 Chen Zhang1
Chen Zhang1 Cassandra L. Miller1
Cassandra L. Miller1 Rakshya Gorkhali1
Rakshya Gorkhali1 Juan Zou1Kenneth Huang1Edward M. Brown2,3
Juan Zou1Kenneth Huang1Edward M. Brown2,3 Jenny J. Yang1*
Jenny J. Yang1*- 1Department of Chemistry, Georgia State University, Atlanta, GA, USA
- 2Center for Diagnostics and Therapeutics, Georgia State University, Atlanta, GA, USA
- 3Division of Endocrinology, Diabetes and Hypertension, Department of Medicine, Brigham and Women's Hospital, Boston, MA, USA
Ca2+-sensing receptors (CaSRs) play a central role in regulating extracellular calcium concentration ([Ca2+]o) homeostasis and many (patho)physiological processes in multiple organs. This regulation is orchestrated by a cooperative response to extracellular stimuli such as small changes in Ca2+, Mg2+, amino acids, and other ligands. In addition, CaSR is a pleiotropic receptor regulating several intracellular signaling pathways, including calcium mobilization and intracellular calcium oscillation. Nearly 200 mutations and polymorphisms have been found in CaSR in relation to a variety of human disorders associated with abnormal Ca2+ homeostasis. In this review, we summarize efforts directed at identifying binding sites for calcium and amino acids. Both homotropic cooperativity among multiple calcium binding sites and heterotropic cooperativity between calcium and amino acid were revealed using computational modeling, predictions, and site-directed mutagenesis coupled with functional assays. The hinge region of the bilobed Venus flytrap (VFT) domain of CaSR plays a pivotal role in coordinating multiple extracellular stimuli, leading to cooperative responses from the receptor. We further highlight the extensive number of disease-associated mutations that have also been shown to affect CaSR's cooperative action via several types of mechanisms. These results provide insights into the molecular bases of the structure and functional cooperativity of this receptor and other members of family C of the G protein-coupled receptors (cGPCRs) in health and disease states, and may assist in the prospective development of novel receptor-based therapeutics.
Introduction
In 1883, Sydney Ringer serendipitously discovered Ca2+ to be essential for the contraction of isolated hearts (Ringer, 1883). After the mid-twentieth century, research on Ca2+ has grown at an exponential rate, and Ca2+ is now considered as a universal signal carrier for biological information (Krebs and Michalak, 2007). Ca2+ controls life and death, as it modulates the process of fertilization as well as apoptosis. Ca2+ was primarily considered as a crucial secondary messenger via rapidly and efficiently regulated changes in intracellular calcium levels, and modulated extensive molecular signaling components through calcium channels, exchangers along with pumps. The discovery of the calcium sensing receptor (CaSR) defined an additional role of extracellular Ca2+ as a first messenger (Smajilovic and Tfelt-Hansen, 2007). It is known that serum Ca2+ concentration can regulate the secretion of parathyroid hormone (PTH) and, therefore, research was pursued to investigate how this process is accomplished. In 1993, Dr. Edward M. Brown cloned CaSR, which is primarily responsible for this type of regulation, from bovine parathyroid gland (Brown et al., 1993). The previously observed cytosolic Ca2+ changes in parathyroid cells, as well as in other in vitro expression systems, were triggered by changes in serum Ca2+ concentration and have been largely proven to be mediated by the CaSR (Nemeth and Scarpa, 1987; Muff et al., 1988). In addition to the key tissues involved in extracellular Ca2+ and Mg2+ homeostasis (e.g., parathyroid, thyroid, kidney, bone), CaSR has also been reported to be present in diverse other, non-homeostatic tissues (e.g., brain, skin, etc.) (Lundgren et al., 1994; Hebert, 1996; Cima et al., 1997; Hinson et al., 1997; Cheng et al., 1998; Kovacs et al., 1998; Chattopadhyay et al., 1999; Brown and MacLeod, 2001; Buchan et al., 2001; Mathias et al., 2001; Hofer and Brown, 2003; Chang and Shoback, 2004; Fudge and Kovacs, 2004; Hofer et al., 2004). To date, extracellular Ca2+ has been shown to be a first messenger via CaSR and 14 other family cGPCRs, including metabotropic glutamate receptors (mGluRs) and γ-aminobutyric acid (GABA)B receptors. As shown in Figure 1, high [Ca2+]o triggers multiple CaSR-regulated intracellular signaling pathways, including Gq∕11 signaling, Gi∕osignaling, Gs signaling, extracellular signal-regulated kinases 1 and 2 (ERK1∕2) signaling, and intracellular calcium mobilization.
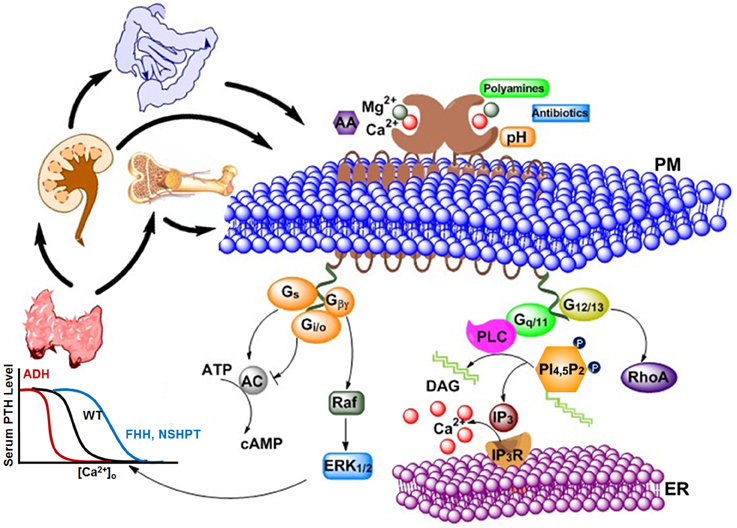
Figure 1. Various types of agonists, including cations, peptides, amino acids, antibiotics, etc., can act on the extracellular calcium-sensing receptor (CaSR) to generate a complex intracellular signaling network. CaSR is also a pleiotropic receptor in its regulation of four G protein-mediated intracellular signaling pathways (Gq∕11, Gi∕o, Gs, and G12∕13). The correlations and crosstalk among different signaling cascades contribute the cooperative responses of intracellular calcium responses as well as parathyroid hormone (PTH) secretion (left) and intracellular calcium responses (right) to extracellular calcium. Bottom left: The sigmoidal relationship between calcium concentration in blood and PTH level in serum is demonstrated. Higher Ca2+ concentration is required for normal level of PTH in patients with familial hypocalciuric hypercalcemia (FHH) or patients with Neonatal Severe Primary Hyperparathyroidism (NSHPT), as the response curve shifts to the right; on the other hand, lower Ca2+ concentration than normal is enough to trigger PTH secretion in patients with autosomal dominant hypocalcemia (ADH). PM, Plasma membrane; ER, Endoplasmic reticulum; AA, arachidonic acid; AC, adenylate cyclase; cAMP, cyclic AMP; DAG, diacylglycerol; ERK1∕2, extracellular-signal-regulated kinase; Gs, Gi∕o,G12∕13, and Gq∕11, subunits of the s-, i-, 12/13, and q-type alpha subunit of heterotrimeric G proteins, respectively; Gβγ, G beta and gamma complex; IP3, inositol-1,4,5-trisphosphate; IP3R, inositol-1,4,5-trisphosphate receptor; PLC, phospholipase C; PI(4,5)P2, phoshatidylinositol-4,5-bisphosphate; RhoA, Ras homolog gene family, member A.
CaSR is comprised of 1078 amino acids, and is encoded by exons 2–7 to encompass a 19 amino acids (AA) N-terminal signal peptide, a ~600 AA extracellular domain (ECD), a cysteine rich domain (CRD), a ~250 AA 7-transmembrane (7TM) domain, and the ~216 AA intracellular domain (ICD) (Pollak et al., 1993; Chikatsu et al., 2000; Hendy et al., 2013). The ECD of CaSR not only plays crucial roles in the sensing of nutrients, such as Ca2+, L-Phe and polypeptides, allowing ligands to modulate CaSR cooperatively, but is also essential for the dimerization of the receptor (Ray et al., 1999; Zhang et al., 2014b). Binding of Ca2+ and other CaSR ligands to the ECD is thought to produce conformational changes in the 7TM domain, introducing alterations in intracellular loops and the C-terminal domain, which further trigger the downstream signaling pathways (Brown and MacLeod, 2001). The intracellular C-terminal domain (residues 863–1078) is quite diverse among species. It participates in controlling the CaSR signaling and its cooperativity, modulating receptor trafficking, expression and desensitization (Gama and Breitwieser, 1998; Bai, 2004; Ward, 2004; Huang et al., 2006).
A unique characteristic of CaSR is the high cooperativity of Ca2+ dependent activation, which tightly controls the secretion of parathyroid hormone when the receptor is exposed to serum Ca2+ concentrations within its responsive range and also regulates the delicate Ca2+ homeostasis within the body as a whole (Brown et al., 1993). Functional cooperativity of CaSR (i.e., based on biological activity determined using functional assays), particularly the functional positive homotropic cooperative response to extracellular calcium, is essential for the receptor's ability to respond over a narrow physiological range of [Ca2+]o (1.1–1.3 mM) (Breitwieser, 2006). CaSR has an estimated Hill coefficient of 3–4 for its regulation of biological processes, such as activating intracellular Ca2+ signaling, inhibiting PTH release in parathyroid cells (Figure 1) and stimulating calcitonin secretion in C-cells (Walter et al., 2013). Other extracellular mineral cations, such as Mg2+, and amino acids, are able to function as agonists and co-agonists to regulate/potentiate the Ca2+-induced activation of the CaSR. L-amino acids, especially aromatics, at physiological conditions potentiate the Ca2+–elicited activation of the CaSR by altering the EC50 values for extracellular calcium evoked intracellular calcium responses via positive heterotropic functional cooperativity (Conigrave et al., 2000; Francesconi and Duvoisin, 2004; Wang et al., 2006). This capacity of CaSRs to integrate both divalent cations and other extracellular stimuli, such as amino acids (Vetter and Lohse, 2002), is a feature shared by other cGPCRs (Wise et al., 1999; Galvez et al., 2000; Gether, 2000; Oldham and Hamm, 2008; Rosenbaum et al., 2009). In this paper, we will review the major discoveries and approaches that have been taken in uncovering the molecular basis of the cooperative responses of CaSR and its impact in understanding molecular bases of diseases (Zhang et al., 2014b). Key determinants contributing to the functional cooperativity of CaSR including the calcium and ligand binding sites, and the connectivity between distant sites on the ECD domain as well as protein expression at the membrane surface and intracellular domain will be discussed.
Challenges
To date, how Ca2+, Mg2+, and amino acids cooperatively modulate intracellular Ca2+ signaling is a long-standing unanswered question mainly due to the lack of a determined CaSR structure. Like other cGPCRs, CaSR functions as a dimer (Bai et al., 1998a, 1999; Pace et al., 1999; Kunishima et al., 2000; Zhang et al., 2001; Bai, 2004; Suzuki et al., 2004) with a very long N-terminus that is predicted to fold into a bilobed ECD (Brown et al., 1993; Hebert and Brown, 1995; Pearce et al., 1996; Hinson et al., 1997; Hofer and Brown, 2003; Hu and Spiegel, 2003; Jingami et al., 2003; Quinn et al., 2004). The ECD has been shown to play an important role in the cooperative responses of the receptor to changes of [Ca2+]o, amino acids, metabolites, and neurotransmitters (Bai, 1999, 2004; Conigrave et al., 2002; Zhang et al., 2002; Chang and Shoback, 2004; Conigrave and Lok, 2004; Mun et al., 2004). Determination of the X-ray structure of the ECD of CaSR is largely hampered by difficulty in crystallization due to heterogeneous and extensive glycosylation (11 potential N-glycosylation sites) and challenges associated with membrane proteins (Yang et al., 2005). Further, Ca2+ and ligand-binding sites with weak binding affinities and rapid off rates are often not occupied in a determined X-ray structure. For example, no bound Ca2+ has been observed in over 30 X-ray structures of the ECD of mGluRs (Kunishima et al., 2000; Tsuchiya et al., 2002; Jiang et al., 2010; Zhang et al., 2014a), despite the clear modulatory effect of [Ca2+]o on this receptor. Furthermore, additional challenges result from the lack of direct binding assays for weak Ca2+-binding, amino acid-binding (Kd ~mM) and limitations in obtaining purified membrane proteins with native conformations (Nagar et al., 1996; Hu and Spiegel, 2003; Magno et al., 2011). The quantification of functional cooperativity with binding cooperativity and visualization of the molecular connectivity in tuning cooperativity requires innovative approaches.
Modeling Structures of the CaSR
Several regions of the CaSR and mGluRs are highly homologous, providing the basis for molecular modeling of the structure of CaSR under various conditions based on the determined crystal structures of mGluRs with and without ligands or agonists/antagonists (Bai, 2004; Hu and Spiegel, 2007; Huang Y. et al., 2007). Various mGluR structures have indicated that agonist binding induces a rearrangement of the dimeric VFT by bringing the two monomers closer together owing to pulling the lower lobes inwards upon the change toward the closed state. This is further evidenced by the addition of gadolinium, which is found to likely stabilize the active form of mGluR1 (Tsuchiya et al., 2002). Recent crystal structures of various mGluRs have been solved that shed light on several possible sequential activation events. Orthosteric ligands bind to the VFT in the dimer, stabilizing the closed conformation to induce an activation signal to the transmembrane domain by the CRD, which then activates G protein-regulated signaling pathways (Kunishima et al., 2000; Jingami et al., 2003; Muto et al., 2007; Wu et al., 2014).
