital hypertrophic and dilated cardiomyopathy. It is only recently, however, that the MYBPC1 gene encoding the slow skeletal isoform (sMyBP-C) has gained attention. Accordingly, during the last 5 years it has been shown that MYBPC1 undergoes extensive exon shuffling resulting in the generation of multiple slow variants, which are co-expressed in different combinations and amounts in both slow and fast skeletal muscles. The sMyBP-C variants are subjected to PKA- and PKC-mediated phosphorylation in constitutive and alternatively spliced sites. More importantly, missense, and nonsense mutations in MYBPC1 have been directly linked with the development of severe and lethal forms of distal arthrogryposis myopathy and muscle tremors. Currently, there is no mammalian animal model of sMyBP-C, but new technologies including CRISPR/Cas9 and xenografting of human biopsies into immunodeficient mice could provide unique ways to study the regulation and roles of sMyBP-C in health and disease.
Introduction
Myosin Binding Protein-C (MyBP-C) comprises a family of accessory proteins expressed in striated muscles, and constitutes 2–4% of the myofibrillar protein mass (Okagaki et al., 1993; Moss et al., 2015) There are three MyBP-C isoforms encoded by different genes; slow (s) skeletal MyBP-C is encoded by MYBPC1 present in human chromosome 12, fast (f) skeletal MyBP-C is encoded by MYBPC2 present in human chromosome 19, and cardiac (c) MyBP-C is encoded by MYBPC3 present in human chromosome 11 (Weber et al., 1993). While cMyBP-C is selectively expressed in cardiac muscle, fMyBP-C and sMyBP-C co-exist in fast and slow twitch muscles at varying amounts (Ackermann and Kontrogianni-Konstantopoulos, 2011b). The three isoforms share structural and sequence homology, primarily consisting of immunoglobulin (Ig), and fibronectin-III (Fn-III) domains, referred from the NH2-terminus to the COOH-terminus as C1-C10; notably, the cardiac isoform contains an additional Ig domain, termed C0 (Figure 1; Ackermann and Kontrogianni-Konstantopoulos, 2010).
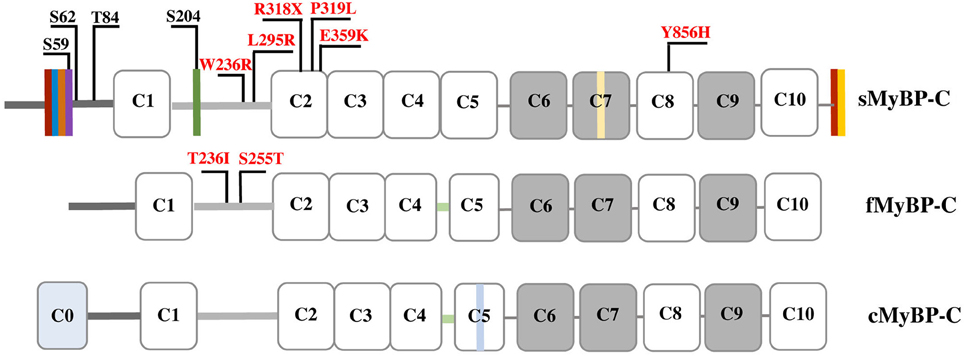
Figure 1. Schematic representation of the three MyBP-C isoforms. Dark and light gray lines correspond to the Pro/Ala-rich region and the M-motif, respectively, while white and gray rectangles represent Ig and Fn-III domains, respectively. Vertical colored boxes in the Pro/Ala-rich region, the M-motif, Fn-III domain C7 and the extreme COOH-terminus of sMyBP-C indicate alternative spliced segments. fMyBP-C and cMyBP-C share a conserved linker region between C4 and C5, denoted in light green. cMyBP-C contains an additional Ig domain, C0, and an isoform-specific insertion in C5 shown in light blue. Phosphorylation sites in the Pro/Ala-rich region and the M-motif of sMyBP-C are indicated in black, and myopathic mutations in sMyBP-C and fMyBP-C in the M-motif and Ig domains C2 and C8 are shown in red.
MyBP-C interacts directly with both thick and thin filaments via its NH2- and COOH-termini. The NH2-terminal domains C1-M-C2 bind to myosin subfragment 2 (S2) (Gruen et al., 1999), while the COOH-terminal C10 domain binds to light meromyosin (LMM) (Okagaki et al., 1993; Harris et al., 2011). The latter interaction is enhanced by binding of domains C8-C10 to titin that is likely instrumental in the periodic arrangement of MyBP-C in the C-zone (Craig and Offer, 1976; Freiburg and Gautel, 1996; Luther et al., 2008; Luther and Craig, 2011). In addition to binding to S2, the NH2-terminal C0-C1-M-C2 region contains (relatively weak) binding sites for actin (Kulikovskaya et al., 2003; Whitten et al., 2008; Ackermann et al., 2009; Shaffer et al., 2009; Orlova et al., 2011; Bhuiyan et al., 2012), although recently domains C6-C10 were suggested to mediate high-affinity binding to actin (Rybakova et al., 2011). The interactions at the NH2-terminus of MyBP-C are highly dynamic and regulated via phosphorylation (Figure 1; Gruen et al., 1999; Sadayappan et al., 2005; Shaffer et al., 2009; Barefield and Sadayappan, 2010). Accordingly, phosphorylation of cMyBP-C within the M-motif accelerates contraction by disrupting binding to myosin, thereby increasing the probability of binding to actin and thus the rate of force development (Sadayappan and de Tombe, 2012; Walcott et al., 2015; Colson et al., 2016; Moss, 2016; Previs et al., 2016).