Figure 2 shows three modeled structures of the ECD of CaSR in different conformational states based on the crystal structure of mGluRs. The ECD of CaSR possesses a featured bilobed VFT structure similar to mGluRs and bacterial periplasmic binding proteins (O'Hara et al., 1993; Ray et al., 1999; Reyes-Cruz et al., 2001). These modeled structures of CaSR derived from several crystal structures of mGluR1 provide molecular views of large conformational variances among different states and therefore provide a potential functional mechanism of CaSR and by extension, of other cGPCRs. The VFT region alternates between closed and open states, determined by the presence of a ligand (e.g., glutamate for mGluR1 or Ca2+ for CaSR), which then causes the signal to be relayed to the 7TM domain. As cGPCRs are only functional as dimers, the dimerization of CaSR is likely to be driven by the hydrophobic interactions between the monomers along the dimer interface. CaSR and mGluR protomers are also covalently linked by several disulfide bridges between the two monomers. A ligand induced rearrangement of the dimeric VFT brings the two monomers closer together by pulling the lower lobes inwards upon the change toward the closed state. Similar to mGluR, the ligand was proposed to interact with both lobes of the VFT to stabilize the closed form by providing additional points of contact between the lobes (Kunishima et al., 2000).
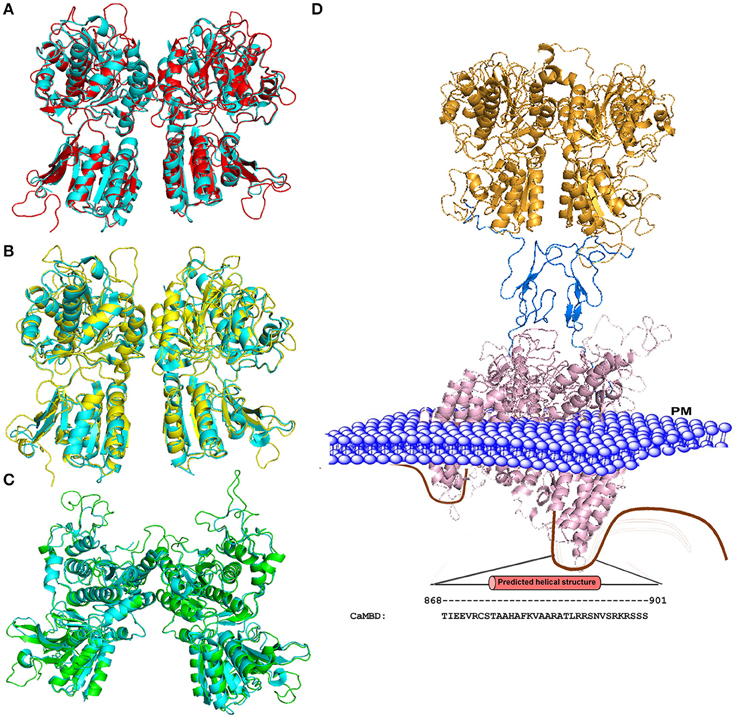
Figure 2. Homology models of the CaSR ECD. (A) Structural alignment of the mGluR1 holo form (cyan; PDB ID 1EWK, mGluR1 with glutamate) with the modeled structure of CaSR (red). (B) Structural alignment of mGluR1 apo form I (cyan, PDB ID 1EWV) with the modeled structure of CaSR (yellow). (C) Structural alignment of mGluR1 apo form II (cyan, PDB ID 1EWT) with the modeled structure of CaSR (green). (D) Homology model of the full CaSR structure. The ECD is based on the crystal structure of CaSR, the cysteine rich domain modeled from mGluR3 (PDB ID 2E4U), and the 7TM domain is modeled from mGluR1 with a ligand (PDB ID 4OR2). The C-terminal of CaSR can interact with protein kinases, ubiquitin ligase, CaM, etc. The region including residues from 868 to 901 is predicted to be CaM binding site. Mutations on the CaM binding site compromise the stability of surface expressed CaSR (Huang et al., 2010). PM: Plasma Membrane.
The homology models of the CaSR 7TM domain was built based on the crystal structure of bovine rhodopsin by several independent research groups (Petrel et al., 2003; Miedlich et al., 2004; Hu et al., 2006; Bu et al., 2008). Miedlich et al. docked the CaSR with the antagonist, NPS 2143, and the calcimimetic, NPS R-568, to the derived 7TM domain model structure and reported a shared pocket composed of residues F668, F684 and E837 (Miedlich et al., 2004). The evidence that there are additional Ca2+-binding sites located at the 7TM domain of the CaSR (7TM) have been provided by different independent groups (Hauache et al., 2000; Hu et al., 2002, 2005). The ECD portion of CaSR, including the CRD linker, has a 30–35% sequence similarity across all mGluRs, while the linker alone has 65–70% similarity (Bai, 2004). Together with a recent determination of structure of the ECD domain of mGluR3 with the cysteine-rich domain and the transmembrane domain of mGluR1 makes it possible to model the full length CaSR shown in Figures 2C,D. Due to high sequence homology, the modeled transmembrane structure provides more structural information than previous modeled 7TM domains based on the crystal structure of bovine rhodopsin (Petrel et al., 2003; Miedlich et al., 2004; Hu et al., 2006; Bu et al., 2008). These modeling and structural studies, therefore, provide important insights into the large conformational changes induced by agonist binding to both mGluR and CaSR. For instance, the ECD is changed from an open status to a closed form, although the calcium binding sites and amino acid binding sites remain elusive due to their weak binding affinities.
Identification of Ca2+ Binding Sites in the ECD and their Homotropic Cooperativity
Since the ECD is responsible for the main binding activity, researchers turned to identifying the binding regions for Ca2+ and amino acids based on studies from homology modeling, mutational studies and the effect of disease related mutations (Silve et al., 2005). To overcome the challenges involved in identifications of calcium binding sites with weak binding affinity of membrane proteins, Huang et al. developed a computational algorithm to be used for the prediction of potential Ca2+ binding pockets based on a statistical analysis of multiple calcium binding proteins (Huang Y. et al., 2007; Kirberger et al., 2008; Huang et al., 2009; Wang et al., 2009, 2010; Zhao et al., 2012). Shown in Figure 3, five potential Ca2+-binding sites were predicted in each monomer of the ECD of the homologous model of CaSR. Site 1 (S147, S170, D190, Y218, and E297) is located within the hinge region between the two lobes of the ECD (Kubo et al., 1998; Silve et al., 2005; Huang Y. et al., 2007). It is interesting to note that E297, S147, and S170 were reported to be important for sensing Ca2+ and/or amino acids (Bai et al., 1999; Mun et al., 2005; Silve et al., 2005). Recently, a novel Ca2+-binding site in the similar hinge region adjacent to the reported L-Glu binding site in mGluR1 was found to synergistically activate the receptor with L-Glu, and enhance the activity of the mGluR1 orthosteric and allosteric ligands (Jiang et al., 2010, 2014). This same phenomenon may also be applicable to other type of cGPCRs.
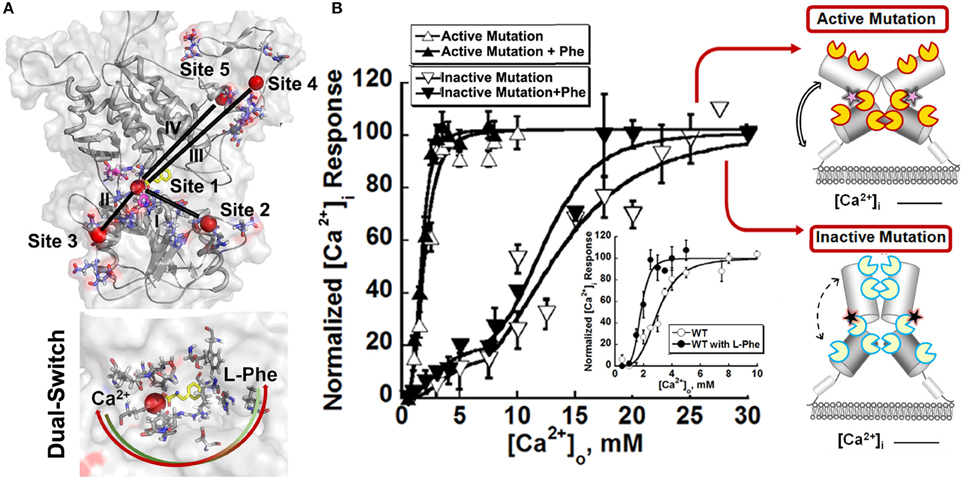
Figure 3. Molecular connectivity and heterotrophic cooperativity. (A) The correlated motions between calcium binding site 1 and the other calcium binding sites, designated by roman numbers (I~V), are mapped onto the CaSR ECD models. The correlation map and parameters for molecular dynamics simulation can be found in the paper (Zhang et al., 2014a). Bottom: The hetero-communication between Ca2+ and an amino acid functions as a dual switch that enhances the function of CaSR by positively impacting multiple Ca2+-binding sites within the ECD. Red sphere: Ca2+. Two headed arrow: both ligands can affect the binding of the other, therefore, the downstream signaling pathways. (B) The intracellular Ca2+ responses of HEK293 cells transiently overexpressing an active mutation or an inactive mutation. Inset: Representative response of WT CaSR to the changes of extracellular Ca2+ in the absence or presence of L-Phe, showing homotropic, and heterotropic cooperativity. Some disease mutations can interrupt the homotropic cooperativity shown as intracellular calcium responses to changes in the extracellular calcium concentration as well as the heterotropic cooperativity in the presence of allosteric modulators (e.g., L-Phe). Open circles: in the absence of L-Phe; Closed circles: in the presence of 5 mM L-Phe. Yellow “pacman”: Ca2+ binding sites in an active receptor; Blue “pacman”: Ca2+ binding sites in an inactive receptor. Pink stars: gain-of-function mutations in the hinge region. Black stars: loss-of-function mutations in the hinge region. Two headed arrows with solid lines: enhanced correlation motions between different Ca2+ binding sites. Two headed arrows with dash lines: impaired correlation motions.
Since the initial prediction, several complementary approaches have been used to provide important insights into how CaSR functions and the behavior of the receptor at the molecular level. First, functional cooperativity contributed by each Ca2+-binding site was determined by site-directed mutagenesis and monitoring intracellular Ca2+ oscillations, IP production and ERK1∕2 activation in living cells. We reported that the predicted Ca2+-binding site 1 within the hinge region of the ECD of CaSR and its interaction with other Ca2+-binding sites within the ECD is essential in tuning functional positive homotropic cooperativity of CaSR caused by changes in [Ca2+]o. Furthermore, molecular dynamic simulations indicated that there is molecular connectivity among the other predicted Ca2+-binding sites with site 1 and that the VFT hinge domain plays a central role in functional cooperativity (Zhang et al., 2014a). These results from various functional studies suggest that cooperative binding of Ca2+ at multiple predicted Ca2+-binding sites of the CaSR likely maximizes its capacity to respond over a narrow physiological range of [Ca2+]o independent of amino acids or other agonists (Bai et al., 1998a; Zhang et al., 2001; Bai, 2004; Figure 2)
The direct binding of Ca2+ to the other predicted Ca2+-binding sites in the ECD of CaSR and related binding cooperativity has been probed by using several methods. Ca2+ binding capabilities of predicted Ca2+-binding Site 3 (residues E224, E228, E229, E 231 E232), and Site 5, which is formed by contiguous Ca2+ binding residues (residues E378, E379, T396, D398, E399), were verified using a grafting approach (Huang Y. et al., 2007). By grafting peptide sequences composed of key predicted Ca2+ ligand binding residues separated by flexible linkers into a non-Ca2+-binding protein, site specific Ca2+ binding capability was determined using a Tb3+-sensitized FRET assay. Huang et al. also applied this approach to probe Ca2+ binding at three additional Ca2+ binding sites and potential binding cooperativity. Three major subdomains of the CaSR containing various numbers of predicted Ca2+ binding sites were expressed and purified. Their Ca2+-binding capabilities were examined using mutagenesis studies combined with various spectroscopic studies utilizing 8-anilino-1-naphthalenesulfonic acid (ANS) fluorescence, intrinsic tryptophan fluorescence spectra and nuclear magnetic resonance (NMR) (Huang et al., 2009). It was striking to observe that subdomain 1 with three binding sites (including sites 1, 2, and 3) exhibited a large Ca2+ dependent conformational change. A binding process with strong and weak Ca2+-binding affinities has been clearly unveiled for subdomain 1 by both fluorescence and NMR studies. These studies not only confirmed the existence of multiple Ca2+ binding sites, but also revealed that the molecular connectivity among multiple metal binding sites is essential for a highly cooperative functional activity (Figure 2).
The complex glycosylated and high mannose CaSR ECD (residues 20–612) were purified from HEK293S and its mutant cell line (Zhang et al., 2014c). Using various spectroscopic methods, it was shown that both form the ECD bound to Ca2+ with a Kd of 3.0–5.0 mM. The local conformational changes of the proteins induced by their interactions with Ca2+ were visualized by NMR with specific 15N Phe-labeled forms of the ECD. These studies also suggest that glycosylation does not affect calcium binding properties.