Although several questions remain unanswered regarding the (patho)physiology of MyBP-C, early and recent work (primarily on the cardiac isoform) has postulated that through its direct binding to both actin and myosin filaments, it contributes to their assembly and stabilization, modulates the cycling of actomyosin crossbridges, and regulates the ATPase activity of myosin (Martyn, 2004; McClellan et al., 2004; de Tombe, 2006; Oakley et al., 2007; Ackermann and Kontrogianni-Konstantopoulos, 2010, 2013; James and Robbins, 2011; Rybakova et al., 2011; Ackermann et al., 2013). Below, we summarize old and new information on slow skeletal MyBP-C, highlight its direct involvement in disease pathogenesis and provide a perspective on its roles and regulation.
sMyBP-C: A Complex Sub-Family Of Proteins Regulated by Phosphorylation
MyBP-C proteins are highly modular consisting of tandem immunoglobulin (Ig) and fibronectin-III (Fn-III) modules interspersed with unique short amino acid segments (Figure 1; Winegrad, 1999). Ig domain C1 is flanked by a ~50 amino acids long Proline/Alanine-rich motif (Pro/Ala-rich motif) and a ~100 amino acids long MyBP-C specific motif, referred to as M-motif (Craig et al., 2014). Single transcripts have been identified for the mammalian cardiac and fast isoforms, which result in proteins of ~140 and ~130 kDa, respectively (Yasuda et al., 1995). sMyBP-C is unique, however, as there are several mammalian variants that have been characterized ranging in size from 126 to 131 kDa (Ackermann et al., 2015b). This size variability results from extensive splicing of small amino acid segments within the Pro/Ala-rich motif, the M-motif, Fn-III domain C7, and the extreme COOH-terminus (Ackermann and Kontrogianni-Konstantopoulos, 2011b). The different sMyBP-C variants are co-expressed in different amounts and combinations in both slow- and fast-twitch skeletal muscles were they co-exist with fMyBP-C (Ackermann and Kontrogianni-Konstantopoulos, 2011b, 2013).
Phosphorylation of cMyBP-C contributes significantly to contractile regulation (Sadayappan et al., 2005; Stelzer et al., 2006; Gresham et al., 2014; Gupta and Robbins, 2014; Gresham and Stelzer, 2016; Mamidi et al., 2016; Moss, 2016; Previs et al., 2016). Contrary to early studies suggesting that sMyBP-C is not subjected to phosphorylation (Gruen et al., 1999), work from our group demonstrated that similar to its cardiac counterpart, sMyBP-C also undergoes phosphorylation (Ackermann and Kontrogianni-Konstantopoulos, 2011a). Interestingly, while phosphorylation of cMyBP-C is restricted to the M-motif, phosphorylation of sMyBP-C takes place primarily in the Pro/Ala-rich motif and to a lesser extent in the M-motif (Ackermann and Kontrogianni-Konstantopoulos, 2011a). In particular, proteomics studies confirmed by the use of phospho-specific antibodies demonstrated that in the Pro/Ala-rich motif Ser-59 and Ser-62 are substrates of PKA, and Thr-84 is substrate of PKC, while in the M-motif Ser-204 is substrate of both PKA and PKC (Figure 1). Ser-59 and Ser-204 reside in alternatively spliced exons 5 and 10, respectively, and are therefore present in select slow variants (Ackermann and Kontrogianni-Konstantopoulos, 2013). Consistent with a purported important role of phosphorylation in the regulation of MyBP-C, the phosphorylation levels of sMyBP-C are differentially altered in relation to different (patho)physiological stressors. Accordingly, the phosphorylation levels of sMyBP-C are significantly reduced in fast-twitch Flexor Digitorum Brevis (FDB) muscle as a function of aging and dystrophy (Ackermann et al., 2015b). Similarly, the phosphorylation levels of sMyBP-C are notably decreased in slow-twitch soleus muscle as a result of aging and dystrophy, but increased in response to fatigue (Ackermann et al., 2015a). Although these observations are interesting, a detailed examination of the effects of individual or combinatorial phosphorylation events in the modulation of the structural and regulatory activities of sMyBP-C is currently lacking. It is therefore expected that future endeavors combining sophisticated in vitro and in vivo approaches will shed light on the role of phosphorylation in the modulation of sMyBP-C.
MYBPC1: A Recent Myopathic Gene
MYBPC3 has garnered much attention over the past several decades due to its prevalent mutational rate leading to congenital hypertrophic and dilated cardiomyopathy (Harris et al., 2011; Santos et al., 2012; Kuster and Sadayappan, 2014; Lynch et al., 2015). It is only recently, however, that mutations in MYBPC1 have been directly associated with inherited myopathies, and specifically with severe and lethal forms of distal arthrogryposis myopathy (Table 1) (Markus et al., 2012; Ha et al., 2013; Li et al., 2015). Contrary to MYBPC3 mutations that mainly result in truncated proteins and function via haploinsufficiency (Marston et al., 2012; Kuster and Sadayappan, 2014; Barefield et al., 2015; Carrier et al., 2015), the currently known MYBPC1 mutations have been suggested to result in poisonous proteins and manifest in a dominant negative manner after incorporation into sarcomeres (Markus et al., 2012; Ha et al., 2013; Li et al., 2015).
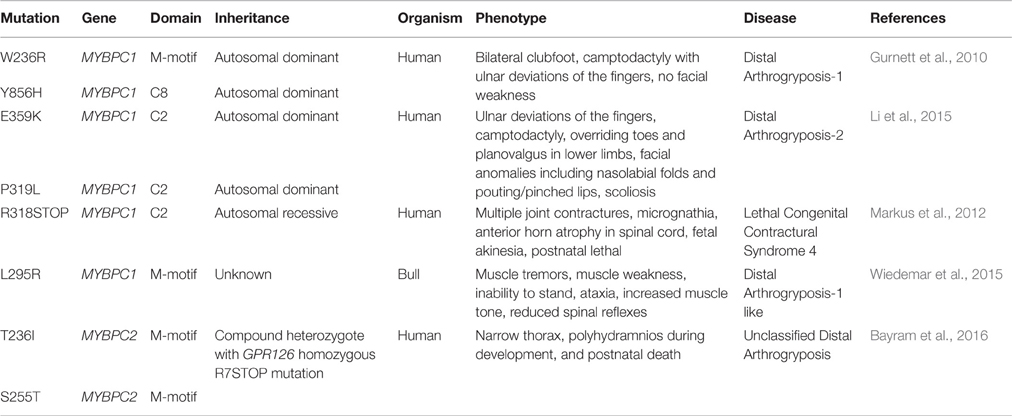
Table 1. Current disease-causing mutations in MYBPC1 and MYBPC2.
Arthrogryposis, also known as arthrogryposis multiplex congenita, is clinically defined by congenital joint contractures or movement restriction in multiple body areas (Bayram et al., 2016). Generally, arthrogryposis occurs as a secondary effect of decreased fetal joint mobility, which can result from abnormalities of the central nervous system, the neuromuscular system, the skeletal system, or connective and cartilage tissue disturbances. (Filges and Hall, 2013; Haliloglu and Topaloglu, 2013; Hall, 2014; Bayram et al., 2016). Distal arthrogryposis (DA) myopathies are a group of autosomal dominant arthrogryposis disorders that mainly involve the distal parts of the limbs (Bamshad et al., 2009). Ten different types of DA have been described to date that share common general features, including a consistent pattern of hand and foot defects, limited involvement of proximal joints and variable expressivity (Bamshad et al., 1996; Krakowiak et al., 1998; Stevenson et al., 2006).
DA type-1 (DA-1) is the most common DA myopathy that affects approximately 1 in 10,000 individuals, and results in contractures limited to the distal muscles of the hands and feet. These include clubfoot, vertical talus, camptodactyly, overriding fingers and ulnar deviations of the fingers with no additional anomalies (Hall, 1985; Klemp and Hall, 1995; Gurnett et al., 2010). DA type-2 (DA-2) is a more severe form of DA, also characterized by contractures of the hands and feet, that is often accompanied by mild to severe craniofacial anomalies and/or scoliosis (Kulkarni et al., 2008; Bamshad et al., 2009). There are two subtypes of DA-2, including DA-2A (Freeman-Sheldon syndrome) and DA-2B (Sheldon-Hall syndrome) (Kulkarni et al., 2008; Bamshad et al., 2009; Li et al., 2015). While individuals with DA-2B Sheldon-Hall syndrome display mild to moderate facial contractures, individuals with DA-2A Freeman-Sheldon syndrome have moderate to severe facial contractures (Beck et al., 2013; Li et al., 2015).
In the last 5 years, dominant missense mutations in MYBPC1 have been linked to the development of both DA-1 and DA-2 (Gurnett et al., 2010; Li et al., 2015). Specifically, missense mutations, W236R and Y856H, located in the M-motif and Ig domain C8, respectively, have been linked to DA-1 (Figure 1; Gurnett et al., 2010). Both of these substitutions are present in constitutively expressed exons and thus are contained in all slow variants (Gurnett et al., 2010; Ackermann et al., 2015b). ATPase staining of biopsies obtained from the distal Abductor Hallucis (AH) foot muscle of DA-1 patients carrying either mutation revealed severe type-I fiber atrophy, although localization of the mutant proteins was unaltered (Gurnett et al., 2010). In vitro binding and actin sliding assays demonstrated that the presence of the W236R and Y856H mutations markedly diminished the ability of the NH2 and COOH termini of sMyBP-C, respectively, to bind actin and myosin, and regulate the formation of actomyosin crossbridges (Ackermann et al., 2013). Examination of the expression levels of mutant sMyBP-C that contained the Y856H or the W236R mutation in human biopsies of AH or gastrocnemius muscles, respectively, revealed that the total amounts of the protein were significantly reduced in AH (~25%), but not gastrocnemius, muscle compared to controls (Ackermann et al., 2015b). Although puzzling, since both DA-1 mutations reside in constitutive exons, this finding is in agreement with the selective effects of DA-1 on distal muscles, and the lack of a myopathic phenotype in proximal muscles. Interestingly, the phosphorylation profile of mutant sMyBP-C containing the Y856H mutation was also altered in the affected AH muscle, whereas the phosphorylation profile of sMyBP-C carrying the W236R mutation was unchanged in gastrocnemius muscle (Ackermann et al., 2015b). Accordingly, use of a panel of phospho-specific antibodies and phos-tag gel electrophoresis revealed that mutant sMyBP-C harboring the Y856H mutation in AH muscle was phosphorylated at all four known residues, however the extent of phosphorylation was decreased by 30–70% for individual phospho-sites, compared to control tissue (Ackermann et al., 2015b).
Recently, two novel autosomal dominant missense mutations in Ig domain C2 of sMyBP-C, P319L and E359K, were linked to the development of DA-2 (Figure 1) (Li et al., 2015). Although a mechanistic understanding of the effect(s) of these mutations is still lacking, it is tempting to speculate that they may affect binding to the S2 portion of myosin and/or actin via induction of an unfavorable conformation (P319L) or altered electrostatic interactions (E359K). Future studies using a combination of biochemical, structural, biophysical and in vivo approaches will address these hypotheses.
In addition to DA-1 and DA-2, MYBPC1 has been directly linked to the development of a neonatal lethal form of arthrogryposis myopathy, referred to as Lethal Congenital Contracture Syndrome type-4 (LCCS-4; Markus et al., 2012). Specifically, an autosomal recessive nonsense mutation in Ig domain C2 of sMyBP-C results in a premature stop codon at amino acid 318 (R318Stop; Figure 1). Given the recessive inheritance of LCCS-4, along with the absence of any phenotypic or functional abnormalities in the heterozygous carriers, it is highly likely that the R318Stop mutation results in loss of sMyBP-C rather than a poisonous truncated protein. Nevertheless, if the mutant protein is indeed expressed, it will lack domains C3-C10 downstream of Ig C2, which contain binding sites for LMM, titin and obscurin (Okagaki et al., 1993; Freiburg and Gautel, 1996; Ackermann et al., 2009).
In addition to mutations in the human MYBPC1 gene that are associated with severe and lethal forms of DA, a new mutation in the bull MYBPC1 gene was recently identified, too (Wiedemar et al., 2015). In particular, a 2-week old female calf presented with muscle tremors since birth, standing difficulty and reduced spinal reflexes (Wiedemar et al., 2015). Whole genome sequencing analysis revealed a de novo missense mutation, L295R, localized in the M-motif following the Ig domain C1, similar to the human W236R mutation. Although the phenotypic manifestation of the L295R mutation is reminiscent of DA-1, it is further accompanied by muscle tremors, which is indicative of a more complex and/or severe myopathy. At this time, a mechanistic understanding of the effects of the L295R mutation is lacking.
As novel mutations in MYBPC1 are being identified in the mammalian genome underscoring its role in skeletal muscle (patho)physiology, it is worth mentioning that recently MYBPC2, encoding fMyBP-C, was also linked to an unclassified, neonatal lethal DA in the form of a compound heterozygote (Bayram et al., 2016). Specifically, a patient presenting with narrow thorax, polyhydramnios during fetal development, and neonatal death was found to possess two missense mutations in MYBPC2, T236I and S255T, located in the M-motif. The same patient also contained an R7Stop homozygous mutation in the GPR126 gene, which encodes a G-protein coupled receptor that regulates neural, cardiac, and ear development (Patra et al., 2014; Bayram et al., 2016). Although mutations in GPR126 have been linked with isolated arthrogryposis multiplex congenital (Ravenscroft et al., 2015), it is likely that the additional mutations in MYBPC2 contributed to the postnatal lethality of the carrier due to accumulating anomalies in motor neurons and muscle structure and function (Bayram et al., 2016).