Identification of Amino Acid Binding Site and Heterotropic Functional Activity
The extracellular calcium induced activation of CaSR is potentiated by L-amino acids to maintain the whole body Ca2+ homeostasis, making the receptor a multimodal and multimetabolic sensor (Francesconi and Duvoisin, 2004; Breitwieser, 2006; Conigrave et al., 2007a). Under physiological conditions, L-amino acids, especially aromatic amino acids (e.g., L-Phe, L-Trp), as well as short aliphatic and small polar amino acids, potentiate the extracellular calcium triggered CaSR activity by altering the EC50 values required for Ca2+-evoked intracellular calcium responses and its functional cooperativity. For example, the EC50 for Ca2+ decreased from 4.2 ± 0.2 to 2.5 ± 0.1 mM in the presence of L-Phe (Conigrave et al., 2000, 2007a; Wang et al., 2006). At the threshold level of extracellular Ca2+ concentration, L-amino acids induced slow intracellular Ca2+ oscillations with a frequency at around 1–2 peaks/min (Breitwieser, 2006). In aggregate, the levels of amino acids in human serum in the fed state are close to those activating the CaSR in vitro (Conigrave et al., 2000, 2007b) and can further enhance functional cooperativity via positive heterotropic cooperativity. The CaSR in cells within the lumen of the gastrointestinal (GI) tract are also activated by L-Phe and other amino acids, which have been long recognized as activators of key digestive processes. Thus, the CaSR enables the GI tract to monitor events relevant to both mineral ion and protein/amino acid metabolism, in addition to its sensing capability in blood and related extracellular fluids (Conigrave et al., 2000; Wang et al., 2006; Broadhead et al., 2011; Liou et al., 2011). Distinctive signaling pathways elicited by amino acids compared with Ca2+ have been demonstrated in quite a few studies (Breitwieser, 2006; Broadhead et al., 2011). Ca2+ activates CaSR though activation of the heterotrimeric GTP binding protein Gq, causing the generation of Ins(1,4,5)P3 (IP3), which binds to the IP3 receptor on the ER, releasing stored Ca2+. On the other hand, L-Phe in the presence of Ca2+ induces coupling to heterotrimeric GTP binding protein G12∕13, leading to the activation of RhoA via filamin A.
Some potential L-Phe binding site residues in the CaSR were identified based on the conserved amino acid residues involved in the binding of glutamate to mGluRs (Zhang et al., 2002). Their studies demonstrated that mutating three adjacent Ser (S169, S170, S171) to Ala eliminated L-Phe potentiated receptor activity measured by [Ca2+]i mobilization using a fluorescence-based cell population assay. Meanwhile, studies from Mun et al. showed the binding site for amino acids was within the VFT domain of CaSR by utilizing CaSR-mGluR chimeric receptor constructs. The receptor lacking the ECD exhibited impaired response to L-amino acids (Mun et al., 2004). The same group showed that two residues, Thr145 and Ser170, could be crucial for sensing L-amino acids, using mutagenesis and fluorimetry studies (Mun et al., 2005).
Recently, an L-Phe binding site adjacent to the previously predicted calcium-binding site 1 based on computational docking results was reported. The potential L-Phe-binding site is composed of residues Leu51, Thr145, Ser170, Tyr218, and Ser272. The residue Tyr218 is involved in both Ca2+ and Phe binding. Extensive mutational studies using both single cell imaging and fluorimetric assays identified the importance of L-Phe-binding pocket for positive heterotropic cooperativity between extracellular Ca2+ and L-Phe in eliciting CaSR-mediated Ca2+ signaling (Zhang et al., 2014a). The frequency of sinusoidal intracellular Ca2+ oscillations in CaSR transfected HEK293 cells not only depend on the extracellular Ca2+, but also affected by the binding of allosteric modulators. For example, the binding of CaSR with Ca2+ alone produces a intracellular Ca2+ oscillations frequency (1.5/min at room temperature) which is different than those produced by Ca2+ with L-amino acid (e.g., L-Phe) (2.2/min) (Breitwieser, 2006; Zhang et al., 2014b). This change in frequency is extremely important for the role of CaSR signal transduction in the many different tissue and organ environments in which it is present. For instance, the Ca2+ oscillations in parathyroid, kidney, intestine and bone are tightly related to maintenance of Ca2+ homeostasis while the Ca2+ signals are processed differently in many epithelial cells and the nervous system (Riccardi, 2002; Breitwieser, 2006).
The hetero-communication between Ca2+ and an amino acid functions as a dual switch that globally enhances functional positive homotropic cooperative activation of CaSR in response to Ca2+ signaling by positively impacting multiple Ca2+-binding sites within the ECD (Figure 3; Zhang et al., 2014a). A direct interaction between the CaSR ECD and L-Phe was finally reported using saturation transfer difference NMR approaches with a determined binding affinity of ~10 mM in the absence of Ca2+ (Zhang et al., 2014c). The study further demonstrated that L-Phe increases the binding affinity of the CaSR ECD for Ca2+. Thus, dual binding of calcium and amino acids at the hinge regions of the bilobed VFTD of the CaSR leads to activation of the receptor with highly cooperative responses to the changes in the extracellular concentrations of these agonists (Figure 4).
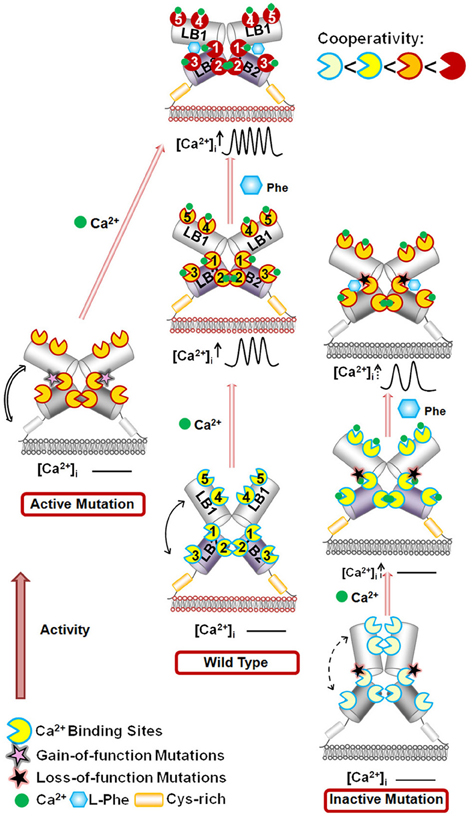
Figure 4. Schematic representation of the mechanisms underlying the effects of the mutations on the CaSR and the modulation of receptor activity by extracellular Ca2+ and L-Phe. Ca2+ and L-Phe modulate the activity as well as the cooperativity of CaSR (the color changes of the receptor from ivory to red indicate an increase in functional activity). Elevating [Ca2+]o, e.g., to 3.0 mM, is proposed to change the basal WT CaSR status into an active form in a positive homotropic cooperative manner and further trigger intracellular Ca2+ oscillations. L-Phe binds to the hinge region between lobe 1 and lobe 2, modulating the receptor together with Ca2+ in a positive heterotropic cooperative way. This could potentiate conversion of the receptor to a “fully active” form associated with a higher frequency of intracellular Ca2+ oscillations and a left-shifted EC50. Loss-of-function CaSR mutants (indicated by ivory color) could cause a disruption of the cooperativity among the various Ca2+-binding sites as shown by impaired correlation motions (dashed arrows). The impaired receptor function and the cross-talk between Ca2+-binding sites can be at least be partially rescued for some mutants by L-Phe (e.g., P221Q). However, if the mutation interferes with the interaction between CaSR and L-Phe, the function of the receptor may not be fully recovered (e.g., L173P). CaSR gain-of-function mutants (left) exhibit enhanced correlated motions (double line arrows) and their activity is not further potentiated by L-Phe, potentially due to a ceiling effect. LB1(2): Lobe 1 or lobe 2 in the VFT domain of CaSR.
Molecular Basis of Disease Related CaSR Mutations
More than 200 mutations of the CaSR that lead to the disorders of Ca2+ homeostasis have been identified (http://www.casrdb.mcgill.ca/) (Hendy et al., 2009). Mutations of CaSR may inhibit receptor activity, lead to an over activation of the receptor, or markedly impair the receptor's structure. The majority of these disease-associated mutations are missense mutations, with a single amino acid substituted, often from one base pair change. Amino acids insertion, deletion, open reading frame shift, and splice-site mutations have also been reported (Hendy et al., 2009). The diseases associated with inactivate receptors include cases of Familial Hypocalciuric Hypercalcemia (FHH) (Ward et al., 2006) and Neonatal Severe Hyperparathyroidism (NSHPT) (Thakker, 2004) while the disorders associated with the CaSR activating mutations include Autosomal Dominant Hypocalcemia (ADH) (Hendy et al., 2000) and Bartter syndrome type V (Watanabe et al., 2002).
Below is the summarized four types of disease mutations based on their distinct mechanisms of action. Type I and type II mutations alter the CaSR's function, especially EC50 or Hill number without altering surface expression or trafficking. Type III and type IV mutations largely affect degree of cooperativity via altering surface expression and trafficking of the protein to the plasma surface (Figure 5, Table 1).
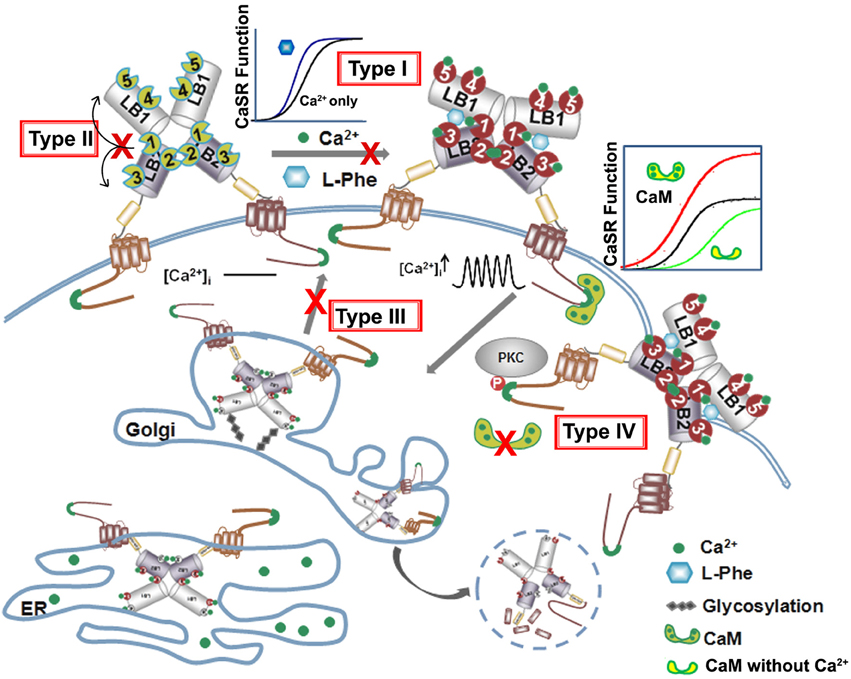
Figure 5. Summary of four types of disease mutations. The CaSR trafficking, expression and surface stabilization contribute to its functional cooperativity. Type I disease associated mutations directly alter key calcium and ligand binding capability at or in close proximity of the predicted ligand binding sites in the ECD. Type II mutations alter CaSR function especially EC50 or Hill number without altering surface expression or trafficking, but affect the molecular connectivity between different ligand binding sites. Type III mutations disrupt the cooperativity via interfering with the receptor cell surface expression and Type IV mutations largely affect potency of cooperativity by altering trafficking and protein stability via interaction with proteins binding to the CaSR intracellular C-tail.

Table 1. Summarized molecular basis of disease related CaSR mutations.
The first type of mutational effect, type I, is via direct alterations in key calcium and ligand binding capability at or in close proximity to the predicted ligand binding sites in the ECD (Figure 5). Hannan et al. found that more than 50% of their newly identified CaSR mutations in patients with FHH, NSHPT, and ADH are within the ECD of the receptor (Hannan et al., 2012). Analysis using the homology modeled structure of the CaSR further revealed that >50% of these missense substitutions are located within 10 Å of one or more proposed calcium-binding sites, indicating that the bilobed VFT domain plays a pivotal role in interacting with Ca2+ and regulating the function of CaSR (Hannan et al., 2012). Intriguingly, more than half of the mutations near Ca2+-binding sites are situated close to Site 1, suggesting the importance of the VFT domain cleft. The molecular connectivity between Site 1 and rest of the calcium-binding sites can possibly facilitate the positive cooperativity of the CaSR (Zhang et al., 2014d).
Through Ca2+ binding site modeling and mutational studies, several key residues for the predicted Ca2+ binding sites in the hinge region of the VFT domain have been identified (Deng et al., 2006; Huang Y. et al., 2007; Huang et al., 2009). Amongst them, more than half of the ECD mutations are near the proposed Ca2+ binding sites, and a few of them are found directly in the potential binding site residues. One ADH mutation (E297D) and several FHH mutations (Y218S, Y218C, and E297K) are within Ca2+ binding site 1. The FHH mutation D215G is located in Ca2+ binding site 2 while two ADH mutations E228Q and E228K are in Site 3. The Ca2+ binding Site 4 embraces residues involved in a carcinoma-associated mutation E350V and an ADH mutation E354A. Mutations on these key binding residues leads to a decrease of the EC50 of Ca2+ in ADH in order to produce PTH, and an increase of EC50 in FHH or NSHPT. This can also lead to an alteration of binding cooperativity between the other Ca2+ binding sites, allowing them to bind more or less effectively as indicated by changes in the Hill number. Several studies have investigated the effect of FHH Ca2+ binding site 1 mutations, such as Y218S (Pearce et al., 1996) and E297K (Bai et al., 1999), both of which led to a loss-of-function with an increase in EC50 for Ca2+ in terms of intracellular Ca2+ mobilization. Additionally, E297K also decreases inositol-1-phosphate (IP1) accumulation, but does not change CaSR cell expression levels as compared to the wild-type (WT) (Bai et al., 1999). ADH mutation E297D, as well as FHH mutation E297K, were analyzed by Silve et al. for IP1 accumulation and expression levels (Silve et al., 2005). Again, they saw no change in cell expression compared to the WT, but they did see an activation shift for E297D, as the EC50 for Ca2+ induced IP stimulation changed from the WT EC50 of 4.30 ± 0.20 to 2.70 ± 0.30 mM, and a loss-of-function shift in EC50 for E297K (Silve et al., 2005).
Additionally, mutations in residues neighboring the binding site also show the same trend in an altered EC50 and cooperativity but with no apparent effect on cell surface expression or trafficking, such as the FHH/NSHPT mutations S171N, R227Q, R227L, S296N, F351V, W352X, C395R, G397R, the ADH mutations E191K, E241K, Q245R, and cancer-associated mutations, S169F and S171G. Mutations on these binding site residues or their neighboring residues would have major effects on either direct binding or conformational direction of the binding site residues, respectively. Pearce et al. looked at two such mutants R227L (NSHPT) and E191K (ADH) (Pearce et al., 1996). R227L increased EC50 for extracellular Ca2+ induced intracellular Ca2+ response to 9.3 mM compared to the WT 4.0 mM, while the E191K mutant decreased EC50 for [Ca2+]o from the WT 3.7 to 2.8 mM. Western blot analysis showed that compared to WT there was no increase or loss in expression (Pearce et al., 1996). Thus, concluding that binding site residues, and their immediate bordering residues, significantly alter the charge or conformation of the binding pocket therefore altering functionality into a gain-of or loss-of-function mutation with a major effect on protein expression, translation, and/or trafficking.
The second type of disease mutations, type II, disrupts molecular connectivity embedded into the hinge region of the ECD (Figures 3, 5). Mutations at the particular region of the N-terminal VFT domain produce either receptor inactivation (L173P, P221Q) or activation (L173F, P221L) related to hypercalcemic or hypocalcemic disorders. We have shown that both L173P and P221Q markedly impair the functional positive cooperativity of the CaSR as reflected by Ca2+–induced intracellular Ca2+ oscillations, IP1 accumulation and extracellular signal-regulated kinases (ERK1∕2) activity. In contrast, L173F and P221L show enhanced responsiveness of these three functional readouts to [Ca2+]o change. Further analysis of the dynamics of the VFT domain mutants using computational simulation studies supports disruption in the correlated motions in the loss-of-function CaSR mutants, while these motions are enhanced in the gain-of-function mutants. WT CaSR was modulated by L-Phe in a heterotropic positively cooperative way, achieving an EC50 for Ca2+ induced intracellular responses similar to those of the two activating mutations. The response of the inactivating P221Q mutant to Ca2+ was partially rescued by L-Phe, illustrating the capacity of the L-Phe binding site to enhance the positive homotropic cooperativity of CaSR. L-Phe had no effect on the other inactivating mutant. Moreover, our results carried out both in silico and within intact cells indicate that residue Leu173, which is adjacent to residues that are part of the L-Phe-binding pocket, exhibited impaired heterotropic cooperativity in the presence of L-Phe. Thus, P221 and L173 are important for the positive homo—and heterotropic cooperative regulation elicited by agonist binding.
The third type of mutations, type III, directly affect CaSR protein surface expression level which, in turn, alters potency or cooperativity. Apparent functional cooperativity and the EC50 for extracellular Ca2+ elicited intracellular response of the CaSR are also determined by the receptor expression level at the plasma membrane (Christopoulos and Kenakin, 2002; Casadó et al., 2007; Huang Y. et al., 2007). Like many other membrane proteins, CaSR's surface expression level is determined by the rate of internalization and folding in the endoplasmic reticulum (ER) and Golgi, and membrane insertion. The CaSR protein is synthesized in the ER and is trafficked intracellularly in the ER and Golgi complex as it undergoes translations in the ER and post-translational modifications before shuttling to the cell surface. Interruptions in this trafficking from translation to cell surface expression, such as those from disease mutations, can lead to loss-of or gain-of-function CaSR on the cell surface, CaSR that is retained in the ER or Golgi, or degraded altogether. Breitwieser's group investigated how these disease mutations affect trafficking as a whole. They identified several classes of mutations, spanning the whole CaSR length, which they categorized based on their ability to traffic to the cell surface and to have their function rescued by the positive allosteric modulator NPS R-568. Class Ib mutations (Q27R, P39A, R66C, G143E, L174R, G549R, C582Y, R638L, S657Y, G670E, P748R, G778D, L849P) interrupt trafficking so substantially that the CaSR is not able to leave the ER, while Class Ia mutations (E297K, C395R, and A804D) similarly remain in the Golgi complex. All Class I mutations fail to traffic to the cell surface. Class II mutations still allows CaSR to reach the cell surface and their function either can be enhanced by NPS R-568 (Class IIa: P55L, R62M, S137P, R185Q, D215R, R227Q, G553R, L650P, R680C, V689M, R795W, V817I) or not (Class IIb: L159P and P798T) (Huang and Breitwieser, 2007; White et al., 2009). Consistent with these observations, Leach et al. reported that FHH mutations S657Y, G670R, G670E, R680H, R680C, M734R, G778D, and V817I significantly decrease cell surface expression compared to the WT; while ADH mutations F821L, V836L, and A843E increase cell surface expression, although the ADH mutation F832S did lead to a 45% decrease in surface expression (Leach et al., 2013).
The 14 cysteines found in the ECD and the cys-rich domain were also found to affect CaSR function and cell surface expression (Bai et al., 1998a; Fan et al., 1998; Zhang et al., 2001). Of these 14 cysteines, 9 have disease mutations associated with them producing ADH, FHH, and various carcinomas. C129S and C131S, the cysteines thought to participate in the homodimerization of CaSR through disulfide bond formation, only led to a decrease of expression to 88 and 97%, respectively, compared to WT CaSR. Whereas, C60S led to a decrease in expression by half, and all other Cys mutations linked to disease led to a decrease to lower than 28% or undetectable levels (Fan et al., 1998). It is clear that the disruption of disulfide formation as well as trafficking have profound effects on functional cooperativity via alterations in both the Hill number and the concentration of active form on the plasma surface.
The fourth mutational effect, type IV, alters CaSR interactions with trafficking and stabilization partners leading to a change in surface expression that further influences CaSR signaling as well as functional cooperativity. Breitwieser et al. have concluded that agonist-driven insertional signaling (ADIS) contributed to the hallmarks of CaSR signaling, including the high degree of cooperativity and the lack of functional desensitization. Additionally, they have shown that the life cycle of the CaSR controls the cellular abundance of CaSR, suggesting an intimate link between trafficking and signaling (Breitwieser, 2012, 2013). As shown in Figure 5 and Table 2, a large number of proteins have been reported to interact at the C-terminal tail, the protein interaction sensitive zone, of the CaSR (Hebert and Brown, 1995; Chakravarti et al., 2012; Brown, 2013). Interactions of these proteins may be important for both CaSR signaling and functional cooperativity. Among these proteins, E3 ubiquitin ligase, dorfin is linked to ubiquitination and degradation of CaSR via binding to C-terminal amino acids 880–968 (Huang et al., 2006) and/or the C-terminal amino acids 920–970 (Zhuang et al., 2012). Zhang et al. investigated the effect of lysosomal mediated degradation of CaSR C-terminus and found that loss of the region 920–970 led to the highest cell expression level and decrease in degradation (Zhuang et al., 2012). Several insertion and deletion mutations in these regions of the C-terminus of CaSR lead to ADH, resulting in reduced degradation and, therefore, over-expression at the cell surface. Additionally, we have reported that the surface expression of CaSR is largely influenced by interaction of calmodulin (CaM) at residues 871–898 at the C-terminal tail of CaSR. Interestingly, multiple disease mutations, such as T876ins, F881L, R886W, and R886P for FHH/NSHPT and S895del for ADH reside at this CaM binding region (Huang et al., 2010). CaM has been reported to bind both the immature and mature forms of CaSR, which suggests its role in modulating anterograde trafficking (Bai et al., 1998b). We have shown that deletion of the CaM binding region significantly abolishes/reduces functional cooperativity of calcium oscillations. Therefore, CaM binding stabilizes the receptor on the cell membrane and thus increases potency of its functional activity (Huang et al., 2010). It is worth pointing out that this region also is involved in phosphorylation and biased signaling (Davey et al., 2012; Leach et al., 2012). Moreover, protein 14-3-3 was reported to interact with the arginine-rich domain of CaSR (890RRxxxxRKR898), which may lead to the retention of CaSR in the ER (Stepanchick et al., 2010; Grant et al., 2011; Arulpragasam et al., 2012). CaSR associated disorders can also be caused by different mechanisms other than direct mutations on the receptor itself. Recent studies by Nesbit et al. and Rogers et al. revealed that disorders with FHH or ADH phenotype can be associated with mutations on partner proteins associated with CaSR-mediated signaling, for instance the loss-of-function mutations of the sigma subunit of adaptor protein-2 (AP2) and mutations on G-protein subunit-α11 (GNA11), (Nesbit et al., 2013; Rogers et al., 2014). Moreover, antibodies against the CaSR can also regulate the function of the receptor (Thakker, 2004; Kemp et al., 2009, 2010).
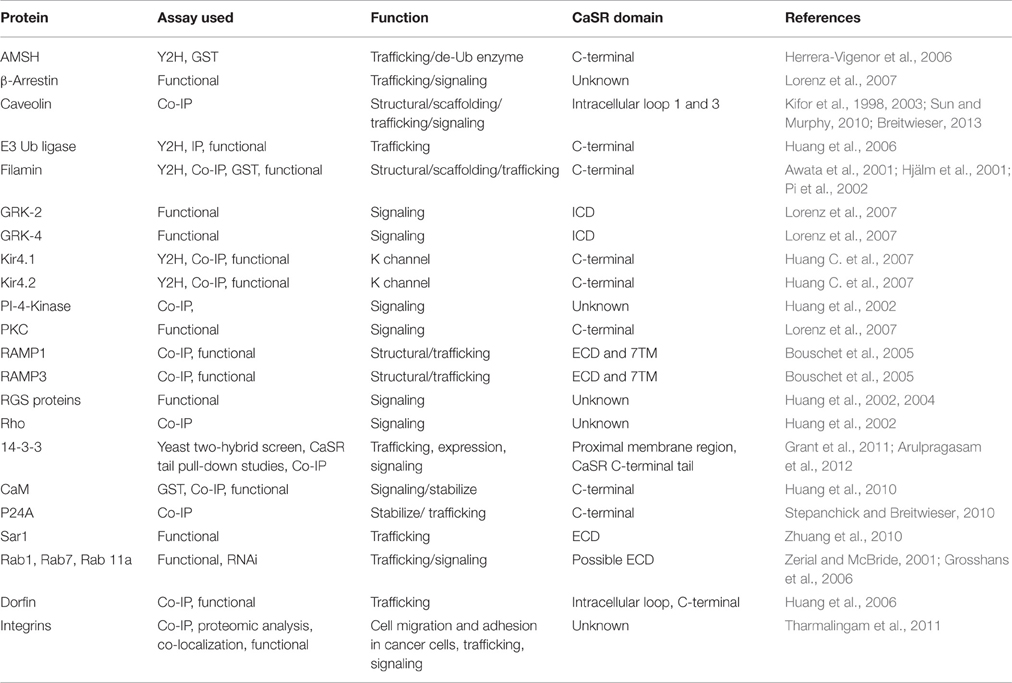
Table 2. The calcium-sensing receptor interacting proteins.
Additional Mechanisms to Alter the Functional Activity of CaSR
There are several possible reasons why a single CaSR could play a major role in various organisms, organs and tissue types at different physiological environments. First, besides Ca2+, other divalent cations such as Mg2+, Ba2+, Mn2+, Ni2+, Sr2+ (Thomsen et al., 2012), and trivalent cations La3+ and Gd3+ can also interact at similar reported calcium binding sites, and/or in their local vicinity, via altered electrostatic interactions of the protein. In general, the higher the positive charge density, the higher the potency as CaSR agonists. The CaSR has a relatively low affinity for Mg2+ with an EC50 at about 10 mM as measured for ligand induced current in oocytes, whereas there has been a reported EC50 of 3 mM for Ca2+ in the same assay (Brown et al., 1993). Mg2+ also has only half the maximal capacity for the production of inositol phosphate and arachidonic acid when stimulated by Ca2+ in CHO cells transiently expressing rat CaSR (Ruat et al., 1996). Consistently, CaSR transiently expressed in HEK293 cells has EC50s for intracellular calcium responses of 15–20 and 3–5 mM for Mg2+ and Ca2+ under physiological conditions, respectively (Bai et al., 1996; Nearing et al., 2002). CaSR ligands are also important for therapeutics, for example, Sr2+ (as a ranelate salt) has been shown to be effective for the treatment of osteoporosis (Kendler et al., 2009). CaSR has also been seen to respond to binding ligands (e.g., Ca2+, Mg2+, L-Phe, etc.) on a pH dependent manner where slightly lower extracellular pH of 7.2 inhibits the binding of the ligand to CaSR, and a higher pH of 7.6 favors the binding process (Campion et al., 2015).