Although at the current time limited, the above studies clearly indicate that mutations in the genes that encode the skeletal MyBP-C proteins (and especially the slow isoform) are intimately associated with the development of severe and lethal myopathies. Obviously, the challenge now lies in deciphering the cell processes that are altered or compromised due to individual mutations using sophisticated and high resolution in vitro approaches and appropriate in vivo models.
In vivo Models of MYBPC1: Perspectives and Endeavors
Over the last four decades, a tremendous emphasis has been placed on the regulation and roles of cMyBP-C due to its direct involvement in congenital heart disease resulting in the generation of multiple animal models (Harris et al., 2002; Sadayappan et al., 2005; Carrier et al., 2015). This is not the case for sMyBP-C (or fMyBP-C). Remarkably though, the direct association of MYBPC1 with the development of severe and lethal forms of DA has tunneled the interest of the scientific community toward the molecular and functional characterization of MYBPC1, too.
A recent study used the zebrafish model and antisense morpholinos to down-regulate the expression of MYBPC1 (Ha et al., 2013). Knock-down zebrafish exhibited severe ventral body curvature, decreased mobility, and early lethality, along with impaired sarcomeric development and reduced number of myofibrils (Ha et al., 2013). Moreover, overexpression of mutant sMyBP-C proteins carrying either of the DA-1 mutations, W236R or Y856H, in zebrafish demonstrated that both mutant proteins exerted a dominant negative effect, resulting in embryos with mild bent body curvature, impaired mobility, and muscles with less tightly compacted fibers compared to controls (Ha et al., 2013).
Apart from the zebrafish model, no mammalian MYBPC1 animal models have been generated yet. Our group has been systematically working on sMyBP-C for the last 5 to 6 years focusing on its molecular characterization, regulation and functional evaluation. Given the unique complexity of MYBPC1 (Ackermann and Kontrogianni-Konstantopoulos, 2010, 2011b, 2013), its early expression during fetal development preceding that of MYBPC2 (Gautel et al., 1998; Kurasawa et al., 1999), and the neonatal lethality of LCCS-4 patients, who most likely lack sMyBP-C, we predict that a constitutive MYBPC1 null model would be postnatal lethal. If this is the case, such a model is still worth generating, since it will highlight the non-redundant roles of sMyBP-C and fMyBP-C, and will allow the study of the structural and regulatory roles of sMyBP-C in myofibrillar assembly and contractility during embryogenesis and (early) postnatal life. Obviously, conditional null models would circumvent the potential neonatal lethality of a constitutive MYBPC1 knock-out allowing the detailed investigation of the roles of sMyBP-C in modulating actomyosin contractility in mature muscles. Along the same lines, knock-in models carrying the DA-1, DA-2, or LSSC-4 mutations are also lacking, limiting our understanding of the effects of the respective mutations to in vitro studies, which although informative, need to be accompanied by in vivo data.
Type II bacterial Clustered Regularly Interspaced Short Palindromic Repeats-associated protein Cas9 (CRISPR-Cas9) mediated genome editing has emerged as a powerful tool for genetic manipulation. Unlike small interfering RNAs (siRNAs) or short hairpin RNAs (shRNAs), the CRISPR-Cas9 system is able to knockout individual gene expression at the genomic level with minimal off-target effects (Zhang et al., 2014; Humphrey and Kasinski, 2015). Conversely, CRISPR/Cas9 technology is also being refined for generating knock-in models to recapitulate disease development (Chu et al., 2015; Tu et al., 2015). As the technology becomes increasingly popular, the generation of constitutive or conditional MYBPC1 knock-out and knock-in mouse models should be feasible in a fairly short amount of time.
In vivo gene transfer (IVGT) followed by electroporation is also an efficient, non-viral method for gene delivery that has been successfully used by several groups (Gehl, 2003; Spanggaard et al., 2012; Hu et al., 2014). Although the mechanisms underlying DNA electrotransfer are not yet fully elucidated, it has been suggested that permeabilization of the cell membrane as well as electrophoretic migration of the DNA are involved (Mir et al., 1999; Bureau et al., 2000; Golzio et al., 2002; Satkauskas et al., 2002). Muscle tissue is a favorable target for gene electrotransfer as it is easily accessible, allowing high-level, long-term transgenic expression (Mir et al., 1999; Lucas and Heller, 2001; Spanggaard et al., 2012). Such an approach could therefore be employed to knock-down (via shRNA technology) or knock-out (via CRISPR-Cas9 technology) MYBPC1 in a muscle-specific manner, circumventing the potential neonatal lethality of a constitutive null model, and enabling the functional examination of different slow variants given that they are expressed in distinct combinations among skeletal muscles. Moreover, IVGT combined with electroporation could be used to overexpress myopathic forms of sMyBP-C. Although the stoichiometry of endogenous to exogenous proteins is an important issue to consider, it could potentially be alleviated by inducible, titratable expression systems. Notably, IVGT experiments are particularly beneficial as the effects of gene knock-down, knock-out or knock-in experiments can be analyzed from the single fiber to the whole animal level. For instance, many groups have taken advantage of permeabilized muscle fiber preparations of human biopsies or pre-clinical mouse models to elucidate the (patho)physiology of cMyBP-C (Harris et al., 2002; Stelzer et al., 2006; James and Robbins, 2011; Wang et al., 2016).
An alternative to generating animal models of muscle disease has emerged in the last few years, entailing the development and propagation of grafts of myopathic or dystrophic human muscle tissue in mice (Riederer et al., 2012; Meng et al., 2014; Sakellariou et al., 2015). This approach has tremendous benefits, given that animal models often fail to replicate the features of human muscle disease. Along these lines, a recent study reported the generation of xenografts from human bicep muscle biopsies of facioscapulohumeral muscular dystrophy (FSHD) patients that were transplanted into the hindlimbs of immunodeficient NOD-Rag1nullIL2rynull mice (Sakellariou et al., 2015). The engrafted human muscle was efficiently regenerated and innervated, and displayed normal contractile properties (Sakellariou et al., 2015). While the xenografting model approach is still being perfected, the largest hurdle is the unavailability of fresh muscle biopsies and the lack of organized biobanks; obviously, this is a major issue that applies to MYBPC1 related myopathies, as well. Nevertheless, the xenografting model could prove to be an extremely useful tool for studying the effects of human myopathies in vivo, since it may recapitulate the course of disease development more faithfully compared to engineered models of C. elegans, zebrafish, or mouse that are commonly used to date.