Second, various classes of agonists, antagonists and drugs can also regulate the activation of CaSR via the ECD. The established homotropic cooperativity and heterotropic co-activation model of CaSR can also explain the regulatory action on CaSR. Three basic peptides including poly-arginine, protamine, and poly-lysine exhibited a dose-dependent inhibition of dopamine-stimulated cAMP accumulation in dispersed bovine parathyroid cells with EC50 values at micromolar or sub-micromolar concentrations (Brown et al., 1991). Studies from Quinn et al. demonstrated alteration of several cellular parameters including [Ca2+]i change, IP production, and the activity of a nonselective cation channel via polyamines. The potency of polyamines was directly proportional to number of positive charges with the order of spermine > spermidine > > putrescine (Quinn et al., 1997). Glutathione and its γ-glutamyl peptides also allosterically modulate the CaSR at a site that appears to be similar to the L-amino acids binding site but with >100-fold higher apparent affinity (Conigrave et al., 2000; Wang et al., 2006; Broadhead et al., 2011). The EC50 values for GSH and the oxidized form glutathione disulfide (GSSG), to induce intracellular Ca2+ mobilization, were in the range of sub-micromolar (0.08 and 0.33 μM, respectively) (Wang et al., 2006). In 2006, using [Ca2+]i mobilization, PTH secretion, as well as intracellular cAMP inhibition, Broadhead et al. showed γ-glutamyl-tripeptides (γ-Glu-Cys-Gly, S-methylglutathione and S-propylglutathione, and dipeptides γ-Glu-Ala and γ-Glu-Cys) to be positive allosteric modulators of CaSR. They also found that the double mutant T145A/S170T had an impaired response to these peptides, indicating a potential peptide binding site at the extracellular domain of CaSR (Broadhead et al., 2011). Recently, a peptide drug AMG 416 has been shown to act as an agonist of the CaSR and is undergoing clinical trials (Walter et al., 2013). Interestingly, the action of AMG 416 is also modeled to be at the hinge region of the Phe-Site1 calcium binding site (Alexander et al., 2015). Mutations on Cys482 at the hinge region lead to impaired peptide activity in both pig and human CaSR (Alexander et al., 2015).
Third, calcium sensitivity could be related with the difference in CaSR sequence observed in various species. In mammals, CaSRs were detected in multiple organ systems besides parathyroid glands (e.g., kidneys, colon, skin, parathyroid, stomach, vascular, bone, etc.), participating in various functions, such as Ca2+ and fluid reabsorption, acid secretion, osteoblast and keratinocyte differentiation, etc. (Tfelt-Hansen and Brown, 2005; Alfadda et al., 2014). The CaSR sequences among major clades exhibit few differences both at the ECD and ICD. The length of the ICD varies greatly among the clades (Herberger and Loretz, 2013). Intriguingly, the sequence of the putative calcium binding sites, especially the Site 1 within the ECD thought to be the main Ca2+-binding domain, is relatively conserved among vertebrates (Huang et al., 2009). On the other hand, residues in the other predicted Ca2+-binding sites differ substantially, which may be an effective way to adjust CaSR sensitivity to a specific physiological environment, i.e., with varying Ca2+, Mg2+, or pH, and also for evolution. For example, CaSR is present in different organs of fish, suggesting it might play a crucial role in sensing the changes in the salinity in surrounding water with calcium concentrations changes from 10 mM in sea water to 0.07–2.0 mM in freshwater. The presence of many alternatively spliced forms of the human CaSR could imply an impact of these altered sequences on the CaSR's Ca2+ sensitivity (Oda et al., 1998, 2000).
Fourth, Ca2+ signaling could be modulated through the formation of CaSR hetero-dimers with other GPCRs or higher order oligomers with non GPCR chaperones in tissues other than parathyroid. Gama et al. showed co-localization of the CaSR and mGluR1α in hippocampal and cerebellar neurons by immunoprecipitation of CaSR from bovine brain. The CaSR became sensitive to glutamate triggering internalization and exhibited altered trafficking via reported hetero-dimerization with GABAB receptors (Gama et al., 2001). Thus, it was speculated that the CaSR would respond to Ca2+ concentration within the synaptic cleft at various levels of synaptic activity. Recent work by Kim et al. revealed that CaSR forms a heteromeric complex with the inhibitory type B γ-aminobutyric acid receptor 1 (GABABR1) in hippocampal neurons (Kim et al., 2011). This study demonstrates a novel receptor interaction, which contributes to ischemic neuron death through CaSR upregulation and GABABR1 downregulation, and feasibility of neuroprotection by concurrently targeting these two receptors. It is also a new way to tailor functional cooperativity and specificity.
Conclusion and Perspective
Recently, the crystal structure of extracellular domain has been first solved by our group. We reported the first CaSR crystal structure with multiple Mg2+ binding sites (Zhang et al., 2016). Consistent to our modeled structure reported earlier, the determined X-ray structure of the ECD shares the same fold of the mGluRs with the VFT motif. Unexpectedly, we also identified a tryptophan derivative L-1,2,3,4-tetrahydronorharman-3-carboxylic acid (TNCA) at the hinge region between the two subdomains where orthosteric ligand binding is thought to occur. We further demonstrated that TNCA binds to CaSR at unusually high affinity and potentiates CaSR activity with Ca2+ and Mg2+ (Zhang et al., 2016). Geng and colleagues subsequently reported X-ray structures of CaSR ECD in both apo form and the Ca2+ binding form. Similar as proposed in earlier studies, CaSR active form contained multiple binding sites for Ca2+. Additional Ca2+ bound sites and binding sites were reported. They also showed that L-Trp bound to the orthosteric agonist-binding site in CaSR active form (Geng et al., 2016). These advances and various pioneer studies since the discovery of CaSR in understanding the cooperative extracellular Ca2+ signaling mediated through CaSR have uncovered important structural and functional characteristics about this receptor. Family cGPCRs all sense extracellular calcium with differing degrees and also exhibit sensitivity to either amino acids, neurotransmitters, or related ligands. The discovered co-activation by calcium and amino acids of CaSR is likely to have a broad impact for the regulation of cGPCRs. However, many questions about CaSR are still unanswered. Though accelerated elucidation of cGPCR structures have been accomplished, the unavailability of the full CaSR structure and largely “invisible loop residues” in the determined structure of cGPCRs limit the next step forward on understanding of molecular mechanism of CaSR. Thus, high resolution structures of CaSR with various forms of agonists and antagonists are essential to gain a better understanding of the underlying mechanism in CaSR regulated physiological functions as well as pathological activities. Equal importance should be directed at studies designed to probe the functional cooperativity of CaSR as it plays a pivotal role in controlling the receptor response within a narrow fluctuation of metal concentration. Family cGPCRs are highly relevant for drug design and have tremendous potential as therapeutic targets because cGPCRs play vital roles in neurotransmitter release and Ca2+ homeostasis. The aforementioned results, along with further determination of the structure of CaSR, will provide great insights into the molecular basis of the structure and function of CaSR and shed new light on other cGPCRs in health and disease states. Thus, further uncovering of the structural and functional mysteries of CaSR could aid the development of novel receptor-based therapeutics to use in the treatment of many different diseases.
Author Contributions
JY, CZ, CM, JZ, and RG contributed to the manuscript's conception and wrote the manuscript. KH performed computational analysis. EB made searches and helped with the manuscript.
Funding
This work was supported by National Institutes of Health Grants GM081749 and EB007268 (to JY), and funds from the Georgia Research Alliance.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Center for Diagnostics and Therapeutics fellowship (to CZ and RG), Brain and Behavior Fellowship (to JZ and CM).
References
Alexander, S. T., Hunter, T., Walter, S., Dong, J., Maclean, D., Baruch, A., et al. (2015). Critical cysteine residues in both the calcium-sensing receptor and the allosteric activator AMG 416 underlie the mechanism of action. Mol. Pharmacol. 88, 853–865. doi: 10.1124/mol.115.098392
Alfadda, T. I., Saleh, A. M., Houillier, P., and Geibel, J. P. (2014). Calcium-sensing receptor 20 years later. Am. J. Physiol. Cell Physiol. 307, C221–C231. doi: 10.1152/ajpcell.00139.2014
Arulpragasam, A., Magno, A. L., Ingley, E., Brown, S. J., Conigrave, A. D., Ratajczak, T., et al. (2012). The adaptor protein 14-3-3 binds to the calcium-sensing receptor and attenuates receptor-mediated Rho kinase signalling. Biochem. J. 441, 995–1006. doi: 10.1042/BJ20111277
Awata, H., Huang, C., Handlogten, M. E., and Miller, R. T. (2001). Interaction of the calcium-sensing receptor and filamin, a potential scaffolding protein. J. Biol. Chem. 276, 34871–34879. doi: 10.1074/jbc.M100775200
Bai, M. (1999). Structure and function of the extracellular calcium-sensing receptor (Review). Int. J. Mol. Med. 4, 115–125. doi: 10.3892/ijmm.4.2.115
Bai, M. (2004). Structure-function relationship of the extracellular calcium-sensing receptor. Cell Calcium 35, 197–207. doi: 10.1016/j.ceca.2003.10.018
Bai, M., Quinn, S., Trivedi, S., Kifor, O., Pearce, S. H., Pollak, M. R., et al. (1996). Expression and characterization of inactivating and activating mutations in the human -sensing receptor. J. Biol. Chem. 271, 19537–19545. doi: 10.1074/jbc.271.32.19537
Bai, M., Trivedi, S., and Brown, E. M. (1998a). Dimerization of the extracellular calcium-sensing receptor (CaR) on the cell surface of CaR-transfected HEK293 cells. J. Biol. Chem. 273, 23605–23610.
Bai, M., Trivedi, S., Kifor, O., Quinn, S. J., and Brown, E. M. (1999). Intermolecular interactions between dimeric calcium-sensing receptor monomers are important for its normal function. Proc. Natl. Acad. Sci. U.S.A. 96, 2834–2839. doi: 10.1073/pnas.96.6.2834
Bai, M., Trivedi, S., Lane, C. R., Yang, Y., Quinn, S. J., and Brown, E. M. (1998b). Protein kinase C phosphorylation of threonine at position 888 in -sensing receptor (CaR) inhibits coupling to Ca2+ store release. J. Biol. Chem. 273, 21267–21275.
Bouschet, T., Martin, S., and Henley, J. M. (2005). Receptor-activity-modifying proteins are required for forward trafficking of the calcium-sensing receptor to the plasma membrane. J. Cell Sci. 118(Pt 20), 4709–4720. doi: 10.1242/jcs.02598
Breitwieser, G. E. (2006). Calcium sensing receptors and calcium oscillations: calcium as a first messenger. Curr. Top. Dev. Biol. 73, 85–114. doi: 10.1016/S0070-2153(05)73003-9
Breitwieser, G. E. (2012). Minireview: the intimate link between calcium sensing receptor trafficking and signaling: implications for disorders of calcium homeostasis. Mol. Endocrinol. 26, 1482–1495. doi: 10.1210/me.2011-1370
Breitwieser, G. E. (2013). The calcium sensing receptor life cycle: trafficking, cell surface expression, and degradation. Best Pract. Res. Clin. Endocrinol. Metab. 27, 303–313. doi: 10.1016/j.beem.2013.03.003
Broadhead, G. K., Mun, H. C., Avlani, V. A., Jourdon, O., Church, W. B., Christopoulos, A., et al. (2011). Allosteric modulation of the calcium-sensing receptor by gamma-glutamyl peptides: inhibition of PTH secretion, suppression of intracellular cAMP levels, and a common mechanism of action with L-amino acids. J. Biol. Chem. 286, 8786–8797. doi: 10.1074/jbc.M110.149724
Brown, E. M. (2013). Role of the calcium-sensing receptor in extracellular calcium homeostasis. Best Pract. Res. Clin. Endocrinol. Metab. 27, 333–343. doi: 10.1016/j.beem.2013.02.006
Brown, E. M., and MacLeod, R. J. (2001). Extracellular calcium sensing and extracellular calcium signaling. Physiol. Rev. 81, 239–297.
Brown, E. M., Gamba, G., Riccardi, D., Lombardi, M., Butters, R., Kifor, O., et al. (1993). Cloning and characterization of an extracellular Ca2+-sensing receptor from bovine parathyroid. Nature 366, 575–580.