Conclusions
Slow skeletal MyBP-C has recently attracted considerable interest primarily due to its direct involvement in the development of severe and lethal forms of distal arthrogryposis myopathy. Contrary to the fast and cardiac isoforms, sMyBP-C comprises a subfamily of proteins with possibly distinct structural and regulatory roles, which are modulated by constitutive and variant-specific phosphorylation events. Given the recent involvement of MYBPC1 in severe and lethal myopathies, we predict that a comprehensive, multidisciplinary evaluation of its regulation and roles in health and disease is in order.
Author Contributions
JG and AK-K drafted, revised and approved the final version of the manuscript.
Funding
This work was supported by NIH/NIAMS (Training Program in Muscle Biology, T32 AR007592-17 to JG), and the Muscular Dystrophy Association (Research Grant 313579 to AK-K).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ackermann, M. A., Hu, L. Y., Bowman, A. L., Bloch, R. J., and Kontrogianni-Konstantopoulos, A. (2009). Obscurin interacts with a novel isoform of MyBP-C slow at the periphery of the sarcomeric M-band and regulates thick filament assembly. Mol. Biol. Cell 20, 2963–2978. doi: 10.1091/mbc.E08-12-1251
Ackermann, M. A., Kerr, J. P., King, B., Ward, C. W., and Kontrogianni-Konstantopoulos, A. (2015a). The phosphorylation profile of myosin binding protein-C slow is dynamically regulated in slow-twitch muscles in health and disease. Sci. Rep. 5:12637. doi: 10.1038/srep12637
Ackermann, M. A., and Kontrogianni-Konstantopoulos, A. (2010). Myosin binding protein-C slow: an intricate subfamily of proteins. J. Biomed. Biotechnol. 2010:652065. doi: 10.1155/2010/652065
Ackermann, M. A., and Kontrogianni-Konstantopoulos, A. (2011a). Myosin binding protein-C slow is a novel substrate for protein kinase A (PKA) and C (PKC) in skeletal muscle. J. Proteome Res. 10, 4547–4555. doi: 10.1021/pr200355w
Ackermann, M. A., and Kontrogianni-Konstantopoulos, A. (2011b). Myosin binding protein-C: a regulator of actomyosin interaction in striated muscle. J. Biomed. Biotechnol. 2011:636403. doi: 10.1155/2011/636403
Ackermann, M. A., and Kontrogianni-Konstantopoulos, A. (2013). Myosin binding protein-C slow: a multifaceted family of proteins with a complex expression profile in fast and slow twitch skeletal muscles. Front. Physiol. 4:391. doi: 10.3389/fphys.2013.00391
Ackermann, M. A., Patel, P. D., Valenti, J., Takagi, Y., Homsher, E., Sellers, J. R., et al. (2013). Loss of actomyosin regulation in distal arthrogryposis myopathy due to mutant myosin binding protein-C slow. FASEB J. 27, 3217–3228. doi: 10.1096/fj.13-228882
Ackermann, M. A., Ward, C. W., Gurnett, C., and Kontrogianni-Konstantopoulos, A. (2015b). Myosin binding protein-C slow phosphorylation is altered in duchenne dystrophy and arthrogryposis myopathy in fast-twitch skeletal muscles. Sci. Rep. 5:13235. doi: 10.1038/srep13235
Bamshad, M., Jorde, L. B., and Carey, J. C. (1996). A revised and extended classification of the distal arthrogryposes. Am. J. Med. Genet. 65, 277–281.
Bamshad, M., Van Heest, A. E., and Pleasure, D. (2009). Arthrogryposis: a review and update. J. Bone Joint Surg. Am. 91(Suppl. 4), 40–46. doi: 10.2106/JBJS.I.00281
Barefield, D., Kumar, M., Gorham, J., Seidman, J. G., Seidman, C. E., de Tombe, P. P., et al. (2015). Haploinsufficiency of MYBPC3 exacerbates the development of hypertrophic cardiomyopathy in heterozygous mice. J. Mol. Cell. Cardiol. 79, 234–243. doi: 10.1016/j.yjmcc.2014.11.018
Barefield, D., and Sadayappan, S. (2010). Phosphorylation and function of cardiac myosin binding protein-C in health and disease. J. Mol. Cell. Cardiol. 48, 866–875. doi: 10.1016/j.yjmcc.2009.11.014
Bayram, Y., Karaca, E., Coban Akdemir, Z., Yilmaz, E. O., Tayfun, G. A., Aydin, H., et al. (2016). Molecular etiology of arthrogryposis in multiple families of mostly Turkish origin. J. Clin. Invest. 126, 762–778. doi: 10.1172/JCI84457
Beck, A. E., McMillin, M. J., Gildersleeve, H. I., Kezele, P. R., Shively, K. M., Carey, J. C., et al. (2013). Spectrum of mutations that cause distal arthrogryposis types 1 and 2B. Am. J. Med. Genet. A 161A, 550–555. doi: 10.1002/ajmg.a.35809
Bhuiyan, M. S., Gulick, J., Osinska, H., Gupta, M., and Robbins, J. (2012). Determination of the critical residues responsible for cardiac myosin binding protein C's interactions. J. Mol. Cell. Cardiol. 53, 838–847. doi: 10.1016/j.yjmcc.2012.08.028
Bureau, M. F., Gehl, J., Deleuze, V., Mir, L. M., and Scherman, D. (2000). Importance of association between permeabilization and electrophoretic forces for intramuscular DNA electrotransfer. Biochim. Biophys. Acta 1474, 353–359. doi: 10.1016/S0304-4165(00)00028-3
Carrier, L., Mearini, G., Stathopoulou, K., and Cuello, F. (2015). Cardiac myosin-binding protein C (MYBPC3) in cardiac pathophysiology. Gene 573, 188–197. doi: 10.1016/j.gene.2015.09.008
Chu, V. T., Weber, T., Wefers, B., Wurst, W., Sander, S., Rajewsky, K., et al. (2015). Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 33, 543–548. doi: 10.1038/nbt.3198
Colson, B. A., Thompson, A. R., Espinoza-Fonseca, L. M., and Thomas, D. D. (2016). Site-directed spectroscopy of cardiac myosin-binding protein C reveals effects of phosphorylation on protein structural dynamics. Proc. Natl. Acad. Sci. U.S.A. 113, 3233–3238. doi: 10.1073/pnas.1521281113