Brown, E. M., Katz, C., Butters, R., and Kifor, O. (1991). Polyarginine, polylysine, and protamine mimic the effects of high extracellular calcium concentrations on dispersed bovine parathyroid cells. J. Bone Miner. Res. 6, 1217–1225. doi: 10.1002/jbmr.5650061112
Buchan, A. M., Squires, P. E., Ring, M., and Meloche, R. M. (2001). Mechanism of action of the calcium-sensing receptor in human antral gastrin cells. Gastroenterology 120, 1128–1139. doi: 10.1053/gast.2001.23246
Bu, L., Michino, M., Wolf, R. M., and Brooks, C. L. III. (2008). Improved model building and assessment of the Calcium-sensing receptor transmembrane domain. Proteins 71, 215–226. doi: 10.1002/prot.21685
Campion, K. L., McCormick, W. D., Warwicker, J., Khayat, M. E., Atkinson-Dell, R., Steward, M. C., et al. (2015). Pathophysiologic changes in extracellular pH modulate parathyroid calcium-sensing receptor activity and secretion via a histidine-independent mechanism. J. Am. Soc. Nephrol. 26, 2163–2171. doi: 10.1681/ASN.2014070653
Casadó, V., Cortés, A., Ciruela, F., Mallol, J., Ferré, S., Lluis, C., et al. (2007). Old and new ways to calculate the affinity of agonists and antagonists interacting with G-protein-coupled monomeric and dimeric receptors: the receptor-dimer cooperativity index. Pharmacol. Ther. 116, 343–354. doi: 10.1016/j.pharmthera.2007.05.010
Chakravarti, B., Chattopadhyay, N., and Brown, E. M. (2012). Signaling through the extracellular calcium-sensing receptor (CaSR). Adv. Exp. Med. Biol. 740, 103–142. doi: 10.1007/978-94-007-2888-2_5
Chang, W., and Shoback, D. (2004). Extracellular Ca2+-sensing receptors–an overview. Cell Calcium 35, 183–196. doi: 10.1016/j.ceca.2003.10.012
Chattopadhyay, N., Ye, C. P., Yamaguchi, T., Kerner, R., Vassilev, P. M., and Brown, E. M. (1999). Extracellular calcium-sensing receptor induces cellular proliferation and activation of a nonselective cation channel in U373 human astrocytoma cells. Brain Res. 851, 116–124. doi: 10.1016/S0006-8993(99)02132-0
Cheng, I., Klingensmith, M. E., Chattopadhyay, N., Kifor, O., Butters, R. R., Soybel, D. I., et al. (1998). Identification and localization of the extracellular calcium-sensing receptor in human breast. J. Clin. Endocrinol. Metab. 83, 703–707. doi: 10.1210/jc.83.2.703
Chikatsu, N., Fukumoto, S., Takeuchi, Y., Suzawa, M., Obara, T., Matsumoto, T., et al. (2000). Cloning and characterization of two promoters for the human calcium-sensing receptor (CaSR) and changes of CaSR expression in parathyroid adenomas. J. Biol. Chem. 275, 7553–7557. doi: 10.1074/jbc.275.11.7553
Christopoulos, A., and Kenakin, T. (2002). G protein-coupled receptor allosterism and complexing. Pharmacol. Rev. 54, 323–374. doi: 10.1124/pr.54.2.323
Cima, R. R., Cheng, I., Klingensmith, M. E., Chattopadhyay, N., Kifor, O., Hebert, S. C., et al. (1997). Identification and functional assay of an extracellular calcium-sensing receptor in Necturus gastric mucosa. Am. J. Physiol. 273(5 Pt 1), G1051–G1060.
Conigrave, A. D., Franks, A. H., Brown, E. M., and Quinn, S. J. (2002). L-amino acid sensing by the calcium-sensing receptor: a general mechanism for coupling protein and calcium metabolism? Eur. J. Clin. Nutr. 56, 1072–1080. doi: 10.1038/sj.ejcn.1601463
Conigrave, A. D., and Lok, H. C. (2004). Activation of renal calcium and water excretion by novel physiological and pharmacological activators of the calcium-sensing receptor. Clin. Exp. Pharmacol. Physiol. 31, 368–371. doi: 10.1111/j.1440-1681.2004.04000.x
Conigrave, A. D., Mun, H. C., and Brennan, S. C. (2007a). Physiological significance of L-amino acid sensing by extracellular Ca2+-sensing receptors. Biochem. Soc. Trans. 35(Pt 5), 1195–1198. doi: 10.1042/BST0351195
Conigrave, A. D., Mun, H. C., and Lok, H. C. (2007b). Aromatic L-amino acids activate the calcium-sensing receptor. J. Nutr. 137(6 Suppl. 1), 1524S–1527S. discussion: 1548S.
Conigrave, A. D., Quinn, S. J., and Brown, E. M. (2000). L-amino acid sensing by the extracellular Ca2+-sensing receptor. Proc. Natl. Acad. Sci. U.S.A. 97, 4814–4819. doi: 10.1073/pnas.97.9.4814
Davey, A. E., Leach, K., Valant, C., Conigrave, A. D., Sexton, P. M., and Christopoulos, A. (2012). Positive and negative allosteric modulators promote biased signaling at the calcium-sensing receptor. Endocrinology 153, 1232–1241. doi: 10.1210/en.2011-1426
Deng, H., Chen, G., Yang, W., and Yang, J. J. (2006). Predicting calcium-binding sites in proteins - a graph theory and geometry approach. Proteins 64, 34–42. doi: 10.1002/prot.20973
Fan, G. F., Ray, K., Zhao, X. M., Goldsmith, P. K., and Spiegel, A. M. (1998). Mutational analysis of the cysteines in the extracellular domain of the human Ca2+ receptor: effects on cell surface expression, dimerization and signal transduction. FEBS Lett. 436, 353–356. doi: 10.1016/S0014-5793(98)01165-X
Felderbauer, P., Klein, W., Bulut, K., Ansorge, N., Dekomien, G., Werner, I., et al. (2006). Mutations in the calcium-sensing receptor: a new genetic risk factor for chronic pancreatitis? Scand. J. Gastroenterol. 41, 343–348. doi: 10.1080/00365520510024214
Francesconi, A., and Duvoisin, R. M. (2004). Divalent cations modulate the activity of metabotropic glutamate receptors. J. Neurosci. Res. 75, 472–479. doi: 10.1002/jnr.10853
Fudge, N. J., and Kovacs, C. S. (2004). Physiological studies in heterozygous calcium sensing receptor (CaSR) gene-ablated mice confirm that the CaSR regulates calcitonin release in vivo. BMC Physiol. 4:5. doi: 10.1186/1472-6793-4-5
Galvez, T., Urwyler, S., Prézeau, L., Mosbacher, J., Joly, C., Malitschek, B., et al. (2000). Ca2+ requirement for high-affinity γ-aminobutyric acid (GABA) binding at GABAB receptors: involvement of serine 269 of the GABABR1 subunit. Mol. Pharmacol. 57, 419–426. doi: 10.1124/mol.57.3.419
Gama, L., and Breitwieser, G. E. (1998). A carboxyl-terminal domain controls the cooperativity for extracellular Ca2+ activation of the human calcium sensing receptor. A study with receptor-green fluorescent protein fusions. J. Biol. Chem. 273, 29712–29718.
Gama, L., Wilt, S. G., and Breitwieser, G. E. (2001). Heterodimerization of calcium sensing receptors with metabotropic glutamate receptors in neurons. J. Biol. Chem. 276, 39053–39059. doi: 10.1074/jbc.M105662200
Geng, Y., Mosyak, L., Kurinov, I., Zuo, H., Sturchler, E., Cheng, T. C., et al. (2016). Structural mechanism of ligand activation in human calcium-sensing receptor. Elife 5:e13662. doi: 10.7554/eLife.13662
Gether, U. (2000). Uncovering molecular mechanisms involved in activation of G protein-coupled receptors. Endocr. Rev. 21, 90–113. doi: 10.1210/edrv.21.1.0390
Grant, M. P., Stepanchick, A., Cavanaugh, A., and Breitwieser, G. E. (2011). Agonist-driven maturation and plasma membrane insertion of calcium-sensing receptors dynamically control signal amplitude. Sci. Signal. 4, ra78. doi: 10.1126/scisignal.2002208
Grosshans, B. L., Ortiz, D., and Novick, P. (2006). Rabs and their effectors: achieving specificity in membrane traffic. Proc. Natl. Acad. Sci. U.S.A. 103, 11821–11827. doi: 10.1073/pnas.0601617103
Hannan, F. M., Nesbit, M. A., Zhang, C., Cranston, T., Curley, A. J., Harding, B., et al. (2012). Identification of 70 calcium-sensing receptor mutations in hyper- and hypo-calcaemic patients: evidence for clustering of extracellular domain mutations at calcium-binding sites. Hum. Mol. Genet. 21, 2768–2778. doi: 10.1093/hmg/dds105
Hauache, O. M., Hu, J., Ray, K., Xie, R., Jacobson, K. A., and Spiegel, A. M. (2000). Effects of a calcimimetic compound and naturally activating mutations on the human Ca2+ receptor and on Ca2+ receptor/metabotropic glutamate chimeric receptors. Endocrinology 141, 4156–4163. doi: 10.1210/endo.141.11.7753
Hebert, S. C. (1996). Extracellular calcium-sensing receptor: implications for calcium and magnesium handling in the kidney. Kidney Int. 50, 2129–2139. doi: 10.1038/ki.1996.539
Hebert, S. C., and Brown, E. M. (1995). The extracellular calcium receptor. Curr. Opin. Cell Biol. 7, 484–492. doi: 10.1016/0955-0674(95)80004-2
Hendy, G. N., Canaff, L., and Cole, D. E. (2013). The CASR gene: alternative splicing and transcriptional control, and calcium-sensing receptor (CaSR) protein: structure and ligand binding sites. Best Pract. Res. Clin. Endocrinol. Metab. 27, 285–301. doi: 10.1016/j.beem.2013.02.009
Hendy, G. N., Guarnieri, V., and Canaff, L. (2009). Calcium-sensing receptor and associated diseases. Prog. Mol. Biol. Transl. Sci. 89, 31–95. doi: 10.1016/S1877-1173(09)89003-0
Hendy, G. N., D'souza-Li, L., Yang, B., Canaff, L., and Cole, D. E. (2000). Mutations of the calcium-sensing receptor (CASR) in familial hypocalciuric hypercalcemia, neonatal severe hyperparathyroidism, and autosomal dominant hypocalcemia. Hum. Mutat. 16, 281–296. doi: 10.1002/1098-1004(200010)16:4<281::AID-HUMU1>3.0.CO;2-A
Herberger, A. L., and Loretz, C. A. (2013). Vertebrate extracellular calcium-sensing receptor evolution: selection in relation to life history and habitat. Comp. Biochem. Physiol. Part D Genomics Proteomics 8, 86–94. doi: 10.1016/j.cbd.2012.12.004
Herrera-Vigenor, F., Hernández-García, R., Valadez-Sánchez, M., Vázquez-Prado, J., and Reyes-Cruz, G. (2006). AMSH regulates calcium-sensing receptor signaling through direct interactions. Biochem. Biophys. Res. Commun. 347, 924–930. doi: 10.1016/j.bbrc.2006.06.169
Hinson, T. K., Damodaran, T. V., Chen, J., Zhang, X., Qumsiyeh, M. B., Seldin, M. F., et al. (1997). Identification of putative transmembrane receptor sequences homologous to the calcium-sensing G-protein-coupled receptor. Genomics 45, 279–289. doi: 10.1006/geno.1997.4943
Hjälm, G., MacLeod, R. J., Kifor, O., Chattopadhyay, N., and Brown, E. M. (2001). Filamin-A binds to the carboxyl-terminal tail of the calcium-sensing receptor, an interaction that participates in CaR-mediated activation of mitogen-activated protein kinase. J. Biol. Chem. 276, 34880–34887. doi: 10.1074/jbc.M100784200
Hofer, A. M., and Brown, E. M. (2003). Extracellular calcium sensing and signalling. Nat. Rev. Mol. Cell Biol. 4, 530–538. doi: 10.1038/nrm1154
Hofer, A. M., Gerbino, A., Caroppo, R., and Curci, S. (2004). The extracellular calcium-sensing receptor and cell-cell signaling in epithelia. Cell Calcium 35, 297–306. doi: 10.1016/j.ceca.2003.10.021
Huang, C., Handlogten, M. E., and Miller, R. T. (2002). Parallel activation of phosphatidylinositol 4-kinase and phospholipase C by the extracellular calcium-sensing receptor. J. Biol. Chem. 277, 20293–20300. doi: 10.1074/jbc.M200831200
Huang, C., Hujer, K. M., Wu, Z., and Miller, R. T. (2004). The Ca2+-sensing receptor couples to Galpha12/13 to activate phospholipase D in Madin-Darby canine kidney cells. Am. J. Physiol. Cell Physiol. 286, C22–C30. doi: 10.1152/ajpcell.00229.2003
Huang, C., Sindic, A., Hill, C. E., Hujer, K. M., Chan, K. W., Sassen, M., et al. (2007). Interaction of the Ca2+-sensing receptor with the inwardly rectifying potassium channels Kir4.1 and Kir4.2 results in inhibition of channel function. Am. J. Physiol. Ren. Physiol. 292, F1073–F1081. doi: 10.1152/ajprenal.00269.2006
Huang, Y., and Breitwieser, G. E. (2007). Rescue of calcium-sensing receptor mutants by allosteric modulators reveals a conformational checkpoint in receptor biogenesis. J. Biol. Chem. 282, 9517–9525. doi: 10.1074/jbc.M609045200
Huang, Y., Niwa, J., Sobue, G., and Breitwieser, G. E. (2006). Calcium-sensing receptor ubiquitination and degradation mediated by the E3 ubiquitin ligase dorfin. J. Biol. Chem. 281, 11610–11617. doi: 10.1074/jbc.M513552200
Huang, Y., Zhou, Y., Castiblanco, A., Yang, W., Brown, E. M., and Yang, J. J. (2009). Multiple Ca2+-binding sites in the extracellular domain of the Ca2+-sensing receptor corresponding to cooperative Ca2+ response. Biochemistry 48, 388–398. doi: 10.1021/bi8014604
Huang, Y., Zhou, Y., Wong, H. C., Castiblanco, A., Chen, Y., Brown, E. M., et al. (2010). Calmodulin regulates Ca2+-sensing receptor-mediated Ca2+ signaling and its cell surface expression. J. Biol. Chem. 285, 35919–35931. doi: 10.1074/jbc.M110.147918
Huang, Y., Zhou, Y., Yang, W., Butters, R., Lee, H. W., Li, S., et al. (2007). Identification and dissection of Ca2+-binding sites in the extracellular domain of Ca2+-sensing receptor. J. Biol. Chem. 282, 19000–19010. doi: 10.1074/jbc.M701096200
Hu, J., Jiang, J., Costanzi, S., Thomas, C., Yang, W., Feyen, J. H., et al. (2006). A missense mutation in the seven-transmembrane domain of the human Ca2+ receptor converts a negative allosteric modulator into a positive allosteric modulator. J. Biol. Chem. 281, 21558–21565. doi: 10.1074/jbc.M603682200
Hu, J., McLarnon, S. J., Mora, S., Jiang, J., Thomas, C., Jacobson, K. A., et al. (2005). A region in the seven-transmembrane domain of the human Ca2+ receptor critical for response to Ca2+. J. Biol. Chem. 280, 5113–5120. doi: 10.1074/jbc.M413403200
Hu, J., Reyes-Cruz, G., Chen, W., Jacobson, K. A., and Spiegel, A. M. (2002). Identification of acidic residues in the extracellular loops of the seven-transmembrane domain of the human Ca2+ receptor critical for response to Ca2+ and a positive allosteric modulator. J. Biol. Chem. 277, 46622–46631. doi: 10.1074/jbc.M207100200
Hu, J., and Spiegel, A. M. (2003). Naturally occurring mutations of the extracellular Ca2+-sensing receptor: implications for its structure and function. Trends Endocrinol. Metab. 14, 282–288. doi: 10.1016/S1043-2760(03)00104-8
Hu, J., and Spiegel, A. M. (2007). Structure and function of the human calcium-sensing receptor: insights from natural and engineered mutations and allosteric modulators. J. Cell. Mol. Med. 11, 908–922. doi: 10.1111/j.1582-4934.2007.00096.x
Jiang, J. Y., Nagaraju, M., Meyer, R. C., Zhang, L., Hamelberg, D., Hall, R. A., et al. (2014). Extracellular calcium modulates actions of orthosteric and allosteric ligands on metabotropic glutamate receptor 1α. J. Biol. Chem. 289, 1649–1661. doi: 10.1074/jbc.M113.507665
Jiang, Y., Huang, Y., Wong, H. C., Zhou, Y., Wang, X., Yang, J., et al. (2010). Elucidation of a novel extracellular calcium-binding site on metabotropic glutamate receptor 1α (mGluR1α) that controls receptor activation. J. Biol. Chem. 285, 33463–33474. doi: 10.1074/jbc.M110.147033
Jingami, H., Nakanishi, S., and Morikawa, K. (2003). Structure of the metabotropic glutamate receptor. Curr. Opin. Neurobiol. 13, 271–278. doi: 10.1016/S0959-4388(03)00067-9
Kemp, E. H., Gavalas, N. G., Akhtar, S., Krohn, K. J., Pallais, J. C., Brown, E. M., et al. (2010). Mapping of human autoantibody binding sites on the calcium-sensing receptor. J. Bone Miner. Res. 25, 132–140. doi: 10.1359/jbmr.090703
Kemp, E. H., Gavalas, N. G., Krohn, K. J., Brown, E. M., Watson, P. F., and Weetman, A. P. (2009). Activating autoantibodies against the calcium-sensing receptor detected in two patients with autoimmune polyendocrine syndrome type 1. J. Clin. Endocrinol. Metab. 94, 4749–4756. doi: 10.1210/jc.2009-1080
Kendler, D. L., Adachi, J. D., Josse, R. G., and Slosman, D. O. (2009). Monitoring strontium ranelate therapy in patients with osteoporosis. Osteoporos. Int. 20, 1101–1106. doi: 10.1007/s00198-009-0886-1
Kifor, O., Diaz, R., Butters, R., Kifor, I., and Brown, E. M. (1998). The calcium-sensing receptor is localized in caveolin-rich plasma membrane domains of bovine parathyroid cells. J. Biol. Chem. 273, 21708–21713. doi: 10.1074/jbc.273.34.21708
Kifor, O., Kifor, I., Moore, F. D., Butters, R. R., Cantor, T., Gao, P., et al. (2003). Decreased expression of caveolin-1 and altered regulation of mitogen-activated protein kinase in cultured bovine parathyroid cells and human parathyroid adenomas. J. Clin. Endocrinol. Metab. 88, 4455–4464. doi: 10.1210/jc.2002-021427
Kim, J. Y., Kim, N., Yenari, M. A., and Chang, W. (2011). Mild hypothermia suppresses calcium-sensing receptor (CaSR) Induction following forebrain ischemia while increasing GABA-B receptor 1 (GABA-B-R1) expression. Transl. Stroke Res. 2, 195–201. doi: 10.1007/s12975-011-0082-4
Kirberger, M., Wang, X., Deng, H., Yang, W., Chen, G., and Yang, J. J. (2008). Statistical analysis of structural characteristics of protein Ca2+-binding sites. J. Biol. Inorg. Chem. 13, 1169–1181. doi: 10.1007/s00775-008-0402-7
Kovacs, C. S., Ho-Pao, C. L., Hunzelman, J. L., Lanske, B., Fox, J., Seidman, J. G., et al. (1998). Regulation of murine fetal-placental calcium metabolism by the calcium-sensing receptor. J. Clin. Invest. 101, 2812–2820. doi: 10.1172/JCI2940
Kubo, Y., Miyashita, T., and Murata, Y. (1998). Structural basis for a Ca2+-sensing function of the metabotropic glutamate receptors. Science 279, 1722–1725.
Kunishima, N., Shimada, Y., Tsuji, Y., Sato, T., Yamamoto, M., Kumasaka, T., et al. (2000). Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature 407, 971–977. doi: 10.1038/35039564
Leach, K., Wen, A., Cook, A. E., Sexton, P. M., Conigrave, A. D., and Christopoulos, A. (2013). Impact of clinically relevant mutations on the pharmacoregulation and signaling bias of the calcium-sensing receptor by positive and negative allosteric modulators. Endocrinology 154, 1105–1116. doi: 10.1210/en.2012-1887
Leach, K., Wen, A., Davey, A. E., Sexton, P. M., Conigrave, A. D., and Christopoulos, A. (2012). Identification of molecular phenotypes and biased signaling induced by naturally occurring mutations of the human calcium-sensing receptor. Endocrinology 153, 4304–4316. doi: 10.1210/en.2012-1449
Liou, A. P., Sei, Y., Zhao, X., Feng, J., Lu, X., Thomas, C., et al. (2011). The extracellular calcium-sensing receptor is required for cholecystokinin secretion in response to L-phenylalanine in acutely isolated intestinal I cells. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G538–G546. doi: 10.1152/ajpgi.00342.2010
Lorenz, S., Frenzel, R., Paschke, R., Breitwieser, G. E., and Miedlich, S. U. (2007). Functional desensitization of the extracellular calcium-sensing receptor is regulated via distinct mechanisms: role of G protein-coupled receptor kinases, protein kinase C and beta-arrestins. Endocrinology 148, 2398–2404. doi: 10.1210/en.2006-1035
Lundgren, S., Hjalm, G., Hellman, P., Ek, B., Juhlin, C., Rastad, J., et al. (1994). A protein involved in calcium sensing of the human parathyroid and placental cytotrophoblast cells belongs to the LDL-receptor protein superfamily. Exp. Cell Res. 212, 344–350. doi: 10.1006/excr.1994.1153
Magno, A. L., Ward, B. K., and Ratajczak, T. (2011). The calcium-sensing receptor: a molecular perspective. Endocr. Rev. 32, 3–30. doi: 10.1210/er.2009-0043
Mathias, R. S., Mathews, C. H., Machule, C., Gao, D., Li, W., and Denbesten, P. K. (2001). Identification of the calcium-sensing receptor in the developing tooth organ. J. Bone Miner. Res. 16, 2238–2244. doi: 10.1359/jbmr.2001.16.12.2238
Miedlich, S. U., Gama, L., Seuwen, K., Wolf, R. M., and Breitwieser, G. E. (2004). Homology modeling of the transmembrane domain of the human calcium sensing receptor and localization of an allosteric binding site. J. Biol. Chem. 279, 7254–7263. doi: 10.1074/jbc.M307191200
Muff, R., Nemeth, E. F., Haller-Brem, S., and Fischer, J. A. (1988). Regulation of hormone secretion and cytosolic Ca2+ by extracellular Ca2+ in parathyroid cells and C-cells: role of voltage-sensitive Ca2+ channels. Arch. Biochem. Biophys. 265, 128–135. doi: 10.1016/0003-9861(88)90378-5
Mun, H. C., Culverston, E. L., Franks, A. H., Collyer, C. A., Clifton-Bligh, R. J., and Conigrave, A. D. (2005). A double mutation in the extracellular Ca2+-sensing receptor's venus flytrap domain that selectively disables L-amino acid sensing. J. Biol. Chem. 280, 29067–29072. doi: 10.1074/jbc.M500002200
Mun, H.-C., Franks, A. H., Culverston, E. L., Krapcho, K., Nemeth, E. F., and Conigrave, A. D. (2004). The venus fly trap domain of the extracellular Ca2+ -sensing receptor is required for L-amino acid sensing. J. Biol. Chem. 279, 51739–51744. doi: 10.1074/jbc.M406164200
Muto, T., Tsuchiya, D., Morikawa, K., and Jingami, H. (2007). Structures of the extracellular regions of the group II/III metabotropic glutamate receptors. Proc. Natl. Acad. Sci. U.S.A. 104, 3759–3764. doi: 10.1073/pnas.0611577104
Nagar, B., Overduin, M., Ikura, M., and Rini, J. M. (1996). Structural basis of calcium-induced E-cadherin rigidification and dimerization. Nature 380, 360–364.
Nearing, J., Betka, M., Quinn, S., Hentschel, H., Elger, M., Baum, M., et al. (2002). Polyvalent cation receptor proteins (CaRs) are salinity sensors in fish. Proc. Natl. Acad. Sci. U.S.A. 99, 9231–9236. doi: 10.1073/pnas.152294399
Nemeth, E. F., and Scarpa, A. (1987). Rapid mobilization of cellular Ca2+ in bovine parathyroid cells evoked by extracellular divalent cations. Evidence for a cell surface calcium receptor. J. Biol. Chem. 262, 5188–5196.
Nesbit, M. A., Hannan, F. M., Howles, S. A., Reed, A. A., Cranston, T., Thakker, C. E., et al. (2013). Mutations in AP2S1 cause familial hypocalciuric hypercalcemia type 3. Nat. Genet. 45, 93–97. doi: 10.1038/ng.2492
Oda, Y., Tu, C. L., Chang, W., Crumrine, D., Komuves, L., Mauro, T., et al. (2000). The calcium sensing receptor and its alternatively spliced form in murine epidermal differentiation. J. Biol. Chem. 275, 1183–1190. doi: 10.1074/jbc.275.2.1183
Oda, Y., Tu, C. L., Pillai, S., and Bikle, D. D. (1998). The calcium sensing receptor and its alternatively spliced form in keratinocyte differentiation. J. Biol. Chem. 273, 23344–23352. doi: 10.1074/jbc.273.36.23344
O'Hara, P. J., Sheppard, P. O., Thogersen, H., Venezia, D., Haldeman, B. A., McGrane, V., et al. (1993). The ligand-binding domain in metabotropic glutamate receptors is related to bacterial periplasmic binding proteins. Neuron 11, 41–52. doi: 10.1016/0896-6273(93)90269-W
Oldham, W. M., and Hamm, H. E. (2008). Heterotrimeric G protein activation by G-protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 9, 60–71. doi: 10.1038/nrm2299
Pace, A. J., Gama, L., and Breitwieser, G. E. (1999). Dimerization of the calcium-sensing receptor occurs within the extracellular domain and is eliminated by Cys –> Ser mutations at Cys101 and Cys236. J. Biol. Chem. 274, 11629–11634. doi: 10.1074/jbc.274.17.11629
Pearce, S. H., Bai, M., Quinn, S. J., Kifor, O., Brown, E. M., and Thakker, R. V. (1996). Functional characterization of calcium-sensing receptor mutations expressed in human embryonic kidney cells. J. Clin. Invest. 98, 1860–1866. doi: 10.1172/JCI118987
Petrel, C., Kessler, A., Maslah, F., Dauban, P., Dodd, R. H., Rognan, D., et al. (2003). Modeling and mutagenesis of the binding site of Calhex 231, a novel negative allosteric modulator of the extracellular Ca2+-sensing receptor. J. Biol. Chem. 278, 49487–49494. doi: 10.1074/jbc.M308010200
Pi, M., Spurney, R. F., Tu, Q., Hinson, T., and Quarles, L. D. (2002). Calcium-sensing receptor activation of rho involves filamin and rho-guanine nucleotide exchange factor. Endocrinology 143, 3830–3838. doi: 10.1210/en.2002-220240
Pollak, M. R., Brown, E. M., Chou, Y. H., Hebert, S. C., Marx, S. J., Steinmann, B., et al. (1993). Mutations in the human Ca2+-sensing receptor gene cause familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Cell 75, 1297–1303. doi: 10.1016/0092-8674(93)90617-Y
Quinn, S. J., Bai, M., and Brown, E. M. (2004). pH Sensing by the calcium-sensing receptor. J. Biol. Chem. 279, 37241–37249. doi: 10.1074/jbc.M404520200
Quinn, S. J., Ye, C. P., Diaz, R., Kifor, O., Bai, M., Vassilev, P., et al. (1997). The Ca2+-sensing receptor: a target for polyamines. Am. J. Physiol. 273(4 Pt 1), C1315–C1323.