Craig, R., Lee, K. H., Mun, J. Y., Torre, I., and Luther, P. K. (2014). Structure, sarcomeric organization, and thin filament binding of cardiac myosin-binding protein-C. Pflugers Arch. 466, 425–431. doi: 10.1007/s00424-013-1426-6
Craig, R., and Offer, G. (1976). The location of C-protein in rabbit skeletal muscle. Proc. R. Soc. Lond. B. Biol. Sci. 192, 451–461. doi: 10.1098/rspb.1976.0023
de Tombe, P. P. (2006). Myosin binding protein C in the heart. Circ. Res. 98, 1234–1236. doi: 10.1161/01.RES.0000225873.63162.c4
Filges, I., and Hall, J. G. (2013). Failure to identify antenatal multiple congenital contractures and fetal akinesia–proposal of guidelines to improve diagnosis. Prenat. Diagn. 33, 61–74. doi: 10.1002/pd.4011
Freiburg, A., and Gautel, M. (1996). A molecular map of the interactions between titin and myosin-binding protein C. Implications for sarcomeric assembly in familial hypertrophic cardiomyopathy. Eur. J. Biochem. 235, 317–323. doi: 10.1111/j.1432-1033.1996.00317.x
Gautel, M., Fürst, D. O., Cocco, A., and Schiaffino, S. (1998). Isoform transitions of the myosin binding protein C family in developing human and mouse muscles: lack of isoform transcomplementation in cardiac muscle. Circ. Res. 82, 124–129. doi: 10.1161/01.RES.82.1.124
Gehl, J. (2003). Electroporation: theory and methods, perspectives for drug delivery, gene therapy and research. Acta Physiol. Scand. 177, 437–447. doi: 10.1046/j.1365-201X.2003.01093.x
Golzio, M., Teissie, J., and Rols, M. P. (2002). Direct visualization at the single-cell level of electrically mediated gene delivery. Proc. Natl. Acad. Sci. U.S.A. 99, 1292–1297. doi: 10.1073/pnas.022646499
Gresham, K. S., Mamidi, R., and Stelzer, J. E. (2014). The contribution of cardiac myosin binding protein-c Ser282 phosphorylation to the rate of force generation and in vivo cardiac contractility. J. Physiol. 592, 3747–3765. doi: 10.1113/jphysiol.2014.276022
Gresham, K. S., and Stelzer, J. E. (2016). The contributions of cardiac myosin binding protein C and troponin I phosphorylation to beta-adrenergic enhancement of in vivo cardiac function. J. Physiol. 594, 669–686. doi: 10.1113/JP270959
Gruen, M., Prinz, H., and Gautel, M. (1999). cAPK-phosphorylation controls the interaction of the regulatory domain of cardiac myosin binding protein C with myosin-S2 in an on-off fashion. FEBS Lett. 453, 254–259. doi: 10.1016/S0014-5793(99)00727-9
Gupta, M. K., and Robbins, J. (2014). Post-translational control of cardiac hemodynamics through myosin binding protein C. Pflugers Arch. 466, 231–236. doi: 10.1007/s00424-013-1377-y
Gurnett, C. A., Desruisseau, D. M., McCall, K., Choi, R., Meyer, Z. I., Talerico, M., et al. (2010). Myosin binding protein C1: a novel gene for autosomal dominant distal arthrogryposis type 1. Hum. Mol. Genet. 19, 1165–1173. doi: 10.1093/hmg/ddp587
Ha, K., Buchan, J. G., Alvarado, D. M., McCall, K., Vydyanath, A., Luther, P. K., et al. (2013). MYBPC1 mutations impair skeletal muscle function in zebrafish models of arthrogryposis. Hum. Mol. Genet. 22, 4967–4977. doi: 10.1093/hmg/ddt344
Haliloglu, G., and Topaloglu, H. (2013). Arthrogryposis and fetal hypomobility syndrome. Handb. Clin. Neurol. 113, 1311–1319. doi: 10.1016/B978-0-444-59565-2.00003-4
Hall, J. G. (1985). Genetic aspects of arthrogryposis. Clin. Orthop. Relat. Res. 194, 44–53. doi: 10.1097/00003086-198504000-00006
Hall, J. G. (2014). Arthrogryposis (multiple congenital contractures): diagnostic approach to etiology, classification, genetics, and general principles. Eur. J. Med. Genet. 57, 464–472. doi: 10.1016/j.ejmg.2014.03.008
Harris, S. P., Bartley, C. R., Hacker, T. A., McDonald, K. S., Douglas, P. S., Greaser, M. L., et al. (2002). Hypertrophic cardiomyopathy in cardiac myosin binding protein-C knockout mice. Circ. Res. 90, 594–601. doi: 10.1161/01.RES.0000012222.70819.64
Harris, S. P., Lyons, R. G., and Bezold, K. L. (2011). In the thick of it: HCM-causing mutations in myosin binding proteins of the thick filament. Circ. Res. 108, 751–764. doi: 10.1161/CIRCRESAHA.110.231670
Hu, J., Cutrera, J., and Li, S. (2014). The impact of non-electrical factors on electrical gene transfer. Methods Mol. Biol. 1121, 47–54. doi: 10.1007/978-1-4614-9632-8_3
Humphrey, S. E., and Kasinski, A. L. (2015). RNA-guided CRISPR-Cas technologies for genome-scale investigation of disease processes. J. Hematol. Oncol. 8, 31. doi: 10.1186/s13045-015-0127-3
James, J., and Robbins, J. (2011). Signaling and myosin-binding protein C. J. Biol. Chem. 286, 9913–9919. doi: 10.1074/jbc.R110.171801
Klemp, P., and Hall, J. G. (1995). Dominant distal arthrogryposis in a Maori family with marked variability of expression. Am. J. Med. Genet. 55, 414–419. doi: 10.1002/ajmg.1320550406
Krakowiak, P. A., Bohnsack, J. F., Carey, J. C., and Bamshad, M. (1998). Clinical analysis of a variant of Freeman-Sheldon syndrome (DA2B). Am. J. Med. Genet. 76, 93–98.
Kulikovskaya, I., McClellan, G., Flavigny, J., Carrier, L., and Winegrad, S. (2003). Effect of MyBP-C binding to actin on contractility in heart muscle. J. Gen. Physiol. 122, 761–774. doi: 10.1085/jgp.200308941
Kulkarni, K. P., Panigrahi, I., Ray, M., and Marwaha, R. K. (2008). Distal arthrogryposis syndrome. Indian J. Hum. Genet. 14, 67–69. doi: 10.4103/0971-6866.44108
Kurasawa, M., Sato, N., Matsuda, A., Koshida, S., Totsuka, T., and Obinata, T. (1999). Differential expression of C-protein isoforms in developing and degenerating mouse striated muscles. Muscle Nerve 22, 196–207.