Ray, K., Hauschild, B. C., Steinbach, P. J., Goldsmith, P. K., Hauache, O., and Spiegel, A. M. (1999). Identification of the cysteine residues in the amino-terminal extracellular domain of the human Ca2+ receptor critical for dimerization. Implications for function of monomeric Ca2+ receptor. J. Biol. Chem. 274, 27642–27650.
Reyes-Cruz, G., Hu, J., Goldsmith, P. K., Steinbach, P. J., and Spiegel, A. M. (2001). Human Ca2+ receptor extracellular domain. Analysis of function of lobe I loop deletion mutants. J. Biol. Chem. 276, 32145–32151. doi: 10.1074/jbc.M102977200
Riccardi, D. (2002). Wellcome Prize Lecture. Cell surface, ion-sensing receptors. Exp. Physiol. 87, 403–411. doi: 10.1111/j.1469-445X.2002.tb00053.x
Ringer, S. (1883). A further contribution regarding the influence of the different constituents of the blood on the contraction of the heart. J. Physiol. 4, 29–42 23. doi: 10.1113/jphysiol.1883.sp000120
Rogers, A., Nesbit, M. A., Hannan, F. M., Howles, S. A., Gorvin, C. M., Cranston, T., et al. (2014). Mutational analysis of the adaptor protein 2 sigma subunit (AP2S1) gene: search for autosomal dominant hypocalcemia type 3 (ADH3). J. Clin. Endocrinol. Metab. 99, E1300–E1305. doi: 10.1210/jc.2013-3909
Rosenbaum, D. M., Rasmussen, S. G., and Kobilka, B. K. (2009). The structure and function of G-protein-coupled receptors. Nature 459, 356–363. doi: 10.1038/nature08144
Ruat, M., Snowman, A. M., Hester, L. D., and Snyder, S. H. (1996). Cloned and expressed rat Ca2+-sensing receptor. J. Biol. Chem. 271, 5972–5975. doi: 10.1074/jbc.271.11.5972
Silve, C., Petrel, C., Leroy, C., Bruel, H., Mallet, E., Rognan, D., et al. (2005). Delineating a Ca2+ binding pocket within the venus flytrap module of the human calcium-sensing receptor. J. Biol. Chem. 280, 37917–37923. doi: 10.1074/jbc.M506263200
Smajilovic, S., and Tfelt-Hansen, J. (2007). Calcium acts as a first messenger through the calcium-sensing receptor in the cardiovascular system. Cardiovasc. Res. 75, 457–467. doi: 10.1016/j.cardiores.2007.03.015
Stepanchick, A., and Breitwieser, G. E. (2010). The cargo receptor p24A facilitates calcium sensing receptor maturation and stabilization in the early secretory pathway. Biochem. Biophys. Res. Commun. 395, 136–140. doi: 10.1016/j.bbrc.2010.03.156
Stepanchick, A., McKenna, J., McGovern, O., Huang, Y., and Breitwieser, G. E. (2010). Calcium sensing receptor mutations implicated in pancreatitis and idiopathic epilepsy syndrome disrupt an arginine-rich retention motif. Cell. Physiol. Biochem. 26, 363–374. doi: 10.1159/000320560
Sun, J., and Murphy, E. (2010). Calcium-sensing receptor: a sensor and mediator of ischemic preconditioning in the heart. Am. J. Physiol. Heart Circ. Physiol. 299, H1309–H1317. doi: 10.1152/ajpheart.00373.2010
Suzuki, Y., Moriyoshi, E., Tsuchiya, D., and Jingami, H. (2004). Negative cooperativity of glutamate binding in the dimeric metabotropic glutamate receptor subtype 1. J. Biol. Chem. 279, 35526–35534. doi: 10.1074/jbc.M404831200
Tfelt-Hansen, J., and Brown, E. M. (2005). The calcium-sensing receptor in normal physiology and pathophysiology: a review. Crit. Rev. Clin. Lab. Sci. 42, 35–70. doi: 10.1080/10408360590886606
Thakker, R. V. (2004). Diseases associated with the extracellular calcium-sensing receptor. Cell Calcium 35, 275–282. doi: 10.1016/j.ceca.2003.10.010
Tharmalingam, S., Daulat, A. M., Antflick, J. E., Ahmed, S. M., Nemeth, E. F., Angers, S., et al. (2011). Calcium-sensing receptor modulates cell adhesion and migration via integrins. J. Biol. Chem. 286, 40922–40933. doi: 10.1074/jbc.M111.265454
Thomsen, A. R., Hvidtfeldt, M., and Brauner-Osborne, H. (2012). Biased agonism of the calcium-sensing receptor. Cell Calcium 51, 107–116. doi: 10.1016/j.ceca.2011.11.009
Tsuchiya, D., Kunishima, N., Kamiya, N., Jingami, H., and Morikawa, K. (2002). Structural views of the ligand-binding cores of a metabotropic glutamate receptor complexed with an antagonist and both glutamate and Gd3+. Proc. Natl. Acad. Sci. U.S.A. 99, 2660–2665. doi: 10.1073/pnas.052708599
Vetter, T., and Lohse, M. J. (2002). Magnesium and the parathyroid. Curr. Opin. Nephrol. Hypertens. 11, 403–410. doi: 10.1097/00041552-200207000-00006
Walter, S., Baruch, A., Dong, J., Tomlinson, J. E., Alexander, S. T., Janes, J., et al. (2013). Pharmacology of AMG 416 (Velcalcetide), a novel peptide agonist of the calcium-sensing receptor, for the treatment of secondary hyperparathyroidism in hemodialysis patients. J. Pharmacol. Exp. Ther. 346, 229–240. doi: 10.1124/jpet.113.204834
Wang, M., Yao, Y., Kuang, D., and Hampson, D. R. (2006). Activation of family C G-protein-coupled receptors by the tripeptide glutathione. J. Biol. Chem. 281, 8864–8870. doi: 10.1074/jbc.M512865200
Wang, X., Kirberger, M., Qiu, F., Chen, G., and Yang, J. J. (2009). Towards predicting Ca2+-binding sites with different coordination numbers in proteins with atomic resolution. Proteins 75, 787–798. doi: 10.1002/prot.22285
Wang, X., Zhao, K., Kirberger, M., Wong, H., Chen, G., and Yang, J. J. (2010). Analysis and prediction of calcium-binding pockets from apo-protein structures exhibiting calcium-induced localized conformational changes. Protein Sci. 19, 1180–1190. doi: 10.1002/pro.394
Ward, B. K., Magno, A. L., Blitvich, B. J., Rea, A. J., Stuckey, B. G., Walsh, J. P., et al. (2006). Novel mutations in the calcium-sensing receptor gene associated with biochemical and functional differences in familial hypocalciuric hypercalcaemia. Clin. Endocrinol. (Oxf). 64, 580–587. doi: 10.1111/j.1365-2265.2006.02512.x
Ward, D. T. (2004). Calcium receptor-mediated intracellular signalling. Cell Calcium 35, 217–228. doi: 10.1016/j.ceca.2003.10.017
Watanabe, S., Fukumoto, S., Chang, H., Takeuchi, Y., Hasegawa, Y., Okazaki, R., et al. (2002). Association between activating mutations of calcium-sensing receptor and Bartter's syndrome. Lancet 360, 692–694. doi: 10.1016/S0140-6736(02)09842-2
White, E., McKenna, J., Cavanaugh, A., and Breitwieser, G. E. (2009). Pharmacochaperone-mediated rescue of calcium-sensing receptor loss-of-function mutants. Mol. Endocrinol. 23, 1115–1123. doi: 10.1210/me.2009-0041
Wise, A., Green, A., Main, M. J., Wilson, R., Fraser, N., and Marshall, F. H. (1999). Calcium sensing properties of the GABA(B) receptor. Neuropharmacology 38, 1647–1656. doi: 10.1016/S0028-3908(99)00119-7
Wu, H., Wang, C., Gregory, K. J., Han, G. W., Cho, H. P., Xia, Y., et al. (2014). Structure of a class C GPCR metabotropic glutamate receptor 1 bound to an allosteric modulator. Science 344, 58–64. doi: 10.1126/science.1249489
Yang, W., Wilkins, A. L., Li, S., Ye, Y., and Yang, J. J. (2005). The effects of Ca2+ binding on the dynamic properties of a designed Ca2+-binding protein. Biochemistry 44, 8267–8273. doi: 10.1021/bi050463n
Zerial, M., and McBride, H. (2001). Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol. 2, 107–117. doi: 10.1038/35052055
Zhang, C., Huang, Y., Jiang, Y., Mulpuri, N., Wei, L., Hamelberg, D., et al. (2014a). Identification of an L-phenylalanine binding site enhancing the cooperative responses of the calcium-sensing receptor to calcium. J. Biol. Chem. 289, 5296–5309. doi: 10.1074/jbc.M113.537357
Zhang, C., Mulpuri, N., Hannan, F. M., Nesbit, M. A., Thakker, R. V., Hamelberg, D., et al. (2014b). Role of Ca2+ and L-Phe in regulating functional cooperativity of disease-associated “toggle” calcium-sensing receptor mutations. PLoS ONE 9:e113622. doi: 10.1371/journal.pone.0113622
Zhang, C., Zhang, T., Zou, J., Miller, C. L., Gorkhali, R., Yang, J. Y., et al. (2016). Structural basis for regulation of human calcium-sensing receptor by magnesium ions and an unexpected tryptophan derivative co-agonist. Sci. Adv. 2:e1600241. doi: 10.1126/sciadv.1600241
Zhang, C., Zhuo, Y., Moniz, H. A., Wang, S., Moremen, K. W., Prestegard, J. H., et al. (2014c). Direct determination of multiple ligand interactions with the extracellular domain of the calcium-sensing receptor. J. Biol. Chem. 289, 33529–33542. doi: 10.1074/jbc.M114.604652
Zhang, X., Zhang, T., Wu, J., Yu, X., Zheng, D., Yang, F., et al. (2014d). Calcium sensing receptor promotes cardiac fibroblast proliferation and extracellular matrix secretion. Cell. Physiol. Biochem. 33, 557–568. doi: 10.1159/000358634
Zhang, Z., Qiu, W., Quinn, S. J., Conigrave, A. D., Brown, E. M., and Bai, M. (2002). Three adjacent serines in the extracellular domains of the CaR are required for L-amino acid-mediated potentiation of receptor function. J. Biol. Chem. 277, 33727–33735. doi: 10.1074/jbc.M200976200
Zhang, Z., Sun, S., Quinn, S. J., Brown, E. M., and Bai, M. (2001). The extracellular calcium-sensing receptor dimerizes through multiple types of intermolecular interactions. J. Biol. Chem. 276, 5316–5322. doi: 10.1074/jbc.M005958200
Zhao, K., Wang, X., Wong, H. C., Wohlhueter, R., Kirberger, M. P., Chen, G., et al. (2012). Predicting Ca2+ -binding sites using refined carbon clusters. Proteins 80, 2666–2679. doi: 10.1002/prot.24149
Zhuang, X., Chowdhury, S., Northup, J. K., and Ray, K. (2010). Sar1-dependent trafficking of the human calcium receptor to the cell surface. Biochem. Biophys. Res. Commun. 396, 874–880. doi: 10.1016/j.bbrc.2010.05.014
Keywords: calcium sensing receptor, cooperativity, amino acids, structure, trafficking, disease mutations
Citation: Zhang C, Miller CL, Gorkhali R, Zou J, Huang K, Brown EM and Yang JJ (2016) Molecular Basis of the Extracellular Ligands Mediated Signaling by the Calcium Sensing Receptor. Front. Physiol. 7:441. doi: 10.3389/fphys.2016.00441
Received: 03 May 2016; Accepted: 16 September 2016;
Published: 30 September 2016.
Edited by:
Enikö Kallay, Medical University of Vienna, AustriaReviewed by:
Michael B. Morris, University of Sydney, AustraliaThomas E. Adrian, United Arab Emirates University, UAE
Copyright © 2016 Zhang, Miller, Gorkhali, Zou, Huang, Brown and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jenny J. Yang, amVubnlAZ3N1LmVkdQ==