Kuster, D. W., and Sadayappan, S. (2014). MYBPC3's alternate ending: consequences and therapeutic implications of a highly prevalent 25 bp deletion mutation. Pflugers Arch. 466, 207–213. doi: 10.1007/s00424-013-1417-7
Li, X., Zhong, B., Han, W., Zhao, N., Liu, W., Sui, Y., et al. (2015). Two novel mutations in myosin binding protein C slow causing distal arthrogryposis type 2 in two large Han Chinese families may suggest important functional role of immunoglobulin domain C2. PLoS ONE 10:e0117158. doi: 10.1371/journal.pone.0117158
Lucas, M. L., and Heller, R. (2001). Immunomodulation by electrically enhanced delivery of plasmid DNA encoding IL-12 to murine skeletal muscle. Mol. Ther. 3, 47–53. doi: 10.1006/mthe.2000.0233
Luther, P. K., Bennett, P. M., Knupp, C., Craig, R., Padrón, R., Harris, S. P., et al. (2008). Understanding the organisation and role of myosin binding protein C in normal striated muscle by comparison with MyBP-C knockout cardiac muscle. J. Mol. Biol. 384, 60–72. doi: 10.1016/j.jmb.2008.09.013
Luther, P. K., and Craig, R. (2011). Modulation of striated muscle contraction by binding of myosin binding protein C to actin. Bioarchitecture 1, 277–283. doi: 10.4161/bioa.1.6.19341
Lynch, T. L. IV Sivaguru, M., Velayutham, M., Cardounel, A. J., Michels, M., Barefield, D., et al. (2015). Oxidative stress in dilated cardiomyopathy caused by MYBPC3 mutation. Oxid. Med. Cell. Longev. 2015:424751. doi: 10.1155/2015/424751
Mamidi, R., Gresham, K. S., Verma, S., and Stelzer, J. E. (2016). Cardiac myosin binding protein-C phosphorylation modulates myofilament length-dependent activation. Front. Physiol. 7:38. doi: 10.3389/fphys.2016.00038
Markus, B., Narkis, G., Landau, D., Birk, R. Z., Cohen, I., and Birk, O. S. (2012). Autosomal recessive lethal congenital contractural syndrome type 4 (LCCS4) caused by a mutation in MYBPC1. Hum. Mutat. 33, 1435–1438. doi: 10.1002/humu.22122
Marston, S., Copeland, O., Gehmlich, K., Schlossarek, S., and Carrier, L. (2012). How do MYBPC3 mutations cause hypertrophic cardiomyopathy? J. Muscle Res. Cell Motil. 33, 75–80. doi: 10.1007/s10974-011-9268-3
Martyn, D. A. (2004). Myosin binding protein-C: structural and functional complexity. J. Mol. Cell. Cardiol. 37, 813–815. doi: 10.1016/j.yjmcc.2004.07.005
McClellan, G., Kulikovskaya, I., Flavigny, J., Carrier, L., and Winegrad, S. (2004). Effect of cardiac myosin-binding protein C on stability of the thick filament. J. Mol. Cell. Cardiol. 37, 823–835. doi: 10.1016/j.yjmcc.2004.05.023
Meng, Y., Sohar, I., Sleat, D. E., Richardson, J. R., Reuhl, K. R., Jenkins, R. B., et al. (2014). Effective intravenous therapy for neurodegenerative disease with a therapeutic enzyme and a peptide that mediates delivery to the brain. Mol. Ther. 22, 547–553. doi: 10.1038/mt.2013.267
Mir, L. M., Bureau, M. F., Gehl, J., Rangara, R., Rouy, D., Caillaud, J. M., et al. (1999). High-efficiency gene transfer into skeletal muscle mediated by electric pulses. Proc. Natl. Acad. Sci. U.S.A. 96, 4262–4267. doi: 10.1073/pnas.96.8.4262
Moss, R. L. (2016). Cardiac myosin-binding protein C: a protein once at loose ends finds its regulatory groove. Proc. Natl. Acad. Sci. U.S.A. 113, 3133–3135. doi: 10.1073/pnas.1602568113
Moss, R. L., Fitzsimmons, D. P., and Ralphe, J. C. (2015). Cardiac MyBP-C regulates the rate and force of contraction in mammalian myocardium. Circ. Res. 116, 183–192. doi: 10.1161/CIRCRESAHA.116.300561
Oakley, C. E., Chamoun, J., Brown, L. J., and Hambly, B. D. (2007). Myosin binding protein-C: enigmatic regulator of cardiac contraction. Int. J. Biochem. Cell Biol. 39, 2161–2166. doi: 10.1016/j.biocel.2006.12.008
Okagaki, T., Weber, F. E., Fischman, D. A., Vaughan, K. T., Mikawa, T., and Reinach, F. C. (1993). The major myosin-binding domain of skeletal muscle MyBP-C (C protein) resides in the COOH-terminal, immunoglobulin C2 motif. J. Cell Biol. 123, 619–626. doi: 10.1083/jcb.123.3.619
Orlova, A., Galkin, V. E., Jeffries, C. M., Egelman, E. H., and Trewhella, J. (2011). The N-terminal domains of myosin binding protein C can bind polymorphically to F-actin. J. Mol. Biol. 412, 379–386. doi: 10.1016/j.jmb.2011.07.056
Patra, C., Monk, K. R., and Engel, F. B. (2014). The multiple signaling modalities of adhesion G protein-coupled receptor GPR126 in development. Receptors Clin. Investig. 1:79. doi: 10.14800/rci.79
Previs, M. J., Mun, J. Y., Michalek, A. J., Previs, S. B., Gulick, J., Robbins, J., et al. (2016). Phosphorylation and calcium antagonistically tune myosin-binding protein C's structure and function. Proc. Natl. Acad. Sci. U.S.A. 113, 3239–3244. doi: 10.1073/pnas.1522236113
Ravenscroft, G., Nolent, F., Rajagopalan, S., Meireles, A. M., Paavola, K. J., Gaillard, D., et al. (2015). Mutations of GPR126 are responsible for severe arthrogryposis multiplex congenita. Am. J. Hum. Genet. 96, 955–961. doi: 10.1016/j.ajhg.2015.04.014
Riederer, I., Negroni, E., Bencze, M., Wolff, A., Aamiri, A., Di Santo, J. P., et al. (2012). Slowing down differentiation of engrafted human myoblasts into immunodeficient mice correlates with increased proliferation and migration. Mol. Ther. 20, 146–154. doi: 10.1038/mt.2011.193
Rybakova, I. N., Greaser, M. L., and Moss, R. L. (2011). Myosin binding protein C interaction with actin: characterization and mapping of the binding site. J. Biol. Chem. 286, 2008–2016. doi: 10.1074/jbc.M110.170605
Sadayappan, S., and de Tombe, P. P. (2012). Cardiac myosin binding protein-C: redefining its structure and function. Biophys. Rev. 4, 93–106. doi: 10.1007/s12551-012-0067-x
Sadayappan, S., Gulick, J., Osinska, H., Martin, L. A., Hahn, H. S., Dorn, G. W. II, et al. (2005). Cardiac myosin-binding protein-C phosphorylation and cardiac function. Circ. Res. 97, 1156–1163. doi: 10.1161/01.RES.0000190605.79013.4d
Sakellariou, P., O'neill, A., Mueller, A. L., Stadler, G., Wright, W. E., Roche, J. A., et al. (2015). Neuromuscular electrical stimulation promotes development in mice of mature human muscle from immortalized human myoblasts. Skelet. Muscle 6:4. doi: 10.1186/s13395-016-0078-6
Santos, S., Marques, V., Pires, M., Silveira, L., Oliveira, H., Lança, V., et al. (2012). High resolution melting: improvements in the genetic diagnosis of hypertrophic cardiomyopathy in a Portuguese cohort. BMC Med. Genet. 13:17. doi: 10.1186/1471-2350-13-17
Satkauskas, S., Bureau, M. F., Puc, M., Mahfoudi, A., Scherman, D., Miklavcic, D., et al. (2002). Mechanisms of in vivo DNA electrotransfer: respective contributions of cell electropermeabilization and DNA electrophoresis. Mol. Ther. 5, 133–140. doi: 10.1006/mthe.2002.0526
Shaffer, J. F., Kensler, R. W., and Harris, S. P. (2009). The myosin-binding protein C motif binds to F-actin in a phosphorylation-sensitive manner. J. Biol. Chem. 284, 12318–12327. doi: 10.1074/jbc.M808850200
Spanggaard, I., Corydon, T., Hojman, P., Gissel, H., Dagnaes-Hansen, F., Jensen, T. G., et al. (2012). Spatial distribution of transgenic protein after gene electrotransfer to porcine muscle. Hum. Gene Ther. Methods 23, 387–392. doi: 10.1089/hgtb.2012.173
Stelzer, J. E., Patel, J. R., and Moss, R. L. (2006). Protein kinase A-mediated acceleration of the stretch activation response in murine skinned myocardium is eliminated by ablation of cMyBP-C. Circ. Res. 99, 884–890. doi: 10.1161/01.RES.0000245191.34690.66
Stevenson, D. A., Carey, J. C., Palumbos, J., Rutherford, A., Dolcourt, J., and Bamshad, M. J. (2006). Clinical characteristics and natural history of Freeman-Sheldon syndrome. Pediatrics 117, 754–762. doi: 10.1542/peds.2005-1219
Tu, Z., Yang, W., Yan, S., Guo, X., and Li, X. J. (2015). CRISPR/Cas9: a powerful genetic engineering tool for establishing large animal models of neurodegenerative diseases. Mol. Neurodegener. 10, 35. doi: 10.1186/s13024-015-0031-x
Walcott, S., Docken, S., and Harris, S. P. (2015). Effects of cardiac Myosin binding protein-C on actin motility are explained with a drag-activation-competition model. Biophys. J. 108, 10–13. doi: 10.1016/j.bpj.2014.11.1852
Wang, J., Ma, Y., Sachs, F., Li, J., and Suchyna, T. M. (2016). GsMTx4-D is a cardioprotectant against myocardial infarction during ischemia and reperfusion. J. Mol. Cell. Cardiol. 98, 83–94. doi: 10.1016/j.yjmcc.2016.07.005
Weber, F. E., Vaughan, K. T., Reinach, F. C., and Fischman, D. A. (1993). Complete sequence of human fast-type and slow-type muscle myosin-binding-protein C (MyBP-C). Differential expression, conserved domain structure and chromosome assignment. Eur. J. Biochem. 216, 661–669. doi: 10.1111/j.1432-1033.1993.tb18186.x
Whitten, A. E., Jeffries, C. M., Harris, S. P., and Trewhella, J. (2008). Cardiac myosin-binding protein C decorates F-actin: implications for cardiac function. Proc. Natl. Acad. Sci. U.S.A. 105, 18360–18365. doi: 10.1073/pnas.0808903105
Wiedemar, N., Riedi, A. K., Jagannathan, V., Drögemöller, C., and Meylan, M. (2015). Genetic Abnormalities in a Calf with Congenital Increased Muscular Tonus. J. Vet. Intern. Med. 29, 1418–1421. doi: 10.1111/jvim.13599
Winegrad, S. (1999). Cardiac myosin binding protein C. Circ. Res. 84, 1117–1126. doi: 10.1161/01.RES.84.10.1117
Yasuda, M., Koshida, S., Sato, N., and Obinata, T. (1995). Complete primary structure of chicken cardiac C-protein (MyBP-C) and its expression in developing striated muscles. J. Mol. Cell. Cardiol. 27, 2275–2286. doi: 10.1016/S0022-2828(95)91731-4
Keywords: MyBP-C slow, MYBPC1, actomyosin crossbridges, phosphorylation, distal arthrogryposis myopathy
Citation: Geist J and Kontrogianni-Konstantopoulos A (2016) MYBPC1, an Emerging Myopathic Gene: What We Know and What We Need to Learn. Front. Physiol. 7:410. doi: 10.3389/fphys.2016.00410
Received: 25 July 2016; Accepted: 31 August 2016;
Published: 14 September 2016.
Edited by:
Jose Renato Pinto, Florida State University, USAReviewed by:
Stuart Campbell, Yale University, USARanganath Mamidi, Case Western Reserve University, USA
Copyright © 2016 Geist and Kontrogianni-Konstantopoulos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aikaterini Kontrogianni-Konstantopoulos, YWtvbnRyb2dpYW5uaUBzb20udW1hcnlsYW5kLmVkdQ==